

Abstract
Dysregulation of expression of functional genes and pathways plays critical roles in the etiology and progression of hepatocellular carcinoma (HCC). Next generation‐based RNA sequencing (RNA‐seq) offers unparalleled power to comprehensively characterize HCC at the whole transcriptome level. In this study, 17 fresh‐frozen HCC samples with paired non‐neoplastic liver tissue from Caucasian patients undergoing liver resection or transplantation were used for RNA‐seq analysis. Pairwise differential expression analysis of the RNA‐seq data was performed to identify genes, pathways, and functional terms differentially regulated in HCC versus normal tissues. At a false discovery rate (FDR) of 0.10, 13% (n = 4335) of transcripts were up‐regulated and 19% (n = 6454) of transcripts were down‐regulated in HCC versus non‐neoplastic tissue. Eighty‐five Kyoto Encyclopedia of Genes and Genomes pathways were differentially regulated (FDR, <0.10), with almost all pathways (n = 83) being up‐regulated in HCC versus non‐neoplastic tissue. Among the top up‐regulated pathways was oxidative phosphorylation (hsa00190; FDR, 1.12E‐15), which was confirmed by Database for Annotation, Visualization, and Integrated Discovery (DAVID) gene set enrichment analysis. Consistent with potential oxidative stress due to activated oxidative phosphorylation, DNA damage‐related signals (e.g., the up‐regulated hsa03420 nucleotide excision repair [FDR, 1.14E‐04] and hsa03410 base excision repair [FDR, 2.71E‐04] pathways) were observed. Among down‐regulated genes (FDR, <0.10), functional terms related to cellular structures (e.g., cell membrane [FDR, 3.05E‐21] and cell junction [FDR, 2.41E‐07], were highly enriched, suggesting compromised formation of cellular structure in HCC at the transcriptome level. Interestingly, the olfactory transduction (hsa04740; FDR, 1.53E‐07) pathway was observed to be down‐regulated in HCC versus non‐neoplastic tissue, suggesting impaired liver chemosensory functions in HCC. Our findings suggest oxidative phosphorylation and the associated DNA damage may be the major driving pathologic feature in HCC.
The cause and progression of hepatocellular carcinoma (HCC) are associated with dysregulation of expression of functional genes and pathways. In this next generation‐based RNA sequencing study, we identified oxidative phosphorylation as the major signal underlying in U.S. Caucasian patients with alcoholic and non‐alcoholic fatty liver disease as major risk factors.

INTRODUCTION
Hepatocellular carcinoma (HCC) represents a leading cause of cancer‐related mortality worldwide, especially in East Asia and sub‐Saharan Africa.[ 1 , 2 , 3 ] Etiologies and pathophysiology of HCC in various ethnicities are fairly different. Hepatitis B virus (HBV) is a major cause of HCC in East Asians, while alcoholic and nonalcoholic fatty liver disease and chronic hepatitis C are the most common etiologies in the US and European populations.[ 4 , 5 ] Although the incidence of HCC is relatively low in the United States, it has more than doubled over the past 2 decades and is anticipated to continue increasing due to a growing number of patients with alcoholic/nonalcoholic steatohepatitis (ASH/NASH) and advanced hepatitis C virus (HCV) infection.[ 1 , 3 ] The development of HCC is considered a multistep process that involves the accumulation of genetic and epigenetic alterations.[ 6 , 7 , 8 , 9 , 10 ]
Over the past decade, microarray‐based gene expression studies have been performed to identify the molecular and genomic mechanisms underlying HCC,[ 8 , 9 , 10 , 11 ] including comparative analysis of cancer versus noncancerous samples,[ 12 ] early stage versus late stage,[ 13 ] good prognosis versus poor prognosis,[ 12 ] and HBV versus HCV infection.[ 14 ] The studies have discovered a number of genetic and genomic landmarks of HCC, including, for example, dysregulation of genes involved in cell‐cycle regulation and the Wnt/beta‐catenin pathway,[ 12 ] cell adhesion molecules involved in cell–cell and cell–matrix interactions,[ 13 ] and genes responsible for detoxification and immune response.[ 14 ] In a more recent large‐sample microarray‐based study on 183 HCC samples recruited from Japan,[ 15 ] genes involved in the G2/M cell‐cycle phase and stem/progenitor markers were identified by comparing two subgroups with differential survival outcomes.[ 15 ]
With the advance of next‐generation sequencing technologies, RNA sequencing (RNA‐seq) has become a powerful tool in defining the transcriptomic changes related to HCC. To date, several RNA‐seq studies have been performed on human HCC samples, predominantly in Asian populations.[ 16 , 17 , 18 , 19 ] Based on RNA‐seq analysis on 10 HBV‐related HCC versus the paired adjacent noncancerous tissue from patients recruited in China, pathways related to cell growth, metabolism, and immune functions were identified.[ 16 ] In another study involving 98 HCC samples from Asian patients, an 85‐gene signature was identified to be up‐regulated in the CD8+ T‐cell‐excluded tumors.[ 18 ] Furthermore, the signature was enriched with genes for collagens, extracellular matrix (ECM), Notch pathway, transforming growth factor beta pathway, and hedgehog pathway, suggesting that elevated fibrosis may be associated with CD8+ T‐cell exclusion in HCC.[ 18 ] A recent trans‐omics study of HCC used a large sample containing a mixture of Caucasians, Asians, African Americans, and other ethnicities.[ 20 ] Risk factors and etiologies of patients with HCC in that study included hepatitis B, hepatitis C, ASH, and NASH.[ 20 ] Because there were no control subjects or normal noncancerous tissue samples involved in the analyses,[ 20 ] case‐control‐based differential expression analyses to identify genes/pathways pertinent to HCC per se were not performed.
Here, we present an RNA‐seq‐based transcriptome‐wide study of HCC from a cohort of the US Caucasian patients. Based on our analyses, we identified, for the first time, up‐regulation of oxidative phosphorylation as the major signal underlying HCC. Interestingly, we also detected signals related to DNA damage in HCC samples, presumably due to oxidative stress caused by oxidative phosphorylation. Our findings provide strong evidence on novel molecular mechanisms for liver carcinogenesis and potential therapeutic targets for HCC treatment.
MATERIALS AND METHODS
Patients and tissue specimens
This study was approved by the institutional review boards of the University of Wisconsin (UW)–Madison. We identified 17 fresh‐frozen HCC samples with paired adjacent non‐neoplastic tissue from the UW–Madison Translational Science Biocore‐Biobank inventory. The inclusion criteria were (1) HCC samples of adult Caucasian patients of both sexes; (2) HCC samples without preoperative chemotherapy or chemoembolization; and (3) HCC samples with available paired adjacent non‐neoplastic tissue. HCC samples of various histologic grades and pathologic staging were included. The diagnosis of HCC was histologically confirmed, and all HCC tumor tissues were assessed by hematoxylin and eosin staining. Only those with a percentage of tumor cells more than 90% and without extensive necrosis were used for the analysis. The clinical and pathologic features of the patients are summarized in Table 1.
TABLE 1.
Patient characteristics
| Patient ID | Sex (F/M) | Age (years) | Alcohol use (Y/N) | Viral hepatitis (Y/N) | BMI (kg/m2) | Obesity (Y/N) | Diabetes (Y/N) | Hypertension (Y/N) | Tumor histology grade | Nontumor liver histology | Liver resection or transplant |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PT1 | F | 86 | Y | Y | 32.9 | Y | Y | Y | Moderately differentiated | Steatosis | Resection |
| PT2 | M | 73 | Y | N | 21.8 | N | N | Y | Moderately Differentiated | Cirrhosis | Transplant |
| PT3 | M | 84 | Y | N | 33.5 | Y | Y | Y | Moderately Differentiated | Cirrhosis | Transplant |
| PT4 | F | 68 | N | Y | 52.6 | Y | Y | Y | Moderately Differentiated | Cirrhosis | Transplant |
| PT5 | F | 78 | N | N | 24.2 | N | N | N | Moderately Differentiated | Cirrhosis | Transplant |
| PT6 | M | 86 | Y | N | 33.1 | Y | Y | Y | Well Differentiated | Steatosis | Resection |
| PT7 | M | 64 | Y | Y | 20.3 | N | N | N | Well Differentiated | Cirrhosis | Transplant |
| PT8 | M | 96 | Y | N | 30.6 | Y | N | Y | Poorly Differentiated | Cirrhosis | Transplant |
| PT9 | F | 65 | Y | Y | 18.6 | N | N | Y | Well Differentiated | Chronic hepatitis | Resection |
| PT10 | M | 76 | Y | N | 38.2 | Y | Y | Y | Well Differentiated | Cirrhosis | Transplant |
| PT11 | M | 84 | Y | N | 22.6 | N | N | Y | Well Differentiated | Cirrhosis | Transplant |
| PT12 | M | 78 | N | Y | 22.8 | N | N | Y | Well to moderately differentiated | Cirrhosis | Transplant |
| PT13 | M | 69 | Y | Y | 38.3 | Y | Y | Y | Moderately Differentiated | Cirrhosis | Transplant |
| PT14 | M | 70 | N | N | 32.6 | Y | Y | Y | Well to moderately differentiated | Steatosis | Transplant |
| PT15 | M | 71 | Y | N | 28.3 | N | Y | Y | Well differentiated | Steatosis | Resection |
| PT16 | M | 65 | Y | N | 34.4 | Y | Y | Y | Moderate to poorly differentiated | Steatosis | Transplant |
| PT17 | M | 68 | Y | N | 35.3 | Y | Y | Y | Moderately differentiated | Steatosis | Resection |
Abbreviations: BMI, body mass index; F, female; M, male; N, no; Y, yes.
RNA preparation and sequencing
Total RNA extractions were performed in the UW–Madison Translational Research Initiatives in Pathology laboratory. The Maxwell 16 LEV simplyRNA Tissue kit (Promega AS1280) was used per kit instructions and was run on a Maxwell 16 MDx automated instrument. RNA samples were transferred to a 96‐well plate and delivered to the UW–Madison Biotechnology Center. Total RNA was assayed for purity and integrity using the NanoDrop One Spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA), respectively. RNA libraries were prepared from samples that met the TruSeq Stranded Total RNA Sample Preparation Guide (15031048 E) input guidelines using the Illumina TruSeq Stranded Total (Gold) RNA Sample Preparation kit (Illumina Inc., San Diego, CA). Libraries were standardized to 2 nM. Paired‐end 150‐base pair sequencing was performed using standard sequencing by synthesis chemistry on an Illumina NovaSeq6000 sequencer. Images were analyzed using the standard Illumina Pipeline, version 1.8.2.
RNA‐seq data analysis
Raw RNA‐seq data were deposited into the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) with accession number GSE184733. RNA‐seq raw data (fastq data) were passed for an overall quality check using the FastQC program (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The transcript quantification analysis of fastq data was performed using the Salmon program (Salmon 0.99.0)[ 21 ] with human reference transcriptome (Homo_sapiens.GRCh38.cdna.all.fa) followed by the Bioconductor’s tximport package,[ 22 ] which generated a raw count matrix with rows as transcript identifications and columns as specific samples.
Differential expression analysis
The count matrix was analyzed using the DESeq2 package[ 23 ] for pairwise differential expression analysis, which produced differentially expressed genes (DEGs) (or transcripts) between HCC and non‐neoplastic control tissue based on a model of negative binomial distribution. Due to the pairwise design at each subject level (i.e., HCC tissue compared with a patient’s own non‐neoplastic liver tissue), a patient’s covariates, such as age, sex, hepatic viral infection, and alcohol use, do not contribute to the differential expression analysis result. Hence, they were not included in the model for differential expression analysis. To adjust for multiple testing, the raw p value of each gene (or transcript) for differential expression was transformed into a false discovery rate (FDR) value, and an FDR < 0.10 was used as a cutoff for statistical significance of differential expression.
To discover differential expression at the pathway level, the Bioconductor package GAGE[ 24 ] was used to identify Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that are differentially regulated in HCC versus non‐neoplastic tissue. The package is based on a meta‐test that summarizes test statistics (for differential expression at the individual gene level) for all genes contained in a pathway. Essentially, a certain pathway’s differential expression is detected if a large number of individual genes in the pathway show the same trend of up‐ or down‐regulation. Each pathway was provided an FDR value for up or down‐regulation. An FDR < 0.10 was used as the threshold for declaring a significantly up‐ or down‐regulated pathway. The Bioconductor package Pathview[ 25 ] was used to visualize a certain KEGG pathway’s differential expression.
To test differential expression with Gene Ontology (GO) terms or other types of functional terms, such as UP_keywords (UniProtKB keywords), we submitted those up‐ or down‐regulated genes (FDR, <0.10) as identified in DESeq2[ 23 ] to the Database for Annotation, Visualization, and Integrated Discovery (DAVID)[ 26 , 27 ] for functional enrichment analysis to identify a list of up‐ or down‐regulated enriched functional terms, respectively. Due to the limit of 3000 as the maximum number of genes that can be submitted for DAVID analysis,[ 26 , 27 ] the top 3000 up‐regulated genes (in terms of FDR) and top 3000 down‐regulated genes (in terms of FDR) were submitted to DAVID for enrichment analysis. Here, the actual cutoff for the submitted up‐regulated genes was an FDR of 0.0150939 (i.e., the 3000th most significant up‐regulated gene’s FDR value), and the actual cutoff for the submitted down‐regulated genes was an FDR of 0.0039512 (i.e., the 3000th most significant down‐regulated gene’s FDR value).
Cibersortx analysis
Tumor‐infiltrating immune cells were enumerated based on the RNA‐seq data by using software Cibersortx[ 28 ] to identify immune cells with differential abundance in HCC versus non‐neoplastic tissue. Cibersort[ 28 ] is a well‐established bioinformatics algorithm to infer the components and proportions of cell types within a tissue based on the RNA‐seq expression data. The underlying computation is based on a matrix decomposition algorithm through linear equations where a gene expression matrix is decomposed into the product of a signature gene matrix and a cell‐type component matrix. The gene expression matrix came from the RNA‐seq data. The signature gene matrix, provided by Cibersort developers, was established through public gene expression data sets on gene expression signatures for specific cell types, such as B and monocytes.[ 28 ] Through matrix computation using machine‐learning techniques, the cell‐type matrix that contains information regarding the proportions of each cell type in a given sample can be inferred.[ 28 ]
To implement the analysis, a submatrix from the RNA‐seq gene expression count matrix that contained all signature genes in the signature matrix provided by Cibersortx was extracted.[ 28 ] The signature matrix contained expression counts of signature genes for 22 distinct human immune cells.[ 19 ] The submatrix was then submitted to the Cibersortx website (https://cibersortx.stanford.edu/) for analysis. The output file is a matrix with rows as samples, columns as different types of immune cells, and each entry as the inferred proportion of a specific immune cell within all the 22 ones in a sample. We then compared the immune cell proportion between HCC and the paired non‐neoplastic samples to determine if the cell proportion was statistically different. The analysis was performed using the R generalized linear model function under a quasibinomial model. Visualization of immune cell proportions in an HCC and its paired non‐neoplastic tissue was done using the ggplot2 R package.[ 29 ]
RESULTS
Patient characteristics
HCC samples and paired non‐neoplastic tissue from 17 patients were used. These patients, all Caucasians, included 4 female patients and 13 male patients, with ages ranging from 64 to 96 years (mean, 75.4; SD, 9.2). Their body mass index (BMI) values ranged from 18.6 to 52.6 kg/m2 (mean, 30.6; SD, 8.52). Based on a cut‐off value of BMI ≥ 30 kg/m2, 10 patients were obese. Additionally, 10 patients had a history of diabetes and 7 were nondiabetic. Fifteen patients had a history of hypertension. Most of the patients (13/17) had a history of alcohol abuse, and most had at least one risk factor for metabolic syndrome (e.g., obesity, diabetes, and hypertension). Six patients had a history of chronic hepatitis C that was treated with antiviral medications; 4 of these patients had concurrent alcohol abuse. In terms of tumor histology grade, 6 patients were categorized as well differentiated, 7 moderately differentiated, 2 well to moderately differentiated, 1 moderately to poorly differentiated, and 1 poorly differentiated. One HCC case demonstrated steatohepatitic features, including macrovesicular steatosis, ballooned hepatocytes, and Mallory‐Denk bodies. In terms of nontumor background liver tissue (the paired non‐neoplastic tissue), 10 patients had cirrhosis, 6 had at least mild steatosis, and 1 had features of chronic hepatitis, including a portal‐based moderate degree of lymphocyte‐rich inflammatory infiltrate. The detailed characteristics of these 17 patients are shown in Table 1.
Identification of DEGs and pathways in tumor versus adjacent non‐neoplastic tissue
Based on differential expression analysis to pairwise comparison of each of the 17 HCC samples with its adjacent non‐neoplastic tissue, a total of 4335 genes were up‐regulated and 6454 genes down‐regulated (FDR, <0.10) (Table S1). Among the up‐regulated genes, the mean fold change was 2.04 for HCC over the paired non‐neoplastic tissue. Among the down‐regulated genes, the mean fold change was 0.30 for HCC over the paired non‐neoplastic tissue.
At the significance level of Bonferroni‐corrected p < 0.05, there were 1906 DEGs, with 827 genes up‐regulated and 1079 genes down‐regulated (Table S2). Among the up‐regulated genes, the average fold change was 2.86 for HCC over the paired non‐neoplastic tissue. Among the down‐regulated genes, the average fold change was 0.14 for HCC over the paired non‐neoplastic tissue.
Based on differential expression analysis at the KEGG pathway level, 83 KEGG pathways were identified to be up‐regulated at FDR < 0.10 and two KEGG pathways were identified to be down‐regulated at FDR < 0.10 (Table S3). Among the top up‐regulated pathways were ribosome (hsa03010) (p = 4.73e‐25; FDR, 7.71e‐23), protein processing in endoplasmic reticulum (hsa04141) (p = 3.64e‐19; FDR, 2.97e‐17), oxidative phosphorylation (hsa00190) (p = 2.06e‐17; FDR, 1.12e‐15), and cell cycle (hsa04110) (p = 4.85e‐13; FDR, 1.98e‐11). Other functionally important pathways that were up‐regulated included DNA damage‐related pathways (e.g., base excision repair [hsa03410] [p = 3.49e‐5; FDR, 2.71e‐4], mismatch repair [hsa03430] [p = 7.65e‐5; FDR, 5.25e‐4], DNA replication [hsa03030] [p = 1.32e‐6; FDR, 1.53e‐5], and p53 signaling pathway [hsa04115] [p = 2.32e‐4; FDR, 1.26e‐3]). The two down‐regulated KEGG pathways (FDR, <0.10) were olfactory transduction (hsa04740) (p = 9.39e‐10; FDR, 1.53e‐7) and calcium signaling pathways (hsa04020) (p = 2.74e‐4; FDR, 0.02). Information on these pathways is shown in Table 2. Additionally, the up‐regulated oxidative phosphorylation pathway and down‐regulated olfactory transduction pathway are illustrated in KEGG plots in Figure 1A,B, respectively.
TABLE 2.
KEGG pathway analysis results
| KEGG pathway names | Direction of regulation | p value | FDR |
|---|---|---|---|
| hsa03010 Ribosome | up‐regulation | 4.73E‐25 | 7.71E‐23 |
| hsa04141 Protein processing in endoplasmic reticulum | up‐regulation | 3.64E‐19 | 2.97E‐17 |
| hsa00190 Oxidative phosphorylation | up‐regulation | 2.06E‐17 | 1.12E‐15 |
| hsa04110 Cell cycle | up‐regulation | 4.85E‐13 | 1.98E‐11 |
| hsa03030 DNA replication | up‐regulation | 1.32E‐06 | 1.53E‐05 |
| hsa03420 Nucleotide excision repair | up‐regulation | 1.33E‐05 | 1.14E‐04 |
| hsa03410 Base excision repair | up‐regulation | 3.49E‐05 | 2.71E‐04 |
| hsa03430 Mismatch repair | up‐regulation | 7.65E‐05 | 5.25E‐04 |
| hsa04115 p53 signaling pathway | up‐regulation | 2.32E‐04 | 1.26E‐03 |
| hsa04740 Olfactory transduction | down‐regulation | 9.39E‐10 | 1.53E‐07 |
| hsa04020 Calcium signaling pathway | down‐regulation | 2.84E‐04 | 2.31E‐02 |
Abbreviations: FDR, false discovery rate; KEGG, Kyoto Encyclopedia of Genes and Genomes.
FIGURE 1.
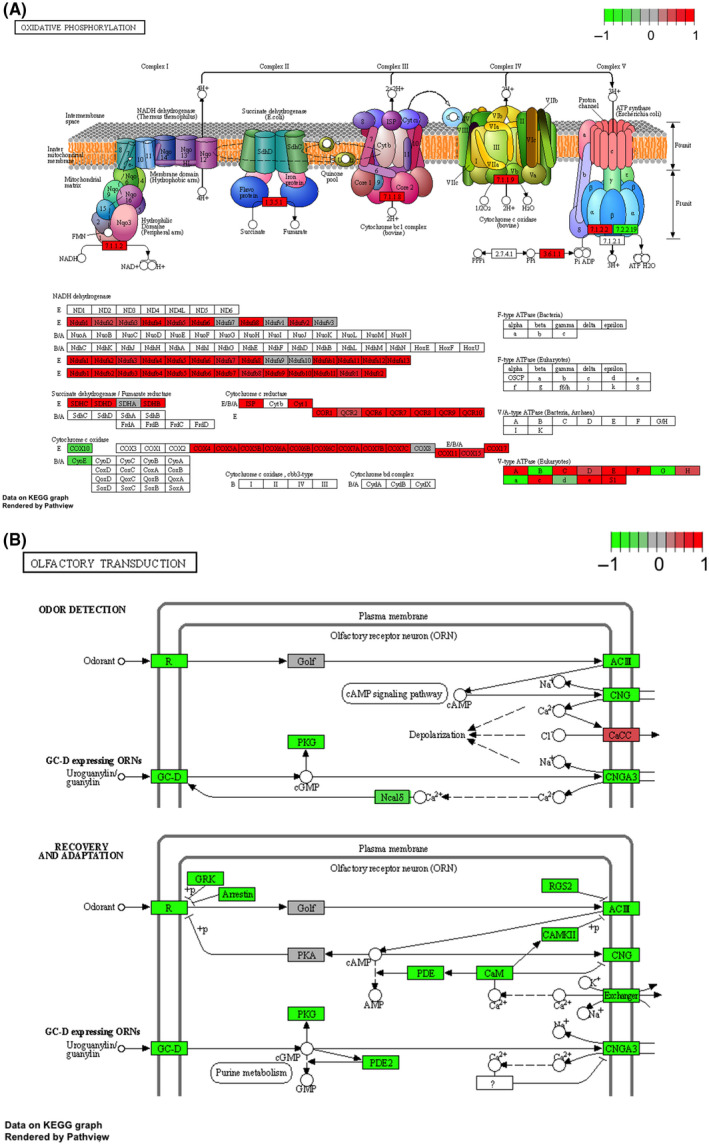
KEGG plots. (A) Up‐regulated oxidative phosphorylation pathway. (B) Down‐regulated olfactory transduction pathway. Up‐ or down‐regulation of genes in HCC versus non‐neoplastic tissue are illustrated with red or green colors, respectively. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; ATPase, adenosine triphosphatase; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; E. coli, Escherichia coli; FMN, flavin mononucleotide; GC‐D, guanylyl cyclase‐D; GRK, G protein‐coupled receptor kinase; KEGG, Kyoto Encyclopedia of Genes and Genomes; NAD+, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; R, receptor
Many genes within the pathways have strong differential expression, which makes the colors of the genes appear to be monotonically red or green with little in‐between dynamic range, as shown in the color scale bar of Figure 1. In the oxidative phosphorylation pathway (Figure 1A), many structural components of the enzyme system, such as reduced nicotinamide adenine dinucleotide (NADH) dehydrogenase, are shown in different colors, which is for better illustration of molecular construction of the enzyme. However, when reading the plot, focus should be placed on the small rectangles with gene symbols inside (the “gene block”), which are the component genes of a pathway.
The transparent gene blocks (those without color) are those genes that were not detected by RNA‐seq analysis (Figure 1). The gene block with colors (red, green, gray, or gray red/green) are those genes that were detected by RNA‐seq analysis. Depending on the color, a gene block may be shown as strongly up‐regulated (red), strongly down‐regulated (green), moderately up‐regulated (gray red), moderately down‐regulated (gray green) or weakly changed/unchanged in expression (gray).
As there are a vast number of olfactory receptor (OR) genes, it is impossible to visualize them individually in a plot showing the overall structure of the pathway. Therefore, for the olfactory transduction pathway (Figure 1B), all the detected genes belonging to ORs map to the receptor (R) block, and their average differential expression is illustrated with appropriate colors. Here in our study, most of the OR genes were down‐regulated. Therefore, the OR gene block (R block) is colored green.
Enrichment analysis of DEGs
DAVID enrichment analysis of the top 3000 up‐regulated genes (with FDR < 0.10) identified more than 250 enriched pathways, gene sets, or functional terms (FDR, <0.10) (Table 3; Table S4). Compared with the GAGE analysis results (Table 2), similar pathways or functional terms were identified. For example, the same KEGG pathway, oxidative phosphorylation (hsa00190), achieved an enrichment FDR of 1.33E‐24 (Table 3). Additionally, multiple functional terms related to DNA damage and repair, such as GO:0042769~DNA damage response and GO:0006283~transcription‐coupled nucleotide‐excision repair, achieved enrichment FDR values of 8.42E‐07 and 7.77E‐05, respectively (Table 3). Also shown enriched were several KEGG pathways related to human diseases, including nonalcoholic fatty liver disease (NAFLD) (hsa04932) (FDR, 6.34E‐12), Parkinson's disease (hsa05012) (FDR, 8.73E‐18), Huntington's disease (hsa05016) (FDR, 3.91E‐15), Alzheimer's disease (hsa05010) (FDR, 1.59E‐14), and functional terms related to mitochondrion, including UP_keywords mitochondrion (FDR, 4.06E‐77), mitochondrion inner membrane (FDR, 1.13E‐36), and respiratory chain (FDR, 7.61E‐19).
TABLE 3.
DAVID enrichment analysis results on up‐regulated genes
| Category | Term | p value | Fold Enrichment | FDR |
|---|---|---|---|---|
| UP_KEYWORDS | Mitochondrion | 1.75E‐79 | 2.55 | 4.06E‐77 |
| UP_KEYWORDS | Mitochondrion inner membrane | 1.22E‐38 | 3.35 | 1.13E‐36 |
| KEGG_PATHWAY | hsa00190:Oxidative phosphorylation | 1.06E‐26 | 3.30 | 1.33E‐24 |
| UP_KEYWORDS | Respiratory chain | 1.64E‐20 | 4.75 | 7.61E‐19 |
| KEGG_PATHWAY | hsa04932:Nonalcoholic fatty liver disease | 2.01E‐13 | 2.44 | 6.34E‐12 |
| KEGG_PATHWAY | hsa05012:Parkinson's disease | 1.04E‐19 | 2.86 | 8.73E‐18 |
| KEGG_PATHWAY | hsa05016:Huntington's disease | 6.20E‐17 | 2.43 | 3.91E‐15 |
| KEGG_PATHWAY | hsa05010:Alzheimer's disease | 3.15E‐16 | 2.52 | 1.59E‐14 |
| UP_KEYWORDS | Ribosomal protein | 7.31E‐71 | 5.12 | 1.13E‐68 |
| UP_KEYWORDS | Cell cycle | 8.70E‐13 | 1.74 | 2.12E‐11 |
| UP_KEYWORDS | Cell division | 1.10E‐09 | 1.84 | 1.96E‐08 |
| UP_KEYWORDS | DNA repair | 7.52E‐06 | 1.70 | 7.40E‐05 |
| UP_KEYWORDS | DNA damage | 1.15E‐05 | 1.62 | 1.11E‐04 |
| GOTERM_BP_DIRECT | GO:0042769~DNA damage response, detection of DNA damage | 4.19E‐09 | 3.97 | 8.42E‐07 |
| GOTERM_BP_DIRECT | GO:0006283~transcription‐coupled nucleotide‐excision repair | 5.80E‐07 | 2.63 | 7.77E‐05 |
| KEGG_PATHWAY | hsa03430:Mismatch repair | 1.09E‐03 | 2.87 | 1.31E‐02 |
| GOTERM_BP_DIRECT | GO:0000715~nucleotide‐excision repair, DNA damage recognition | 4.40E‐05 | 3.67 | 4.08E‐03 |
| UP_KEYWORDS | Antioxidant | 2.13E‐05 | 5.09 | 1.93E‐04 |
| GOTERM_BP_DIRECT | GO:0000302~response to reactive oxygen species | 3.91E‐04 | 2.67 | 2.51E‐02 |
| GOTERM_BP_DIRECT | GO:0098869~cellular oxidant detoxification | 2.31E‐05 | 2.41 | 2.32E‐03 |
Abbreviations: DAVID, Database for Annotation, Visualization, and Integrated Discovery; FDR, false discovery rate; GOTERM, Gene Ontology term; KEGG, Kyoto Encyclopedia of Genes and Genomes.
DAVID enrichment analysis of the top 3000 down‐regulated genes (with FDR < 0.10) identified more than 350 enriched gene sets or functional terms (enrichment FDR, <0.10) (Table 4; Table S5). Key enriched functional terms included cell membrane (UP_keywords: cell membrane, FDR of 1.68E‐37) and cell junction (UP_keywords: cell junction, FDR of 1.99E‐12). Other enriched functional terms were those related to immune response, such as GO:0003823~antigen binding (FDR, 7.91E‐17), GO:0050776~regulation of immune response (FDR, 4.32E‐15), GO:0006898~receptor‐mediated endocytosis (FDR, 2.78E‐12), and GO:0050853~B cell receptor signaling pathway (FDR, 1.88E‐11).
TABLE 4.
DAVID enrichment analysis results on down‐regulated genes
| Category | Term | p value | Fold Enrichment | FDR |
|---|---|---|---|---|
| UP_KEYWORDS | Cell membrane | 1.45E‐39 | 1.60 | 1.68E‐37 |
| GOTERM_CC_DIRECT | GO:0005886~plasma membrane | 2.66E‐39 | 1.48 | 1.80E‐36 |
| UP_KEYWORDS | Cell junction | 7.35E‐14 | 1.82 | 1.99E‐12 |
| UP_KEYWORDS | Immunoglobulin domain | 1.34E‐52 | 3.02 | 6.19E‐50 |
| GOTERM_MF_DIRECT | GO:0003823~antigen binding | 5.12E‐20 | 3.93 | 7.91E‐17 |
| GOTERM_BP_DIRECT | GO:0050776~regulation of immune response | 2.78E‐18 | 2.98 | 4.32E‐15 |
| GOTERM_BP_DIRECT | GO:0006898~receptor‐mediated endocytosis | 3.42E‐15 | 2.73 | 2.78E‐12 |
| GOTERM_BP_DIRECT | GO:0006956~complement activation | 3.60E‐15 | 3.70 | 2.78E‐12 |
| GOTERM_BP_DIRECT | GO:0050853~B‐cell receptor signaling pathway | 3.13E‐14 | 4.43 | 1.88E‐11 |
| GOTERM_MF_DIRECT | GO:0034987~immunoglobulin receptor binding | 8.89E‐10 | 5.29 | 4.20E‐07 |
| GOTERM_MF_DIRECT | GO:0004896~cytokine receptor activity | 3.86E‐06 | 3.61 | 5.96E‐04 |
Abbreviations: DAVID, Database for Annotation, Visualization, and Integrated Discovery; FDR, false discovery rate; GOTERM, Gene Ontology term; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Cibersort analysis on tumor‐infiltrating immune cells
Based on analysis, several types of immune cell proportions were different at significance (p < 0.05) or marginal significance level (p < 0.10) in HCC versus non‐neoplastic tissue, as shown in Table 5. Except for macrophages and resting mast cells, all other immune cells, including monocytes, naive and memory B cells, CD4 T cells, activated mast cells, and eosinophils, were observed to be decreased in HCC versus non‐neoplastic tissue. The proportions of these cells in each patient in HCC versus non‐neoplastic tissue are illustrated in Figure 2.
TABLE 5.
Cibersort analysis on tumor‐infiltrating immune cells in HCC versus non‐neoplastic tissue
| Cell type | Change of proportion in HCC versus non‐neoplastic tissue | p value |
|---|---|---|
| Monocyte | Decreased | 0.009 |
| Macrophages.M2 | Increased | 0.03 |
| Macrophages.M1 | Increased | 0.007 |
| Mast.cells.resting | Increased | 0.01 |
| B.cells.naive | Decreased | 0.03 |
| T.cells.CD4.naive | Decreased | 0.06 |
| B.cells.memory | Decreased | 0.005 |
| Mast.cells.activated | Decreased | 0.01 |
| Eosinophils | Decreased | 0.09 |
Abbreviation: HCC, hepatocellular carcinoma.
FIGURE 2.
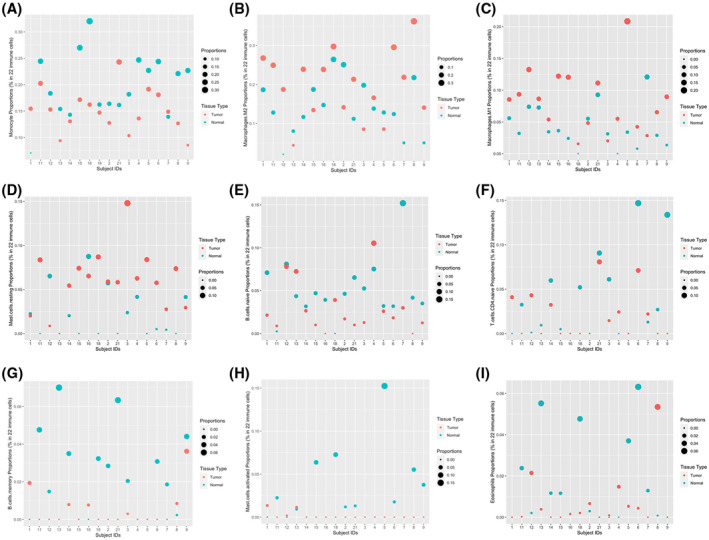
Comparison of proportions of infiltrating immune cells in HCC versus non‐cancerous tissue for each patient. (A) Monocyte proportions. (B) Macrophage.M2 proportions
DISCUSSION
Early transcriptomic studies have identified general mechanisms and some key signatures associated with HCC development. Most such studies used patients with diverse underlying liver diseases and specifically focused on patients with HBV and HCV infections.[ 16 , 17 , 18 , 19 ] Underlying chronic liver diseases may have a significant impact on the molecular genetic mechanisms leading to HCC development. From a clinical perspective, this may partially account for the variation in findings among transcriptomic studies where patients had different background liver diseases. Our study was performed on a US Caucasian cohort whose major etiologies for HCC were alcoholic and/or nonalcoholic fatty liver disease. The majority (13/17) of our study subjects had a history of alcohol usage (Table 1), and the up‐regulated genes from the subjects’ HCC versus non‐neoplastic tissue were enriched with the NAFLD pathway (hsa04932) (Table 3). In our cohort, only 6 out of 17 patients had a history of chronic hepatitis C that was under control by medication (Table 1), demonstrating that viral hepatitis was not the major contributing factor for HCC in this cohort of subjects.
Overall, we achieved highly significant results, as manifested by >10,000 DEGs at FDR < 0.10 (Table S1). Even at a stringent and overconservative significance level (i.e., Bonferroni‐corrected p < 0.0) >1900 genes were observed to be differentially expressed. At levels of pathways, gene sets, or functional terms, a total of 85 KEGG pathways were differentially expressed at FDR < 0.10 (Table S3), more than 250 gene sets or functional terms were observed to be enriched in the up‐regulated genes (FDR, <0.10) (Table S4), and more than 350 gene sets or functional terms were observed to be enriched in the down‐regulated genes (FDR, <0.10) (Table S5). Such a large number of significant DEGs, pathways, gene sets and functional terms indicate a strikingly distinctive transcriptomics landscape of HCC compared to the neighboring non‐neoplastic tissue.
Our major findings have not been reported in previous transcriptomics analyses of HCC. One of the key signals is the up‐regulation of the oxidative phosphorylation pathway (hsa00190) in HCC versus non‐neoplastic tissue (Table 2; Figure 1); this was identified in both GAGE analyses (FDR, 1.12E‐15; Table 2) and DAVID enrichment analysis of the top 3000 up‐regulated genes (FDR, 1.33E‐24; Table 3) with highly significant FDR values. Oxidative phosphorylation is an important biochemical process generating energy in the form of adenosine triphosphate. While essential to energy metabolism, oxidative phosphorylation produces reactive oxygen species (ROS) (e.g., superoxide and hydrogen peroxide), which may damage key cellular components, such as DNA, proteins, and lipids. Excessive damage caused by ROS leads to oxidative stress. Consistent with such damages, several up‐regulated functional terms related to DNA damage have been detected (Table 2); these include nucleotide excision repair (hsa03420) (FDR, 1.14E‐04), base excision repair (hsa03410) (FDR, 2.71E‐04), mismatch repair (hsa03430) (FDR, 5.25E‐04), and the p53 signaling pathway (hsa04115) (FDR, 1.26E‐03), as identified in GAGE analysis. Such DNA damage signals were also detected in DAVID enrichment analysis of up‐regulated genes (Table 3), for example, GO:0042769~DNA damage response (FDR, 8.42E‐07), UP_keywords DNA damage (FDR, 1.11E‐04), and UP_keywords DNA repair (FDR, 7.40E‐05). Enrichment of up‐regulated genes was also found with several terms related to oxidative stress, such as antioxidant (FDR, 1.93E‐04), GO:0098869~cellular oxidant detoxification (FDR, 2.32E‐03), and GO:0000302~response to ROS (FDR, 2.51E‐02) (Table 3).
To date, limited data have been available for the importance of oxidative phosphorylation to HCC pathogenesis. A recent study of in silico analysis of The Cancer Genome Atlas data set has discovered that oxidative phosphorylation and ROS pathways were more enriched in a subset of patients with a low survival outcome than those with a better survival outcome,[ 30 ] which is consistent with our findings of increased oxidative phosphorylation in HCC versus in non‐neoplastic tissue. Activation of oxidative phosphorylation was also observed in HCC cells that were resistant to doxorubicin, a chemotherapy medication to treat cancer,[ 31 ] and in liver cancer stem cells (as compared to regular liver cancer cells), which suggests oxidative phosphorylation as a required condition for maintaining the stemness of liver cancer cells.[ 32 ] The above studies revealed oxidative phosphorylation as a potential therapeutic target for HCC. Our study is the first to report activated oxidative phosphorylation as a transcriptomic signature of HCC. Notably, studies have also shown that oxidative phosphorylation is up‐regulated in other types of malignancies, such as breast cancer[ 33 , 34 , 35 ] and classical Hodgkin lymphoma.[ 36 ] Moreover, Ras‐driven pancreatic ductal adenocarcinoma stem cells had a strong reliance on oxidative phosphorylation for survival and high sensitivity to oxidative phosphorylation inhibitors.[ 37 ]
The observed DNA damage signals in HCC liver tissue provide another important potential mechanism for liver carcinogenesis. DNA damage is a major mechanism underlying the development of human cancers because damaged DNA may be replicated before repair, giving rise to somatic mutations and altered proteins.[ 38 ] Notably in our study, in addition to DNA damage‐related pathways, cell‐cycle and DNA replication pathways were also up‐regulated in HCC versus non‐neoplastic tissue (Tables 2 and 3), which are in line with the findings from previous transcriptomic studies of HCC.[ 11 , 39 , 40 ] The increased cell cycle and DNA replication activities, possibly as a compensatory mechanism to replace and repair damaged cells, may further replicate and propagate the somatic mutations generated from DNA damage, which further enhances carcinogenesis.
Among the down‐regulated genes, there was an enrichment of functional terms related to cell membrane (FDR, 1.68E‐37), GO:0005886~plasma membrane (FDR, 1.80E‐36), and cell junction (FDR, 1.99E‐12) (Table 4), suggesting compromised formation of cellular structures in HCC at the transcriptome level. Significant enrichment of down‐regulated genes in HCC versus non‐neoplastic tissue was also found for functional terms related to immune response, such as immunoglobulin domain (FDR, 6.19E‐50), GO:0003823~antigen binding (FDR, 7.91E‐17), GO:0050776~regulation of immune response (FDR, 4.32E‐15), and GO:0050853~B‐cell receptor signaling pathway (FDR, 1.88E‐11) (Table 4). This may suggest an impaired immune environment inside the HCC tissue, which is supported by the Cibersort analysis result showing decreased proportions of multiple types of infiltrating immune cells, including monocytes, naive and memory B cells, CD4 T cells, activated mast cells, and eosinophils in HCC versus non‐cancerous tissue (Table 5; Figure 2). Other studies also found a relevance of the immune environment to HCC subclassification and prognosis.[ 41 , 42 ] For example, HCC subtypes with immunodeficient and immunosuppressive features were identified by clustering immune cells in the HCC microenvironment.[ 41 ]
As another novel finding, GAGE analysis identified olfactory transduction (hsa04740; FDR, 1.53E‐07) (Table 2; Figure 2) as the most significantly down‐regulated KEGG pathway in HCC versus normal tissue, which is also one of the only two significantly down‐regulated pathways at FDR < 0.10 (Table 2; Table S3). This finding is somewhat unexpected due to the liver tissue analyzed here. However, a recent study highlights the importance of ORs as part of chemosensory functions of the liver[ 43 ] and the important roles of receptors in hepatic homeostasis. In that study,[ 43 ] expression of genes for multiple ORs and signaling pathways was detected in the murine liver, and two ORs (Olfr177 and Olfr57) were shown to be able to respond to known ligands, suggesting their active roles as receptors. As shown in another study, hepatic Olfr43, an OR expressed in the liver, regulates hepatic lipid accumulation and adiposity through the cyclic adenosine monophosphate response element‐binding protein (CREB)–hes family bHLH transcription factor 1 (HES1)–peroxisome proliferator‐activated receptor gamma (PPARγ) signaling axis in mice.[ 44 ] In our study, a total of 13 OR genes were detected, the majority (9/13) of which were down‐regulated, and all the down‐regulated OR genes achieved an FDR < 0.10 according to DESeq analysis (Table S6). Importantly, the top three OR genes with the highest mean expression levels (OR52I1, OR7A5, OR9H1P) were all down‐regulated. Given the important roles of hepatic homeostasis as exerted by ORs,[ 44 , 45 , 46 ] the down‐regulation of multiple OR genes and the olfactory transduction pathway (hsa04740) in HCC versus non‐neoplastic tissue, as detected in our study, may represent another key potential mechanism for liver carcinogenesis.
In summary, this is an RNA‐seq study of HCC in a US Caucasian cohort with fatty liver disease as the main etiology. Through this study, several new potential mechanistic features of HCC were identified. These include activated oxidative phosphorylation, DNA damage, impaired immune environment, as well as a deteriorated liver chemosensory system. Our study provides important information and novel insights into the pathogenesis and potential therapeutic targets of HCC.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
Yongjun Liu and Yao‐Zhong Liu conceived the study, directed the research, analyzed and interpreted the data, and drafted, revised, and approved the manuscript. David Al‐Adra and Matthew Yeh provided help in interpreting data and editing the manuscript. All authors read and approved the final manuscript.
Supporting information
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
ACKNOWLEDGMENTS
This work was supported by Departmental Research and Development funding from the Department of Pathology at UW–Madison. We thank the UW Translational Science Biocore BioBank for providing the HCC patient samples. We are also thankful to the UW Translational Research Initiatives in Pathology laboratory, supported by the UW Department of Pathology and Laboratory Medicine, for its services. We thank Ms. Michelle Chi for editing the manuscript.
Liu Y, Al‐Adra DP, Lan R, Jung G, Li H, Yeh MM, et al. RNA sequencing analysis of hepatocellular carcinoma identified oxidative phosphorylation as a major pathologic feature. Hepatol Commun. 2022;6:2170–2181. 10.1002/hep4.1945
Funding information
Department of Pathology at the University of Wisconsin–Madison: Departmental research and development funds
Contributor Information
Yongjun Liu, Email: liu945@wisc.edu.
Yao‐Zhong Liu, Email: yliu8@tulane.edu.
DATA AVAILABILITY STATEMENT
The data sets are currently deposited in the NCBI GEO with accession number GSE184733.
REFERENCES
- 1. Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–16. [DOI] [PubMed] [Google Scholar]
- 2. El‐Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–27. [DOI] [PubMed] [Google Scholar]
- 3. Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGlynn KA, Petrick JL, El‐Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73(Suppl 1):4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwong AJ, Kim WR, Lake JR, Smith JM, Schladt DP, Skeans MA, et al. OPTN/SRTR 2019 annual data report: liver. Am J Transplant. 2021;21(Suppl 2):208–315. [DOI] [PubMed] [Google Scholar]
- 6. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. [DOI] [PubMed] [Google Scholar]
- 7. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. [DOI] [PubMed] [Google Scholar]
- 8. Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouzé E, Blanc J‐F, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol. 2017;67:727–38. [DOI] [PubMed] [Google Scholar]
- 9. Calderaro J, Ziol M, Paradis V, Zucman‐Rossi J. Molecular and histological correlations in liver cancer. J Hepatol. 2019;71:616–30. [DOI] [PubMed] [Google Scholar]
- 10. Zucman‐Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149:1226–39.e4. [DOI] [PubMed] [Google Scholar]
- 11. Maass T, Sfakianakis I, Staib F, Krupp M, Galle PR, Teufel A. Microarray‐based gene expression analysis of hepatocellular carcinoma. Curr Genomics. 2010;11:261–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu XR, Huang J, Xu ZG, Qian BZ, Zhu ZD, Yan Q, et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci U S A. 2001;98:15089–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nam SW, Park JY, Ramasamy A, Shevade S, Islam A, Long PM, et al. Molecular changes from dysplastic nodule to hepatocellular carcinoma through gene expression profiling. Hepatology. 2005;42:809–18. [DOI] [PubMed] [Google Scholar]
- 14. Iizuka N, Oka M, Yamada‐Okabe H, Mori N, Tamesa T, Okada T, et al. Comparison of gene expression profiles between hepatitis B virus‐ and hepatitis C virus‐infected hepatocellular carcinoma by oligonucleotide microarray data on the basis of a supervised learning method. Cancer Res. 2002;62:3939–44. [PubMed] [Google Scholar]
- 15. Shimada S, Mogushi K, Akiyama Y, Furuyama T, Watanabe S, Ogura T, et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine. 2019;40:457–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang Q, Lin B, Liu H, Ma XI, Mo F, Yu W, et al. RNA‐Seq analyses generate comprehensive transcriptomic landscape and reveal complex transcript patterns in hepatocellular carcinoma. PLoS One. 2011;6:e26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang YI, Pan J, Chen D, Zheng J, Qiu F, Li F, et al. Identification and functional analysis of differentially expressed genes in poorly differentiated hepatocellular carcinoma using RNA‐seq. Oncotarget. 2017;8:35973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okrah K, Tarighat S, Liu B, Koeppen H, Wagle MC, Cheng G, et al. Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol. 2018;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pan QI, Long X, Song L, Zhao D, Li X, Li D, et al. Transcriptome sequencing identified hub genes for hepatocellular carcinoma by weighted‐gene co‐expression analysis. Oncotarget. 2016;7:38487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cancer Genome Atlas Research Network . Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–41 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods. 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soneson C, Love MI, Robinson MD. Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Res. 2015;4:1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo W, Friedman MS, Shedden K, Hankenson KD, Woolf PJ. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway‐based data integration and visualization. Bioinformatics. 2013;29:1830–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang DW , Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 28. Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer‐Verlag; 2016. [Google Scholar]
- 30. Hoki T, Katsuta E, Yan L, Takabe K, Ito F. Low DMT1 expression associates with increased oxidative phosphorylation and early recurrence in hepatocellular carcinoma. J Surg Res. 2019;234:343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu LI, Zhao J, Cao K, Liu X, Cai H, Wang J, et al. Oxidative phosphorylation activation is an important characteristic of DOX resistance in hepatocellular carcinoma cells. Cell Commun Signal. 2018;16:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu G, Luo Q, Li H, Liu Q, Ju Y, Song G. Increased oxidative phosphorylation is required for stemness maintenance in liver cancer stem cells from hepatocellular carcinoma cell line HCCLM3 cells. Int J Mol Sci. 2020;21:5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitaker‐Menezes D, Martinez‐Outschoorn UE, Flomenberg N, Birbe R, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zacksenhaus E, Shrestha M, Liu JC, Vorobieva I, Chung PED, Ju Y, et al. Mitochondrial OXPHOS induced by RB1 deficiency in breast cancer: implications for anabolic metabolism, stemness, and metastasis. Trends Cancer. 2017;3:768–79. [DOI] [PubMed] [Google Scholar]
- 35. Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, et al. RB1 deficiency in triple‐negative breast cancer induces mitochondrial protein translation. J Clin Invest. 2016;126:3739–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Birkenmeier K, Dröse S, Wittig I, Winkelmann R, Käfer V, Döring C, et al. Hodgkin and Reed‐Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. Int J Cancer. 2016;138:2231–46. [DOI] [PubMed] [Google Scholar]
- 37. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sánchez N, Marchesini M, et al. Oncogene ablation‐resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Basu AK. DNA damage, mutagenesis and cancer. Int J Mol Sci. 2018;19:970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoshida Y, Moeini A, Alsinet C, Kojima K, Villanueva A. Gene signatures in the management of hepatocellular carcinoma. Semin Oncol. 2012;39:473–85. [DOI] [PubMed] [Google Scholar]
- 40. Woo HG, Park ES, Thorgeirsson SS, Kim YJ. Exploring genomic profiles of hepatocellular carcinoma. Mol Carcinog. 2011;50:235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang QI, Lou YU, Yang J, Wang J, Feng J, Zhao Y, et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut. 2019;68:2019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179:829–45 e20. [DOI] [PubMed] [Google Scholar]
- 43. Kurtz R, Steinberg LG, Betcher M, Fowler D, Shepard BD. The sensing liver: localization and ligands for hepatic murine olfactory and taste receptors. Front Physiol. 2020;11:574082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu C, Thach TT, Kim YJ, Lee SJ. Olfactory receptor 43 reduces hepatic lipid accumulation and adiposity in mice. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:489–99. [DOI] [PubMed] [Google Scholar]
- 45. Wu C, Hwang SH, Jia Y, Choi J, Kim Y‐J, Choi D, et al. Olfactory receptor 544 reduces adiposity by steering fuel preference toward fats. J Clin Invest. 2017;127:4118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu C, Jia Y, Lee JH, Kim Y, Sekharan S, Batista VS, et al. Activation of OR1A1 suppresses PPAR‐gamma expression by inducing HES‐1 in cultured hepatocytes. Int J Biochem Cell Biol. 2015;64:75–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Data Availability Statement
The data sets are currently deposited in the NCBI GEO with accession number GSE184733.