

Abstract
Bacterial diversity in unimproved and improved grassland soils was assessed by PCR amplification of bacterial 16S ribosomal DNA (rDNA) from directly extracted soil DNA, followed by sequencing of ∼45 16S rDNA clones from each of three unimproved and three improved grassland samples (A. E. McCaig, L. A. Glover, and J. I. Prosser, Appl. Environ. Microbiol. 65:1721–1730, 1999) or by denaturing gradient gel electrophoresis (DGGE) of total amplification products. Semi-improved grassland soils were analyzed only by DGGE. No differences between communities were detected by calculation of diversity indices and similarity coefficients for clone data (possibly due to poor coverage). Differences were not observed between the diversities of individual unimproved and improved grassland DGGE profiles, although considerable spatial variation was observed among triplicate samples. Semi-improved grassland samples, however, were less diverse than the other grassland samples and had much lower within-group variation. DGGE banding profiles obtained from triplicate samples pooled prior to analysis indicated that there was less evenness in improved soils, suggesting that selection for specific bacterial groups occurred. Analysis of DGGE profiles by canonical variate analysis but not by principal-coordinate analysis, using unweighted data (considering only the presence and absence of bands) and weighted data (considering the relative intensity of each band), demonstrated that there were clear differences between grasslands, and the results were not affected by weighting of data. This study demonstrated that quantitative analysis of data obtained by community profiling methods, such as DGGE, can reveal differences between complex microbial communities.
Bacteria play a central role in the rhizosphere, which is a complex and dynamic environment that varies temporally, spatially, and with different agricultural practices that are likely to influence the bacterial community. However, the relationships among nutrient cycling, plant physiology, plant diversity, and bacterial community structure are not well understood. Molecular analysis of bacterial diversity in terrestrial ecosystems (4, 12, 27, 39) most frequently involves retrieval of 16S rRNA gene sequences by PCR amplification of extracted and purified nucleic acids, using broad-range or group-specific primer sets, along with subsequent analysis by cloning and characterization of clones by sequencing (3, 17, 22) or restriction fragment length polymorphism analysis (15, 21, 39). Alternatively, fingerprinting of total PCR products may be carried out by using, for example, amplified ribosomal DNA (rDNA) restriction analysis (28, 34), length heterogeneity PCR (30), single-strand conformation polymorphism (19, 31), and terminal restriction fragment length polymorphism (20, 37). The most frequently used community fingerprinting methods are denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (11, 13, 16, 26), which separate sequences on the basis of differences in denaturing properties, and hence migration distances, in chemical and temperature gradients, respectively. Fingerprinting methods allow more rapid comparison of samples and are generally used to detect shifts in populations over time and/or under different environmental conditions.
The cloning approach has provided lists of sequence percentages or restriction fragment length polymorphism classes, along with their relative amounts in libraries. Quantification of data recovered in rDNA libraries is limited by the restricted number of clones that can feasibly be screened, but data have been used to calculate indices of diversity (21, 22). In contrast, fingerprinting techniques are more amenable to quantification; for example, they can be used to compare the presence and relative intensities of individual bands in DGGE gels and to calculate changes in their relative intensities (24, 36), to calculate diversity indices (9, 14), and to perform cluster analysis of banding patterns (8, 10). With both of these approaches, however, care must be taken in relating findings to in situ community structure, as accurate quantification may be impaired by biases introduced during DNA extraction, PCR, or cloning.
Bacterial populations in grassland soils at Sourhope, Scotland, have been characterized by 16S rRNA gene sequence analysis of isolated cultures and analysis of 16S rDNA clone libraries obtained from DNA extracted from soil (22, 23). The aims of this study were to compare cloning and fingerprinting approaches and to exploit the potential for greater replication and quantification provided by DGGE analysis to assess differences between bacterial communities in these soils. Three grassland types were compared by using DGGE, while unimproved and improved soils were compared by cloning.
MATERIALS AND METHODS
Soil samples from three characteristic grassland types, designated unimproved, semi-improved, and improved, were collected from Sourhope Research Station in the Borders Region, Scotland, as part of the Scottish Executive Rural Affairs Department MICRONET program (http://www.scri.sari.ac.uk/MICRONET/Default.html). The unimproved site was classified as a Festuca ovina-Agrostis capillaris-Galium saxatile grassland, while the semi-improved grassland also had a Holcus lanatus-Trifolium repens subcommunity. Neither of these grasslands had received fertilizer treatments, and both had been grazed by sheep throughout the year. The improved grassland, classified as a Lolium perenne-Cynosurus cristatus grassland, was fertilized three times per year and had also been grazed by sheep. The improved grassland was originally unimproved grassland that was cultivated and seeded with a L. perenne-T. repens mixture in 1982. The soil physicochemical conditions, a detailed vegetation analysis, and sampling of this site have been reported elsewhere (6, 22).
Total soil DNA was extracted by C. D. Clegg (Scottish Crop Research Institute, Invergowrie, United Kingdom) by freeze-thawing (5), and PCR amplification of 16S rRNA genes for cloning and sequence analysis was carried out with primers Bf and 1390r, as described by McCaig et al. (22). Products for DGGE analysis were amplified with primers p3 and p2 (25), which amplify a 194-bp fragment of the 16S rRNA gene, including the variable V3 region, and include a 40-bp GC clamp at the 5′ end of p3. Amplification reactions were performed as described previously for primers Bf and 1390r, except that Taq polymerase was obtained from Bioline, London, United Kingdom, and the cycling parameters for amplification were as follows: 95°C for 5 min, followed by 10 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s, 25 cycles of 92°C for 30 s (95°C is not necessary to denature ∼200-bp products and the lower temperature preserves enzyme activity), 55°C for 30 s, and 72°C for 45 s, and a final incubation at 72°C for 10 min. Construction of clone libraries and analysis of 275 16S rDNA clones were performed as described by McCaig et al. (22). Clone data were used to calculate richness, the Shannon diversity index, evenness, and dominance (22), and clones with >97% sequence similarity were clustered into operational taxonomic units (OTUs).
Products obtained with the DGGE primers were purified by adding 10 μl of phenol and 10 μl of chloroform-isoamyl alcohol (24:1); to remove bovine serum albumin, the tubes were briefly vortexed and centrifuged at 10,000 × g for 5 min, and aqueous layers were transferred into clean tubes. DGGE analysis was carried out with the DCode universal mutation detection system (Bio-Rad). Polyacrylamide gels (8% Acrylogel 2.6 solution; BDH Laboratory Supplies, Poole, United Kingdom) with a 40% (2.8 M urea–16% [vol/vol] formamide) to 60% (4.2 M urea–24% [vol/vol] formamide) vertical denaturing gradient were poured by using a gradient former (Fisher Scientific UK, Loughborough, United Kingdom) and a peristaltic pump (5 ml min−1). Gels were poured onto the hydrophilic side of Gelbond PAG film (FMC BioProducts, Rockland, Maine) that was hydrophobically bonded to the small glass plate, in order to facilitate handling of gels during staining procedures. Approximately 50 ng of each PCR product was loaded, and the gels were electrophoresed for 16 h at 75 V and 60°C. The gels were fixed overnight (10% ethanol, 0.5% glacial acetic acid, 89.5% H2O) prior to silver staining and were then incubated with shaking in freshly prepared staining solution (0.2% [wt/vol]) silver nitrate) for 20 min; this was followed by incubation in fresh developing solution (0.1 mg of sodium borohydride ml−1 in 1.5% [wt/vol] NaOH–0.4% [vol/vol] formaldehyde) until bands appeared. The gels were then fixed for 10 min in 0.75% (wt/vol) Na2CO3, preserved in ethanol-glycerol preservative (25% ethanol, 10% glycerol, 65% H2O) for at least 15 min, and stored in sealed plastic bags. The gels were scanned (GT-9600 scanner; Epson UK, Hemel Hempstead, United Kingdom) by using Presto! PageManager for Epson software (version 4.00.01; NewSoft Technology Corp., Fremont, Calif.). Phoretix one-dimensional gel analysis software (version 4.00; Phoretix International, Newcastle upon Tyne, United Kingdom) was used to determine the intensity and relative position of each band compared to a composite lane, created by the software, of all sample lanes. To correct for variations in DNA loading between lanes, the total band intensity for each lane was normalized to that of the lane with the lowest DNA loading. The faintest band on the gel prior to this normalization was assumed to be at the limit of detection, and all bands below this band were ignored. The intensity of each band was then calculated by determining the proportion of the total band intensity in a particular lane, and the resulting normalized data (weighted data), along with a simple binary matrix describing the presence and absence of bands at each position (unweighted data), were used in subsequent analyses.
DGGE banding data were used to estimate the four diversity indices calculated from the cloning data by treating each band as an individual OTU and using the number of bands as an indicator of richness. The Shannon diversity index, evenness, and dominance (29, 32, 33) were calculated from the number of bands present and the relative intensities of the bands in each lane. Similarity coefficients for pairwise comparisons of DGGE gel lanes were calculated from both unweighted and weighted data. The unweighted data were treated in two ways. First, a band-matching coefficient was calculated by using the approach described above for the clone data, in order to allow direct comparison of the strategies. The similarity matrix obtained was designated unweighted matrix 1 (UM1). In contrast to this approach, in which similarity was assessed on the basis of matching only OTUs, a second approach (unweighted matrix 2 [UM2]) was adopted, in which the presence or the absence of bands at the same position in two lanes was considered a band match. In this case, therefore, similarity values were calculated by SAB = MAB/N, where MAB is the number of matches (i.e., the number of bands present or absent in both lane A and lane B for each possible band position) and N is the number of band positions (i.e., the number of bands in the composite lane). Using the intensity data, each band was weighted according to the magnitude of the difference in the relative intensities of the bands at the same position in two lanes. This procedure was carried out by using positions where a match was assigned if one or both lanes contained a band (weighted matrix 1 [WM1]) and where absence in both lanes was also considered a match (weighted matrix 2 [WM2]). Paired band weights (WABi) were calculated by:
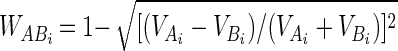 |
where VAi and VBi are the relative intensities of the ith bands in samples A and B, respectively, for positions where VAi − VBi ≠ 0. When VAi = VBi, then WABi = 0 for WM1 and WABi = 1 for WM2. Similarity was then calculated by:
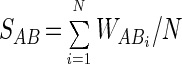 |
For WM1, N is the number of positions at which a band occurs in one or both lanes (i.e., analogous to UM1). For WM2, N is the total number of band positions (i.e., analogous to UM2). Principal-coordinate analysis was carried out for UM1, UM2, WM1, and WM2 by using Genstat for Windows, 4th ed. (The Numerical Algorithms Group Ltd., Oxford, United Kingdom). Multivariate analysis was also carried out directly with the original unweighted and weighted data by first reducing the data to six principal components and then performing canonical variate analysis (CVA) using Genstat for Windows. Sample groupings were specified prior to the CVA; i.e., for this study data were grouped as unimproved, semi-improved, and improved. CVA finds linear combinations of variates that maximize the ratio of between-group variation to within-group variation. In order to test the validity of this approach, data were randomly grouped into three sets of triplicates prior to CVA.
RESULTS
Samples from triplicate unimproved and improved grassland plots were compared directly by sequencing 16S rDNA clones or by DGGE, and an additional three samples from an intermediate, semi-improved grassland were analyzed only by DGGE. Grassland-specific patterns could not be detected by visual comparison of DGGE profiles (Fig. 1), although specific patterns may have been masked by small variations between replicate samples that were evident, particularly in the unimproved grassland sample lanes. The estimates of richness from the clone data (37.8 to 42.0 and 37.3 to 41.0 for unimproved and improved soils, respectively) were generally close to the number of clones sequenced (45 to 48) due to the low library coverage achieved (7 to 16%) (22) and were not significantly different for improved and unimproved grassland samples. Richness was assessed by determining the number of DGGE bands detected after correction for differences in DNA loading (the mean values were 44.7, 39.0, and 42.0 for unimproved, semi-improved, and improved plots, respectively) and was significantly greater in unimproved grassland samples than in semi-improved grassland samples (P = 0.055, as determined by Student's t test). After DGGE data were pooled, both unimproved and improved grassland samples showed greater richness than semi-improved grassland samples (77 bands for unimproved and improved grassland samples, 65 bands for semi-improved grassland samples). For all but one sample, the number of DGGE bands exceeded the number of clone types (37 to 42 clone types and 38 to 46 DGGE bands in unimproved and improved grassland samples), but there were more clone types after data were pooled (∼113 clone types, compared to 77 DGGE bands). This is probably explained by the limited resolution of DGGE and the probability that single bands from a complex bacterial PCR product comprise more than one OTU.
FIG. 1.

DGGE analysis of 16S rRNA genes amplified from DNA extracted from replicate samples (lanes 1, 2, and 3) of unimproved, semi-improved, and improved grassland soils using eubacterial primers (25).
The Shannon diversity index, evenness, and dominance values calculated by using either clone libraries or DGGE profiles did not differ significantly for the different grassland types. For both approaches, however, there was considerable variation within triplicate samples, making treatment differences difficult to detect. A slight decrease in evenness was observed for pooled DGGE data with improved grassland samples compared to unimproved grassland samples (0.890 to 0.879), which, given the similar values for richness for these grasslands, may have led to the slight decrease in the Shannon diversity index (1.68 to 1.66). Similarly, the dominance values for pooled DGGE data were slightly higher for improved grassland samples than for unimproved grassland samples (0.033 compared to 0.029), although both of the differences were small. The clone data showed a similar trend, although to a lesser extent. DGGE analysis of semi-improved grassland samples revealed a very different bacterial community structure than both unimproved and improved grasslands, as reflected by lower values for the Shannon diversity index (1.58, 1.66, and 1.68 for semi-improved, unimproved, and improved grassland samples, respectively), richness (65, 77, and 77, respectively), and evenness (0.869, 0.890, and 0.879, respectively) and a higher dominance value (0.038, 0.029, and 0.033, respectively). However, while semi-improved grassland samples had significantly lower richness values than unimproved grassland samples (P = 0.055), no other comparisons were statistically significant.
The similarity coefficients were considerably higher for DGGE profiles than for clone libraries and had an approximately 10-fold-greater range of values on average (0.53 compared to 0.06). This is because very few OTUs were found in more than one library due to the low coverage; thus, these data are probably not a true reflection of the similarity between grasslands. For DGGE of semi-improved grassland samples, the within-group similarity was greater than the similarity between semi-improved and improved grassland samples (0.58 compared to 0.53; P = 0.12, as determined by Student's t test. This reduced within-group variability may also be partially responsible for the lower Shannon diversity index, evenness, and richness values and greater dominance values for the semi-improved grassland samples compared to the values for the samples from the other two grasslands. The within-group similarities for the other grasslands, however, were comparable to the between-group similarities. This may have been due to a combination of low numbers of replicates and high spatial variation, and it is possible, therefore, that analysis of a higher number of replicate samples may allow better discrimination between grasslands. Analysis of similarity matrices prepared from weighted (WM1 and WM2) and unweighted (UM1 and UM2) DGGE data by principal-coordinate analysis (Fig. 2) did not distinguish the three grassland types; i.e., no grassland-dependent clustering was observed, although PC1 and PC2 represented only 17 to 18 and 15 to 16% of the variation, respectively. No clear pattern of clustering was observed, and in general, the position of points relative to each other remained consistent regardless of the type of analysis. A high degree of similarity between two of the semi-improved grassland samples, SH1 and SH2, was also seen throughout, reflecting the low within-group variation observed in this group. Dendrograms also showed that there was a high level of similarity between these two samples, but the relationship between other samples varied depending upon the algorithm used (data not shown), indicating that there was a lack of support for differences between grasslands. Neither weighting data nor the method of calculating similarity matrices affected the outcome of the analysis. Similarly, using the squares of the weights calculated for WM1 and WM2, thereby emphasizing large differences in intensity, did not affect the outcome of the analysis (data not shown).
FIG. 2.

Principal-coordinate analyses of similarity matrices produced from unweighted and weighted DGGE banding data from triplicate samples of three grassland soils. Calculation of matrices is described in Materials and Methods. (a) UM1; (b) UM2; (c) WM1; (d) WM2. PC1 represents 18.5, 18.2, 16.7, and 17.3% of the variation for panels a to d, respectively, and PC2 represents 16.0, 16.5, 15.2, and 15.1% of the variation for panels a to d, respectively. The consistency of clustering of samples SH1 and SH2, as described in the text, is indicated.
CVA of both original sets of data (i.e., binary matrix and intensity data) clearly separated the three grasslands (Fig. 3a and b, respectively), particularly for the unweighted data, and CV1 accounted for 99.9 and 96.5% of the variation. The band loadings indicated that many bands were cumulatively responsible for the separation of the groups and that, in general, different bands were responsible for the separation in the unweighted and weighted data. If this separation were due to the statistical approach used rather than to real differences between the sample types, then the clustering into three assigned groups would be expected regardless of how the data were grouped. Separation was not observed when the three assigned groups contained a single replicate from each grassland (Fig. 3c and d) but was observed when each group contained two replicates from one grassland and a single replicate from another grassland (Fig. 3e and f). In conclusion, therefore, CVA of the DGGE data did discriminate among the three grassland types.
FIG. 3.

Ordination of canonical variates (CV1 and CV2) produced from multivariate analysis of DGGE banding data from triplicate samples of three grassland soils. (a, c, and e) Analysis of an unweighted matrix; (b, d, and f) analysis of the corresponding weighted data. Panels a and b show the true grouping of the original data into unimproved, semi-improved, and improved soils, while panels c to f show analysis of data that were randomly grouped prior to analysis (see Materials and Methods). The points on all graphs indicate the grassland types, and the three random groups of data for each set are circled in panels a to d and f; in panel e only two sets of data are clearly marked. CV1 accounts for 99.9, 96.5, 73.9, 89.2, 94.0, and 98.2% of the variation in panels a to f, respectively, and CV2 accounts for 0.1, 3.5, 26.1, 10.8, 6.0, and 1.8% of the variation, respectively.
DISCUSSION
The hypothesis that was tested by comparison of the three vegetation types and management regimens was that high nutrient input and lower plant diversity in improved grasslands lead to a less diverse bacterial community than the community in unimproved grasslands, with the semi-improved grassland community being intermediate. Analysis of diversity indices and similarity coefficients for clone data indicated that there was no difference between unimproved and improved grasslands. While this finding can also be attributed to poor coverage of libraries (22), spatial variation of triplicate plots may have concealed differences between grasslands, and similar approaches have demonstrated that there are differences in ammonia oxidizer populations in sediment and soil (35) and in communities located at different distances from plant roots (21). Variation was also observed in DGGE profiles of triplicate samples from all three grassland types, but despite this, quantification of diversity and multivariate analysis of DGGE banding data did reveal differences in community structure among the three grassland types. Diversity, as calculated from pooled DGGE data, was lower in improved grassland samples than in unimproved grassland samples due to a decrease in evenness, possibly because of selection for particular bacteria. CVA also provided evidence that the communities were different, but the complexity of the DGGE data prevented identification of distinguishing bands. Similarity indices indicated that semi-improved grassland samples were more similar to improved grassland samples than to unimproved grassland samples, suggesting that there is some progression of bacterial communities during soil improvement. This hypothesis was supported by community DNA cross-hybridization analysis but not by CVA of the same samples (7), although the variation between DNA melting profiles was significant for semi-improved grassland replicates, while diversity and variability were considerably lower in the semi-improved grasslands than in the other two grasslands. DNA hybridization, however, analyzes both prokaryotic DNA and eukaryotic DNA.
The differences between sequence analysis of randomly selected clones and DGGE analysis may have been due to the use of different primer sets for amplifying products for cloning and DGGE, although the same region of the 16S rDNA (i.e., the V3 region) was compared in both approaches. While more than 100 clone types were obtained from each grassland, only 65 to 77 bands were detected by DGGE, demonstrating the lower resolution of this method. Furthermore, clones were grouped into OTUs when >97% similarity was observed, while DGGE can potentially separate sequences with only one base difference (i.e., >99% similarity). If the clone libraries in this study were also assessed at this stringent level, the number of OTUs rose to ∼130, emphasizing the restricted resolution of DGGE gels. As with cloning, only the more abundant sequences are generally detected by DGGE, although selection of low-abundance sequences by chance may occur with cloning, while DGGE is constrained by resolution and detection limits of staining. In addition, comigration of different sequences to the same gel position reduces the observed number of bands, and smears may comprise several bands. Nevertheless, DGGE analysis was more discriminatory and more rapid and is a relatively inexpensive method for providing broad qualitative and quantitative comparisons of large numbers of samples.
Although quantification of DGGE data is possible, care should be taken in interpreting results. Most analyses performed to date have been done on simple communities, including ammonia oxidizer communities, (24, 36), maize fermentations (1), wastewater reactors (18), relatively simple fermentation reactors, and activated sludge (1, 2, 9). Analysis of more complex soil communities is less common, although cluster analyses have been carried out by construction of similarity matrices (generally unweighted), followed by construction of dendrograms (8, 10, 14). This is comparable to the principal-coordinate analyses performed in this study, but the output is presented in a different format. In our study, principal-coordinate analysis did not discriminate between populations in different grasslands. Similarly, while Juck et al. (14) observed discrimination between soils from different geographical locations, unpolluted and polluted soils from the same location could not be separated. In contrast, Duineveld et al. (8) used cluster analysis to distinguish between 16S rRNA and rDNA profiles and between chrysanthemum root tip and root base rDNA populations at four different time points. Correspondence analysis was also used by Yang and Crowley (38) to demonstrate the effect of plant iron nutrient status on the bacterial community in the barley rhizosphere. Variation was reduced in both rhizosphere experiments described above through the use of mesocosms containing homogenized soil and uniform growth conditions, thus emphasizing any treatment effect, and the great spatial variation observed in our replicates may have masked changes due to differences in agricultural practice. No difference was observed between analysis results for similarity matrices when weighted or unweighted data were used, possibly due to the high degree of evenness for all samples, which resulted in approximately equal weights for all bands. CVA was more sensitive and discriminated among the populations in the three grassland types despite spatial variation, and interestingly, greater separation of populations was observed when the unweighted data were used. While the other methods reduced the complex banding patterns to small, relatively simple numbers, representing either a single lane (i.e., diversity indices) or a comparison between two lanes (i.e., similarity coefficients), this more sophisticated approach looked at the overall patterns of variation across all of the data, determining the influence of individual bands in the separation of samples, and was able to detect subtle differences which the other methods could not detect. Additionally, CVA is a subjective statistical method that is designed to maximize between-group differences, although in this study randomization of DGGE data demonstrated that statistical separation of groups did represent inherent differences between profiles for the three grasslands. Multivariate approaches, therefore, may be useful in future studies in which complex patterns are produced and in which subtle changes in community structure are expected. In conclusion, quantification and multivariate analysis of DGGE banding patterns enabled distinction between bacterial communities from three grassland types, whereas cloning and sequence analysis could not do this. Sequence analysis was limited to ∼137 clones for each grassland type, and it is likely that discrimination would have been achieved if higher numbers of clones had been screened. DGGE, however, has a significantly greater capacity for routine and rapid analysis of multiple samples and, in combination with other environmental data, provides a basis for more comprehensive ecological studies.
ACKNOWLEDGMENTS
We thank C. D. Campbell (Macaulay Land Use Research Institute, Aberdeen, United Kingdom) and D. A. Elston (Biomathematics and Statistics Scotland, Macaulay Land Use Research Institute) for assistance with statistical analysis of DGGE data.
This work was carried out as part of the MICRONET project funded by the Scottish Executive Rural Affairs Department (SERAD).
REFERENCES
- 1.Ampe F, Miambi E. Cluster analysis, richness and biodiversity indexes derived from denaturing gradient gel electrophoresis fingerprints of bacterial communities demonstrate that traditional maize fermentations are driven by the transformation process. Int J Food Microbiol. 2000;60:91–97. doi: 10.1016/s0168-1605(00)00358-5. [DOI] [PubMed] [Google Scholar]
- 2.ben Omar N, Ampe F. Microbial community dynamics during production of the Mexican fermented maize dough pozol. Appl Environ Microbiol. 2000;66:3664–3673. doi: 10.1128/aem.66.9.3664-3673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borneman J, Skroch P W, O'Sullivan K M, Palus J A, Rumjanek N G, Jansen J L, Nienhuis J, Triplett E W. Molecular microbial diversity of an agricultural soil in Wisconsin. Appl Environ Microbiol. 1996;62:1935–1943. doi: 10.1128/aem.62.6.1935-1943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borneman J, Triplett E W. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl Environ Microbiol. 1997;63:2647–2653. doi: 10.1128/aem.63.7.2647-2653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clegg C D, Ritz K, Griffiths B S. Direct extraction of microbial community DNA from humified upland soils. Lett Appl Microbiol. 1997;25:30–33. doi: 10.1046/j.1472-765x.1997.00166.x. [DOI] [PubMed] [Google Scholar]
- 6.Clegg C D, Ritz K, Griffiths B S. Broad-scale analysis of soil microbial community DNA from upland grasslands. Antonie van Leeuwenhoek Int. J Gen Mol Microbiol. 1998;73:9–14. doi: 10.1023/a:1000545804190. [DOI] [PubMed] [Google Scholar]
- 7.Clegg C D, Ritz K, Griffiths B S. %G+C profiling and cross hybridisation of microbial DNA reveals great variation in below-ground community structure in UK upland grasslands. Appl Soil Ecol. 2000;14:125–134. [Google Scholar]
- 8.Duineveld B M, Kowalchuk G A, Keijzer A, van Elsas J D, van Veen J A. Analysis of bacterial communities in the rhizosphere of chrysanthemum via denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA as well as DNA fragments coding for 16S rRNA. Appl Environ Microbiol. 2001;67:172–178. doi: 10.1128/AEM.67.1.172-178.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichner C A, Erb R W, Timmis K N, Wagner-Dobler I. Thermal gradient gel electrophoresis analysis of bioprotection from pollutant shocks in the activated sludge microbial community. Appl Environ Microbiol. 1999;65:102–109. doi: 10.1128/aem.65.1.102-109.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Fantroussi S, Verschuere L, Verstraete W, Top E M. Effect of phenylurea herbicides on soil microbial communities estimated by analysis of 16S rRNA gene fingerprints and community-level physiological profiles. Appl Environ Microbiol. 1999;65:982–988. doi: 10.1128/aem.65.3.982-988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Felske A, Akkermans A D L, De Vos W M. Quantification of 16S rRNAs in complex bacterial communities by multiple competitive reverse transcription-PCR in temperature gradient gel electrophoresis fingerprints. Appl Environ Microbiol. 1998;64:4581–4587. doi: 10.1128/aem.64.11.4581-4587.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felske A, Wolterink A, Van Lis R, Akkermans A D L. Phylogeny of the main bacterial 16S rRNA sequences in Drentse A grassland soils (The Netherlands) Appl Environ Microbiol. 1998;64:871–879. doi: 10.1128/aem.64.3.871-879.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heuer H, Krsek M, Baker P, Smalla K, Wellington E M H. Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol. 1997;63:3233–3241. doi: 10.1128/aem.63.8.3233-3241.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Juck D, Charles T, Whyte L G, Greer C W. Polyphasic microbial community analysis of petroleum hydrocarbon-contaminated soils from two northern Canadian communities. FEMS Microbiol Ecol. 2000;33:241–249. doi: 10.1111/j.1574-6941.2000.tb00746.x. [DOI] [PubMed] [Google Scholar]
- 15.Jurgens G, Saano A. Diversity of soil Archaea in boreal forest before and after clear-cutting and prescribed burning. FEMS Microbiol Ecol. 1999;29:205–213. [Google Scholar]
- 16.Kowalchuk G A, Stephen J R, DeBoer W, Prosser J I, Embley T M, Woldendorp J W. Analysis of ammonia-oxidizing bacteria of the beta subdivision of the class Proteobacteria in coastal sand dunes by denaturing gradient gel electrophoresis and sequencing of PCR-amplified 16S ribosomal DNA fragments. Appl Environ Microbiol. 1997;63:1489–1497. doi: 10.1128/aem.63.4.1489-1497.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuske C R, Barns S M, Busch J D. Diverse uncultivated bacterial groups from soils of the arid southwestern United States that are present in many geographic regions. Appl Environ Microbiol. 1997;63:3614–3621. doi: 10.1128/aem.63.9.3614-3621.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaPara T M, Nakatsu C H, Pantea L, Alleman J E. Phylogenetic analysis of bacterial communities in mesophilic and thermophilic bioreactors treating pharmaceutical wastewater. Appl Environ Microbiol. 2000;66:3951–3959. doi: 10.1128/aem.66.9.3951-3959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee D H, Noh S A, Kim C K. Development of molecular biological methods to analyze bacterial species diversity in freshwater and soil ecosystems. J Microbiol. 2000;38:11–17. [Google Scholar]
- 20.Liu W T, Marsh T L, Cheng H, Forney L J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol. 1997;63:4516–4522. doi: 10.1128/aem.63.11.4516-4522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marilley L, Vogt G, Blanc M, Aragno M. Bacterial diversity in the bulk soil and rhizosphere fractions of Lolium perenne and Trifolium repens as revealed by PCR restriction analysis of 16S rDNA. Plant Soil. 1998;198:219–224. [Google Scholar]
- 22.McCaig A E, Glover L A, Prosser J I. Molecular analysis of bacterial community structure and diversity in unimproved and improved upland grass pastures. Appl Environ Microbiol. 1999;65:1721–1730. doi: 10.1128/aem.65.4.1721-1730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCaig A E, Grayston S J, Prosser J I, Glover L A. Impact of cultivation on characterisation of species composition of soil bacterial communities. FEMS Microbiol Ecol. 2001;35:37–48. doi: 10.1111/j.1574-6941.2001.tb00786.x. [DOI] [PubMed] [Google Scholar]
- 24.McCaig A E, Phillips C J, Stephen J R, Kowalchuk G A, Harvey S M, Herbert R A, Embley T M, Prosser J I. Nitrogen cycling and community structure of proteobacterial beta-subgroup ammonia-oxidizing bacteria within polluted marine fish farm sediments. Appl Environ Microbiol. 1999;65:213–220. doi: 10.1128/aem.65.1.213-220.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muyzer G, Dewaal E C, Uitterlinden A G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S ribosomal RNA. Appl Environ Microbiol. 1993;59:695–700. doi: 10.1128/aem.59.3.695-700.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakatsu C H, Torsvik V, Ovreas L. Soil community analysis using DGGE of 16S rDNA polymerase chain reaction products. Soil Sci Soc Am J. 2000;64:1382–1388. [Google Scholar]
- 27.Nusslein K, Tiedje J M. Characterization of the dominant and rare members of a young Hawaiian soil bacterial community with small-subunit ribosomal DNA amplified from DNA fractionated on the basis of its guanine and cytosine composition. Appl Environ Microbiol. 1998;64:1283–1289. doi: 10.1128/aem.64.4.1283-1289.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ovreas L, Torsvik V. Microbial diversity and community structure in two different agricultural soil communities. Microb Ecol. 1998;36:303–315. doi: 10.1007/s002489900117. [DOI] [PubMed] [Google Scholar]
- 29.Pielou E C. The measurement of diversity in different types of biological collections. J Theor Biol. 1966;13:131–144. [Google Scholar]
- 30.Ritchie N J, Schutter M E, Dick R P, Myrold D D. Use of length heterogeneity PCR and fatty acid methyl ester profiles to characterize microbial communities in soil. Appl Environ Microbiol. 2000;66:1668–1675. doi: 10.1128/aem.66.4.1668-1675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwieger F, Tebbe C C. A new approach to utilize PCR-single-strand-conformation polymorphism for 16S rRNA gene-based microbial community analysis. Appl Environ Microbiol. 1998;64:4870–4876. doi: 10.1128/aem.64.12.4870-4876.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon C E, Weaver W. The mathematical theory of communication. Urbana: University of Illinois Press; 1963. [Google Scholar]
- 33.Simpson E H. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- 34.Smit E, Leeflang P, Wernars K. Detection of shifts in microbial community structure and diversity in soil caused by copper contamination using amplified ribosomal DNA restriction analysis. FEMS Microbiol Ecol. 1997;23:249–261. [Google Scholar]
- 35.Stephen J R, McCaig A E, Smith Z, Prosser J I, Embley T M. Molecular diversity of soil and marine 16S rRNA gene sequences related to beta-subgroup ammonia-oxidizing bacteria. Appl Environ Microbiol. 1996;62:4147–4154. doi: 10.1128/aem.62.11.4147-4154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stephen J R, Kowalchuk G A, Bruns M A V, McCaig A E, Phillips C J, Embley T M, Prosser J I. Analysis of beta-subgroup proteobacterial ammonia oxidizer populations in soil by denaturing gradient gel electrophoresis analysis and hierarchical phylogenetic probing. Appl Environ Microbiol. 1998;64:2958–2965. doi: 10.1128/aem.64.8.2958-2965.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tiedje J M, Asuming-Brempong S, Nusslein K, Marsh T L, Flynn S J. Opening the black box of soil microbial diversity. Appl Soil Ecol. 1999;13:109–122. [Google Scholar]
- 38.Yang C H, Crowley D E. Rhizosphere microbial community structure in relation to root location and plant iron nutritional status. Appl Environ Microbiol. 2000;66:345–351. doi: 10.1128/aem.66.1.345-351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou J Z, Davey M E, Figueras J B, Rivkina E, Gilichinsky D, Tiedje J M. Phylogenetic diversity of a bacterial community determined from Siberian tundra soil DNA. Microbiology. 1997;143:3913–3919. doi: 10.1099/00221287-143-12-3913. [DOI] [PubMed] [Google Scholar]