

Abstract
Different pathogenic variants in the fibrillin‐1 gene (FBN1) cause Marfan syndrome and acromelic dysplasias. Whereas the musculoskeletal features of Marfan syndrome involve tall stature, arachnodactyly, joint hypermobility, and muscle hypoplasia, acromelic dysplasia patients present with short stature, brachydactyly, stiff joints, and hypermuscularity. Similarly, pathogenic variants in the fibrillin‐2 gene (FBN2) cause either a Marfanoid congenital contractural arachnodactyly or a FBN2‐related acromelic dysplasia that most prominently presents with brachydactyly. The phenotypic and molecular resemblances between both the FBN1 and FBN2‐related disorders suggest that reciprocal pathomechanistic lessons can be learned. In this review, we provide an updated overview and comparison of the phenotypic and mutational spectra of both the “tall” and “short” fibrillinopathies. The future parallel functional study of both FBN1/2‐related disorders will reveal new insights into how pathogenic fibrillin variants differently affect the fibrillin microfibril network and/or growth factor homeostasis in clinically opposite syndromes. This knowledge may eventually be translated into new therapeutic approaches by targeting or modulating the fibrillin microfibril network and/or the signaling pathways under its control.
Keywords: fibrillin‐1, fibrillin‐2, fibrillinopathies, pathophysiology, skeletal dysplasia
The paradigm of opposing phenotypes for both FBN1 and FBN2 suggests shared pathomechanisms.
1. INTRODUCTION
Fibrillin microfibrils provide mechanical and functional support to human cells, tissues, and organs. The fibrillinopathies, a diverse group of connective tissue disorders, are caused by pathogenic variants in the fibrillin‐1 (FBN1) and fibrillin‐2 (FBN2) genes, respectively, encoding FBN1 and FBN2. A spectrum of disorders with opposite phenotypic features has been linked to pathogenic variants in FBN1, for which the divergent pathophysiological mechanisms remain largely elusive. Recent evidence suggests that this paradigm of opposing phenotypes is recapitulated in FBN2. In this review, we provide an updated overview of the structure, expression, and function of the different fibrillins, compare the phenotypic and mutational spectra of FBN1‐ and FBN2‐related disorders and discuss the state‐of‐the‐art and challenges of current fundamental mechanistic research. In this way, we aim to provide more insights into the underlying mechanisms of the phenotypically distinct fibrillinopathies and guide future research.
2. THE FIBRILLINOPATHIES: A SPECTRUM OF DISORDERS WITH OPPOSING PHENOTYPES
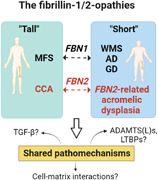
Different pathogenic variants in the FBN1 cause Marfan syndrome (MFS, MIM# 154700) and the acromelic dysplasias, respectively. Remarkably, both disorders present with opposing skeletal (“tall” vs. “short”) and cardiovascular phenotypes. Interestingly, pathogenic variants in FBN2 can cause either a Beals–Hecht syndrome/Marfanoid congenital contractural arachnodactyly (CCA, MIM# 121050) or an acromelic dysplasia that most prominently presents with brachydactyly, suggesting that this paradigm of opposing skeletal phenotypes is recapitulated in FBN2 (Figure 1, Table 1). In the next section, we will explore the phenotypic state‐of‐the‐art of the “tall” and “short” fibrillin‐1/2‐opathies.
Figure 1.
Pathogenic variants in fibrillin‐1 (FBN1) and fibrillin‐2 (FBN2) can cause “tall” and “short” fibrillinopathies.
Table 1.
Overview of the “tall” and “short” fibrillinopathies.
| Disorder | Inheritance (gene) | Skeletal features | Cardiovascular features | Opthalmological features | Other features | Location variant in FBN1/FBN2 |
|---|---|---|---|---|---|---|
| “Tall” fibrillinopathies | ||||||
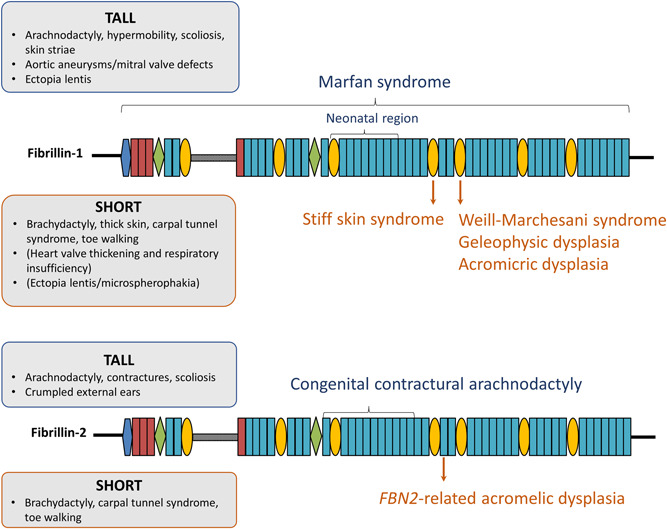
| Marfan syndrome | Autosomal dominant (FBN1) | Tall stature, arachnodactyly, scoliosis, pectus deformities, contractures, hypermobility of joints, muscle hypoplasia, pes planus | Thoracic aortic aneurysms and dissections, mitral/tricuspid valve prolapse | Ectopia lentis | Pneumothorax, skin striae, long narrow face, malar hypoplasia, micrognathia, retrognathia | All over gene, “neonatal region”: TB3‐cbEGF18 |
| Congenital contractural arachnodactyly (Beals–Hecht syndrome; distal arthrogryposis type 9) | Autosomal dominant (FBN2) | Tall stature, arachnodactyly, (kypho)scoliosis, pectus deformities, contractures, muscle hypoplasia | Mild cardiovascular involvement | Long, narrow face, highly arched palate, micrognathia, crumpled external ears | cbEGF10–cbEGF20 | |
| “Short” fibrillinopathies | ||||||
| Weill‐Marchesani syndrome | Autosomal dominant (FBN1) and autosomal recessive (ADAMTS10, ADAMTS17, LTBP2) | Short stature, brachydactyly, joint stiffness and contractures, toe walking, muscular build, early‐onset carpal tunnel syndrome | Ectopia lentis, microspherophakia | Thick skin, maxillary hypoplasia | TB5 domain, TB1‐EGF4 domain | |
| Geleophysic dysplasia | Autosomal dominant (FBN1, LTBP3), autosomal recessive (ADAMTSL2) | Short stature, brachydactyly, joint stiffness and contractures, toe walking, early onset carpal tunnel syndrome | Progressive thickening of cardiac valves | Thick skin, tracheal stenosis, respiratory insufficiency, full cheeks, a short nose, hypertelorism, a thin upper lip (“happy face”) | TB5 domain | |
| Acromicric dysplasia | Autosomal dominant (FBN1, LTBP3) | Short stature, brachydactyly, joint stiffness and contractures, pseudomuscular build, notch femoral head, early onset carpal tunnel syndrome | Thick skin, round face, a bulbous nose, thick lips, short palpebral fissures, hoarse voice | TB5 domain | ||
| Stiff skin syndrome | Autosomal dominant (FBN1) | Relative short stature, brachydactyly, joint stiffness and contractures, diffuse entrapment neuropathy including carpal tunnel syndrome | Thick skin, cutaneous nodules | TB4 domain | ||
| FBN2‐related acromelic dysplasia | Autosomal dominant (FBN2) | Relative short stature, brachydactyly, toe walking, early onset carpal tunnel syndrome | Short palpebral fissures | cbEGF23 | ||
2.1. Phenotypic comparison of the “tall” fibrillinopathies
MFS is an autosomal dominant disorder caused by heterozygous variants in FBN1. It is characterized by thoracic aortic aneurysm and dissection (most commonly at the level of the sinus of valsalva), mitral valve defects, tall stature, arachnodactyly, scoliosis, muscle hypoplasia, and ectopia lentis (Keane & Pyeritz, 2008; Sakai et al., 2016). The revised Ghent nosology stipulates that one of the following four criteria needs to be fulfilled to formally establish a diagnosis of MFS: (1) the presence of ectopia lentis and aortic root dilatation, (2) the presence of aortic root dilatation and a pathogenic FBN1 variant, (3) the presence of aortic root dilatation and a high “systemic” score (≥7), which is based on the presence of other cardiovascular and ocular manifestations of MFS and findings in other organs (skeleton, dura, lungs, skin), or (4) the presence of ectopia lentis and a FBN1 variant that previously has been associated with aortic disease (Loeys, Dietz, et al., 2010).
CCA or Beals–Hecht syndrome (now called Distal Arthrogryposis type 9 or DA9) is an autosomal dominant disorder that has similar phenotypic features as MFS but is caused by heterozygous pathogenic variants in FBN2. It is characterized by tall stature, arachnodactyly, progressive scoliosis, contractures, and crumpled ears (Hecht & Beals, 1972; Viljoen, 1994). In a fraction of CCA patients, cardiovascular abnormalities such as aortic root dilatation and mitral valve prolapse have been reported (Callewaert et al., 2009; Carmical et al., 1999; Gupta et al., 2002; Takeda et al., 2015). Although extremely rare, a few CCA patients with aortic dissection have been reported (Takeda et al., 2015). A clinical scoring system for the diagnosis of CCA has recently been described (Meerschaut et al., 2020).
2.2. Phenotypic comparison of the “short” fibrillinopathies
Interestingly, heterozygous pathogenic variants in FBN1 and FBN2 can either result in conditions presenting with tall stature and arachnodactyly or disorders displaying short stature and brachydactyly. Weill‐Marchesani syndrome (WMS), geleophysic dysplasia (GD), and acromicric dysplasia (AD) are classified under the group of acromelic dysplasias in the latest nosology of genetic skeletal disorders (Mortier et al., 2019). These skeletal dysplasias are characterized by short stature and shortening of the limbs, mainly in the acromelic segments (brachydactyly). Although pathogenic variants in other genes (ADAMTS10, ADAMTS17, ADAMTSL2, LTBP2, LTBP3, see Table 1) have also been linked to these disorders, all three phenotypes can be caused by pathogenic variants in the FBN1 gene. The FBN1‐related acromelic dysplasias share short stature, brachydactyly, joint stiffness, contractures, and thick skin as common features. However, some disorder‐specific clinical features have been suggested. In WMS, severe eye abnormalities such as microspherophakia and ectopia lentis are present (Faivre, Dollfus, et al., 2003). Distinguishing GD features are progressive thickening of the cardiac valves, tracheal stenosis, respiratory insufficiency, toe walking, and a “happy” face characterized by full cheeks, a short nose, hypertelorism, and a thin upper lip (Faivre, Dollfus, et al., 2003; Sakai & Keene, 2019). A round face, a bulbous nose and thick lips, a hoarse voice, and some specific radiological features such as a notch of the femoral head are rather typical for AD (Kochhar et al., 2013; Sakai & Keene, 2019). Interestingly, in all three acromelic dysplasias, early‐onset carpal tunnel syndrome (CTS) has been reported. The frequency of CTS in acromelic dysplasia patients seems to range between 25% and 66% (Faivre, Gorlin, et al., 2003; Globa et al., 2018; Klein et al., 2014; Marzin et al., 2020). Another FBN1‐related disorder that is distinct from the acromelic disorders but in which relative short stature has been described is stiff skin syndrome (SSKS, MIM# 184900), an autosomal dominant congenital form of scleroderma. The fibrotic skin of SSKS patients limits joint mobility and causes flexion contractures (Esterly & Mckusick, 1971). Additional features of SSKS include cutaneous nodules, relative short stature and again, diffuse entrapment neuropathies (including CTS) due to local compression. In one patient, also ectopia lentis was seen (Loeys, Gerber, et al., 2010).
We recently identified a new fibrillin‐2‐opathy characterized by brachydactyly, early‐onset CTS and a peculiar facial appearance with rather short palpebral fissures (Peeters et al., 2020). Relative short stature (mean height SDS = −1.17) and short Achilles' tendons resulting in toe walking during childhood was seen in a proportion of the affected individuals. Interestingly, these features resemble the phenotypic characteristics of the FBN1‐related acromelic dysplasias. However, CTS was more common, and presented earlier in life in the new fibrillin‐2‐opathy. Although only one family has been reported so far, this new FBN2‐related disorder seems to complement the spectrum of fibrillin‐2‐opathies with an acromelic dysplasia‐like phenotype.
3. THE FIBRILLIN FAMILY OF PROTEINS
3.1. Fibrillin protein structure
Fibrillins are large (350 kDa), cysteine‐rich glycoproteins that assemble into beaded structures in the extracellular matrix (ECM) of connective tissues, called microfibrils (Sakai et al., 1986). The fibrillin proteins are encoded by three genes, that is, FBN1 (chromosome 15q15‐21.3), FBN2 (chromosome 5q23‐31), and FBN3 (chromosome 19p13.3‐13.2), and have a highly conserved domain architecture (Piha‐Gossack et al., 2012). At the amino acid level, a sequence homology of 61%–69% is seen between the three human fibrillins, with the highest homology between FBN1 and FBN2 (Sakai & Keene, 2019). Fibrillins are organized in highly repetitive domains (Figure 2). The most prominent domain is the epidermal growth factor (EGF) domain, which is present 47 times in FBN1 and FBN2 and 46 times in FBN3. Most of the EGF domains (43 out of 46–47) contain a EGF consensus sequence for calcium binding (Asp/Asn‐X‐Asp/Asn‐Glu/Gln‐X‐Asp/Asn‐X‐Tyr/Phe) and are referred to as calcium‐binding EGF (cbEGF) domains, where X indicates a variable number of amino acid residues (Handford et al., 1991). Furthermore, fibrillins contain seven 8‐cystein or transforming growth factor beta (TGF‐β) binding‐like (TB) domains and two hybrid domains. These domains are quite unique to the human proteome and are only seen in fibrillins and latent TGF‐β binding proteins (LTBPs) (Robertson et al., 2011). The three fibrillins also contain a characteristic domain immediately following the first TB domain, which is proline‐rich in FBN1, glycine‐rich in FBN2, and proline/glycine‐rich in FBN3. The proline‐rich region in FBN1 is suggested to be a hinge region, enabling folding of the proteins into microfibrils (Baldock et al., 2001). The N‐terminal and the C‐terminal parts of fibrillins contain a 4‐cysteine motif and a 2‐cysteine motif, respectively. Both the N‐ and C‐terminal domains of the fibrillins also contain tribasic consensus sequences (Arg‐X‐Lys/Arg‐Arg) for furin cleavage, which are important for processing profibrilin to fibrillin. The N‐terminal consensus sequences of FBN1 and FBN2 are located between amino acid positions 41–45 and 74–77, respectively, whereas the C‐terminal sequences are located between amino acid positions 2728–2731 and 2776–2779, respectively.
Figure 2.
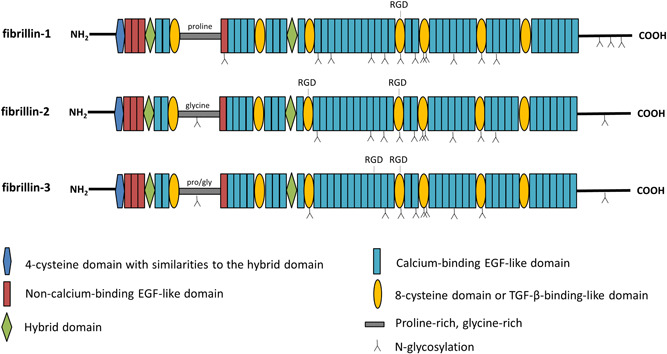
Domain organization of fibrillin‐1 (FBN1), fibrillin‐2 (FBN2), and fibrillin‐3 (FBN3). The fibrillin domain organization consists of a 4‐cysteine motif with similarities to the hybrid domain, four non‐calcium‐binding epidermal growth factor (EGF)‐like domains, 43 calcium‐binding EGF‐like domains, seven 8‐cystein or transforming growth factor beta (TGF‐β) binding‐like (TB) domains, and two hybrid domains. N‐glycosylation sites and Arg‐Gly‐Asp (RGD) motifs for integrin binding are depicted.
Cysteine residues within the different domain types are well conserved between the three fibrillin proteins. The sulfhydryl groups present in cysteines are known to form disulfide bonds with one another. Most of these disulfide bridges are formed within domains and are important to maintain intradomain stability and folding. The (cb)EGF domains contain six highly conserved cysteine residues that form three disulfide bridges in a 1–3, 2–4, 5–6 arrangement (Campbell & Bork, 1993; Downing et al., 1996). The TB domains on the other hand have four intramolecular disulfide bonds in a 1–3, 2–6, 4–7, 5–8 pattern (Lee et al., 2004; Yuan et al., 1997). Similar to the latter domains, the two hybrid domains contain four disulfide bridges but in a 1–3, 2–5, 4–6, 7–8 arrangement. Finally, the N‐terminal part contains two disulfide bridges in a 1–3, 2–4 pattern (Bork et al., 1996; Yadin et al., 2013).
Even though the different fibrillins are very similar with respect to protein domain organization, they differ in other structural aspects. Besides having the characteristic proline‐, glycine‐, or proline/glycine‐rich region after the first TB domain, they also differ in the number and the position of N‐glycosylation sites; FBN1 has 15 N‐glycoslyation sites, FBN2 12, and FBN3 10 (Sakai & Keene, 2019). N‐glycosylation sites play key roles in protein folding, stability, and protein–protein interaction (Hart, 1992). Furthermore, the fibrillins have discrete numbers of Arg‐Gly‐Asp (RGD) motifs for integrin binding, which are important for cell adhesion and normal ECM production (Jovanovic et al., 2008). While all fibrillins contain an RGD‐site in the TB4 domain, only FBN2 and FBN3 have a second RGD‐site in the TB3 and cbEGF18 domain, respectively.
3.2. Fibrillin microfibril assembly
Although the vast majority of studies investigating fibrillin processing have been focusing on FBN1, all fibrillins are predicted to be processed in a similar manner. After secretion, pro‐fibrillins multimerize at the cell surface to assemble into microfibrils, a process that is mediated by cell surface heparan sulfate proteoglycans (HSPG) (Tiedemann et al., 2001). Further multimerization is initiated by cleavage of the C‐ and N‐terminal propeptides by furin (Milewicz et al., 1995). Next, fibrillins form head‐to‐tail and lateral disulfide‐mediated interactions (Ashworth et al., 1999; Trask et al., 1999). In addition, nonreducing lateral cross‐links between glutamines and lysines are formed to provide extra strength to the microfibrillar network (Qian & Glanville, 1997; Thurmond & Trotter, 1996). All these interactions eventually lead to the formation of mature microfibrils. Immunolocalization studies in fetal tissues showed that FBN1 either coassembles with other FBN1 molecules to form a homopolymeric microfibril or coassembles with FBN2 into a heteropolymeric microfibril (Charbonneau et al., 2003; Lin et al., 2002). Interestingly, homopolymeric interactions of FBN2 proteins were not seen (Charbonneau et al., 2003; Corson et al., 2004). Immunolocalization studies showed that FBN3 is localized to microfibrils but it remains unknown whether FBN3 interacts with other fibrillins (Corson et al., 2004).
3.3. Fibrillin expression
A partially overlapping spatiotemporal expression pattern has been reported for the three fibrillins. It has been shown that FBN1 expression persists during life, while the expression of FBN2 and FBN3 seems to drop significantly after birth (Corson et al., 2004; H. Zhang et al., 1995). The observation that FBN2 is mainly expressed during early development has led to the assumption that it is less important after birth. However, since FBN2 makes up the inner core of microfibrils in postnatal life, its role after birth may be underestimated (Charbonneau et al., 2010). Protein expression studies may fail in detecting FBN2 after birth because the protein is hidden in the core of the postnatal microfibrils. Caution is therefore warranted when performing, comparing, and/or interpreting immunohistochemical analyses.
In general, the three fibrillins are expressed in the same organs during development, including the skin, lungs, heart, aorta, kidneys, and nerves (Quondamatteo et al., 2002; Sabatier et al., 2011). Exceptions are the ciliary zonules in the eyes, where primarily FBN1 is found and the liver, in which no expression of FBN3 is seen (Sabatier et al., 2011). However, differences in the distribution of fibrillins have been observed in specific organs. For example, whereas expression of FBN1 and FBN2 proteins is seen in the outer fibrous layer of the perichondrium, FBN3 is primarily found in the inner chondrogenic layer. Furthermore, while FBN1 and FBN3 are both present in the hypertrophic zone of hyaline cartilage, FBN2 is absent in this part of the growth plate. Apart from the perichondrium and hyaline cartilage, differences in fibrillin distribution have been reported in the developing lungs and kidneys (Sabatier et al., 2011).
Interestingly, a few species‐specific differences in expression of FBN1 and FBN2 have been reported. While murine ciliary zonules are composed of both Fbn1 and Fbn2 (Beene et al., 2013), only FBN1 is seen in human zonules (Hubmacher et al., 2014). Furthermore, FBN3 does not exist in rodents (Corson et al., 2004), suggesting that this protein is not essential for all mammalian life. Therefore, and because no disease in humans is unequivocally associated with FBN3 mutations, we will focus this review on FBN1 and FBN2.
3.4. Fibrillin microfibril function and its associated protein network
In elastic tissues such as lungs, blood vessels, skin, and ligaments, microfibrils serve as a scaffold for elastin deposition and modification during elastic fiber formation (Jones et al., 1980; Kewley et al., 1978; Ross & Bornstein, 1969). In nonelastic tissues such as the ciliary zonule and cornea, tendon, perichondrium, and renal glomerulus, microfibrils provide tensile strength (Kumaratilake et al., 1989; Sakai et al., 1986; Sterzel et al., 2000). These discrete architectural roles are reflected by the microfibrils' tissue‐specific properties. It has been shown that both the microfibril bead morphology as well as the proteolytic susceptibility of microfibrils differ between tissues (Eckersley et al., 2018).
Besides exerting a structural role, microfibrils are involved in the control of cell signaling pathways through storage and activation of growth factors, including TGF‐β, bone morphogenic proteins (BMPs), and growth differentiating factors (GDFs) (Sengle et al., 2008). Most fibrillin‐related studies that have been done so far focused on their role in the regulation of the TGF‐β signaling pathway. The TGF‐β cytokines (TGF‐β1, TGF‐β2, and TGF‐β3) are encoded by three genes (TGFB1, TGFB2, and TGFB3). These cytokines are involved in numerous cellular processes such as growth, differentiation, and apoptosis, as well as in immune system regulation and tissue homeostasis. Mature TGF‐β forms a complex with latency associated proteins (LAPs), called the small latent TGF‐β complex (SLC) (Gentry et al., 1988; Lawrence et al., 1984). In turn, these LAPs covalently bind LTBPs (LTBP1, LTBP3, LTBP4) to form a large latent complex (LLC). LTBPs are members of the fibrillin superfamily and, although much smaller, have similar structural properties as fibrillins. Through binding of the SLC with LTBPs, TGF‐β is held in an inactive state (Annes et al., 2003; Koli et al., 2001; Miyazono et al., 1988) (Figure 3). The LLC interacts with ECM components, mostly fibrillins (Isogai et al., 2003), to sequester TGF‐β ligands into the ECM and, as such, controls their biological availability. Activation of the TGF‐β pathway through release of TGF‐β from the LLC is initiated by different mechanisms, including proteolytic cleavage by plasmin or matrix metalloproteinases (MMP2 and MMP9) (Sato & Rifkin, 1989; Yu & Stamenkovic, 2000), activation by thrombospondin (which is upregulated during wound healing) (Schultz‐Cherry & Murphy‐Ullrich, 1993) or conformational alterations of the LLC‐mediated by integrin binding (αvβ6, upregulated during wounding/inflammation), and the ensuing force‐depending activation (Annes et al., 2003; Hinz, 2013; Shi et al., 2011). Binding of active TGF‐β to TGFBRII receptors initiates phosphorylation of the TGFBRI receptors. Further downstream signaling either involves the canonical or noncanonical pathway. In the canonical pathway, receptor‐regulated SMADs (R‐SMADs; SMAD2 or SMAD3) are phosphorylated after which a complex with SMAD4 is formed. This complex is then translocated into the nucleus and activates transcription of TGF‐β target genes. The noncanonical TGF‐β signaling pathway activates ERK, JNK, and p38 via mitogen‐activated protein (MAPK) kinases, eventually also stimulating transcription of TGF‐β target genes.
Figure 3.
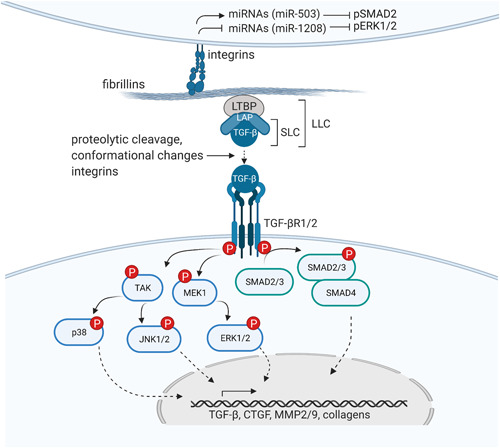
Fibrillins control transforming growth factor beta (TGF‐β) signaling. Through the sequestration of the large latent complex (LLC), consisting of the small latent complex (SLC) and TGF‐β ligands, and the interactions with integrins on the cell surface, fibrillins control TGF‐β signaling. Both the noncanonical (or SMAD‐independent) pathway (blue) and the canonical (SMAD‐dependent) pathway (green) are depicted. Figure created with BioRender.com.
Fibrillins do not only control TGF‐β signaling via their interaction with LTBPs. As previously mentioned, they also interact with cells via RGD‐binding integrin receptors. For FBN, it has been shown that this interaction inhibits TGF‐β signaling through the regulation of miRNA expression. MiR‐503, one of the miRNAs that is upregulated upon fibrillin‐1 RGD‐integrin interaction, has been shown to downregulate TGF‐β2 and reduce pSMAD2 activity (Zeyer et al., 2019). Furthermore, recent evidence suggests that fibrillin‐1 RGD‐integrin interaction inhibits miR‐1208 expression, an inhibitor of noncanonical TGF‐β signaling (R. M. Zhang et al., 2021).
Apart from LTBPs, fibrillin microfibrils interact with several other ECM proteins. These molecules are known to mediate the assembly of microfibrils and/or contribute to their (tissue‐specific) functionality, both structurally and functionally. Key binding proteins of FBN1 include FBN2, MAGPs, MFAPs, ADAMTS(L)s, fibulins, fibronectin, chondroitin/HSPG, integrins, aggrecan, versican, hyaluronan, perlecan, syndecan‐4, tropoelastin, lysyl oxidase (Thomson et al., 2019). Limited studies focused on interaction partners of FBN2 so far, although interactions with FBN1, MFAP5, MFAP2, and ADAMTS17 have been reported (Hubmacher et al., 2017). To date, however, most interaction studies use recombinant FBN1/FBN2 protein fragments because of the propensity of full‐length fibrillin to aggregate. Since the affinity for certain proteins and polysaccharides (including heparan sulfate) tend to change upon multimerization of fibrillins (Sabatier et al., 2014), caution is warranted when interpreting protein‐interaction studies using recombinant fragments.
4. MOLECULAR COMPARISON OF THE “TALL” AND “SHORT” FIBRILLIN‐1/2‐OPATHIES
4.1. Mutational spectrum and genotype–phenotype correlations of the “tall” fibrillinopathies
Zooming in on the mutation spectrum underlying MFS and CCA, both commonalities and discordances can be observed. In both disorders, pathogenic variants have been described that introduce or replace a cysteine residue in the cbEGF domains. Cysteine‐affecting variants have been shown to interfere with normal intradomain or interdomain disulfide bridging, thereby affecting normal protein folding. Fibrillin misfolding affects protein secretion and/or causes an increased proteolytic susceptibility by anomalous exposure of cryptic cleavage sites and/or decreased calcium binding (Reinhardt et al., 1997; Suk et al., 2004; Vollbrandt et al., 2004), ultimately leading to a weakened microfibril scaffold. Other pathogenic missense variants in FBN1 or FBN2, respectively causing MFS or CCA, have been shown to change critical residues in the cbEGF consensus sequence for calcium binding, thereby again affecting normal calcium binding (Rao et al., 1995). In MFS, missense variants that do not affect cysteine or calcium binding residues as well as nonsense variants, splice site variants, (partial) FBN1 deletions, duplications, and small insertions have been described (Booms et al., 2000; Caputi et al., 2002; Collod‐Beroud et al., 2003; Dietz et al., 1991; Hilhorst‐Hofstee et al., 2011; Liu et al., 2001). Similar to MFS (partial) deletions of FBN2 and splice site variants have also been reported in CCA patients (Gupta et al., 2002). In addition, a pathogenic variant that introduces an additional glycosylation site in cbEGF9 has been identified in one CCA patient (Gupta et al., 2002). A missense variant that creates an additional N‐glycosylation site in FBN1 has also been reported in a patient with severe MFS (Lonnqvist et al., 1996). Excessive N‐glycosylation has been shown to severely affect microfibril formation (Lonnqvist et al., 1996).
Few strong genotype–phenotype correlations have been described for both MFS and CCA. Pathogenic variants in the so‐called “neonatal” region of FBN1 (exons 25–33 encoding TB3‐cbEGF18), have been shown to cause a more severe form of MFS that often results in neonatal death, referred to as neonatal MFS (nMFS) (Maeda et al., 2016). However, the use of the term “nMFS” is discouraged as this term does not adequately represent a specific subset of individuals with truly distinguishing characteristics (Dietz, 2001 [Updated February 17, 2022]). Furthermore, there are exceptions and some cases of this “early onset” and “rapidly progressive” MFS are caused by pathogenic variants outside this particular region (Universal Mutation Database (UMD)‐FBN1) (Collod‐Beroud et al., 2003). Pathogenic variants causing this more severe type of MFS have been shown to drastically alter proteolytic sensitivity, protein secretion, and heparin/heparan sulfate binding (Kirschner et al., 2011; Maeda et al., 2016). In addition, multiple studies report a strong link between cysteine creating or replacing variants and ectopia lentis (Loeys et al., 2001; Rommel et al., 2005; Schrijver et al., 2002). Furthermore, some studies suggest that MFS patients with haploinsufficient FBN1 variants generally have a higher aortic risk than MFS patients with dominant negative FBN1 variants (Arnaud et al., 2021; Baudhuin et al., 2015; Franken et al., 2015, 2016). However, these findings are contradicted by a recent study in a pediatric MFS cohort showing that dominant negative variants in exons 26–49 result in a more severe aortic phenotype than patients with haploinsufficient variants or dominant negative variants in other exons (Meester et al., 2021). This study also noticed a clear gradient in the proportion of ectopia lentis according to the location of the dominant negative FBN1 variant, with the highest prevalence of lens dislocation in the N‐terminal region (Meester et al., 2021). In contrast to the findings on aortic risk, skeletal features including pectus excavatum and tall stature are more pronounced in patients carrying haploinsufficient variants (Meester et al., 2021). Similarly, another study in adult MFS patients reported an association between premature stop codon variants and a higher risk of severe scoliosis and tall stature (Arnaud et al., 2021). Finally, it has been shown that pathogenic variants in exon 64 of FBN1 can cause a specific subtype of MFS called marfanoid–progeroid–lipodystrophy syndrome (MFLS, MIM# 616914), a disorder mainly characterized by congenital lipodystrophy, tall stature, arachnodactyly, and progeroid appearance (Romere et al., 2016). MFLS variants are clustered around the C‐terminal furin cleavage site of profibrillin (amino acids 2728–2731), leading to a mutant, truncated profibrillin protein and reduced levels of the 140‐amino‐acid long C‐terminal cleavage product of profibrillin, called asprosin. Reduced levels of the asprosin hormone affect normal adiposity and cause metabolic dysregulation in patients (Duerrschmid et al., 2017; Romere et al., 2016).
Unlike MFS‐causing variants, which are spread across the FBN1 gene, most CCA‐causing variants cluster between exon 23 and exon 34 (cbEGF10–cbEGF20) of FBN2 (Gupta et al., 2002). This specific region of FBN2 roughly corresponds to the “neonatal” region of FBN1. Though less common, pathogenic CCA variants outside this region (e.g., in exon 17 and exon 21) have also been described (Callewaert et al., 2009; Meerschaut et al., 2020). For CCA, a few reports suggest that intragenic deletions (Lavillaureix et al., 2017) and splice‐site alterations in the central region of FBN2 that result in in‐frame exon skipping (Wang et al., 1996), cause a more severe form of CCA (Callewaert et al., 2009), but further confirmation is warranted. Although most CCA patients carry dominant negative variants, also a few haploinsufficient FBN2 variants, including whole gene deletions and a nonsense variant, have been described (Courtens et al., 1998; Inbar‐Feigenberg et al., 2014; Kloth et al., 2021).
In both disorders, an extensive inter‐ and intrafamilial variability in age at onset and phenotypic severity is seen. The mechanisms underlying this variability are largely unknown. For FBN1 this most likely involves the variable expression level of the other normal allele (Aoyama et al., 1995; Aubart et al., 2015; Hutchinson et al., 2003) or other genetic modifiers (Aubart et al., 2018).
4.2. Mutational spectrum and genotype–phenotype correlations of the “short” fibrillinopathies
The short fibrillin‐1‐opathies, WMS, AD, and GD, are all caused by pathogenic variants in the heparin‐binding TB5 domain of FBN1. A deletion of exons 9–11 (coding for TB1‐EGF4), which abolishes a binding site for ADAMTSL proteins, has been described in one family with WMS (Sengle et al., 2012). SSKS, on the other hand, is caused by pathogenic cysteine‐replacing or introducing variants in the TB4 domain of FBN1. The pathogenic variant causing the FBN2‐related acromelic dysplasia (NM_001999.4: c.5009T>G; p.(Phe1670Cys)) is located in the cbEGF23 of FBN2, a domain adjacent to TB4.
Apart from the observation that nearly all these “acromelic” variants cluster within the TB4‐TB5 region of FBN1/FBN2, one additional genotype‐phenotype correlation has been described for the “short” fibrillin‐1‐opathies. A recent study on variants causing AD/GD phenotypes showed that variants involving a cysteine in FBN1‐TB5 were more often associated with a severe phenotype with life‐threatening heart valve disease compared to other FBN1‐TB5 variants (Marzin et al., 2020).
5. THE MECHANISMS UNDERLYING THE OPPOSING PHENOTYPES
The observation that opposing phenotypes may result from heterozygous pathogenic variants in the same gene (either FBN1 or FBN2) is intriguing and deserves further discussion. Different (and sometimes conflicting) disease mechanisms have been proposed to underlie this phenomenon. Whereas loss of the structural integrity or stability of microfibrils (both due to haplo‐insufficiency or dominant negative mechanisms) is suggested to underlie MFS, altered cell–matrix interactions to the TB4–TB5 region are thought to be the primary defect driving the acromelic phenotypes (Jensen et al., 2015; Peeters et al., 2020). However, to date, there is still no consensus on the precise pathophysiology, suggesting that it is more complex and may involve more than one mechanism.
5.1. Cell–matrix interactions
Only a few studies have investigated cell–matrix interactions. Cain et al. showed that FBN1‐TB5 variants causing WMS, AD, and GD (often cysteine‐replacing or ‐creating variants) disrupt pericellular interactions with heparin/heparan sulfate (Cain et al., 2012). These interactions have been shown to be important for microfibril assembly and cell adhesion. Interestingly, pathogenic variants, including cysteine‐affecting missense variants in the TB5 domain of FBN1 have also been identified in at least about a dozen MFS patients without features of acromelic dysplasia. It remains to be determined whether these variants affect heparan sulfate interactions to the TB5 domain. However, pathogenic variants in the so‐called “neonatal” middle region of FBN1 (exons 24–32), causing a severe form of MFS, have also been shown to affect heparan sulfate‐FBN1 interactions (Kirschner et al., 2011). These findings suggest that impaired heparin/heparan sulfate binding to TB5 on itself fails to explain the paradox of opposing phenotypes and that other mechanisms may be involved.
Another mechanism related to disturbed cellular sensing involve alterations in integrin‐binding. The SSKS variant (p.(Trp1570Cys)) in the TB4 domain of FBN1, a domain containing the RGD site for integrin binding, has been shown to inhibit the FBN1‐integrin (i.e., α5β1, αvβ5, and αvβ6) interaction, affecting normal cell adhesion and cell spreading (Del Cid et al., 2019). Similarly, the pathogenic variant (NM_001999.4: c.5009T>G; p.(Phe1670Cys)) causing the “short” fibrillin‐2‐opathy is located in the cbEGF23 of FBN2, a domain adjacent to the TB4 domain that also contains an RGD site. Altered cell adhesion and cell spreading to mutant FBN2 protein fragments was shown for this variant (Peeters et al., 2020). Although SSKS and the “short” fibrillin‐2‐opathy present with milder skeletal phenotypes, these findings may suggest that proper integrin‐binding is required for normal growth. It remains to be determined whether integrin‐binding is affected in the “tall” fibrillinopathies and/or whether more complex mechanisms, such as the ratio of normal microfibrils versus integrin content, have an impact on the overall growth phenotype.
5.2. Clues from the fibrillin protein network: The LTBPs and ADAMTS(L)s
More insights into the pathomechanisms for the acromelic disorders have been obtained by the identification of pathogenic variants in other genes coding for ECM proteins that bind to fibrillin microfibrils.
The LTBP protein family is evolutionarily related to the fibrillins and is characterized by a highly similar domain organization. Most LTBPs, including LTBP1, LTBP3, and LTBP4, have been shown to sequester latent TGF‐β in the ECM (Robertson et al., 2015). Pathogenic variants in the LTBPs have been associated with skeletal dysplasias that have similar—or even identical—clinical features to the FBN1‐ and FBN2‐related phenotypes. Heterozygous pathogenic variants in LTBP3 cause GD (GPHYSD2, MIM# 614185) with an indistinguishable phenotype from FBN1‐related GD. A diminished and disorganized microfibril network was found in tissues of GD patients with pathogenic variants in LTBP3, suggesting that this protein is important for the proper formation of microfibrils (McInerney‐Leo et al., 2016). Although it has been shown that LTBP3 does not bind to the C‐terminal of FBN1, it remains unknown whether it interacts with other parts of FBN1 (or FBN2), including the N‐terminal part. Autosomal recessive variants in LTBP3 cause Short Stature syndrome (DASS, MIM# 601216). Patients with DASS syndrome have amelogenesis imperfecta, brachydactyly, and a short trunk due to platyspondyly (Verloes et al., 1996). Furthermore, some DASS patients also exhibit mitral valve prolapse and aortic (root) dilation (Guo et al., 2018). The pathomechanisms underlying both LTBP3‐related disorders remain largely unknown. Based on the nature of the described variants, it is suggested that gain‐of‐function (missense) variants give rise to acromelic phenotypes while truncating and loss‐of‐function variants give rise to aortic phenotypes (Verstraeten et al., 2020). However, more research is required to confirm this statement.
Bi‐allelic pathogenic variants in the FBN1‐binding region of LTBP2 cause an autosomal recessive form of WMS (WMS3, MIM# 614819). Pathogenic variants in LTBP2 have been shown to affect normal LTBP2 secretion and/or the ability to bind FBN1 (Inoue et al., 2014). Again, disruption of the stability of fibrillin microfibrils was shown (Haji‐Seyed‐Javadi et al., 2012). These findings suggests that, in addition to LTBP3, also LTBP2 is involved in normal microfibril functioning in the ECM.
Another LTBP‐related disorder includes cutis laxa type IIE syndrome (ARCL2E, MIM# 619451). Patients with cutis laxa type IIE syndrome have generalized cutis laxa, inguinal hernias, craniofacial dysmorphology, short stature, brachydactyly, and mild cardiac defects (Pottie et al., 2021). Cutis laxa type IIE is caused by autosomal recessive truncating variants in LTBP1.
The ADAMTS(L) family consists of proteins that function in normal tissue development and homeostasis. Some of them, including ADAMTS10, have a catalytic domain and perform proteolytic activities in the ECM (Mead & Apte, 2018). Interaction studies showed that the deletion of exons 9‐11 of FBN1 (coding for TB1‐EGF4 and found in a WMS family) abolishes the binding site for several ECM proteins including ADAMTSL2, ‐3, and ‐6 (Sengle et al., 2012). Furthermore, pathogenic variants in ADAMTS10 and ADAMTS17 have been shown to cause autosomal recessive WMS (WMS1, MIM# 277600 and WMS4, MIM# 613195). In cultured fibroblasts derived from WMS patients with pathogenic variants in ADAMTSL10, reduced FBN1 microfibril formation was seen (Kutz et al., 2011). In the eye of a homozygous mouse model in which the Adamts10 protein is truncated, excessive fibrillin microfibrils were observed that were primarily composed of Fbn2 (Mularczyk et al., 2018). Also, in patient‐derived fibroblasts of WMS patients with pathogenic variants in ADAMTS17, decreased FBN1 microfibrils and collagen type 1 deposition was observed (Karoulias et al., 2020). Furthermore, mutant ADAMTS17 suppresses FBN2 incorporation in microfibrils by transcriptional downregulation of Fbn2 mRNA expression in cultured fibroblasts (Hubmacher et al., 2017). In contrast to pathogenic variants in ADAMTS10 and ADAMTS17, ADAMTSL2 mutations cause an autosomal recessive form of GD (GPHYSD1, MIM# 231050). Increased Fbn2 staining was also found in the lungs of Adamtsl2 knock‐out mice but, in contrast to what has been shown for ADAMTS17, transcriptional regulation of Fbn2 was not altered in these mice (Hubmacher et al., 2015). These findings suggest that ADAMTS(L) proteins regulate the ratio of FBN1 and FBN2 incorporation into microfibrils, and that an imbalance towards (or gain‐of‐function of) FBN2 may contribute to the development of WMS and GD (Hubmacher et al., 2015; Sakai & Keene, 2019). However, more research is required to further test this hypothesis.
Based on the functional characterization of several Cre‐specific Adamtsl2 knockout mice, several hypotheses have emerged as to which cell types or tissues are implicated in decreased longitudinal bone growth in GD. In the growth plates of chondrocyte‐specific (Col2a1‐Cre) knockout Adamtsl2 mice, an impaired microfibrillar network, increased chondrocyte proliferation and impaired chondrocyte differentiation was observed, suggesting that that chondrocyte dysfunction is the main driver of short stature in GD (Delhon et al., 2019). However, these chondrocyte‐specific growth plate abnormalities could not be confirmed in a limb‐specific (Prx1‐Cre) Adamtsl2 knockout mouse model (Hubmacher et al., 2019). Surprisingly, however, impaired skeletal growth was also observed in a tendon‐ and ligament‐specific (Scx‐Cre) Adamtsl2 knockout mouse, suggesting that the growth impairment may arise secondary to shortened tendons/ligaments rather than intrinsically by chondrocyte‐ or growth plate abnormalities. Supporting this hypothesis, two reports state that tendon‐ and ligament‐specific (Scx‐Cre) Fbn1 deletion, as a model for MFS, results in increased longitudinal growth in mice (Hubmacher et al., 2019; Smaldone et al., 2018). However, more research is required to support this hypothesis.
To date, only a few pathogenic ADAMTS(L) variants have been associated with thoracic aortic aneurysms in humans. One patient with GD and a compound heterozygous variant in ADAMTSL2 developed several arterial aneurysms including a thoracic aortic aneurysm (Legare et al., 2018). In addition, it was recently shown that heterozygous variants in THSD4, which encodes the ADAMTSL6, predispose to hereditary thoracic aortic aneurysms (Elbitar et al., 2021). Skeletal involvement was limited in these patients. Introducing the THSD4 variants in transfected HEK293 cells led to haploinsufficiency or reduced assembly of FBN1 microfibrils. Thsd4 +/− mice showed progressive thoracic aortic aneurysm formation. Medial degeneration and disruption of ECM was seen upon histological examination of aortic samples from a THSD4 patient and from Thsd4 +/− mice. Furthermore, increased TGF‐β signaling was observed in patient samples (Elbitar et al., 2021).
In conclusion, FBN1, FBN2, the LTBPs, and the ADAMTS(L)s seem to interact in a common network to establish normal tissue homeostasis and growth. Alterations in this fibrillin protein network may cause acromelic disorders. It remains to be determined to what extent intrinsic growth plate abnormalities and/or extrinsic factors such as soft tissue defects affect normal bone growth in the fibrillinopathies. The growth phenotypes of the FBN2‐related disorders are milder compared to the fibrillin‐1‐opathies, suggesting that FBN2 has a less important role in longitudinal bone growth. One possible explanation could be that FBN1 takes over the function of FBN2 postnatally, which could explain the less severe FBN2‐related growth phenotypes. However, more research is required to confirm this statement. Despite the number of cases described so far being limited, there is also evidence for a role of the ADAMTS(L)/LTBP/FBN1/FBN2 network in thoracic aortic aneurysms.
5.3. Effects on cell signaling pathways
The effects of pathogenic variants causing the “tall” and “short” fibrillinopathies on cell signaling pathways are not fully understood. Studies in MFS mouse models that recapitulate the human MFS phenotype, demonstrated that increased TGF‐β signaling plays a key role in the progression of aortic aneurysm (Habashi et al., 2006). However, other studies have suggested that TGF‐β can also be protective in specific stages of aortic development (Li et al., 2014; Wei et al., 2017). Elevated TGF‐β signaling has also been found in the mitral valves of MFS mice (Ng et al., 2004). Furthermore, studies of inducted pluripotent stem cell (iPSC)‐derived chondrocyte pellets of MFS patients suggest that increased TGF‐β signaling promotes linear growth in the growth plates of the tubular bones in MFS patients (Quarto et al., 2012). In contrast to MFS mouse models, no Fbn2 animal model fully recapitulates the clinical characteristics of CCA (Table S1) (Chaudhry et al., 2001; Miller et al., 2010; Nistala et al., 2010). Syndactyly, muscle weakness, and transient contractures are the main findings in Fbn2 knockout mice (Miller et al., 2010). Furthermore, also reduced bone mass and a reduced length of bones have been reported in Fbn2− /− mice (Nistala et al., 2010). Although Fbn2 knockout mice do not exhibit the full CCA phenotype, results from these mouse models suggest tissue‐specific roles of FBN2 in growth factor regulation. While the weakened muscles may be caused by altered BMP signaling (Arteaga‐Solis et al., 2001; Sengle et al., 2015), the low bone mass in Fbn2−/− mice mice was attributed to elevated TGF‐β signaling (Nistala et al., 2010).
Similar to what has been observed in MFS, elevated TGF‐β signaling has been reported in the “short” fibrillinopathies, including in the skin and/or fibroblasts of patients with FBN1‐related AD and GD (McInerney‐Leo et al., 2016), ADAMTSL2‐related AD (Le Goff et al., 2008), SSKS (Loeys, Gerber, et al., 2010), and in the carpal tissues of patients with the recently reported “short” fibrillin‐2‐opathy (Peeters et al., 2020). In contrast, normal TGF‐β signaling was reported in fibroblasts of WMS patients and a WMS mouse model (Fbn1 WMdelta, Table S1), both carrying the exon 9–11 deletion (or in mice exon 8–10) in FBN1 (Sengle et al., 2012), and in AD/GD patients with pathogenic variants in LTBP3 (McInerney‐Leo et al., 2016).
The observation that elevated TGF‐β signaling has been reported in tissues of both “tall” and “short” fibrillinopathies suggests that altered TGF‐β signaling may not fully explain the paradigm of opposing phenotypes either. It remains unknown whether the functional outcome of dysregulation of the TGF‐β pathway is dependent on its contextual (i.e., time‐dependent and/or tissue‐specific) microenvironment and/or other cell‐signaling pathways, and what its effect may be in the pathogenesis of the tall and short fibrillinopathies.
Apart from TGF‐β, fibrillin microfibrils sequester other growth factors, including BMPs and GDFs (Sengle et al., 2008). An important role of these growth factors in abnormal skeletal growth is illustrated by the fact that autosomal recessive variants in GDF5 and its receptor gene BMPR1B cause acromesomelic chondrodysplasias, a particular form of short stature known as short‐limb dwarfism (Demirhan et al., 2005; Thomas et al., 1997). In addition, heterozygous variants in GDF5, BMPR1B, and BMP2 cause different types of isolated brachydactylies (BDC, MIM# 113100 and BDA2, MIM# 112600). Furthermore, the fact that pathogenic variants in SMAD4 cause Myhre syndrome (MYHRS, MIM# 139210), another acromelic dysplasia, further suggests involvement of TGF‐β and BMP signaling in the acromelic dysplasias as the SMAD4 protein is a central player in both pathways. However, as most studies to date have mainly focused on the TGF‐β pathway, the exact role of the BMP/GDF pathway in the pathophysiology of the fibrillinopathies remains largely unknown.
6. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Although differences in protein structure and spatio‐temporal expression have been reported, FBN1 and FBN2 seem to co‐operate to provide mechanical and functional support to cells, tissues, and organs, especially during development. Consistent with their important and divergent role in the ECM, an increasing number of connective tissue disorders has been linked to pathogenic variants in the FBN1 and FBN2 genes. For FBN1, a paradigm of opposing phenotypes has been described (MFS vs. AD, GD, WMS), which seems to be recapitulated in FBN2 (CCA vs. the FBN2‐related acromelic dysplasia). To date, the pathophysiological mechanisms underlying these contrasting clinical syndromes remain largely unknown. The current phenotypic and molecular analogy between both “tall” and “short” FBN1 and FBN2‐related disorders are highly suggestive for the existence of shared pathomechanisms. Although it remains unclear which cell types are primarily involved, the few functional studies that have focused on FBN2 (or ADAMTS(L)s) to date point mainly towards the involvement of disturbed fibrillin‐cell interactions, changes in the FBN1/FBN2 ratio (gain‐of‐function of FBN2?) and changes in TGF‐β signaling in the pathophysiology of the acromelic phenotypes. More research is required to further prove this model and to determine whether these (or a combination of these) mechanisms are also underlying the “tall” fibrillino‐1/2‐pathies.
The identification of additional patients and the subsequent reporting of variants with a precise description of the patient's phenotypes in databases (UMD‐FBN1, UMD‐FBN2, LOVD, HGMD) are important to unveil additional genotype‐phenotype correlations in the future. Furthermore, the creation of additional specific knock‐in Fbn1 (for AD) and knock‐in Fbn2 mouse models (for CCA and AD) will be necessary to achieve more insights in the involved pathomechanisms underlying the opposing phenotypes. Additionally, since some differences in spatiotemporal expression of Fbn1 and Fbn2 have been reported in mice compared to humans, the parallel study of the disease mechanisms in human cellular models of relevant cell types, such as iPSC‐derived chondrocytes/tenocytes and iPSC‐vascular smooth muscle cells, will be essential. More specifically, a multiomics approach (transcriptomic, proteomic and, especially, interactomics) in murine and human cellular models may reveal further insights into how the different FBN1 or FBN2 mutations affect cell–matrix interactions, the fibrillin protein network (FBN1, FBN2, ADAMTS(L)s, LTBPs) as well as growth factors switches between phenotypically opposite fibrillinopathies. This knowledge may eventually be translated into new therapeutic approaches by targeting or modulating the fibrillin microfibril network and/or the signaling pathways over which it exerts control.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
S. Peeters (12X5422N) and J. A. N. Meester (12X8520N) are postdoctoral FWO (Fund for Scientific Research Flanders) fellows. B. L. Loeys holds a consolidator grant from the European Research Council (Genomia—ERC‐COG‐2017‐771945).
Peeters, S. , De Kinderen, P. , Meester, J. A. N. , Verstraeten, A. , & Loeys, B. L. (2022). The fibrillinopathies: New insights with focus on the paradigm of opposing phenotypes for both FBN1 and FBN2 . Human Mutation, 43, 815–831. 10.1002/humu.24383
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Annes, J. P. , Munger, J. S. , & Rifkin, D. B. (2003). Making sense of latent TGFbeta activation. Journal of Cell Science, 116(Pt 2), 217–224. 10.1242/jcs.00229 [DOI] [PubMed] [Google Scholar]
- Aoyama, T. , Francke, U. , Gasner, C. , & Furthmayr, H. (1995). Fibrillin abnormalities and prognosis in Marfan syndrome and related disorders. American Journal of Medical Genetics, 58(2), 169–176. 10.1002/ajmg.1320580216 [DOI] [PubMed] [Google Scholar]
- Arnaud, P. , Milleron, O. , Hanna, N. , Ropers, J. , Ould Ouali, N. , Affoune, A. , Langeois, M. , Eliahou, L. , Arnoult, F. , Renard, P. , Michelon‐Jouneaux, M. , Cotillon, M. , Gouya, L. , Boileau, C. , & Jondeau, G. (2021). Clinical relevance of genotype‐phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genetics in Medicine, 23, 1296–1304. 10.1038/s41436-021-01132-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga‐Solis, E. , Gayraud, B. , Lee, S. Y. , Shum, L. , Sakai, L. , & Ramirez, F. (2001). Regulation of limb patterning by extracellular microfibrils. Journal of Cell Biology, 154(2), 275–281. 10.1083/jcb.200105046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth, J. L. , Kelly, V. , Wilson, R. , Shuttleworth, C. A. , & Kielty, C. M. (1999). Fibrillin assembly: Dimer formation mediated by amino‐terminal sequences. Journal of Cell Science, 112(Pt 20), 3549–3558. [DOI] [PubMed] [Google Scholar]
- Aubart, M. , Gazal, S. , Arnaud, P. , Benarroch, L. , Gross, M. S. , Buratti, J. , Boland, A. , Meyer, V. , Zouali, H. , Hanna, N. , Milleron, O. , Stheneur, C. , Bourgeron, T. , Desguerre, I. , Jacob, M. P. , Gouya, L. , Génin, E. , Deleuze, J. F. , … Boileau, C. (2018). Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. European Journal of Human Genetics, 26(12), 1759–1772. 10.1038/s41431-018-0164-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubart, M. , Gross, M. S. , Hanna, N. , Zabot, M. T. , Sznajder, M. , Detaint, D. , Gouya, L. , Jondeau, G. , Boileau, C. , & Stheneur, C. (2015). The clinical presentation of Marfan syndrome is modulated by expression of wild‐type FBN1 allele. Human Molecular Genetics, 24(10), 2764–2770. 10.1093/hmg/ddv037 [DOI] [PubMed] [Google Scholar]
- Baldock, C. , Koster, A. J. , Ziese, U. , Rock, M. J. , Sherratt, M. J. , Kadler, K. E. , Shuttleworth, C.A. , & Kielty, C.M. (2001). The supramolecular organization of fibrillin‐rich microfibrils. Journal of Cell Biology, 152(5), 1045–1056. 10.1083/jcb.152.5.1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudhuin, L. M. , Kotzer, K. E. , & Lagerstedt, S. A. (2015). Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genetics in Medicine, 17(3), 177–187. 10.1038/gim.2014.91 [DOI] [PubMed] [Google Scholar]
- Beene, L. C. , Wang, L. W. , Hubmacher, D. , Keene, D. R. , Reinhardt, D. P. , Annis, D. S. , Mosher, D. F. , Mecham, R. P. , Traboulsi, E. I. , & Apte, S. S. (2013). Nonselective assembly of fibrillin 1 and fibrillin 2 in the rodent ocular zonule and in cultured cells: Implications for Marfan syndrome. Investigative Ophthalmology and Visual Science, 54(13), 8337–8344. 10.1167/iovs.13-13121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booms, P. , Tiecke, F. , Rosenberg, T. , Hagemeier, C. , & Robinson, P. N. (2000). Differential effect of FBN1 mutations on in vitro proteolysis of recombinant fibrillin‐1 fragments. Human Genetics, 107(3), 216–224. 10.1007/s004390000368 [DOI] [PubMed] [Google Scholar]
- Bork, P. , Downing, A. K. , Kieffer, B. , & Campbell, I. D. (1996). Structure and distribution of modules in extracellular proteins. Quarterly Review of Biophysics, 29(2), 119–167. http://www.ncbi.nlm.nih.gov/pubmed/8870072 [DOI] [PubMed] [Google Scholar]
- Cain, S. A. , McGovern, A. , Baldwin, A. K. , Baldock, C. , & Kielty, C. M. (2012). Fibrillin‐1 mutations causing Weill‐Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS One, 7, (11), e48634. 10.1371/journal.pone.0048634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert, B. L. , Loeys, B. L. , Ficcadenti, A. , Vermeer, S. , Landgren, M. , Kroes, H. Y. , De Paepe, A. M. , Yaron, Y. , Pope, M. , Foulds, N. , Boute, O. , Galán, F. , Kingston, H. , Van der Aa, N. , Salcedo, I. , Swinkels, M. E. , Wallgren‐Pettersson, C. , Gabrielli, O. , De Backer, J. , & De Paepe, A. M. (2009). Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: Report of 14 novel mutations and review of the literature. Human Mutation, 30(3), 334–341. 10.1002/humu.20854 [DOI] [PubMed] [Google Scholar]
- Campbell, I. D. , & Bork, P. (1993). Epidermal growth factor‐like modules. Current Opinion in Structural Biology, 3(3), 385–392. 10.1016/S0959-440x(05)80111-3 [DOI] [Google Scholar]
- Caputi, M. , Kendzior, R. J., Jr. , & Beemon, K. L. (2002). A nonsense mutation in the fibrillin‐1 gene of a Marfan syndrome patient induces NMD and disrupts an exonic splicing enhancer. Genes and Development, 16(14), 1754–1759. 10.1101/gad.997502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmical, S. G. , Gupta, P. , Milewicz, D. M. , & Putnam, E. A. (1999). FBN2 mutations identified in congenital contractural arachnodactyly patients with aortic root dilatation. American Journal of Human Genetics, 65(4), A6. [Google Scholar]
- Charbonneau, N. L. , Dzamba, B. J. , Ono, R. N. , Keene, D. R. , Corson, G. M. , Reinhardt, D. P. , & Sakai, L. Y. (2003). Fibrillins can co‐assemble in fibrils, but fibrillin fibril composition displays cell‐specific differences. Journal of Biological Chemistry, 278(4), 2740–2749. 10.1074/jbc.M209201200 [DOI] [PubMed] [Google Scholar]
- Charbonneau, N. L. , Jordan, C. D. , Keene, D. R. , Lee‐Arteaga, S. , Dietz, H. C. , Rifkin, D. B. , Ramirez, F. , & Sakai, L. Y. (2010). Microfibril structure masks fibrillin‐2 in postnatal tissues. Journal of Biological Chemistry, 285(26), 20242–20251. 10.1074/jbc.M109.087031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry, S. S. , Gazzard, J. , Baldock, C. , Dixon, J. , Rock, M. J. , Skinner, G. C. , Steel, K. P. , Kielty, C. M. , & Dixon, M. J. (2001). Mutation of the gene encoding fibrillin‐2 results in syndactyly in mice. Human Molecular Genetics, 10(8), 835–843. 10.1093/hmg/10.8.835 [DOI] [PubMed] [Google Scholar]
- Collod‐Béroud, G. , Le Bourdelles, S. , Ades, L. , Ala‐Kokko, L. , Booms, P. , Boxer, M. , Child, A. , Comeglio, P. , De Paepe, A. , Hyland, J.C. , Holman, K. , Kaitila, I. , Loeys, B. , Matyas, G. , Nuytinck, L. , Peltonen, L. , Rantamaki, T. , Robinson, P. , Steinmann, B. , … Boileau, C. (2003). Update of the UMD‐FBN1 mutation database and creation of an FBN1 polymorphism database. Human Mutation, 22(3), 199–208. 10.1002/humu.10249 [DOI] [PubMed] [Google Scholar]
- Corson, G. M. , Charbonneau, N. L. , Keene, D. R. , & Sakai, L. Y. (2004). Differential expression of fibrillin‐3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics, 83(3), 461–472. 10.1016/j.ygeno.2003.08.023 [DOI] [PubMed] [Google Scholar]
- Courtens, W. , Tjalma, W. , Messiaen, L. , Vamos, E. , Martin, J. J. , Van Bogaert, E. , Keersmaekers, G. , Meulyzer, P. , & Wauters, J. (1998). Prenatal diagnosis of a constitutional interstitial deletion of chromosome 5 (q15q31.1) presenting with features of congenital contractural arachnodactyly. American Journal of Medical Genetics, 77(3), 188–197. https://www.ncbi.nlm.nih.gov/pubmed/9605585 [PubMed] [Google Scholar]
- Del Cid, J. S. , Reed, N. I. , Molnar, K. , Liu, S. , Dang, B. , Jensen, S. A. , DeGrado, W. , Handford, P. A. , Sheppard, D. , & Sundaram, A. B. (2019). A disease‐associated mutation in fibrillin‐1 differentially regulates integrin‐mediated cell adhesion. Journal of Biological Chemistry, 294(48), 18232–18243. 10.1074/jbc.RA119.011109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhon, L. , Mahaut, C. , Goudin, N. , Gaudas, E. , Piquand, K. , Le Goff, W. , Cormier‐Daire, V. , & Le Goff, C. (2019). Impairment of chondrogenesis and microfibrillar network in Adamtsl2 deficiency. FASEB Journal, 33(2), 2707–2718. 10.1096/fj.201800753RR [DOI] [PubMed] [Google Scholar]
- Demirhan, O. , Türkmen, S. , Schwabe, G. C. , Soyupak, S. , Akgül, E. , Tastemir, D. , Karahan, D. , Mundlos, S. , & Lehmann, K. (2005). A homozygous BMPR1B mutation causes a new subtype of acromesomelic chondrodysplasia with genital anomalies. Journal of Medical Genetics, 42(4), 314–317. 10.1136/jmg.2004.023564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz, H. (2001). FBN1‐related Marfan syndrome. In Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Gripp K. W., Mirzaa G. M., & Amemiya A. (Eds.). GeneReviews® [Internet]. University of Washington, Seattle;1993‐2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1335/ [PubMed] [Google Scholar]
- Dietz, H. C. , Cutting, G. R. , Pyeritz, R. E. , Maslen, C. L. , Sakai, L. Y. , Corson, G. M , Puffenberger, E. G. , Hamosh, A. , Nanthakumar, E. J. , Curristin, S. M. , Stetten, G. , Meyers, D. A. , & Francomano, C. A. (1991). Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, 352(6333), 337–339. 10.1038/352337a0 [DOI] [PubMed] [Google Scholar]
- Downing, A. K. , Knott, V. , Werner, J. M. , Cardy, C. M. , Campbell, I. D. , & Handford, P. A. (1996). Solution structure of a pair of calcium‐binding epidermal growth factor‐like domains: Implications for the Marfan syndrome and other genetic disorders. Cell, 85(4), 597–605. 10.1016/S0092-8674(00)81259-3 [DOI] [PubMed] [Google Scholar]
- Duerrschmid, C. , He, Y. , Wang, C. , Li, C. , Bournat, J. C. , Romere, C. , Saha, P. K. , Lee, M. E. , Phillips, K. J. , Jain, M. , Jia, P. , Zhao, Z. , Farias, M. , Wu, Q. , Milewicz, D. M. , Sutton, V. R. , Moore, D. D. , Butte, N. F. , Krashes, M. J. , … Chopra, A. R. (2017). Asprosin is a centrally acting orexigenic hormone. Nature Medicine, 23(12), 1444–1453. 10.1038/nm.4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley, A. , Mellody, K. T. , Pilkington, S. , Griffiths, C. , Watson, R. , O'Cualain, R. , Baldock, C. , Knight, D. , & Sherratt, M. J. (2018). Structural and compositional diversity of fibrillin microfibrils in human tissues. Journal of Biological Chemistry, 293(14), 5117–5133. 10.1074/jbc.RA117.001483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbitar, S. , Renard, M. , Arnaud, P. , Hanna, N. , Jacob, M. P. , Guo, D. C. , Tsutsui, K. , Gross, M. S. , Kessler, K. , Tosolini, L. , Dattilo, V. , Dupont, S. , Jonquet, J. , Langeois, M. , Benarroch, L. , Aubart, M. , Ghaleb, Y. , Abou Khalil, Y. , Varret, M. , … Abifadel, M. (2021). Pathogenic variants in THSD4, encoding the ADAMTS‐like 6 protein, predispose to inherited thoracic aortic aneurysm. Genetics in Medicine, 23(1), 111–122. 10.1038/s41436-020-00947-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterly, N. B. , & Mckusick, V. A. (1971). Stiff skin syndrome. Pediatrics, 47(2), 360–369. [PubMed] [Google Scholar]
- Faivre, L. , Dollfus, H. , Lyonnet, S. , Alembik, Y. , Mégarbané, A. , Samples, J. , Gorlin, R.J. , Alswaid, A. , Feingold, J. , Le Merrer, M. , Munnich, A. , & Cormier‐Daire, V. (2003). Clinical homogeneity and genetic heterogeneity in Weill‐Marchesani syndrome. American Journal of Medical Genetics. Part A, 123A(2), 204–207. 10.1002/ajmg.a.20289 [DOI] [PubMed] [Google Scholar]
- Faivre, L. , Gorlin, R. J. , Wirtz, M. K. , Godfrey, M. , Dagoneau, N. , Samples, J. R. , Le Merrer, M. , Collod‐Beroud, G. , Boileau, C. , Munnich, A. , & Cormier‐Daire, V. (2003). In frame fibrillin‐1 gene deletion in autosomal dominant Weill‐Marchesani syndrome. Journal of Medical Genetics, 40(1), 34–36. 10.1136/jmg.40.1.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken, R. , den Hartog, A. W. , Radonic, T. , Micha, D. , Maugeri, A. , van Dijk, F. S. , Meijers‐Heijboer, H.E. , Timmermans, J. , Scholte, A.J. , van den Berg, M.P. , Groenink, M. , Mulder, B.J. , Zwinderman, A.H. , de Waard, V. , & Pals, G. (2015). Beneficial outcome of Losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circulation: Cardiovascular Genetics, 8(2), 383–388. 10.1161/CIRCGENETICS.114.000950 [DOI] [PubMed] [Google Scholar]
- Franken, R. , Groenink, M. , de Waard, V. , Feenstra, H. M. , Scholte, A. J. , van den Berg, M. P. , Pals, G. , Zwinderman, A. H. , Timmermans, J. , & Mulder, B. J. (2016). Genotype impacts survival in Marfan syndrome. European Heart Journal, 37(43), 3285–3290. 10.1093/eurheartj/ehv739 [DOI] [PubMed] [Google Scholar]
- Gentry, L. E. , Lioubin, M. N. , Purchio, A. F. , & Marquardt, H. (1988). Molecular events in the processing of recombinant type 1 pre‐pro‐transforming growth factor beta to the mature polypeptide. Molecular and Cellular Biology, 8(10), 4162–4168. http://www.ncbi.nlm.nih.gov/pubmed/3185545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globa, E. , Zelinska, N. , & Dauber, A. (2018). The clinical cases of geleophysic dysplasia: One gene, different phenotypes. Case Reports in Endocrinology, 2018, 8212417. 10.1155/2018/8212417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D. C. , Regalado, E. S. , Pinard, A. , Chen, J. , Lee, K. , Rigelsky, C. , Milewicz, D. M. , Zilberberg, L. , Hostetler, E. M. , Aldred, M. , Wallace, S. E. , Prakash, S. K. , University of Washington Center for Mendelian Genomics, Leal, S. M. , Bamshad, M. J. , Nickerson, D. A. , Natowicz, M. , Rifkin, D. B. , & Milewicz, D. M. (2018). LTBP3 pathogenic variants predispose individuals to thoracic aortic aneurysms and dissections. American Journal of Human Genetics, 102(4), 706–712. 10.1016/j.ajhg.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P. A. , Putnam, E. A. , Carmical, S. G. , Kaitila, I. , Steinmann, B. , Child, A. , Danesino, C. , Metcalfe, K. , Berry, S. A. , Chen, E. , Delorme, C. V. , Thong, M. K. , Adès, L. C. , & Milewicz, D. M. (2002). Ten novel FBN2 mutations in congenital contractural arachnodactyly: Delineation of the molecular pathogenesis and clinical phenotype. Human Mutation, 19(1), 39–48. 10.1002/Humu.10017 [DOI] [PubMed] [Google Scholar]
- Habashi, J. P. , Judge, D. P. , Holm, T. M. , Cohn, R. D. , Loeys, B. L. , Cooper, T. K. , Myers, L. , Klein, E. C. , Liu, G. , Calvi, C. , Podowski, M. , Neptune, E. R. , Halushka, M. K. , Bedja, D. , Gabrielson, K. , Rifkin, D. B. , Carta, L. , Ramirez, F. , Huso, D. L. , & Dietz, H. C. (2006). Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science, 312(5770), 117–121. 10.1126/science.1124287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji‐Seyed‐Javadi, R. , Jelodari‐Mamaghani, S. , Paylakhi, S. H. , Yazdani, S. , Nilforushan, N. , Fan, J. B. , Klotzle, B. , Mahmoudi, M. J. , Ebrahimian, M. J. , Chelich, N. , Taghiabadi, E. , Kamyab, K. , Boileau, C. , Paisan‐Ruiz, C. , Ronaghi, M. , & Elahi, E. (2012). LTBP2 mutations cause Weill‐Marchesani and Weill‐Marchesani‐like syndrome and affect disruptions in the extracellular matrix. Human Mutation, 33(8), 1182–1187. 10.1002/humu.22105 [DOI] [PubMed] [Google Scholar]
- Handford, P. A. , Mayhew, M. , Baron, M. , Winship, P. R. , Campbell, I. D. , & Brownlee, G. G. (1991). Key residues involved in calcium‐binding motifs in EGF‐like domains. Nature, 351(6322), 164–167. 10.1038/351164a0 [DOI] [PubMed] [Google Scholar]
- Hart, G. W. (1992). Glycosylation. Current Opinion in Cell Biology, 4(6), 1017–1023. 10.1016/0955-0674(92)90134-x [DOI] [PubMed] [Google Scholar]
- Hecht, F. , & Beals, R. K. (1972). "New" syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. Pediatrics, 49(4), 574–579. http://www.ncbi.nlm.nih.gov/pubmed/4552107 [PubMed] [Google Scholar]
- Hilhorst‐Hofstee, Y. , Hamel, B. C. , Verheij, J. B. , Rijlaarsdam, M. E. , Mancini, G. M. , Cobben, J. M. , Giroth, C. , Ruivenkamp, C. A. , Hansson, K. B. , Timmermans, J. , Moll, H. A. , Breuning, M. H. , & Pals, G. (2011). The clinical spectrum of complete FBN1 allele deletions. European Journal of Human Genetics, 19(3), 247–252. 10.1038/ejhg.2010.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz, B. (2013). It has to be the alphav: Myofibroblast integrins activate latent TGF‐beta1. Nature Medicine, 19(12), 1567–1568. 10.1038/nm.3421 [DOI] [PubMed] [Google Scholar]
- Hubmacher, D. , Reinhardt, D. P. , Plesec, T. , Schenke‐Layland, K. , & Apte, S. S. (2014). Human eye development is characterized by coordinated expression of fibrillin isoforms. Investigative Ophthalmology and Visual Science, 55(12), 7934–7944. 10.1167/iovs.14-15453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher, D. , Schneider, M. , Berardinelli, S. J. , Takeuchi, H. , Willard, B. , Reinhardt, D. P. , Haltiwanger, R. S. , & Apte, S. S. (2017). Unusual life cycle and impact on microfibril assembly of ADAMTS17, a secreted metalloprotease mutated in genetic eye disease. Scientific Reports, 7, 41871. 10.1038/srep41871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher, D. , Taye, N. , Balic, Z. , Thacker, S. , Adams, S. M. , Birk, D. E. , Schweitzer, R. , & Apte, S. S. (2019). Limb‐ and tendon‐specific Adamtsl2 deletion identifies a role for ADAMTSL2 in tendon growth in a mouse model for geleophysic dysplasia. Matrix Biology, 82, 38–53. 10.1016/j.matbio.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubmacher, D. , Wang, L. W. , Mecham, R. P. , Reinhardt, D. P. , & Apte, S. S. (2015). Adamtsl2 deletion results in bronchial fibrillin microfibril accumulation and bronchial epithelial dysplasia—A novel mouse model providing insights into geleophysic dysplasia. Disease Models & Mechanisms, 8(5), 487–499. 10.1242/dmm.017046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Gene Mutation Database (HGMD) . http://www.hgmd.cf.ac.uk/ac/index.php
- Hutchinson, S. , Furger, A. , Halliday, D. , Judge, D. P. , Jefferson, A. , Dietz, H. C. , Firth, H. , & Handford, P. A. (2003). Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: A potential modifier of phenotype? Human Molecular Genetics, 12(18), 2269–2276. 10.1093/hmg/ddg241 [DOI] [PubMed] [Google Scholar]
- Inbar‐Feigenberg, M. , Meirowitz, N. , Nanda, D. , Toi, A. , Okun, N. , & Chitayat, D. (2014). Beals syndrome (congenital contractural arachnodactyly): Prenatal ultrasound findings and molecular analysis. Ultrasound in Obstetrics and Gynecology, 44(4), 486–490. 10.1002/uog.13350 [DOI] [PubMed] [Google Scholar]
- Inoue, T. , Ohbayashi, T. , Fujikawa, Y. , Yoshida, H. , Akama, T. O. , Noda, K. , Horiguchi, M. , Kameyama, K. , Hata, Y. , Takahashi, K. , Kusumoto, K. , & Nakamura, T. (2014). Latent TGF‐beta binding protein‐2 is essential for the development of ciliary zonule microfibrils. Human Molecular Genetics, 23(21), 5672–5682. 10.1093/hmg/ddu283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai, Z. , Ono, R. N. , Ushiro, S. , Keene, D. R. , Chen, Y. , Mazzieri, R. , Charbonneau, N. L. , Reinhardt, D. P. , Rifkin, D. B. , & Sakai, L. Y. (2003). Latent transforming growth factor beta‐binding protein 1 interacts with fibrillin and is a microfibril‐associated protein. Journal of Biological Chemistry, 278(4), 2750–2757. 10.1074/jbc.M209256200 [DOI] [PubMed] [Google Scholar]
- Jensen, S. A. , Iqbal, S. , Bulsiewicz, A. , & Handford, P. A. (2015). A microfibril assembly assay identifies different mechanisms of dominance underlying Marfan syndrome, stiff skin syndrome and acromelic dysplasias. Human Molecular Genetics, 24(15), 4454–4463. 10.1093/hmg/ddv181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, C. J. , Sear, C. H. , & Grant, M. E. (1980). An ultrastructural study of fibroblasts derived from bovine ligamentum nuchae and their capacity for elastogenesis in culture. Journal of Pathology, 131(1), 35–53. 10.1002/path.1711310104 [DOI] [PubMed] [Google Scholar]
- Jovanovic, J. , Iqbal, S. , Jensen, S. , Mardon, H. , & Handford, P. (2008). Fibrillin‐integrin interactions in health and disease. Biochemical Society Transactions, 36, 257–262. 10.1042/Bst0360257 [DOI] [PubMed] [Google Scholar]
- Karoulias, S. Z. , Beyens, A. , Balic, Z. , Symoens, S. , Vandersteen, A. , Rideout, A. L. , Dickinson, J. , Callewaert, B. , & Hubmacher, D. (2020). A novel ADAMTS17 variant that causes Weill‐Marchesani syndrome 4 alters fibrillin‐1 and collagen type I deposition in the extracellular matrix. Matrix Biology, 88, 1–18. 10.1016/j.matbio.2019.11.001 [DOI] [PubMed] [Google Scholar]
- Keane, M. G. , & Pyeritz, R. E. (2008). Medical management of Marfan syndrome. Circulation, 117(21), 2802–2813. 10.1161/CIRCULATIONAHA.107.693523 [DOI] [PubMed] [Google Scholar]
- Kewley, M. A. , Williams, G. , & Steven, F. S. (1978). Studies of elastic tissue formation in the developing bovine ligamentum nuchae. Journal of Pathology, 124(2), 95–101. 10.1002/path.1711240205 [DOI] [PubMed] [Google Scholar]
- Kirschner, R. , Hubmacher, D. , Iyengar, G. , Kaur, J. , Fagotto‐Kaufmann, C. , Brömme, D. , Bartels, R. , & Reinhardt, D. P. (2011). Classical and neonatal Marfan syndrome mutations in fibrillin‐1 cause differential protease susceptibilities and protein function. Journal of Biological Chemistry, 286(37), 32810–32823. 10.1074/jbc.M111.221804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, C. , Le Goff, C. , Topouchian, V. , Odent, S. , Violas, P. , Glorion, C. , & Cormier‐Daire, V. (2014). Orthopedics management of acromicric dysplasia: Follow up of nine patients. American Journal of Medical Genetics, Part A, 164(2), 331–337. 10.1002/ajmg.a.36139 [DOI] [PubMed] [Google Scholar]
- Kloth, K. , Neu, A. , Rau, I. , Hülsemann, W. , Kutsche, K. , & Volk, A. E. (2021). Severe congenital contractural arachnodactyly caused by biallelic pathogenic variants in FBN2. European Journal of Medical Genetics, 64(3), 104161. 10.1016/j.ejmg.2021.104161 [DOI] [PubMed] [Google Scholar]
- Kochhar, A. , Kirmani, S. , Cetta, F. , Younge, B. , Hyland, J. C. , & Michels, V. (2013). Similarity of geleophysic dysplasia and Weill‐Marchesani syndrome. American Journal of Medical Genetics. Part A, 161A(12), 3130–3132. 10.1002/ajmg.a.36147 [DOI] [PubMed] [Google Scholar]
- Koli, K. , Saharinen, J. , Hyytiainen, M. , Penttinen, C. , & Keski‐Oja, J. (2001). Latency, activation, and binding proteins of TGF‐beta. Microscopy Research and Technique, 52(4), 354–362. [DOI] [PubMed] [Google Scholar]
- Kumaratilake, J. S. , Gibson, M. A. , Fanning, J. C. , & Cleary, E. G. (1989). The tissue distribution of microfibrils reacting with a monospecific antibody to MAGP, the major glycoprotein antigen of elastin‐associated microfibrils. European Journal of Cell Biology, 50(1), 117–127. http://www.ncbi.nlm.nih.gov/pubmed/2693088 [PubMed] [Google Scholar]
- Kutz, W. E. , Wang, L. W. , Bader, H. L. , Majors, A. K. , Iwata, K. , Traboulsi, E. I. , Sakai, L. Y. , Keene, D. R. , & Apte, S. S. (2011). ADAMTS10 protein interacts with fibrillin‐1 and promotes its deposition in extracellular matrix of cultured fibroblasts. Journal of Biological Chemistry, 286(19), 17156–17167. 10.1074/jbc.M111.231571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavillaureix, A. , Heide, S. , Chantot‐Bastaraud, S. , Marey, I. , Keren, B. , Grigorescu, R. , Jouannic, J. M. , Gelot, A. , Whalen, S. , Héron, D. , & Siffroi, J. P. (2017). Mosaic intragenic deletion of FBN2 and severe congenital contractural arachnodactyly. Clinical Genetics, 92(5), 556–558. 10.1111/cge.13062 [DOI] [PubMed] [Google Scholar]
- Lawrence, D. A. , Pircher, R. , Kryceve‐Martinerie, C. , & Jullien, P. (1984). Normal embryo fibroblasts release transforming growth factors in a latent form. Journal of Cellular Physiology, 121(1), 184–188. 10.1002/jcp.1041210123 [DOI] [PubMed] [Google Scholar]
- Lee, S. S. , Knott, V. , Jovanović, J. , Harlos, K. , Grimes, J. M. , Choulier, L. , Mardon, H. J. , Stuart, D. I. , & Handford, P. A. (2004). Structure of the integrin binding fragment from fibrillin‐1 gives new insights into microfibril organization. Structure, 12(4), 717–729. 10.1016/j.str.2004.02.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legare, J. M. , Modaff, P. , Strom, S. P. , Pauli, R. M. , & Bartlett, H. L. (2018). Geleophysic dysplasia: 48 year clinical update with emphasis on cardiac care. American Journal of Medical Genetics. Part A, 176(11), 2237–2242. 10.1002/ajmg.a.40377 [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , Morice‐Picard, F. , Dagoneau, N. , Wang, L. W. , Perrot, C. , Crow, Y. J. , Bauer, F. , Flori, E. , Prost‐Squarcioni, C. , Krakow, D. , Ge, G. , Greenspan, D. S. , Bonnet, D. , Le Merrer, M. , Munnich, A. , Apte, S. S. , & Cormier‐Daire, V. (2008). ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS‐like proteins in TGF‐beta bioavailability regulation. Nature Genetics, 40(9), 1119–1123. 10.1038/ng.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiden Open Variant Database (LOVD) . https://www.lovd.nl/
- Li, W. , Li, Q. , Jiao, Y. , Qin, L. , Ali, R. , Zhou, J. , & Tellides, G. (2014). Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. Journal of Clinical Investigation, 124(2), 755–767. 10.1172/JCI69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, G. , Tiedemann, K. , Vollbrandt, T. , Peters, H. , Batge, B. , Brinckmann, J. , & Reinhardt, D. P. (2002). Homo‐ and heterotypic fibrillin‐1 and ‐2 interactions constitute the basis for the assembly of microfibrils. Journal of Biological Chemistry, 277(52), 50795–50804. 10.1074/jbc.M210611200 [DOI] [PubMed] [Google Scholar]
- Liu, W. , Schrijver, I. , Brenn, T. , Furthmayr, H. , & Francke, U. (2001). Multi‐exon deletions of the FBN1 gene in Marfan syndrome. BMC Medical Genetics, 2, 11. 10.1186/1471-2350-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys, B. , Nuytinck, L. , Delvaux, I. , De Bie, S. , & De Paepe, A. (2001). Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin‐1 gene FBN1 because of suspected Marfan syndrome. Archives of Internal Medicine, 161(20), 2447–2454. 10.1001/archinte.161.20.2447 [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Dietz, H. C. , Braverman, A. C. , Callewaert, B. L. , De Backer, J. , Devereux, R. B. , Hilhorst‐Hofstee, Y. , Jondeau, G. , Faivre, L. , Milewicz, D. M. , Pyeritz, R. E. , Sponseller, P. D. , Wordsworth, P. , & De Paepe, A. M. (2010). The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics, 47(7), 476–485. 10.1136/jmg.2009.072785 [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Gerber, E. E. , Riegert‐Johnson, D. , Iqbal, S. , Whiteman, P. , McConnell, V. , Chillakuri, C. R. , Macaya, D. , Coucke, P.J. , De Paepe, A. , Judge, D. P. , Wigley, F. , Davis, E. C. , Mardon, H. J. , Handford, P. , Keene, D. R. , Sakai, L. Y. , & Dietz, H. C. (2010). Mutations in fibrillin‐1 cause congenital scleroderma: Stiff skin syndrome. Science Translational Medicine, 2(23), 23ra20. 10.1126/scitranslmed.3000488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lönnqvist, L. , Karttunen, L. , Rantamäki, T. , Kielty, C. , Raghunath, M. , & Peltonen, L. (1996). A point mutation creating an extra N‐glycosylation site in fibrillin‐1 results in neonatal Marfan syndrome. Genomics, 36(3), 468–475. 10.1006/geno.1996.0492 [DOI] [PubMed] [Google Scholar]
- Maeda, J. , Kosaki, K. , Shiono, J. , Kouno, K. , Aeba, R. , & Yamagishi, H. (2016). Variable severity of cardiovascular phenotypes in patients with an early‐onset form of Marfan syndrome harboring FBN1 mutations in exons 24‐32. Heart and Vessels, 31(10), 1717–1723. 10.1007/s00380-016-0793-2 [DOI] [PubMed] [Google Scholar]
- Marzin, P. , Thierry, B. , Dancasius, A. , Cavau, A. , Michot, C. , Rondeau, S. , Baujat, G. , Phan, G. , Bonnière, M. , Le Bourgeois, M. , Khraiche, D. , Pejin, Z. , Bonnet, D. , Delacourt, C. , & Cormier‐Daire, V. (2020). Geleophysic and acromicric dysplasias: Natural history, genotype‐phenotype correlations, and management guidelines from 38 cases. Genetics in Medicine, 23, 331–340. 10.1038/s41436-020-00994-x [DOI] [PubMed] [Google Scholar]
- McInerney‐Leo, A. M. , Le Goff, C. , Leo, P. J. , Kenna, T. J. , Keith, P. , Harris, J. E. , Steer, R. , Bole‐Feysot, C. , Nitschke, P. , Kielty, C. , Brown, M. A. , Zankl, A. , Duncan, E. L. , & Cormier‐Daire, V. (2016). Mutations in LTBP3 cause acromicric dysplasia and geleophysic dysplasia. Journal of Medical Genetics, 53(7), 457–464. 10.1136/jmedgenet-2015-103647 [DOI] [PubMed] [Google Scholar]
- Mead, T. J. , & Apte, S. S. (2018). ADAMTS proteins in human disorders. Matrix Biology, 71‐72, 225–239. 10.1016/j.matbio.2018.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerschaut, I. , De Coninck, S. , Steyaert, W. , Barnicoat, A. , Bayat, A. , Benedicenti, F. , Berland, S. , Blair, E.M. , Breckpot, J. , de Burca, A. , Destrée, A. , García‐Miñaúr, S. , Green, A.J. , Hanna, B.C. , Keymolen, K. , Koopmans, M. , Lederer, D. , Lees, M. , Longman, C. , … Callewaert, B. (2020). A clinical scoring system for congenital contractural arachnodactyly. Genetics in Medicine, 22(1), 124–131. 10.1038/s41436-019-0609-8 [DOI] [PubMed] [Google Scholar]
- Meester, J. , Peeters, S. , Van Den Heuvel, L. , Vandeweyer, G. , Fransen, E. , Cappella, E. , Dietz, H. C. , Forbus, G. , Gelb, B. D. , Goldmuntz, E. , Hoskoppal, A. , Landstrom, A. P. , Lee, T. , Mital, S. , Morris, S. , Olson, A. K. , Renard, M. , Roden, D. M. , Singh, M. N. , … Loeys, B. L. (2022). Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort. Genetics in Medicine. S1098‐3600 (21)05474‐05475. 10.1016/j.gim.2021.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewicz, D. M. , Grossfield, J. , Cao, S. N. , Kielty, C. , Covitz, W. , & Jewett, T. (1995). A mutation in FBN1 disrupts profibrillin processing and results in isolated skeletal features of the Marfan syndrome. Journal of Clinical Investigation, 95(5), 2373–2378. 10.1172/JCI117930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, G. , Neilan, M. , Chia, R. , Gheryani, N. , Holt, N. , Charbit, A. , Wells, S. , Tucci, V. , Lalanne, Z. , Denny, P. , Fisher, E. M. , Cheeseman, M. , Askew, G. N. , & Dear, T. N. (2010). ENU mutagenesis reveals a novel phenotype of reduced limb strength in mice lacking fibrillin 2. PLoS One, 5(2), e9137. 10.1371/journal.pone.0009137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono, K. , Hellman, U. , Wernstedt, C. , & Heldin, C. H. (1988). Latent high molecular weight complex of transforming growth factor beta 1. Purification from human platelets and structural characterization. Journal of Biological Chemistry, 263(13), 6407–6415. http://www.ncbi.nlm.nih.gov/pubmed/3162913 [PubMed] [Google Scholar]
- Mortier, G. R. , Cohn, D. H. , Cormier‐Daire, V. , Hall, C. , Krakow, D. , Mundlos, S. , Nishimura, G. , Robertson, S. , Sangiorgi, L. , Savarirayan, R. , Sillence, D. , Superti‐Furga, A. , Unger, S. , & Warman, M. L. (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. American Journal of Medical Genetics. Part A, 179(12), 2393–2419. 10.1002/ajmg.a.61366 [DOI] [PubMed] [Google Scholar]
- Mularczyk, E. J. , Singh, M. , Godwin, A. , Galli, F. , Humphreys, N. , Adamson, A. D. , Mironov, A. , Cain, S. A. , Sengle, G. , Boot‐Handford, R. P. , Cossu, G. , Kielty, C. M. , & Baldock, C. (2018). ADAMTS10‐mediated tissue disruption in Weill‐Marchesani syndrome. Human Molecular Genetics, 27(21), 3675–3687. 10.1093/hmg/ddy276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, C. M. , Cheng, A. , Myers, L. A. , Martinez‐Murillo, F. , Jie, C. , Bedja, D. , Gabrielson, K. L. , Hausladen, J. M. , Mecham, R. P. , Judge, D.P. , & Dietz, H. C. (2004). TGF‐beta‐dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. Journal of Clinical Investigation, 114(11), 1586–1592. 10.1172/JCI22715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistala, H. , Lee‐Arteaga, S. , Smaldone, S. , Siciliano, G. , Carta, L. , Ono, R. N. , Sengle, G. , Arteaga‐Solis, E. , Levasseur, R. , Ducy, P. , Sakai, L. Y. , Karsenty, G. , & Ramirez, F. (2010). Fibrillin‐1 and ‐2 differentially modulate endogenous TGF‐beta and BMP bioavailability during bone formation. Journal of Cell Biology, 190(6), 1107–1121. 10.1083/jcb.201003089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man (OMIM) . https://omim.org/
- Peeters, S. , Decramer, A. , Cain, S. A. , Houpt, P. , Verstreken, F. , Noyez, J. , Hermans, C. , Jacobs, W. , Lammens, M. , Fransen, E. , Kumar, A.A. , Vandeweyer, G. , Loeys, B. , Van Hul, W. , Baldock, C. , Boudin, E. , & Mortier, G. (2020). Delineation of a new fibrillino‐2‐pathy with evidence for a role of FBN2 in the pathogenesis of carpal tunnel syndrome. Journal of Medical Genetics, 58, 778–782. 10.1136/jmedgenet-2020-107085 [DOI] [PubMed] [Google Scholar]
- Piha‐Gossack, A. , Sossin, W. , & Reinhardt, D. P. (2012). The evolution of extracellular fibrillins and their functional domains. PLoS One, 7(3), e33560. 10.1371/journal.pone.0033560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottie, L. , Adamo, C. S. , Beyens, A. , Lütke, S. , Tapaneeyaphan, P. , De Clercq, A. , Salmon, P.L. , De Rycke, R. , Gezdirici, A. , Gulec, E.Y. , Khan, N. , Urquhart, J.E. , Newman, W.G. , Metcalfe, K. , Efthymiou, S. , Maroofian, R. , Anwar, N. , Maqbool, S. , Rahman, F. , … Callewaert, B. (2021). Bi‐allelic premature truncating variants in LTBP1 cause cutis laxa syndrome. American Journal of Human Genetics, 108(12), 2386–2388. 10.1016/j.ajhg.2021.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, R. Q. , & Glanville, R. W. (1997). Alignment of fibrillin molecules in elastic microfibrils is defined by transglutaminase‐derived cross‐links. Biochemistry, 36(50), 15841–15847. 10.1021/bi971036f [DOI] [PubMed] [Google Scholar]
- Quarto, N. , Leonard, B. , Li, S. , Marchand, M. , Anderson, E. , Behr, B. , Francke, U. , Reijo‐Pera, R. , Chiao, E. , & Longaker, M. T. (2012). Skeletogenic phenotype of human Marfan embryonic stem cells faithfully phenocopied by patient‐specific induced‐pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America, 109(1), 215–220. 10.1073/pnas.1113442109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quondamatteo, F. , Reinhardt, D. P. , Charbonneau, N. L. , Pophal, G. , Sakai, L. Y. , & Herken, R. (2002). Fibrillin‐1 and fibrillin‐2 in human embryonic and early fetal development. Matrix Biology, 21(8), 637–646. 10.1016/s0945-053x(02)00100-2 [DOI] [PubMed] [Google Scholar]
- Rao, Z. , Handford, P. , Mayhew, M. , Knott, V. , Brownlee, G. G. , & Stuart, D. (1995). The structure of a Ca(2+)‐binding epidermal growth factor‐like domain: its role in protein‐protein interactions. Cell, 82(1), 131–141. http://www.ncbi.nlm.nih.gov/pubmed/7606779 [DOI] [PubMed] [Google Scholar]
- Reinhardt, D. P. , Ono, R. N. , & Sakai, L. Y. (1997). Calcium stabilizes fibrillin‐1 against proteolytic degradation. Journal of Biological Chemistry, 272(2), 1231–1236. http://www.ncbi.nlm.nih.gov/pubmed/8995426 [DOI] [PubMed] [Google Scholar]
- Robertson, I. , Jensen, S. , & Handford, P. (2011). TB domain proteins: Evolutionary insights into the multifaceted roles of fibrillins and LTBPs. Biochemical Journal, 433(2), 263–276. 10.1042/BJ20101320 [DOI] [PubMed] [Google Scholar]
- Robertson, I. B. , Horiguchi, M. , Zilberberg, L. , Dabovic, B. , Hadjiolova, K. , & Rifkin, D. B. (2015). Latent TGF‐beta‐binding proteins. Matrix Biology, 47, 44–53. 10.1016/j.matbio.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romere, C. , Duerrschmid, C. , Bournat, J. , Constable, P. , Jain, M. , Xia, F. , Saha, P.K. , Del Solar, M. , Zhu, B. , York, B. , Sarkar, P. , Rendon, D.A. , Gaber, M.W. , LeMaire, S. A. , Coselli, J. S. , Milewicz, D. M. , Sutton, V. R. , Butte, N. F. , Moore, D. D. , & Chopra, A. R. (2016). Asprosin, a fasting‐induced glucogenic protein hormone. Cell, 165(3), 566–579. 10.1016/j.cell.2016.02.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel, K. , Karck, M. , Haverich, A. , von Kodolitsch, Y. , Rybczynski, M. , Müller, G. , Singh, K. K. , Schmidtke, J. , & Arslan‐Kirchner, M. (2005). Identification of 29 novel and nine recurrent fibrillin‐1 (FBN1) mutations and genotype‐phenotype correlations in 76 patients with Marfan syndrome. Human Mutation, 26(6), 529–539. 10.1002/humu.20239 [DOI] [PubMed] [Google Scholar]
- Ross, R. , & Bornstein, P. (1969). The elastic fiber. I. The separation and partial characterization of its macromolecular components. Journal of Cell Biology, 40(2), 366–381. http://www.ncbi.nlm.nih.gov/pubmed/5812469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier, L. , Djokic, J. , Hubmacher, D. , Dzafik, D. , Nelea, V. , & Reinhardt, D. P. (2014). Heparin/heparan sulfate controls fibrillin‐1, ‐2 and ‐3 self‐interactions in microfibril assembly. FEBS Letters, 588(17), 2890–2897. 10.1016/j.febslet.2014.06.061 [DOI] [PubMed] [Google Scholar]
- Sabatier, L. , Miosge, N. , Hubmacher, D. , Lin, G. , Davis, E. C. , & Reinhardt, D. P. (2011). Fibrillin‐3 expression in human development. Matrix Biology, 30(1), 43–52. 10.1016/j.matbio.2010.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, L. Y. , & Keene, D. R. (2019). Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biology, 80, 6–13. 10.1016/j.matbio.2018.09.005 [DOI] [PubMed] [Google Scholar]
- Sakai, L. Y. , Keene, D. R. , & Engvall, E. (1986). Fibrillin, a new 350‐kD glycoprotein, is a component of extracellular microfibrils. Journal of Cell Biology, 103(6, Pt 1), 2499–2509. http://www.ncbi.nlm.nih.gov/pubmed/3536967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, L. Y. , Keene, D. R. , Renard, M. , & De Backer, J. (2016). FBN1: The disease‐causing gene for Marfan syndrome and other genetic disorders. Gene, 591(1), 279–291. 10.1016/j.gene.2016.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, Y. , & Rifkin, D. B. (1989). Inhibition of endothelial cell movement by pericytes and smooth muscle cells: Activation of a latent transforming growth factor‐beta 1‐like molecule by plasmin during co‐culture. Journal of Cell Biology, 109(1), 309–315. http://www.ncbi.nlm.nih.gov/pubmed/2526131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijver, I. , Liu, W. , Odom, R. , Brenn, T. , Oefner, P. , Furthmayr, H. , & Francke, U. (2002). Premature termination mutations in FBN1: Distinct effects on differential allelic expression and on protein and clinical phenotypes. American Journal of Human Genetics, 71(2), 223–237. 10.1086/341581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz‐Cherry, S. , & Murphy‐Ullrich, J. E. (1993). Thrombospondin causes activation of latent transforming growth factor‐beta secreted by endothelial cells by a novel mechanism. Journal of Cell Biology, 122(4), 923–932. http://www.ncbi.nlm.nih.gov/pubmed/8349738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengle, G. , Carlberg, V. , Tufa, S. F. , Charbonneau, N. L. , Smaldone, S. , Carlson, E. J. , Ramirez, F. , Keene, D. R. , & Sakai, L. Y. (2015). Abnormal activation of BMP signaling causes myopathy in Fbn2 null mice. PLoS Genetics, 11(6), e1005340. 10.1371/journal.pgen.1005340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengle, G. , Charbonneau, N. L. , Ono, R. N. , Sasaki, T. , Alvarez, J. , Keene, D. R. , Bächinger, H.P. , & Sakai, L. Y. (2008). Targeting of bone morphogenetic protein growth factor complexes to fibrillin. Journal of Biological Chemistry, 283(20), 13874–13888. 10.1074/jbc.M707820200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengle, G. , Tsutsui, K. , Keene, D. R. , Tufa, S. F. , Carlson, E. J. , Charbonneau, N. L. , Ono, R.N. , Sasaki, T. , Wirtz, M.K. , Samples, J. R. , Fessler, L. I. , Fessler, J. H. , Sekiguchi, K. , Hayflick, S. J. , & Sakai, L. Y. (2012). Microenvironmental regulation by fibrillin‐1. PLoS Genetics, 8(1), e1002425. 10.1371/journal.pgen.1002425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, M. , Zhu, J. , Wang, R. , Chen, X. , Mi, L. , Walz, T. , & Springer, T. A. (2011). Latent TGF‐beta structure and activation. Nature, 474(7351), 343–349. 10.1038/nature10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaldone, S. , del Solar, A.‐P. N. , & Ramirez F, M. (2018). Tendon‐dependent control of longitudinal bone growth [Abstract]. Orthopaedic Proceedings, 97‐B(Suppl_11). [Google Scholar]
- Sterzel, R. B. , Hartner, A. , Schlötzer‐Schrehardt, U. , Voit, S. , Hausknecht, B. , Doliana, R. , Colombatti, A. , Gibson, M.A. , Braghetta, P. , & Bressan, G. M. (2000). Elastic fiber proteins in the glomerular mesangium in vivo and in cell culture. Kidney International, 58(4), 1588–1602. 10.1046/j.1523-1755.2000.00320.x [DOI] [PubMed] [Google Scholar]
- Suk, J. Y. , Jensen, S. , McGettrick, A. , Willis, A. C. , Whiteman, P. , Redfield, C. , & Handford, P. A. (2004). Structural consequences of cysteine substitutions C1977Y and C1977R in calcium‐binding epidermal growth factor‐like domain 30 of human fibrillin‐1. Journal of Biological Chemistry, 279(49), 51258–51265. 10.1074/jbc.M408156200 [DOI] [PubMed] [Google Scholar]
- Takeda, N. , Morita, H. , Fujita, D. , Inuzuka, R. , Taniguchi, Y. , Imai, Y. , Komuro, I. , Hirata Y., Komuro I. (2015). Congenital contractural arachnodactyly complicated with aortic dilatation and dissection: Case report and review of literature. American Journal of Medical Genetics. Part A, 167A(10), 2382–2387. 10.1002/ajmg.a.37162 [DOI] [PubMed] [Google Scholar]
- Thomas, J. T. , Kilpatrick, M. W. , Lin, K. , Erlacher, L. , Lembessis, P. , Costa, T. , Tsipouras, P. , & Luyten, F. P. (1997). Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nature Genetics, 17(1), 58–64. 10.1038/ng0997-58 [DOI] [PubMed] [Google Scholar]
- Thomson, J. , Singh, M. , Eckersley, A. , Cain, S. A. , Sherratt, M. J. , & Baldock, C. (2019). Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Seminars in Cell and Developmental Biology, 89, 109–117. 10.1016/j.semcdb.2018.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond, F. , & Trotter, J. (1996). Morphology and biomechanics of the microfibrillar network of sea cucumber dermis. Journal of Experimental Biology, 199(Pt 8), 1817–1828. http://www.ncbi.nlm.nih.gov/pubmed/9319729 [DOI] [PubMed] [Google Scholar]
- Tiedemann, K. , Batge, B. , Muller, P. K. , & Reinhardt, D. P. (2001). Interactions of fibrillin‐1 with heparin/heparan sulfate, implications for microfibrillar assembly. Journal of Biological Chemistry, 276(38), 36035–36042. 10.1074/jbc.M104985200 [DOI] [PubMed] [Google Scholar]
- Trask, T. M. , Ritty, T. M. , Broekelmann, T. , Tisdale, C. , & Mecham, R. P. (1999). N‐terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: A possible first step in microfibril assembly. Biochemical Journal, 340(Pt 3), 693–701. http://www.ncbi.nlm.nih.gov/pubmed/10359653 [PMC free article] [PubMed] [Google Scholar]
- Universal Mutation Database (UMD)‐FBN1 . http://www.umd.be/FBN1
- Universal Mutation Database (UMD)‐FBN2 . http://www.umd.be/FBN2
- Verloes, A. , Jamblin, P. , Koulischer, L. , & Bourguignon, J. P. (1996). A new form of skeletal dysplasia with amelogenesis imperfecta and platyspondyly. Clinical Genetics, 49(1), 2–5. 10.1111/j.1399-0004.1996.tb04315.x [DOI] [PubMed] [Google Scholar]
- Verstraeten, A. , Meester, J. , Peeters, S. , Mortier, G. , & Loeys, B. (2020). Chondrodysplasias and aneurysmal thoracic aortopathy: An emerging tale of molecular intersection. Trends in Molecular Medicine, 26(8), 783–795. 10.1016/j.molmed.2020.05.004 [DOI] [PubMed] [Google Scholar]
- Viljoen, D. (1994). Congenital contractural arachnodactyly (Beals syndrome). Journal of Medical Genetics, 31(8), 640–643. http://www.ncbi.nlm.nih.gov/pubmed/7815423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollbrandt, T. , Tiedemann, K. , El‐Hallous, E. , Lin, G. , Brinckmann, J. , John, H. , Bätge, B. , Notbohm, H. , & Reinhardt, D. P. (2004). Consequences of cysteine mutations in calcium‐binding epidermal growth factor modules of fibrillin‐1. Journal of Biological Chemistry, 279(31), 32924–32931. 10.1074/jbc.M405239200 [DOI] [PubMed] [Google Scholar]
- Wang, M. , Clericuzio, C. L. , & Godfrey, M. (1996). Familial occurrence of typical and severe lethal congenital contractural arachnodactyly caused by missplicing of exon 34 of fibrillin‐2. American Journal of Human Genetics, 59(5), 1027–1034. http://www.ncbi.nlm.nih.gov/pubmed/8900230 [PMC free article] [PubMed] [Google Scholar]
- Wei, H. , Hu, J. H. , Angelov, S. N. , Fox, K. , Yan, J. , Enstrom, R. , Smith, A. , & Dichek, D. A. (2017). Aortopathy in a mouse model of Marfan syndrome is not mediated by altered transforming growth factor beta signaling. Journal of the American Heart Association, 6(1), e004968. 10.1161/JAHA.116.004968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadin, D. A. , Robertson, I. B. , McNaught‐Davis, J. , Evans, P. , Stoddart, D. , Handford, P. A. , Jensen, S. A. , & Redfield, C. (2013). Structure of the fibrillin‐1 N‐terminal domains suggests that heparan sulfate regulates the early stages of microfibril assembly. Structure, 21(10), 1743–1756. 10.1016/j.str.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q. , & Stamenkovic, I. (2000). Cell surface‐localized matrix metalloproteinase‐9 proteolytically activates TGF‐beta and promotes tumor invasion and angiogenesis. Genes and Development, 14(2), 163–176. http://www.ncbi.nlm.nih.gov/pubmed/10652271 [PMC free article] [PubMed] [Google Scholar]
- Yuan, X. , Downing, A. K. , Knott, V. , & Handford, P. A. (1997). Solution structure of the transforming growth factor beta‐binding protein‐like module, a domain associated with matrix fibrils. EMBO Journal, 16(22), 6659–6666. 10.1093/emboj/16.22.6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyer, K. A. , Zhang, R. M. , Kumra, H. , Hassan, A. , & Reinhardt, D. P. (2019). The fibrillin‐1 RGD integrin binding site regulates gene expression and cell function through microRNAs. Journal of Molecular Biology, 431(2), 401–421. 10.1016/j.jmb.2018.11.021 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Hu, W. , & Ramirez, F. (1995). Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. Journal of Cell Biology, 129(4), 1165–1176. 10.1083/jcb.129.4.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. M. , Zeyer, K. A. , Odenthal, N. , Zhang, Y. , & Reinhardt, D. P. (2021). The fibrillin‐1 RGD motif posttranscriptionally regulates ERK1/2 signaling and fibroblast proliferation via miR‐1208. FASEB Journal, 35(5), e21598. 10.1096/fj.202100282R [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.