

Abstract
Alcohol use is a leading cause of chronic liver disease worldwide, and changes in the microbiome associated with alcohol use contribute to patients’ risk for liver disease progression. Less is known about the effects of alcohol use on the intestinal viral microbiome (virome) and interactions between bacteriophages and their target bacteria. We studied changes in the intestinal virome of 62 clinically well‐characterized patients with alcohol use disorder (AUD) during active alcohol use and after 2 weeks of alcohol abstinence, by extracting virus‐like particles and performing metagenomic sequencing. We observed decreased abundance of Propionibacterium, Lactobacillus, and Leuconostoc phages in patients with active AUD when compared with controls, whereas after 2 weeks of alcohol abstinence, patients with AUD demonstrated an increase in the abundance of Propionibacterium, Lactobacillus, and Leuconostoc phages. The intestinal virome signature was also significantly different in patients with AUD with progressive liver disease, with increased abundance of phages targeting Enterobacteria and Lactococcus species phages compared with patients with AUD with nonprogressive liver disease. By performing moderation analyses, we found that progressive liver disease is associated with changes in interactions between some bacteriophages and their respective target bacteria. In summary, active alcohol use and alcohol‐associated progressive liver disease are associated with changes in the fecal virome, some of which are partially reversible after a short period of abstinence. Progression of alcohol‐associated liver disease is associated with changes in bacteriophage–bacteria interactions.
The intestinal virome of patients with alcohol use disorder (AUD) is significantly different from controls. Abstinence from alcohol in patients with AUD is also associated with significant differences in the intestinal virome, trending toward that of controls. Patients with progressive liver disease as determined by liver stiffness measurement and serum caspase‐cleaved and intact cytokeratin 18 (CK18‐M65) have a different intestinal virome signature than those with nonprogressive disease, which may be in part due to changes in bacteriophage–bacteria correlations with liver disease progression.
INTRODUCTION
Alcohol‐associated liver disease is a leading cause of liver disease worldwide. Alcohol‐associated liver disease initially manifests as steatosis and can progress to steatohepatitis, steatofibrosis, and cirrhosis.[ 1 ] The cornerstone of managing alcohol‐associated liver disease is abstinence from alcohol, which can improve steatosis in as little as 2–4 weeks.[ 2 , 3 ] Conversely, continued alcohol consumption greatly increases patients’ risk for disease progression.[ 4 ] The pathogenesis of alcohol‐induced liver injury is not completely understood, but changes in the intestinal microbiome are considered an additional risk factor for progression of alcohol‐associated liver disease.
Chronic heavy alcohol use is associated with dysbiosis, with a higher relative abundance of the phylum Proteobacteria and a lower relative abundance of the phylum Bacteroidetes and family Ruminococcaceae, including the genus Faecalibacterium.[ 5 , 6 , 7 ] Increases in the family Ruminococcaceae and species Lactobacillus and Bifidobacterium were observed after alcohol abstinence, whereas patients with alcohol‐associated liver disease demonstrated lower abundance of species within the Lactobacillaceae and Bacteroidaceae families.[ 6 , 7 , 8 ] Patients with alcohol‐associated liver disease also showed reduced levels of short‐chain fatty acid‐producing bacteria, such as Faecalibacterium prausnitzii, Coprococcus species, and members of the Lachnospiraceae families, which may lead to dysregulation of gut integrity and health.[ 9 , 10 , 11 ] Progression to cirrhosis is associated with increased abundance of oral commensals that can cause opportunistic infections, such as Streptococcus, Veillonella, and Prevotella species as well as endotoxin‐producing members of the Enterobacteriaceae family.[ 9 , 12 ]
Less is known about changes in the intestinal virome in alcohol‐associated liver disease. In one study, fecal samples from patients with alcohol‐associated liver disease and especially with alcohol‐associated hepatitis contained significantly more mammalian viruses, such as those from the Parvoviridae and Herpesviridae families, than controls.[ 13 ] In addition to mammalian viruses, bacteriophages targeting Escherichia, Enterobacteria, and Enterococcus species were overrepresented in patients with alcohol‐associated hepatitis compared with controls while Lactococcus and Parabacteroides phages were underrepresented. Alterations in the intestinal virome can modulate the mammalian–bacterial interaction in a variety of ways. Lytic bacteriophages kill their hosts by causing lysis to release virions, while lysogenic phages integrate into the host genome and can supply bacteria with genes involved in toxin, polysaccharide, and carbohydrate metabolism or modulate bacterial antigenicity.[ 14 , 15 , 16 ] The composition of the microbiome community can be altered by phage–bacterial interactions or vice versa, and this crosstalk can be modulated by disease states. Here, we evaluate how alcohol use disorder (AUD) and subsequent abstinence affect the intestinal virome, how these changes relate to liver disease progression, and how the viral–bacterial interaction is affected by liver disease.
MATERIALS AND METHODS
Patient cohort
Our patient cohort and study design has been described in detail.[ 17 , 18 ] In brief, patients who were actively drinking and with a diagnosis of alcohol dependence (n = 62) were admitted for elective alcohol rehabilitation at St. Luc University Hospital in Brussels, Belgium, from April 2017 to January 2019. All patients were heavy drinkers consuming over 60 g of alcohol per day (self‐reported consumption) for more than 1 year. They followed a highly standardized and controlled 3‐week detoxification and rehabilitation program that included a 7‐day hospitalization at the start and end of the treatment program, during which they received a standardized hospital diet. On the day of admission, FibroScan (Echosense, Paris, France) with controlled attenuation parameter (CAP) was performed and a fasting blood sample was collected. Stool samples were collected from the first bowel movement after admission. In the patients with AUD, FibroScan was repeated and blood and stool samples were again collected following 2 weeks of abstinence. Patients were excluded from the study if they used antibiotics, probiotics, or prebiotics during the 2 months preceding enrollment, were receiving immunosuppressive medications, or suffered from diabetes, inflammatory bowel disease, known liver disease of any other etiology, or clinically significant cardiovascular, pulmonary, or renal comorbidities. The AUD patients were compared to healthy volunteers matched for sex, age, and body mass index (BMI) who drank less than 20 g of alcohol per day. Data from 36 patients with AUD at the active alcohol use time point have been reported in a prior study.[ 13 ]
Serum biomarkers
Patient blood samples were tested for standard biochemical serum studies, including aspartate and alanine aminotransferases (AST, ALT), gamma‐glutamytransferase (GGT), and alkaline phosphatase (ALP) at the clinical laboratory associated with St. Luc University Hospital. Additionally, serum intact cytokeratin 18 (CK18‐M65) was measured using the CK18‐M65 enzyme‐linked immunosorbent assay kit (TECOmedical AG, Sissach, Switzerland) according to the manufacturer’s instructions.[ 18 ]
Bacterial DNA extraction, 16S ribosomal RNA sequencing, and read analysis
The 16S ribosomal RNA gene sequencing of human stool samples and processing of 16S sequence reads with MOTHUR‐based 16S analysis workflow was performed as described.[ 19 ] Raw sequence reads are available for download in the National Center for Biotechnology Information Sequence Read Archive associated with Bioproject PRJNA786875.
Virome preparation and metagenomic sequencing
Viral nucleic acids were extracted from fecal samples, reverse transcribed, and subjected to metagenomic sequencing using the Novel enrichment technique of VIRomes (NetoVIR) protocol with minor modifications as described.[ 13 , 20 , 21 ] Briefly, human stool samples were resuspended in phosphate‐buffered saline and sequentially filtered using a 0.8‐µm polyethersulfone filter (Sartorius, Goettingen, Germany). Any remaining DNA that was not encapsidated was degraded by treating with a mixture of benzonase (EMD Millipore, Billerica, MA) and micrococcal nuclease (New England Biolabs, Beverly, MA), followed by ethylenediaminetetraacetic acid inactivation of deoxyribonucleases. The remaining supernatant was subjected to lysis, and virome DNA and RNA were extracted using the QIAamp Viral RNA mini kit without carrier RNA (Qiagen, Hilden, Germany). Amplification was performed using a modified Complete Transcriptome Amplification kit (WTA2) protocol from Sigma‐Aldrich (St. Louis, MO). Library preparation was performed using an adjusted protocol for the Nextera XT DNA Library Preparation kit from Illumina. The size of amplified viral products was determined using a high‐sensitivity DNA kit on a bioanalyzer (Agilent Technologies, Palo Alto, CA), and concentration was measured by the High Sensitivity Double Stranded DNA kit on a Qubit Fluorometer (Thermo Fisher Scientific, Foster City, CA). The sterile water control contained no detectable DNA, indicating no contamination of exogenous DNA during the analysis. Viral DNA from each sample was pooled into equimolar proportions and sequenced on the Illumina platform at the University of California San Diego (UCSD) Institute for Genomic Medicine Genomics Center.
Virome analysis
Raw sequence reads were processed as described.[ 13 , 21 ] Briefly, raw sequence reads were deduplicated using Clumpify (https://sourceforge.net/projects/bbmap/) followed by trimming and filtering for low‐quality and contaminating human reads using Kneaddata[ 22 ] with the GRCh38_v25 human genome reference. Reads were aligned and assigned taxonomy by using the PathSeq pipeline (distributed in the Genome Analysis Toolkit [GATK], version 4.1.3.0) with default settings.[ 23 , 24 ] An in‐house Perl script was made (pathseq2taxsummary.pl) to convert PathSeq concatenated scores .txt files into a MOTHUR33‐style .taxsummary file. The Perl script is available at https://github.com/JCVenterInstitute/pathseq2taxsummary. Read counts, allowing ambiguity, were imported into R (R Foundation for Statistical Computing, Vienna, Austria), and data were normalized.
Statistical analysis
For parametric data (e.g., serum markers), the Student t test was used for comparison between two groups and the one‐way analysis of variance with Tukey’s post hoc test was used for three or more groups. Results were expressed as mean and SD for each continuous outcome, if not stated otherwise. For nonparametric data (e.g., microbiome data), the Mann‐Whitney U test/Wilcoxon rank‐sum test was used for comparison between two groups and the Kruskal‐Wallis test with Dunn’s post hoc test was used for three or more groups. All statistical tests were two sided. The respective statistical test was unpaired for controls versus subjects with AUD and paired for AUD active versus AUD abstinent. Relative abundances for further analyses were calculated within each virus category (phages versus mammalian viruses) at the species level. Single phages were analyzed at the species level and summarized according to their hosts. Bray‐Curtis dissimilarity matrices were used for principal coordinate analysis (PCoA) to identify differences in the relative abundance of all phages grouped according to their hosts. p values were determined by permutational multivariate analysis of variance while adjusting for potentially confounding factors. Linear discriminant effect‐size analysis (LEfSe) was performed to determine the features most likely to account for differences between groups.[ 25 ] To assess for significant changes in bacterial–viral correlations with liver disease progression, we performed generalized estimating equation modeling to identify variables with a moderating effect on the bacteria–phage association while accounting for the variables sex, age, and BMI. We chose a generalized estimating equation model rather than a regular regression model because the generalized estimating equation model requires less stringent distributional assumption of the data when providing inference.[ 26 ] Visualization of the effect of increasing CK18‐M65 on the phage–bacteria correlation coefficient was performed using time‐varying coefficients linear modeling. p ≤ 0.05 was considered statistically significant. Statistical analysis was performed using R statistical software (version 4.0.3; R Foundation for Statistical Computing).
RESULTS
Changes in the virome in the AUD population
The study population consisted of 62 patients with AUD and 16 controls. The study population was similar in terms of age, sex, and BMI. Laboratory parameters obtained at the start of the study showed significant differences in AST, ALT, GGT, ALP, bilirubin, albumin, and creatinine between the controls and patients with active AUD (Table 1). There were significant differences in their fecal viromes, specifically in the composition of bacteriophage species, as demonstrated by PCoA analysis comparing controls with patients with AUD regardless of alcohol use status (Figure 1A). LEfSe was applied to identify the features most likely to account for differences between the two groups. This revealed 18 bacteriophages more abundant in the control population, including eight bacteriophages targeting Propionibacterium, five targeting Enterobacteria, and the rest targeting Salmonella, Lactobacillus, Cronobacter, Escherichia, and Leuconostoc (Figure 1B). Of the bacteriophage species that were significantly more abundant in the AUD population, two bacteriophages targeted Streptococcus and two targeted Lactococcus (Figure 1B–F).
TABLE 1.
Demographic and laboratory parameters of the study population
| Characteristics | Control (n = 16) | Alcohol use disorder (n = 62) | p value |
|---|---|---|---|
| Age (years), n = 78 | 40.8 ± 12.3 | 44.4 ± 11.9 | 0.303 |
| Sex (male), n (%), n = 78 | 13 (81.3) | 44 (71.0) | 0.385 |
| BMI (kg/m2), n = 78 | 23.9 ± 3.8 | 24.3 ± 3.8 | 0.676 |
| AST (IU/L), n = 70 | 18.4 ± 5.1 | 67.5 ± 64.7 | <0.001 |
| ALT (IU/L), n = 70 | 11.3 ± 3.6 | 53.3 ± 42.3 | <0.001 |
| GGT (IU/L), n =69 | 23.0 ± 13.0 | 208.5 ± 291.9 | <0.001 |
| ALP (IU/L), n = 68 | 45.8 ± 22.5 | 79.1 ± 33.6 | 0.003 |
| Bilirubin (mg/dL), n = 70 | 0.2 ± 0.2 | 0.6 ± 0.4 | <0.001 |
| Albumin (g/dL), n = 67 | 4.4 ± 0.1 | 4.6 ± 0.4 | 0.009 |
| INR, n = 61 | n/a | 0.98 ± 0.11 | n/a |
| Creatinine (mg/dL), n = 70 | 0.97 ± 0.18 | 0.80 ± 0.14 | 0.030 |
| Platelet count (109/L), n = 61 | n/a | 228.1 ± 79.3 | n/a |
Values presented are mean ± SD. The number of subjects for which data were available is indicated in the first column.
Abbreviations: ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; GGT, gamma‐glutamyltransferase; INR, international normalized ratio; n/a, not applicable.
FIGURE 1.
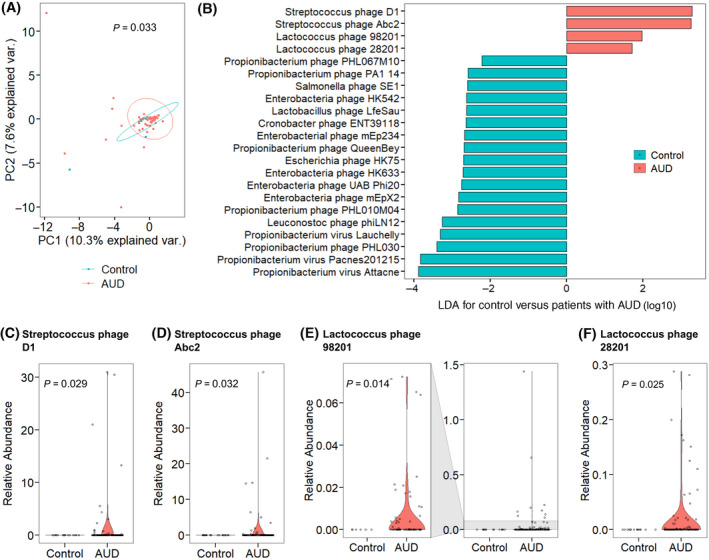
Patients with AUD have differences in their fecal virome compared with control subjects. (A) PCoA of fecal bacteriophages grouped by target bacteria species in controls (n = 16) and patients with AUD (n = 62 active alcohol use and 56 after abstinence). Axes represent the two most discriminating axes using the Bray‐Curtis distance metric. The p value was determined by permutational multivariate analysis of variance. (B) Linear discriminant analysis of bacteriophage species in patients with AUD versus controls. (C–F) Relative abundance of species (C) Streptococcus phage D1, (D) Streptococcus phage Abc2, (E) Lactococcus phage 98201, and (F) Lactococcus phage 28201. Axes are magnified to the left in (E) for better resolution of data. Abbreviations: AUD, alcohol use disorder; LDA, linear discriminant analysis; PC, principal component; PCoA, principal coordinate analysis; var., variance
Impact of abstinence on the fecal virome in AUD
After abstaining from alcohol for 2 weeks, patients with AUD experienced significant reductions in hepatic steatosis as measured by CAP, with corresponding significant decline in liver cell necrosis and apoptosis marker CK18‐M65[ 27 ] (Table 2). The fecal virome was significantly different between controls and patients with AUD actively using alcohol and after 2 weeks of abstinence from alcohol, as demonstrated by PCoA analysis (Figure 2A). LEfSe identified that phages targeting specific Lactococcus, Leuconostoc, and Streptococcus species and those targeting Propionibacterium and Lactobacillus species as a whole were more abundant in patients with AUD after abstinence (Figure 2B–H). The proportion of actively drinking patients with bacteriophages targeting Lactobacillus bacteria was also significantly lower than in patients after abstinence (Figure 2I). Further, compared to both control subjects and patients with AUD who were abstinent, there was a significantly smaller proportion of patients who were actively drinking with Propionibacterium phages (Figure 2J). The relative abundance of Propionibacterium phages was also significantly different across these three groups (Kruskal‐Wallis, p = 0.002), with significantly lower abundance in patients with AUD who were actively drinking compared to both control subjects (p < 0.001) and patients with AUD who were abstinent (p = 0.005).
TABLE 2.
Clinical characteristics of the study population
| Characteristics | Control (n = 16) | AUD active (n = 62) | AUD abstinent (n = 56) | p value |
|---|---|---|---|---|
| CAP, n = 92 | n/a | 285.1 ± 57.9 | 244.2 ± 59.2 | 0.003 |
| LSM (kPa), n = 92 | n/a | 7.67 ± 9.71 | 9.75 ± 12.20 | 0.418 |
| CK18‐M65 (U/L), n = 114 | 183.5 ± 64.5 | 469.5 ± 387.3 | 295.1 ± 255.2 | *0.006 |
Values presented are mean ± SD. The number of subjects for which data were available is indicated in the first column.
Abbreviations: AUD, alcohol use disorder; CAP, controlled attenuation parameter; CK18‐M65, caspase‐cleaved and intact cytokeratin 18; LSM, liver stiffness measurement; n/a, not applicable.
Post hoc p values for CK18‐M65: control versus AUD active, p < 0.001; control versus AUD abstinent, p = 0.014; AUD active versus AUD abstinent, p = 0.006.
FIGURE 2.
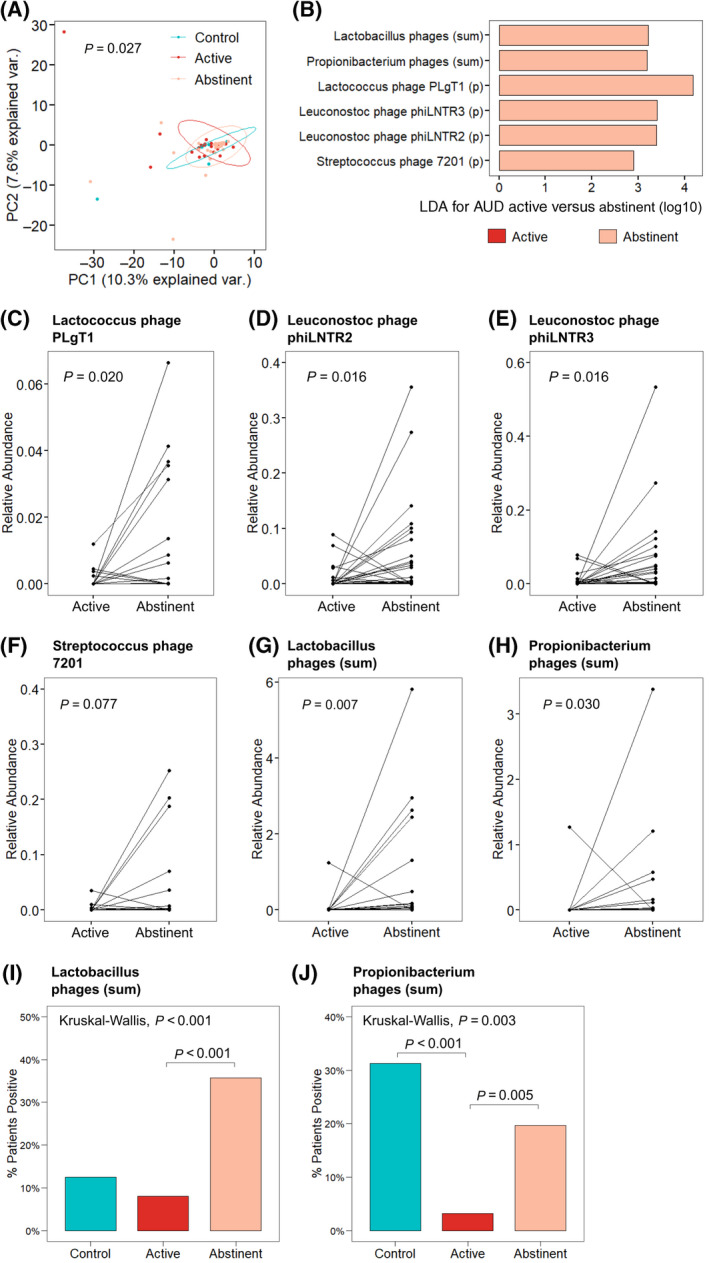
Abstinence from alcohol use changes the composition of the fecal virome in patients with AUD. (A) PCoA of bacteriophages grouped by host bacteria species in controls (n = 18), patients with AUD during active alcohol use (n = 62), and patients with AUD after abstinence from alcohol (n = 56). Axes represent the two most discriminating axes using the Bray‐Curtis distance metric. The p value was determined by permutational multivariate analysis of variance. (B) LDA of the sum of bacteriophages grouped by target bacteria species (sum) and individual bacteriophage species in patients with AUD during active alcohol use versus patients with AUD after abstinence from alcohol. (C–F) Relative abundance of bacteriophage species (C) Lactococcus phage PLgT1, (D) Leuconostoc phage phiLNTR2, (E) Leuconostoc phage phiLNTR3, and (F) Streptococcus phage 7201 in paired patients with AUD during active alcohol use versus abstinence. (G,H) Relative abundance of the sum of bacteriophages targeting bacteria species (G) Lactobacillus and (H) Propionibacterium in paired patients with AUD (n = 56). (I,J) Percentage of patients positive for bacteriophages targeting (I) Lactobacillus and (J) Propionibacterium bacteria in controls, patients with AUD actively using alcohol, and after abstinence. Abbreviations: AUD, alcohol use disorder; LDA, linear discriminant analysis; PC, principal component; (p), individual bacteriophage species; PCoA, principal coordinate analysis; var., variance
Progression of liver disease is associated with differences in the fecal virome
CK18 is a liver‐specific cell‐damage marker and is significantly increased in patients with AUD compared with healthy controls.[ 18 , 27 ] In conjunction with a liver stiffness measurement (LSM) >7.9 kPa, a cut‐off value of 416 U/L for CK18‐M65 can be used to help differentiate patients with no or nonprogressive liver disease (simple steatosis) from those with progressive liver disease (steatohepatitis or steatofibrosis).[ 18 ] Patients with progressive liver disease had significantly more steatosis as measured by CAP; additionally, they demonstrated significant changes in laboratory parameters other than CK18‐M65, including elevations in AST, ALT, GGT, and ALP (Table 3). The fecal virome was significantly different between patients with AUD with nonprogressive and progressive liver disease, as demonstrated by PCoA analysis (Figure 3A). Phages targeting Enterobacteria and Lactococcus species were more abundant in patients with AUD with progressive liver disease (Figure 3B–E). Correlation analysis between CK18‐M65 and the group of phages with Streptococcus as host demonstrated a positive correlation between the two variables, with higher relative abundance of Streptococcus phages in patients with higher CK18‐M65 (Figure 3F). Using random forest classification to extract characteristics most important in discriminating between patients with AUD with nonprogressive versus progressive liver disease, we found that aside from CK18‐M65 and LSM (used to stratify nonprogressive versus progressive liver disease) and CAP (a known direct marker of steatosis), the relative abundance of phages targeting Lactococcus and Parabacteroides play an important role in differentiating the two populations (Figure 3G).
TABLE 3.
Clinical characteristics of the patients with AUD stratified by nonprogressive and progressive liver disease
| Characteristics | Nonprogressive (n = 77) | Progressive (n = 43) | p value |
|---|---|---|---|
| Age (years), n = 120 | 44.5 ± 11.5 | 47.7 ± 13.1 | 0.181 |
| Sex (male), n (%), n = 120 | 52 (67.5) | 31 (72.1) | 0.604 |
| BMI (kg/m2), n = 120 | 24.0 ± 3.8 | 25.0 ± 4.0 | 0.191 |
| AST (IU/L), n = 120 | 48.8 ± 44.1 | 104.1 ± 78.3 | <0.001 |
| ALT (IU/L), n = 120 | 41.4 ± 37.4 | 76.9 ± 43.0 | <0.001 |
| GGT (IU/L), n = 118 | 124.4 ± 138.2 | 376.5 ± 401.7 | <0.001 |
| ALP (IU/L), n = 116 | 73.3 ± 15.8 | 90.5 ± 49.1 | 0.031 |
| Bilirubin (mg/dL), n = 120 | 0.5 ± 0.3 | 0.7 ± 0.5 | 0.101 |
| Albumin (g/dL), n = 116 | 4.7 ± 0.4 | 4.6 ± 0.5 | 0.276 |
| INR, n = 118 | 0.97 ± 0.08 | 1.02 ± 0.14 | 0.045 |
| Creatinine (mg/dL), n = 120 | 0.81 ± 0.15 | 0.80 ± 0.13 | 0.715 |
| Platelet count (109/L), n = 118 | 235.5 ± 72.9 | 203.2 ± 95.9 | 0.096 |
| CAP, n = 101 | 256.0 ± 55.2 | 292.3 ± 65.1 | 0.005 |
| LSM (kPa), n = 101 | 4.77 ± 1.15 | 13.97 ± 15.21 | <0.001 |
| CK18‐M65 (U/L), n = 114 | 205.5 ± 112.4 | 673.5 ± 378.0 | <0.001 |
Values presented are mean ± SD. The number of subjects for which data were available is indicated in the first column. The laboratory values are all from T1 (active use).
Abbreviations: ALT, alanine aminotransferase; ALP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; CAP, controlled attenuation parameter; CK18‐M65, caspase‐cleaved and intact cytokeratin 18; GGT, gamma‐glutamyltransferase; INR, international normalized ratio; LSM, liver stiffness measurement.
FIGURE 3.
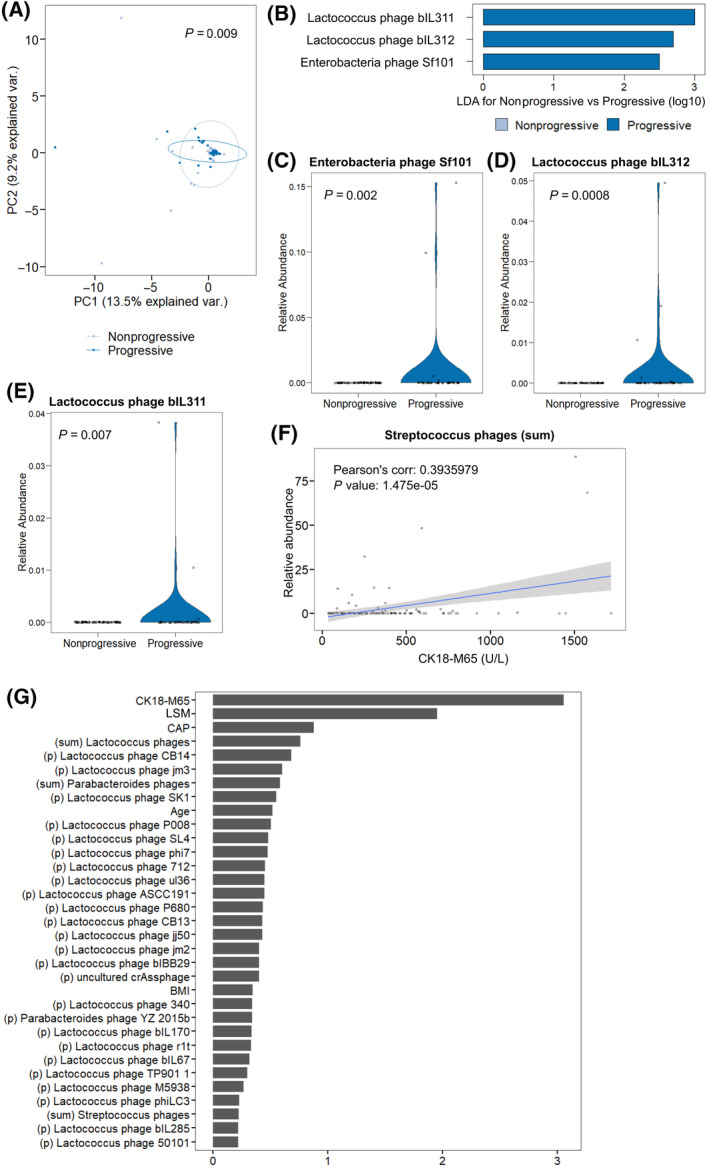
Progressive liver disease as defined by CK18‐M65 (cutoff, 416 U/L) is associated with differences in fecal virome composition. (A) PCoA of bacteriophages grouped by host bacteria species in patients with AUD with nonprogressive (n = 77) and progressive (n = 43) liver disease. Axes represent the two most discriminating axes using the Bray‐Curtis distance metric. The p value was determined by permutational multivariate analysis of variance. (B) LDA of individual bacteriophage species in patients with AUD with nonprogressive versus progressive liver disease. (C–E) Relative abundance of species (C) Enterobacteria phage Sf101, (D) Lactococcus phage bIL312, and (E) Lactococcus phage bIL311. (F) Pearson’s correlation of the sum of bacteriophages targeting Streptococcus bacteria species versus CK18‐M65 of patients with AUD at the time of stool sample collection. (G) Random forest feature selection, including relative abundance of viral taxa at the species level and grouped by target bacteria species, together with selected clinical features to discriminate between nonprogressive and progressive liver disease. Abbreviations: AUD, alcohol use disorder; BMI, body mass index; CAP, controlled attenuation parameter; CK18‐M65, caspase‐cleaved and intact cytokeratin 18; corr., correlation; LDA, linear discriminant analysis; (p) individual bacteriophage species; PC, principal component; PCoA, principal coordinate analysis; var., variance
Changes in bacterial–viral correlations seen in patients with progressive liver disease
To better understand phage–bacteria interaction in patients with AUD in the context of liver disease progression, we used patients’ serum CK18‐M65 values as a continuous marker for severity of liver disease.[ 28 ] We ran moderation analyses using bacteria as the response and corresponding phage and CK18‐M65 values as the predictors and additionally included the interaction term between the bacteriophage and CK18‐M65 by using a generalized estimating equation model (suitable for correlated data). We discovered that among the bacterial species tested, Enterobacteria, Escherichia, and Streptococcus are the only bacteria and phage pairs for which the interaction term is significant (p = 6 × 10−7, p = 0.018, and p = 0.0003, respectively). In order to better visualize this moderation role of CK18‐M65, we also ran a varying coefficient model and presented the change of the fitted coefficients across the level of (scaled) CK18‐M65 levels (Figure 4).
FIGURE 4.
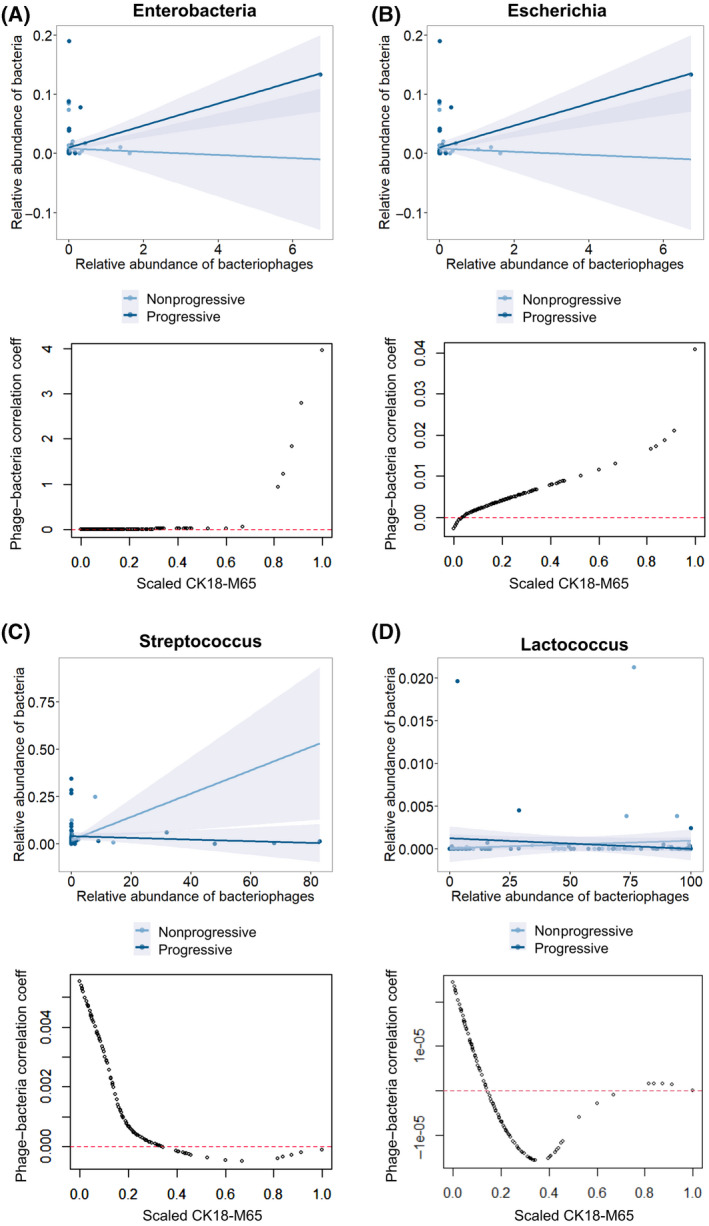
Phage–host interactions in patients with AUD are affected by liver disease progression. (A–D) Plot of correlation analysis between relative abundance of bacteriophages grouped by target bacteria species versus their respective target bacteria (above) for patients with AUD with nonprogressive versus progressive liver disease. Below shows plot of correlation coefficients of the relative abundance of respective bacteriophages and their target bacteria against scaled measured CK18‐M65 for each patient with AUD. (A) Enterobacteria, (B) Escherichia, (C) Streptococcus, and (D) Lactococcus species. Abbreviations: AUD, alcohol use disorder; CK18‐M65, caspase‐cleaved and intact cytokeratin 18; coeff, coefficient
The correlation coefficient between Enterobacteria phages and Enterobacteria bacteria grew increasingly positive with progressive liver disease as did the correlation coefficient between Escherichia phages and Escherichia bacteria (Figure 4A,B). This is reflective of coincidently higher levels of Enterobacteria and Escherichia bacteria, with higher levels of Enterobacteria and Escherichia phage in patients with AUD with progressive liver disease. Conversely, the correlation coefficient between Streptococcus phage and Streptococcus bacteria became increasingly negative with progressive liver disease (Figure 4C). In other words, in patients with AUD with increasingly progressive liver disease, the fecal abundance of Streptococcus phages grew while the abundance of Streptococcus bacteria was concurrently much lower. Progressive liver disease did not have a significant effect on the correlation between other phage–bacteria species pairs, such as Lactococcus (Figure 4D).
DISCUSSION
Here, we studied the changes in the intestinal virome associated with AUD as well as the effects of alcohol abstinence. Interestingly, we identified decreased abundance of various Propionibacterium, Lactobacillus, and Leuconostoc phages in the fecal viromes of patients with active AUD when compared to controls, and conversely, the abundance of Leuconostoc phages and bacteriophages targeting Lactobacillus and Propionibacterium species as a group increased in patients with AUD after 2 weeks of alcohol abstinence. These findings suggest that, with sobriety, the intestinal virome in patients with AUD trends toward the virome community composition of control subjects. Interestingly, in the only other existing study on changes in the intestinal virome associated with alcohol‐associated liver disease, Leuconostoc phage was also shown to be more abundant in mild liver disease (lower Model for End‐Stage Liver Disease score) compared with more severe liver disease.[ 13 ] Our findings suggest that alcohol‐associated changes in the fecal virome may be partially reversible after a short period of abstinence.
In this study, we also found that progression of liver disease not only was associated with differences in phage abundance but also in phage–bacteria interactions. Interestingly, using generalized estimating equation modeling, liver disease progression was found to significantly affect the correlation between phages and bacteria for Enterobacteria, Escherichia, and Streptococcus species. Specifically, progressive liver disease was associated with coincident increase in Streptococcus phages and decrease in Streptococcus bacteria. This could imply that in patients with more significant liver disease, Streptococcus phages are more likely to have a lytic effect on bacterial hosts. Environmental changes, such as antibiotics,[ 29 ] bile salts,[ 30 ] and intestinal inflammation,[ 31 ] are reported examples that can trigger prophage induction and resumption of the lytic cycle. Perhaps changes in bile acid secretion or inflammatory cytokines secondary to progression in liver disease can induce phage lysis of Streptococcus bacteria. Another recent study also found that phage and bacterial correlations centered on Streptococcus species were affected by progression of liver disease, treatment with rifaximin, and hospitalization.[ 32 ] Conversely, the correlation coefficient between Enterobacteria phage and Enterobacteria bacteria and Escherichia phage and Escherichia bacteria became increasingly positive with progressive liver disease, reflective of coincident high levels of bacteria with high levels of phage. Both antagonistic and mutualistic relationships between phages and their respective bacterial hosts have been described in different environments, including intestinal and oral.[ 33 , 34 ] In adverse/hostile environments, lysogeny enhances both phage and bacterial host survival.[ 35 ] Temperate phages can help confer beneficial traits on their bacterial hosts, such as introducing virulence factor production or altering metabolism, through genetic integration or transduction.[ 36 , 37 ] In patients with AUD with progressive liver disease, one hypothesis might be that an increased abundance of lysogenic Enterobacteria and Escherichia phages could be facilitating the growth of respective bacterial species, which are known to be pathogenic. Another possible explanation could be that the dense population of Enterobacteria and Escherichia bacteria in patients with progressive liver disease spurs growth of corresponding phages as in a predator–prey relationship.
In summary, we demonstrate partial reversibility of an alcohol‐associated fecal virome after abstinence and a virome signature associated with progressive liver disease. Our findings raise some interesting questions regarding how liver disease might affect the phage–bacterial relationship. Ultimately, longitudinal collection from a larger patient cohort over time will provide more insight into the interactions between phages and bacteria and how these interactions are modified by the liver disease process or vice versa.
CONFLICTS OF INTEREST
Bernd Schnabl has been consulting for Ferring Research Institute, Gelesis, HOST Therabiomics, Intercept Pharmaceuticals, Mabwell Therapeutics, Patara Pharmaceuticals, and Takeda; his institution, UCSD, has received research support from Axial Biotherapeutics, BiomX, CymaBay Therapeutics, NGM Biopharmaceuticals, Prodigy Biotech, and Synlogic Operating Company; he is founder of Nterica Bio. UCSD has filed several patents with Bernd Schnabl as inventor related to this work.
ETHICS STATEMENT
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institution’s human research and ethical committee (Université Catholique de Louvain, Brussels, Belgium; B403201422657), and patients were enrolled after written informed consent was obtained.[ 17 , 18 ] We followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) criteria for reporting cohort studies.
ACKNOWLEDGMENTS
Cynthia Hsu is supported by T32 DK007202. This study was supported in part by Deutsche Forschungsgemeinschaft (German Research Foundation) fellowship #LA 4286/1‐1; the Clinical and Translational Research Fellowship in Liver Disease by the American Association for the Study of Liver Diseases Foundation to Sonja Lang; National Institutes of Health grant K12 HD85036 to Phillipp Hartmann; the Fond National de Recherche Scientifique Belgium grants #FRS‐FNRS J.0146.17 and #T.0217.18; the Fédération Wallonie‐Bruxelles Action de Recherche Concertée #ARC18‐23/092 to Peter Stärkel; National Institutes of Health (NIH) grants #R01 AA24726, #R37 AA020703, #U01 AA026939, and #U01 AA026939‐04S1; the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development Award #BX004594; a Biocodex Microbiota Foundation Grant to Bernd Schnabl; and services provided by NIH centers #P50 AA011999 and the San Diego Digestive Diseases Research Center #P30 DK120515.
Hsu CL, Zhang X, Jiang L, Lang S, Hartmann P, Pride D, et al. Intestinal virome in patients with alcohol use disorder and after abstinence. Hepatol Commun. 2022;6:2058–2069. 10.1002/hep4.1947
Funding information
Deutsche Forschungsgemeinschaft, Fellowship Award Number: LA 4286/1‐1. American Association for the Study of Liver Diseases, Clinical and Translational Research Fellowship in Liver Disease. National Institutes of Health, Grant Numbers: K12 HD85036, R01 AA24726, R37 AA020703, U01 AA026939, U01 AA026939‐04S1, P50 AA011999. Fond National de Recherche Scientifique Belgium, Grant Numbers: FRS‐FNRS J.0146.17, T.0217.18. Fédération Wallonie‐Bruxelle, Action de Recherche Concertée Number: ARC18‐23/092. Biomedical Laboratory Research and Development Service of the VA Office of Research and Development, Award Number: BX004594. Biocodex Microbiota Foundation Grant. San Diego Digestive Diseases Research Center, Grant Number: P30 DK120515
REFERENCES
- 1. Lieber CS. Alcoholic fatty liver: its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol. 2004;34:9–19. [DOI] [PubMed] [Google Scholar]
- 2. Gianni E, Forte P, Galli V, Razzolini G, Bardazzi G, Annese V. Prospective evaluation of liver stiffness using transient elastography in alcoholic patients following abstinence. Alcohol Alcohol. 2017;52:42–7. [DOI] [PubMed] [Google Scholar]
- 3. Tang‐Barton P, Vas W, Weissman J, Salimi Z, Patel R, Morris L. Focal fatty liver lesions in alcoholic liver disease: a broadened spectrum of CT appearances. Gastrointest Radiol. 1985;10:133–7. [DOI] [PubMed] [Google Scholar]
- 4. Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346:987–90. [DOI] [PubMed] [Google Scholar]
- 5. Bjørkhaug ST, Aanes H, Neupane SP, Bramness JG, Malvik S, Henriksen C, et al. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes. 2019;10:663–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. 2012;302:G966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Stärkel P, et al. Intestinal permeability, gut‐bacterial dysbiosis, and behavioral markers of alcohol‐dependence severity. Proc Natl Acad Sci U S A. 2014;111:E4485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–72. [DOI] [PubMed] [Google Scholar]
- 9. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao LI, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513:59–64. [DOI] [PubMed] [Google Scholar]
- 10. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60:940–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis. 2015;33:338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, Puri P, Sterling RK, et al. Randomised clinical trial: Lactobacillus GG modulates gut microbiome, metabolome and endotoxemia in patients with cirrhosis. Aliment Pharmacol Ther. 2014;39:1113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang LU, Lang S, Duan YI, Zhang X, Gao B, Chopyk J, et al. Intestinal virome in patients with alcoholic hepatitis. Hepatology. 2020;72:2182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodriguez‐Valera F, Martin‐Cuadrado A‐B, Rodriguez‐Brito B, Pašić L, Thingstad TF, Rohwer F, et al. Explaining microbial population genomics through phage predation. Nat Rev Microbiol. 2009;7:828–36. [DOI] [PubMed] [Google Scholar]
- 15. Davies MR, Broadbent SE, Harris SR, Thomson NR, van der Woude MW. Horizontally acquired glycosyltransferase operons drive salmonellae lipopolysaccharide diversity. PLoS Genet. 2013;9:e1003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verma NK, Brandt JM, Verma DJ, Lindberg AA. Molecular characterization of the O‐acetyl transferase gene of converting bacteriophage SF6 that adds group antigen 6 to Shigella flexneri. Mol Microbiol. 1991;5:71–5. [DOI] [PubMed] [Google Scholar]
- 17. Hartmann P, Lang S, Zeng S, Duan YI, Zhang X, Wang Y, et al. Dynamic changes of the fungal microbiome in alcohol use disorder. Front Physiol. 2021;12:699253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maccioni L, Gao B, Leclercq S, Pirlot B, Horsmans Y, De Timary P, et al. Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes. 2020;12:1782157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duan YI, Llorente C, Lang S, Brandl K, Chu H, Jiang LU, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conceicao‐Neto N, Yinda KC, Van Ranst M, Matthijnssens J. NetoVIR: modular approach to customize sample preparation procedures for viral metagenomics. Methods Mol Biol. 2018;1838:85–95. [DOI] [PubMed] [Google Scholar]
- 21. Lang S, Demir M, Martin A, Jiang LU, Zhang X, Duan YI, et al. Intestinal virome signature associated with severity of nonalcoholic fatty liver disease. Gastroenterology. 2020;159:1839–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McIver LJ, Abu‐Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, et al. bioBakery: a meta'omic analysis environment. Bioinformatics. 2018;34:1235–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RGW, Getz G, et al. PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat Biotechnol. 2011;29:393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Walker MA, Pedamallu CS, Ojesina AI, Bullman S, Sharpe T, Whelan CW, et al. GATK PathSeq: a customizable computational tool for the discovery and identification of microbial sequences in libraries from eukaryotic hosts. Bioinformatics. 2018;34:4287–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang W, He H, Tu XM. Applied categorical and count data analysis. Boca Raton: CRC Press; 2012. [Google Scholar]
- 27. Mueller S, Nahon P, Rausch V, Peccerella T, Silva I, Yagmur E, et al. Caspase‐cleaved keratin‐18 fragments increase during alcohol withdrawal and predict liver‐related death in patients with alcoholic liver disease. Hepatology. 2017;66:96–107. [DOI] [PubMed] [Google Scholar]
- 28. Wei X, Wei H, Lin W, Hu Z, Zhang J. Cell death biomarker M65 is a useful indicator of liver inflammation and fibrosis in chronic hepatitis B: a cross‐sectional study of diagnostic accuracy. Medicine (Baltimore). 2017;96:e6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. Quinolone antibiotics induce Shiga toxin‐encoding bacteriophages, toxin production, and death in mice. J Infect Dis. 2000;181:664–70. [DOI] [PubMed] [Google Scholar]
- 30. Hernandez SB, Cota I, Ducret A, Aussel L, Casadesus J. Adaptation and preadaptation of Salmonella enterica to bile. PLoS Genet. 2012;8:e1002459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Diard M, Bakkeren E, Cornuault JK, Moor K, Hausmann A, Sellin ME, et al. Inflammation boosts bacteriophage transfer between Salmonella spp. Science. 2017;355:1211–5. [DOI] [PubMed] [Google Scholar]
- 32. Bajaj JS, Sikaroodi M, Shamsaddini A, Henseler Z, Santiago‐Rodriguez T, Acharya C, et al. Interaction of bacterial metagenome and virome in patients with cirrhosis and hepatic encephalopathy. Gut. 2021;70:1162–73. [DOI] [PubMed] [Google Scholar]
- 33. Edlund A, Santiago‐Rodriguez TM, Boehm TK, Pride DT. Bacteriophage and their potential roles in the human oral cavity. J Oral Microbiol. 2015;7:27423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sausset R, Petit MA, Gaboriau‐Routhiau V, De Paepe M. New insights into intestinal phages. Mucosal Immunol. 2020;13:205–15. Erratum in: Mucosal Immunol. 2020;13:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stewart FM, Levin BR. The population biology of bacterial viruses: why be temperate. Theor Popul Biol. 1984;26:93–117. [DOI] [PubMed] [Google Scholar]
- 36. Obeng N, Pratama AA, Elsas JDV. The significance of mutualistic phages for bacterial ecology and evolution. Trends Microbiol. 2016;24:440–9. [DOI] [PubMed] [Google Scholar]
- 37. Davies EV, Winstanley C, Fothergill JL, James CE. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol Lett. 2016;363:fnw015. [DOI] [PubMed] [Google Scholar]