

Abstract
Open‐shell non‐alternant polycyclic hydrocarbons (PHs) are attracting increasing attention due to their promising applications in organic spintronics and quantum computing. Herein we report the synthesis of three cyclohepta[def]fluorene‐based diradicaloids (1–3), by fusion of benzo rings on its periphery for the thermodynamic stabilization, as evidenced by multiple characterization techniques. Remarkably, all of them display a very narrow optical energy gap (E g opt=0.52–0.69 eV) and persistent stability under ambient conditions (t 1/2=11.7–33.3 h). More importantly, this new type of diradicaloids possess a low‐lying triplet state with an extremely small singlet–triplet energy gap, as low as 0.002 kcal mol−1, with a clear dependence on the molecular size. This family of compounds thus offers a new route to create non‐alternant open‐shell PHs with high‐spin ground states, and opens up novel possibilities and insights into understanding the structure–property relationships.
Keywords: Azulenes, Fused-Ring Systems, Magnetic Properties, Open-Shell Diradicaloids, Polycyclic Hydrocarbons
Three open‐shell non‐alternant polycyclic hydrocarbons (PHs) based on the skeleton of cyclohepta[def]fluorene are synthesized via an efficient benzannulation stabilization strategy. The obtained persistent singlet diradicaloids have a narrow optical energy gap (0.52–0.69 eV). Moreover, variable‐temperature EPR measurements indicate that all of them have a low‐lying triplet state with an extremely small singlet–triplet energy gap.
Persistent open‐shell polycyclic hydrocarbons (PHs) with high‐spin ground state are potential candidates for application in organic electronic and spintronic devices on account of their unique electronic and magnetic properties as well as intriguing quantum properties and phenomena.[ 1 , 2 , 3 , 4 , 5 ] One general design principle to access the triplet ground state in quinonoid PHs is to extend the π‐conjugation for achieving an extremely small highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap, which is essential to stabilize the triplet state.[ 4 , 6 ] However, this extremely low HOMO–LUMO splitting can only be realized in a relatively large π‐system, making the synthesis tedious along with the solubility issues of the precursors. [4] On the other hand, the introduction of non‐alternant units (i.e., azulene, pentalene, heptalene)[ 7 , 8 , 9 , 10 , 11 ] in the PH‐based carbon skeleton seems to be a more promising strategy to realize the low HOMO–LUMO splitting, as compared to alternant π‐systems. Non‐alternant PHs,[ 9 , 12 ] also hold a very fundamental interest because the lack of investigation still leaves room for uncertainty regarding their spin ground state. Most of non‐alternant PHs can be drawn as a resonance Kekulé structure, suggesting a singlet ground state, but the extension of Ovchinnikov rule [13] to some non‐alternant PHs predicts a triplet ground state (S=1, S is spin multiplicity of the ground state). Theory indicates that such open‐shell non‐alternant PHs usually have a singlet ground state with a low‐lying exited triplet state, for which the two spin states are nearly degenerated in their respective equilibrium geometries, leading to a bistability phenomenon. [14]
As a representative example, cyclohepta[def]fluorene (g), the last unknown representative of seven non‐alternant pyrene isomers (a–g in Figure 1a), [15] was early predicted to be a “singlet‐triplet bistability” molecule with a triplet ground state and small singlet‐triplet energy gap (ΔE ST) of 1.7 kcal mol−1. [16] The diradical structure of cyclohepta[def]fluorene was expected to be more stable than its Kekulé structure as the result of gaining two more Clar sextets in the open‐shell form. According to the extended use of the Ovchinnikov rule, cyclohepta[def]fluorene was also proposed to have a triplet ground state (S=1). However, the synthesis of pristine cyclohepta[def]fluorene and its derivatives is quite challenging because of their intrinsic instability. [17] Early synthetic attempts toward cyclohepta[def]fluorene have been so far unsuccessful, except for its dianion form. [18a] During the submission of our work, Yasuda et al. reported the synthesis of the triaryl derivatives of g, which exhibited singlet ground states, contrary to previous theoretical predictions for g. [18b] Therefore, the experimental access and the intrinsic properties of this kind of non‐alternative PHs remain elusive due to the lack of efficient synthetic strategy.
Figure 1.
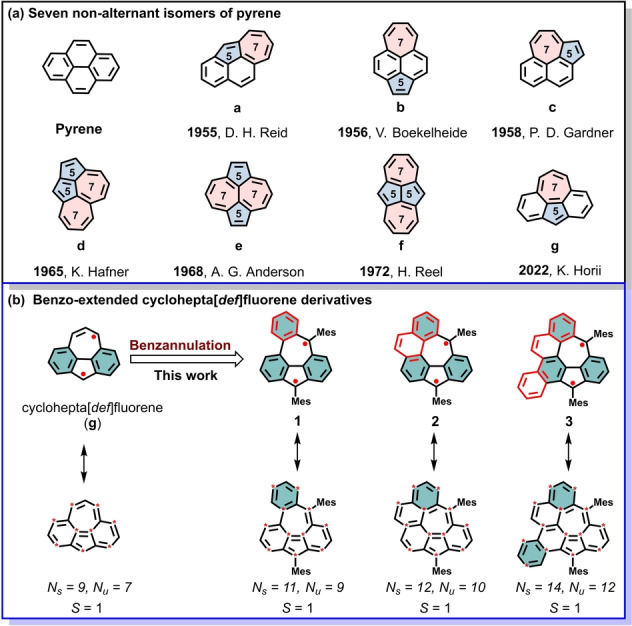
Molecular structures of a) seven non‐alternant isomers of pyrene, and b) benzo‐extended cyclohepta[def]fluorene derivatives in this work: benzo[5,6]cyclohepta[1,2,3,4‐def]fluorene (1), indeno[6,7,1,2‐ghij]pleiadene (2) and benzo[4,5]indeno[6,7,1,2‐ghij]pleiadene (3). Spin nature of ground state (S) is anticipated by the extension of Ovchinnikov rule (S=(N s−N u)/2, N s and N u represent number of starred and unstarred atoms, respectively). Clar's sextet is donated by cyan‐colored benzenoid ring in the resonance structure.
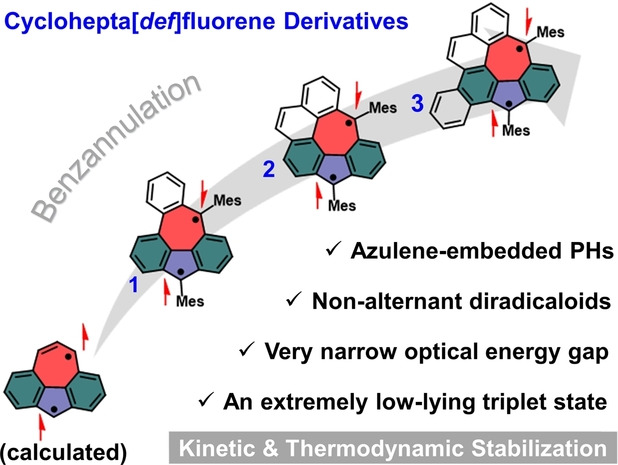
Herein, we demonstrate the synthesis of three unprecedented derivatives of cyclohepta[def]fluorene, that is, benzo[5,6]cyclohepta[1,2,3,4‐def]fluorene (1), indeno[6,7,1,2‐ghij]pleiadene (2) and benzo[4,5]indeno[6,7,1,2‐ghij]pleiadene (3), by fusion of benzo rings to its periphery. The azulene core was built up in the latter stage, instead of using azulene or its derivatives as the starting compounds. [19] To realize this strategy, three key dialdehyde‐intermediates (9, 12 and 14) with different numbers of benzo rings were firstly synthesized, which were then converted into dihydro‐precursors (4, 5 and 6) bearing the pentagon‐heptagon pair. Further oxidative dehydrogenation of 4–6 with 2,3‐dichloro‐5,6‐dicyano‐1,4‐benzoquinone (DDQ) yielded the desired targets (1–3) containing cyclohepta[def]fluorene skeleton, respectively. The successful formation of azulene‐embedded diradicaloids 1–3 was clearly confirmed by high‐resolution matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry (HR MALDI‐TOF MS), UV/Vis‐NIR and electron paramagnetic resonance (EPR) analysis. Notably, these three derivatives show persistent stability under ambient conditions (half‐life time (t 1/2)=11.7, 23.8 and 33.3 h for 1–3, respectively), which can be attributed to both the increased spin delocalization by the additional fused benzo rings (thermodynamic stabilization) and the introduction of sterically bulky side groups at the most reactive sites (kinetic stabilization). [20] Compounds 1–3 exhibit long‐wavelength electronic absorption with a very narrow optical energy gap of 0.54, 0.52 and 0.69 eV, respectively. Moreover, variable temperature EPR studies indicate that 1–3 all have a singlet ground state with a low‐lying triplet state. An extremely small ΔE ST is derived to be −0.04, −0.015 and −0.002 kcal mol−1 for 1–3 based on EPR, respectively, which gradually decreases upon increasing the number of the external fused benzo rings. This study provides more insights into both synthesis and properties of the unprecedented open‐shell non‐alternant PHs with a low‐lying triplet state or even high‐spin state.
Our synthetic strategy toward benzo‐fused cyclohepta[def]fluorene derivatives 1–3 is illustrated in Scheme 1, in which the azulene core was built up in the latter stage based on the resultant dihydro‐precursors with 5/7 pair (4–6) that are derived from dialdehyde intermediates (9, 12 and 14). According to the Clar's rule of the aromatic sextet, 1–3 all have more aromatic sextets in their open‐shell form than in their closed‐shell resonance form, providing additional aromatic stabilization energy and contribute to their diradical character (Figure 1b). Theoretical calculations show that the maximum spin density sites of these three cyclohepta[def]fluorene derivatives are located at the carbon atom of the five‐ and seven‐membered rings (Figure S18 and S20); therefore, mesityl groups are intentionally introduced to kinetically block these most reactive sites.
Scheme 1.
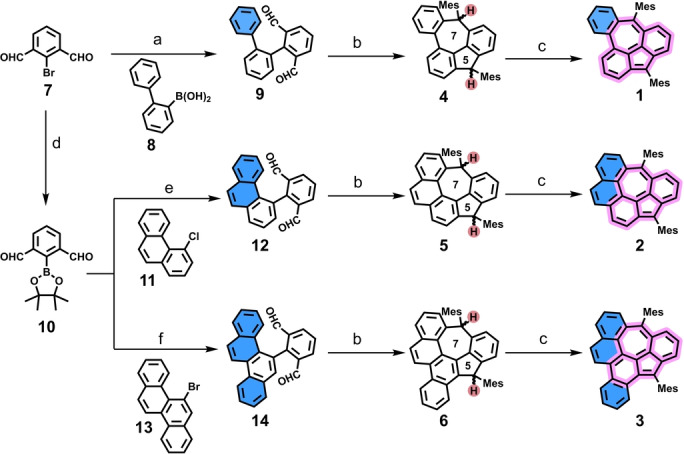
Synthetic route towards the non‐alternant diradicaloids 1–3. Reagents and conditions: a) Pd(PPh3)4, K2CO3, dioxane/H2O, 95 °C, 36 h, 81 %. b) (i) 2‐mesitylmagnesium bromide, THF, RT, overnight; (ii) BF3⋅OEt2, CH2Cl2, RT, 40 min; 61 % for 4, 72 % for 5, 70 % for 6 in two steps. c) 3 equiv DDQ, chlorobenzene for 1 and toluene for 2 and 3, RT, 30 min, quant. d) B2Pin2, Pd(PPh3)2Cl2, KOAc, 1,4‐dioxane, 90 °C, overnight, 71 %. e) Pd(OAc)2, SPhos, K3PO4, toluene/EtOH/H2O, 80 °C, 40 h, 34 %. f) Pd2(dba)3, SPhos, K3PO4, toluene/THF, 100 °C, 36 h, 70 %.
For the synthesis of 1, (1,1′ : 2′,1′′‐terphenyl)‐2,6‐dicarbaldehyde (9) was firstly obtained by Suzuki coupling between the commercially available 2‐bromoisophthalaldehyde (7) and (1,1′‐biphenyl)‐2‐ylboronic acid (8) in 81 % yield. Afterwards, compound 9 was treated with excess 2‐mesitylmagnesium bromide to give the diol, which was then subjected to Friedel–Crafts alkylation promoted by BF3⋅OEt2 to afford the dihydro‐precursor 4 with a yield of 61 % in two steps. Compound 4 was fully characterized by HR MALDI‐TOF MS, NMR spectroscopy (Table S9 and Figure S46–S50), and single crystal analysis. The single crystal of 4 was obtained by slow evaporation of its dichloromethane/methanol solution. From the two identified isomers, only the cis‐isomer of 4 was observed in the crystal structure (Figure 2a). The attempted dehydrogenation of 4 with p‐chloranil did not give the target mass as only partial dehydrogenation byproduct was identified by MALDI‐TOF MS. In order to ensure the complete dehydrogenation, the treatment with stronger oxidant DDQ in anhydrous and oxygen‐free chlorobenzene afforded the target benzocyclohepta[def]fluorene derivative 1. It was found that 1 decomposed very quickly in a silica gel or aluminum oxide column. Instead, using a flash bio‐beads SX12 column (eluted by anhydrous and oxygen‐free toluene under argon) enabled us to remove most of the unreacted DDQ and its reduced side‐product. The obtained compound 1 was then directly used for the further characterizations. HR MALDI‐TOF MS of 1 shows only one dominant peak with isotopic distribution pattern consistent with the simulated spectrum, revealing its well‐defined molecular composition (Figure 2d). Time‐dependent UV/Vis spectra show that 1 has a persistent stability under ambient conditions with a t 1/2=11.7 h (Figure S9).
Figure 2.

Top view and side view of the X‐ray crystallographic structure (ORTEP drawing with at the 50 % probability level) of dihydro‐precursors a) 4, b) 5 and c) 6. HR MALDI‐TOF MS of d) 1, e) 2 and f) 3. Inset: experimental (red solid line) and simulated (blue solid line) isotopic distribution patterns of the mass peak.
Encouraged by the successful synthesis of 1, we anticipate that additional benzo‐annulation will further delocalize the spin distribution, mostly giving more stable derivatives through thermodynamic stabilization. Thus, the synthesis toward π‐extended 2 and 3 through the similar synthetic strategies was carried out. Compounds 2‐(4,4,5,5‐tetramethyl‐1,3,2‐dioxaborolan‐2‐yl)isophthalaldehyde (10), [21] 4‐chlorophenanthrene (11) [22] and 5‐bromochrysene (13) [5b] were firstly synthesized through multi‐step procedures. Subsequently, 2‐(phenanthren‐4‐yl) isophthalaldehyde (12) or 2‐(chrysen‐5‐yl) isophthalaldehyde (14) were synthesized by Suzuki coupling of 11 or 13 with 10 in 34 % and 70 % yield, respectively. Similar to the synthesis of 4, dihydrogen‐precursor 5 and 6 were also synthesized in 72 % and 70 % yield in two steps. Both compounds were formed as mixtures of geometric isomers as proved by 1H and 13C NMR spectroscopy (Table S10, S11 and Figure S51–S60). Their chemical identities were unambiguously confirmed by NMR and X‐ray crystallographic analysis. Single‐crystal X‐ray analysis illustrated that the main skeletons of 4–6 all contain a pentagon‐heptagon pair (Figure 2a–c). [23] After treatment of 5 or 6 with 3 equiv DDQ, the diradicaloid 2 or 3 were successfully synthesized, respectively, which were confirmed by HR‐MALDI‐TOF MS (Figure 2e, f). The 1H NMR spectra of 1–3 did not show resonance signal in the aromatic region at room temperature and no any clear signal appeared even when the temperature was cooled below −50 °C (Figure S64–S68), indicating their open‐shell diradical feature. Upon further benzo‐annulation, the half‐life time of π‐extended analogues 2 and 3 increased to 23.8 h and 33.3 h, respectively (Figure S10 and S11). However, due to the high instability, attempts to grow single crystals for diradicaloids 1–3 were not successful despite the enormous efforts. It is worth to mention that 1–3 can be easily decomposed by protic solvent, such as methanol and ethanol. For instance, by adding methanol to the CH2Cl2 solution of 1–3, the peak of product [P]+ disappeared while a new peak of [P+MeO]+ appeared (Figure S61–S63), as monitored by MALDI‐TOF MS, which provide an indirect proof for their open‐shell nature. [24]
Next, the optoelectronic properties of 1–3 were investigated with UV‐vis‐NIR spectroscopy. As shown in Figure S8, their dihydrogen‐precursors 4–6 only showed UV/Vis absorption below 450 nm. In sharp contrast, the UV‐vis‐NIR absorption spectra of 1–3 in CH2Cl2 solution all exhibited extremely low‐energy light absorption band with the maximum absorption wavelength (λ max) at 1700 nm, 1860 nm, and 1220 nm, respectively (Figure 3). These weak and lowest‐energy absorptions are derived from their open‐shell characters. [25] The optical energy gap (E g opt) of 1–3 are estimated to be 0.54, 0.52 and 0.69 eV, respectively, from the onset of their UV‐vis‐NIR absorption. In addition, time‐dependent DFT calculations suggest the lowest energy HOMO→LUMO transition at λ max of 1767.7 nm (oscillator strength, f=0.05080), 1813.2 nm (oscillator strength, f=0.03340) and 1302.5 nm (oscillator strength, f=0.05100) for 1–3, respectively, which are consistent with their experimental UV/Vis spectra (Figure S21, S23 and S25).
Figure 3.

UV/Vis‐NIR absorption spectra of diradicaloids 1–3. The spectra were recorded in dry and degassed CH2Cl2 (1.4×10−3 M) at room temperature. Inset shows the magnified view of NIR region. The background absorbance at ca. 2250 nm and 2370 nm may arise from the overtone of CH vibration of the solvent.
To clarify the electronic ground state of diradicaloids 1–3, we performed continuous‐wave electron paramagnetic resonance (cw‐EPR) measurements. All three molecules presented a strong signal both in powder and toluene solution, indicating an open‐shell electronic structure. The powder spectra are featureless and do not show half‐field lines, which is common for the strongly delocalized graphenoid radicals.[ 7b , 26 ] The solution spectra exhibit splitting due to the hyperfine coupling of edge hydrogens, which can be extracted by simulations (Figure S15–S17 and Table S2). In addition, EPR spectra of 1–3 are directly compared and the width of the spectra should be related to the strength of the hyperfine coupling (Figure 4a, b), which is a fingerprint of the delocalization of the spin density over the whole molecule. From the chemical structure of 1–3, compound 1 has the minimum number of benzene rings, 2 and 3 follows. This is matched in what we see in Figure 4a, b: 1 has the widest spectrum, 2 is the middle, and 3 is the narrowest. Moreover, variable‐temperature (VT) EPR measurements of 1–3 are carried out at 5–293 K, in which their signal intensity gradually increases upon cooling (Figure 4c, e, g). However, the signal x T of 1–3 increases as the temperature increased, plateauing already at very low temperatures (below 50 K), as shown in Figure 4d, f, h. Thus, compounds 1–3 have low‐lying triplet states which determine the magnetic properties at room temperature. Careful fitting of the data by using the Bleaney–Bowers equation [27] gives a ΔE ST 1 of −0.04 kcal mol−1 for 1 and ΔE ST 2 of −0.015 kcal mol−1 for 2, while for 3 the temperature resolution is not enough to observe the difference and we can only provide an upper limit ΔE ST 3 ≈−0.002 kcal mol−1. The trend ΔE ST 1 >ΔE ST 2 >ΔE ST 3 can be interpreted in terms of molecular size and spin delocalization. Since the electronic structures of all three molecules are fully delocalized, the delocalization of the spin density is correlated with the extension of the molecular backbone. The strength of the exchange‐coupling constant (J, or −ΔE ST) can be understood in terms of the overlap of electron wavefunction, in which the smaller overlap gives a larger J value. Therefore, the smallest molecule 1 has the largest J, while the largest molecule 3 has the smallest J, with 2 representing in between.
Figure 4.

Continuous‐wave electron paramagnetic resonance (cw‐EPR) spectra of 1–3. a) Room temperature, powder; b) room temperature, in toluene solution. Temperature dependence of the EPR signal in powder c) 1, e) 2 and g) 3. Integrated signal multiplied by temperature and fitting with Bleaney–Bowers’ equation d) 1, f) 2 and h) 3. Inset: Spin density surfaces calculated by DFT PBE0/EPR‐II in the triplet state, isovalue 0.005.
To gain deep insight into the electronic structure of 1–3, DFT calculations at the B3LYP/6‐31G(d) (GD3BJ) level of theory by using the Gaussian 16 program package were performed. As predicted by the extension of Ovchinnikov rule (Figure 1b), the most stable ground state of 1–3 are estimated to be triplet state since their minimum number of spin frustrations is equal to 1 (S=1). According to the single‐point energy calculation, 1 and 2 demonstrate triplet ground state with the ΔE ST value of 1.93 and 0.873 kcal mol−1, respectively. While for 3, the most stable ground state is predicted to be singlet state and the ΔE ST value is −1.67 kcal mol−1. The different electronic structures between 1–3 can be explained by the more effective π‐conjugation for 3 that enhances the stability of its singlet ground state. [28] On the other hand, the differences between the experiment and calculated ΔE ST value are all quite small, which mostly due to the imperfect energy calculation of the broken‐symmetry singlet state by the UB3LYP function. [29] To analyze the aromaticity of 1–3, nucleus‐independent chemical shift (NICS) [30] and anisotropy of the induced current density (ACID) [31] calculations at the UB3LYP/6‐311+G(d,2p) and UB3LYP/6‐31G(d) level of theory were performed, respectively (Figure S22, S24 and S26). The five‐ and seven‐membered rings are more antiaromatic than other benzenoid rings as indicated by their large positive NICS (1) ZZ‐avg values. These are consistent with their ACID plots that display a counterclockwise diatropic ring current of the π‐electrons along the azulene unit.
In conclusion, we successfully opened up the path to the synthesis of open‐shell non‐alternant PHs, demonstrating three novel PHs based on the skeleton of cyclohepta[def]fluorene through an efficient stabilization strategy by benzannulation. [32] The formations of 1–3 are clearly validated by HR‐MALDI‐TOF MS analysis and UV/Vis‐NIR spectroscopy. Besides, the obtained persistent diradcialoids have the long‐wavelength electronic absorption with an extremely narrow optical energy gap (0.54, 0.52 and 0.69 eV for 1–3, respectively). Moreover, we demonstrate the role of external fused benzo rings in stabilizing the triplet ground state onto the non‐alternant skeleton of cyclohepta[def]fluorenes. VT EPR results indicate that 1–3 have a singlet ground state with an extremely small singlet‐triplet energy gap. These results validate the incorporation of non‐alternant units into benzenoid PHs as a promising strategy for designing open‐shell PHs with very low singlet–triplet energy gaps. High spins state can now be envisaged by chemical tuning; these developments are ongoing in our laboratory, together with the creation of multistable molecules that involve states of higher spin multiplicities.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
This research was financially supported by the EU Graphene Flagship (Graphene Core 3, 881603), ERC Consolidator Grant (T2DCP, 819698), the Center for Advancing Electronics Dresden (cfaed), H2020‐EU.1.2.2.—FET Proactive Grant (LIGHT‐CAP, 101017821), the DFG‐SNSF Joint Switzerland‐German Research Project (EnhanTopo, No. 429265950), the EU (ERC‐CoG‐MMGNRs‐773048), EPSRC (EP/N509711/1, EP/R042594/1, EP/L011972/1), and the Royal Society (URF and Grant). F. Wu gratefully acknowledges funding from the China Scholarship Council. The authors gratefully acknowledge the GWK support for funding this project by providing computing time through the Center for Information Services and HPC (ZIH) at TU Dresden. Diffraction data have been collected on BL14.2 at the BESSY II electron storage ring operated by the Helmholtz‐Zentrum Berlin; we appreciate the help and support of Dr. Manfred Weiss and his team during the experiments at BESSY II. We also thank Prof. Dr. Martin Baumgarten (Max Planck Institute for Polymer Research, Mainz), Dr. Zichao Li and Dr. Shengqiang Zhou (Helmholtz‐Zentrum Dresden‐Rossendorf, Dresden) for the helpful discussions. Open Access funding enabled and organized by Projekt DEAL.
F. Wu, J. Ma, F. Lombardi, Y. Fu, F. Liu, Z. Huang, R. Liu, H. Komber, D. I. Alexandropoulos, E. Dmitrieva, T. G. Lohr, N. Israel, A. A. Popov, J. Liu, L. Bogani, X. Feng, Angew. Chem. Int. Ed. 2022, 61, e202202170; Angew. Chem. 2022, 134, e202202170.
Contributor Information
Dr. Ji Ma, Email: ji.ma@tu-dresden.de.
Prof. Dr. Lapo Bogani, Email: lapo.bogani@materials.ox.ac.uk.
Prof. Dr. Xinliang Feng, Email: xinliang.feng@tu-dresden.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Nishide H., Adv. Mater. 1995, 7, 937–941; [Google Scholar]
- 1b. Ratera I., Veciana J., Chem. Soc. Rev. 2012, 41, 303–349; [DOI] [PubMed] [Google Scholar]
- 1c. Wang Z.-Y., Dai Y.-Z., Ding L., Dong B.-W., Jiang S.-D., Wang J.-Y., Pei J., Angew. Chem. Int. Ed. 2021, 60, 4594–4598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4644–4648; [Google Scholar]
- 1d. Shu C., Zhang H., Olankitwanit A., Rajca S., Rajca A., J. Am. Chem. Soc. 2019, 141, 17287–17294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Ma H., Liu C., Zhang C., Jiang Y., J. Phys. Chem. A 2007, 111, 9471–9478; [DOI] [PubMed] [Google Scholar]
- 2b. Rajca S., Rajca A., J. Am. Chem. Soc. 1995, 117, 9172–9179; [Google Scholar]
- 2c. Tang S., Zhang L., Ruan H., Zhao Y., Wang X., J. Am. Chem. Soc. 2020, 142, 7340–7344. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wang K., Liu P., Zhang F., Xu L., Zhou M., Nakai A., Kato K., Furukawa K., Tanaka T., Osuka A., Song J., Angew. Chem. Int. Ed. 2021, 60, 7002–7006; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 7078–7082; [Google Scholar]
- 3b. Zeng Z., Sung Y. M., Bao N., Tan D., Lee R., Zafra J. L., Lee B. S., Ishida M., Ding J., López Navarrete J. T., Li Y., Zeng W., Kim D., Huang K.-W., Webster R. D., Casado J., Wu J., J. Am. Chem. Soc. 2012, 134, 14513–14525; [DOI] [PubMed] [Google Scholar]
- 3c. Dressler J. J., Zhou Z., Marshall J. L., Kishi R., Takamuku S., Wei Z., Spisak S. N., Nakano M., Petrukhina M. A., Haley M. M., Angew. Chem. Int. Ed. 2017, 56, 15363–15367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15565–15569. [Google Scholar]
- 4. McMasters D. R., Wirz J., J. Am. Chem. Soc. 2001, 123, 238–246. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Lombardi F., Lodi A., Ma J., Liu J., Slota M., Narita A., Myers W. K., Müllen K., Feng X., Bogani L., Science 2019, 366, 1107–1110; [DOI] [PubMed] [Google Scholar]
- 5b. Lombardi F., Ma J., Alexandropoulos D. I., Komber H., Liu J., Myers W. K., Feng X., Bogani L., Chem 2021, 7, 1363–1378; [Google Scholar]
- 5c. Mishra S., Catarina G., Wu F., Ortiz R., Jacob D., Eimre K., Ma J., Pignedoli C. A., Feng X., Ruffieux P., Fernández-Rossier J., Fasel R., Nature 2021, 598, 287–292; [DOI] [PubMed] [Google Scholar]
- 5d. Mishra S., Beyer D., Eimre K., Kezilebieke S., Berger R., Gröning O., Pignedoli C. A., Müllen K., Liljeroth P., Ruffieux P., Feng X., Fasel R., Nat. Nanotechnol. 2020, 15, 22–28. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Longuet-Higgins H. C., J. Chem. Phys. 1950, 18, 265–274; [Google Scholar]
- 6b. Hoffmann R., J. Am. Chem. Soc. 1968, 90, 1475–1485. [Google Scholar]
- 7.
- 7a. Konishi A., Horii K., Shiomi D., Sato K., Takui T., Yasuda M., J. Am. Chem. Soc. 2019, 141, 10165–10170; [DOI] [PubMed] [Google Scholar]
- 7b. Liu J., Mishra S., Pignedoli C. A., Passerone D., Urgel J. I., Fabrizio A., Lohr T. G., Ma J., Komber H., Baumgarten M., Corminboeuf C., Berger R., Ruffieux P., Müllen K., Fasel R., Feng X., J. Am. Chem. Soc. 2019, 141, 12011–12020; [DOI] [PubMed] [Google Scholar]
- 7c. Mishra S., Lohr T. G., Pignedoli C. A., Liu J., Berger R., Urgel J. I., Müllen K., Feng X., Ruffieux P., Fasel R., ACS Nano 2018, 12, 11917–11927; [DOI] [PubMed] [Google Scholar]
- 7d. Ma J., Fu Y., Dmitrieva E., Liu F., Komber H., Hennersdorf F., Popov A. A., Weigand J. J., Liu J., Feng X., Angew. Chem. Int. Ed. 2020, 59, 5637–5642; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5686–5691. [Google Scholar]
- 8.
- 8a. Zhang X.-S., Huang Y.-Y., Zhang J., Meng W., Peng Q., Kong R., Xiao Z., Liu J., Huang M., Yi Y., Chen L., Fan Q., Lin G., Liu Z., Zhang G., Jiang L., Zang D., Angew. Chem. Int. Ed. 2020, 59, 3529–3533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3557–3561; [Google Scholar]
- 8b. Yang X., Rominger F., Mastalerz M., Angew. Chem. Int. Ed. 2019, 58, 17577–17582; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17741–17746; [Google Scholar]
- 8c. Fei Y., Fu Y., Bai X., Du L., Li Z., Komber H., Low K.-H., Zhou S., Phillips D. L., Feng X., Liu J., J. Am. Chem. Soc. 2021, 143, 2353–2360. [DOI] [PubMed] [Google Scholar]
- 9. Konishi A., Yasuda M., Chem. Lett. 2021, 50, 195–212. [Google Scholar]
- 10.
- 10a. Hafner K., Süss H. U., Angew. Chem. Int. Ed. Engl. 1973, 12, 575–577; [Google Scholar]; Angew. Chem. 1973, 85, 626–628; [Google Scholar]
- 10b. Bally T., Chai S., Neuenschwander M., Zhu Z., J. Am. Chem. Soc. 1997, 119, 1869–1875. [Google Scholar]
- 11.
- 11a. H. J. Dauben Jr. , Bertelli D. J., J. Am. Chem. Soc. 1961, 83, 4659–4660; [Google Scholar]
- 11b. Lindner H. J., Kitschke B., Angew. Chem. Int. Ed. Engl. 1976, 15, 106–107; [Google Scholar]; Angew. Chem. 1976, 88, 123–124. [Google Scholar]
- 12.
- 12a. Tobe Y., Chem. Rec. 2015, 15, 86–96; [DOI] [PubMed] [Google Scholar]
- 12b. Chaolumen, Stepek I. A., Yamada K. E., Ito H., Itami K., Angew. Chem. Int. Ed. 2021, 60, 23508–23532; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 23700–23724. [Google Scholar]
- 13. Ovchinnikov A. A., Theor. Chim. Acta 1978, 47, 297–304. [Google Scholar]
- 14.
- 14a. Guihery N., Maynau D., Jean-Paul Malrieu A., New J. Chem. 1998, 22, 281–286; [Google Scholar]
- 14b. Dias J. R., Mol. Phys. 2013, 111, 735–751. [Google Scholar]
- 15.
- 15a. Boekelheide V., Vick G. K., J. Am. Chem. Soc. 1956, 78, 653–658; [Google Scholar]
- 15b. Gardner P. D., Wulfman C. E., Osborn C. L., J. Am. Chem. Soc. 1958, 80, 143–148; [Google Scholar]
- 15c. Jun-ichi A., Bull. Chem. Soc. Jpn. 1978, 51, 3540–3543; [Google Scholar]
- 15d. Hafner K., Fleischer R., Fritz K., Angew. Chem. Int. Ed. Engl. 1965, 4, 69–70; [Google Scholar]; Angew. Chem. 1965, 77, 42–43; [Google Scholar]
- 15e. Reel H., Vogel E., Angew. Chem. Int. Ed. Engl. 1972, 11, 1013–1014; [Google Scholar]; Angew. Chem. 1972, 84, 1064–1066; [Google Scholar]
- 15f. A. G. Anderson Jr. , MacDonald A. A., Montana A. F., J. Am. Chem. Soc. 1968, 90, 2993–2994. [Google Scholar]
- 16.
- 16a. Nendel M., Goldfuss B., Houk K. N., Hafner K., Grieser U., Theor. Chem. Acc. 1999, 102, 397–400; [Google Scholar]
- 16b. Baumgartner P., Weltin E., Wagnière G., Heilbronner E., Helv. Chim. Acta 1965, 48, 751–764. [Google Scholar]
- 17.
- 17a. Munday R., Sutherland I. O., Chem. Commun. 1967, 12, 569–570; [Google Scholar]
- 17b. Munday R., Sutherland I. O., J. Chem. Soc. C 1969, 10, 1427–1434. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Grieser U., Hafner K., Chem. Ber. 1994, 127, 2307–2314; [Google Scholar]
- 18b. Horii K., Kishi R., Nakano M., Shiomi D., Sato K., Takui T., Konishi A., Yasuda M., J. Am. Chem. Soc. 2022, 144, 3370–3375. [DOI] [PubMed] [Google Scholar]
- 19. Das S., Wu J., Org. Lett. 2015, 17, 5854–5857. [DOI] [PubMed] [Google Scholar]
- 20. Tang B., Zhao J., Xu J.-F., Zhang X., Chem. Sci. 2020, 11, 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y.-K., Plattner J. J., Easom E. E., Waterson D., Ge M., Li Z., Li L., Jian Y., Tetrahedron Lett. 2011, 52, 3909–3911. [Google Scholar]
- 22. Matsubara S., Koga Y., Segawa Y., Murakami K., Itami K., Nat. Catal. 2020, 3, 710–718. [Google Scholar]
- 23.Deposition Numbers 2111792 (for 4), 2111790 (for 5) and 2111791 (for 6) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 24. Yang X., Zhang D., Liao Y., Zhao D., J. Org. Chem. 2020, 85, 5761–5770. [DOI] [PubMed] [Google Scholar]
- 25. Shimizu A., Kishi R., Nakano M., Shiomi D., Sato K., Takui T., Hisaki I., Miyata M., Tobe Y., Angew. Chem. Int. Ed. 2013, 52, 6076–6079; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6192–6195. [Google Scholar]
- 26.
- 26a. Wang Q., Gopalakrishna T. Y., Phan H., Herng T. S., Dong S., Ding J., Chi C., Angew. Chem. Int. Ed. 2017, 56, 11415–11419; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11573–11577; [Google Scholar]
- 26b. Ma J., Zhang K., Schellhammer K. S., Fu Y., Komber H., Xu C., Popov A. A., Hennersdorf F., Weigand J. J., Zhou S., Pisula W., Ortmann F., Berger R., Liu J., Feng X., Chem. Sci. 2019, 10, 4025–4031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c. Zeng W., Phan H., Herng T. S., Gopalakrishna T. Y., Aratani N., Zeng Z., Yamada H., Ding J., Wu J., Chem 2017, 2, 81–92. [Google Scholar]
- 27. Bleaney B., Bowers K. D., Proc. R. Soc. London Ser. A 1952, 214, 451–465. [Google Scholar]
- 28. Li G., Matsuno T., Han Y., Wu S., Zou Y., Jiang Q., Isobe H., Wu J., Angew. Chem. Int. Ed. 2021, 60, 10326–10333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10414–10421. [Google Scholar]
- 29.
- 29a. Hajgató B., Huzak M., Deleuze M. S., J. Phys. Chem. A 2011, 115, 9282–9293; [DOI] [PubMed] [Google Scholar]
- 29b. Shee J., Arthur E. J., Zhang S., Reichman D. R., Friesner R. A., J. Chem. Theory Comput. 2019, 15, 4924–4932. [DOI] [PubMed] [Google Scholar]
- 30. Schleyer P. V. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J., J. Am. Chem. Soc. 1996, 118, 6317–6318. [DOI] [PubMed] [Google Scholar]
- 31. Herges R., Geuenich D., J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar]
- 32.During the peer-review process of our manuscript (submission date: February 9), Yasuda et al. reported the synthesis of triaryl derivatives of cyclohepta[def]fluorene (publication date: February 21, see Ref. [18b]).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.