

Abstract
The development of targeted therapies for the treatment of myelofibrosis highlights a unique issue in a field that has historically relied on symptom relief, rather than survival benefit or modification of disease course, as key response criteria. There is, therefore, a need to understand what constitutes disease modification of myelofibrosis to advance appropriate drug development and therapeutic pathways. Here, the authors discuss recent clinical trial data of agents in development and dissect the potential for novel end points to act as disease modifying parameters. Using the rationale garnered from latest clinical and scientific evidence, the authors propose a definition of disease modification in myelofibrosis. With improved overall survival a critical outcome, alongside the normalization of hematopoiesis and improvement in bone marrow fibrosis, there will be an increasing need for surrogate measures of survival for use in the early stages of trials. As such, the design of future clinical trials will require re‐evaluation and updating to incorporate informative parameters and end points with standardized definitions and methodologies.
Keywords: bone marrow fibrosis, disease modification, myelofibrosis, myelofibrosis pathophysiology, targeted therapy
Short abstract
There is a need to understand and define disease modification in myelofibrosis. This review examines the latest clinical data of targeted therapies for myelofibrosis and proposes a definition of disease modification.
A Unique Clinical Challenge in Myelofibrosis
Myelofibrosis (MF) is primarily driven by constitutive activation of the Janus kinase (JAK)/signal transducers and activators of transcription (STAT) pathway. The first approved targeted therapy class for MF, JAK inhibitors (JAKi) have demonstrated amelioration of some key disease symptoms but have otherwise failed to provide survival benefits. This has created a standard for disease response in MF that focuses on symptom relief rather than benefit to progression‐free or overall survival (PFS and OS, respectively) or disease modification. This review aims to evaluate, based on clinical and scientific evidence, what constitutes disease modification in MF, together with how this may be measured clinically. The latest developing clinical trial data will be discussed, alongside how these data may inform disease modification, in an attempt to refocus future clinical trial design and MF patient care on potential disease cure and patient survival outcomes.
Introduction to Myelofibrosis
MF pathology includes myeloproliferation, inflammation, bone marrow fibrosis (BMF), extra‐medullary hematopoiesis (EMH), splenomegaly, anemia, thrombocytopenia, and constitutional symptoms. 1 , 2 , 3 The median OS of patients with primary MF is ~6 years, and is influenced by a variety of clinical and genetic features including age, mutations, bone marrow fibrosis (BMF), and treatment history. 4 , 5 , 6
Allogeneic stem cell transplantation (allo‐SCT) is currently the only curative option for MF, but its suitability is limited to a minority. 7 The JAKi ruxolitinib (JAK1/2i) and fedratinib (JAK2i) are currently the only approved treatments for patients with MF. 8 , 9 Despite the efficacy demonstrated by these JAKi in reducing splenomegaly and constitutional symptoms across a spectrum of MF patient subgroups, 10 , 11 , 12 , 13 , 14 , 15 JAKi exert little effect on BMF, driver mutation allele frequency (MAF), leukemic transformation or OS. Most patients ultimately experience ruxolitinib failure, leading to treatment discontinuation with limited alternative options. 2 , 16 There is, therefore, a need for novel treatment options with overt disease‐modifying activity.
Greater understanding of MF pathophysiology has unveiled multiple non‐JAK targets, leading to the development of several novel agents. 17 However, the lack of an accepted definition and assessment of disease modification is a key hurdle to progressing patient care. In particular, the absence of consistent and standardized parameters by which modification can be assessed limits the impact of emerging data and inadvertently promotes the development of agents that may not provide significant benefit to patients. Furthermore, the lack of coordination across clinical trial end points and clinical practice precludes the inter‐trial and ‐agent comparisons required to optimize treatment pathways.
As with any rapidly evolving field, it is important to acknowledge that many of the clinical trials discussed here are ongoing, and that many of the data that informed our rationale when defining disease modification are immature. The definition we propose will undoubtedly evolve and mature in line with the emergence of future data.
Myelofibrosis Disease Course
Disease Pathogenesis
Constitutive activation of the JAK/STAT pathway may be facilitated by driver mutations that confer a fitness advantage. 2 The resultant clonally expanded megakaryocytes cluster in the bone marrow (BM) where they are infiltrated by neutrophils. This results in a “cytokine storm” that generates an inflammatory BM microenvironment, stimulating fibrosis and angiogenesis (Fig. 1; Supporting Table 1). 1 , 3 , 6 , 18 , 19 As such, BMF is a proximal manifestation of disease biology, stemming from the underlying molecular dysregulation. As a direct result of the dysregulated BM microenvironment, hematopoietic progenitor cells subsequently migrate to sites of EMH, resulting in progressive splenomegaly. 2 , 20 JAKi alone do not robustly impact BMF, reverse abnormal hematopoiesis, or target the aberrant stem cell niche, suggesting that dysregulated JAK signaling is not the only driving factor of MF disease etiology.
Figure 1.
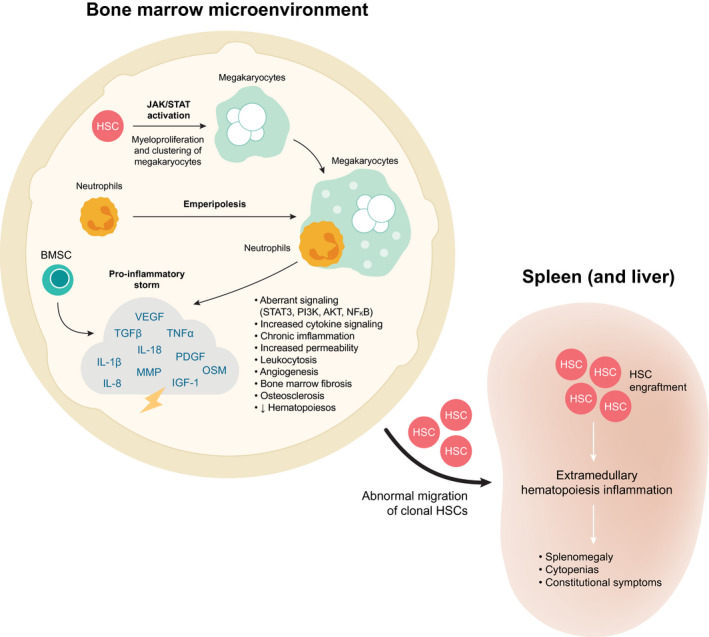
Overview of disease pathology. 3 , 6 , 18 , 73 , 77 , 78 , 79 BMSC indicates bone marrow stromal cell; FGF, fibroblast growth factor; HSC, hematopoietic stem cell; IGF‐1, insulin‐like growth factor‐1; IL, interleukin; JAK/STAT, Janus kinase/signal transducer and activator of transcription; MMP, matrix metalloproteinases; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; OSM, oncostatin M; OPG, osteoprotegerin; PDGF, platelet‐derived growth factor; PI3K, phosphoinositide 3‐kinase; TGF‐β, transforming growth factor β; TNF‐α, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
Monitoring Disease Progression
MF has a highly variable disease course, ranging from an indolent, asymptomatic disease, to BM failure or leukemic transformation, 3 , 17 with somatic mutations playing a central role in determining disease risk. 2 , 3 , 21 Progression is often clinically determined by worsening splenomegaly or leukemic transformation. 13
Novel, targeted therapies may have the potential to interrupt, or even reverse, the disease trajectory, possibly returning the BM microenvironment to a pre‐disease state (Fig. 2). 22 , 23 However, data are required to inform potential modifiers and to understand the impact of these on PFS and OS. This may serve to accelerate therapeutic developments and facilitate clinical decision‐making.
Figure 2.
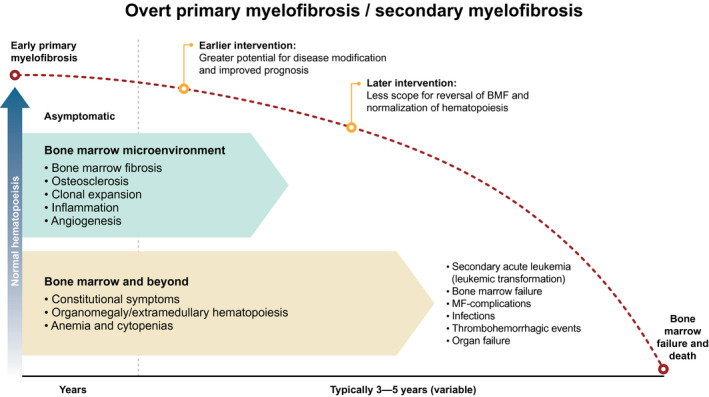
Natural history of myelofibrosis and potential time points for intervention. The red dotted line represents the decline in normal hematopoiesis along the natural course of disease. 1 , 2 , 23 , 79 BMF indicates bone marrow fibrosis; MF, myelofibrosis.
As mentioned, allo‐SCT is the only potential cure for patients with MF and thus determination of true remission and positioning of a patient for SCT is an important goal. To this end, measurable residual disease assessment (with the aim of clonal remission) is now recommended in several other hematological malignancies where it forms the backbone of patient management. 24 , 25 , 26 , 27 Molecular responses are commonly included in clinical trial designs and may be considered representative of disease modification. As experimental therapies begin to emerge for MF, similar responses may become a new hallmark of treatment and it will be important to define end points and their assessments accordingly. This approach will be of particular significance in the future, when commonly used agents begin to modify disease course and are capable of inducing remission and positioning patients for SCT with greater frequency.
Myelofibrosis Clinical Trials
Clinical Trial End Points
Given the role of the JAK/STAT pathway in driving MF pathology, JAK was an obvious initial therapeutic focus. As such, JAKi were the first approved targeted therapy class for MF, and although they have minimal impact on survival, they have been highly effective at controlling splenomegaly and constitutional symptoms. This has resulted in SVR, and total symptom score (TSS) becoming standard end points in MF trials (Supporting Table 2), 11 , 12 as reflected in the International Working Group for Myelofibrosis Research and Treatment and European Leukemia Net response criteria for myelofibrosis. 13 These symptom and quality‐of‐life based measures have remained the primary end points for MF treatment trials due to the lack of definitive pathological and/or biochemical criteria to determine MF progression or disease modification. This approach has culminated in a standard for disease response in MF that focuses on symptom relief rather than benefit to PFS, OS, or modification of disease course.
Recently, the number of clinical trial end points has expanded alongside emerging agents, including an increased use of patient reported outcomes (PROs) in parallel with OS, PFS, MAF, cytokine modulation, event‐free or leukemia‐free survival (failure to transform to leukemia), transfusion independence (TI), and reduction in BMF. 28 However, many of these present their own challenges, often due to a lack of standardized definitions. 29 , 30 , 31
Latest Clinical Trials of Novel Agents
Here, we discuss the key therapies that begin to inform how disease modification may be defined, with focus on potential disease‐modifying effects and the challenges of defining disease modification. Although many ongoing trials lack novel end points, these should not be overlooked. Importantly, they highlight the need for greater standardization between trial designs as MF research evolves. Early evidence of a progression toward nontraditional primary end points is encouraging, 32 , 33 and defining key modifiers will help to establish greater uniformity.
An overview of selected targets is presented in Figure 3, with the most advanced clinical trial data summarized in Table 1. Although a comprehensive analysis of all novel therapies is beyond the scope of this review, an overview of the most encouraging clinical trial data to emerge for novel therapies in phase 2/3 development for MF is presented in Supporting Table 3, and Supporting Table 4 lists ongoing trials of promising agents in early development.
Figure 3.
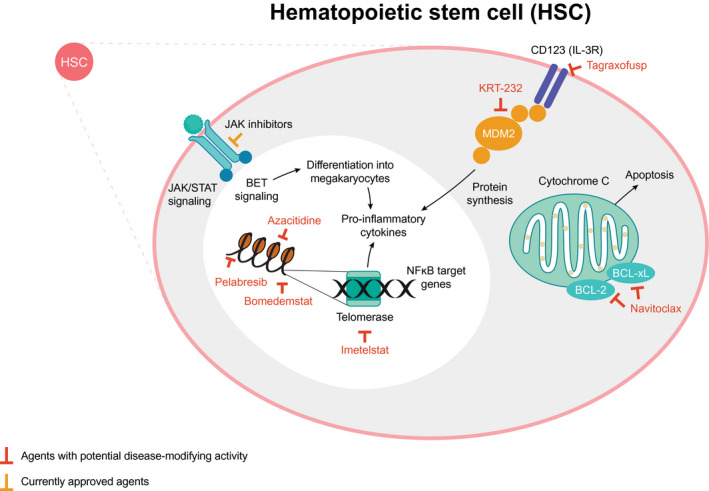
Novel and potentially disease‐modifying therapeutic targets in myelofibrosis. 80 , 81 , 82 BCL indicates B‐cell lymphoma; BET, bromodomain and extra‐terminal motif; IL, interleukin; JAK/STAT, Janus kinase/signal transducers and activators of transcription; MDM2, mouse double minute 2; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; R, receptor.
TABLE 1.
Novel Agents in Development for MF With Potential for Disease‐Modifying Activity
| Study Name/No. | Drug | MoA | Phase/Status | Population | Comparator | Primary End Point | BMF | Mutation Burden | Survival | Notes |
|---|---|---|---|---|---|---|---|---|---|---|
| Epigenetic modulation | ||||||||||
| NCT02158858 (MANIFEST) 40 , 41 , 43 , 44 , 63 , 74 | Pelabresib ± Rux | BET ± JAK1/2 inhibition | 1/2 Ongoing |
Arm 1: JAKi‐experienced (pelabresib) Arm 2: JAKi‐experienced (pelabresib + Rux) Arm 3: JAKi‐naive (pelabresib + Rux) N ≈ 271 |
Arm 1: SVR35 at wk 24: 24% TD → TI: 21% Arm 2: SVR35 at wk 24: 21% TD → TI: 36% Arm 3: SVR35 at wk 24: 67% |
1 Grade improvement: 33% | NR | NR | Several cytokines suppressed with mono and combination therapy | |
| NCT04603495 (MANIFEST‐2) 38 | Pelabresib + Rux | BET ± JAK1/2 inhibition | 3 Ongoing | JAKi‐ and BETi‐naive, N ≈ 310 | Placebo + Rux | SVR35 at wk 24 | ||||
| NCT03136185 46 | Bomedemstat | LSD1 inhibitor | 2 Ongoing | Rux‐naive and ‐experienced, N = 89 | None | AEs: 89% | 1 Grade improvement: 17% (stable 66%) | Reduction in MAF in driver and HMR mutations: 42% (stable in 44%) | NR | MAF reduction correlated with SVR and/or TSS |
| NCT01787487 61 | Azacitidine + Rux | HMA + JAK1/2 inhibition | 2 | Rux‐naive, azacitidine‐ naive, N = 60 | None | ORR: 74% | Improvement in BM morphology: 61% | NR | Median OS: not reached | |
| Hematopoietic stem cell compartment/bone marrow microenvironment | ||||||||||
| NCT03662126 (BOREAS) 60 | Navtemadlin | MDM2 inhibition | 2/3 Ongoing | JAKi‐experienced, N = 113 | None | NR | ≥1 Grade improvement: 27% | Reduction in MAF ≥20%: 34% (complete reduction 29%) | NR | Reduced circulating CD34+ cells and TNF‐α; MAF, BMF, CD34+, and TNF‐α correlated with SVR and/or TSS |
| NCT04640532 | Navtemadlin ± TL‐895 | MDM2 inhibition + tyrosine kinase inhibition | 1/2 Ongoing | JAKi‐experienced, N ≈ 116 | None | MTD/MAD; RP2D; SVR35 at wk 24 | — | — | — | |
| NCT04485260 59 | Navtemadlin + Rux | MDM2 inhibition + JAK1/2 inhibition | 1b/2 Ongoing | Rux‐experienced, N ≈ 78 | None | RP2D | — | — | Secondary end points: OS, PFS, LFS | |
| NCT02268253 63 | Tagraxofusp | Anti‐CD123 | 1/2 Ongoing | JAKi‐experienced, N ≈ 130 | None | AEs; RR: 60% SD | NR | NR | Median OS: 31 mo | |
| Apoptosis | ||||||||||
| NCT03222609 (REFINE) 31 , 51 | Navitoclax ± Rux | BCL inhibition ± JAK1/2 inhibition | 2 Ongoing | Rux‐experienced, N ≈ 174 | None | SVR35 at wk 24: 27% (dual therapy) | ≥1 Grade improvement: 33% | >10% reduction in driver gene MAF: 46% | Median OS: not reached | Changes in MF‐associated cytokines were correlated with SV changes |
| NCT04472598 (TRANSFORM‐1) 52 | Navitoclax + Rux | BCL inhibition + JAK1/2 inhibition | 3 Ongoing | JAKi‐naive, N ≈ 230 | Placebo | SVR35 at wk 24 | Secondary end point | — | Additional end points: OS, LFS, PFS | |
| NCT04468984 (TRANSFORM‐2) 53 | Navitoclax + Rux | BCL inhibition + JAK1/2 inhibition | 3 Ongoing | JAKi‐experienced, N ≈ 330 | BAT | SVR35 at wk 24 | Secondary end point | — | Additional end points: OS, LFS, PFS | |
| Telomerase | ||||||||||
| NCT04576156 (MYF3001) | Imetelstat | Telomerase inhibition | 3 Ongoing | JAKi‐experienced, N ≈ 320 | BAT | OS | Secondary end point | — | — | Biomarker and mutation analyses will be performed |
| NCT02426086 (IMbark) 56 | Imetelstat | Telomerase inhibition | 2 Complete | JAKi‐experienced, N = 59 | None | SVR35 at wk 24: 10.2% ≥50%; reduction in TSS at wk 24: 32.2% | Improvement: 40.5% | Reduction in driver mutation MAF: 42.1% | Median OS: 29.9 mo | |
| Immune therapies | ||||||||||
| NCT01178281 (RESUME) 75 | Pomalidomide | IMiD | 3 Complete | N = 252 | Placebo | RBC‐TI ≥84 days within 6 mo: 16% vs 16% (P = 1.00) | NR | NR | NR | |
| NCT01644110 (POMINC/MPNSG‐0212) 76 | Pomalidomide + Rux | IMiD + JAK1/2 inhibition | 1/2 Ongoing | Rux‐naive and ‐experienced, N ≈ 90 | None | CI, 18% (low dose pom); 20% (high dose pom) | NR | NR | NR | |
| NCT03069326 67 | Thalidomide + Rux | IMiD + JAK1/2 inhibition | 2 Ongoing | Rux‐experienced, N ≈ 65 | None | ORR, 60% | NR | NR | NR | Significant increase in platelet count after cycle 3 vs baseline (P < .01); platelet count increase in 75% of patients with thrombocytopenia |
Abbreviations: ACVR, activin A receptor; AE, adverse event; BAT, best available therapy; BCL, B‐cell lymphoma; BET, bromodomain and extra‐terminal; BM, bone marrow; BMF, bone marrow fibrosis; CI, clinical improvement; CRP, C‐reactive protein; ELN‐IWG, European LeukemiaNet international working group; Hb, hemoglobin; HMA, hypomethylating agent; HMR, high molecular risk; HR, hazard ratio; IL, interleukin; IMiD, immunomodulatory; IRAK, interleukin 1 receptor associated kinase; JAK, Janus kinase; JAKi, JAK inhibitor; LFS, leukemia‐free survival; LSD, lysine‐specific histone demethylase; MAD, maximum administered dose; MAF, mutant allele frequency; MDM2, mouse double minute 2 homolog; MF, myelofibrosis; MoA, mechanism of action; MTD, maximum tolerated dose; ORR, overall response rate; OS, overall survival; PD‐1; programmed cell death protein 1; PFS, progression‐free survival; PI3K, phosphoinositide 3‐kinase; NR, not reported; QD, once daily; QW, once weekly; RBC, red blood cell; RP2D, recommended phase 2 dose; Rux, ruxolitinib; SD, stable disease; SVR35, ≥35% spleen volume reduction from baseline; TD, transfusion dependence; TGF, transforming growth factor; TNF‐α, tumor necrosis factor alpha; TI, transfusion independence; TSS, total symptom response
Pelabresib
MF progenitor cells exhibit altered gene regulation via nuclear factor–κB (NF‐κB) pathway activation that may sustain the inflammation associated with disease progression and transformation via aberrant cytokine signaling. 34 , 35 Bromodomain and extra‐terminal motif inhibitors (BETi) have been developed to exploit this, with the aim of attenuating NF‐κB signaling and suppressing cytokine release. 36 Pelabresib is the most advanced BETi, with recruitment currently underway in phase 2 and 3. 37 , 38 , 39 The phase 2 trial, MANIFEST, included 3 arms: 1) pelabresib as monotherapy in JAKi‐experienced patients; 2) as “add‐on” to ruxolitinib in patients with inadequate response to ruxolitinib; and 3) in combination with ruxolitinib in JAKi‐naive patients. Interim data from MANIFEST have demonstrated improvement in BMF of ≥1 grade in 33% of patients across the arms (21%, 41%, and 33% in arms 1, 2, and 3, respectively). 40 Translational studies have also reported broad clinical responses regardless of baseline mutational status, with pelabresib treatment associated with significant reduction of several cytokines in ruxolitinib‐naive and ruxolitinib‐experienced patients, increased erythroid progenitors, and improved megakaryocyte histology. With survival data yet to be reported, these data suggest disease‐modifying potential. 40 , 41 The primary trial end point, ≥35% SVR from baseline (SVR35) at week 24, was achieved by 24%, 21%, and 67% of patients, respectively. 42 , 43 , 44 This suggests that potentially disease‐modifying activity may be largely independent of prior therapy or treatment order, unlike SVR that demonstrated an association with treatment history. Although any definitive association between reduced BMF and disease modification remains to be characterized, these data are supportive of BMF as a potential indicator of disease modification.
The encouraging activity reported from JAKi‐naive patients in MANIFEST support the assessment of JAKi combination therapy in the first‐line setting and will be further explored in MANIFEST‐2, where JAKi‐naive patients will be randomized to pelabresib plus ruxolitinib or placebo plus ruxolitinib. Bone marrow morphology and proinflammatory cytokine modulation will also be explored alongside the primary end point of SVR35. 38
Bomedemstat
Hematopoiesis is dependent on the epigenetic modifier lysine‐specific demethylase 1 (LSD1), which has a specific role in megakaryocyte maturation. 45 Bomedemstat was developed as an irreversible inhibitor of LSD1 and is under investigation in the phase 1/2 setting. Interim data have reported improvement in BMF of ≥1 grade for 17% of evaluable patients and reductions in MAF in driver and HMR mutations in 42% of patients. MAF reduction was found to correlate with improvements in spleen volume and/or TSS. 46 Although any correlation with survival end points remains to be determined, these data further support the need for BMF and/or mutation‐focused short‐term end points to inform their utility in defining disease modification. Serving to bolster this assertion, spleen volume and TSS reduction or stability were also observed in the vast majority of these patients.
Navitoclax
Targeting the B‐cell lymphoma‐2 (BCL‐2) antiapoptotic pathway has been remarkably successful in chronic lymphocytic leukemia and acute myeloid leukemia. 47 , 48 Preclinically, JAK2V617F mutated CD34+ HSC exhibit apoptotic resistance through overexpression of BCL‐2 family proteins. 49 , 50 Navitoclax is a BCL‐2/BCL‐xL inhibitor that is undergoing phase 2 investigation as monotherapy and in combination with ruxolitinib in the REFINE study. Interim analyses have demonstrated improvement in BMF of ≥1 grade for 33% (11/33) of patients and >10% reduction in driver gene MAF in 46% (12/26) of patients. The clinical impact of these data remains to be determined—median OS was not reached at a median follow‐up of 105 weeks—and again reflects the need for further understanding and standardization of such end points. Alongside these, SVR35 at any time was achieved by 44% of patients (27% at week 24), which was durable regardless of HMR. 31 , 51 Furthermore, direct correlations were observed between several MF‐associated cytokines and changes in spleen volume. 31 Phase 3 recruitment is now ongoing to investigate navitoclax plus ruxolitinib versus placebo plus ruxolitinib in JAK2i‐naive patients (TRANSFORM‐1) 52 and versus best available therapy (BAT) in JAK2i‐experienced patients (TRANSFORM‐2). The traditional primary end point of SVR35 will be explored alongside additional analyses of BMF and time‐to‐event survival measures. 53
Imetelstat
Telomerase is upregulated in many cancers, and CD34+ hematopoietic cells in MPNs are characterized by shortened telomeres. 54 Preclinical models of the competitive telomerase inhibitor, imetelstat, have demonstrated selective inhibition of pre‐leukemic stem cell transformation via downregulation of hTERT and decreased ADAR1 activity. 55 In the phase 2 study, IMbark, 41% of patients experienced reversal of BMF and 42% had reduced MAF of driver mutations, both of which correlated with improved OS. Median OS was 29.9 months. 56 Cytogenetic analyses within IMbark have demonstrated the selective targeting of malignant cells by imetelstat, further supporting the potential for disease‐modifying activity. 57 Interestingly, improvements in the primary end points of SVR35 and TSS at week 24 were modest (10% and 32%, respectively), 56 and not correlated BMF or MAF, clearly presenting the need for more informative end points to be adopted across MF clinical trials. Imetelstat is now under further evaluation versus BAT for patients refractory to JAKi in the phase 3 trial MYF3001, which will evaluate impact on malignant clones leading to disease modification alongside a primary end point of OS. This deviation from traditional end points reflects the reform required across clinical trials to better understand the full potential of emerging therapies.
Navtemadlin
Inhibition of mouse double minute 2 (MDM2) drives selective depletion of JAK2V617F mutated stem cells. 37 , 58 Navtemadlin, a potent MDM2 inhibitor is undergoing assessment as monotherapy and combination therapy. 59 Interim data from the phase 2/3 BOREAS trial of navtemadlin monotherapy in JAKi relapsed/refractory (R/R) patients are encouraging, with improved BMF observed in 27% (12/45) of patients and stable scores in 51%. Best driver gene reduction ≥20% was reported in 34% (22/65) of patients and 29% (19/65) had a complete MAF reduction below the limit of detection. Furthermore, navtemadlin was associated with reduced levels of circulating CD34+ cells and tumor necrosis factor α (TNF‐α). Each of these parameters correlated with SVR, and an association was also observed between fibrosis scores and mutational burden. 60 As navtemadlin is investigated in phase 3, correlations between these potentially disease‐modifying parameters and survival outcomes are eagerly awaited.
Azacitidine
Azacitidine is a hypomethylating agent currently undergoing testing as “add‐on” to ruxolitinib in JAKi‐naive patients. Although interim data are encouraging in this single arm study, with improvement in BM morphology reported for 61% of patients, and an overall response rate of 74%, the additional benefit of azacitidine remains to be fully determined. However, it is noteworthy that median OS had not been reached after a median follow‐up of 35 months. 61
Tagraxofusp
Tagraxofusp is CD123‐directed cytotoxin selected for testing in MF as CD123 is an established marker of leukemic stem cells. 62 Interim results of a phase 2 study were modest, with spleen responses observed in 45% of patients at 24 weeks and a median OS of 31 months, the latter of which may prove indicative of disease‐modifying activity. 63
Immune therapies
Allo‐SCT is an immune therapy that represents the only definitively disease‐modifying option currently available, and successful transplant generally leads to the reversal of BMF. 64 The atypical and dysregulated immune environment associated with MF provides a variety of additional immune targets. Interferon (IFN)‐based regimens have shown promise, with a survival benefit of pegylated IFN‐α2 demonstrated in the long‐term follow‐up of patients with intermediate or high‐risk disease, whereby the median OS of 89 months was longer than expected and accompanied by a reduction in JAK2V617F burden. 65 , 66 Additional immune therapies including immunomodulatory drugs and checkpoint inhibitors are undergoing investigation in combination with ruxolitinib. 67 , 68
Finally, the JAK/STAT pathway remains an attractive target and several JAKi are in development. Although there is no evidence to suggest these modify disease course, JAKi are ideal candidates for combination therapy and many trials are examining novel agents in combination with ruxolitinib.
Readers should exercise caution when interpreting the above data, as many of these trials are ongoing; data are preliminary and often based on small numbers with incomplete follow‐up. However, these early reports are encouraging, provide initial rationale for defining disease modifying parameters, and hold promise that significant evolution of the MF treatment landscape is on the horizon.
Proposed Definition Of Disease Modification In Myelofibrosis
The current benchmark for curative therapy in MF is allo‐SCT, which replaces disease through healthy repopulation of the stem cell compartment. When considering disease modification, a treatment is not expected to replace disease, but rather alter the disease biology, with the aim of reversing disease trajectory. As such, true disease modification is difficult to measure without standardized assessment or consensus recommendation to guide clinical evaluation. This makes it critical for any definition of disease modification to represent true modifiers and mechanisms of improvement, rather than resultant downstream effects.
With consideration of the current knowledge and the rationale provided by available clinical trial data, we propose the following definition of disease‐modifying activity (Table 2):
Disease modification in MF is defined as therapy that exerts a clinically meaningful impact on survival outcomes and/or restoration of normal hematopoiesis in conjunction with improvement in bone marrow fibrosis through a substantial and durable reduction in the clonal burden of disease.
TABLE 2.
Proposed Parameters, With Rationale, Considered as Evidence of Disease Modification
| Parameters | Rationale | Supporting Data From Novel Agents | Limitations |
|---|---|---|---|
| Primary outcomes of disease modification | |||
| OS |
|
|
|
| Event‐, progression‐ or leukemia‐free survival | |||
| Key modifiers | |||
| BMF |
Improved BMF and correlation to OS reported by:
Correlation with survival outcomes remain to be determined |
|
|
| Clonal disease/mutational burden |
Reduced MAF and correlation to OS reported by:
Reductions in MAF reported by:
Correlation with survival outcomes remain to be determined |
|
|
| Cytokine modulation |
Reductions in key cytokine exression demonstrated by: |
|
Abbreviations: allo‐SCT, allogenic stem cell transplant; BMF, bone marrow fibrosis; MAF, mutation allele frequency; MF, myelofibrosis; OS, overall survival.
It seems inevitable that achieving disease modification will also lead to beneficial downstream effects such as the elimination of symptoms and splenomegaly and improved PROs.
Discussion and Future Perspectives
The treatment landscape of MF has remained almost static for a decade but is set to evolve rapidly as understanding of the molecular pathogenesis of MF sheds light on novel therapeutic targets and the possibility of selectively depleting the malignant HSC compartment (Fig. 3). 17
As novel treatment strategies emerge, their optimal use and place in the treatment paradigm will need to be determined. Ascertaining the appropriate timing of interventions to maximize the potential for disease modification will be key. Trials tend to take place in the heavily pretreated setting; however, it is logical that the greatest impact of disease modifying treatment will be observed when initiated early, before clonal evolution (Fig. 2).
Despite the limited disease‐modifying activity of JAKi, the JAK/STAT pathway remains a pivotal feature of MF pathology and it is unlikely that JAK inhibition will be relinquished. Rather, synergy between inhibitors of JAK and non‐JAK targets may positively impact disease modification, optimizing clinical responses. Treatment strategies that combine JAKi with novel agents, especially given the current reliance on JAKi to control disease symptoms, are likely to feature heavily as clinical trial programs develop.
Several agents and combinations undergoing study in MF are suggestive of disease modification. Although interpretation across trials is limited by the lack of control arms and disparities in the definition and recruitment of patients with ruxolitinib failure, together with inconsistent trial designs, available data are beginning to inform recommendations for defining and measuring disease‐modifying activity. Emerging data from novel end points will facilitate this evolution and we await the correlative assessments between modifiers and OS. Future trials that can demonstrate significant correlation between reduction in BMF grade or reduction in clonal disease burden with increased median PFS or OS will provide evidence for the first wave of MF treatments that modify disease.
These exciting developments highlight the need for new surrogate measures and study end points, particularly for use in the early stages of trials before survival read outs, to help identify the most promising new approaches and accelerate their approval. As the landscape evolves, it will be important to standardize such parameters across clinical trials to better define the potential for disease modification and facilitate inter‐trial comparability. Here, we present our recommendation for how disease modification of MF should be defined, informed by current knowledge and available data.
Looking to the future, assessment of disease burden within the HSC compartment may emerge, not only as a defining end point of disease modification, but a measure that begins to align MF with other hematological malignancies in which disease modification is already established. Additionally, baseline characteristics or genetic factors that may impact the potential for disease modification will need to be identified and understood in terms of what may be achieved.
In summary, the possibility of disease modification has the potential to revolutionize clinical practice and treatment decision‐making for patients with MF. As novel end points begin to emerge, it will be important to re‐evaluate clinical trial designs, and potentially redefine disease modification, adding new end points to survival outcomes, to ensure the true potential for disease modification and MF therapy is realized. Standardized definitions and assessments are needed across clinical trials, along with the inclusion of patients with newly diagnosed disease, where the greater potential for disease modification may lie.
Funding Support
AbbVie, Inc, sponsored the study and provided final review and approval of the manuscript.
Conflict of Interest Disclosure
Naveen Pemmaraju had consulting or advisory roles with Celgene, Stemline, Incyte, Novartis, Mustang Bio, Roche Diagnostics, and LFB; received honoraria from Celgene, Stemline, Incyte, Novartis, Mustang Bio, Roche Diagnostics, and LFB; received grants and/or funding from Affymetrix and Sager Strong Foundation; and had board memberships (noncompensated) with Dan's House of Hope (board of directors) and HemOnc Times/Oncology Times (board member and editor‐in‐chief). Srdan Verstovsek had consulting or advisory roles with Celgene, Constellation Pharmaceuticals, Incyte, Novartis, Pragmatist, and Sierra and received research funding from Blueprint Medicines, Celgene, CTI BioPharma Corp, Genentech, Gilead Sciences, Incyte, Novartis, NS Pharma, Promedior, and Roche. Ruben Mesa had consulting roles with Constellation, La Jolla Pharmaceutical Company, Novartis, Pharma, and Sierra Oncology and received research funding (institutional) from AbbVie, Celgene, Constellation, CTI, Genotech Pharma, Incyte, Promedior, and Samus. Vikas Gupta provided consultancy for BMS‐Celgene, AbbVie, Pfizer, Roche, Constellation Pharma, Sierra Oncology, and Novartis; served on advisory committees for BMS‐Celgene, Sierra Oncology, and Novartis; and received research funding from Incyte and Novartis. Jacqueline S. Garcia had consulting/advisory roles with AbbVie, Takeda, and Astellas and received research funding (institutional) from AbbVie, Genentech, Pfizer, Prelude, and Astra Zeneca. Joseph M. Scandura served on consulting/advisory boards for AbbVie, Constellation, and SDP Oncology and received research support from AbbVie and Constellation. Stephen T. Oh served on consulting/advisory boards for Novartis, Kartos Therapeutics, CTI BioPharma, Celgene/Bristol‐Myers Squibb, Disc Medicine, Blueprint Medicines, PharmaEssentia, Constellation, Geron, AbbVie, Sierra Oncology, and Incyte. Francesco Passamonti had advisory board roles with AbbVie, BMS, Novartis, Janssen, and Astellas and received speaker fees from AbbVie, BMS, and Novartis. Konstanze Döhner reported consulting/advisory roles with and honoraria from AbbVie, Celgene/BMS, Novartis, CTI BioPharma Corp, and Roche. Adam J. Mead received honoraria for consulting and speaker fees from Novartis, Celgene/BMS, AbbVie, CTI, Sierra Oncology, Karyopharm, Sensyn, Incyte, Galecto, Pfizer and Gilead; received research funding from Celgene/BMS, Novartis, and Galecto; and is a cofounder and equity holder in Alethiomics, Ltd, a spin‐out company from the University of Oxford.
Author Contributions
All authors had access to relevant data and participated in the writing, review, and approval of the manuscript.
Supporting information
Supplementary Material
Writing support was provided by Fiona Powell, PhD, associated with Fishawack Health, and Ryan Bourgo, PhD, of Fishawack Health, funded by AbbVie.
References
- 1. Schieber M, Crispino JD, Stein B. Myelofibrosis in 2019: moving beyond JAK2 inhibition. Blood Cancer J. 2019;9:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrison CN, Schaap N, Mesa RA. Management of myelofibrosis after ruxolitinib failure. Ann Hematol. 2020;99:1177‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nasillo V, Riva G, Paolini A, et al. Inflammatory microenvironment and specific T cells in myeloproliferative neoplasms: immunopathogenesis and novel immunotherapies. Int J Mol Sci. 2021;22:1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Al‐Khabori M, Al‐Zadjali S, Al Noumani I, et al. Overall survival in patients with JAK2 and ASXL1 positive myeloproliferative neoplasms. Blood. 2019;134:5383. [Google Scholar]
- 5. Vannucchi AM, Barbui T, Cervantes F, et al. Philadelphia chromosome–negative chronic myeloproliferative neoplasms: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2015;26(suppl 5):v85‐99. [DOI] [PubMed] [Google Scholar]
- 6. Agarwal A, Morrone K, Bartenstein M, Zhao ZJ, Verma A, Goel S. Bone marrow fibrosis in primary myelofibrosis: pathogenic mechanisms and the role of TGF‐beta. Stem Cell Investig. 2016;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gagelmann N, Ditschkowski M, Bogdanov R, et al. Comprehensive clinical‐molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood. 2019;133:2233‐2242. [DOI] [PubMed] [Google Scholar]
- 8. NCCN Clinical Practice Guidelines in Oncology: Myeloproliferative Neoplasms. Version 1.2021. National Comprehensive Cancer Network. Published April 13, 2021. [Google Scholar]
- 9. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649‐656. [PubMed] [Google Scholar]
- 10. Verstovsek S, Mesa RA, Gotlib J, et al. The clinical benefit of ruxolitinib across patient subgroups: analysis of a placebo‐controlled, phase III study in patients with myelofibrosis. Br J Haematol. 2013;161:508‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verstovsek S, Mesa RA, Gotlib J, et al. A double‐blind, placebo‐controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366:799‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrison C, Kiladjian JJ, Al‐Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787‐798. [DOI] [PubMed] [Google Scholar]
- 13. Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working Group‐Myeloproliferative Neoplasms Research and Treatment (IWG‐MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122:1395‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1:643‐651. [DOI] [PubMed] [Google Scholar]
- 15. Harrison CN, Schaap N, Vannucchi AM, et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: an updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am J Hematol. 2020;95:594‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verstovsek S, Talpaz M, Ritchie E, et al. A phase I, open‐label, dose‐escalation, multicenter study of the JAK2 inhibitor NS‐018 in patients with myelofibrosis. Leukemia. 2017;31:393‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bankar A, Gupta V. Investigational non‐JAK inhibitors for chronic phase myelofibrosis. Expert Opin Investig Drugs. 2020;29:461‐474. [DOI] [PubMed] [Google Scholar]
- 18. Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013;24:133‐145. [DOI] [PubMed] [Google Scholar]
- 19. Fisher DAC, Fowles JS, Zhou A, Oh ST. Inflammatory pathophysiology as a contributor to myeloproliferative neoplasms. Front Immunol. 2021;12:683401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song MK, Park BB, Uhm JE. Understanding splenomegaly in myelofibrosis: association with molecular pathogenesis. Int J Mol Sci. 2018;19:898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferreira Cristina S, Polo B, Lacerda JF. Somatic mutations in Philadelphia chromosome–negative myeloproliferative neoplasms. Semin Hematol. 2018;55:215‐222. [DOI] [PubMed] [Google Scholar]
- 22. Venugopal S, Mascarenhas J. Current clinical investigations in myelofibrosis. Hematol/Oncol Clin N Am. 2021;35:353‐373. [DOI] [PubMed] [Google Scholar]
- 23. Savona MR. Are we altering the natural history of primary myelofibrosis? Leuk Res. 2014;38:1004‐1012. [DOI] [PubMed] [Google Scholar]
- 24. Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745‐2760. [DOI] [PubMed] [Google Scholar]
- 25. Mahon FX, Etienne G. Deep molecular response in chronic myeloid leukemia: the new goal of therapy? Clin Cancer Res. 2014;20:310‐322. [DOI] [PubMed] [Google Scholar]
- 26. Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moreau P, San Miguel J, Sonneveld P, et al. Multiple myeloma: ESMO clinical practice guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2017;28:iv52‐iv61. [DOI] [PubMed] [Google Scholar]
- 28. Kosiorek HE, Dueck AC, Mascarenhas J, Mesa RA, Hoffman R, Langlais BT. Clinical trial design features of myelofibrosis trials during the last decade: comprehensive review of Clinicaltrials.gov data 2010‐2019. Blood. 2020;136(suppl 1):37. [Google Scholar]
- 29. Ianotto JC, Boyer‐Perrard F, Gyan E, et al. Efficacy and safety of pegylated‐interferon alpha‐2a in myelofibrosis: a study by the FIM and GEM French cooperative groups. Br J Haematol. 2013;162:783‐791. [DOI] [PubMed] [Google Scholar]
- 30. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pemmaraju N, Garcia JS, Potluri J, et al. The addition of navitoclax to ruxolitinib demonstrates efficacy within different high‐risk populations in patients with relapsed/refractory myelofibrosis. Blood. 2020;136(suppl 1):49‐50. [Google Scholar]
- 32. Drexler B, Passweg JR, Tzankov A, et al. The sympathomimetic agonist mirabegron did not lower JAK2‐V617F allele burden, but restored nestin‐positive cells and reduced reticulin fibrosis in patients with myeloproliferative neoplasms: results of phase II study SAKK 33/14. Haematologica. 2019;104:710‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verstovsek S, Talpaz M, Wadleigh M, et al. A randomized, double blind phase 2 study of 3 different doses of PRM‐151 in patients with myelofibrosis who were previously treated with or ineligible for ruxolitinib. HemaSphere. 2019;3(suppl 1):367. [Google Scholar]
- 34. Fisher DAC, Miner CA, Engle EK, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK‐STAT, MAP kinase, and NFkappaB signaling. Leukemia. 2019;33:1978‐1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fisher DAC, Malkova O, Engle EK, et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia. 2017;31:1962‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33:29‐43 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mascarenhas J, Lu M, Kosiorek H, et al. Oral idasanutlin in patients with polycythemia vera. Blood. 2019;134:525‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marcellino BK, Verstovsek S, Mascarenhas J. The myelodepletive phenotype in myelofibrosis: clinical relevance and therapeutic implication. Clin Lymphoma Myeloma Leuk. 2020;20:415‐421. [DOI] [PubMed] [Google Scholar]
- 39. Harrison CN, Patriarca A, Mascarenhas J, et al. Preliminary report of MANIFEST, a phase 2 study of CPI‐0610, a bromodomain and extraterminal domain inhibitor (BETi), in combination with ruxolitinib, in JAK inhibitor (JAKi) treatment naive myelofibrosis patients. Blood. 2019;134:4164‐4164. [Google Scholar]
- 40. Keller P, Cui J, Mertz J, et al. BET inhibitor pelabresib decreases inflammatory cytokines, improves bone marrow fibrosis and function, and demonstrates clinical response irrespective of mutation status in myelofibrosis patients. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 41. Kremyanskaya M, Mascarenhas J, Palandri F, et al. Pelabresib (CPI‐0610) monotherapy in patients with myelofibrosis – update of clinical and translational data from the ongoing MANIFEST trial. Blood. 2021;138(suppl 1):141. [Google Scholar]
- 42. Talpaz M, Rampal RK, Verstovsek S, et al. CPI‐0610, a bromodomain and extraterminal domain protein (BET) inhibitor, as monotherapy in advanced myelofibrosis patients refractory/intolerant to JAK inhibitor: update from phase 2 MANIFEST study. Abstract presented at: 63rd ASH Annual Meeting and Exposition; December 5, 2020; Atlanta, GA.
- 43. Kremyanskaya M, Mascarenhas J, Patriarca A, et al. Clinical benefit of pelabresib (CPI‐0610) in combination with ruxolitinib in JAK inhibitor treatment naive myelofibrosis patients: Interim efficacy subgroup analysis from arm 3 of MANIFEST PH2 study. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 44. Verstovsek S, Mascarenhas J, Kremyanskaya M, et al. CPI‐0610, bromodomain and extraterminal domain protein (BET) inhibitor, as ‘add‐on’ to ruxolitinib (RUX), in advanced myelofibrosis patients with suboptimal response: update of MANIFEST phase 2 study. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 45. Sprussel A, Schulte JH, Weber S, et al. Lysine‐specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia. 2012;26:2039‐2051. [DOI] [PubMed] [Google Scholar]
- 46. Gill H, Yacoub A, Pettit KM, et al. A Phase 2 Study of the LSD1 inhibitor Img‐7289 (bomedemstat) for the treatment of advanced myelofibrosis. Blood. 2021;138(suppl 1):139. [Google Scholar]
- 47. Kater AP, Seymour JF, Hillmen P, et al. Fixed duration of venetoclax‐rituximab in relapsed/refractory chronic lymphocytic leukemia eradicates minimal residual disease and prolongs survival: post‐treatment follow‐up of the MURANO phase III study. J Clin Oncol. 2019;37:269‐277. [DOI] [PubMed] [Google Scholar]
- 48. Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo‐controlled trial. Blood. 2020;135:2137‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Freitas RM, da Costa Maranduba CM. Myeloproliferative neoplasms and the JAK/STAT signaling pathway: an overview. Rev Bras Hematol Hemoter. 2015;37:348‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tognon R, Gasparotto EP, Neves RP, et al. Deregulation of apoptosis‐related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in essential thrombocythemia and myelofibrosis. J Hematol Oncol. 2012;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harrison C, Garcia J, Somervaille T, et al. Navitoclax and ruxolitinib for patients with myelofibrosis and JAK inhibitor experience: response duration in phase 2 study. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 52. Potluri J, Harb J, Masud AA, Hutti JE. A phase 3, double‐blind, placebo‐controlled, randomized study evaluating navitoclax in combination with ruxolitinib in patients with myelofibrosis (TRANSFORM‐1). Blood. 2020;136:4.32614961 [Google Scholar]
- 53. Dilley K, Harb J, Jalaluddin M, Hutti JE, Potluri J. A phase 3, open‐label, randomized study evaluating the efficacy and safety of navitoclax plus ruxolitinib versus best available therapy in patients with relapsed/refractory myelofibrosis (TRANSFORM‐2). Blood. 2020;136:8.32614959 [Google Scholar]
- 54. Ruella M, Salmoiraghi S, Risso A, et al. Telomere shortening in Ph‐negative chronic myeloproliferative neoplasms: a biological marker of polycythemia vera and myelofibrosis, regardless of hydroxycarbamide therapy. Exp Hematol. 2013;41:627‐634. [DOI] [PubMed] [Google Scholar]
- 55. Ma W, Balaian L, Mondala P, et al. Imetelstat inhibits telomerase and prevents propagation of ADAR1‐activated myeloproliferative neoplasm and leukemia stem cells. Blood. 2020;136:140771. [Google Scholar]
- 56. Mascarenhas J, Komrokji RS, Palandri F, et al. Randomized, single‐blind, multicenter phase ii study of two doses of imetelstat in relapsed or refractory myelofibrosis. J Clin Oncol. 2021;39:2881‐2892. [DOI] [PubMed] [Google Scholar]
- 57. Mascarenhas J, Komrokji RS, Cavo M, et al. Potential disease‐modifying activity of imetelstat demonstrated by reduction in cytogenetically abnormal clones and mutation burden leads to clinical benefits in relapsed/refractory myelofibrosis patients. Blood. 2020;136(suppl 1):39‐40. [Google Scholar]
- 58. Lu M, Xia L, Li Y, Wang X, Hoffman R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon alpha 2a target JAK2V617F‐positive progenitor and stem cells. Blood. 2014;124:771‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mascarenhas J, Vannucchi AM, Mead AJ, et al. An open‐label, global, multicenter, phase 1b/2 study of KRT‐232, a first‐in‐class, oral small‐molecule inhibitor of murine double minute 2 (MDM2), combined with ruxolitinib in patients who have myelofibrosis and a suboptimal response to ruxolitinib. Blood. 2020;136:44‐45. [Google Scholar]
- 60. Vachani P, Lange A, Garcia Delgado R, et al. Potential disease‐modifying activity of navtemadlin (KRT‐232), a first‐in‐class MDM2 inhibitor, correlates with clinical benefits in relapsed/refractory myelofibrosis (MF). Blood. 2021;138(suppl 1):3581. [Google Scholar]
- 61. Masarova L, Verstovsek S, Bose P, et al. Phase 2 study of ruxolitinib (RUX) in combination with 5‐azacitidine (AZA) in patients (pts) with myelofibrosis. Blood. 2019;134:1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin‐3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777‐1784. [DOI] [PubMed] [Google Scholar]
- 63. Pemmaraju N, Gupta V, Ali H, et al. A multicenter phase 1/2 clinical trial of tagraxofusp, a CD123‐targeted therapy, in patients with poor‐risk primary and secondary myelofibrosis. Blood. 2020;136(suppl 1):39‐40. [Google Scholar]
- 64. Tamari R, Mughal TI, Rondelli D, et al. Allo‐SCT for myelofibrosis: reversing the chronic phase in the JAK inhibitor era? Bone Marrow Transplant. 2015;50:628‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ianotto JC, Chauveau A, Boyer‐Perrard F, et al. Benefits and pitfalls of pegylated interferon‐alpha2a therapy in patients with myeloproliferative neoplasm‐associated myelofibrosis: a French intergroup of myeloproliferative neoplasms (FIM) study. Haematologica. 2018;103:438‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sorensen AL, Mikkelsen SU, Knudsen TA, et al. Ruxolitinib and interferon‐alpha2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica. 2020;105:2262‐2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rampal RK, Verstovsek S, Devlin SM, et al. Safety and efficacy of combined ruxolitinib and thalidomide in patients with myelofibrosis: a phase II study. Blood. 2019;134:4163. [Google Scholar]
- 68. Ross D, Heidel F, Perkins A, et al. Adore: a randomized, open‐label, phase 1/2 open‐platform study evaluating safety and efficacy of novel ruxolitinib combinations in patients with myelofibrosis. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 69. Lekovic D, Gotic M, Perunicic‐Jovanovic M, et al. Contribution of comorbidities and grade of bone marrow fibrosis to the prognosis of survival in patients with primary myelofibrosis. Med Oncol. 2014;31:869. [DOI] [PubMed] [Google Scholar]
- 70. Gianelli U, Vener C, Bossi A, et al. The European Consensus on grading of bone marrow fibrosis allows a better prognostication of patients with primary myelofibrosis. Mod Pathol. 2012;25:1193‐1202. [DOI] [PubMed] [Google Scholar]
- 71. Zhao L‐P, Daltro De Oliveira R, Marcault C, et al. SF3B1 mutations in the driver clone increase the risk of evolution to myelofibrosis in patients with myeloproliferative neoplasms (MPN). Blood. 2020;136:1.32430499 [Google Scholar]
- 72. Pozdnyakova O, Hasserjian RP, Verstovsek S, Orazi A. Impact of bone marrow pathology on the clinical management of Philadelphia chromosome–negative myeloproliferative neoplasms. Clin Lymphoma Myeloma Leuk. 2015;15:253‐261. [DOI] [PubMed] [Google Scholar]
- 73. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129:667‐679. [DOI] [PubMed] [Google Scholar]
- 74. Verstovsek S, Kremyanskaya M, Mascarenhas J, et al. Pelabresib (CPI‐0610) improved anemia associated with myelofibrosis: Interim results from manifest phase 2 study. Paper presented at: EHA2021 Virtual Congress; June 4‐17, 2021.
- 75. Tefferi A, Al‐Ali HK, Barosi G, et al. A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm‐associated myelofibrosis and RBC‐transfusion dependence. Leukemia. 2017;31:1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stegelmann F, Koschmieder S, Isfort S, et al. Updated results from the German Mpnsg‐0212 combination trial: ruxolitinib plus pomalidomide in myelofibrosis with anemia. Blood. 2019;134:672. [Google Scholar]
- 77. Gangat N, Tefferi A. Myelofibrosis biology and contemporary management. Br J Haematol. 2020;191:152‐170. [DOI] [PubMed] [Google Scholar]
- 78. Longhitano L, Li Volti G, Giallongo C, et al. The role of inflammation and inflammasome in myeloproliferative disease. J Clin Med. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mughal TI, Vaddi K, Sarlis NJ, Verstovsek S. Myelofibrosis‐associated complications: pathogenesis, clinical manifestations, and effects on outcomes. Int J Gen Med. 2014;7:89‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Morsia E, Gangat N. Myelofibrosis: challenges for preclinical models and emerging therapeutic targets. Expert Opin Ther Targets. 2021;25:211‐222. [DOI] [PubMed] [Google Scholar]
- 81. Fenaux P, Kiladjian JJ, Platzbecker U. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood. 2019;133:790‐794. [DOI] [PubMed] [Google Scholar]
- 82. Tremblay D, Mascarenhas J. Next generation therapeutics for the treatment of myelofibrosis. Cells. 2021;10:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material