

SUMMARY
TDP-43 mediates proper Stathmin-2 (STMN2) mRNA splicing, and STMN2 protein is reduced in the spinal cord of most patients with amyotrophic lateral sclerosis (ALS). To test the hypothesis that STMN2 loss contributes to ALS pathogenesis, we generated constitutive and conditional STMN2 knockout mice. Constitutive STMN2 loss results in early-onset sensory and motor neuropathy featuring impaired motor behavior and dramatic distal neuromuscular junction (NMJ) denervation of fast-fatigable motor units, which are selectively vulnerable in ALS, without axon or motoneuron degeneration. Selective excision of STMN2 in motoneurons leads to similar NMJ pathology. STMN2 knockout heterozygous mice, which better model the partial loss of STMN2 protein found in patients with ALS, display a slowly progressive, motor-selective neuropathy with functional deficits and NMJ denervation. Thus, our findings strongly support the hypothesis that STMN2 reduction owing to TDP-43 pathology contributes to ALS pathogenesis.
In brief
STMN2 is regulated by TDP-43, and its levels are decreased in many patients with ALS. Krus et al. demonstrate that loss of mouse Stmn2 is sufficient to induce ALS-relevant pathology, including progressive motor neuropathy with preferential loss of distal neuromuscular junctions of fast-fatigable motor units.
Graphical Abstract
INTRODUCTION
Loss of STMN2 expression is hypothesized to contribute to the pathogenesis of amyotrophic lateral sclerosis (ALS). ALS is a neurodegenerative disease characterized by progressive loss of motor function, respiratory failure, and death 3–4 years after symptom onset (van Es et al., 2017). Almost all ALS cases, both familial and sporadic, have nuclear loss and/or cytoplasmic inclusions of the RNA-binding protein transactive response DNA-binding protein (TDP-43) in motor neurons and other cells (Ling et al., 2013). Two recent landmark studies identified STMN2 mRNA as the most downregulated transcript in human motor neurons upon TDP-43 knockdown or dysfunction and demonstrated that Stathmin-2 (STMN2) protein levels are decreased in human ALS-affected spinal cords (Klim et al., 2019; Melamed et al., 2019). These findings imply that most patients with ALS will have decreased levels of STMN2 protein and raise the question as to whether or not this loss of STMN2 contributes to ALS pathogenesis. While STMN2 transcript is a major target of TDP-43, TDP-43 also regulates many other transcripts, some of which likely contribute to ALS pathogenesis (Ma et al., 2022; Brown et al., 2022). Individually, each key target could play a role in creating the overall phenotype that arises from TDP-43 loss of function. Here we generate constitutive and conditional Stmn2 mouse mutants to test the hypothesis that a decrease in the levels of STMN2 contributes to ALS pathology.
In human neurons, TDP-43 binds to STMN2 pre-mRNAs and represses the inclusion of a cryptic exon that results in a prematurely polyadenylated STMN2 transcript. With the loss of TDP-43, truncated transcript levels increase with a concomitant loss of the normal transcript, thereby decreasing the amount of functional STMN2 protein in the cell (Klim et al., 2019; Melamed et al., 2019). Importantly, the TDP-43-dependent regulation of STMN2 is not observed in mice, because cryptic exons are not conserved between mice and humans (Ling et al., 2015). Thus, mouse models with TDP-43 dysfunction do not feature a concurrent loss of STMN2 and only partially mimic an ALS phenotype. Moreover, Stmn2-mutant mice have not yet been described, so the in vivo consequences of STMN2 loss are unknown.
While an in vivo analysis of STMN2 function is lacking, the molecular and cellular functions of STMN2 are well studied. STMN2 is a nervous system-specific stathmin family member (Anderson and Axel, 1985; Ozon et al., 1997) that binds tubulin dimers to regulate microtubule stability and influences growth cone dynamics to regulate axon outgrowth (Riederer et al., 1997; Di Paolo et al., 1997). In injured neurons, STMN2 is upregulated and localizes in regenerating growth cones (Shin et al., 2014). In addition, STMN2 is an axonal maintenance factor—the loss of STMN2 promotes degeneration of injured sensory axons, while overexpression of stabilized STMN2 delays axon degeneration after injury (Shin et al., 2012). Interestingly, knockdown of the only stathmin ortholog in Drosophila motor neurons leads to neuromuscular junction (NMJ) degeneration and motor axon retraction (Graf et al., 2011), while knockout of STMN1, an STMN2 ortholog, in mice leads to a slowly progressive peripheral neuropathy (Liedtke et al., 2002), a disease of axon loss. Hence, the function of stathmins as axonal/synaptic maintenance factors is evolutionarily conserved. In addition to its postulated role in ALS, the truncated STMN2 transcript is increased in frontotemporal dementia (Prudencio et al., 2020), and STMN2 transcript levels are decreased in Alzheimer’s disease and Parkinson’s disease (Mathys et al., 2019; Wang et al., 2019). Indeed, the viral-mediated knockdown of STMN2 in the mouse substantia nigra results in dopaminergic neuron death and motor deficits (Wang et al., 2019). Thus, the role of stathmin family members in axon and NMJ maintenance is consistent with the model that STMN2 is a functionally relevant target of TDP-43 whose loss contributes to ALS pathology and likely other neurodegenerative disorders.
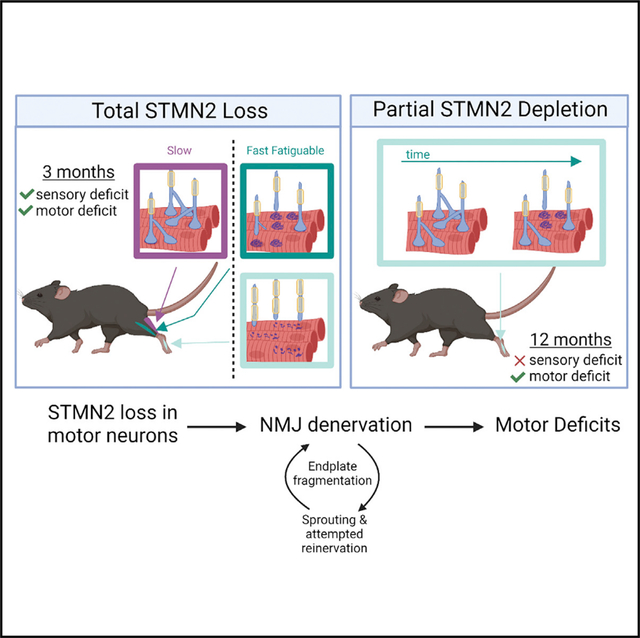
Here we test the hypothesis that in vivo STMN2 depletion results in axon and/or NMJ instability and contributes to ALS pathology. We generated Stmn2 constitutive and conditional knockout mice and find that STMN2 is required for motor and sensory system function. Stmn2 deletion results in a severe motor and sensory neuropathy with behavioral defects, decreased compound muscle action potentials, NMJ denervation, and intraepidermal nerve fiber loss. The motor neuropathy in the Stmn2 KO mouse is most prominent in more distal NMJs, and predominantly affects the fast-fatigable motor units as is seen in ALS (Dengler et al., 1990). Conditional deletion of Stmn2 in choline acetyltransferase (ChAT)-expressing neurons results in motor defects and NMJ degeneration, indicating a cell-autonomous role for STMN2 in motor neurons. The cryptic splicing observed in patients with ALS with TDP-43 pathology results in a partial loss of STMN2, with postmortem ALS patient spinal cords showing low, but variable, levels of STMN2 (Klim et al., 2019). To more closely model the partial loss of STMN2 protein observed in patients with ALS, we examined Stmn2+/− heterozygous mice. These mice exhibit a progressive, distal motor neuropathy with no observed sensory defect. Moreover, loss of this single copy of Stmn2 results in distal NMJ denervation, an early feature of ALS pathology. These findings demonstrate that STMN2 loss is sufficient to replicate some aspects of ALS disease progression and pathology in vivo, and strongly support the hypothesis that the reduction of STMN2 protein levels owing to TDP-43 dysfunction contributes to ALS pathogenesis.
RESULTS
STMN2 promotes microtubule polymerization and axon outgrowth
Previous genetic analyses of STMN2 used overexpression or knockdown models, not genetic deletion. To investigate the effects of STMN2 loss in vivo, we engineered a constitutive knockout (KO) mouse using CRISPR/Cas9-mediated genome editing. The Stmn2 gene contains 5 exons spanning approximately 55 kb. Just over 1.2 kb were deleted in our model including exon 3, which encodes a large part of the tubulin-binding stathmin-like domain of STMN2 and parts of the surrounding introns (Figure 1A). This deletion was identified by PCR and subsequently confirmed by DNA sequencing. Heterozygous Stmn2 mutant mice were then bred to produce Stmn2 KO animals. Loss of STMN2 protein was confirmed by western blot (Figure 1B).
Figure 1. Constitutive Stmn2 KO results in delayed microtubule polymerization and axon outgrowth.
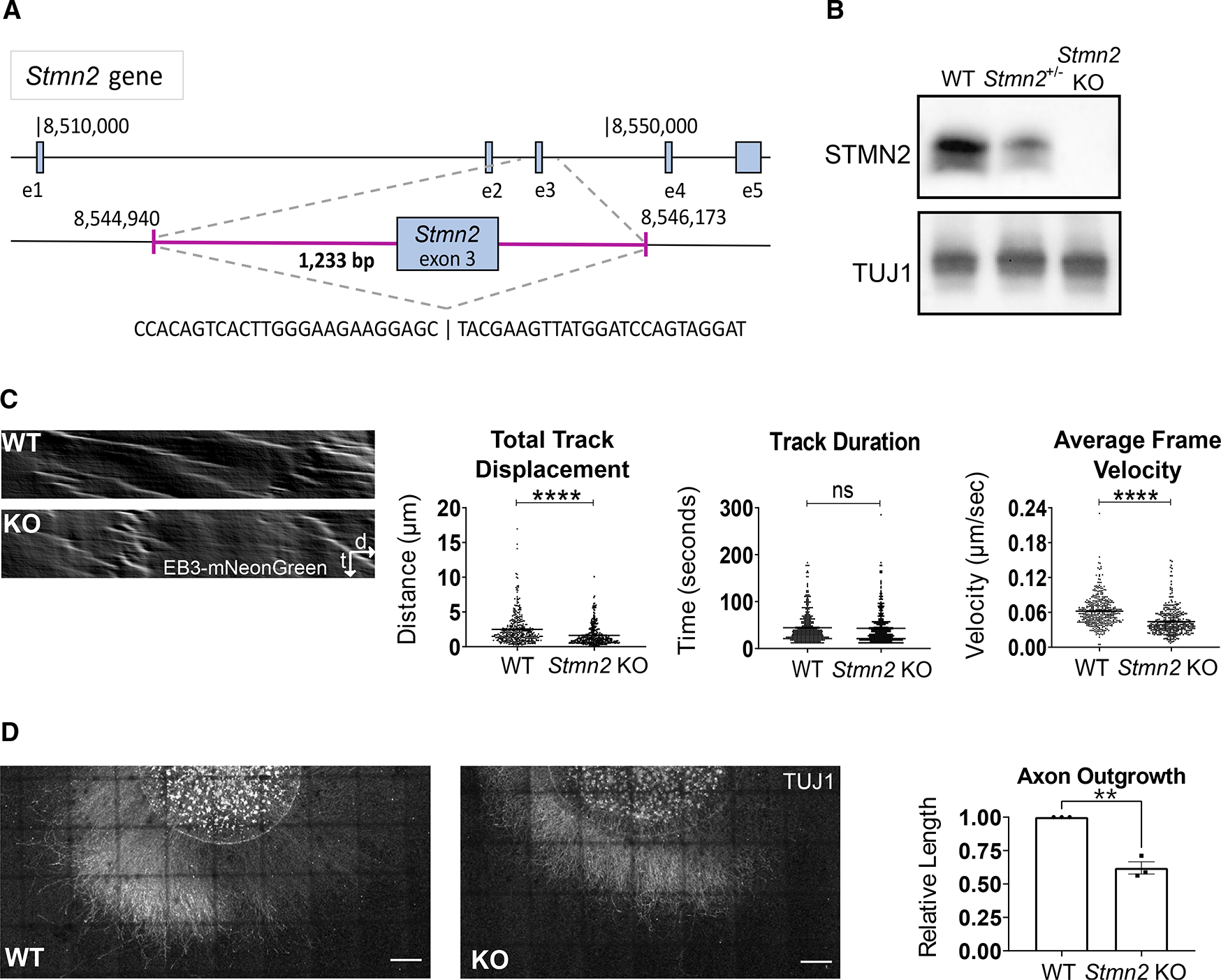
(A) Schematic of Stmn2 gene deletion region. The magenta area represents the deleted region.
(B) STMN2 protein in brain lysates from 3-month-old mice heterozygous and homozygous for Stmn2 deletion allele.
(C) Representative kymographs of EB3-mNeonGreen movement from WT and Stmn2 KO embryonic DRG neurons. Quantified to the right is total track displacement (μm), track duration (s), and average frame velocity (μm/s).
(D) Representative images of spot culture axon length on DIV3. Relative axon length quantified to the right. Scale bar, 500 μm. All data are presented as mean ± SEM. Statistical significance was determined by the Student unpaired t-test. ns, not significant. **p < 0.01, ****p < 0.0001.
To examine STMN2 loss in individual neurons, we first performed in vitro studies. In biochemical assays, STMN2 binds tubulin heterodimers to promote microtubule catastrophe (Riederer et al., 1997). In neuronal cultures, it regulates microtubule morphology in growth cones (Morii et al., 2006). To assess the effects of STMN2 loss on tubulin polymerization dynamics in a cellular context, we used lentivirus to express mNeonGreen-tagged EB3 (Chertkova et al., 2017), a microtubule plus-end binding protein, in wild-type (WT) and Stmn2 KO dorsal root ganglion (DRG) neurons collected from E13.5 embryos. Compared with microtubules in WT neurons, microtubules in Stmn2 KO neurons show a similar track length, but a shorter track displacement, indicating that the rate of tubulin polymerization is slower in the absence of STMN2 (Figure 1C). This demonstrated defect in tubulin polymerization, along with prior STMN2 knockdown studies showing decreased axon outgrowth (Morii et al., 2006), led us to examine axon outgrowth in our Stmn2 KO neurons. We cultured embryonic DRG neurons for 3 days and then measured the length of the axon halo surrounding the cell bodies. We found that axon outgrowth from Stmn2 KO neurons was on average 35% shorter than the outgrowth from WT neurons (Figure 1D). Hence, STMN2 is required for normal microtubule polymerization and axon outgrowth, but is not essential for either process. Having identified molecular and cellular defects in Stmn2 KO neurons, we next explored the in vivo requirement for STMN2.
Stmn2 KO mice display reduced perinatal viability
The Stmn2 KO mice were first assessed by examining whether STMN2 was required for development. To explore viability, we determined the genotypes of progeny from Stmn2+/− heterozygous animals 1 day before birth (E18.5) and at 3 weeks of age. At E18.5, Stmn2 KO embryos comprised approximately 25% of progeny; however, by 3 weeks of age there was a deviation from Mendelian ratios; only 8% were homozygous for the Stmn2 KO allele (Figure 2A). The genotype distribution was similar for male and female progeny (p = 0.09), with both sexes exhibiting some perinatal lethality. While perinatal death is increased, we observed very little premature death after weaning. Indeed, the surviving homozygous Stmn2 KO mice seem to be grossly normal and do not exhibit obvious changes in mobility or paralysis up to 1 year of age. Hence, Stmn2 KO embryos are viable in utero, but have reduced perinatal fitness.
Figure 2. Total STMN2 loss causes perinatal lethality as well as sensory and motor deficits.
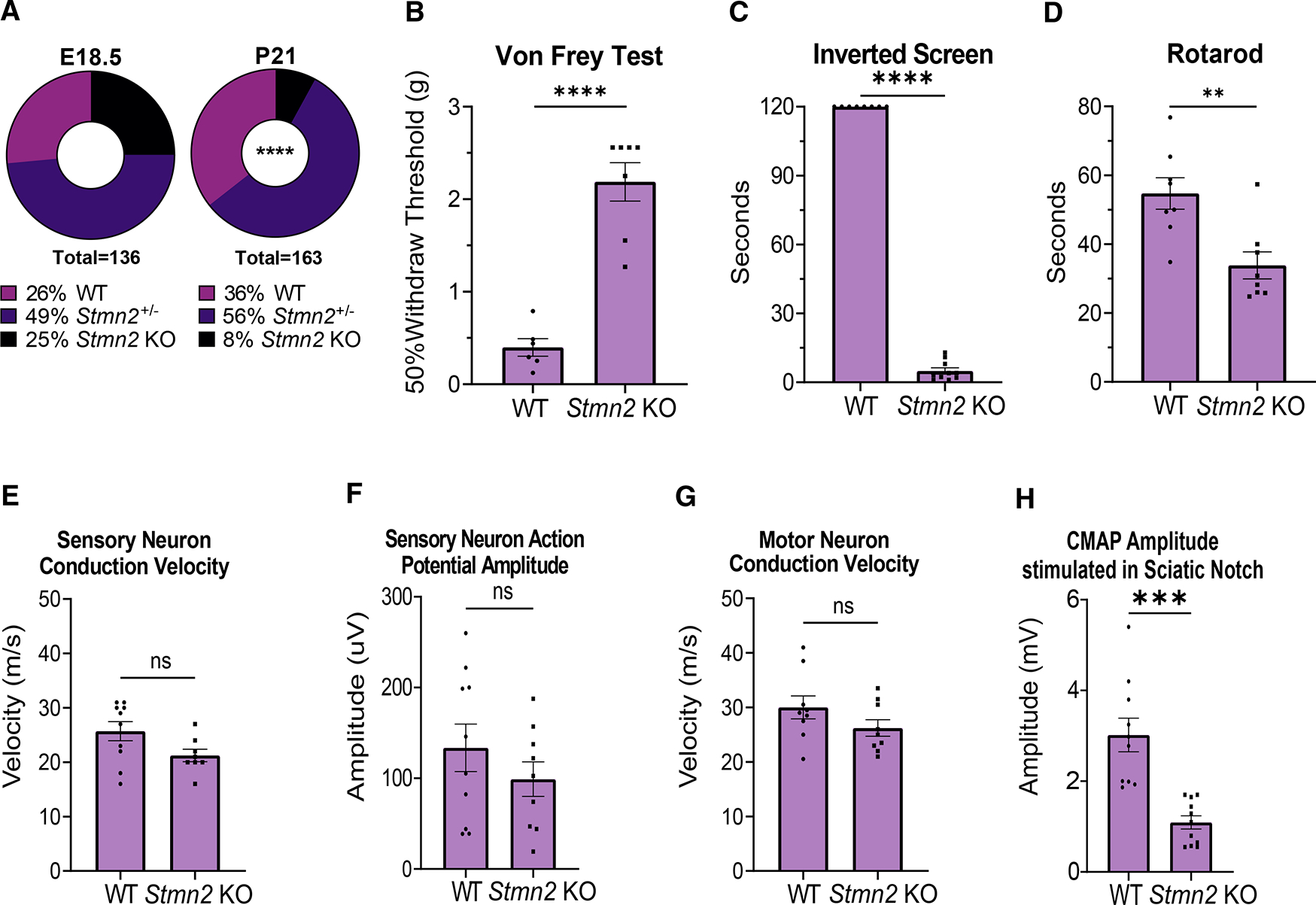
(A) Genotype distribution of E18.5 embryos (n = 136) and P21 mice (n = 163). Statistical significance was determined by the χ2 test for goodness of fit.
(B) Average 50% hind paw withdrawal threshold when force (grams) applied (WT n = 6, KO n = 7).
(C and D) (C) Latency time (s) to fall from an inverted screen (WT n = 8, KO n = 10; max. 120 s), and (D) length of time (s) on an accelerating rotarod before falling (WT n = 8, KO n = 8), for WT and Stmn2 KO mice.
(E and F) (E) Average sensory nerve conduction velocity and (F) Action potential amplitude for WT (n = 10) and Stmn2 KO mice (n = 8,9).
(G and H) (G) Average motor neuron conduction velocity (WT and KO n = 9) and (H) Compound muscle action potential (CMAP) amplitude stimulated in the sciatic notch for WT (n = 9,10) and Stmn2 KO (n = 9,11) mice. All data reported here are from 3-month-old mice. Unless otherwise stated, statistical significance was determined by the Student unpaired t-test. All data are presented as mean ± SEM. ns, not significant. **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figures S1 and S2.
The defects observed in axon outgrowth in vitro led us to hypothesize that Stmn2 KO embryos might show defects in peripheral nerves. We focused on the phrenic nerve, as a loss of diaphragm innervation would be incompatible with life. Analysis of whole mount preparations stained for the axonal protein neurofilament revealed that innervation of the diaphragm is grossly normal in the Stmn2 KO embryos. To assess NMJ development, we imaged post-synaptic acetylcholine receptors (AChR), axons, and synaptic vesicles. We found diaphragm innervation to be very similar in WT and Stmn2 KO embryos, with presynaptic terminals well apposed to post-synaptic AChR clusters, with no significant differences in the numbers of primary and secondary neurites, length of primary branches, or number and size of AChR clusters. Stmn2 KO animals do have a mild decrease in secondary neurite length compared with WT animals (Figures S1A and S1B). While other developmental defects may occur in Stmn2 KO animals that die soon after birth, we conclude that peripheral nerve outgrowth and innervation of the diaphragm are grossly normal and so are not the cause of perinatal lethality.
STMN2 has three close protein orthologs, STMN1, STMN3, and STMN4. We investigated whether any of these proteins were upregulated in surviving 3-month-old Stmn2 KO mice to explain why some mice survive to adulthood. We performed a western blot analysis of brain tissue from WT, Stmn2 KO, and Stmn2+/− mice, but did not find significant differences in these orthologs in Stmn2 mutants (Figure S2). Hence, we have no evidence that ortholog compensation explains the viability of the surviving Stmn2 KO animals.
Stmn2 KO mice exhibit distal motor and sensory neuropathy
If the loss of STMN2 contributes to ALS pathology, then we would predict motor defects in the adult Stmn2 KO animals. To explore the requirement for STMN2 in the adult nervous system, we assayed sensory and motor behavior in 3-month-old Stmn2 KO mice. Although the mice seem to be healthy with grossly normal behavior, we found they have profoundly impaired mechanosensory perception (assessed by the Von Frey test) and grip strength (assessed by the inverted screen test) (Figures 2B and 2C). The mutant mice also display decreased motor coordination on the rotarod, consistent with prior findings using viral-mediated Stmn2 knockdown in the mouse midbrain (Wang et al., 2019) (Figure 2D). We next performed electrophysiological assays of motor and sensory fibers, which revealed normal conduction velocities in both sensory and motor nerves in Stmn2 KO mice, suggesting normal axon myelination (Figures 2E and 2G). Similarly, the amplitude of the sensory nerve action potential measured in the tail is normal in Stmn2 KO mice, indicating that axon loss is minimal in sensory nerves. In contrast, there is a large reduction in the compound motor action potential (CMAP) of Stmn2 KO mice compared with control animals after stimulation at the sciatic notch and recording at the foot (Figures 2F and 2H). Defects in CMAP likely reflect either axon loss or NMJ denervation, consistent with the motor deficits in these mice.
To determine if peripheral axons are lost, we counted myelinated axons in nerve sections. The sciatic, femoral, and sural nerves from Stmn2 KO mice all have a normal axon density at 3 and 12 months of age (Figures 3A and S3A). These represent mixed (sciatic), primarily motor (femoral), and primarily sensory (sural) nerves, indicating that constitutive STMN2 deletion does not cause axon degeneration within the nerve of either fiber type. Consistent with the preservation of motor axons, we observed no decrease in motor neuron number in the lumbar spinal cord of Stmn2 KO animals, indicating that STMN2 loss does not cause motor neuron death (Figure S3B).
Figure 3. STMN2 deletion results in IENF loss and distal NMJ denervation.
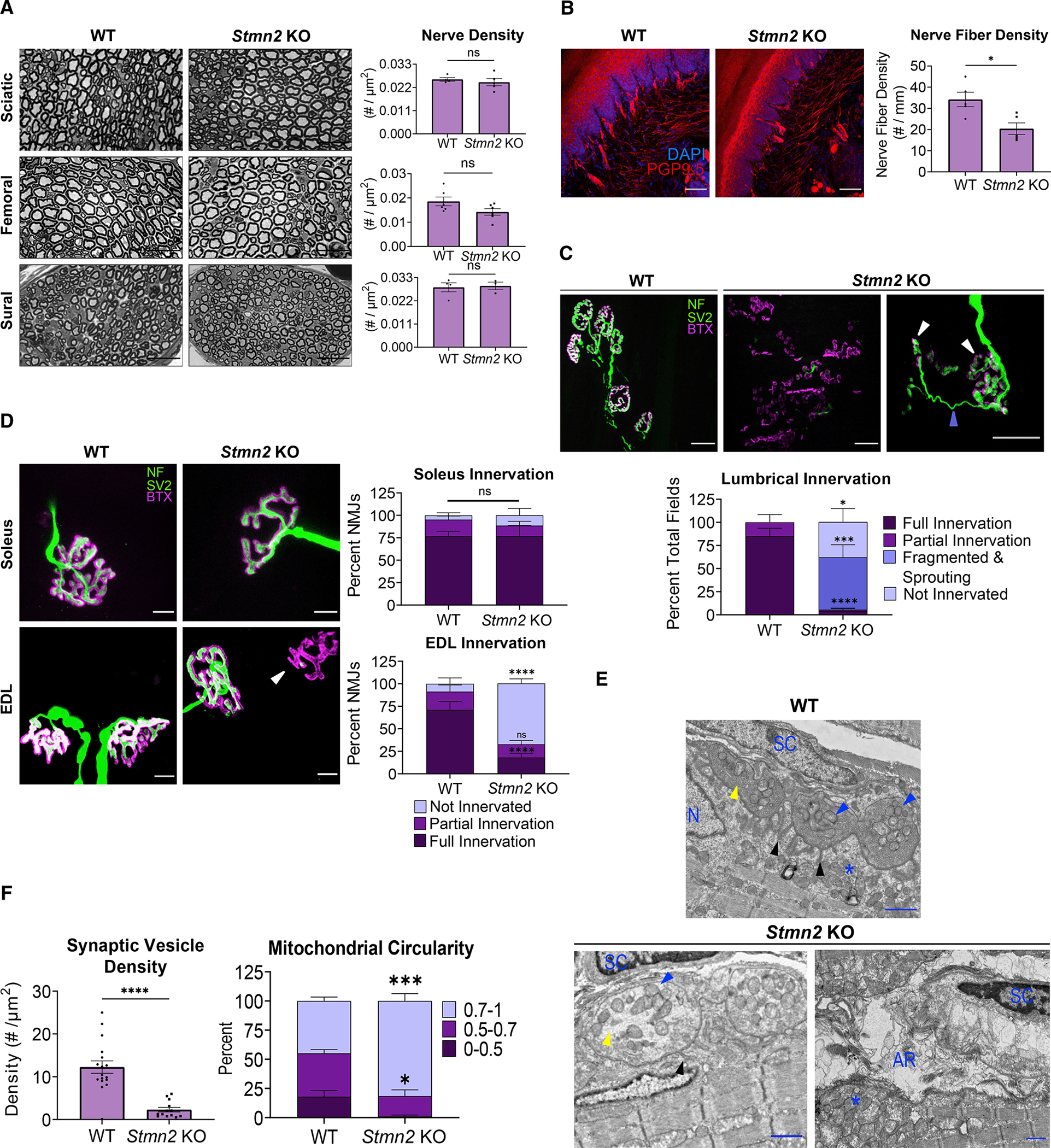
(A) Representative images of sciatic, femoral, and sural nerves in 3-month-old WT (n = 4, 6, and 4, respectively) and Stmn2 KO (n = 5, 6, and 3, respectively) animals with quantification of nerve density (#/mm2) shown to the right. Scale bar, 25 μm. Statistical significance determined by the Student unpaired t-test.
(B) IENF density of footpad skin of 3-month-old WT (n = 5) and Stmn2 KO (n = 5) mice. Nerve fiber density quantified as the number of nerves per millimeter of basement membrane. Scale bar, 50 μm. Statistical significance determined by the Student unpaired t-test.
(C) Representative images from lumbrical muscles of 3-month-old WT and Stmn2 KO mice. NMJs visualized by neurofilament/SV2 (green) and bungarotoxin (magenta). The white arrow shows fragmented AChR clusters, and the periwinkle arrow indicates sprout from an adjacent NMJ. Quantification is based on the innervation of a region of AChR clusters contained within a 20× field. Scale bar, 25 μm. (WT animals n = 7, KO n = 10).
(D) Representative images from soleus and EDL muscles from 3-month-old WT (n = 3–4) and Stmn2 KO (n = 4) mice. Scale bar, 10 μm. Quantification of percent NMJs of each innervation status on the right. Arrow highlights denervated endplate.
(E) Representative electron micrographs of NMJs from 3-month-old WT and Stmn2 KO lumbricals. Scale bar, 1 μm. Black arrow = junctional folds, blue arrow = presynaptic mitochondria, yellow arrow = synaptic vesicles (or lack thereof). *Post-synaptic mitochondrial clustering. AR, axon remnants; N, myocyte nuclei; SC, Schwann cell.
(F) Quantification of synaptic vesicle density in the presynapse (#/μm2) (WT n = 17, KO n = 13 synapses). Statistical significance determined by the unpaired Student’s t test. Mitochondrial Circularity quantified on the right (mitochondria of WT and KO n = 3 synapses). “1” represents a perfect circle. Unless otherwise stated, statistical significance was determined using two-way ANOVA with Sidak’s multiple comparisons test. All data are presented as mean ± SEM. ns, not significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S3.
The lack of axonal loss coupled with the robust behavioral abnormalities in Stmn2 KO mice prompted us to examine the most distal units of both the motor and sensory systems. We first analyzed the intraepidermal nerve fiber (IENF) density in the footpad using PGP9.5 immunostaining and counted the fibers that crossed the basement membrane (Ebenezer et al., 2007). In Stmn2 KO mice, IENF density (number per millimeter of dermis) in the mouse footpad is significantly decreased (Figure 3B). Thus, we can conclude that STMN2 loss results in a distal sensory neuropathy affecting small unmyelinated nerve fibers in the footpad skin.
With predominantly motor deficits in ALS, we were most interested in evaluating the motor synapses, which have been implicated in early ALS pathology (Fischer et al., 2004; Dadon-Nachum et al., 2011). We examined NMJs from lumbrical muscles in the hind feet. In samples from WT mice, we observe clustered bungarotoxin-labelled AChR forming characteristic post-synaptic endplates that are consistently co-localized with SV2-labelled presynapses. Strikingly, in the Stmn2 KO mouse, the NMJ is severely disorganized with little innervation of AChRs and substantial endplate fragmentation. The individual AChR clusters were so aberrant that we could not quantify percent innervation because it was not possible to determine the relationship between the residual AChR clusters and presynaptic axons and terminals. We uniformly observe that AChR clusters seem to be fragmented, with many small clusters that do not form the characteristic pretzel morphology of WT AChR clusters. When assessing fields of these AChR clusters, we would either observe (A) a complete absence of any presynaptic elements, (B) occasional motor axons in the vicinity of some AChR clusters, or (C) apparently innervated endplate fragments with a dramatic presynaptic sprouting phenotype. Such sprouting is diagnostic of denervation of the endplate and subsequent reinnervation by regenerating fibers from adjacent NMJs (Brown and Ironton, 1978). Hence, the endplate fragmentation and presynaptic sprouting observed in Stmn2 KO animals is consistent with presynaptic degeneration (Figure 3C).
In patients with ALS, there is preferential degeneration of fast-fatigable (FF) motor units (Dengler et al., 1990). Lumbrical muscles are primarily composed of FF motor units; thus, we investigated whether other muscles primarily innervated by FF motoneurons are also susceptible to denervation in Stmn2 KO mice. The extensor digitorum longus (EDL) is a fast twitch muscle located on the lateral side of the lower leg. The soleus muscle, which primarily consists of slow twitch muscle fibers, is located on the posterior portion of the lower leg at roughly the same distal position as the EDL and is relatively spared in many mouse models of ALS (Frey et al., 2000; Sharma et al., 2016; Spiller et al., 2016; Ebstein et al., 2019; Korobeynikov et al., 2022). Neither of these muscles showed endplate fragmentation, so we were able to quantify individual NMJs. For blinded quantification, we defined fully innervated NMJs as those with complete apposition of AChRs and synaptic vesicles, partially innervated as those with some presynapse apposition but with some exposed AChRs as well, and un-innervated as those with endplates but no presynapse present. Examining these muscles, we found denervation at the NMJs of the Stmn2 KO EDL muscle, with more than one-half of all post-synaptic AChR clusters uninnervated. In contrast, denervation is not observed in the soleus muscle (Figure 3D). While there is dramatic denervation of the EDL, the pathology is less severe than the endplate fragmentation present in the more distal lumbrical muscles. Taken together, we conclude that the absence of STMN2 results in a motor neuropathy with NMJ defects that preferentially affect distal, FF motor units.
With such a striking phenotype in lumbrical muscles, we next performed transmission electron microscopy (TEM) to explore the ultrastructural basis of the NMJ defects in 3-month-old Stmn2 KO animals. In WT animals, NMJs are abundant, with clear apposition of presynaptic terminals opposite post-synaptic endings with characteristic junctional folds. These WT presynaptic terminals are densely occupied by synaptic vesicles and mitochondria, the post-synapse exhibits mitochondrial clustering near the edge of the myocyte and synapse, and Schwann cells are present nearby (Figure 3E). The micrographs of NMJs from Stmn2 KO mice are consistent with the observations from the immunofluorescence images—it is very difficult to identify NMJs on these muscles. Occasionally, putative remnants of post-synaptic specializations are observed, with myocyte nuclei at the edge of muscle fibers and a cluster of mitochondria nearby; however, there is rarely a recognizable presynaptic terminal. Instead, there are only remnants of an axon terminal with an occasional Schwann cell present. Those few NMJs that are still intact exhibit more circular mitochondria and synaptic vesicle depletion in the presynaptic terminal. In these cases, Schwann cells are present near the NMJ and the post-synapse seems to be more organized with some mitochondrial clustering at the edge of the myocyte (Figures 3E and 3F). These ultrastructural findings are consistent with the behavioral, electrophysiological, and gross anatomical defects described above, all highlighting that STMN2 is required for maintenance of distal motor synapses.
Stmn2 KO mouse motor phenotype is not due to SARM1 activation
We previously demonstrated that STMN2 is an axonal maintenance factor whose overexpression delays axon degeneration following injury (Shin et al., 2012). Furthermore, STMN2 co-localizes with the potent axonal maintenance factor NMNAT2 in transport vesicles, and it is co-regulated with NMNAT2 by MAPK stress signaling and, when overexpressed with NMNAT2, leads to synergistic axon protection (Summers et al., 2018, 2020; Walker et al., 2017). Importantly, NMNAT2 inhibits SARM1, the central executioner of axon degeneration (Osterloh et al., 2012; Gilley et al., 2015; Figley et al., 2021). We therefore hypothesized that STMN2 regulates NMNAT2 levels and that STMN2 loss causes the observed neurodegenerative phenotypes by decreasing axonal NMNAT2 and promoting SARM1 activation.
To obtain large enough cell numbers for the biochemical and metabolic analysis necessary to test our hypothesis, we used CRISPR to knockdown Stmn2 in cultured DRG neurons. We developed highly effective guide RNAs against Stmn2 and used lentiviruses to express them in neurons from Cas9 transgenic mice. Contrary to our hypothesis, CRISPR/Cas9 KD of Stmn2 in cultured neurons does not change axonal NMNAT2 levels as measured by western blot (Figures 4A and 4B). Levels of cADPR, a specific biomarker of SARM1 activity (Essuman et al., 2017; Sasaki et al., 2020), are not significantly different in axonal lysates in neurons expressing STMN2 vs. scrambled control gRNAs (Figure 4C), demonstrating that loss of STMN2 does not activate SARM1 in vitro.
Figure 4. Phenotypes associated with STMN2 loss are not mediated by SARM1 activation.
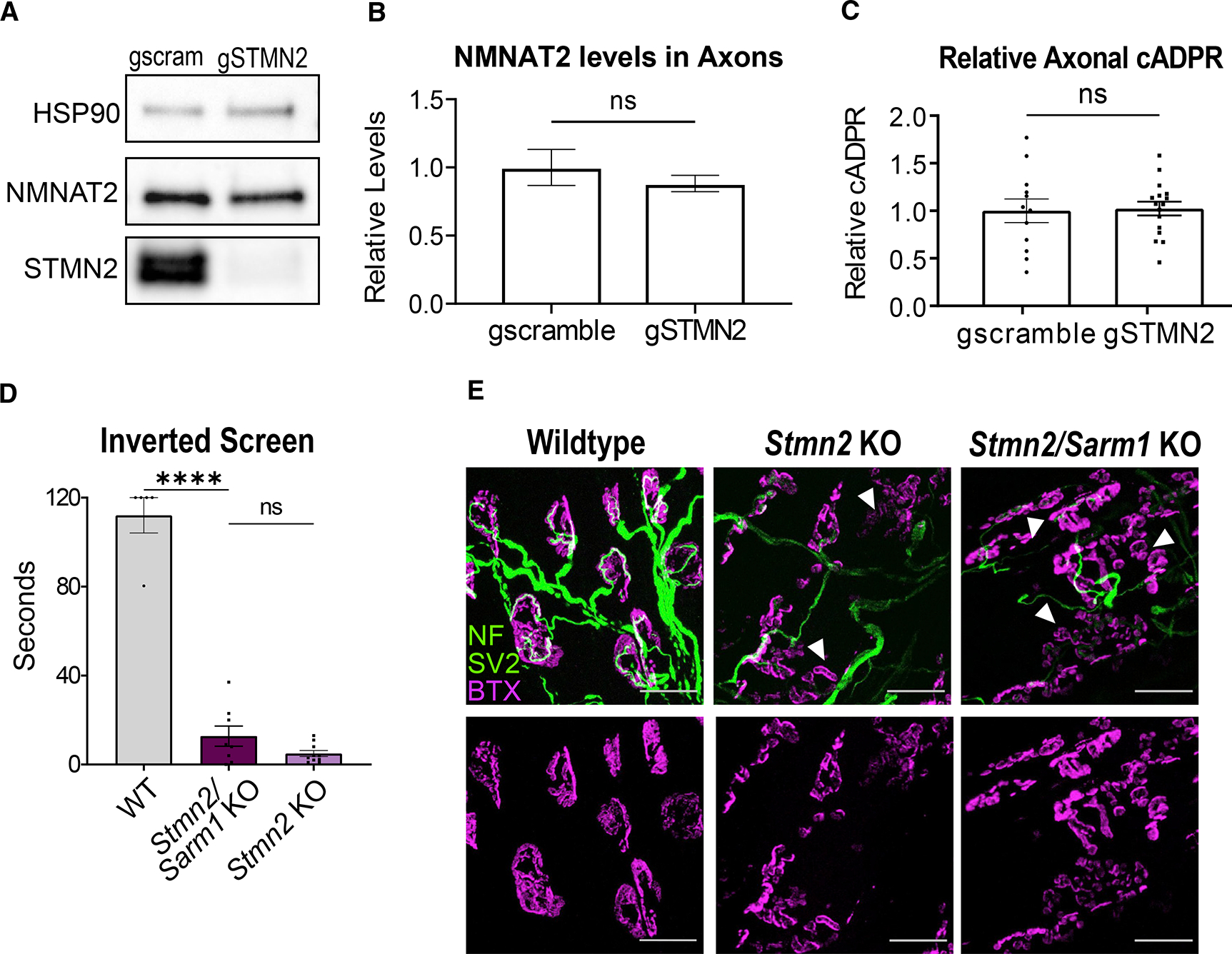
(A) Representative western blots of NMNAT2 and STMN2 using axonal lysates from neurons derived from Cas9 transgenic mice infected with lentivirus expressing Stmn2 or scrambled control gRNAs.
(B) Relative NMNAT2 protein band intensity (n = 4 experiments).
(C) Average relative cADPR levels from axonal lysates from neurons derived from Cas9 transgenic mice infected with lentivirus expressing Stmn2 (n = 16) or scrambled control (n = 12) gRNAs. Statistical significance determined by the Student unpaired t-test.
(D) Latency time to fall from an inverted screen (max. 120s) for WT (n = 5), Stmn2/Sarm1 KO (n = 8), and Stmn2 KO (n = 10, from Figure 2) animals. Statistical significance determined by one-way ANOVA with Tukey’s multiple comparison test.
(E) Representative images of lumbrical NMJs from WT, Stmn2/Sarm1 KO, and Stmn2 KO mice. NMJs visualized by neurofilament/SV2 (green) and bungarotoxin (magenta). Arrows point to fragmented and denervated AChR clusters. Scale bar, 25 μm. All data are presented as mean ± SEM. ns, not significant. ****p < 0.0001.
To test the contribution of SARM1 to STMN2 phenotypes in vivo, we generated mice doubly mutant for Sarm1 and Stmn2 KO and assessed their motor phenotypes. At 3 months of age, Stmn2/Sarm1 KO animals display the same severe motor function impairment as Stmn2 KO mice (Figure 4D). Moreover, presynaptic and post-synaptic elements of lumbrical NMJs from Stmn2/Sarm1 KO animals were indistinguishable from those of Stmn2 KO mice, including endplate fragmentation and many regions of partial innervation (Figure 4E). Therefore, we conclude that SARM1 is not required for the motor phenotype of Stmn2 KO mice. Moreover, these data suggest that STMN2 does not regulate the activity of SARM1, but instead promotes axonal maintenance via a parallel pathway.
NMJ denervation and fragmentation is due to STMN2 loss in motor neurons
In patients with ALS, TDP-43 dysfunction leads to the loss of STMN2 protein in motor neurons, and STMN2 constitutive KO mice have motor defects and NMJ degeneration. This suggests that STMN2 is required in the motor neuron. To test the role of STMN2 in motor neurons, we selectively deleted Stmn2 by crossing a floxed Stmn2 allele (an independent Stmn2 allele; see STAR Methods) (Figures 5A and S4A) with ChAT-Cre, which expresses Cre-recombinase in motor neurons. Excision is highly efficient, as staining in the spinal cord reveals a clear reduction of STMN2+ motor neurons of the Cre expressing mice (Figures S4C and 4D). The deleterious effects of STMN2 loss in cholinergic neurons is evident as there is a significant deviation from Mendelian ratios (p = 0.019) owing to loss of the ChAT-Cre+/Stmn2f/f progeny (Figure S4B). To determine if STMN2 loss in motor neurons induces motor neuropathy, we tested the mice on the inverted screen. Indeed, the mice positive for ChAT-cre show significant defects in hangtime (Figure 5B). We next examined the effects of motor neuron-specific STMN2 loss on the structure of the NMJ. We stained lumbrical muscles for presynaptic and post-synaptic elements in ChAT-Cre+/Stmn2f/f mice and found that the NMJs closely resemble those in Stmn2 KO mice where most lumbrical muscle fibers are partially denervated with highly fragmented post-synaptic endplates (Figure 5C). Hence, STMN2 deletion in motor neurons recapitulates the NMJ phenotype found in constitutive Stmn2 KO mice. These data do not exclude the potential involvement of other cell types, but, importantly, they demonstrate that there is a cell-autonomous requirement for STMN2 in motoneurons and that the loss of STMN2 in motoneurons is sufficient to cause a distal motor neuropathy.
Figure 5. Stmn2 KO in motor neurons causes motor pathology.
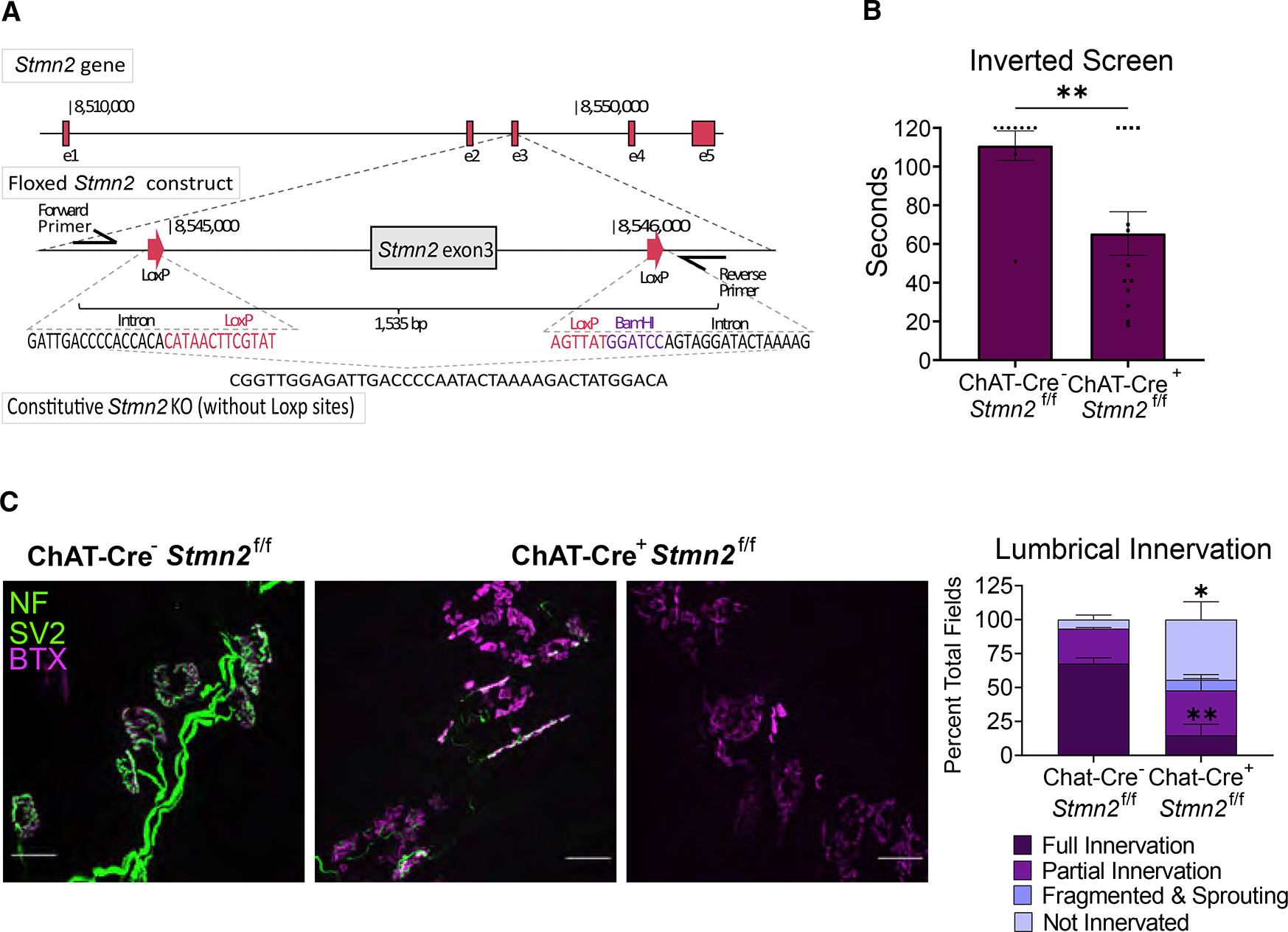
(A) Schematic of floxed Stmn2 allele.
(B) Latency time to fall from an inverted screen (max. 120 s) for ChAT-Cre−/Stmn2f/f (n = 9) and ChAT-Cre+/Stmn2f/f (n = 13) mice at 3 months of age. Statistical significance was determined by the Student unpaired t-test.
(C) Representative images of NMJs from 3-month-old ChAT-Cre−/Stmn2f/f (n = 3) and ChAT-Cre+/Stmn2f/f (n = 6) lumbrical muscles. NMJs were visualized by neurofilament/SV2 (green) and bungarotoxin (magenta) staining. Scale bar, 25 μm. Quantification is to the right. All data are presented as mean ± SEM. *p < 0.05, **p < 0.01. See also Figure S4.
Stmn2+/− heterozygous mice exhibit a progressive motor neuropathy
In both human neurons and patients with TDP-43 dysfunction, there is a decrease rather than complete absence of STMN2 (Klim et al., 2019; Melamed et al., 2019). Thus, to better model the partial loss of STMN2 that occurs in most cases of ALS, we studied the Stmn2+/− heterozygous mice that show an approximately 50% decrease in STMN2 protein (Figure 1B). Stmn2+/− heterozygous mice display normal motor function as young adults, but develop a slowly progressive motor weakness by 1 year of age (Figure 6A). In contrast, 1-year-old Stmn2+/− heterozygous mice behave normally in the Von Frey test (Figure 6B) and rotarod (Figure 6C). Hence, the partial loss of STMN2 protein results in a progressive and motor-selective behavioral defect.
Figure 6. Partial STMN2 depletion results in progressive, distal motor neuropathy.
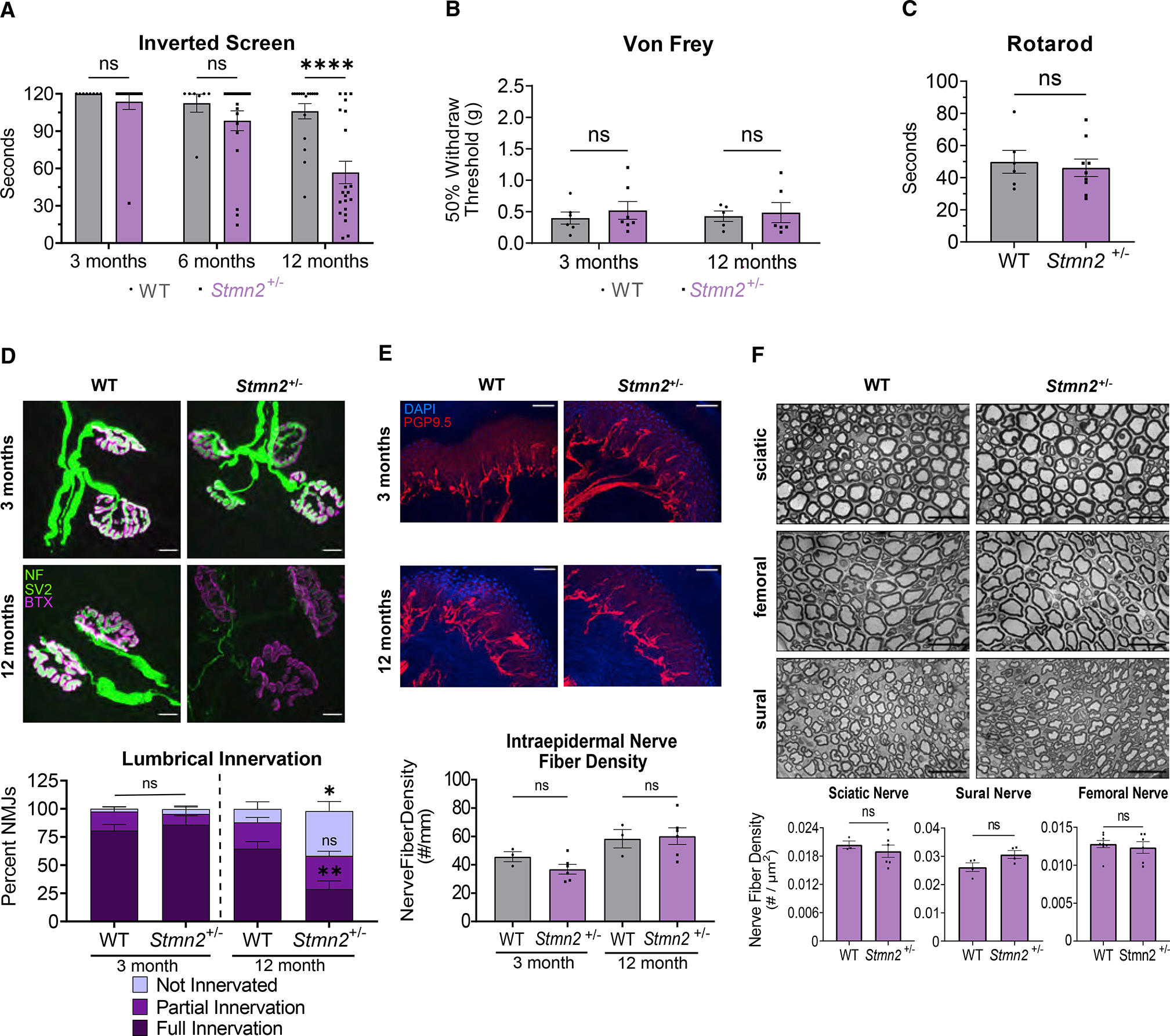
(A) Latency time to fall from an inverted screen (max. 120 s) for WT and Stmn2+/− mice at 3 (n = 8 and 14), 6 (n = 7 and 20), and 12 (n = 17 and 21) months of age.
(B) Average 50% hindpaw withdrawal threshold when force (grams) applied to 3- and 12-month-old WT (n = 6 and 5) and Stmn2+/− (n = 7 and 6) mice.
(C) Time to fall (s) from an accelerating rod for 12-month-old WT and Stmn2+/− mice. Statistical significance was determined by the Student unpaired t-test.
(D) Lumbrical NMJs visualized by neurofilament/SV2 (green) and bungarotoxin (magenta) staining. Scale bar, 10 μm. Quantification is below. Three months, WT and KO n = 4; 12 months, WT n = 5, KO n = 8.
(E) IENF labelled with anti-PGP9.5 and DAPI. Scale bar, 50 μm. (WT n = 3, KO n = 6).
(F) Representative images of sciatic (WT n = 3, KO n = 6), femoral (WT n = 8, KO n = 6), and sural nerves (WT and KO n = 4) in 12-month-old WT and Stmn2+/− animals with quantification of nerve density (#/μm2) shown underneath. Scale bar, 25 μm. Statistical significance determined by the Student unpaired t-test. Unless otherwise noted, statistical significance was determined by two-way ANOVA with Sidak’s multiple comparisons test. All data are presented as mean ± SEM. ns, not significant. *p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S5.
We next investigated the pathological basis for the motor dysfunction in 1-year-old Stmn2+/− heterozygous mice. We focused on innervation of the lumbrical muscles, which were the most dramatically affected in Stmn2 KO mice. Consistent with the absence of a motor phenotype at 3 months of age, staining Stmn2+/− heterozygous and littermate control lumbrical muscle NMJs demonstrates post-synaptic AChR clusters with the classic pretzel morphology and well-apposed presynaptic terminals. Hence, NMJ development and maintenance appear normal in young adults. At 1 year of age, post-synaptic AChR clustering still seems to be very similar to WT at the lumbrical muscle NMJs of Stmn2+/− heterozygous mice. Excitingly, however, the 1-year-old Stmn2+/− heterozygous mice exhibit denervation of lumbrical NMJs with a nearly four-fold increase in the fraction of fully denervated post-synaptic AChR clusters (Figure 6D). Denervation occurs in roughly contiguous regions that are adjacent to well innervated portions of the same muscle (Figure S5A), likely reflecting the degeneration of an axon branch. This phenotype is less severe than in the full Stmn2 KO as the structure of the post-synapse is maintained, likely reflecting the ability of lower levels of STMN2 protein to provide some synaptic maintenance function. Consistent with this finding, we observe no obvious synaptic denervation in the more proximal EDL muscle of 12-month-old mice (Figure S5B), a muscle that also shows less dramatic denervation in the constitutive Stmn2 KO. Constitutive Stmn2 KO leads to loss of IENFs, so we tested whether this phenotype was also induced with partial loss of STMN2. We examined the footpads of control and Stmn2+/− mice and observed no difference in IENF density, demonstrating that motor endings are selectively vulnerable to a decrease in STMN2 levels (Figure 6E). Finally, we observe no difference in the number of axons in the sural, sciatic, or femoral nerves of Stmn2+/− heterozygous mice, consistent with findings in the full Stmn2 KO mice (Figure 6F). Taken together, these findings demonstrate that a decease in STMN2 protein levels leads to a progressive, distal-predominant motor neuropathy with NMJ denervation. These data strongly support the hypothesis that a decrease in STMN2 protein levels contributes to ALS pathology and suggests that the restoration of normal STMN2 protein levels in patients with ALS with TDP-43 pathology could promote synaptic maintenance and motor function.
DISCUSSION
TDP-43 pathology occurs in almost all patients with ALS, inspiring efforts to understand the consequences of TDP-43 dysfunction (Prasad et al., 2019). Two recent landmark studies identified STMN2 as the most dysregulated transcript in TDP-43-deficient human motor neurons. They demonstrated that TDP-43 represses the inclusion of a cryptic exon in STMN2 mRNA and found that STMN2 protein levels are decreased in most spinal cords of patients with ALS. These findings led to the compelling hypothesis that the loss of STMN2 is an important contributor to ALS pathology (Melamed et al., 2019; Klim et al., 2019). Here we test this hypothesis by generating and analyzing constitutive and conditional Stmn2 KO mice. We find that constitutive STMN2 loss induces an early motor and sensory neuropathy with distal NMJ denervation and concomitant motor defects. FF motor synapses, which are preferentially susceptible in ALS (Dengler et al., 1990), are also preferentially lost in these mice. STMN2 is required in motor neurons for synaptic maintenance, as selective excision of Stmn2 in ChAT-expressing neurons triggers motor neuropathy and NMJ degeneration. To better model the partial loss of STMN2 protein that occurs in ALS, we examined Stmn2 heterozygous mice. Excitingly, this moderate reduction in Stmn2 expression is sufficient to cause a slowly progressive, motor-selective neuropathy with NMJ denervation. These findings strongly support the hypothesis that decreased STMN2 levels contribute to ALS pathology and raise the hope that increasing the levels of STMN2 proteins in motor neurons will provide therapeutic benefit for patients with ALS.
STMN2 loss in vivo recapitulates aspects of ALS neuromuscular pathology
ALS is a complex neurodegenerative disease with progressive loss of both upper and lower motor neurons. Symptoms often begin with distal motor weakness and neuromuscular degeneration, with a gradual progression to more proximal muscles (van Es et al., 2017). The Stmn2 KO mouse displays some aspects of this ALS pathology. First, STMN2 loss leads to distal weakness, as these mice have almost no capacity to hang on to the inverted screen, and yet have grossly normal gait reflecting the function of more proximal muscles. Second, Stmn2 KO mice have distal-predominant NMJ loss. In the hind paw lumbrical muscles, we observe dramatic denervation with AChR cluster fragmentation. The more proximal EDL muscle shows less severe denervation, with denervated post-synaptic endplates retaining their classic pretzel-shaped morphology. Third, the Stmn2 KO mice show selective loss of the FF motor units, which generally innervate fast twitch muscles. The EDL, a predominantly fast twitch muscle, displays extensive denervation, while the slow-twitch soleus muscle shows no denervation. The selective vulnerability of FF motor units is seen both in patients with ALS (Dengler et al., 1990) and in mouse ALS models carrying pathogenic mutations in FUS (Sharma et al., 2016; Korobeynikov et al., 2022), SOD1 (Frey et al., 2000), and TDP-43 (Spiller et al., 2016; Ebstein et al., 2019). Finally, the Stmn2 KO mice display sensory defects. While ALS is, of course, a predominantly motor disease, patients with ALS can have sensory abnormalities as well. These symptoms can be accompanied by the loss of IENFs (Riancho et al., 2021), consistent with the deficits we observe in Stmn2 KO mice. While Stmn2 deletion recapitulates important hallmarks of ALS, the mouse model does not exhibit axon loss or motor neuron death by 1 year of age.
An analysis of the Stmn2 heterozygote revealed two additional aspects of the STMN2 phenotype that are consistent with ALS pathology. First, this more modest decrease in STMN2 protein results in a progressive motor neuropathy. As young adults, these mice perform normally on the inverted screen test, but by 1 year of age show clear weakness. Similarly, young adult Stmn2 heterozygous mice have fully innervated NMJs at their distal lumbrical muscles, yet have lost many of these synapses by 1 year of age. Second, Stmn2 heterozygous mice reveal a motor selective neuropathy. In ALS, the motor system is preferentially affected. The Stmn2 heterozygous mice show only motor deficits, with no loss of IENFs or sensory abnormalities. Taken together, the phenotypes of the heterozygous and homozygous Stmn2 mutants are consistent, demonstrating that STMN2 is required for the maintenance of distal NMJs and imply that the dose of STMN2 is important, with the homozygote displaying an earlier and more severe disorder.
ALS is a highly heterogenous disease despite the nearly uniform presence of TDP-43 inclusions, indicating that there must be disease-modifying factors. Thus, the dosage sensitivity of the STMN2 loss-of-function phenotype has important implications for ALS pathogenesis and treatment. It implies that ALS disease progression may be influenced by the degree of STMN2 loss that results from TDP-43 dysfunction. More dramatic decreases in STMN2 could be associated with earlier age of onset and a more rapid progression of motor dysfunction and denervation. If so, then factors regulating STMN2 expression levels such as STMN2 sequence variants (Theunissen et al., 2021) or natural variation in STMN2 transcription may be important modifiers of the ALS disease course. We previously demonstrated that the neuronal stress kinase dual leucine zipper kinase (DLK) is a key regulator of STMN2 protein turnover, with the activation of DLK leading to more rapid loss of axonal STMN2 (Summers et al., 2020). Interestingly, DLK is itself implicated in ALS pathogenesis, with activation of DLK stress signaling occurring in both ALS mouse models and, likely, in human patients (Le Pichon et al., 2017). If the DLK stress response were activated in TDP-43 dysfunctional neurons, it would lead to an even more dramatic loss of STMN2 from the axon. While there has been much interest in DLK as a therapeutic target for ALS, a recent clinical trial of a DLK inhibitor unfortunately failed owing to its poor safety profile (Katz et al., 2022).
While DLK inhibition is likely not a viable approach for augmenting STMN2 function in ALS, the dosage sensitivity of the mouse phenotypes implies that boosting STMN2 levels could be therapeutic. One attractive approach is to use antisense oligonucleotides (ASOs) to block the cryptic splice site in neurons afflicted with dysfunctional TDP-43 (Klim et al., 2021). Indeed, the development of ASOs targeting STMN2 splicing is underway. Alternatively, STMN2 gene therapy could be considered. In addition to boosting STMN2 levels, it is also worth exploring whether Stathmin-2 orthologs could provide therapeutic benefit. While we did not see ortholog upregulation in Stmn2 KO mice, there still may be partial redundancy among the four stathmin paralogs, all of which are highly expressed in neurons (Ozon et al., 1998). Indeed, Stmn1 mutants have a late-onset central and peripheral axonopathy, implying that STMN1 is also an axon maintenance factor (Liedtke et al., 2002). Furthermore, neuronal loss of the only Drosophila stathmin protein triggers NMJ degeneration (Graf et al., 2011), indicating that NMJ maintenance is a fundamental function of stathmin proteins rather than a unique attribute of STMN2. If so, then the upregulation of any of the other STMNs could provide a therapeutic benefit. Finally, TDP-43 pathology occurs in multiple neurodegenerative diseases. For example, TDP-43 inclusions have been seen in over half of individuals with Alzheimer’s disease and the presence of inclusions corresponds with greater cognitive impairment (Nag et al., 2018; Meneses et al., 2021; Wilson et al., 2013). TDP-43 inclusions are also found in approximately 50% of patients with frontotemporal dementia (Cairns et al., 2007). Importantly, the aberrant splice form of STMN2 is present in brains from patients with FTD (Prudencio et al., 2020), and STMN2 is decreased in the brains of patients with Alzheimer’s disease (Mathys et al., 2019). Hence, methods for boosting stathmin function may also be beneficial in those debilitating diseases.
STMN2 has been well described to regulate microtubule dynamics through the binding of tubulin heterodimers to promote microtubule catastrophe (Reiderer et al., 1997). During development, this allows for growth cone guidance and axon outgrowth (Morii et al., 2006). However, the mechanism by which STMN2 promotes synaptic maintenance after development is unknown. STMN2 is an axon maintenance factor in cultured neurons (Shin et al., 2012). Consistent with our current in vivo findings, this STMN2 loss was not sufficient to induce axon loss in vitro; however, STMN2 did promote the stability of injured axons. Furthermore, STMN2 protein is co-regulated with the potent axonal maintenance factor and SARM1 inhibitor, NMNAT2 (Summers et al., 2018; Shin et al., 2012; Summers et al., 2020; Geisler et al., 2019; Walker et al., 2017). SARM1 is the central executioner of pathological axon degeneration (reviewed in Figley and DiAntonio, 2020), and rare activating mutations in SARM1 are enriched in patients with ALS (Gilley et al., 2021; Bloom et al., 2022). Therefore, we hypothesized that STMN2 acts in concert with NMNAT2 to inhibit SARM1. However, here we show that the loss of STMN2 does not regulate NMNAT2 levels or promote SARM1 activation, and the deletion of SARM1 does not suppress the STMN2 motor neuropathy phenotype in double-mutant mice. Hence, STMN2 must function in a parallel pathway to support axon and synapse survival. We previously demonstrated in both Drosophila and in cultured mammalian sensory neurons that stathmin and Stathmin-2 are required for general axon transport and mitochondrial transport, respectively (Graf et al., 2011; Shin et al., 2012). Here we show that in Stmn2 KO cultured neurons microtubule dynamics are disrupted, and electron micrographs revealed damaged mitochondria, such as is seen in ALS (Atsumi, 1981). Mutations associated with ALS disrupt axonal transport (Castellanos-Montiel et al., 2020), and we suggest that this disruption of microtubule-based fast axonal transport may be the mechanism by which STMN2 deficiency disrupts the NMJ. Dysfunctional mitochondria could result from axonal transport deficits and could further potentiate NMJ instability.
This study demonstrates that the loss of STMN2 recapitulates important aspects of ALS pathology. The finding that STMN2 loss does not replicate all aspects of ALS pathology is expected, as TDP-43 regulates the splicing of many neuronal transcripts. Indeed, two exciting new studies demonstrated TDP-43 represses cryptic exon inclusion in UNC13A transcripts (Ma et al., 2022; Brown et al., 2022). UNC13A is an important synaptic protein, and a non-coding SNP in UNC13A that affects splicing efficiency is significantly enriched among individuals with sporadic ALS (van Es et al., 2009). Likely, the combined depletion of STMN2, UNC13A, and other TDP-43 targets would result in a more complete model of ALS-like phenotypes. Moreover, TDP-43 has functions beyond regulating splicing, and its pathological inclusion in cytosolic aggregates may cause additional gain-of-function phenotypes (Prasad et al., 2019). While this model of TDP-43-dependent pathology suggests that combination treatments may be necessary to ameliorate ALS, the demonstration that STMN2 is a functionally relevant target is an important step forward for the development of rational treatments for this devastating disease.
While this article was under review, Guerra San Juan et al. (Guerra San Juan et al., 2022) described a genetically distinct Stmn2 KO mouse and their findings are in excellent agreement with ours. Both studies showed that the loss of STMN2 impairs microtubule dynamics, and that constitutive STMN2 loss causes early motor deficits with denervation of skeletal muscle and endplate fragmentation and without spinal cord motor neuron loss. Each study also described unique, but complementary, findings. Guerra San Juan et al. found that STMN2 is more highly expressed in the FF motor neurons, while we demonstrated that loss of STMN2 leads to preferential degeneration of these FF motor units. Guerra San Juan et al. also demonstrated that denervation results in muscle atrophy, that Stmn2 loss does not induce inflammation in the spinal cord, and that germline expression of human STMN2 rescues behavioral and pathological defects in the Stmn2 KO mouse. Our study demonstrates a role for Stmn2 in sensory neurons, explores ultrastructural defects at mutant NMJs, excludes a role of SARM1 in the observed degeneration, shows that Stmn2 is required in motor neurons, and, most important, demonstrates that partial depletion of STMN2 is sufficient to cause progressive, motor-selective defects and NMJ denervation. The only apparent discrepancy between the studies was our finding that two genetically distinct STMN2 mutants showed some perinatal lethality, which was not observed by Guerra San Juan et al. Taken together, the two studies strongly support the hypothesis that the loss of STMN2 expression owing to TDP-43 dysfunction contributes to ALS pathology.
Limitations of the study
First, while this study demonstrated that a decrease of the TDP-43 target STMN2 is sufficient to cause ALS-relevant pathology, STMN2 is not the only transcript dysregulated in patients with TDP-43-associated ALS, and TDP-43 dysfunction may also cause gain-of-function effects. Hence, we only assessed the role of STMN2 loss in isolation, not in the full context that occurs within the human disease. Second, we did not perform rescue experiments with STMN2 re-expression in adults, which could address whether phenotypes are reversible after symptom onset. Third, by using genetic alleles of Stmn2 we were limited to studying the effect of either a 50% or 100% loss of STMN2, whereas patients with TDP-43 dysfunction likely experience a steady decline of STMN2 as pathology progresses in each affected neuron. Finally, we demonstrated that degeneration in the Stmn2 KO mice is not due to SARM1, but we did not identify the degenerative mechanism.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Aaron DiAntonio (diantonio@wustl.edu).
Materials availability
Requests of material can be directed to the lead contact, Aaron DiAntonio (diantonio@wustl.edu). All material will be available after the completion of a Material transfer agreement (MTA).
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of Stmn2 KO mice
All experiments were performed in accordance with the protocols of the Institutional Animal Care and Use Committee of Washington University in St. Louis and the guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were housed on a 12-h light dark cycle with less than 5 mice per cage and with water and food available at all times. Male and female mice were used for all experiments. The creation of the floxed Stmn2 allele by the Genome Engineering and Stem Cell Center (GESC@MGI) via CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/Cas9-mediated genetic modification (Chen et al., 2011) was previously described (Sentmanat et al., 2022). In brief, guide RNAs (gRNAs) were designed corresponding to the introns flanking exon 3 and single-stranded oligodeoxynucleotides were designed with a loxP site and a BamHI site inserted directly in the gRNA cleavage sites, flanked by 60 bases of homology on each side. Transgenic C57BL/6N founders were generated by single-cell embryo electroporation. The constitutive KO allele was generated spontaneously in this process and confirmed by next generation sequencing. Stmn2 KO mice were continuously produced via Stmn2+/− mating pairs as Stmn2 KO mice would not breed. Stmn2 KO mouse experiments were conducted at 3 months of age. Stmn2+/− experiments were conducted at ages indicated in figure.
Additional mouse models
Sarm1 KO mice were a gift from Macro Colonna in the Department of Pathology and Immunology, Washington University School of Medicine in St. Louis (Szretter et al., 2009). Sarm1 KO mice were crossed to Stmn2+/− to produce Sarm1+/−Stmn2+/− mice. Trans-heterozygous mice were then mated to produce Sarm1/Stmn2 dKO mice and WT littermate controls. The ChAT-cre mice were a gift from Timothy Miller in the Department of Neurology, Washington University School of Medicine in St. Louis but are commercially available through Jackson Laboratory. Founder mice heterozygous for the floxed allele were initially crossed back to a C57BL6/N mouse. Progeny (F1) with the floxed allele were then bred according to the breeding scheme found in Figure S4 to produce ChAT-Cre + Stmn2f/f mice.
Mouse dorsal root ganglia neuron cultures
Embryonic DRG spot cultures were prepared as described by Gerdts et al. (Gerdts et al., 2013). Briefly, DRG neurons were collected from Cas9 knock-in embryos (Jackson Laboratory) or WT and Stmn2 KO littermates on embryonic day 12.5–13.5, genotyped, stored in Hibernate-A Medium (Thermo Fischer) overnight, and cultured the following day. Neurons from both male and female embryos were used and pooled during experiments. DRG neurons were cultured on plates coated with poly-D-lysine and laminin in neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 50 ng/mL nerve growth factor (Harlan Laboratories), and 1 uM 5-fluoro-2′-deoxyuridine (Sigma). The neurons were kept at 5% CO2 and 37°C over the course of the experiment and half-volume media changes were done every 3–4 days. Polymerization tracking studies were conducted on DIV6-7. Axon outgrowth assays were conducted on DIV3. Cell lysates were collected for Western blot and mass spectrometry on DIV7.
METHOD DETAILS
Lentivirus transduction & plasmids
EB3-mNeonGreen was cloned into the FCIV plasmid. EB3-mNeonGreen was a gift from Dorus Gadella (Addgene plasmid # 98881). Guide RNAs were cloned into a lentiguide vector. LentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid # 52963). Sequences for STMN2 guide RNAs are: AGGTGAAGCAGATCAACAAC and GAAGAAAGACCTGTCTCTGG. The sequence for the scramble guide is: CGCGGCAGCCGGTAGCTATG. Lentiviruses were generated as previously described (Araki et al., 2004). Virus was added to Cas9 DRG cultures on DIV3. Virus was added to WT and Stmn2 KO cultures on DIV1-2, achieving ~100% transduction efficiency in DRG neurons.
Cellular imaging
Live cell microtubule polymerization
Spot cultures were plated on FluoroDishes (World Precision Instruments, FD35-100) and transduced with the EB3-mNeonGreen lentivirus on DIV1-2. Live cell images were taken on a Nikon Spinning Disk Confocal Microscope every 3 s for 5 min. Kymographs were produced with Kymolyzer (Basu et al., 2020) and analyzed with KymoButler (Jakobs et al., 2019).
Axon outgrowth
Spot cultures were grown in glass 4-well chamber slides (Fischer Scientific, 08-774-25) so that WT and KO cultures were in adjacent wells. On DIV 3, cells were fixed for 15 min with 4% PFA. The cells were washed with PBS, blocked in 10% NGS 0.1% Triton X-100 in PBS (PBS-T), and incubated with anti-TUJ1 primary antibody diluted in blocking buffer overnight at 4°C. The following day they were washed with PBS-T and incubated with Alexa Fluoro 488 conjugated goat anti-rabbit secondary (1:1000, Invitrogen, A11034) for one hour at room temperature. Afterwards, they were washed and mounted with Vectashield mounting media. Images were taken on a Leica DMI 4000B Confocal Microscope. The distance between the perimeter of each cell spot and the outer perimeter of its axon halo were measured in four different locations in each image and averaged. Stmn2 KO axon lengths are normalized to WT axon length in the same experiment.
Western Blot
Cell and brain lysates were resolved using SDS polyacrylamide gel electrophoresis (PAGE) on 4–20% Mini-Protean Gel (BioRad), followed by immunoblotting for NMNAT2 (1:100 anti-NMNAT2 (B-10) mouse monoclonal IgG1, sc-515206), STMN1 (1:1000 Anti-Stathmin 1 Rabbit Monoclonal Antibody, ab52630), STMN2 (1:1000 anti-SCG10, Shin et al., 2012), STMN3 (1:2000 Anti-STMN3 Rabbit Polyclonal Antibody, 11311-1-AP), STMN4 (1:1000 Anti-STMN4 Rabbit Polyclonal, 12027-1-AP), HSP90 (1:1000 Anti-HSP90 Rabbit IgG, C45G5), and TUJ1(anti-B3 tubulin, 1:10000 Sigma-Aldrich, T2200), and visualized using standard chemiluminescence. Band intensity was quantified using ImageJ. Blots were normalized to their respective loading controls and then normalized to the control sample.
Mass spectrometry
Sciatic nerves were homogenized in 160 uL of cold 50% MeOH in water using a sonicator and then centrifuged (15000×g, 4°C, 10 min). The supernatant was transferred to a new tube with 50 μL chloroform. The mixture was shaken vigorously and centrifuged again (15000×g, 4°C, 10 min). This chloroform extraction was repeated twice. The clear aqueous phase was transferred to a new tube, lyophilized, and stored at −80°C until measurement. Lyophilized samples were reconstituted with 5mM ammonium formate (50μL, Sigma) and centrifuged for 10 min at 12000×g. Supernatant was transferred to the sample plate, and metabolite measurements were acquired as previously described (Sasaki et al., 2016). Axonal metabolites were collected on DIV7 by removing the cell body spot from the well and extracting metabolites as previously described (Sasaki et al., 2020).
Behavioral assays
Von frey assay
Von Frey assay was performed according to the up-down method (Chaplan et al., 1994). Briefly, mice were habituated individually in plexiglass boxes on a wire mesh screen. They were given four hours to rest before testing. The plantar surface of their hind paw was stimulated for 2 s with a filament, beginning at a bending force of 0.32 g. If withdraw occurred, a filament that delivered less force would be used next. If withdraw did not occur, a filament of greater force would be used. Stimulation occurred four more times after the first change in response. The 50% withdraw threshold was calculated (Dixon, 1980), and results from left and right hind paws were averaged for each mouse. Mice were allowed to rest between trials, and mice were always tested at the same time of day.
Inverted screen assay
Mice were placed on a wire mesh screen and the screen was inverted. Each mouse underwent 3 trials with five-minute rest periods in between, and the latency to fall for each mouse was recorded. If the mouse was still hanging on the screen at 120 s, they were taken off the screen and 120 s was recorded. The average of the three trials was taken.
Rotarod assay
The mice were trained to walk on a rotarod (Panlab, LE8205) at a constant speed for five minutes the day prior to testing. On the day of testing, they were trained for another five minutes. Each mouse then underwent five trials on the rotarod that started at 2 rpm and accelerated 1 rpm every 3 s to a maximum of 24 rpm, with five minutes of rest in between trials. Latency to fall was recorded and the average of the five trials was taken.
Nerve electrophysiology
Compound muscle action potentials (CMAPs) were acquired using a Viking Quest electromyography device (Nicolet) as previously described (Beirowski et al., 2011). Briefly, mice were anesthetized, then a stimulating electrode was placed in the sciatic notch and a recording electrode was placed in the foot. Supramaximal stimulation was used for CMAPs. SNAPs were acquired using the same device, as previously described (Geisler et al., 2016). Mice were anesthetized and electrodes were placed subcutaneously with the stimulating electrode placed in the tail tip 30 mm distal from the recording electrode that was placed into the base of the tail. A ground electrode was placed in between the two. Supramaximal stimulation was used for SNAPs.
Spinal cord
Spinal cords were dissected and embedded in O.C.T., twenty-five-micron thick sections cut on a cryostat and mounted onto slides. Slides were dried at room temperature overnight, incubated in 0.1% Triton X- in PBS for 30 min, blocked in 4% BSA 1% Triton X- in PBS for 30 min, and incubated overnight at 4°C with goat anti-ChAT (1:100, Millipore Sigma-Aldrich, AB144P) and anti-STMN2 (1:100, rabbit anti-SCG10, Shin et al., 2012) in blocking buffer. The next day, slides were washed with 0.1% Triton X-3 times, incubated with Cy3 anti-goat (1:250, Jackson Immunoresearch, 705-166-147) and Alexa Fluoro-488 goat anti-rabbit IgG (H + L) (Invitrogen A21121) in wash solution for 2 h at room temperature, then washed in PBS and mounted in Vectashield with DAPI. Images of motor neurons in the lumbar spinal cord were taken on a Zeiss Axio Imager Z2 Fluorescence Microscope with ApoTome 2. Cell bodies were counted and averaged across 3 or more sections. For quantification of STMN2, motor neurons were identified by ChAT expression and the percent of STMN2+ motor neurons was calculated.
Nerve structural analysis
Sciatic, sural, and femoral nerves were processed as previously described (Geisler et al., 2016). Briefly, nerves were fixed in 3% glutaraldehyde in 0.1 mL PBS overnight at 4°C, washed and stained with 1% osmium tetroxide (Sigma Aldrich) overnight at 4°C. Nerves were washed and dehydrated in a serial gradient of 50% to 100% ethanol. Nerves were then incubated in 50% propylene oxide/50% ethanol, then 100% propylene oxide. Following that, nerves were incubated in Araldite resin/propylene oxide solutions in 50:50, 70:30, 90:10 ratios for 24 h, and subsequently embedded in 100% Araldite resin solution (Araldite: DDSA: DMP30, 12:9:1, Electron Microscopy Sciences) and baked at 60°C overnight. Semithin 400–600 nm sections were cut using a Leica EM UC7 Ultramicrotome, placed on microscopy slides, and stained with Toluidine blue. Staining and quantification were performed as previously described. All quantification was performed blinded (Sasaki et al., 2018).
IENF density quantification
Intraepidermal nerve fiber (IENF) staining and quantification was performed as previously described (Geisler et al., 2016). Briefly, the footpad skin was placed in fresh picric acid fixative overnight at 4°C. The following day the samples were washed in PBS and placed into 30% sucrose for over 24 h. Samples were embedded in O.C.T. and sectioned using a cryostat in 50 micron sections and placed floating in cryoprotectant (30% sucrose and 33% ethylene glycol in PBS) and stored at −20°C. Free floating sections were then washed with PBS, blocked in 5% normal goat serum 0.3% Triton X- in PBS, then incubated in blocking solution with PGP9.5 antibody (1:1000, AB1761, Millipore) overnight at 4°C. The following day the free-floating sections were washed with PBS-T, incubated with anti-rabbit-Cy3 (Invitrogen, A10520, 1:500) for 2 h at room temperature, washed again with PBS-T, and mounted in Vectashield with DAPI.
The footpads were imaged on a Leica DMI 4000B confocal microscope using a 20× objective. Z-stacks were acquired through the whole sample and maximal projection was applied. Density was quantified by the number of PGP9.5 + axons that crossed the basement membrane and normalized to the length of the basement membrane. Densities were averaged over 3 sections per animal. Imaging and analysis were performed blinded to genotype.
NMJ analysis
Dissected muscles were incubated in 2% Triton X- in PBS for 30 min, blocked with 5% BSA 1% Triton X- for 30 min, and incubated overnight at 4°C in blocking buffer with primary antibodies against neurofilament and SV2 (2H3, 1:100, DHSB AB2314897; SV2, 1:200, DSHB AB2315387). The following day, the samples were washed in PBS, incubated in 1% Triton X- in PBS with FITC rabbit anti-mouse IgG1 (1:400, Invitrogen A21121) and Alexa fluoro-568 conjugated α-bungarotoxin (1:500, Biotium 00006) for 3 h at room temperature. Muscles were washed in PBS 3 times for 15 min and were mounted with Vectashield Mounting medium.
To analyze NMJ morphology, z-stack images were obtained using a confocal microscope and maximal projection was applied. Due to the severe disruption of endplate morphology observed in Stmn2 KO and ChAT-cre+/Stmn2f/f lumbrical muscles, for those experiments innervation status of lumbrical muscles was determined according to entire images of areas containing postsynaptic endplates. Each 20× field was categorized as 1) fully innervated, 2) partially innervated, 3) sprouting with endplate disruption, or 4) not innervated. Lumbricals from Stmn2+/− mice and non-lumbrical muscles from all mice had preserved endplate morphology; thus, innervation status in these experiments was quantified by assessing individual NMJs. Individual NMJs were categorized as 1) fully innervated, 2) partially innervated, or 3) not innervated. Over 50 NMJs were evaluated per animal for soleus and EDL muscles. Images shown are representative images taken with a 63× oil immersion lens. Researchers were blinded to genotype during imaging and image analysis.
Embryonic tissue
Embryos were collected at embryonic day 18.5–19.5 and incubated decapitated in 4% PFA overnight. The following day, embryos were rinsed with PBS. Heads were placed in 30% sucrose for multiple days to ensure equilibration, then embedded in O.C.T. 18-micron sections were cut using a cryostat and collected on slides. Sections were stained according to the same protocol as adult spinal cords using anti-2H3 (1:300, DHSB AB2314897) and Alexa fluoro-568 anti-mouse (1:250, Thermo Scientific, A-21124). Images were taken using a 20× lens on a Zeiss Axio Scan Z1 slide scanner. Diaphragms were dissected from the body and stained free floating utilizing the protocol described above for adult muscles. Diaphragm images were taken using a 20× objective along the endplate band in the lateral anterior portion of the muscle. ImageJ was used for quantification from two images per embryo.
Transmission electron microscopy
Mice were perfused with 4% PFA and whole feet immersed in 3% glutaraldehyde for 3 days. Lumbrical muscles were then dissected out, rinsed with 0.1M phosphate buffer, placed in 1% osmium tetroxide overnight at 4°C, subsequently washed with 0.1M phosphate buffer 3 times, and dehydrated. Dehydration consisted of 30-min incubations with increasing concentrations of acetone in water: 50%, 70%, 90%, and finally three times in 100% acetone. Muscles were further incubated in Spurr:acetone prepared using the Spurr Resin kit (Electron Microscopy Science, 14300), at ratios of 1:2, 1:1, 3:1 and finally 100% Spurr, with each incubation lasting 24 h at room temperature. Lumbricals were then embedded in 100% Spurr and polymerized at 65°C for 48 h. Thin sections were cut by the Washington University Core for Cellular Imaging (WUCCI) and placed on screens for imaging. A JEOL JEM-1400Plus transmission electron microscope was used for imaging. 6–10 images were taken per sample. Presynapse mitochondrial circularity was quantified using the formula Circularity = 4π*area/perimeter2, as described (Abbade et al., 2020). As the value approaches 0.0, it indicates an increasingly elongated polygon. A circularity value of 1.0 indicates a perfect circle. Additionally, synaptic vesicles in the presynapse were counted, and the synaptic vesicle density was calculated by dividing the number of vesicles by the area. Only mitochondria and synaptic vesicles from intact axons were quantified. The researcher was blinded to genotype during image analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are reported as means ± standard error of the mean (SEM). Statistics were calculated with the aid of Prism 9 (Graphpad) software. For between group comparisons, one-way and two-way ANOVA were used with post-hoc Holm-Sidak multiple comparison or post-hoc Tukey’s multiple comparison tests and unpaired t-tests when appropriate. Two-tailed significance tests were used with p < 0.05 considered statistically significant. For animal studies, n represents the number of animals used. For cell studies, n represents the number of individual experiments. For all studies, n is either reported in the figure legend or displayed in the figure via individual points on the graph.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| TUJ1 | Sigma-Aldrich | T2200 |
| Alexa Fluoro 488 conjugated goat anti-rabbit secondary | Invitrogen | A11034 |
| anti-NMNAT2 (B-10) mouse monoclonal IgG1 | Santa Cruz | sc-515206 |
| Anti-Stathmin 1 Rabbit Monoclonal Antibody | Abcam | ab52630 |
| anti-SCG10 | Shin et al. (2012) | N/A |
| Anti-STMN3 Rabbit Polyclonal Antibody | Proteintech | 11311-1-AP |
| Anti-STMN4 Rabbit Polyclonal | Proteintech | 12027-1-AP |
| Anti-HSP90 Rabbit IgG | Cell Signaling Technologies | C45G5 |
| anti-ChAT goat antibody | Millipore Sigma-Aldrich | AB144P |
| Cy3 anti-goat | Jackson Immunoresearch | 705-166-147 |
| Alexa Fluoro-488 goat anti-rabbit IgG (H + L) | Invitrogen | A21121 |
| PGP9.5 antibody | Millipore | AB1761 |
| Goat anti-Rabbit IgG (H + L) Cross-Adsorbed Secondary Antibody, Cyanine3 | Invitrogen | A10520 |
| 2H3 | DHSB | AB2314897 |
| SV2 | DSHB | AB2315387 |
| Alexa fluoro-568 anti-mouse | Thermo Scientific | A-21124 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Toluidine Blue O | Fisher Scientific | T16125 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | 11873580001 |
| Laminin, Mouse, natural | Sigma-Aldrich | 23017015 |
| Ammonium formate | Sigma-Aldrich | 70221-100G-F |
| Osmium Tetroxide | Sigma-Aldrich | 251755-10mL |
| Nerve growth factor (NGF) | Sigma-Aldrich | N6009-4X25UG |
| Neurobasal Medium | ThermoFisher | 21103049 |
| Penicillin-Streptomycin (10,000 U/mL) | ThermoFisher | 15140122 |
| Poly-D-lysine hydrobromide | Sigma-Aldrich | P7280 |
| 5-Fluoro-2′-deoxyuridine (FUDR) | Sigma-Aldrich | F0503 |
| B-27 Supplement, serum free | ThermoFisher | 17504044 |
| D-(+)-Glucose solution | Sigma-Aldrich | G8644 |
| Paraformaldehyde, 32% solution | Electron Microscopy Sciences | 15714-S |
| Fetal Bovine Serum, Heat Inactivated | ThermoFisher | 10500064 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA Protein Assay Kit | ThermoFisher | 23227 |
| Araldite 502 epoxy resin solution | Electron Microscopy Science | 13900 |
| Spurr Resin kit | Electron Microscopy Science | 14300 |
| Rotarod Assay | Panlab | LE8205 |
| Viking Quest Electromyography Device | Nicolet | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Stmn2 KO | This paper | N/A |
| Mouse: Stmn2 floxed | This paper | N/A |
| Mouse: Rosa26-LSL-Cas9 knockin Cas9 mouse | The Jackson Laboratory | JAX:024857 |
| Mouse: ChAT-IRES-Cre | The Jackson Laboratory | JAX:006410 |
| Mouse: SARM1 B6.129X1-Sarm1tm1Aidi/J | The Jackson Laboratory | JAX:018069 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Guide RNA targeting STMN2 Sequence #1: AGGTGAAGCAGATCAACAAC | This paper | N/A |
| Guide RNA targeting STMN2 Sequence #2: GAAGAAAGACCTGTCTCTGG | This paper | N/A |
| Scramble guide RNA sequence: CGCGGCAGCCGGTAGCTATG | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| FCIV- NC vector | Essuman et al. (2017) | N/A |
| Plasmid: EB3-mNeonGreen | Chertkova et al. (2017) | Addgene #98881 |
| Plasmid: LentiGuide-Puro | Sanjana et al. (2014) | Addgene #52963 |
|
| ||
| Software and algorithms | ||
|
| ||
| Graph Pad Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Kymolyzer | Basu et al. (2020) | https://github.com/ThomasSchwarzLab/KymolyzerCodes |
| KymoButler | Jakobs et al. (2019) | https://www.wolframcloud.com/objects/deepmirror/Projects/KymoButler/KymoButlerForm |
| ImageJ | Schneider et al. (2012) | RRID:SCR_003070 |
|
| ||
| Other | ||
|
| ||
| FluoroDishes | World Precision Instruments | FD35-100 |
| 4-well Chamber Slides | Fischer Scientific | 08-774-25 |
Highlights.
Stmn2 knockout results in motor deficits and distal NMJ denervation
There is a cell-autonomous requirement for STMN2 in motoneurons
Partial STMN2 loss causes progressive, motor-selective neuropathy with NMJ loss
Findings suggest that STMN2 loss contributes to ALS pathogenesis
ACKNOWLEDGMENTS
The authors thank members of the DiAntonio and Milbrandt laboratories for their thoughtful discussions on the study. We also thank Cassidy Menendez, Rachel McClarney, Alicia Neiner, Sylvia Johnson, Xiaolu Sun, Kelli Simburger, Matthew Figley, John Palucki, Yo Sasaki, and Liya Yuan for their technical support and/or technical training and advice. Thank you to Margaret Hayne for her guidance on writing and making figures. We also thank the Washington University Core for Cellular Imaging (WUCCI) for their technical support, expertise, and training on the spinning disk microscope and transmission electron microscope. We thank the Genome Engineering & Stem Cell Center (GESC@MGI) at Washington University for generating the Stmn2 mice. This work was supported by National Institutes of Health grants (R01NS119812 to A.J.B., A.D., and J.M., R01NS087632 to A.D. and J.M., R37NS065053 to A.D., and RF1AG013730 to J.M.) and an ALS Finding a Cure Grant to A.D. and A.J.B. This work was also supported by the Needleman Center for Neurometabolism and Axonal Therapeutics, Washington University Institute of Clinical and Translational Sciences which is, in part, supported by the NIH/National Center for Advancing Translational Sciences (NCATS), CTSA grant #UL1 TR002345. L.D. is funded by F32NS117784.
Footnotes
DECLARATION OF INTERESTS
A.D. and J.M. are co-founders, scientific advisory board members, and shareholders of Disarm Therapeutics, a wholly owned subsidiary of Eli Lilly. A.J.B. is a consultant to Disarm Therapeutics. The authors have no other competing conflicts or financial interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111001.
REFERENCES
- Abbade J, Klemetti MM, Farrell A, Ermini L, Gillmore T, Sallais J, Tagliaferro A, Post M, and Caniggia I (2020). Increased placental mitochondrial fusion in gestational diabetes mellitus: an adaptive mechanism to optimize feto-placental metabolic homeostasis? BMJ Open Diabetes. Res Care. 8, e000923. 10.1136/bmjdrc-2019-000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ, and Axel R (1985). Molecular probes for the development and plasticity of neural crest derivatives. Cell 42, 649–662. 10.1016/0092-8674(85)90122-9. [DOI] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, and Milbrandt J (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Atsumi T (1981). The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 55, 193–198. 10.1007/bf00691318. [DOI] [PubMed] [Google Scholar]
- Basu H, Ding L, Pekkurnaz G, Cronin M, and Schwarz TL (2020). Kymolyzer, a semi-autonomous kymography tool to analyze intracellular motility. Curr. Protoc. Cell Biol 87, e107. 10.1002/cpcb.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Gustin J, Armour SM, Yamamoto H, Viader A, North BJ, Michán S, Baloh RH, Golden JP, Schmidt RE, et al. (2011). Sir-two-homolog 2 (Sirt2) modulates peripheral myelination through polarity protein Par3/atypical protein kinase C (aPKC) signaling. Proc. Natl. Acad. Sci. U S A 108, E952–E961. 10.1073/pnas.1104969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AJ, Mao X, Strickland A, Sasaki Y, Milbrandt J, and DiAntonio A (2022). Constitutively active SARM1 variants that induce neuropathy are enriched in ALS patients. Mol. Neurodegener. 17, 1. 10.1186/s13024-021-00511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FCY, Masino L, Qi YA, et al. (2022). TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137. 10.1038/s41586-022-04436-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, and Ironton R (1978). Sprouting and regression of neuromuscular synapses in partially denervated mammalian muscles. J. Physiol. 278, 325–348. 10.1113/jphysiol.1978.sp012307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL 3rd, Schneider JA, Kretzschmar HA, et al. (2007). TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 171, 227–240. 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos-Montiel MJ, Chaineau M, and Durcan TM (2020). The neglected genes of ALS: cytoskeletal dynamics impact synaptic degeneration in ALS. Front. Cell. Neurosci. 14, 594975. 10.3389/fncel.2020.594975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Huang Y, Gjoka M, Duda K, Taunton J, Collingwood TN, Frodin M, and Davis GD (2011). High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods 8, 753–755. 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertkova AO, Mastop M, Postma M, van Bommel N, van der Niet S, Batenburg KL, Joosen L, Gadella TWJ, Okada Y, and Goedhart J (2017). Robust and bright genetically encoded fluorescent markers for highlighting structures and compartments in mammalian cells. Preprint at bioRxiv. 10.1101/160374. [DOI] [Google Scholar]
- Dadon-Nachum M, Melamed E, and Offen D (2011). The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 43, 470–477. 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Dengler R, Konstanzer A, Küther G, Hesse S, Wolf W, and Strupplerdr A (1990). Amyotrophic lateral sclerosis: macro-EMG and twitch forces of single motor units. Muscle Nerve 13, 545–550. 10.1002/mus.880130612. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Lutjens R, Osen-Sand A, Sobel A, Catsicas S, and Grenningloh G (1997). Differential distribution of stathmin and SCG10 in developing neurons in culture. J. Neurosci. Res. 50, 1000–1009. . [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980). Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462. 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Ebenezer GJ, McArthur JC, Thomas D, Murinson B, Hauer P, Polydefkis M, and Griffin JW (2007). Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain 130, 2703–2714. 10.1093/brain/awm199. [DOI] [PubMed] [Google Scholar]
- Ebstein SY, Yagudayeva I, and Shneider NA (2019). Mutant TDP-43 causes early-stage dose-dependent motor neuron degeneration in a TARDBP knockin mouse model of ALS. Cell Rep. 26, 364–373.e4. 10.1016/j.celrep.2018.12.045. [DOI] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, and Milbrandt J (2017). The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343.e5. 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, and DiAntonio A (2020). The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 63, 59–66. 10.1016/j.conb.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, Malde AK, Jia X, Luo Z, Saikot FK, et al. (2021). SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 109, 1118–1136.e11. 10.1016/j.neuron.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD, et al. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, and Caroni P (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542. 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Höke A, Milbrandt J, and DiAntonio A (2019). Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight 4, e129920. 10.1172/jci.insight.129920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, and DiAntonio A (2016). Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139, 3092–3108. 10.1093/brain/aww251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, and Milbrandt J (2013). Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33, 13569–13580. 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Jackson O, Pipis M, Estiar MA, Al-Chalabi A, Danzi MC, van Eijk KR, Goutman SA, Harms MB, Houlden H, et al. (2021). Enrichment of SARM1 alleles encoding variants with constitutively hyperactive NADase in patients with ALS and other motor nerve disorders. Elife 10, e70905. 10.7554/eLife.70905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Orsomando G, Nascimento-Ferreira I, and Coleman MP (2015). Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 10, 1974–1981. 10.1016/j.celrep.2015.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Heerssen HM, Wright CM, Davis GW, and DiAntonio A (2011). Stathmin is required for stability of the Drosophila neuromuscular junction. J. Neurosci. 31, 15026–15034. 10.1523/JNEUROSCI.2024-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra San Juan I, Nash LA, Smith KS, Leyton-Jaimes MF, Qian M, Klim JR, Limone F, Dorr AB, Couto A, Pintacuda G, et al. (2022). Loss of mouse Stmn2 function causes motor neuropathy. Neuron 110, 1671–1688.e6. 10.1016/j.neuron.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs MA, Dimitracopoulos A, and Franze K (2019). KymoButler, a deep learning software for automated kymograph analysis. Elife 8, e42288. 10.7554/eLife.42288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JS, Rothstein JD, Cudkowicz ME, Genge A, Oskarsson B, Hains AB, Chen C, Galanter J, Burgess BL, Cho W, et al. (2022). A Phase 1 study of GDC-0134, a dual leucine zipper kinase inhibitor, in ALS. Ann. Clin. Transl. Neurol. 9, 50–66. 10.1002/acn3.51491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klim JR, Pintacuda G, Nash LA, Guerra San Juan I, and Eggan K (2021). Connecting TDP-43 pathology with neuropathy. Trends Neurosci. 44, 424–440. 10.1016/j.tins.2021.02.008. [DOI] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179. 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, Jafar-Nejad P, and Shneider NA (2022). Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 28, 104–116. 10.1038/s41591-021-01615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pichon CE, Meilandt WJ, Dominguez S, Solanoy H, Lin H, Ngu H, Gogineni A, Sengupta Ghosh A, Jiang Z, Lee SH, Maloney J, et al. (2017). Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 9, eaag0394. 10.1126/scitranslmed.aag0394. [DOI] [PubMed] [Google Scholar]
- Liedtke W, Leman EE, Fyffe RE, Raine CS, and Schubart UK (2002). Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am. J. Pathol. 160, 469–480. 10.1016/S0002-9440(10)64866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, and Wong PC (2015). TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655. 10.1126/science.aab0983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, and Cleveland DW (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, et al. (2022). TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 603, 124–130. 10.1038/s41586-022-04424-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed Z, López-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190. 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneses A, Koga S, O’Leary J, Dickson DW, Bu G, and Zhao N (2021). TDP-43 pathology in alzheimer’s disease. Mol. Neurodegener. 16, 84. 10.1186/s13024-021-00503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morii H, Shiraishi-Yamaguchi Y, and Mori N (2006). SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. J. Neurobiol. 66, 1101–1114. 10.1002/neu.20295. [DOI] [PubMed] [Google Scholar]
- Nag S, Yu L, Boyle PA, Leurgans SE, Bennett DA, and Schneider JA (2018). TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer’s disease. Acta Neuropathol. Commun. 6, 33. 10.1186/s40478-018-0531-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozon S, Byk T, and Sobel A (1998). SCLIP: a novel SCG10-like protein of the stathmin family expressed in the nervous system. J. Neurochem. 70, 2386–2396. 10.1046/j.1471-4159.1998.70062386.x. [DOI] [PubMed] [Google Scholar]
- Ozon S, Maucuer A, and Sobel A (1997). The stathmin family – molecular and biological characterization of novel mammalian proteins expressed in the nervous system. Eur. J. Biochem. 248, 794–806. 10.1111/j.1432-1033.1997.t01-2-00794.x. [DOI] [PubMed] [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, and Patel BK (2019). Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12, 25. 10.3389/fnmol.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus JM, Shi J, Heckman MG, Spiegel MR, Cook C, et al. (2020). Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Invest. 130, 6080–6092. 10.1172/jci139741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riancho J, Paz-Fajardo L, and López de Munaín A (2021). Clinical and preclinical evidence of somatosensory involvement in amyotrophic lateral sclerosis. Br. J. Pharmacol. 178, 1257–1268. 10.1111/bph.15202. [DOI] [PubMed] [Google Scholar]
- Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lütjens R, Lütjens R, Catsicas S, and Grenningloh G (1997). Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proc. Natl. Acad. Sci. U S A 94, 741–745. 10.1073/pnas.94.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Engber TM, Hughes RO, Figley MD, Wu T, Bosanac T, Devraj R, Milbrandt J, Krauss R, and DiAntonio A (2020). cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp. Neurol. 329, 113252. 10.1016/j.expneurol.2020.113252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Hackett AR, Kim S, Strickland A, and Milbrandt J (2018). Dysregulation of NAD+ metabolism induces a Schwann cell dedifferentiation program. J. Neurosci. 38, 6546–6562. 10.1523/JNEUROSCI.3304-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, and Milbrandt J (2016). NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 5, e19749. 10.7554/eLife.19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentmanat MF, White JM, Kouranova E, and Cui X (2022). Highly reliable creation of floxed alleles by electroporating single-cell embryos. BMC Biol. 20, 31. 10.1186/s12915-021-01223-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, and Shneider NA (2016). ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 7, 10465. 10.1038/ncomms10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Geisler S, and DiAntonio A (2014). Dynamic regulation of SCG10 in regenerating axons after injury. Exp. Neurol. 252, 1–11. 10.1016/j.expneurol.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, and DiAntonio A (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U S A 109, E3696–E3705. 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller KJ, Cheung CJ, Restrepo CR, Kwong LK, Stieber AM, Trojanowski JQ, and Lee VMY (2016). Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J. Neurosci. 36, 7707–7717. 10.1523/JNEUROSCI.1457-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Frey E, Walker LJ, Milbrandt J, and DiAntonio A (2020). DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol. Neurobiol. 57, 1146–1158. 10.1007/s12035-019-01796-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers DW, Milbrandt J, and DiAntonio A (2018). Palmitoylation enables MAPK-dependent proteostasis of axon survival factors. Proc. Natl. Acad. Sci. U S A 115, E8746–E8754. 10.1073/pnas.1806933115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, and Diamond MS (2009). The immune adaptor molecule SARM modulates tumor necrosis factor Alpha production and microglia activation in the brainstem and restricts west nile virus pathogenesis. J. Virol. 83, 9329–9338. 10.1128/jvi.00836-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen F, Anderton RS, Mastaglia FL, Flynn LL, Winter SJ, James I, Bedlack R, Hodgetts S, Fletcher S, Wilton SD, Laing NG, MacShane M, Needham M, Saunders A, Mackay-Sim A, Melamed Z, Ravits J, Cleveland DW, and Akkari PA (2021). Novel STMN2 variant linked to amyotrophic lateral sclerosis risk and clinical phenotype. Front. Aging Neurosci. 13, 658226. 10.3389/fnagi.2021.658226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, et al. (2009). Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 41, 1083–7. 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, and van den Berg LH (2017). Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. 10.1016/S0140-6736(17)31287-4. [DOI] [PubMed] [Google Scholar]
- Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, and DiAntonio A (2017). MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 6, e22540. 10.7554/eLife.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Wang M, Song WM, Shen Q, McKenzie A, Choi I, Zhou X, Pan PY, Yue Z, and Zhang B (2019). The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun. 10, 5234. 10.1038/s41467-019-13144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, Yu L, Trojanowski JQ, Chen EY, Boyle PA, Bennett DA, and Schneider JA (2013). TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 70, 1418. 10.1001/jamaneurol.2013.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.