

Abstract
Seed inoculation with beneficial microorganisms has gained importance as it has been proven to show biostimulant activity in plants, especially in terms of abiotic/biotic stress tolerance and plant growth promotion, representing a sustainable way to ensure yield stability under low input sustainable agriculture. Nevertheless, limited knowledge is available concerning the molecular and physiological processes underlying the root‐inoculant symbiosis or plant response at the root system level. Our work aimed to integrate the interrelationship between agronomic traits, rhizosphere microbial population and metabolic processes in roots, following seed treatment with either arbuscular mycorrhizal fungi (AMF) or Plant Growth‐Promoting Rhizobacteria (PGPR). To this aim, maize was grown under open field conditions with either optimal or reduced nitrogen availability. Both seed treatments increased nitrogen uptake efficiency under reduced nitrogen supply revealed some microbial community changes among treatments at root microbiome level and limited yield increases, while significant changes could be observed at metabolome level. Amino acid, lipid, flavone, lignan, and phenylpropanoid concentrations were mostly modulated. Integrative analysis of multi‐omics datasets (Multiple Co‐Inertia Analysis) highlighted a strong correlation between the metagenomics and the untargeted metabolomics datasets, suggesting a coordinate modulation of root physiological traits.
1. INTRODUCTION
The world population is predicted to reach 9.7 billion by 2050 (DeSa, 2015) and, consequently, global demand for food and other agricultural products is expected to rise by 50% (FAO, 2017), thus requiring a marked increase in agricultural productivity. Cereal production in the world is projected to reach 3054 Mt in 2029, and maize yield is expected to increase the most (+193 Mt) (OECD/FAO, 2021). Maize represents the second most important cereal globally in terms of acreage and the first one for production, predominantly cultivated in the USA and used to feed livestock and humans, in addition to bioethanol (FAOSTAT, 2019).
Agriculture worldwide will need to increase its yields to meet the growing demand also for maize‐based products (OECD/FAO, 2021). With this regard, intensive cropping systems will require sustainable approaches to avoid the negative externalities normally linked to the over‐use of chemical inputs, such as biodiversity loss and water/soil degradation (Panfili et al., 2019). In this framework, the use of plant biostimulants in agriculture, recently normed by the Regulation (EU) 2019/1009, may represent a sustainable tool to ensure yield stability under low input sustainable agriculture. Recently, positive advances have been found for plant microbial biostimulant application, reflecting advantageous implications for many agronomic and physiological crop traits (Guerrieri et al., 2020; Guerrieri et al., 2021; Rouphael et al., 2020). The most promising microbial biostimulants include arbuscular mycorrhizal fungi (AMF), Trichoderma spp., and plant growth‐promoting rhizobacteria (PGPR) such as Azotobacter ssp., Rhizobium ssp. and Azospirillum ssp. The inoculation of beneficial microorganisms has gained remarkable importance, having been proven to show a biostimulant activity in plants, especially in terms of abiotic/biotic stress alleviation (Sangiorgio et al., 2020), growth promotion (Nacoon et al., 2020), and improvement of food functional quality (Ganugi et al., 2021a, 2021b). The putative mechanisms of biostimulation include the provision of enzymatic activities and/or release of small molecules and peptides (in turn affecting nutrients uptake), the release of antimicrobials or quorum‐sensing compounds, and the modulation of plant root architecture (Fiorentino et al., 2018; Lucini et al., 2019; Raaijmakers & Mazzola, 2012; Saia et al., 2020).
Despite the consensus on their effects, limited knowledge is available to date concerning the molecular and physiological processes underlying plant‐inoculants symbiosis or regarding plant response at root system level under field conditions. The latter information is relevant given the pivotal role played by roots in coping with drought, nutrient deficiencies, toxicants, and soil compaction (Ryan et al., 2016). Notwithstanding, the plant dependence on its microbiota across all development stages is also known, with several microbial strains thriving in the rhizosphere that can affect plant health and productivity, as well as resistance to both abiotic and biotic stresses (Colla et al., 2017). The interrelationships between root agronomic traits, microbiota and metabolites remain largely unstudied, and the assessment of the metabolomic changes in roots is necessary to fully understand the tripartite interaction between roots, soil and microorganisms underlying the biostimulant activity (Rouphael et al., 2020).
The lack of information about the interplay mechanisms between roots and beneficial microorganisms can be related to the complex and dynamic processes involved. However, the recent development of new tools, including the integration of multiple omics datasets, has paved the way for a deeper understanding of plant‐microbe interactions. Multivariate approaches such as Multiple Co‐inertia Analysis (MCIA) have been proposed to identify co‐relationships between multiple high dimensional datasets, based on a covariance optimization criterion (Min & Long, 2020). Nevertheless, despite the advances in this field, this approach remained limited to human and food studies (Afshari et al., 2020; Meng et al., 2014).
The present study aims at integrating root metabolomics and rhizosphere metagenomics to investigate the interrelationships between agronomic traits, rhizosphere bacterial community and root metabolic processes in maize, following seed treatment with either AMF or PGPR. To this object, MCIA has been used for data integration, combining agronomic trait data, metagenomics analysis, and untargeted metabolomics analyses.
2. MATERIALS AND METHODS
2.1. Experimental site, treatments, and crop management
The field experiment started in June 2020, using maize (Zea mays L.), and was conducted over the entire cropping season (until October 2020) at the CERZOO experimental station of Università Cattolica del Sacro Cuore, in Piacenza, Northern Italy (45°00′21.6″N, 9°42′27.1″E; altitude 68 m a.s.l.). The local climate is temperate (Cfa as Köppen classification), with an average annual temperature of 13.2 °C and annual precipitation of 837 mm. During the experiment, climatic data were collected from an automated meteorological station positioned close to the field (Figure S1). The soil is a fine, mixed, mesic Udertic Haplustalf according to Soil Taxonomy (Soil Survey Staff, 2014). Soil properties in the top 30 cm soil layer (mean values ± standard deviation) were: organic matter 27.1 ± 0.5 g kg−1; pH (H2O) 7.5 ± 0.1; bulk density 1.36 ± 0.5 g cm−3; sand 136 ± 15 g kg−1; silt 448 ± 33 g kg−1; clay 416 ± 20 g kg−1; soil total N 1.7 ± 0.1 g kg−1; soil N‐NO3 − 8.3 ± 0.2 mg kg−1; soil N‐NH4 + 3.3 ± 0.3 mg kg−1; available P (Olsen) 41.4 ± 2.0 mg kg−1; exchangeable K (NH4 + Ac) 197 ± 16 mg kg−1; and cation exchange capacity 26.2 ± 0.9 cmol+ kg−1.
A split‐plot (SP) design was arranged to test maize responses to two commercially available biostimulant‐based treatments, consisting of seed dressing with either the AMF‐based product Aegis Sym irriga (Rhizoglomus irregulare BEG72 and Funneliformis mosseae BEG234, 700 sp g−1 each species), or the PGPR formulation Bactrium (Bacillus megaterium BM77 e BM06), all from Atens, Agrotecnologias Naturales SL (Tarragona, Spain). The seed dressing was homogenized with the maize seeds using an automatic mixer. The rate of seed dressing applied to the maize seed (around 88,000 seeds ha−1) was 2 kg ha−1 for Aegis Sym irriga, and 2 kg ha−1 for Bactrium, according to the manufacturer's label specifications. Then, a commercially available protein hydrolysate (Trainer, from Hello Nature SpA, Rivoli Veronese, Italy) was applied at the V6‐V7 phenological stage by foliar spraying application. The main factor in the SP design was the biostimulant treatment, with three levels: (1) T1, no seed treatment as the Control; (2) T2, treatment with AMF; and (3) T3, treatment with the B. megaterium PGPR formulation. The secondary factor was the chemical N‐fertilization rate (Urea 46%‐N), with two levels: (i) 230 kg N ha−1 as the 100% N‐fertilization, and (ii) 160 kg N ha−1 as the 70% N‐fertilization). The 100% N‐fertilization rate was estimated according to the N balance, considering crop and soil‐climate variables (Sainju, 2017). The number of replicates was three (three blocks), with a total of 18 sub‐plots. The elemental sub‐plot size was 160 m2 (25 m long and 8 m wide).
The previous crop was maize. Tillage operations (i.e., 30 cm subsoiling followed by 15 cm rotary harrowing) were conducted during the previous winter–spring season (between November 2019 and April 2020). Maize (hybrid LG Aaoitgeiz, FAO 400) was planted on June 16, 2020, with 75 cm spacing between rows. The N‐fertilizer was applied once at the V4–V5 stage (July 17, 2020) and was incorporated into the soil during distribution. To prevent water stress, maize was sprinkler‐irrigated four times at doses of 25, 35, 35, and 40 mm. A detailed description of irrigation water doses estimation from the maize crop evapotranspiration, crop coefficients (Kc) calculation, and crop irrigation requirements (CIR) is reported in Fiorini et al. (2020). The field was treated with 0.4 L ha−1 of the pre‐emergence herbicide Adengo Xtra (Isoxaflutole 19.1% + Thiencarbazone‐methyl 7.6% + Cyprosulfamide 12.7%) to control weeds. Maize was harvested on October 8, 2020.
2.2. Maize yield and N‐uptake efficiency
Yield components of the maize crop were assessed by manually harvesting a 10 m2 area per plot. Plants collected were weighed and separated into grain and stover for biomass determination. Then, a 100 g sub‐sample of each grain and stover sample was oven‐dried at 65°C until constant weight to measure relative water content. Nitrogen (N) concentration for each sub‐sample was determined by Dumas combustion method with an elemental analyzer varioMax C:N (VarioMax C:NS, Elementar). N‐uptake in grain and stover was calculated by multiplying grain and stover yield by their N concentration. N‐uptake efficiency (NUpE; kg kg−1) was calculated as the ratio of total plant N‐uptake to N‐supply according to López‐Bellido and López‐Bellido (2001).
2.3. Root sampling and analyses
Maize roots were sampled at anthesis, BBCH 69, on September 2, 2020 (79 days after sowing), following the procedures previously reported in Maris et al., 2021. Briefly, a self‐constructed “Shelby” tube sampler of 7 cm diameter was pushed into the soil to collect an intact 0–15 cm soil core. Sampling occurred in each plot, at two positions on the perpendicular of the crop row: 0 cm (on the row, i.e., close to the base of the sampled plant but not including the maize stalk) and at 37.5 cm (mid‐row). Then, root biomass was extracted from the surrounding soil by washing in a hydraulic sieving‐centrifugation device. After extraction, roots were scanned at 600 dpi with the scanner Epson Expression 10000xl, equipped with a double light source to avoid roots overlapping. The determination of Root Length Density (RLD, cm cm−3) and root diameter was performed using the software winRHIZO Reg 2012. The Diameter Class Length (DCL, mm cm−3) was then calculated for very fine (0.0–0.075 mm), fine (0.075–0.2 mm), medium (0.2–1.0 mm), and coarse (>1 mm) diameters for the whole soil profile, as adapted from Fiorini et al. (2018). After the analysis, the root samples were oven‐dried at 65°C until constant weight to measure root dry biomass. Root dry weight (RDW, mg cm−3) was then computed as the ratio between the root dry biomass and the initial volume of the soil sample.
2.4. Sampling and UHPLC/QTOF‐MS untargeted metabolomic analysis
Additional root samples were collected (on September 2, 2020) from each sub‐plot and isolated from the surrounding soil following the procedures reported above. Then, clean roots were ground with liquid nitrogen using pestle and mortar and extracted, as previously reported by Rouphael et al. (2020). In brief, a 2 g aliquot was extracted in 10 ml of 0.1% formic acid in 80% aqueous methanol using an Ultra‐Turrax (Ika T‐25, Staufen) and successively centrifuged (12,000 g). Untargeted metabolomics was concomitantly next carried out using a UHPLC liquid chromatography system and a quadrupole‐time‐of‐flight mass spectrometer equipped with an electrospray ionization source (UHPLC/QTOF), according to Ganugi et al. (2021a, 2021b). To this aim, a 1290 LC system was coupled to a G6550 quadrupole‐time‐of‐flight mass spectrometer (Agilent Technologies). A water‐acetonitrile reverse phase gradient elution (6–94% acetonitrile in 34 min) and positive polarity SCAN acquisition (range 100–1200 m/z) were used for chromatography and electrospray mass spectrometry, respectively. Profinder B.07 software (Agilent Technologies) and the comprehensive database PlantCyc 9.6 (Schläpfer et al., 2017) were used for compound annotation by combining both the monoisotopic accurate mass and isotopic pattern (i.e., isotope spacing and ratio) and adopting a mass accuracy tolerance of <5 ppm, as previously described (Ceccarelli et al., 2021). As referred by COSMOS Metabolomics Standards Initiative (Salek et al., 2015), a putative Level 2 annotation was achieved. Thereafter, annotated features were filtered by frequency to retain compounds in at least 75% of replicates within at least one treatment.
2.5. Rhizosphere bacterial community diversity
A third sampling was carried out from each sub‐plot to analyze the rhizosphere bacterial community. The soil particles adhering to roots, following vigorous shaking, were carefully and aseptically collected using forceps at the time of sampling into the Whirl‐Pak (Nasco) sample bags and frozen until further analysis at −20 °C. The whole soil DNA was then extracted using the DNeasy PowerSoil Kit (Ref 12888–100, QIAGEN) according to the manufacturer's protocol. The extracted DNA of the bacterial community associated with plant roots was amplified by primers 343F (5′‐TACGGRAGGCAGCAG‐3′) and 802R (5′‐TACNVGGGTWTCTAATCC‐3′) targeting the hypervariable V3 and V4 regions of the bacterial 16S rRNA gene (Maris et al., 2021). A two‐step nested‐PCR was applied, and conditions used for reaction mix and amplification experiments were described by Vasileiadis et al. (2015). The PCR products of the second step for all samples were multiplexed in a single pool in equimolar amounts based on the QuBit quantification data. The PCR products pool was then purified using the solid phase reverse immobilization method of the AgencourtAMPure XP kit (Beckman Coulter) and sequenced at Fasteris SA (Geneva, Switzerland). The TruSeq rDNA sample preparation kit (Illumina Inc.) was used for the amplicon library preparation, while the sequencing reaction was performed with a MiSeq Illumina instrument (Illumina Inc.) with V3 chemistry, generating 300 bp paired‐end reads.
The raw sequences data have been deposited in the Sequence Read Archive of NCBI (accession number being assigned).
3. STATISTICAL ANALYSES
3.1. Maize yield, N‐uptake efficiency and root traits
A two‐way analysis of variance (ANOVA) was performed with a mixed effect model on (1) grain yield and grain N‐uptake, (2) stover biomass and stover N‐uptake, and (3) NUpE. Maize treatment (T) and N‐fertilization (N) were selected as fixed factors, while the plot block was the random effect of the mixed model. A repeated‐measures ANOVA was conducted with a mixed effect model on root traits (diameter class length, DCL; root length density, RLD; and root dry weight, RDW) selecting T, N and distance from the row (D) as fixed factors and block as a random effect. The distribution of measured variables was checked for normality using the Shapiro–Wilk test and homogeneity of variances with the Levene's test. Data were log‐transformed prior to analysis when the ANOVA assumptions were violated; then, Shapiro–Wilk and Levene's tests were performed again on log‐transformed data. Treatment means were compared using Tukey's honestly significant difference (HSD) at p‐value <0.05. Statistical analyses were conducted using R 4.0.3. (R Core Team, 2020) with nlme (Pinheiro et al., 2013) and multcomp (Hothorn et al., 2007) packages.
3.2. Metabolomics
The metabolomics dataset was elaborated using Mass Profiler Professional B12.6 (Agilent Technologies, Santa Clara, CA) following log2 transformation, normalization and baselining against the median abundance of each compound (Miras‐Moreno et al., 2020). Metabolomic patterns across treatments were naively investigated through unsupervised hierarchical cluster analysis (Euclidean distance, Ward's linkage) based on fold change (FC) values. The dataset was then imported into SIMCA 13 (Umetrics) to perform the supervised orthogonal projections to latent structures discriminant analysis (OPLS‐DA), whose model was successively cross‐validated (CV‐ANOVA) and inspected for outliers (Hotelling's T2). Thereafter, model parameters, including the goodness‐of‐fit (R2Y) and goodness‐of‐prediction (Q2Y), were recorded, and a permutation test (N = 200) was carried out to exclude overfitting. The variable importance in projection (VIP), calculated as a weighted sum of the squared correlations between the OPLS‐DA components and the original variables, was used to select the most discriminant compounds (VIP ≥1.3). Finally, to achieve information on the regulation of these discriminant compounds, a Fold‐Change (FC) analysis (FC ≥ 1) was carried out, and the resulting values were exported to the PlantCyc Pathway Tools software (Karp et al., 2009) for biological and biochemical interpretations.
3.3. Metagenomics
High‐throughput sequencing data filtering, multiplexing and preparation for concomitant statistical analyses were carried out as previously detailed (Połka et al., 2015; Vasileiadis et al., 2015). Paired‐reads were assembled to reconstruct the full V3‐V4 amplicons with the “pandaseq” script (Masella et al., 2012), allowing a maximum of 2 mismatches and at least 30 bp of overlap between the read pairs. Samples demultiplexing was then carried out using the Fastx‐toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) and. Mothur v.1.32.1 (Schloss et al., 2009) was applied to remove sequences with large homopolymers (≥10), sequences that did not align within the targeted V3‐V4 region, chimeric sequences out of quality criteria (Edgar et al., 2011) and sequences that were not classified as bacterial ones, after alignment against the Mothur version of the RDP training data set. The resulting high‐quality sequences were analyzed with Mothur and R 4.0 (Team, 2012) was used for the following two main approaches: the operational taxonomic unit (OTU) and the taxonomy‐based approach. For the OTU approach, sequences were first aligned against the SILVA reference aligned database for bacteria (Pruesse et al., 2007) using the NAST algorithm and a kmer approach (DeSantis et al., 2006; Schloss, 2010), and then clustered at the 3% distance using the average linkage algorithm. Canonical correspondence analysis (CCA) was applied to assess the significance of different treatments on the analyzed diversity of the samples with the statistical analysis using the same software. OTUs having a sum of their abundances across all samples of more than 0.1% of the total were grouped into a single “rare OTUs” group. For the taxonomy based analyses, sequences were classified into taxa using an amended version of the Greengenes database (McDonald et al., 2012). Alpha diversity indices results were subjected to the one‐way analysis of variances (ANOVA) and the means were statistically compared by the least significant differences (LSD) test at p < 0.05 threshold using CoStat Statistical Software (Version 6400, CoHort Software).
3.4. Integrative analysis of maize roots traits, microbiome, and metabolome datasets
Multiple Co‐inertia Analysis (MCIA) was performed with the omicade4 package in Rstudio software (4.0.2 version) to identify the co‐relationships between multiple high‐dimensional datasets, which contained the same samples and were obtained through the agronomic, metagenomic, and metabolomic analyses. Initially, OTUs and metabolites with zero values in more than 90% of the samples were removed. Successively, a principal component (PCA) was applied to each multidimensional‐omics dataset separately, transforming data into comparable lower dimensional spaces. Finally, as previously reported by Afshari et al. (2020), the variance structures were combined into a single analysis to find a new axis on which the omics datasets could be projected by maximizing the square covariance, according to Afshari et al. (2020). Different shapes were used to represent the three datasets in the MCIA graphical outputs (Figure 7), while different colors were adopted to highlight the treatments. Each shape was connected by lines whose length was proportional to the divergence between the data derived from the same sample.
FIGURE 7.
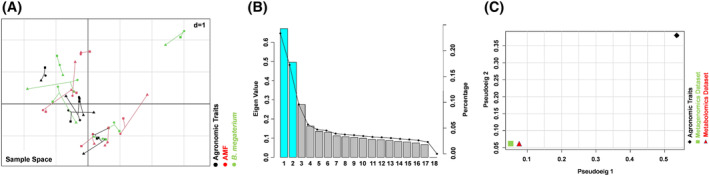
MCIA projection plot. (A) The first two axes of MCIA represents metabolomics, microbial community and agronomic traits of the maize root samples. Different shapes (diamond: Agronomic traits; triangle: Metagenomics dataset; square: Metabolomics dataset) represent the different variables connected by lines, the length of these lines is proportional to the divergence between the datasets. Lines for each sample are joined at a common point, at which the covariance derived from the MCIA analysis is maximal. Color shows the different thesis (black: Control; red: AMF‐based product; green: B. megaterium PGPR formulation). (B) Pseudo‐eigenvalue space representing the percentage of variance explained by the first twoof the MCIA components. (C) Pseudo‐eigenvalues space of all datasets for maize roots, showing overall co‐structure between three datasets and shows which dataset contributes more to the total variance
3.5. Correlation analysis
The correlation between metabolites and OTUs was calculated using Pearson's rank correlation analysis, performed in Rstudio with the rcorr function of the Hmisc package (Harrell Jr & Harrell Jr, 2019). The first 50 more discriminant metabolites and OTUs were selected for the analysis using the VIP approach to identify the most significant inter‐omic correlations following the biostimulant treatments. Differences were considered significant when p‐value <0.05.
4. RESULTS
4.1. Maize yield and N‐uptake efficiency
Grain yield and grain N‐uptake were not affected by the factors or the interaction between factors (Table 1). Conversely, stover biomass and stover N‐uptake were significantly affected by N‐fertilization (N) (Table 1), with 100% N‐fertilization having higher values than 70% N‐fertilization for both parameters. NUpE was significantly affected by N and the interaction maize treatment (T) × N (Table 1). Overall, 70% N‐fertilization had higher NUpE than 100% N‐fertilization. More in detail, the interaction T × N showed that both T2 and T3 had higher NUpE than T1 under 70% N‐fertilization level, while no difference between treatments occurred under the 100% fertilization level (Figure 1).
TABLE 1.
Analysis of variance on grain yield, grain N‐uptake, stover biomass, stover N‐uptake, and N‐uptake efficiency (NUpE) as affected by maize treatment (T) and N‐fertilization (N)
| Grain yield (mg ha−1) | Grain N‐uptake (kg ha−1) | Stover biomass (mg ha−1) | Stover N‐uptake (kg ha−1) | NUpE (kg kg−1) | |
|---|---|---|---|---|---|
| Maize treatment (T) | 0.7745 | 0.8601 | 0.9702 | 0.6650 | 0.6767 |
| N‐fertilization (N) | 0.1352 | 0.5767 | 0.0305 | 0.0271 | <0.0001 |
| T × N | 0.5338 | 0.1106 | 0.4580 | 0.7329 | 0.0150 |
FIGURE 1.
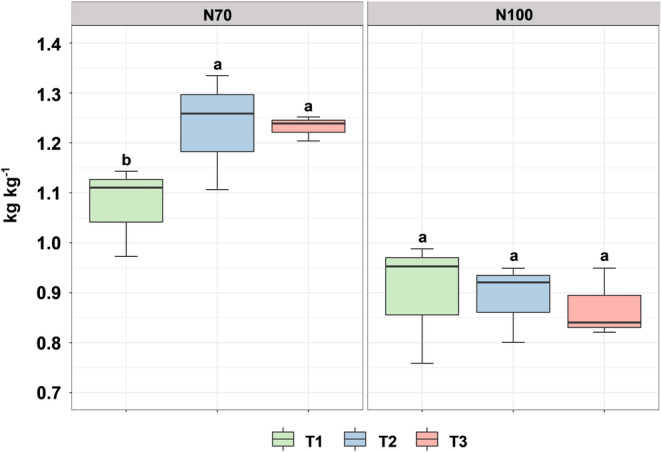
N‐uptake efficiency (NUpE) as affected by the interaction maize treatment (T1: Control; T2: AMF‐based product; T3: B. megaterium. PGPR formulation) × N‐fertilization (70: 70% N‐fertilization and 100: 100% N‐fertilization). Letters indicate significant differences between T levels (p < 0.05), within the same N‐fertilization level
4.2. Root traits
Root Length Density (RLD, cm cm−3) and diameter class length (DCL, mm cm−3) for very fine, fine, medium, and coarse diameters were significantly affected by the distance from the row (D) (Table 2), with the level 0 cm distance (on the row) being higher than the 37.5 cm distance (mid‐row; data not shown). Conversely, no significant effect of T and N, as well as of the interactions T × N, T × D, N × D, and T × N × D was found (Table 2).
TABLE 2.
Analysis of variance on diameter class length (DCL) for very fine (Ø = 0.00–0.075 mm), fine (Ø = 0.075–0.2 mm), medium (Ø = 0.2–1.0 mm) and coarse (Ø ≥ 1.0 mm) root diameter classes, root length density (RLD), and root dry weight (RDW), as affected by maize treatment (T), N‐fertilization (N), and distance from the row (D)
| DCL (cm cm−3) | RLD (cm cm−3) | RDW (mg cm−3) | ||||
|---|---|---|---|---|---|---|
| Very fine Ø = 0.00–0.075 mm | Fine Ø = 0.075–0.2 mm | Medium Ø = 0.2–1.0 mm | Coarse Ø > 1.0 mm | |||
| p‐value | ||||||
| Maize treatment (T) | 0.5387 | 0.3065 | 0.6752 | 0.6825 | 0.5645 | 0.0056 |
| N‐fertilization (N) | 0.949 | 0.8191 | 0.405 | 0.647 | 0.6565 | 0.0495 |
| Distance from the row (D) | <0.0001 | <0.0001 | 0.0007 | <0.0001 | <0.0001 | <0.0001 |
| T × N | 0.4331 | 0.6952 | 0.5865 | 0.691 | 0.6599 | 0.8674 |
| T × D | 0.582 | 0.2066 | 0.9131 | 0.466 | 0.7435 | 0.0002 |
| N × D | 0.9449 | 0.2863 | 0.9286 | 0.6731 | 0.8144 | 0.0651 |
| T × N × D | 0.8165 | 0.9497 | 0.4761 | 0.6933 | 0.8054 | 0.7087 |
Root dry weight (RDW, mg cm−3) was significantly affected by T, N, D, and the interaction T × D (Table 2). Overall, RDW was: (i) higher in T3 than T2 and T1, (ii) higher with 100% N‐fertilization than with 70% N‐fertilization, and (iii) higher at the 0 cm distance than at the 37.5 cm distance to the maize row. More in detail, a significant T × D interaction showed that T3 had higher RDW than T2 and T1 at the 0 cm distance, while not at the 37.5 cm distance (Table 3).
TABLE 3.
Root dry weight (RDW; mg cm−3), as affected by levels of maize treatment (T), N‐fertilization (N), distance from the row (D), and interaction T × D
| Source of variation | Maize treatment (T) | N‐fertilization (N) | Distance from the row (D) (cm) | RDW (mg cm−3) | |
|---|---|---|---|---|---|
| Maize treatment (T) | T1 | 0.67 | b | ||
| T2 | 0.56 | b | |||
| T3 | 1.00 | a | |||
| N‐fertilization (N) | 70% N‐fertilization | 0.64 | b | ||
| 100% N‐fertilization | 0.85 | a | |||
| Distance from the row (D) | 0 | 1.38 | a | ||
| 37.5 | 0.10 | b | |||
| T × D | T1 | 0 | 1.22 | b | |
| T2 | 0 | 1.03 | b | ||
| T3 | 0 | 1.90 | a | ||
| T1 | 37.5 | 0.11 | a | ||
| T2 | 37.5 | 0.09 | a | ||
| T3 | 37.5 | 0.11 | a | ||
Note: Different letters indicate statistically significant differences between means within the same source of variation. Letters in T × D indicate differences between T levels, within the same D level.
4.3. Metabolomic profiling of maize roots
Overall, UHPLC/QTOF untargeted metabolomics allowed annotating 3830 putative compounds (provided in Table S1, together with respective abundances, composite mass spectra and retention time). Initially, the fold change‐based unsupervised hierarchical cluster analysis (HCA) was carried out to describe similarities and dissimilarities across treatments based on metabolic profiles. The first analysis, carried out also considering the fertilization regime, showed that the microbial treatment provided a hierarchically stronger effect on maize root metabolome, with replications related to fertilization regime being not discriminated within each treatment (Figure S2). Therefore, the level of fertilization was not considered as a clustering factor in the following clustering to specifically point out differences related to seed treatments (Figure 2). This latter analysis highlighted two main clusters, which separated the treatment and highlighted as T3 provided the most distinctive metabolomic signature.
FIGURE 2.
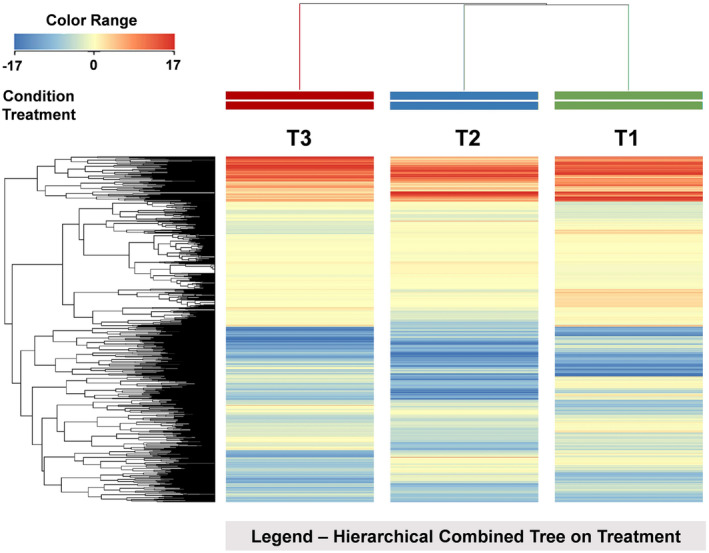
Unsupervised hierarchical cluster analysis of maize roots metabolomics profiles, obtained by UHPLC‐ESI/QTOF‐MS untargeted analysis, as a function of the seed treatment with biostimulants. A fold‐change based heatmap was built and samples were clustered according to Ward's algorithm, based on Euclidean distances. T1: Control; T2: AMF‐based product; T3: B. megaterium PGPR formulation
Successively, to investigate the contribution of the different metabolites for treatment discrimination, a supervised Orthogonal Projection of Latent Structures Discriminant Analysis (OPLS‐DA) was performed. The OPLS‐DA score plot (Figure 3) showed a clear differentiation between the three treatments, with goodness‐of‐fit (R2Y = 0.981), goodness‐of‐prediction (Q2Y = 0.424), adequate cross‐validation parameters (CV‐ANOVA; p = 0.035) and without overfitting as provided via permutation testing.
FIGURE 3.
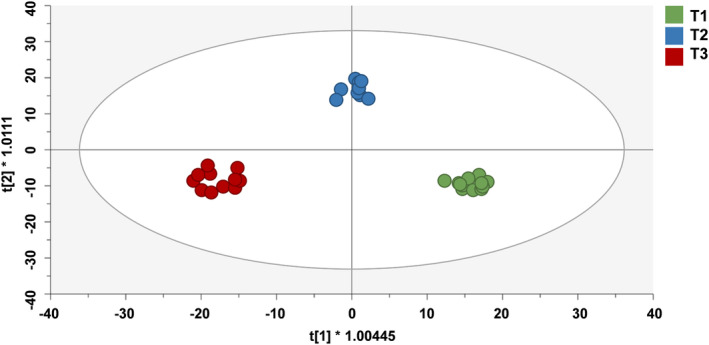
Orthogonal projections to latent structures discriminant analysis (OPLS‐DA) score plot for maize root metabolomic following seed treatment with microbial biostimulants. T1: Control; T2: AMF‐based product; T3: B. megaterium PGPR formulation
Thereafter, the variables importance in the projection (VIP) approach was used to select the compounds having the highest discrimination potential (VIP score >1.3) in the prediction model. The VIP approach allowed identifying 323 compounds (provided in Table S2). Therein, a diversity of metabolites, including mainly amino acids, lipids, flavones, lignans phenylpropanoids, and hormones, was represented (Figure 4).
FIGURE 4.
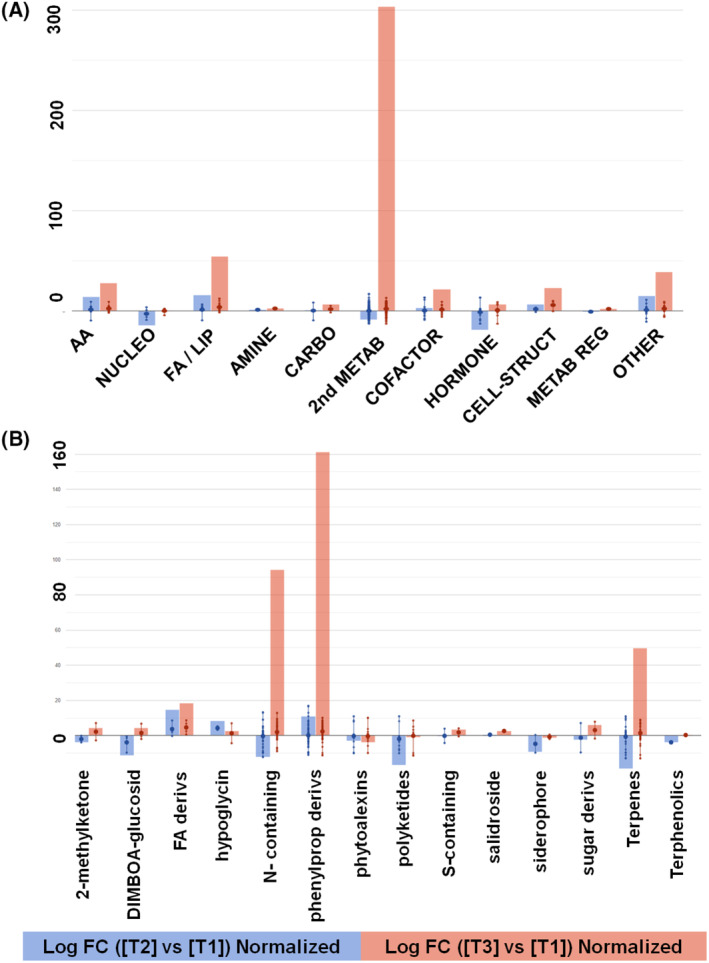
Maize roots metabolic processes (A) and the relative details of secondary metabolism (B) as affected by AMF‐based product (T2) or B. megaterium PGPR formulation (T3), compared with untreated control (T1). The VIP compounds were subjected to fold change analysis (FC ≥ 1), and the resulting values were loaded into the PlantCyc pathway tool (https://www.plantcyc.org/). The x‐axis represents each set of metabolic subcategories, while the y‐axis corresponds to the cumulative log fold change (FC). The large dots represent the average (mean) of all FCs for the different metabolites in the class, while the small dots represent the individual log FC. aa, amino acids; carbo, carbohydrate; derivs, derivative; FA/Lip, fatty acid/lipid; sec met, secondary metabolite; struct, structure; syn, synthesis; metab reg, metabolic regulator; nucleo, nucleotide
Among amino acids, N‐acetyl‐L‐glutamic acid, L‐serine, S‐methylmethionine, cystathionine, and L‐tyrosine contents were up‐accumulated by T2 and T3 treatments, even if the strongest elicitation could be observed following T3 inoculation. The same trend was observed for lipids, which appeared to be increased in both T2 and T3, but more markedly T3 inoculation. In detail, oleoyl‐CoA, (9,12,15)‐linolenic acid, cis,cis‐octadeca‐9,12‐dienoyl‐CoA, 3R‐hydroxy‐lesqueroloyl‐CoA 3‐oxo‐auricoloyl‐CoA, stearyl‐CoA, icosanoyl‐CoA, (8Z,11Z,14Z,17Z)‐3‐oxoicosatetraenoyl‐CoA, icosanoyl‐CoA and linoleoyl‐CoA were accumulated.
On the contrary, secondary metabolism appeared to be differently modulated by the two microbial seed treatments. Particularly, N‐ and S‐containing compound concentrations, including N‐hydroxy‐L‐valine, 1‐O‐feruloyl‐β‐D‐glucose, dihydrochelerythrine and the glucosinolate related (E)‐1‐(L‐cystein‐S‐yl)‐N‐hydroxy‐ω‐(methylsulfanyl)heptan‐1‐imine and 6‐(methylsulfanyl)hexyl‐desulfoglucosinolate were down‐accumulated in T2 samples and strongly increased in T3 roots. Similarly, T3 treatment elicited terpenoids such as gypsogenin‐28‐beta‐D‐glucoside, 4′‐hydroxyadonixanthin, (20S)‐ginsenoside Rh2 and (2Z,6Z)‐farnesyl diphosphate, which appeared to be decreased under T2 treatment. Nevertheless, this opposite trend was not observed for phenylpropanoids compounds, which showed a general increase in both treatments, compared with the control, even if this accumulation was remarkably highlighted with T3. Among this class, curcumin, deguelin, isovitexin 7‐O‐xylosyl 2″‐O‐arabinoside, afrormosin‐7‐O‐glucoside‐6″‐O‐malonate, (2S)‐sakuranetin and cyanidin 3‐O‐[2″‐O‐(xylosyl)‐6″‐O‐(p‐coumaroyl) glucoside] 5‐O‐malonylglucoside showed the highest increase.
Finally, concerning phytohormone profiles, lower amounts of N6‐dimethylallyladenine and gibberellin A12‐aldehyde were observed in T2‐treated samples, whereas accumulation of 5‐deoxystrigol, the cytokinins dihydrozeatin‐9‐N‐glucoside‐O‐glucoside and N6‐dimethylallyladenine, and jasmonate was elicited in T3 roots.
4.4. Diversity of rhizosphere bacterial community
Bacterial community diversity in the rhizosphere samples were analyzed for α‐ and β‐ diversity, respectively, by a total number of observed species (Sobs), Chao's and Simpson's indexes and Shannon's evenness, and by taxonomic comparison of all samples through hierarchical clustering of bacterial communities in samples. Although suggestive trends and differences were present, the overall distinction of the impact of treatments bt α‐diversity analysis was not as significant as in the case of root metabolomics analysis of the present study. Simpson's evenness index indicated a slight decrease in biodiversity for the T2 and T3 treatments (Figure 5A), with the latter being lower than Control and T2. The impact of reduced fertilization levels, 70% versus 100%, remained insignificant across treatments (Figure 5B). However, both results remained statistically insignificant as there were no significant differences among treatments according to one‐way ANOVA followed by LSD test at p < 0.05 threshold.
FIGURE 5.
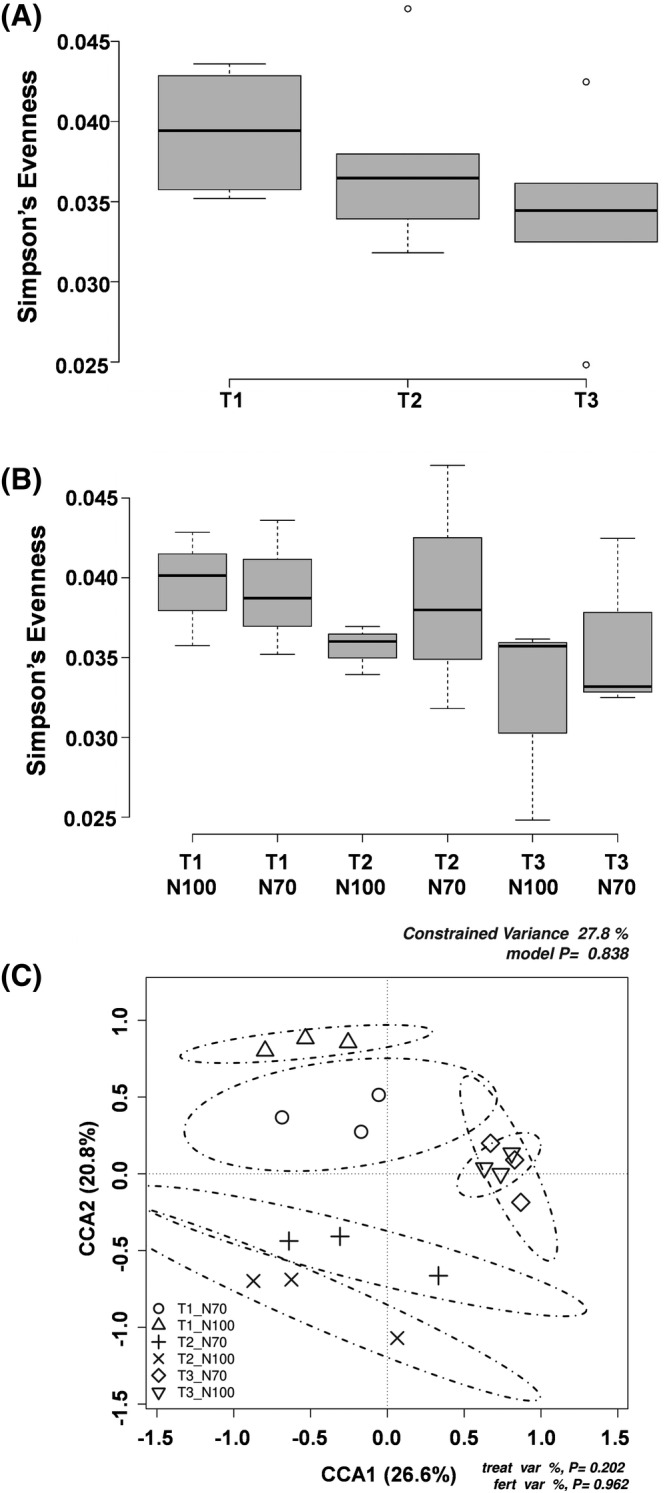
Bacterial community diversity in the rhizosphere samples through Simpson's index and canonical correspondence analyses (CCA). (A) Simpson's evenness for biostimulant treatment; (B) Simpson's evenness for biostimulant and nitrogen treatments; (C) Canonical correspondence analyses (CCAs) on the impact of the biostimulant and nitrogen treatments on the structure of bacterial communities (A and B were subjected to one‐way ANOVA at p ≤ 0.05 according to LSD, absence of significance letters indicate that there were no significant differences between treatments. Dedicated p values are indicated on the right upper and left lower corners of the “C”)
Furthermore, Canonical Correspondence Analysis (CCA) was carried out to evaluate the impact of the treatments on the clustering of bacterial communities. CCA results (model p = 0.838, constrained variance = 27.8%) suggest, in agreement with those of biodiversity indexes, differences between treatments Control, T2 and T3 (p = 0.202) and the insignificant impact of the fertilization regimes (70% vs. 100%, p = 0.962) (Figure 5C).
Taxonomic comparison of all samples through hierarchical clustering of bacterial communities at the family level across all samples used in this study, too, indicated a trend where fertilization regimes remained at the level of impact that was not significant to overall results. Interestingly, taxonomical clustering indicated some clustering of the T3 group with few exceptions, whereas T2 was often found clustering with Control (T1) (Figure 6).
FIGURE 6.
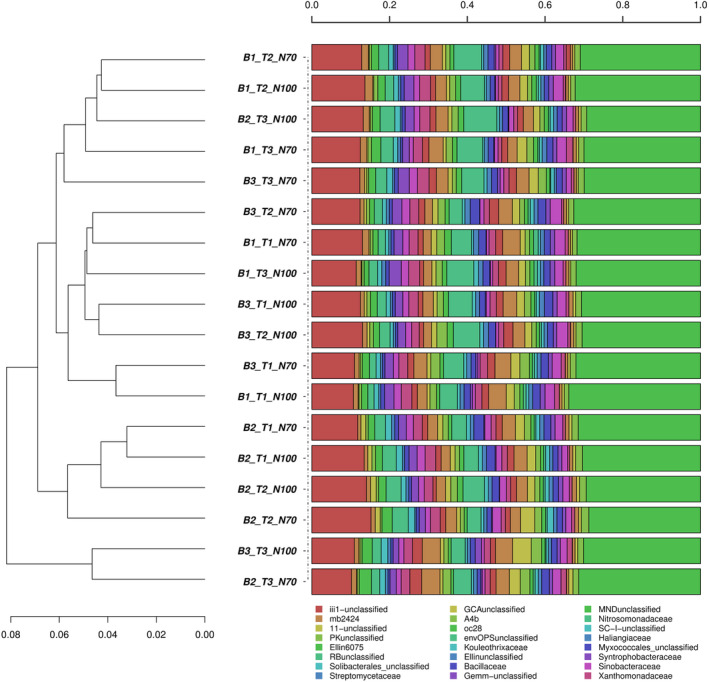
Taxonomic comparison of all samples through hierarchical clustering of bacterial communities at the family level
4.5. MCIA analysis
Multiple co‐inertia analysis (MCIA) was used to determine whether inter‐omic relationships existed between the three datasets (agronomic traits, metagenomics and UHPLC/QTOF‐MS untargeted metabolomics). Figures 7A and 8A show the projections of maize root samples onto the first two and three principal components (PCs) of MCIA, which accounted for approximately 25, 18 and 10% of the variation, respectively (Figures 7B and 8B). The pairwise RV (R‐vector) coefficient, which is the multivariate generalization of the squared Pearson correlation coefficient, indicated higher global similarity between metabolomics and metagenomics datasets (RV score for core = 0.85) compared with the similarities between the metagenomic dataset and the agronomic traits (RV score for core = 0.48), and between the metabolomics dataset and the agronomic traits (RV score for core = 0.46).
FIGURE 8.
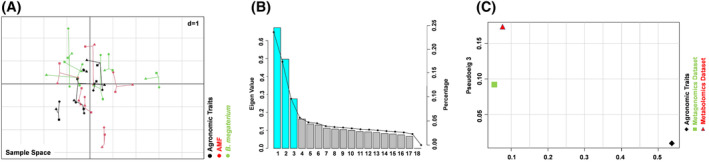
MCIA projection plot. (A) The first and the third axes of MCIA represents metabolomics, microbial community and agronomic traits of the maize root samples. Different shapes (diamond: Agronomic traits; triangle: Metagenomics dataset; square: Metabolomics dataset) represent the different variables connected by lines, the length of these lines is proportional to the divergence between the datasets. Lines for each sample are joined at a common point, at which the covariance derived from the MCIA analysis is maximal. Color shows the different thesis (black: Control; red: AMF‐based product; green: B. megaterium PGPR formulation). (B) Pseudo‐eigenvalue space representing the percentage of variance explained by the first three of the MCIA components. (C) Pseudo‐eigenvalues space of all datasets for maize roots, showing overall co‐structure between three datasets and shows which dataset contributes more to the total variance
4.6. Correlation analysis
Since significant inter‐omic correlations were found between maize root microbiome and metabolome, Pearson's rank correlation analysis was performed between VIP metabolites and VIP OTUs (listed in Table S3). The correlation coefficients are provided in Table S4.
5. DISCUSSION
Bacterial and mycorrhizal biostimulants are known to hold the potential to improve agronomical and physiological traits in crops, especially under stress conditions (Rouphael & Colla, 2020). The present study indicated that using the tested bio‐stimulants did not impair the rhizosphere soil bacteria biodiversity in treated plants. This can be attributed to the fact that the microbial treatments accounted for a small portion of the microbial diversity in the rhizo‐microbiome (Nuzzo et al., 2020). Nonetheless, it must also be considered that the changes in the microbiome are the consequence of centuries of coevolution and that plants actively seek microbial interactions (Durán et al., 2018; Kwak et al., 2018). On the other hand, the selection of a functionally positive community at the rhizosphere level can improve plant fitness (Liu et al., 2020), a process that typically involves root exudation processes (Carvalhais et al., 2015). Indeed, it has been proposed that plant crop species and nutrients were the main drivers of change (Armada et al., 2018). In light of these considerations, it is not surprising that our results highlighted moderate differences in root microbiome under the different treatments. Regarding the non‐significant impact of 30% less N fertilization on soil rhizomicrobiome, our results are in accordance with Maris et al., 2021.
The complex dialog between plants and rhizomicrobiome generally paves the way for the active recruitment of specific microorganisms providing benefits to plants, a process that may induce changes in rhizosphere microbial biodiversity (Bandyopadhyay et al., 2017). In turn, plant pattern‐recognition receptors (PRRs) at the plasma membrane level are activated by root‐microbe interaction in the rhizosphere and can trigger intracellular processes at the root level (Teixeira et al., 2019). Microbe‐associated molecular patterns (MAMPs) are among the most studied molecular processes being elicited in plants following host‐microbiota interaction(s), mostly in the framework of induced systemic resistance or, more generally, plant defense (Pieterse et al., 2014). However, plant response to microbial colonization has been proposed to be much wider than defense mechanisms, including direct and indirect effects related to the promotion of plant growth and fitness and tolerance to abiotic stresses (Kumar et al., 2018; Oleńska et al., 2020). This agrees with our findings, where a broad metabolic reprogramming was observed, with secondary metabolite biosynthesis affected by T2 and T3 treatments. Although limited, the impact of metabolic reprogramming was visible through trends observed in the reprogramming of the microbial communities at soil rhizomicrobiome, hinting at previously cited interlinkages between rhizomicrobiome and metabolites. Statistically insignificant differences at the alpha diversity level were contrasted with the changes in whole community ecology, especially on the community composition level. However, the limited differences in rhizosphere biodiversity between treatments can be attributed to the occurrence of the above‐mentioned interactions directly between plants and specific microorganisms (possibly including microbial dressing treatments), instead of the whole bacterial community of the rhizosphere, in accordance with Dal Cortivo et al., (2020). Surprisingly, the metabolomics signatures of maize roots differed as a function of the treatment, even though 79 days passed from seed dressing to root sampling and despite trials being carried out in open fields with non‐sterilized agricultural soil. This indicates that the treatments could modify root biochemical processes in a rather persistent manner. The ability of the root microbiome to affect root morphology (Bardgett et al., 2014; Pervaiz et al., 2020) and exudation patterns (Iannucci et al., 2021; Zuluaga et al., 2021) (and vice versa) corroborates our findings, where a broad reprogramming of metabolic processes could be observed in maize plant roots. Despite some positive trends could be observed, such modulation was translated into only a limited yield increase, irrespective of the nitrogen availability level. Dal Cortivo et al., (2020) found similar results, with small grain yield increases (1–4%) when either microbial or fungal consortia were applied to wheat seeds.
Nevertheless, it can be interestingly observed that the treatments induced significant changes at the functional level and that root metabolome and rhizosphere population were highly correlated. With this regard, a correlation of 0.84 (as provided by MCIA) under field conditions is indicative of a rather strong link between the two omic profiles. This coordinate modulation of root physiological traits was not translated into yield increase, probably because the concentration of nutrients into the soil was per se relatively high even when the 30% decrease of N‐fertilizer was applied or because root performance was increased following our biostimulant treatments. Nevertheless, our results showed that (1) both biostimulants had higher NUpE with 70% N‐fertilization level, and (2) B. megaterium PGPR led to increased maize root biomass, mostly due to an increase of root development on the crop row. Given these findings and the well‐recognized biostimulant effect of the treatments applied, we can postulate that either more severe nitrogen starvation status or other abiotic stresses such as drought and/or low nutrients (other than N) concentrations and/or high temperatures might have helped in highlighting the beneficial effects of the treatments considered.
Notwithstanding, plant response to seed treatment persisted at anthesis, thus paving the way towards a set of beneficial aspects in maize production that go far beyond the direct effects of exogenously applied biostimulant microorganisms. Plant beneficial rhizospheric microorganisms are known to increase nutrient use efficiency (Meena et al., 2017). However, we observed the possible mechanisms underlying the coordinate modulation of root metabolome and rhizomicrobiome. It would be aleatory to identify a main player between plants and microorganisms, and the intricate series of interactions occurring at the rhizosphere should be considered instead. In fact, from one side, plant root exudates include chemotaxis compounds and are known to shape the microbial community (Pérez‐Jaramillo et al., 2016; van Dam & Bouwmeester, 2016), but it is also important to consider that microorganisms produce signaling compounds that are perceived by roots (Mendes et al., 2014; Zancarini et al., 2013). Together with the exchange of chemical messengers or functional metabolites, the microorganisms perceived by roots are also able to interact with root receptors directly (Poole, 2017), thus eliciting specific biochemical responses in the plant cells, and some symbionts are even endophytes. The elucidation of such intricate agroecological crosstalk is very complicated because of technical limitations in sampling and analysis (Escolà Casas & Matamoros, 2021) and the need to track temporal and spatial dynamics (van Dam & Bouwmeester, 2016). However, it can be noted that under our experimental conditions, the microbial species showing the higher correlation to root metabolome (provided in Table S4) are well‐known beneficial rhizobacteria. In particular, Actinobacteria (such as Gemmatimonas, Gaiella, and Solilubrobacter spp.), and some Acidobacteria and Chloroflexi spp. have been reported to provide positive functions at the rhizosphere level, particularly under stress conditions (Akinola et al., 2021; Khan et al., 2020; Lazcano et al., 2021; Yue et al., 2020). In particular, some Acidobacteria have been reported to possess the genetic capability to support nitrate, nitrite and nitric oxide reduction and to release serine endopeptidases, hence having the potential to improve nitrogen uptake (Kalam et al., 2020). Chloroflexi spp. are beneficial PGPR that have been linked to plant root growth promotion. Similarly, Actinobacteria can promote plant growth under adverse conditions and contribute to fixing atmospheric nitrogen (Yadav et al., 2018; Yue et al., 2020) and are reported to be increased by AMF (Agnolucci et al., 2019). Noteworthy, Gemmatimonadetes and Gaiella spp. play a key role in plant abiotic stress (Khan et al., 2020; Yue et al., 2020), and both have been specifically linked to the level of N‐nitrate levels in maize (Akinola et al., 2021). Among the root metabolites showing the strongest correlation with rhizomicrobiota, flavonoids (sakuranetin and 2′,4,4′,6′‐tetrahydroxychalcone) and isoflavonoids (vestitone and 7,2,4,2′‐tetrahydroxy‐4′,5′‐methylenedioxyisoflav‐3‐ene) were the most represented, followed by the hydroxycinnamic phenolic cinnamoyl‐beta‐D‐glucoside, and the phenolic glycoside salicin. Among non‐phenolics, lipids (a diacylglycerol, a decaprenylbenzoate ubiquinol intermediate and 3‐oxoicosatetraenoyl‐CoA), D‐glucono‐1,5‐lactone, phlormethylbutanophenone (a 2‐acylphloroglucinol) and amino acids intermediates could be found. Plant roots may exudate up to 20% of their photosynthate, in the attempt of shaping the root microbiome (Poole, 2017). The molecular processes involved in this chemotaxis are largely unknown, even though literature agrees that root exudation patterns are paramount in the tripartite soil–root–microbiome interaction. Despite no specific pathways for root exudation have been identified to date, our results indicate that the root microbiome may represent an upstream process in the modulation of root exudation.
This coordinate modulation of rhizomicrobiome and root metabolomic signature, linked to higher NUpE with reduced N‐fertilization level, implies that biostimulants could be particularly suitable in less suited soils, in arid and semi‐arid regions, with poor soil quality, and where N‐fertilization is a limiting factor (e.g., organic farming). However, since in many temperate areas across the world, the climate is changing rapidly (Zhongming et al., 2021) and the European Commission (EU) recently set ambitious goals for reducing fertilizer use significantly at the field level (Schebesta et al., 2020), using effective tools to mitigate yield losses by increase nutrient use efficiency will become more important in a greater proportion of arable land across the world.
6. CONCLUSIONS
Improved yields are required to meet the food demand of an increasing population. Until now, this need has been highly dependent on chemical inputs, and more sustainable approaches are needed. In this framework, beneficial microorganisms are gaining popularity because of the multiple effects they may play several functional roles in plants. Here we show that both the fungal and the PGPR seed treatments were able to increase nitrogen uptake efficiency under low nitrogen availability without compromising yields. This point is of paramount practical importance since it indicates that these biostimulants may support agricultural production in a sustainable manner, under a reduced input farming perspective. Both the biostimulant treatments induced a coordinate modulation of root metabolome and rhizomicrobiome, although with differences between mycorrhiza and PGPR treatments. Nonetheless, such coordinate modulation could be observed several weeks after seeding, supporting the involvement of the biostimulants in the improved maize performance we observed. The effects observed involved the positive modulation of several beneficial rhizosphere microorganisms, possibly involving indirect effects mediated by root exudation patterns. This latter point is of relevance and deserves further ad hoc investigation.
Supporting information
FIGURE S1. Graphical representation of experimental station climatic data
FIGURE S2. Unsupervised hierarchical cluster analysis of maize roots metabolomics profiles, obtained by UHPLC‐ESI/QTOF‐MS untargeted analysis, as a function of biostimulant and nitrogen treatments
TABLE S1. Maize root metabolites revealed with the untargeted UHPLC‐ESI/QTOF‐MS analysis
TABLE S2. VIP compounds in maize roots
TABLE S3. List of VIP metabolites and Vip Otus selected for the correlation analysis
TABLE S4. Pearson's rank correlation coefficients between VIP metabolites and VIP OTUs. Positive correlations with p value <0.05 were displayed in blue while negative correlations with p value <0.05 were displayed in red
ACKNOWLEDGMENTS
The project was funded by the Emilia‐Romagna region, Italy, in the framework of the Regional Development Program PSR 2014–2020, Measure 16.1.01—Focus Area 4B (project 5150325—INBIOS: Sviluppo di un approccio INtegrato a base di BIOStimolanti per la sostenibilità delle produzioni agrarie). The authors thank the “Romeo ed Enrica Invernizzi” Foundation (Milan, Italy) for the kind support to the metabolomic facility at Università Cattolica del Sacro Cuore. Open Access Funding provided by Universita Cattolica del Sacro Cuore within the CRUI‐CARE Agreement.
Ganugi, P. , Fiorini, A. , Ardenti, F. , Caffi, T. , Bonini, P. , Taskin, E. et al. (2022) Nitrogen use efficiency, rhizosphere bacterial community, and root metabolome reprogramming due to maize seed treatment with microbial biostimulants. Physiologia Plantarum, 174(2), e13679. Available from: 10.1111/ppl.13679
Edited by J. Flexas
Funding information INBIOS, Grant/Award Number: 5150325; Emilia‐Romagna region
[Correction added on 16 May 2022: CRUI‐CARE funding statement has been added.]
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Afshari, R. , Pillidge, C.J. , Read, E. , Rochfort, S. , Dias, D.A. , Osborn, A.M. et al. (2020) New insights into cheddar cheese microbiota‐metabolome relationships revealed by integrative analysis of multi‐omics data. Scientific Reports, 10, 1–13. 10.1038/s41598-020-59617-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnolucci, M. , Palla, M. , Cristani, C. , Cavallo, N. , Giovannetti, M. , de Angelis, M. et al. (2019) Beneficial plant microorganisms affect the endophytic bacterial communities of durum wheat roots as detected by different molecular approaches. Frontiers in Microbiology, 10, 2500. 10.3389/fmicb.2019.02500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinola, S.A. , Ayangbenro, A.S. & Babalola, O.O. (2021) Metagenomic insight into the community structure of maize‐rhizosphere bacteria as predicted by different environmental factors and their functioning within plant proximity. Microorganisms, 9(7), 1419. 10.3390/microorganisms9071419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armada, E. , Leite, M.F.A. , Medina, A. , Azcón, R. & Kuramae, E.E. (2018) Native bacteria promote plant growth under drought stress condition without impacting the rhizomicrobiome. FEMS Microbiology Ecology, 94, 1–13. 10.1093/femsec/fiy092 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay, P. , Bhuyan, S.K. , Yadava, p.K. , Varma, A. & Tuteja, N. (2017) Emergence of plant and rhizospheric microbiota as stable interactomes. Protoplasma, 254, 617–626. 10.1007/s00709-016-1003-x [DOI] [PubMed] [Google Scholar]
- Bardgett, R.D. , Mommer, L. & de Vries, F.T. (2014) Going underground: root traits as drivers of ecosystem processes. Trends in Ecology & Evolution, 29, 692–699. 10.1016/j.tree.2014.10.006 [DOI] [PubMed] [Google Scholar]
- Carvalhais, L.C. , Dennis, p.G. , Badri, D.V. , Kidd, B.N. , Vivanco, J.M. & Schenk, p.M. (2015) Linking Jasmonic acid signaling, root exudates, and rhizosphere microbiomes. Molecular Plant‐Microbe Interactions, 28, 1049–1058. 10.1094/MPMI-01-15-0016-R [DOI] [PubMed] [Google Scholar]
- Ceccarelli, A.V. , Miras‐Moreno, B. , Buffagni, V. , Senizza, B. , Pii, Y. , Cardarelli, M. et al. (2021) Foliar application of different vegetal‐derived protein hydrolysates distinctively modulates tomato root development and metabolism. Plants, 10(2), 326. 10.3390/plants10020326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla, G. , Hoagland, L. , Ruzzi, M. , Cardarelli, M. , Bonini, P. , Canaguier, R. et al. (2017) Biostimulant action of protein hydrolysates: unraveling their effects on plant physiology and microbiome. Frontiers in Plant Science, 8, 1–14. 10.3389/fpls.2017.02202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Cortivo, C. , Ferrari, M. , Visioli, G. , Lauro, M. , Fornasier, F. , Barion, G. et al. (2020) Effects of seed‐applied biofertilizers on rhizosphere biodiversity and growth of common wheat (Triticum aestivum L.) in the field. Frontiers in Plant Science, 11, 1–14. 10.3389/fpls.2020.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSA, U. 2015. World population projected to reach 9.7 billion by 2050. United Nations Department of Economic and Social Affairs.
- DeSantis, T.Z. , Hugenholtz, P. , Keller, K. , Brodie, E.L. , Larsen, N. , Piceno, Y.M. et al. (2006) NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Research, 34, W394–W399. 10.1093/nar/gkl244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durán, P. , Thiergart, T. , Garrido‐Oter, R. , Agler, M. , Kemen, E. , Schulze‐Lefert, P. et al. (2018) Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell, 175, 973–983.e14. 10.1016/j.cell.2018.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. , Haas, B.J. , Clemente, J.C. , Quince, C. & Knight, R. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27, 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escolà Casas, M. & Matamoros, V. (2021) Analytical challenges and solutions for performing metabolomic analysis of root exudates. Trends in Environmental Analytical Chemistry, 31, e00130. 10.1016/j.teac.2021.e00130 [DOI] [Google Scholar]
- FAO . (2017) The future of food and agriculture: trends and challenges. Annual Report, 296, 1–180. [Google Scholar]
- FAOSTAT Statistical Database , 2019. Food and Agriculture Organization of the United Nations
- Fiorentino, N. , Ventorino, V. , Woo, S.L. , Pepe, O. , de Rosa, A. , Gioia, L. et al. (2018) Trichoderma‐based biostimulants modulate rhizosphere microbial populations and improve N uptake efficiency, yield, and nutritional quality of leafy vegetables. Frontiers in Plant Science, 9, 1–15. 10.3389/fpls.2018.00743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini, A. , Boselli, R. , Amaducci, S. & Tabaglio, V. (2018) Effects of no‐till on root architecture and root‐soil interactions in a three‐year crop rotation. European Journal of Agronomy, 99, 156–166. 10.1016/j.eja.2018.07.009 [DOI] [Google Scholar]
- Fiorini, A. , Boselli, R. , Maris, S.C. , Santelli, S. , Ardenti, F. , Capra, F. et al. (2020) May conservation tillage enhance soil C and N accumulation without decreasing yield in intensive irrigated croplands? Results from an eight‐year maize monoculture. Agriculture, Ecosystems and Environment, 296, 106926. 10.1016/j.agee.2020.106926 [DOI] [Google Scholar]
- Ganugi, P. , Martinelli, E. & Lucini, L. (2021b) Microbial biostimulants as a sustainable approach to improve the functional quality in plant‐based foods: a review. Current Opinion in Food Science, 41, 217–223. 10.1016/j.cofs.2021.05.001 [DOI] [Google Scholar]
- Ganugi, P. , Miras‐Moreno, B. , Garcia‐Perez, P. , Lucini, L. & Trevisan, M. (2021a) Concealed metabolic reprogramming induced by different herbicides in tomato. Plant Science, 303, 110727. 10.1016/j.plantsci.2020.110727 [DOI] [PubMed] [Google Scholar]
- Guerrieri, M.C. , Fanfoni, E. , Fiorini, A. , Trevisan, M. & Puglisi, E. (2020) Isolation and screening of extracellular PGPR from the rhizosphere of tomato plants after long‐term reduced tillage and cover crops. Plants, 9, 668. 10.3390/plants9050668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrieri, M.C. , Fiorini, A. , Fanfoni, E. , Tabaglio, V. , Cocconcelli, p.S. , Trevisan, M. et al. (2021) Integrated genomic and greenhouse assessment of a novel plant growth‐promoting Rhizobacterium for tomato plant. Frontiers in Plant Science, 12, 1–16. 10.3389/fpls.2021.660620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell Jr, F. E. , Harrell Jr, M. F. E. , 2019. Package ‘hmisc’. CRAN2018, 2019, 235–236.
- Hothorn, T. , Bretz, F. , Westfall, P. , Heiberger, R. , 2007. The multcomp Package. R Doc.
- Iannucci, A. , Canfora, L. , Nigro, F. , de Vita, P. & Beleggia, R. (2021) Relationships between root morphology, root exudate compounds and rhizosphere microbial community in durum wheat. Applied Soil Ecology, 158, 103781. 10.1016/j.apsoil.2020.103781 [DOI] [Google Scholar]
- Kalam, S. , Basu, A. , Ahmad, I. , Sayyed, R.Z. , el Enshasy, H.A. , Dailin, D.J. et al. (2020) Recent understanding of soil acidobacteria and their ecological significance: a critical review. Frontiers in Microbiology, 11, 2712. 10.3389/fmicb.2020.580024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, p.D. , Paley, S.M. , Krummenacker, M. , Latendresse, M. , Dale, J.M. , Lee, T.J. et al. (2009) Pathway tools version 13.0: integrated software for pathway/genome informatics and systems biology. Briefings in Bioinformatics, 11, 40–79. 10.1093/bib/bbp043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A.L. , Asaf, S. , M Abed, R.M. , Ning Chai, Y. , N Al‐Rawahi, A. , Mohanta, T.K. et al. (2020) Rhizosphere microbiome of arid land medicinal plants and extra cellular enzymes contribute to their abundance. Microorganisms, 8(2), 213. 10.3390/microorganisms8020213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, A. , Singh, V.K. , Tripathi, V. , Singh, p.P. & Singh, A.K. (2018) Plant growth‐promoting rhizobacteria (PGPR): perspective in agriculture under biotic and abiotic stress. In: Crop improvement through microbial biotechnology. Amsterdam: Elsevier, pp. 333–342. [Google Scholar]
- Kwak, M.J. , Kong, H.G. , Choi, K. , Kwon, S.K. , Song, J.Y. , Lee, J. et al. (2018) Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nature Biotechnology, 36, 1100–1116. 10.1038/nbt.4232 [DOI] [PubMed] [Google Scholar]
- Lazcano, C. , Boyd, E. , Holmes, G. , Hewavitharana, S. , Pasulka, A. & Ivors, K. (2021) The rhizosphere microbiome plays a role in the resistance to soil‐borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Scientific Reports, 11, 3188. 10.1038/s41598-021-82768-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Brettell, L.E. , Qiu, Z. & Singh, B.K. (2020) Microbiome‐mediated stress resistance in plants. Trends in Plant Science, 25, 733–743. 10.1016/j.tplants.2020.03.014 [DOI] [PubMed] [Google Scholar]
- López‐Bellido, R.J. & López‐Bellido, L. (2001) Efficiency of nitrogen in wheat under Mediterranean conditions: effect of tillage, crop rotation and N fertilization. Field Crops Research, 71, 31–46. 10.1016/S0378-4290(01)00146-0 [DOI] [Google Scholar]
- Lucini, L. , Colla, G. , Miras Moreno, M.B. , Bernardo, L. , Cardarelli, M. , Terzi, V. et al. (2019) Inoculation of Rhizoglomus irregulare or Trichoderma atroviride differentially modulates metabolite profiling of wheat root exudates. Phytochemistry, 157, 158–167. 10.1016/j.phytochem.2018.10.033 [DOI] [PubMed] [Google Scholar]
- Maris, S.C. , Capra, F. , Ardenti, F. , Chiodini, M.E. , Boselli, R. , Taskin, E. et al. (2021) Reducing N fertilization without yield penalties in maize with a commercially available seed dressing. Agronomy, 11, 1–19. 10.3390/agronomy11030407 [DOI] [Google Scholar]
- Masella, A.P. , Bartram, A.K. , Truszkowski, J.M. , Brown, D.G. & Neufeld, J.D. (2012) PANDAseq: paired‐end assembler for illumina sequences. BMC Bioinformatics, 13, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, D. , Price, M.N. , Goodrich, J. , Nawrocki, E.P. , DeSantis, T.Z. , Probst, A. et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. The ISME Journal, 6, 610–618. 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meena, V.S. , Meena, S.K. , Verma, J.P. , Kumar, A. , Aeron, A. , Mishra, p.K. et al. (2017) Plant beneficial rhizospheric microorganism (PBRM) strategies to improve nutrients use efficiency: a review. Ecological Engineering, 107, 8–32. 10.1016/j.ecoleng.2017.06.058 [DOI] [Google Scholar]
- Mendes, L.W. , Kuramae, E.E. , Navarrete, A.A. , van Veen, J.A. & Tsai, S.M. (2014) Taxonomical and functional microbial community selection in soybean rhizosphere. The ISME Journal, 8, 1577–1587. 10.1038/ismej.2014.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, C. , Kuster, B. , Culhane, A.C. & Gholami, A.M. (2014) A multivariate approach to the integration of multi‐omics datasets. BMC Bioinformatics, 15, 1–13. 10.1186/1471-2105-15-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, E.J. & Long, Q. (2020) Sparse multiple co‐inertia analysis with application to integrative analysis of multi‐omics data. BMC Bioinformatics, 21, 1–12. 10.1186/s12859-020-3455-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miras‐Moreno, B. , Corrado, G. , Zhang, L. , Senizza, B. , Righetti, L. , Bruni, R. et al. (2020) The metabolic reprogramming induced by sub‐optimal nutritional and light inputs in soilless cultivated green and red butterhead lettuce. International Journal of Molecular Sciences, 21, 1–16. 10.3390/ijms21176381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacoon, S. , Jogloy, S. , Riddech, N. , Mongkolthanaruk, W. , Kuyper, T.W. & Boonlue, S. (2020) Interaction between phosphate solubilizing bacteria and arbuscular mycorrhizal fungi on growth promotion and tuber inulin content of Helianthus tuberosus L. Scientific Reports, 10, 1–10. 10.1038/s41598-020-61846-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzzo, A. , Satpute, A. , Albrecht, U. & Strauss, S.L. (2020) Impact of soil microbial amendments on tomato rhizosphere microbiome and plant growth in field soil. Microbial Ecology, 80, 398–409. 10.1007/s00248-020-01497-7 [DOI] [PubMed] [Google Scholar]
- OECD/FAO (2021) OECD‐FAO Agricultural Outlook: 2021–2030. Paris: OECD Publishing. 10.1787/agr-outl-dataen. [DOI]
- Oleńska, E. , Małek, W. , Wójcik, M. , Swiecicka, I. , Thijs, S. & Vangronsveld, J. (2020) Beneficial features of plant growth‐promoting rhizobacteria for improving plant growth and health in challenging conditions: a methodical review. Science of the Total Environment, 743, 140682. 10.1016/j.scitotenv.2020.140682 [DOI] [PubMed] [Google Scholar]
- Panfili, I. , Bartucca, M.L. , Marrollo, G. , Povero, G. & del Buono, D. (2019) Application of a plant biostimulant to improve maize (Zea mays) tolerance to metolachlor. Journal of Agricultural and Food Chemistry, 67, 12164–12171. 10.1021/acs.jafc.9b04949 [DOI] [PubMed] [Google Scholar]
- Pérez‐Jaramillo, J.E. , Mendes, R. & Raaijmakers, J.M. (2016) Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Molecular Biology, 90, 635–644. 10.1007/s11103-015-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz, Z.H. , Contreras, J. , Hupp, B.M. , Lindenberger, J.H. , Chen, D. , Zhang, Q. et al. (2020) Root microbiome changes with root branching order and root chemistry in peach rhizosphere soil. Rhizosphere, 16, 100249. 10.1016/j.rhisph.2020.100249 [DOI] [Google Scholar]
- Pieterse, C.M.J. , Zamioudis, C. , Berendsen, R.L. , Weller, D.M. , van Wees, S.C.M. & Bakker, p.A.H.M. (2014) Induced systemic resistance by beneficial microbes. Annual Review of Phytopathology, 52, 347–375. 10.1146/annurev-phyto-082712-102340 [DOI] [PubMed] [Google Scholar]
- Pinheiro, J. , Bates, D. , DebRoy, S. & Sarkar, D. (2013) R Development Core Team (2012) nlme: linear and nonlinear mixed effects models. R package version 3.1‐103.
- Połka, J. , Rebecchi, A. , Pisacane, V. , Morelli, L. & Puglisi, E. (2015) Bacterial diversity in typical Italian salami at different ripening stages as revealed by high‐throughput sequencing of 16S rRNA amplicons. Food Microbiology, 46, 342–356. 10.1016/j.fm.2014.08.023 [DOI] [PubMed] [Google Scholar]
- Poole, P. (2017) Shining a light on the dark world of plant root‐microbe interactions. Proceedings of the National Academy of Sciences of the United States of America, 114, 4281–4283. 10.1073/pnas.1703800114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse, E. , Quast, C. , Knittel, K. , Fuchs, B.M. , Ludwig, W. , Peplies, J. et al. (2007) SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Research, 35, 7188–7196. 10.1093/nar/gkm864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers, J.M. & Mazzola, M. (2012) Diversity and natural functions of antibiotics produced by beneficial and plant pathogenic bacteria. Annual Review of Phytopathology, 50, 403–424. 10.1146/annurev-phyto-081211-172908 [DOI] [PubMed] [Google Scholar]
- Rouphael, Y. & Colla, G. (2020) Editorial: biostimulants in agriculture. Frontiers in Plant Science, 11, 1–7. 10.3389/fpls.2020.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouphael, Y. , Lucini, L. , Miras‐Moreno, B. , Colla, G. , Bonini, P. & Cardarelli, M. (2020) Metabolomic responses of maize shoots and roots elicited by combinatorial seed treatments with microbial and non‐microbial biostimulants. Frontiers in Microbiology, 11, 1–13. 10.3389/fmicb.2020.00664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, p.R. , Delhaize, E. , Watt, M. & Richardson, A.E. (2016) Plant roots: understanding structure and function in an ocean of complexity. Annals of Botany, 118, 555–559. 10.1093/aob/mcw192 [DOI] [Google Scholar]
- Saia, S. , Aissa, E. , Luziatelli, F. , Ruzzi, M. , Colla, G. , Ficca, A.G. et al. (2020) Growth‐promoting bacteria and arbuscular mycorrhizal fungi differentially benefit tomato and corn depending upon the supplied form of phosphorus. Mycorrhiza, 30, 133–147. 10.1007/s00572-019-00927-w [DOI] [PubMed] [Google Scholar]
- Sainju, U.M. (2017) Determination of nitrogen balance in agroecosystems. MethodsX, 4, 199–208. 10.1016/J.MEX.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salek, R.M. , Neumann, S. , Schober, D. , Hummel, J. , Billiau, K. , Kopka, J. et al. (2015) COordination of standards in MetabOlomicS (COSMOS): facilitating integrated metabolomics data access. Metabolomics, 11, 1587–1597. 10.1007/s11306-015-0810-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiorgio, D. , Cellini, A. , Donati, I. , Pastore, C. , Onofrietti, C. & Spinelli, F. (2020) Facing climate change: application of microbial biostimulants to mitigate stress in horticultural crops. Agronomy, 10, 794. 10.3390/agronomy10060794 [DOI] [Google Scholar]
- Schebesta, H. & Candel, J.J. (2020) Game‐changing potential of the EU's Farm to Fork Strategy. Nature Food, 1(10), 586–588. 10.1038/s43016-020-00166-9 [DOI] [PubMed] [Google Scholar]
- Schläpfer, P. , Zhang, P. , Wang, C. , Kim, T. , Banf, M. , Chae, L. et al. (2017) Genome‐wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiology, 173, 2041–2059. 10.1104/pp.16.01942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, p.D. (2010) The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene‐based studies. PLoS Computational Biology, 6, e1000844. 10.1371/journal.pcbi.1000844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, p.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. et al. (2009) Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soil Survey Staff . (2014) Keys to soil taxonomy. Soil Conservation Service, 12, 410. [Google Scholar]
- Team, R. Core . (2012) R: a language and environment for statistical computing. Austria: R Foundation for Statistical Computing. http://www.R-project.org [Google Scholar]
- Team, R. Core . (2020) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. www.R-project.org/ [Google Scholar]
- Teixeira, p.J.P. , Colaianni, N.R. , Fitzpatrick, C.R. & Dangl, J.L. (2019) Beyond pathogens: microbiota interactions with the plant immune system. Current Opinion in Microbiology, 49, 7–17. 10.1016/j.mib.2019.08.003 [DOI] [PubMed] [Google Scholar]
- van Dam, N.M. & Bouwmeester, H.J. (2016) Metabolomics in the rhizosphere: tapping into belowground chemical communication. Trends in Plant Science, 21, 256–265. 10.1016/j.tplants.2016.01.008 [DOI] [PubMed] [Google Scholar]
- Vasileiadis, S. , Puglisi, E. , Trevisan, M. , Scheckel, K.G. , Langdon, K.A. , McLaughlin, M.J. et al. (2015) Changes in soil bacterial communities and diversity in response to long‐term silver exposure. FEMS Microbiology Ecology, 91, fiv114. 10.1093/femsec/fiv114 [DOI] [PubMed] [Google Scholar]
- Yadav, A.N. , Verma, P. , Kumar, S. , Kumar, V. , Kumar, M. , Sugitha, T.C.K. et al. (2018) Actinobacteria from rhizosphere: molecular diversity, distributions, and potential biotechnological applications. In: New and future developments in microbial biotechnology and bioengineering. Amsterdam: Elsevier, pp. 13–41. [Google Scholar]
- Yue, Y. , Shao, T. , Long, X. , He, T. , Gao, X. , Zhou, Z. et al. (2020) Microbiome structure and function in rhizosphere of Jerusalem artichoke grown in saline land. Science of the Total Environment, 724, 138259. 10.1016/j.scitotenv.2020.138259 [DOI] [PubMed] [Google Scholar]
- Zancarini, A. , Lépinay, C. , Burstin, J. , Duc, G. , Lemanceau, P. , Moreau, D. et al. (2013) Combining molecular microbial ecology with ecophysiology and plant genetics for a better understanding of plant‐microbial Communities' interactions in the rhizosphere. Molecular Microbial Ecology of the Rhizosphere, 1, 69–86. [Google Scholar]
- Zhongming, Z. , Linong, L. , Xiaona, Y. , Wangqiang, Z. & Wei, L. (2021) AR6 Climate Change 2021: The Physical Science Basis.
- Zuluaga, M.Y. , Milani, K.M.L. , Miras‐Moreno, M.B. , Lucini, L. , Valentinuzzi, F. , Mimmo, T. et al. (2021) The adaptive metabolomic profile and functional activity of tomato rhizosphere are revealed upon PGPB inoculation under saline stress. Environmental and Experimental Botany, 189, 104552. 10.1016/j.envexpbot.2021.104552 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Graphical representation of experimental station climatic data
FIGURE S2. Unsupervised hierarchical cluster analysis of maize roots metabolomics profiles, obtained by UHPLC‐ESI/QTOF‐MS untargeted analysis, as a function of biostimulant and nitrogen treatments
TABLE S1. Maize root metabolites revealed with the untargeted UHPLC‐ESI/QTOF‐MS analysis
TABLE S2. VIP compounds in maize roots
TABLE S3. List of VIP metabolites and Vip Otus selected for the correlation analysis
TABLE S4. Pearson's rank correlation coefficients between VIP metabolites and VIP OTUs. Positive correlations with p value <0.05 were displayed in blue while negative correlations with p value <0.05 were displayed in red
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.