

Abstract

FtmOx1 is a nonheme iron (NHFe) endoperoxidase, catalyzing three disparate reactions, endoperoxidation, alcohol dehydrogenation, and dealkylation, under in vitro conditions; the diversity complicates its mechanistic studies. In this study, we use two substrate analogues to simplify the FtmOx1-catalyzed reaction to either a dealkylation or an alcohol dehydrogenation reaction for structure–function relationship analysis to address two key FtmOx1 mechanistic questions: (1) Y224 flipping in the proposed COX-like model vs α-ketoglutarate (αKG) rotation proposed in the CarC-like mechanistic model and (2) the involvement of a Y224 radical (COX-like model) or a Y68 radical (CarC-like model) in FtmOx1-catalysis. When 13-oxo-fumitremorgin B (7) is used as the substrate, FtmOx1-catalysis changes from the endoperoxidation to a hydroxylation reaction and leads to dealkylation. In addition, consistent with the dealkylation side-reaction in the COX-like model prediction, the X-ray structure of the FtmOx1•CoII•αKG•7 ternary complex reveals a flip of Y224 to an alternative conformation relative to the FtmOx1•FeII•αKG binary complex. Verruculogen (2) was used as a second substrate analogue to study the alcohol dehydrogenation reaction to examine the involvement of the Y224 radical or Y68 radical in FtmOx1-catalysis, and again, the results from the verruculogen reaction are more consistent with the COX-like model.
Keywords: FtmOx1, nonheme iron enzymes, endoperoxidation, hydroxylation, dehydrogenation, X-ray structure
Introduction
Numerous peroxy-containing metabolites have been isolated from various organisms1−4 and show diverse biological activities, including antimalarial, antitumor, and antimicrobial activities.5,6 One of the best known peroxy-containing metabolites is artemisinin, which is a front-line drug for the treatment of 200–300 million malaria cases each year.5,7−9 Despite its vital therapeutic value, the biosynthetic pathway for incorporation of the endoperoxide into artemisinin remains elusive.9,10 To date, very few endoperoxidases have been reported, including iodide peroxidase,11 cyclooxygenase (COX),12 FtmOx1,13,14 and NvfI.15,16
For decades, the heme-containing endoperoxidase in prostaglandin G2 (PGG2) biosynthesis, prostaglandin H synthase (PGHS or COX), was the only well-characterized endoperoxidase.17−19 COX catalyzes a complicated transformation by incorporating both endoperoxide and peroxide functionalities into arachidonic acid to produce PGG2 (Scheme S1); the reaction has two cycles, the activation and catalytic cycles.19 A tyrosyl radical (Y•) is formed in the activation cycle and abstracts a hydrogen atom from the arachidonic acid substrate to produce a substrate-based radical, which initiates the incorporation of both endoperoxide and peroxide functionalities to produce PGG2. In contrast to the heme-containing COX, FtmOx1 in verruculogen biosynthesis (1 → 2, Scheme 1a)13,14 and NvfI in novofumigatonin biosynthesis (asnovolin A → fumigitomoid A, Scheme 1b)15,16 are α-ketoglutarate (αKG)-dependent mononuclear nonheme iron (αKG-NHFe) enzymes.20−24 FtmOx1 and NvfI are also very distinct from one another in both structural and biochemical aspects (Scheme 1a vs b).15,16
Scheme 1. Two αKG-NHFe Enzyme-Catalyzed Endoperoxidation Reactions.

FtmOx1-catalyzed conversion of fumitremorgin B (1) to verruculogen (2) and the crystal structure of FtmOx1•FeII•αKG binary complex (pdb entry 4Y5S) indicated that FtmOx1 is a distal-type αKG-NHFe enzyme.
NvfI-catalyzed conversion of asnovolin A to fumigatonoid A and the crystal structure of NvfI•FeII•N-oxalylglycine•asnovolin A ternary complex (pdb entry 7ENB) indicated that NvfI is a proximal-type αKG-NHFe enzyme.
Key mechanistic features for the proximal- and distal-type of αKG-NHFe enzymes, which are reflected in the COX-like and CarC-like mechanistic models proposed for FtmOx1-catalysis.
In addition to the most common type of hydroxylation reaction,25−27 αKG-NHFe enzymes also catalyze many nonhydroxylation reactions, including desaturation,28 ring formation,29 decarboxylation-assisted desaturation,30,31 sequential desaturation and epoxidation,32−34 ring expansion,35 halogenation,36 carbon skeleton rearrangements,37 endoperoxidation,14,16 and αKG decarboxylation to form ethylene.38−42 Among the ∼60 X-ray crystal structures of αKG-NHFe enzymes in the Protein Data Bank (PDB), two subclasses exist (the distal- and proximal-types).43 A vast majority of αKG-NHFe enzymes have a conserved His-X-Asp/Glu-Xn-His (2-His-1-carboxylate) motif, in which His and Glu/Asp residues serve as the iron-coordinating ligands,21,44 and αKG is a bidentate ligand in either the distal type (e.g., FtmOx1 with pdb entry 4Y5S in Scheme 1a) or the proximal type (e.g., NvfI with pdb entry 7ENB in Scheme 1b).43,45 Based on phylogenetic tree analysis, FtmOx1 and NvfI belong to two different subclasses.14,16,27,45−55
Structurally, NvfI is a member of the proximal type of αKG-NHFe enzymes. In the NvfI•FeII•N-oxalylglycine•asnovolin A ternary complex, the C1 carboxylate of αKG is trans to the first histidine of the 2-His-1-carboxylate motif (e.g., His135 in Scheme 1b),16 and the O2 binding site is next to the substrate (e.g., species A-1 in Scheme 1c).16,56 Therefore, in NvfI (mechanistic model detailed in Scheme S2), the FeIV=O (A-2 in Scheme 1c) is in line with the substrate pocket, which allows FeIV=O to abstract a hydrogen atom directly from the substrate to generate a substrate-based radical. The substrate-based radical then reacts with O2 to incorporate the endoperoxide functionality. In the end, oxygen rebound completes the reaction cycle by hydroxylation (Schemes 1b and S2), which explains that NvfI-catalysis does not need an extra reductant (e.g., ascorbate, Scheme 1a vs b).15,16
FtmOx1 is a distal-type αKG-NHFe enzyme. In the FtmOx1•FeII•αKG binary complex (pdb entry 4Y5S), the C1 carboxylate of αKG is opposite to the second histidine of the 2-His-1-carboxylate motif (e.g., His205 in Scheme 1a),14,43 and the O2 binding site points away from the substrate binding pocket (species B-1 in Scheme 1c). The resulting FeIV=O (species B-2 in Scheme 1c) might be offline relative to the substrate binding pocket. In addition, in the FtmOx1•FeII• αKG binary complex (Scheme 1a), Y224 is positioned right next to the metallo-center, which is reminiscent of Y•-involving catalysis in COX (Scheme S1).17−19,57,58 The corresponding tyrosine residue is absent in NvfI (Scheme 1a vs b).16 Biochemically, FtmOx1-catalysis is also very different from NvfI-catalysis (Schemes 2 vs S2).14−16 Under single-turnover conditions and in the absence of any reductant (e.g., ascorbate), verruculogen (2) is only the minor product, and 13-oxo-verruculogen (3) (Scheme 2) is the major product.14 To explain the production of 3 as the dominant product in FtmOx1 single-turnover reactions, a COX-like mechanistic model was proposed (Schemes 2a and S3a),14 and it has three key features: (1) FeIV=O species (B-2, Scheme 2a) oxidizes the active site Y224 to Y224•, which then serves as the catalytic species for endoperoxidation (B-2 → B-6, Scheme 2a); (2) Y224• regenerated in species B-7 is also responsible for the C13-OH dehydrogenation (B-7 → compound 3, Scheme 2a); (3) If Y224 is mutated or rotates to an alternative position, oxygen rebound may occur, and FtmOx1-catalysis may change to a typical hydroxylation reaction (for more details, see Scheme S3a).
Scheme 2. Key Features of the COX-Like and CarC-Like Mechanistic Models for FtmOx1-Catalysis.
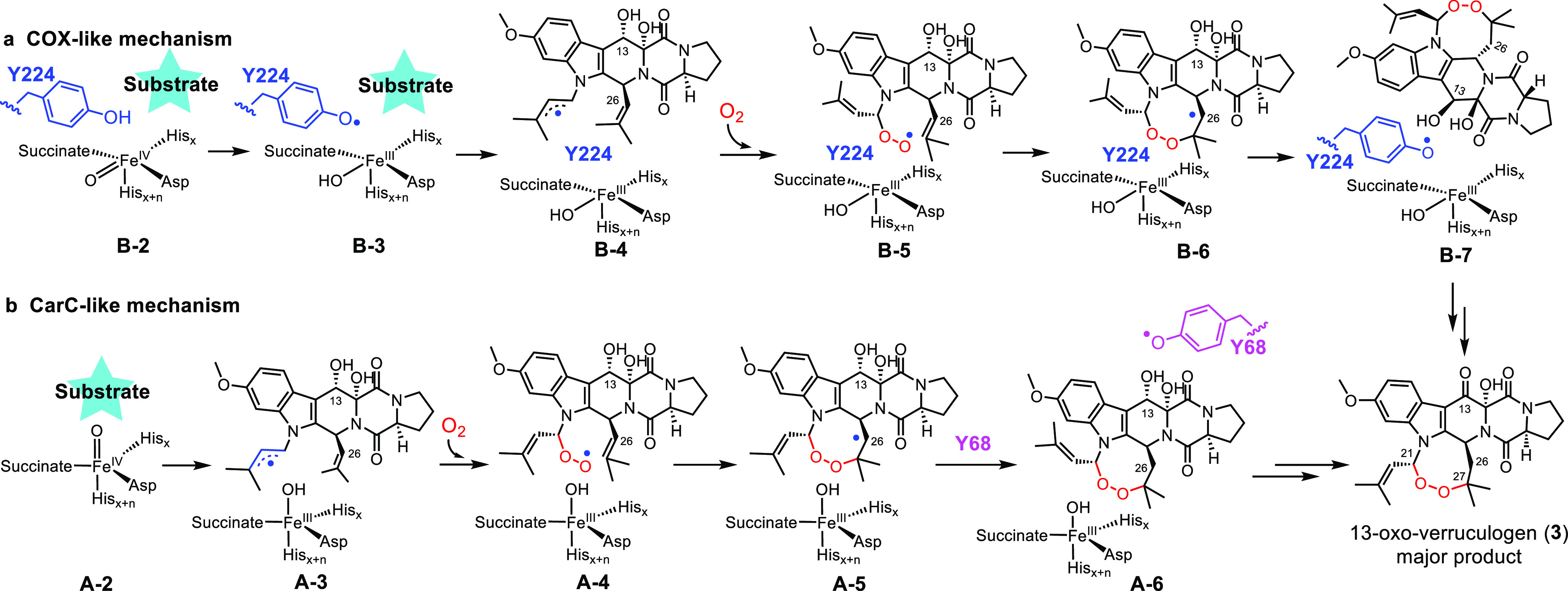
In the FtmOx1 COX-like mechanistic model, there are four key features: (1) B-1 → A-1 or B-2 → A-2 rotation is not required (Scheme 1c); (2) Y224 is next to the metallo-center and Y224• is the key to the endoperoxidation reaction by preventing the oxygen rebound, which would lead to the hydroxylation side-reaction; (3) Y224• regenerated in species B-7 is also responsible for the C13-OH dehydrogenation reaction to produce 3 as the major product; (4) If Y224 is mutated or rotates to an alternative position, FtmOx1 may catalyze a hydroxylation reaction (for more details, see Scheme S3a).
In the FtmOx1 CarC-like mechanistic model, there are four key features: (1) the prerequisite step in FtmOx1-catalysis is the B-1 → A-1 or B-2 → A-2 rotation (Scheme 1c); (2) FeIV=O directly abstracts a hydrogen atom from the substrate C21-position (species A-2 → A-3 conversion); (3) the substrate C26-radical is quenched by Y68 to produce Y68• (A-5 → A-6); (4) Y68• (species A-6) is responsible for the C13-OH dehydrogenation reaction to produce 3 as the major product. More details for these two models are shown in Scheme S3b. Structurally, the other distinctive difference between the COX-like model and the CarC-like model is that Y224 is right next to the metallo-center and is located between the metallo-center and the substrate. However, the metallo-center and Y68 are on the two opposite sides of the substrate.
Upon request from a reader, Nature editorially retracted the 2015 manuscript due to the potential misinterpretation of the electron density associated with pdb entry 4ZON.59 The observed electron density in pdb entry 4ZON modeled with the substrate fumitremorgin B (1) could be modeled alternatively using buffers and a string of H2O molecules. Along with the pdb entry 4ZON electron density discussion, several other mechanistic hypotheses have also been suggested, which we further consider here. It was suggested that because of uncertainties associated with the substrate binding mode in the crystal structure (PDB entry 4ZON), the αKG binding mode may be debatable too. As a result, no protein-based radical species is needed for FtmOx1-catalysis because, upon substrate binding, αKG or the FeIV=O species may rotate (B-1 → A-1 or B-2 → A-2,Scheme 1c). After such a rotation, FeIV=O (A-2, Scheme 1c) could directly oxidize the substrate to a substrate-based radical, which then reacts with O2 to incorporate the endoperoxide functionality (for more details, see Scheme S4). This αKG rotation model could not explain the production of 13-oxo-verruculogen (3) as the dominant product under single-turnover conditions without ascorbate. To address this shortcoming, an additional proposal revised the αKG rotation model, suggesting that FeIV=O is regenerated by oxidizing an FeIII–OH species using the substrate-based organic radical, and subsequently, the regenerated FeIV=O catalyzes 2 → 3 conversion to complete the reaction cycle (see Scheme S4 for the complete mechanistic model). Because the proposed FeIV=O regeneration from an organic radical is energetically unfavorable relative to its competing oxygen rebound pathway,60 a third version of the αKG rotation model, the CarC-like model, was suggested (Schemes 2b and S3b).61 In the latest CarC-like model, a protein-based radical (Y•) is invoked, along with four key features (Scheme 2b): (1) the αKG or FeIV=O rotation is a prerequisite step (B-1 → A-1 or B-2 → A-2, Scheme 1c); (2) FeIV=O (A-2) directly abstracts a hydrogen atom from the substrate C21-position to initiate the reaction (A-2 → A-3, Scheme 2b); (3) a substrate C26-radical is quenched by Y68 to produce Y68• (A-5 → A-6); and, (4) Y68• (A-6) is responsible for the C13-OH dehydrogenation reaction to produce 3 as the product (see Scheme S3b for more details).
In summary, the FtmOx1 mechanistic debates in the last few years have led to two mechanistic models: the COX-like and the CarC-like models. These two models are different in a few aspects: (1) the αKG or FeIV=O rotation is a prerequisite step in the CarC-like model, while it is not a part of the COX-like model; (2) in the COX-like model, Y224• initiates the formation of a substrate-based radical (Scheme 2a), while in the CarC-like model, Y68 quenches the substrate-based radical (Scheme 2b); (3) the COX-like model also suggests that upon mutation of Y224 or with Y224 rotation to alternative positions, FtmOx1-catalysis may change from endoperoxidation to hydroxylation. Because FtmOx1 catalyzes three different reactions (endoperoxidation, dealkylation, and alcohol dehydrogenation, Scheme S3a) under in vitro conditions, kinetic, structural, and spectroscopic results may originate from a combination of these reactions, rendering their interpretation an extremely challenging task. In this study, using substrate analogues, we simplify the FtmOx1-catalyzed reaction to predominantly one of these three reactions, which serves as a starting point for structure–function correlation analysis. The results from both 13-oxo-fumitremorgin B (7) and verruculogen (2) are more consistent with the COX-like model (Scheme 2a).14
Methods
Semisynthesis of 13-Oxo-fumitremorgin B (7)
To a solution of fumitremorgin B (1, 5 mg, 10.4 mmol) in acetone (2.5 mL) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 23.7 mg, 104 mmol, 10 equiv) at 0 °C. The reaction was monitored by thin-layer chromatography. After stirring for 8 h, the reaction mixture was filtered using a 0.22 μm membrane and analyzed by liquid chromatography–mass spectrometry (LC–MS). The reaction mixture was concentrated and dissolved in 1 mL acetonitrile and then purified by semiprep-high-performance liquid chromatography (HPLC) running with the ACN-H2O eluting program (During 0–15 min, ACN was increased from 10% to 99% in 0.1% HCOOH-containing H2O and then the column was eluting for 5 min with 99% ACN in 0.1% HCOOH-containing H2O) at 4 mL/min using a C18 column (Titank, 10 × 250 mm, 5 μm) to obtain 13-oxo-fumitremorgin B (7, tR = 14.8 min, 3.3 mg, 65% yield in one step). 13-oxo-fumitremorgin B (7): pale-yellow powder; 1H and 13C NMR data, see Table S2; HR-MS (ESI+) detected at m/z 478.2320, calcd for [M + H]+ C27H32N3O5: m/z 478.2336.
Anaerobic Purification of FtmOx1
In a typical anaerobic purification of FtmOx1, ∼30 g wet cell paste was resuspended in 50 mL of anaerobic buffer (100 mM Tris–HCl, 50 mM NaCl, pH 7.5) in an anaerobic Coy chamber (Coy Laboratory Products, Inc., Grass Lake, USA). Lysozyme (5 mg/mL final concentration), EDTA (5 mM final concentration), and ascorbate (5 mM final concentration) were then added into the cell suspension, and the mixture was incubated on ice with gentle stirring for 30 min. The cells were disrupted by sonication (40 cycles of 3 s bursts) using a VCX130 ultrasonic processor (SONICS & MATERIALS, Inc., Newtown, USA). The supernatant and the cell debris were anaerobically separated by centrifugation at 4 °C for 60 min at 12,000 rpm. The resulting supernatant was mixed with 30 mL of Strep-Tactin resin (IBA Lifesciences GmbH, Göttingen, Germany) and incubated on ice for 30 min. Then, the column was washed with washing buffer (100 mM Tris–HCl and 50 mM NaCl, pH 7.5) until the OD260 readout of the eluate was less than 0.05. The FtmOx1 protein was eluted with elution buffer (2.5 mM desthiobiotin in 100 mM Tris–HCl and 50 mM NaCl, pH 7.5). The eluted protein was concentrated to be ∼12 mg/mL, flash-frozen with liquid nitrogen, and stored at −80 °C. From 31 g of wet cell paste, ∼300 mg of protein was obtained.
FtmOx1•CoII•αKG•7 Complex Formation, Crystallization, and Data Collection
To facilitate the formation of a stable ternary complex of FtmOx1 with 13-oxo-fumitremorgin B (7) and αKG, they were mixed and preincubated at room temperature for 1 h before being used in setting up crystallization trays. In addition, DMSO was also included at a final concentration of 1–1.5% v/v to improve the substrate solubility. The FtmOx1•CoII•αKG•7 mixture, as well as FtmOx1•CoII, was screened under various crystallization conditions. Crystallization screenings were carried out using the sitting-drop vapor-diffusion method by mixing the protein samples with an equal volume of reservoir solution at room temperature. Crystallization hits from initial screens were then optimized by systematically varying the pH, individual component concentrations, and the presence of additives and detergents. The best crystals of the ternary complex and FtmOx1•CoII were obtained in 0.1 M MES/Imidazole pH 6.5, 20% PEG 500 MME, 10% PEG 20,000, 0.03 M MgCl2, and 0.03 M CaCl2. The FtmOx1•CoII solution yields a thick, plate-shaped crystal after 3 days, while thin, plate-like crystals were visible after 7 days for the ternary complexes. Crystals were harvested for data collection and cryoprotected with a 25% (v/v) solution of glycerol mixed with crystallization solution before being flash-cooled in liquid nitrogen. Diffraction datasets were collected on the beamline BL19U1 at the Shanghai Synchrotron Radiation Facility (SSRF) at 100 K. Crystallographic datasets were integrated and scaled using HKL3000.62 The crystal parameters and the data collection statistics are summarized in Table S8.
Large-Scale Enzymatic Conversion of 13-Oxo-fumitremorgin B (7) into 12-Hydroxy-13-oxo-fumitremorgin C (5)
A 20 mL reaction was conducted in order to isolate sufficient products for spectroscopic characterization. The anaerobic reaction mixture (10 mL, in 100 mM Tris–HCl, pH 7.5) contained 280 μM 13-oxo-fumitremorgin B (7), 140 μM FtmOx1 containing 168 μM FeII, 336 μM αKG, and 420 μM ascorbate. The reaction mixture was sealed and initiated by adding 10 mL of oxygen-saturated buffer (1.2 mM of oxygen) and incubated for 30 min at room temperature. The resulting reaction mixture contained a final concentration of 140 μM 7, 70 μM FtmOx1 containing 84 μM FeII, 168 μM αKG, and 210 μM ascorbate. The enzymatic reaction was quenched by adding 20 mL dichloromethane, the precipitated protein was removed by centrifugation at 13,000 g for 10 min, and the dichloromethane layer was carefully separated for further analysis. The reaction mixture was extracted once more using dichloromethane. The combined dichloromethane layers were concentrated by rotatory evaporation in vacuo to give the crude extract. The crude extract was further purified by semipreparative RP-HPLC using a C18 column (Titank, 10 × 250 mm, 5 μm) with a flow rate of 4.0 mL/min and an isocratic elution (ACN was 30% in 0.1% HCOOH-containing H2O for 20 min and then the column was eluted for 5 min with 99% ACN in 0.1% HCOOH-containing H2O) to obtain 12-hydroxy-13-oxo-fumitremorgin C (5, 1.0 mg, tR = 9.88 min).
For single-turnover reactions using different substrates (fumitremorgin B, 13-oxo-fumitremorgin B, and verruculogen), detailed reaction conditions are described in Supplementary Text.
Results and Discussion
Several Different Reactions Catalyzed by FtmOx1 Using Fumitremorgin B (1) as the Substrate
In our initial FtmOx1 biochemical characterization,14 we reported that, under single-turnover conditions and in the absence of extra reductants (e.g., ascorbate), 13-oxo-verruculogen (3) is the major product (Figure 1a). However, Dunham et al.61 observed a significant level of dealkylation even for the wild-type FtmOx1 and the amount of dealkylation product 4 could be as high as 40% of the product mixture. To explain the differences between these data sets, we first re-evaluated the FtmOx1 experimental conditions. In the Dunham report, the protein overexpression, purification conditions, and reaction conditions are very different from those we have employed. In our study, to ensure the production of high-quality protein, FtmOx1 protein was produced as a Strep-tagged protein and was anaerobically purified within a few hours. In studies conducted by Dunham et al., His-tagged FtmOx1 was purified by Ni-NTA chromatography.61 Then, the His-tag was removed by TEV protease in a 16–18 h incubation, followed by passage of the protein through a second Ni-NTA affinity column, size-exclusion chromatography, and a buffer exchange process. Therefore, before undertaking more detailed characterizations, we carefully re-examined the relevant set of FtmOx1 reactions under a few different conditions. In addition, to ensure the reproducibility, experiments performed in the Liu laboratory were also repeated in Zhang and Yan laboratories.
Figure 1.
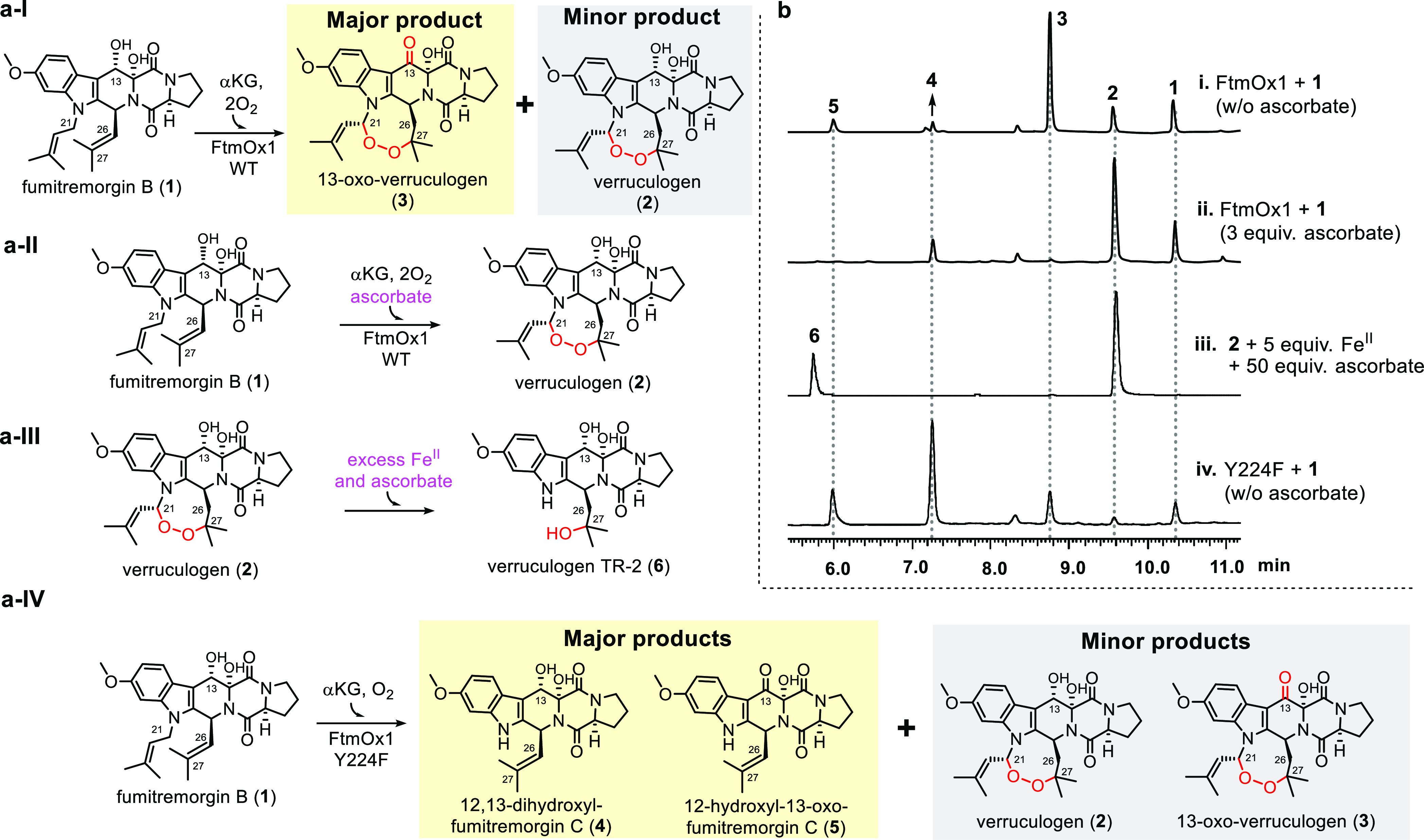
FtmOx1-catalyzed reaction using fumitremorgin B (1) as the substrate. (a) Four different FtmOx1-catalysis outcomes (I–IV) under slightly different conditions observed for the wild-type FtmOx1 enzyme and its Y224F variant; (b) HPLC profiles (i–iv) of reactions under single-turnover conditions from wild-type FtmOx1 and the FtmOx1 Y224F variant.14 The reaction mixture contains FtmOx1 or Y224F variant (70 μM), FeII (63 μM), fumitremorgin B (70 μM), αKG (63 μM), and O2 (480 μM). Trace i, using wild-type FtmOx1 in the absence of ascorbate; Trace ii, using wild-type FtmOx1 in presence of ascorbate; Trace iii, nonenzymatic endoperoxide decomposition in the presence of FeII and ascorbate; Trace iv, using Y224F FtmOx1.
For the biochemical characterization of FtmOx1, we purified fumitremorgin B (1, 35 mg) from the crude extract of Aspergillus fumigatus MF029 cultured in 16 kg rice medium at 28 °C for 30 days (Supplementary Text). We first repeated FtmOx1 single-turnover studies and, indeed, under single-turnover conditions and in the absence of ascorbate, 13-oxo-verruculogen (3) is the major product and verruculogen (2) is the minor product (Figure 1a-I). As shown in the HPLC profile (trace i of Figure 1b), 13-oxo-verruculogen 3 and verruculogen 2 are produced at a ratio of 4.1:1 and indeed 13-oxo-verruculogen 3 is the dominant product. Therefore, our initial FtmOx1 biochemical characterization reports were reproduced,14 and the dealkylation product 4 is generated at a very low level under our protein purification and reaction conditions (trace i of Figure 1b). Consistent with our previous report,14 under single-turnover conditions and in the absence of ascorbate, FtmOx1 catalyzes two distinct, sequential transformations, that is, endoperoxidation between C21 and C27 and alcohol dehydrogenation that converts C13-OH to C13-keto (1 → 2 → 3, Figure 1a-I), forming 13-oxo-verruculogen (3) as the major product and verruculogen (2) as a minor product.14 When ascorbate is included in the FtmOx1 reactions, the outcome changes drastically: verruculogen (2) instead of 13-oxo-verruculogen (3) is the dominant product (trace ii of Figure 1b). In addition, during product purification and characterization, we noticed that the reaction workup must be handled carefully because, in the presence of ascorbate, neither endoperoxide 2 nor 3 is stable. For example, when verruculogen (2) is mixed with ascorbate (50 equiv) and FeII (5 equiv) under aerobic conditions, Fenton chemistry leads to the decomposition of 2 and the resulting dealkylation product is verruculogen TR-2 (6, trace iii, Figure 1b). Therefore, FtmOx1 reactions carried out in the presence of ascorbate need to be handled carefully to minimize the formation of nonenzymatic side products. In the characterization of NvfI, Matsuda et al.15 also reported that the endoperoxidation reaction has to be handled very carefully. Otherwise, side reactions for NvfI-catalyzed ortho-esterification will occur. This FeII-mediated cleavage and rearrangement of endoperoxides have also been reported in other systems, for example, artemisinin.63
When Y224 is mutated to F224, the product profile (trace iv, Figure 1b) is completely different from that in wild-type FtmOx1. The dominant products are the dealkylation products 4 and 5, which account for 51.3% and 21% of the Y224F variant product mixture, respectively. We have isolated and characterized at least two more minor products (compounds 2 and 3, Figure 1a-IV). In the FtmOx1 Y224F variant, the endoperoxidation products are minor products, while the major pathway is now dealkylation (1 → 4 or 5, Figure 1a-IV), which are the decomposition products from the fumitremorgin B C21 hydroxylation reaction. Therefore, in the Y224F variant, the major reaction in FtmOx1-catalysis switches from endoperoxidation to a hydroxylation. The products (1–6) have been isolated, and structures were assigned based on 1H NMR, 13C NMR, and high-resolution mass spectrometry, and they are consistent with the previous structural assignments (Figures S1–S6 and Tables S1–S6).14
Using 13-Oxo-fumitremorgin B (7) as the FtmOx1 Substrate Predominantly Leads to a Dealkylation Reaction
Consistent with our initial report14 and now reproduced herein by three different laboratories, FtmOx1-catalysis is an extremely complicated system (Figure 1) involving at least three different reactions (Figure 1a): endoperoxidation (1 → 2), alcohol dehydrogenation (2 → 3), and dealkylation as a result of a hydroxylation reaction (1 → 4). Because of the inherent activities of endoperoxides, reactions in the presence of ascorbate need to be handled carefully because Fenton chemistry could introduce another impediment into the elucidation of this challenging reaction system. These features add a significant level of complication to FtmOx1 mechanistic studies. To address this challenge, we shifted our focus to FtmOx1 substrate analogues to simplify the FtmOx1 catalytic system to limit the course to predominantly only one of the reactions listed in Figure 1a. The first substrate analogue we characterized was 13-oxo-fumitremorgin B (7, Figure 2a). Compound 7 was selected for two reasons. First, in compound 7, the C13-position is already oxidized to a keto moiety, thus eliminating the alcohol dehydrogenation reaction (2 → 3). Second, if indeed a planar structure for fumitremorgin B (1) is favored in the FtmOx1 active site as suggested in the CarC-like model,61 compound 7 may bind better due to the sp2 hybridization at its C13-keto group in comparison to the sp3 hybridized C13-OH group in fumitremorgin B (1).
Figure 2.
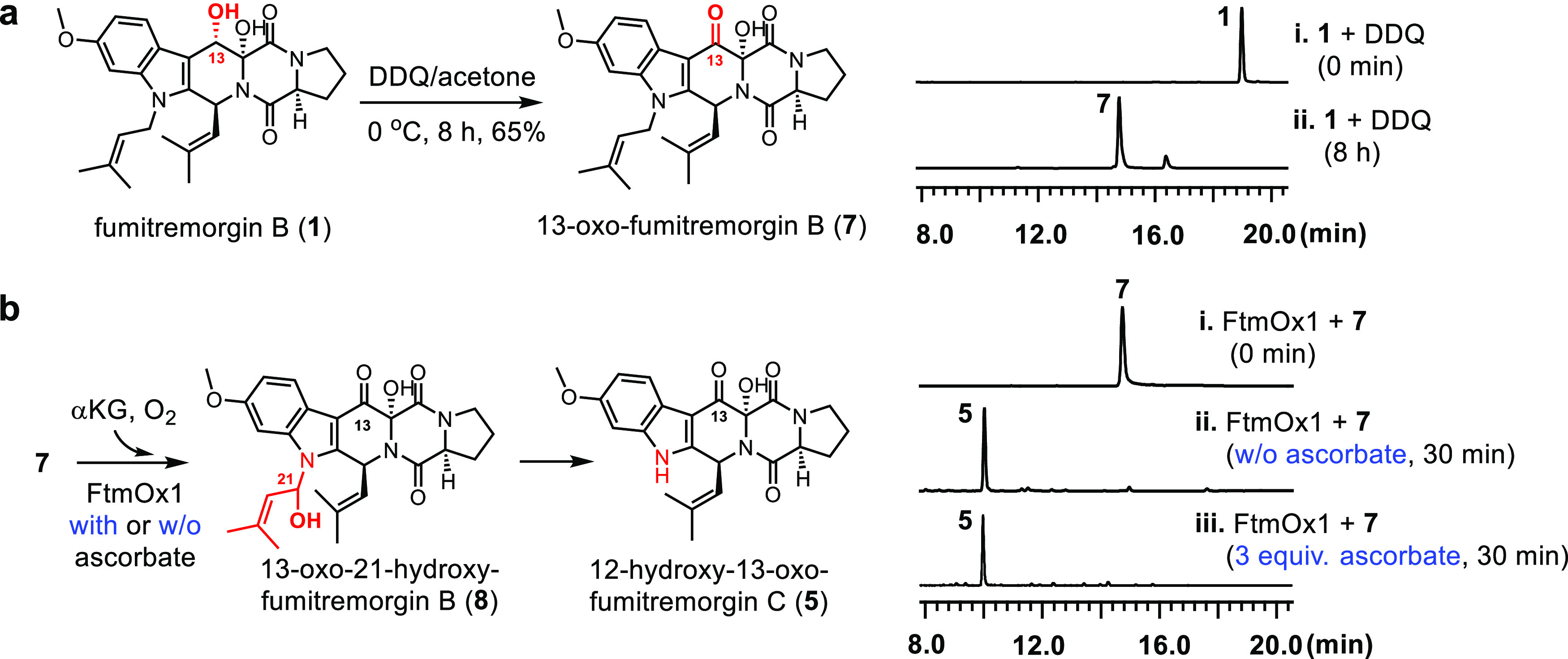
Using 13-oxo-fumitremorgin B (7) as an alternative FtmOx1 substrate. (a) Semisynthesis of 13-oxo-fumitremorgin B (7) by DDQ oxidation (left panel) and HPLC profiles (right panel). (b) FtmOx1-catalyzed C21 hydroxylation of 7, and dealkylation of 8 to 5 (left panel) and HPLC profiles in the absence (ii) and in the presence (iii) of ascorbate (right panel).
13-Oxo-fumitremorgin B (7, Figure 2a) was synthesized by a semisynthetic approach. Fumitremorgin B (1) was purified from A. fumigatus MF029 fermentation materials. After fumitremorgin B (1) was obtained, its C13-OH position was selectively oxidized by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).64 The semisynthetic route was successful, and 13-oxo-fumitremorgin B (7) was successfully purified with a yield of 65% (Figure 2a). 13-Oxo-fumitremorgin B (7) was carefully characterized by a combination of HR-ESI-MS and 1H NMR and 13C NMR spectroscopy methods (Figure S7 and Table S7), and the results were consistent with the previously reported data.65
Compound 7 was then examined as an FtmOx1 substrate under single-turnover conditions without ascorbate. Notably, the HPLC profiles (traces i and ii, Figure 2b) indicate that the predominant reaction (>95%) is the N-dealkylation of 7 to generate 12-hydroxy-13-oxo-fumitremorgin C (5, Figures 2b, S5 and Table S5).61,66 For compound 7, the endoperoxidation reaction observed in the wild-type substrate 1 is barely detectable (<5%). In addition, 12-hydroxy-13-oxo-fumitremorgin C (5) is most likely the decomposition product of 21-hydroxy-13-oxo-fumitremorgin B (8) (Figures 2b and S8), which is the result of hydroxylation of compound 7 at its C21-position. Therefore, FtmOx1-catalysis shifts from a complicated system in the wild-type substrate 1 (Figure 1) to a simplified system in substrate analogue 7, in which the dominant reaction is dealkylation (>95% of the product mixture, Figure 2b). This enables further detailed structure–function correlation analysis. In the case of fumitremorgin B (1), when ascorbate is included, it completely changes the reaction profile (Figure 1). Therefore, we have also repeated the reaction of compound 7 in the presence of ascorbate (trace iii, Figure 2b). In the presence of 3 equiv of ascorbate (related to protein), dealkylation product 5 is still the dominant product. The inclusion of extra reductants does not affect the FtmOx1-catalysis outcomes when 7 is the substrate, which differs from that of the fumitremorgin B (1) reactions reported in Figure 1.
Structural Characterization of the FtmOx1•CoII•αKG• 7 Ternary Complex
After the discovery of a much more simplified reaction system using substrate analogue 7 (Figure 2), we also solved the structure of the FtmOx1•CoII•αKG•7 ternary complex by X-ray crystallography. To avoid enzymatic turnovers during crystallization, we replaced FeII with CoII and obtained the crystal structure of the FtmOx1•CoII•αKG•7 ternary complex at a resolution of 2.87 Å, for which the overall architecture is similar to that of FtmOx1 (pdb entry 4Y5T) and the FtmOx1•FeII•αKG (pdb entry 4Y5S) complex. Each FtmOx1 monomer of the ternary complex maintains a “jelly-roll” fold14 and the FtmOx1 dimer forms by an internal twofold symmetry operator with each active site including residues from both monomers (Figure S9). After model building and refinement, the electron density as well as the composite omit map clearly suggested the existence of 7 in the FtmOx1 active site (Figures 3a and S10), with a Real Space Correlation Coefficient (RSCC) ranging from 0.84 to 0.93 in each chain, indicating that compound 7 fits well with the electron density and is built with high occupancies (0.81 to 0.86).
Figure 3.

Structure of the FtmOx1•CoII•αKG•7 ternary complex and the proposed FtmOx1-catalyzed dealkylation model using 13-oxo-fumitremorgin B (7) as the substrate. (a) Omit map (mFo-DFc) with substrate 7 at the FtmOx1 active site. The map was contoured at 3.0 rmsd. (b) FtmOx1 active site, shown in electrostatic surface mode, with key residues shown as sticks. (c) Flipped Y224 at the FtmOx1 active site. Two alternative positions of Y224 from the FtmOx1•CoII•αKG•7 ternary complex are shown as sticks, and Y224 from the FtmOx1•FeII•αKG binary complex is shown as pink stick. A red arrow indicates the flip of Y224 upon substrate 7 binding. (d) Mechanistic model for the dealkylation reaction when 13-oxo-fumitremorgin B (7) is the substrate.
The FtmOx1 substrate binding pocket could be further divided into two subpockets, the polar subpocket and the hydrophobic subpocket (Figure 3b). αKG occupies the polar subpocket and compound 7 occupies the hydrophobic pocket. The same as the FtmOx1•FeII•αKG binary complex (Scheme 1b),14 αKG is in the distal-mode in the FtmOx1•CoII•αKG•7 ternary complex (Figure 3b). The αKG rotation suggested in the CarC-like model (B-1 → A-1, Scheme 1c) was not observed.43,61 An alternative explanation in the CarC-like model is that FeIV=O species may rotate (B-2 → A-2, Scheme 1c). When the information presented in the subsequent section related to the flipping of Y224 predicted from the COX-like model is taken into account, the FeIV=O rotation in the CarC-like model is not needed to explain the dealkylation reaction either. In comparison with the structure of the FtmOx1•FeII•αKG binary complex (pdb entry 4Y5S), several loops are pushed toward the metallo-center with an average distance of 2–3 Å, including residues 68–72, 120–130, 171–186, and 205–215 in one FtmOx1 monomer, and residues 261′–270′ from the adjacent FtmOx1 monomer (Figure S11). There are very few polar interactions between FtmOx1 and compound 7. The metallo-center and Y68 are separated by compound 7 (Figure 3b,c), while the Y68 hydroxyl group forms a hydrogen bond with the C11-keto of 7 (Figure S12), suggesting that Y68 may function in positioning the substrate. Notably, Y68 is fully solvent-exposed (Y68 is proposed to act as a radical carrier in the CarC-like model,55,61 which will be discussed in a following section). When comparing the structures of the FtmOx1•FeII•αKG binary complex (pdb entry 4Y5S) and the FtmOx1•CoII•αKG•7 ternary complex (pdb entry 7WSB), the most notable difference is that Y224 indeed adopts different conformations in these two structures. In the FtmOx1•FeII•αKG binary complex, Y224 forms a hydrogen bond with the metal-coordinating water, which is presumably the O2 binding site. Consistent with the proposed role of Y224• in FtmOx1-catalyzed endoperoxidation in the COX-like model (Schemes 2a and S3a), upon Y224 mutation (e.g., Y224F and Y224A), FtmOx1-catalysis changes from the endoperoxidation in the wild-type enzyme to predominantly dealkylation in these Y224 variants. These biochemical data from the Liu laboratory14 have been replicated in Zhang and Yan laboratories (Figure 1). Moreover, in the FtmOx1•CoII•αKG•7 ternary complex, Y224 in those chains where compound 7 was built with higher occupancy and better density indeed rearranges by rotating ∼92° to a new position that is 8.8 Å away from the metallo-center (Figure 3c), rather than the 3.6 Å distance from Y224 to the metallo-center in the original position (Figure 3c). This Y224 structural rearrangement allows the substrate prenyl groups to directly access the metallo-center.
The potential importance of the Y224 structural rearrangement is critical to resolution of the current mechanistic debates regarding FtmOx1. Although it has been a consensus view that the COX-like model could successfully explain the production of 3 instead of 2 as the major product under single-turnover conditions in the absence of ascorbate (Scheme 2a), explaining the dealkylation reaction in the COX-like model was a point of contention. Dunham et al. reported the presence of as high as 40% dealkylation product even in the wild-type FtmOx1 reaction mixture.61 In our study, while the dealkylation level is low (Figure 1b, traces i and ii), it does exist. To explain the dealkylation reaction, the COX-like model suggests that Y224 could exist in an alternative conformation by flipping to other orientations (B-1 → B-2′ or B-2″, Figure 3d and Scheme S3a), which creates space for the substrate to directly access the metallo-center (B-3″, Figure 3d). As described above, this conformational change has now been observed in a series of structures. With that change in the Y224 position, the FeIV=O could abstract a hydrogen atom from the C21 position of the substrate analogue 7 directly (B-3″ → B-4″, Figure 3d). The subsequent oxygen rebound completes the reaction cycle to produce C21-hydroxylation product 8 (Figure 3d), which decomposes spontaneously to the dealkylation product 5 (Figure 3d). Here, we have provided experimental evidence in support of this hypothesis (Schemes 2a and S3a): (1) Y224 indeed can exist in an alternative conformation as evidenced by structural characterization of the 13-oxo-fumitremorgin B (7) bound complex (Scheme 1a vs Figure 3); (2) upon flipping of Y224 to an alternative position to create space for the substrate to directly access the metallo-center, the reaction does change from endoperoxidation to hydroxylation, which then decomposes to the observed major dealkylation product (Figure 2); (3) upon Y224 mutation, the variant forms of the enzyme also catalyze the dealkylation as their major reaction (Figure 1). Notably, the proposed αKG rotation suggested in the CarC-like model is yet to be observed by crystallography or through other means.
While the present manuscript was being prepared, Wu et al.55 reported the crystal structure of the FtmOx1•FeII•αKG•1 ternary complex (pdb entry 7ETK). In this section, we present a comparative analysis of the FtmOx1•CoII•αKG•7 ternary complex (pdb entry 7WSB) and the FtmOx1•FeII•αKG•1 ternary complex (pdb entry 7ETK). The general architecture of these two complexes is almost identical (Figure S13). αKG in both the structures takes the same conformation and no αKG rotation relative to the FtmOx1•FeII•αKG binary complex (pdb entry 4Y5S) predicted by the CarC-like model is evident in either structure (Figures 4a and S14). The A, B, and C rings of 1 and 7 (refer to Scheme 1a for the ring labeling system) are almost superimposable. The two prenyl arms of 1 and 7 in these two ternary complexes are also located at a similar position. The distances between the metallo-center and the C21-carbon in 1 and 7 are 4.6 and 4.8 Å (Figure 4a), respectively. The oxygens at the C13-positions in 1 and 7 are also positioned very close to each other, with a distance of ∼0.5 Å. Relative to 1, the D and E rings of 7 bend toward the adjacent FtmOx1 monomer by about 18° (Figure 4a). Because the D and E rings are located in the outside region of the substrate binding pocket and are solvent-exposed, such a slight difference in D and E rings may not be directly related to the observed chemistry. The high degree of similarities between the FtmOx1•FeII•αKG•1 and FtmOx1•CoII•αKG•7 ternary complexes strongly suggests that, when Y224 rotates to an alternative position in this FtmOx1•FeII•αKG•1 complex, FtmOx1-catalysis changes to the dealkylation side-reaction (extended Data Figure 7 in our initial manuscript14) and as predicted in the minor pathway of the COX-like model (Scheme S3a).
Figure 4.
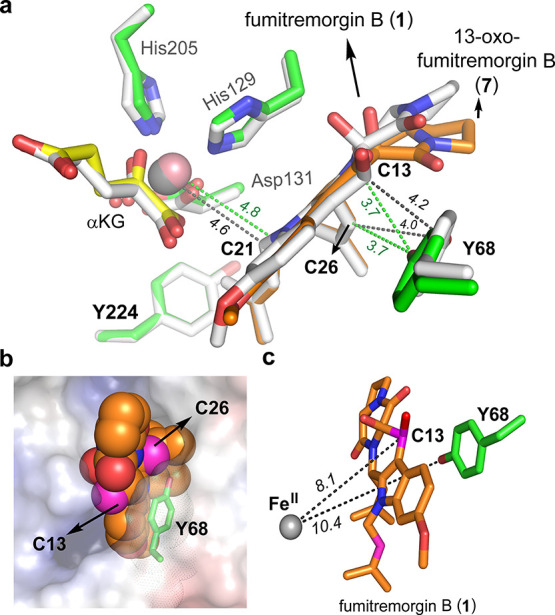
More structural information of FtmOx1 ternary complexes. (a) Active site comparison between the FtmOx1•CoII•αKG• 7 complex and the FtmOx1•FeII•αKG•1 complex (pdb entry 7ETK).55 The FtmOx1•FeII•αKG•1 complex is shown in white. The distances between key positions of the compounds and protein residues are highlighted with black dashed lines. (b) FtmOx1•FeII•αKG•1 complex structure shown in electrostatic mode (generated by APBS), 1 is shown as a sphere, while C13 and C26 are colored in magenta. Y68 is solvent-exposed and is shown as a stick with dots on the surface to highlight the solvent accessibility. (c) Position of Y68 in FtmOx1•FeII•αKG•1 complex structure. The metallo-center and Y68 are on the opposite sides of the substrate. In addition, the C13 position of compound 1 is not adjacent to the Fe-center and the distance between them is ∼8.1 Å. In addition, Y68, the substrate C13 and C26 positions are all solvent-exposed.
In the last few years, FtmOx1-catalysis has also been examined by computational methods. Wang et al.67 investigated FtmOx1-catalysis by a combination of quantum mechanics and molecular mechanics (QM/MM) calculations using crystal structures (pdb entry 4Y5S and 4ZON) as the starting point. Their results suggest that the energy barrier for FeIV=O oxidation of Y224 is 9.1 kcal·mol–1 (COX-like model). In contrast, FeIV=O oxidation of the C21 position of fumitremorgin B’s has an energy barrier of 33.9 kcal·mol–1 (CarC-like model). Miłaczewska et al.,68 re-examined FtmOx1-catalysis based on docking, molecular dynamics simulation, and density functional theory (DFT) calculations. Because of the low substrate occupancy in 4ZON, in the calculation by Miłaczewska et al., they have also modeled the substrate fumitremorgin B in two different conformations (bent or planar). Miłaczewska et al. suggested that a planar structure for fumitremorgin B might be more favored. Consistent with the conclusion of Wang et al.,67 the results reported by Miłaczewska et al. suggest that the activation energy barrier for FeIV=O oxidation of Y224 is 4.1 kcal·mol–1 (COX-like model), while the suggested FeIV=O rotation has a higher energy barrier of 13.0 kcal·mol–1 (CarC-like model). In studies by Miłaczewska et al.,68 the authors have also examined the Y224A variant and their calculation indicated that, in this variant, with the access of substrate to the metallo-center, the energy barrier for the abstraction of substrate C21 hydrogen by FeIV=O species is reduced to 9.5 kcal·mol–1.
Based on the FtmOx1•FeII•αKG•1 ternary complex (pdb entry 7ETK), Wu et al.55 also conducted QM/MM calculations. In the FtmOx1•FeII•αKG•1 ternary complex, because Y224 flips to an alternative position, the Y224 oxidation pathway was not considered in the report by Wu et al. Based on the QM/MM calculation, Wu et al. suggested that dealkylation is thermodynamically favored, while endoperoxidation is kinetically favored in a high O2 concentration. In our experiments described above, for the reaction using compound 7 as the substrate, dealkylation is the predominant reaction. In addition, for the Y224F variant, the major products are also the dealkylation products.
Using Verruculogen 2 as the FtmOx1 Substrate
As discussed above, FtmOx1 catalysis is a very complicated system and it catalyzes three different reactions (endoperoxidation, alcohol dehydrogenation, and dealkylation). In the calculation by Wu et al.,55 the authors suggested that an FtmOx1•FeII•αKG•1 ternary complex (pdb entry 7ETK) could explain all three reactions, while our biochemical and structural studies using substrate analogue 7 clearly indicated that this structure is most likely associated only with the dealkylation reaction. In addition, Y68 and the C26 and C13-positions of 1 are all fully solvent-exposed (Figure 4b). As a result, a C26-based radical (A-5, Scheme 2b) could be quenched by ascorbate directly, which eliminates the need for Y68• as the intermediate step in the CarC-like model. In the study by Miłaczewska et al.,68 using DFT calculations, direct reduction of the substrate radical by ascorbate has also been suggested. Furthermore, in the FtmOx1 single-turnover reaction in the absence of ascorbate, the predominant product is 13-oxo-verruculogen (3, Figure 1). In the structural and theoretical calculation by Wu et al., Y68 along with the C26 and C13 positions of 1 in the FtmOx1•FeII•αKG•1 ternary complex are all solvent-exposed (Figure 4b). In the proposed A-4 → A-6 → compound 3 transformations of the CarC-like model (Schemes 2b and S3b), the protection of these radicals from quenching to produce compound 3 in such high yield (>80% of the product mixture, trace i in Figure 1b) is not apparent. As another note, the substrate C13 position is completely separated from the metallo-center and their distance is ∼8.1 Å (Figure 4c). After considering all these factors, it becomes apparent that to further differentiate between the COX-like and CarC-like models (Scheme 2), whether Y224• or Y68• is responsible for the observed 2 → 3 transformation needs to be examined. In our COX-like mechanistic model, Y224 is next to the metallo-center and Y224 is located between the metallo-center and the substrate. Y224• at the active site has been suggested to be responsible for the 2 → 3 transformation (Schemes 2a and S3a). In contrast, Wu et al.55 suggested that Y68• is responsible for the 2 → 3 transformation (Schemes 2b and S3b) as explained by the CarC-like model.61 Because Y68 and the metallo-center are located on two opposite sides of the substrate (Figures 3 and 4), the CarC-like model suggests that Y68• is a downstream intermediate of the substrate-based C21 and C26 radicals (Scheme 2b). If the CarC-like model is followed, FtmOx1 will not accept verruculogen (2) as an alternative substrate (Figure 5a-II prediction) because the endoperoxide between C21 and C27 is formed already and the radical propagation chain to Y68• in the CarC-like model is blocked already in verruculogen (2). In contrast, in the COX-like model, Y224 is close to the substrate C21, C26, and C13-positions (Figure 5a-I prediction) and Y224 is also next to the metallo-center. Therefore, FeIV=O could oxidize Y224 to Y224•. As a result, the COX-like model predicts that verruculogen (2) will be an FtmOx1 substrate. Based on these key differences, we decided to examine whether verruculogen (2) could serve as an FtmOx1 substrate.
Figure 5.

Further differentiation between COX-like and CarC-like models using verruculogen (2) as the substrate. (a) Predicted reaction outcomes for verruculogen (2) according to the COX-like and CarC-like models. (b) Reaction outcomes: reaction result (left panel) and the HPLC profiles and FtmOx1-catalyzed oxidation of 2 into 13-oxo-verruculogen (3, right panel).
Similar to the case of fumitremorgin B (1) production, we purified verruculogen (2) from the crude extract of A. fumigatus MF029 and fully characterized the purified verruculogen (2) by 1H NMR, 13C NMR, and high-resolution mass spectrometry (Figure S2 and Table S2). The FtmOx1 reaction using verruculogen (2) as the substrate was characterized under single-turnover conditions in the absence of ascorbate by following a protocol similar to that used in our fumitremorgin B (1) studies (Figure 1). The HPLC trace (Figure 5b) clearly indicates that FtmOx1 does accept 2 as the substrate, and 13-oxo-verruculogen (3, Figure S3 and Table S3) was obtained as the product as predicted by the COX-like model. The production of 13-oxo-verruculogen 3 from verruculogen 2 is clearly at odds with the CarC-like model because the CarC-like model will suggest that verruculogen 2 is not an FtmOx1 substrate (Figure 5).
Conclusions
In the last few years, significant efforts have been invested in characterizing FtmOx1 and its variants using an interdisciplinary approach, including biochemical characterization under several different conditions, kinetic and spectroscopic characterization of intermediates trapped, and crystallographic as well as computational investigations.13,14,55,61,67,68 Information obtained in the last few years clearly indicates that FtmOx1-catalysis is a very complicated system with three different reactions: endoperoxidation, alcohol dehydrogenation, and dealkylation (Figure 1). For kinetic, spectroscopic, and structural information obtained from the previous studies, one of the biggest challenges is that when fumitremorgin B (1) is used as the substrate, the results obtained might be a combination from all of these three reactions. Furthermore, as shown in Figure 1, FtmOx1 reaction outcomes may vary depending on the quality of the material and variation of reaction conditions. Because of all these factors, interpretation of FtmOx1 kinetic, spectroscopic, and structural information is very challenging, and this is likely the key reason for all the extensive mechanistic discussion.
To address the challenges faced in FtmOx1 mechanistic studies, in this study, we leveraged the use of substrate analogues to simplify FtmOx1-catalysis from the three reactions (Figure 1) down to one dominant reaction (Figures 2 and 5). As predicted by the COX-like model, Y224 does have two conformations (one close to the metallo-center and one rotating away from the metallo-center). Upon Y224 mutation, the reaction changes from endoperoxidation in the wild-type FtmOx1 to dealkylation predominantly in these variants (e.g., Y224F in Figure 1), a result that is consistent with the proposed role of Y224• in FtmOx1-catalyzed endoperoxidation. In QM/MM and DFT calculations reported by the studies of both Liu and Miłaczewska and their co-workers,67,68 when Y224 is next to the metallo-center, FeIV=O oxidation of Y224 to Y224• is an energetically more favored pathway than the pathway involving a direct hydrogen atom abstraction by FeIV=O from the C21 position of fumitremorgin B.67
In the FtmOx1•CoII•αKG•7 ternary complex, Y224 flips to a position that is far away from the metallo-center to allow the substrate to directly access the metallo-center. As predicted by the COX-like model, associated with this Y224 flipping, FtmOx1-catalysis indeed changes from endoperoxidation to dealkylation (a side-reaction in wild-type FtmOx1, Scheme S3a). In DFT calculations reported by Miłaczewska et al.68 using Y224A variant, it significantly reduces the energy barrier for the direct hydrogen atom abstraction by FeIV=O from the C21 position of fumitremorgin B. In fact, in QM/MM studies reported by Wu et al.,55 because they are based on the FtmOx1•FeII•αKG•1 ternary complex where Y224 is located in a position similar to that in the FtmOx1•CoII•αKG•7 ternary complex, FeIV=O is accessible by the substrate. Their calculation results indicate that the hydroxylation pathway (leading to dealkylation) is thermodynamically favored. In the reaction of FtmOx1 with substrate analogue 7, because the predominant reaction observed is dealkylation, it is highly likely that FtmOx1•FeII•αKG•1 ternary complex reported by Wu et al. is the one responsible for the dealkylation side-reaction (minor reaction) observed in the wild-type enzyme when fumitremorgin B (1) is used as the substrate.
To explain the observed production of 3 as the dominant product in the 1 → 2 → 3 sequential transformation under single-turnover conditions (Figure 1), Y224• is suggested in the COX-like model, while Y68• is proposed in the CarC-like model (Scheme 2a). However, because these two tyrosine residues are on the two opposite sides of the substrate with Y224 on the metallo-center side and Y68 on the side opposite to the metallo-center, the demonstration of verruculogen (2) as an alternative FtmOx1 substrate provides another line of evidence supporting the COX-like model. Considering the observed interactions between Y68 and substrate/substrate analogues, Y68 might be important for substrate binding and positioning. The calculation studies by Miłaczewska et al. have also suggested that Y68 might be important for both substrate positioning and ascorbate binding to explain the reaction outcome observed in Figure 1a-II.70
In summary, in this study, using substrate analogues, we simplified the FtmOx1-catalysis from a very complicated system with three reactions (endoperoxidation, alcohol dehydrogenation, and dealkylation) to predominantly either the dealkylation or an alcohol dehydrogenation reaction. These simplified systems will serve as excellent models for future FtmOx1 mechanistic characterization using kinetic, spectroscopic, and structural methods.
Acknowledgments
We thank Profs. Yisong Guo, Sean J. Elliott, John Caradonna, Yan Zhang, Deborah Perlstein, and Dr. Andrew Weitiz for critical comments on the manuscript. The authors thanked facility team members in Shanghai Synchrotron Radiation Facility (SSRF), China, for X-ray crystal data collection.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacsau.2c00248.
Fermentation and compound purification conditions, single-turnover reaction under various combinations and under different conditions, supplementary mechanistic discussion and schemes, spectroscopic data for substrate, substrate analogues, and products isolated from the enzymatic reactions, as well as additional crystal structural information. The structure of the FtmOx1•CoII•αKG•7 ternary complex (pdb entry 7WSB) can be accessed through https://www.pdbus.org/ (PDF)
Author Contributions
P.L., L.Z., and X.L. designed the study. X.L., G.Z., X.W., R.C., H.S., and N.N. performed the semisynthesis and biochemical experiments. W.Y., X.W., J.W., and Y.W. did the crystallization and structure analysis. G.Z., K.W., H.L., and X.X. did the fermentation and purification of substrates. P.L., L.Z., X.L., C.C., and W.Y. analyzed the data and prepared the manuscript with inputs from all authors. All authors have given approval to the final version of the manuscript. G.Z., W.Y., X.W., and R.C. contributed equally.
This work was supported by the National Natural Science Foundation of China (31720103901), the National Key Research and Development Program of China (2020YFA090032 and 2019YFA0906201), the National Natural Science Foundation of China (21977029 and 81903529), China Postdoctoral Science Foundation funded project (2019M661403), the Open Project Funding of the State Key Laboratory of Bioreactor Engineering, the 111 Project (B18022), and Shanghai Science and Technology Commission (18JC1411900). The authors thank staffs from BL02U1 and BL19U1 beamline at Shanghai Synchrotron Radiation Facility (SSRF), China, for their assistance in X-ray crystal data collection. This work is supported in part by the National Institutes of Health (GM140040 to P.L. and R24 GM134210 to C.E.C.).
The authors declare no competing financial interest.
Supplementary Material
References
- Casteel D. A. Peroxy Natural Products. Nat. Prod. Rep. 1999, 16, 55–73. 10.1039/a705725c. [DOI] [PubMed] [Google Scholar]
- Tamez-Fernández J. F.; Melchor-Martínez E. M.; Ibarra-Rivera T. R.; Rivas-Galindo V. M. Plant-derived Endoperoxides: Structure, Occurrence, and Bioactivity. Phytochem. Rev. 2020, 19, 827–864. 10.1007/s11101-020-09687-4. [DOI] [Google Scholar]
- Liu D.; Liu J. Peroxy Natural Products. Nat. Prod. Bioprospect. 2013, 3, 161–206. 10.1007/s13659-013-0042-7. [DOI] [Google Scholar]
- Casteel D. A. Peroxy Natural Products. Nat. Prod. Rep. 1992, 9, 289–312. 10.1039/np9920900289. [DOI] [PubMed] [Google Scholar]
- Dembitsky V. M. Bioactive Peroxides as Potential Therapeutic Agents. Eur. J. Med. Chem. 2008, 43, 223–251. 10.1016/j.ejmech.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Dembitsky V. M. Bioactive Fungal Endoperoxides. Med. Mycol. 2015, 1, 5. 10.21767/2471-8521.100005. [DOI] [Google Scholar]
- Chaturvedi D.; Goswami A.; Saikia P. P.; Barua N. C.; Rao P. G. Artemisinin and Its Derivatives: a Novel Class of Anti-malarial and Anti-cancer Agents. Chem. Soc. Rev. 2010, 39, 435–454. 10.1039/b816679j. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Chang W. C.; Xiao Y.; Liu H. W.; Liu P. Methylerythritol Phosphate Pathway of Isoprenoid Biosynthesis. Annu. Rev. Biochem. 2013, 82, 497–530. 10.1146/annurev-biochem-052010-100934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddon C. J.; Westfall P. J.; Pitera D. J.; Benjamin K.; Fisher K.; McPhee D.; Leavell M. D.; Tai A.; Main A.; Eng D.; Polichuk D. R.; Teoh K. H.; Reed D. W.; Treynor T.; Lenihan J.; Fleck M.; Bajad S.; Dang G.; Dengrove D.; Diola D.; Dorin G.; Ellens K. W.; Fickes S.; Galazzo J.; Gaucher S. P.; Geistlinger T.; Henry R.; Hepp M.; Horning T.; Iqbal T.; Jiang H.; Kizer L.; Lieu B.; Melis D.; Moss N.; Regentin R.; Secrest S.; Tsuruta H.; Vazquez R.; Westblade L. F.; Xu L.; Yu M.; Zhang Y.; Zhao L.; Lievense J.; Covello P. S.; Keasling J. D.; Reiling K. K.; Renninger N. S.; Newman J. D. High-level Semi-Synthetic Production of the Potent Antimalarial Artemisinin. Nature 2013, 496, 528–532. 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- Lévesque F.; Seeberger P. H. Continuous-flow Synthesis of the Anti-malaria Drug Artemisinin. Angew. Chem., Int. Ed. Engl. 2012, 51, 1706–1709. 10.1002/anie.201107446. [DOI] [PubMed] [Google Scholar]
- Johnson M. A.; Croteau R. Biosynthesis of Ascaridole: Iodide Peroxidase-Catalyzed Synthesis of a Monoterpene Endoperoxide in Soluble Extracts of Chenopodium Ambrosioides Fruit. Arch. Biochem. Biophys. 1984, 235, 254–266. 10.1016/0003-9861(84)90274-1. [DOI] [PubMed] [Google Scholar]
- Smith W. L.; DeWitt D. L.; Garavito R. M. Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu. Rev. Biochem. 2000, 69, 145–182. 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Steffan N.; Grundmann A.; Afiyatullov S.; Ruan H.; Li S. M. FtmOx1, a Non-heme Fe(II) and Alpha-Ketoglutarate-Dependent Dioxygenase, Catalyses the Endoperoxide Formation of Verruculogen in Aspergillus fumigatus. Org. Biomol. Chem. 2009, 7, 4082–4087. 10.1039/b908392h. [DOI] [PubMed] [Google Scholar]
- Yan W.; Song H.; Song F.; Guo Y.; Wu C. H.; Her A. S.; Pu Y.; Wang S.; Naowarojna N.; Weitz A.; Hendrich M. P.; Costello C. E.; Zhang L.; Liu P.; Zhang Y. J. Endoperoxide Formation by an Alpha-Ketoglutarate-Dependent Mononuclear Non-haem Iron Enzyme. Nature 2015, 527, 539–543. 10.1038/nature15519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matsuda Y.; Bai T.; Phippen C. B. W.; Nodvig C. S.; Kjaerbolling I.; Vesth T. C.; Andersen M. R.; Mortensen U. H.; Gotfredsen C. H.; Abe I.; Larsen T. O. Novofumigatonin Biosynthesis Involves a Non-Heme Iron-Dependent Endoperoxide Isomerase for Orthoester Formation. Nat. Commun. 2018, 9, 2587–2596. 10.1038/s41467-018-04983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Zhai R.; Ushimaru R.; Matsuda Y.; Abe I. Molecular Insights Into the Endoperoxide Formation by Fe(II)/Alpha-KG-Dependent Oxygenase NvfI. Nat. Commun. 2021, 12, 4417–4427. 10.1038/s41467-021-24685-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Mechanism of Free Radical Oxygenation of Polyunsaturated Fatty Acids by Cyclooxygenases. Chem. Rev. 2003, 103, 2239–2304. 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- van der Donk W. A.; Tsai A. L.; Kulmacz R. J. The Cyclooxygenase Reaction Mechanism. Biochemistry 2002, 41, 15451–15458. 10.1021/bi026938h. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; van der Donk W. A. Protein Radicals in Enzyme Catalysis. Chem. Rev. 1998, 98, 705–762. 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- Hausinger R. P. FeII/Alpha-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- Clifton I. J.; McDonough M. A.; Ehrismann D.; Kershaw N. J.; Granatino N.; Schofield C. J. Structural Studies on 2-Oxoglutarate Oxygenases and Related Double-Stranded Beta-Helix Fold Proteins. J. Inorg. Biochem. 2006, 100, 644–669. 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–350. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol. 2008, 4, 186–193. 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Hoffart L. M.; Krebs C.; Martin Bollinger J. Kinetic Dissection of the Catalytic Mechanism of Taurine/a-Ketoglutarate: Dioxygenase (TauD) from Escherichia coli. Biochemistry 2005, 44, 8138–8147. 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- Grzyska P. K.; Appelman E. H.; Hausinger R. P.; Proshlyakov D. A. Insight into the Mechanism of an Iron Dioxygenase by Resolution of Steps Following the FeIV=O Species. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3982–3987. 10.1073/pnas.0911565107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M.; Chang W. c.; Matthews M. L.; Martinie R. J.; Boal A. K.; Krebs C., Mechanisms of 2-Oxoglutarate-Dependent Oxygenases: The Hydroxylation Paradigm and Beyond. In RSC Metallobiology, 3rd ed.; Hausinger R. P.; Schofield C. J., Eds.; Royal Society of Chemistry, 2015; Vol. 3. [Google Scholar]
- Clifton I. J.; Doan L. X.; Sleeman M. C.; Topf M.; Suzuki H.; Wilmouth R. C.; Schofield C. J. Crystal Structure of Carbapenem Synthase (CarC). J. Biol. Chem. 2003, 278, 20843–20850. 10.1074/jbc.M213054200. [DOI] [PubMed] [Google Scholar]
- Brauer A.; Beck P.; Hintermann L.; Groll M. Structure of the Dioxygenase AsqJ: Mechanistic Insights into a One-Pot Multistep Quinolone Antibiotic Biosynthesis. Angew. Chem., Int. Ed. Engl. 2016, 55, 422–426. 10.1002/anie.201507835. [DOI] [PubMed] [Google Scholar]
- Chen T. Y.; Chen J.; Tang Y.; Zhou J.; Guo Y.; Chang W. C. Pathway from N-alkylglycine to Alkylisonitrile Catalyzed by Iron(II) and 2-Oxoglutarate-Dependent Oxygenases. Angew. Chem., Int. Ed. Engl. 2020, 59, 7367–7371. 10.1002/anie.201914896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. L.; Tang Y.; Yu C. P.; Sanyal D.; Jia X.; Liu X.; Guo Y.; Chang W. C. Mechanistic Investigation of Oxidative Decarboxylation Catalyzed by Two Iron(II)- and 2-Oxoglutarate-Dependent Enzymes. Biochemistry 2018, 57, 1838–1841. 10.1021/acs.biochem.8b00115. [DOI] [PubMed] [Google Scholar]
- Liao H. J.; Li J.; Huang J. L.; Davidson M.; Kurnikov I.; Lin T. S.; Lee J. L.; Kurnikova M.; Guo Y.; Chan N. L.; Chang W. C. Insights into the Desaturation of Cyclopeptin and Its C3 Epimer Catalyzed by a Non-heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chem., Int. Ed. Engl. 2018, 57, 1831–1835. 10.1002/anie.201710567. [DOI] [PubMed] [Google Scholar]
- Li J.; Liao H. J.; Tang Y.; Huang J. L.; Cha L.; Lin T. S.; Lee J. L.; Kurnikov I. V.; Kurnikova M. G.; Chang W. C.; Chan N. L.; Guo Y. Epoxidation Catalyzed by the Nonheme Iron(II)- and 2-Oxoglutarate-Dependent Oxygenase, AsqJ: Mechanistic Elucidation of Oxygen Atom Transfer by a Ferryl Intermediate. J. Am. Chem. Soc. 2020, 142, 6268–6284. 10.1021/jacs.0c00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q.; Liu Y.; Chen L.; Xu M.; Naowarojna N.; Lee N.; Chen L.; Zhu D.; Hong X.; Deng Z.; Liu P.; Zhao C. Biochemical Characterization of a Multifunctional Mononuclear Nonheme Iron Enzyme (PtlD) in Neopentalenoketolactone Biosynthesis. Org. Lett. 2019, 21, 7592–7596. 10.1021/acs.orglett.9b02872. [DOI] [PubMed] [Google Scholar]
- Valegard K.; van Scheltinga A. C. T.; Lloyd M. D.; Hara T.; Ramaswamy S.; Perrakis A.; Thompson A.; Lee H. J.; Baldwin J. E.; Schofield C. J.; Hajdu J.; Andersson I. Structure of a Cephalosporin Synthase. Nature 1998, 394, 805–809. 10.1038/29575. [DOI] [PubMed] [Google Scholar]
- Weichold V.; Milbredt D.; van Pee K. H. Specific Enzymatic Halogenation from the Discovery of Halogenated Enzymes to Their Applications in vitro and in vivo. Angew. Chem., Int. Ed. 2016, 55, 6374–6389. 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- Matsuda Y.; Awakawa T.; Wakimoto T.; Abe I. Spiro-Ring Formation is Catalyzed by a Multifunctional Dioxygenase in Austinol Biosynthesis. J. Am. Chem. Soc. 2013, 135, 10962–10965. 10.1021/ja405518u. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Davis K. M.; Shoda T. K. C.; Blaesi E. J.; Boal A. K.; Krebs C.; Bollinger J. M. An Iron(IV)-Oxo Intermediate Initiating L-Arginine Oxidation but not Ethylene Production by the 2-Oxoglutarate-Dependent Oxygenase, Ethylene-Forming Enzyme. J. Am. Chem. Soc. 2021, 143, 2293–2303. 10.1021/jacs.0c10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J.; Lu J.; Lai W. Mechanistic Insights into a Non-Heme 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Selectivity of Ethylene-Formation Versusl-Arg Hydroxylation. Phys. Chem. Chem. Phys. 2019, 21, 9957–9968. 10.1039/c9cp00794f. [DOI] [PubMed] [Google Scholar]
- Li M.; Martinez S.; Hausinger R. P.; Emerson J. P. Thermodynamics of Iron(II) and Substrate Binding to the Ethylene-Forming Enzyme. Biochemistry 2018, 57, 5696–5705. 10.1021/acs.biochem.8b00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson N.; Persson K. O.; Larsson C.; Norbeck J. Comparative Sequence Analysis and Mutagenesis of Ethylene Forming Enzyme (EFE) 2-Oxoglutarate/Fe(II)-Dependent Dioxygenase Homologs. BMC Biochem. 2014, 15, 22–29. 10.1186/1471-2091-15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez S.; Fellner M.; Herr C. Q.; Ritchie A.; Hu J.; Hausinger R. P. Structures and Mechanisms of the Non-Heme Fe(II)- and 2-Oxoglutarate-Dependent Ethylene-Forming Enzyme: Substrate Binding Creates a Twist. J. Am. Chem. Soc. 2017, 139, 11980–11988. 10.1021/jacs.7b06186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S. S.; Naowarojna N.; Cheng R.; Liu X.; Liu P. Recent Examples of Alpha-Ketoglutarate-Dependent Mononuclear Non-Haem Iron Enzymes in Natural Product Biosyntheses. Nat. Prod. Rep. 2018, 35, 792–837. 10.1039/c7np00067g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aik W.; McDonough M. A.; Thalhammer A.; Chowdhury R.; Schofield C. J. Role of the Jelly-Roll Fold in Substrate Binding by 2-Oxoglutarate Oxygenases. Curr. Opin. Struct. Biol. 2012, 22, 691–700. 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Hangasky J. A.; Taabazuing C. Y.; Valliere M. A.; Knapp M. J. Imposing Function down a (Cupin)-Barrel: Secondary Structure and Metal Stereochemistry in the AlphaKG-Dependent Oxygenases. Metallomics 2013, 5, 287–301. 10.1039/c3mt20153h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss M.; Honda Malca S.; Buller R. Exploring the Biocatalytic Potential of Fe/Alpha-Ketoglutarate-Dependent Halogenases. Chemistry 2020, 26, 7336–7345. 10.1002/chem.201905752. [DOI] [PubMed] [Google Scholar]
- Nakamura H.; Matsuda Y.; Abe I. Unique Chemistry of Non-Heme Iron Enzymes in Fungal Biosynthetic Pathways. Nat. Prod. Rep. 2018, 35, 633–645. 10.1039/c7np00055c. [DOI] [PubMed] [Google Scholar]
- Abe I. Nonheme Iron- and 2-Oxoglutarate-Dependent Dioxygenases in Fungal Meroterpenoid Biosynthesis. Chem. Pharm. Bull. 2020, 68, 823–831. 10.1248/cpb.c20-00360. [DOI] [PubMed] [Google Scholar]
- Kal S.; Que L. Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad that Generate High-Valent Oxoiron Oxidants. J. Biol. Inorg. Chem. 2017, 22, 339–365. 10.1007/s00775-016-1431-2. [DOI] [PubMed] [Google Scholar]
- Huang X.; Groves J. T. Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C-H Activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg M.; Borowski T. Oxoferryl Species in Mononuclear Non-Heme Iron Enzymes: Biosynthesis, Properties and Reactivity from a Theoretical Perspective. Coord. Chem. Rev. 2013, 257, 277–289. 10.1016/j.ccr.2012.03.047. [DOI] [Google Scholar]
- McDonough M. A.; Loenarz C.; Chowdhury R.; Clifton I. J.; Schofield C. J. Structural Studies on Human 2-Oxoglutarate Dependent Oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. 10.1016/j.sbi.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Galonic Fujimori D.; Walsh C. T.; Bollinger J. M. Jr. Non-Heme Fe(IV)-Oxo Intermediates. Acc. Chem. Res. 2007, 40, 484–492. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehntop K. D.; Emerson J. P.; Que L. The 2-His-1-Carboxylate Facial Triad: a Versatile Platform for Dioxygen Activation by Mononuclear Non-heme Iron(II) Enzymes. J. Biol. Inorg. Chem. 2005, 10, 87–93. 10.1007/s00775-005-0624-x. [DOI] [PubMed] [Google Scholar]
- Wu L.; Wang Z.; Cen Y.; Wang B.; Zhou J. Structural Insight into the Catalytic Mechanism of the Endoperoxide Synthase FtmOx1. Angew. Chem., Int. Ed. Engl. 2022, 61, e202112063 10.1002/anie.202112063. [DOI] [PubMed] [Google Scholar]
- O’Brien J. R.; Schuller D. J.; Yang V. S.; Dillard B. D.; Lanzilotta W. N. Substrate-Induced Conformational Changes in Escherichia coli Taurine/a-Ketoglutarate Dioxygenase and Insight into the Oligomeric Structure. Biochemistry 2003, 42, 5547–5554. [DOI] [PubMed] [Google Scholar]
- Dietz R.; Nastainczyk W.; Ruf H. H. Higher oxidation states of prostaglandin H synthase. Rapid Electronic Spectroscopy Detected Two Spectral Intermediates during the Peroxidase Reaction with Prostaglandin G2. Eur. J. Biochem. 1988, 171, 321–328. 10.1111/j.1432-1033.1988.tb13793.x. [DOI] [PubMed] [Google Scholar]
- Tsai A. L.; Kulmacz R. J. Prostaglandin H Synthase: Resolved and Unresolved Mechanistic Issues. Arch. Biochem. Biophys. 2010, 493, 103–124. 10.1016/j.abb.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W.; Song H.; Song F.; Guo Y.; Wu C. H.; Her A. S.; Pu Y.; Wang S.; Naowarojna N.; Weitz A.; Hendrich M. P.; Costello C. E.; Zhang L.; Liu P.; Zhang Y. J. Retraction Note: Endoperoxide Formation by an Alpha-Ketoglutarate-Dependent Mononuclear Non-Haem Iron Enzyme. Nature 2021, 593, 612. 10.1038/s41586-021-03527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser S. P. Trends in Substrate Hydroxylation Reactions by Heme and Nonheme Iron(IV)-Oxo Oxidants Give Correlations between Intrinsic Properties of the Oxidant with Barrier Height. J. Am. Chem. Soc. 2010, 132, 1087–1097. 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]
- Dunham N. P.; Del Rio Pantoja J. M.; Zhang B.; Rajakovich L. J.; Allen B. D.; Krebs C.; Boal A. K.; Bollinger J. M. Jr. Hydrogen Donation but not Abstraction by a Tyrosine (Y68) during Endoperoxide Installation by Verruculogen Synthase (FtmOx1). J. Am. Chem. Soc. 2019, 141, 9964–9979. 10.1021/jacs.9b03567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: the Integration of Data Reduction and Structure Solution - from Diffraction Images to an Initial Model in Minutes. Acta Crystallogr., Sect. D 2006, 62, 859–866. 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- Yaremenko I. A.; Vil V. A.; Demchuk D. V.; Terent’ev A. O. Rearrangements of Organic Peroxides and Related Processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. 10.3762/bjoc.12.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M.; Kodato S.; Hongu M.; Kawate T.; Hino T. Total Synthesis of Fumitremorgin B. Tetrahedron Lett. 1986, 27, 6217–6220. 10.1016/S0040-4039(00)85436-2. [DOI] [Google Scholar]
- Fujimoto H.; Fujimaki T.; Okuyama E.; Yamazaki M. Immunosuppressive Constituents from an Ascomycete, Sordaria gondaensis. Mycotoxins 2000, 50, 93–99. 10.2520/myco1975.50.93. [DOI] [Google Scholar]
- Guengerich F. P.; Okazaki O.; Seto Y.; Macdonald T. L. Radical Cation Intermediates in N-Dealkylation Reactions. Xenobiotica 1995, 25, 689–709. 10.3109/00498259509061886. [DOI] [PubMed] [Google Scholar]
- Wang X.; Su H.; Liu Y. Insights into the Unprecedented Epoxidation Mechanism of Fumitremorgin B Endoperoxidase (FtmOx1) from Aspergillus fumigatus by QM/MM Calculations. Phys. Chem. Chem. Phys. 2017, 19, 7668–7677. 10.1039/C7CP00313G. [DOI] [PubMed] [Google Scholar]
- Milaczewska A.; Borowski T. On the Reaction Mechanism of an Endoperoxide Ring Formation by Fumitremorgin B Endoperoxidase. The Right Arrangement Makes a Difference. Dalton Trans. 2019, 48, 16211–16221. 10.1039/c9dt02581b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.