

Summary
Upon fertilization, embryos undergo chromatin reprogramming and genome activation, however, the mechanisms that regulate these processes are poorly understood. Here, we generated a triple mutant for Nanog, Pou5f3, and Sox19b (NPS) in zebrafish and found that NPS pioneer chromatin opening at >50% of active enhancers. NPS regulate acetylation across core histones at enhancers and promoters, and their function in gene activation can be bypassed by recruiting histone acetyltransferase to individual genes. NPS pioneer chromatin opening individually, redundantly, or additively depending on sequence context and we show that high nucleosome occupancy facilitates NPS pioneering activity. Nucleosome position varies based on the input of different transcription factors (TFs), providing a flexible platform to modulate pioneering activity. Altogether, our results illuminate the sequence of events during genome activation and offer a conceptual framework to understand how pioneer factors interpret the genome and integrate different TF inputs across cell types and developmental transitions.
Keywords: Chromatin reprogramming, genome activation, histone acetylation, pioneer transcription factors, chromatin accessibility, nucleosome occupancy
In Brief
Miao et al. report that transcription factors Nanog, Pou5f3, and Sox19b (NPS) initiate chromatin opening, and regulate histone acetylation to activate the silent genome in zebrafish. The ability of NPS to initiate chromatin opening is facilitated by high nucleosome occupancy, providing framework to understand how pioneer factors interpret the genome.
Graphical Abstract
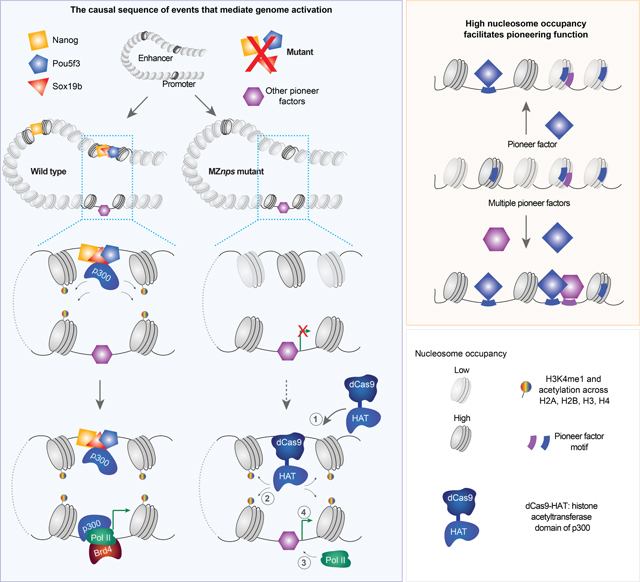
Introduction
Upon fertilization, embryos are transcriptionally silent and then undergo genome-wide chromatin reprogramming to initiate zygotic genome activation (ZGA) and gain transient totipotency (Iwafuchi-Doi and Zaret, 2014; Ladstätter and Tachibana, 2019). This maternal-to-zygotic transition (MZT) marks the transfer of regulatory control from the maternal program to the zygotic program (Giraldez, 2010; Tadros and Lipshitz, 2009; Vastenhouw et al., 2019; Yartseva and Giraldez, 2015). Studies have identified a small subset of individual TFs that function during ZGA (Vastenhouw et al., 2019), however the larger regulatory network of TFs that orchestrate chromatin remodeling, their interdependency, and how they trigger genome activation are still poorly understood.
Genome activation and zygotic transcription coincide with epigenetic remodeling of the chromatin and an increase in chromatin accessibility (Blythe and Wieschaus, 2016; Eckersley-Maslin et al., 2018; Gao et al., 2018; Li et al., 2018; Liu et al., 2018; Lu et al., 2016; Murphy et al., 2018; Vastenhouw et al., 2019; Wu et al., 2016; Wu et al., 2018; Xia et al., 2019), however the causal relationship between these events is poorly understood (Schulz and Harrison, 2019; Vastenhouw et al., 2019). Two models have been proposed to explain transcriptional activation (Chan et al., 2019; Liu et al., 2020; Schulz and Harrison, 2019; Vastenhouw et al., 2019). In one, epigenetic remodeling of the genome increases chromatin accessibility and compatibility for TF binding and gene activation (Samata et al., 2020; Schulz and Harrison, 2019; Vastenhouw et al., 2019). Consistent with this model, biochemical studies have revealed that acetylation reduces the affinity between DNA and nucleosomes (Cary et al., 1982; Garcia-Ramirez et al., 1995; Tse et al., 1998), acetylated chromatin is often more accessible (Hebbes et al., 1994; Krajewski and Becker, 1998); and histone H4 lysine 16 acetylation (H4K16ac) present in the oocyte has been shown to regulate local chromatin accessibility before ZGA in flies (Samata et al., 2020). In the other model, pioneer factors, which bind nucleosomal DNA to promote chromatin accessibility, facilitate the recruitment of other TFs and epigenetic regulators, which then mediate transcriptional activation (Iwafuchi-Doi, 2019; Iwafuchi-Doi and Zaret, 2016). In support of this model, ZGA activators such as Zelda in Drosophila, FoxH1 in Xenopus, OCT4 in human, and NFY proteins in mice function as pioneer factors during early development (Cirillo et al., 2002; Iwafuchi-Doi and Zaret, 2014). Consistent with both models, TFs are associated with histone H3 lysine 27 acetylation (H3K27ac), which correlates with regions of accessible chromatin and active transcription (Creyghton et al., 2010; Wang et al., 2008) and coincides with ZGA in mice, zebrafish, and flies (Chan et al., 2019; Dahl et al., 2016; Li et al., 2014; Sato et al., 2019; Zhang et al., 2018). However, we do not understand the causal relationship between chromatin accessibility, TF binding, histone acetylation, and genome activation.
In zebrafish, pioneer factors Nanog, Pouf5f3, and Sox19b (here NPS), the mammalian homologs of which are reprogramming factors, are required for ZGA (Lee et al., 2013; Leichsenring et al., 2013; Pálfy et al., 2020; Veil et al., 2019). However, the number of regions bound by pioneer factors is much smaller than the widespread occurrence of their consensus motifs, and even for those bound regions only a small fraction of them are opened by these pioneer factors, suggesting pioneering activity is modulated by co-factors and sequence context (Larson et al., 2021; Zaret, 2020). Understanding how the genomic context regulates pioneering function in vivo is fundamental to decode how pioneer TFs interpret the genome across different cell types and developmental transitions. This has been challenging, given that multiple pioneer factors co-bind many regions across the genome (Larson et al., 2021; Zaret, 2020), and the pioneering activity of each factor can be masked by other TFs in individual loss-of-function studies (Larson et al., 2021; Pálfy et al., 2020; Veil et al., 2019). Here, we address this challenge by generating a triple maternal-zygotic nanog−/−;pou5f3−/−;sox19b−/− zebrafish mutant (MZnps) to investigate how pioneering specificity is regulated and identify the mechanisms by which NPS reprogram, remodel, and activate the zygotic genome.
Results
NPS regulate chromatin accessibility and histone modifications during ZGA
Pluripotency factors NANOG, OCT4, and SOX2 co-occupy many regulatory regions and co-regulate the transcription of many genes in human and mouse embryonic stem (ES) cells (Boyer et al., 2005; Chen et al., 2008; Loh et al., 2006; Wang et al., 2012). Similarly, zebrafish homologs Nanog, Pouf5f3 and Sox19b (NPS) occupy thousands of open chromatin regions (Figures 1A and S1A) and are required for ZGA and early development (Lee et al., 2013; Leichsenring et al., 2013). However, the way these factors work together to remodel and reprogram the zygotic genome in vivo remained unexplored due to the technical challenge of simultaneously eliminating all three factors. To address this, we generated a triple maternal-zygotic (MZ) mutant: nanog−/−;pou5f3−/−;sox19b−/− (MZnps), which lacks maternally-inherited NPS mRNA and is deficient in zygotic expression of NPS (Figures 1B, S1B, and S1C). MZnps embryos arrest at the sphere stage prior to gastrulation (4 hours post fertilization, 4 hpf) (Figures 1B and S1D), reminiscent of a block in zygotic transcription (Kane et al., 1996). Analysis of the MZnps nascent transcriptome using Click-iT-seq (Chan et al., 2019) revealed that 37% of zygotic genes (822 out of 2,240) were downregulated more than two-fold compared to wild-type embryos (Figure 1C). Consistent with this, RNA polymerase II (Pol II) ChIP-seq revealed a concomitant loss of Pol II occupancy across the gene body and promoters of these genes (Figure S1E), suggesting NPS recruit Pol II, rather than release paused Pol II. Gene ontology analysis (Ge et al., 2019; Mi et al., 2019a; Mi et al., 2019b) revealed that genes downregulated in MZnps embryos were enriched for gene-specific transcriptional regulators and developmental processes, including embryonic morphogenesis, formation of primary germ layers, and cell fate commitment (Figures S1F and S1G). The developmental arrest and impaired ZGA in MZnps embryos could be rescued by temporarily restoring Nanog and Pouf5f3 via injection of their mRNAs (Figures 1B and S1H), and rescued mutant embryos developed into fertile adults, suggesting that NPS are dispensable for adult life and gametogenesis. Thus, the MZnps mutant is a tractable model to investigate how a cohort of TFs work together to remodel, reprogram, and activate the zygotic genome.
Figure 1. Nanog, Pou5f3, and Sox19b are required for chromatin remodeling and histone modification during ZGA.

(A) Venn diagram showing significant overlap of NPS binding at all accessible regions (P < 1×10−100 Fisher’s exact test). (B) Micrographs of zebrafish embryos. At 6 hpf, wild-type (WT) embryos develop to the shield stage, while MZnps embryos arrest at the sphere stage and resemble those treated with α-amanitin (a Pol II inhibitor). Co-injection of nanog and pou5f3 mRNAs (Rescue) rescues development of MZnps embryos at 6 hpf and 24 hpf. (C) Biplots comparing nascent transcriptomes of MZnps and WT embryos using Click-iT-seq. (D) Heatmaps showing NPS binding, Pol II binding, Omni-ATAC, and H3K4me1, H3K4me3, and H3K27ac levels at N/P/S-bound active enhancer and promoter regions of zygotic genes. Heatmaps are centered at TSSs (promoters) or Omni-ATAC peak summits (active enhancers). Regions are ranked by the average Pol II binding signal within 1kb of the TSS of associated zygotic genes in WT embryos. (E) Representative genome tracks of Pol II binding, NPS binding, accessibility (Omni-ATAC), and H3K4me1, H3K4me3, and H3K27ac levels in WT and MZnps embryos. The dashed box highlights the klf17 promoter, which remains accessible in MZnps embryos. (F) Stacked bar plot showing the percentage of differentially affected accessible chromatin regions in MZnps embryos compared to WT. (G-H) Left: Confocal imaging showing the miR-430 locus with nascent transcription using Click-iT imaging (G) or H3K27ac immunofluorescence (H) in WT and MZnps embryos. Right: quantification of signal at the miR-430 locus. miR-430 was labeled with dCas9-GFP and sgRNAs targeting the locus. Nuclei are outlined with a dashed line based on DAPI signal. Triptolide is a Pol II inhibitor. Scale bar: 5 μm. See also Figure S1.
To determine the causal relationship between NPS binding, changes in chromatin accessibility and histone modifications during ZGA, we surveyed chromatin accessibility (using Omni-ATAC) and profiles of H3K4me1, H3K4me3, H3K27ac (using ChIP-seq) in wild-type and MZnps embryos at 4 hpf (Figures 1D–1F and S1I-S1L). We identified 55,970 high-confidence accessible regions (Figure S1I; P < 10−8 in three replicates, MACS2 (Zhang et al., 2008)) including 17,993 at promoters (defined as the region within 500 bp of the transcription start site (TSS) of annotated transcripts) and a further 16,142 accessible regions marked by H3K27ac, suggestive of active enhancers (Figure 1F). In MZnps embryos, over half of active enhancers (8,274 out of 16,142) showed a significant decrease in chromatin accessibility (FDR < 0.01, DESeq2 (Love et al., 2014)), most of which (6,860 out of 8,274) were strongly bound by Nanog, Pou5f3, or Sox19b (N/P/S; FDR = 0.05, MACS2). In contrast, although 56% of promoters (10,129/17,993) were bound by N/P/S, only 5.1% (914 out of 17,993) showed a significant decrease in accessibility in MZnps embryos (Figure 1F). Promoter accessibility was largely maintained even for NPS strong targets (N/P/S-bound genes that are strongly downregulated in MZnps embryos) despite the loss of Pol II ChIP-seq signal (Figures 1D and 1E). Altogether, these results indicate that NPS establish chromatin accessibility at most of active enhancers during the MZT, while promoter accessibility is largely regulated by other factors.
We investigated whether NPS regulate histone modifications associated with enhancer and promoter function. H3K4me3 is associated with active promoters (Calo and Wysocka, 2013; Schulz and Harrison, 2019; Vastenhouw et al., 2019) and we observed a strong reduction of H3K4me3 at promoters of genes downregulated in MZnps embryos (Figures 1D and 1E), consistent with the dependency of H3K4me3 deposition on transcription (Zhang et al., 2018). H3K4me1 is often associated with primed enhancers and is enriched at promoters (Calo and Wysocka, 2013). H3K4me1 marks the two nucleosomes flanking open enhancers in wild-type embryos, while in MZnps embryos H3K4me1 is dramatically reduced at enhancers associated with NPS strong targets (Figure 1D). These data suggest that NPS enhance H3K4me1 deposition at the flanking nucleosomes.
H3K27ac correlates with accessible regions and often marks active enhancers and promoters, and its deposition coincides with ZGA (Calo and Wysocka, 2013; Schulz and Harrison, 2019; Vastenhouw et al., 2019). Loss of NPS reduced H3K27ac in MZnps embryos at enhancers and promoters associated with NPS strong targets, such as klf17 (Figures 1D–1F), suggesting that NPS binding precedes H3K27ac. Loss of NPS reduced enhancer accessibility but had little effect on promoter accessibility. These results provide a clear contrast between chromatin accessibility and H3K27ac at enhancers and promoters, suggesting that H3K27ac is not absolutely required for chromatin accessibility. To confirm the dependency of H3K27ac on NPS, we performed immunostaining of the miR-430 locus, the first zygotically transcribed locus (Chan et al., 2019; Heyn et al., 2014), which is NPS-dependent (Figure 1G). We observed that H3K27ac was enriched at the miR-430 locus, and that this enrichment was lost in MZnps embryos (Figure 1H). Reduction of H3K27ac correlates with reduction in transcription (Figure 1D; r=0.57 for promoters, r=0.61 for enhancers and P < 10−77), suggesting that H3K27ac could be a critical step by which NPS regulates ZGA. Altogether, these results demonstrate that NPS play critical roles in pioneering chromatin opening at over half of active enhancers and in regulating histone modifications; while H3K27ac is not absolutely required for chromatin opening, it may be a mechanism by which NPS regulates genome activation.
NPS regulate acetylation across core histones
Next, we investigated whether loss of NPS affects histone acetylation on other lysine residues. We used ChIP-seq to compare histone acetylation in wild-type and MZnps embryos across the four canonical core histones (H3K9ac, H3K14ac, H3K18ac, H3K36ac, H4K8ac, H4K16ac, H2BK5ac, H2BK15ac, and H2AK5ac) and histone H2A variant H2A.Z (H2A.ZK4/7ac). We observed a strong reduction of histone acetylation across the four canonical core histones in the MZnps embryos at both enhancers and promoters of NPS strong targets (Figures 2A, S2A, and S2B). Reduction in acetylation was strongly correlated with the reduction in transcription (Figure 2B). While H4K16ac has been shown to regulate promoter accessibility in flies (Samata et al., 2020), our results reveal that promoters remained accessible in the absence of H4K16ac, suggesting that H4K16ac is not absolutely required for chromatin accessibility during ZGA in zebrafish. To further test whether chromatin accessibility depends on histone acetylation, we treated wild-type embryos with chemical inhibitors of the histone acetylation writer p300 (SGC-CBP30 (Hay et al., 2014)) or reader Brd4 (JQ1 (Chan et al., 2019; Filippakopoulos et al., 2010)) and assessed chromatin accessibility using Omni-ATAC at 4 hpf. Neither inhibitor blocked chromatin opening (Figures S2C and S2D), further suggesting that histone acetylation is not the main driver of chromatin opening during ZGA. These results suggest that NPS regulate histone acetylation at both enhancers and promoters across core histones during ZGA.
Figure 2. Nanog, Pou5f3, and Sox19b are required for histone acetylation across core histones and for recruitment of p300 and Brd4.

(A) Heatmaps showing histone acetylation levels across histones at N/P/S-bound active enhancers and promoters of differentially affected zygotic genes. Heatmaps are centered at TSSs (promoters) or Omni-ATAC peak summits (active enhancers) and ranked by Pol II binding intensity within 1kb of the TSS of the associated zygotic genes in wild-type (WT) embryos. (B) Heatmap showing the correlation between changes in transcription and changes in histone acetylation marks in MZnps/WT for N/P/S-bound active enhancers and promoters of zygotic genes. H2A.ZK4/7ac-H2A.Z ratio represents the ratio between H2A.ZK4/7ac and H2A.Z. (C) Heatmaps showing p300 and Brd4 levels at N/P/S-bound active enhancers and promoters of differentially affected zygotic genes. Heatmaps are centered and ranked as in (A). inhibitor: Pol II inhibitors (triptolide + flavopiridol). (D) Representative genome tracks of Pol II, p300, Brd4, and NPS binding, Omni-ATAC and histone acetylation across core histones in WT and MZnps embryos. See also Figure S2.
NPS recruit p300a and Brd4 during early development
Next, we asked whether NPS regulate histone acetylation by recruiting the histone acetyltransferase p300 to enhancers and promoters. ChIP-seq analysis revealed that p300a is enriched at active enhancers and promoters, but also at gene bodies and transcription end sites (TESs) (Figures 2C, S2E-S2I). Across these sites, p300a is largely depleted for NPS target genes in MZnps mutant embryos (Figure 2C). For all N/P/S-bound enhancers and promoters associated with zygotic genes, 621 regions lost p300a in MZnps embryos, suggesting that NPS are required to recruit p300a; while 635 regions maintained similar p300a levels, suggesting that other TFs function partially redundantly with NPS at those sites. p300 might be recruited through a direct interaction with NPS or indirectly through co-factors (Fang et al., 2014). At gene bodies and TESs, p300a had a similar enrichment pattern to Pol II, however this localization was not associated with a marked increase in histone acetylation (Figure S2F), thus we hypothesized that these contacts between p300a and gene bodies/TESs are mediated by the transcription machinery. To test this, we treated wild-type embryos with Pol II inhibitors (triptolide to block Pol II recruitment and flavopiridol to block Pol II release) and observed that p300a is lost at gene bodies and TESs, and reduced at promoters, but maintained at enhancers (Figures 2C, 2D, S2E-S2G). Together, these results suggest that NPS recruit p300a to their binding sites (primarily at enhancers and some promoters), and that Pol II facilitates p300a binding at promoters and may carry it along with the transcription machinery during transcription.
BRD4 is a histone acetylation reader (Filippakopoulos et al., 2012; Shi and Vakoc, 2014) that recruits the Mediator complex and p-TEFB (Cho et al., 2018; Gibson et al., 2019; Itzen et al., 2014; Sabari et al., 2018; Yang et al., 2008) to promote Pol II recruitment and transcriptional initiation and elongation in ES cells (Hnisz et al., 2013; Whyte et al., 2013). In wild-type embryos Brd4 ChIP-seq profile resembled that of Pol II occupancy, with accumulation primarily at promoters and to a lesser extent at gene bodies (Figure S2F). In MZnps embryos, however, Brd4 was substantially reduced across both promoters and gene bodies of NPS target genes (Figures 2C, 2D, and S2E), suggesting that Brd4 recruitment requires NPS. This is consistent with the reduction of p300a and histone acetylation in the absence of NPS (Figures 2C and 2D). Altogether, these results reveal that NPS are required for the recruitment of p300 and Brd4. Based on these results, we propose a model where NPS activate the zygotic genome by recruiting histone acetyltransferases, such as p300, which acetylate histone tails at enhancers and promoters; Brd4 then binds to acetylated histones and promotes Pol II recruitment and transcriptional activation; transcription then translocates part of this complex across the gene body, resulting in contacts of p300 and Brd4 with the chromatin across these regions.
Histone acetyltransferase can bypass NPS function
To test whether NPS activate the zygotic genome by recruiting p300, we asked if restoring histone acetylation at either promoters or enhancers could rescue gene activation in the absence of NPS (Figure 3A). We fused the histone acetyltransferase core domain (HAT) of human p300 (Hilton et al., 2015) to a catalytically dead Cas9 (Chan et al., 2019) (dCas9-HAT) and used sgRNAs to target this complex to NPS strong targets. We used qRT-PCR, RNA in situ hybridization, RNA-seq, Omni-ATAC, and H3K27ac CUT&TAG (Kaya-Okur et al., 2019) to determine how dCas9-HAT affects transcription, accessibility, and histone acetylation at target genes. Several lines of evidence reveal that restoring histone acetylation can rescue NPS function and induce transcriptional activation. First, dCas9-HAT targeting asb11 or her5 restored H3K27ac and transcription of each target gene in MZnps embryos, but had no effect on a control gene that was not targeted by these sgRNAs (klf17) (Figures 3B–3D and S3A-S3C). This effect was specific to acetyltransferase activity, because targeting a catalytically dead HAT (dHAT, D1399Y) (Hilton et al., 2015) did not activate gene expression or increase H3K27ac (Figures 3B–3D and S3A-S3C). RNA in situ hybridization for asb11 and her5 revealed that both genes are transcribed at high levels in large clonal patterns (Figure 3C), likely due to the mosaic nature of the rescue caused by microinjection. Second, dCas9-dHAT restored chromatin accessibility, but not H3K27ac or transcription of the target gene in MZnps embryos (i.e. asb11, Figures 3D, S3C, and S3D). These results indicate that transcription occurs when histone acetylation is restored at the promoter, and that opening of promoters or enhancers is not sufficient to activate transcription during ZGA. Third, targeting dCas9-HAT to the enhancer upstream of the her5 locus also restored transcription of the neighboring gene her11 (Figure 3D), suggesting that a local increase in histone acetylation is sufficient to recruit Pol II and restore transcription at nearby promoters.
Figure 3. Histone acetyltransferase activity partially rescue transcription of NPS strong targets.

(A) Schematic for testing whether recruiting histone acetyltransferase activity can bypass NPS in activating transcription. (B) qPCR analysis showing the relative expression of asb11 and her5, normalized by actb1. When targeted, asb11 and her5 transcription was rescued by dCas9-HAT in MZnps embryos (two-tailed unpaired t-test, ns: not significant, ** P <0.01, *** P <0.001). sgRNAsasb11 injected embryos were used as a control for her5 expression analysis, and vice versa. sgRNAsasb11: sgRNAs targeting asb11. sgRNAsher5: sgRNAs targeting her5. (C) RNA in situ hybridization analysis of asb11 and her5 in wild-type (WT) and MZnps embryos. sgRNA targeting asb11 and her5 were injected separately. (D) Genome tracks showing that accessibility, H3K27ac, and transcription were restored at asb11 and her5 in MZnps embryos by the dCas9-HAT system. Transcription was activated for her11, a gene near her5. Dashed boxes highlight the sgRNA targeting regions. Arrows show regions where H3K27ac is rescued. sgRNA targeting asb11 and her5 were co-injected. (E) Model illustrating the sequence of events of NPS-mediated genome activation. See also Figure S3.
Our analysis revealed that targeting enhancers with dCas9-HAT caused acetylation of the promoter and vice versa, suggesting that spatial proximity between the enhancers and promoters is maintained in the absence of NPS. For example, targeting dCas9-HAT to asb11 promoter increased H3K27ac at upstream and downstream enhancers which were also acetylated in wild-type embryos (Figure 3D). Conversely, for five out of seven genes (asb11, her5, apela, klf17 and vox), targeting NPS-bound enhancers rescued promoter acetylation and transcription (Figures S3C and S3D). In one case (has2), targeting the NPS-bound distal enhancer (>10 kb from the promoter) did not restore acetylation at the promoter nor transcriptional activation (Figure S3D), indicating that histone acetylation at enhancers needs to extend to the promoter region to activate transcription. Conversely, genes (i.e., ddx5, Figure S3E) that lost histone acetylation at NPS-bound enhancers but not at promoters remained transcriptionally active in MZnps mutants. Thus, recruiting histone acetyltransferase activity at enhancers or promoters can substitute NPS function, and is a critical step through which NPS regulate ZGA (Figure 3E).
Identification of TFs regulating chromatin accessibility and their dependency of NPS
To identify the TFs that regulate chromatin accessibility during ZGA and their dependency on NPS, we first analyzed the maternally expressed TFs that are highly translated pre-ZGA, using ribosome footprinting (RFP) data at 2 hpf (Bazzini et al., 2012), and found 188 sequence specific TFs (RFP >5 rpkm) (Table S1). Next, we compared the chromatin accessibility profile in wild-type and MZnps (DESeq2 (Love et al., 2014); FDR < 0.01) and performed motif enrichment analysis (McLeay and Bailey, 2010) across three different groups: regions that lost accessibility in MZnps, and are thus dependent on NPS (Group 1; 7,813 sites); regions with partially reduced accessibility in MZnps embryos (Group 2; 6,665 sites); and regions that maintained accessibility and hence are independent of NPS for chromatin opening (Group 3; 9,371 sites) (Figure 4A). We identified multiple TF motifs that were significantly enriched in regions independent of NPS pioneering activity (Group 2 and 3), including BBX, PATZ1, KLF9, SRF, CLOCK, IRF2, STAT1, SPZ1, E2F2, and ATF1 (Figure 4B). Promoters that were accessible independent of NPS (Group 3) were significantly enriched for SP1, KLF9, FOXK1, PATZ1, NFY, CEBPZ, and ATF1 motifs. Finally, EOMES, YY1, LEF1, TCF3, and TBP were significantly enriched in regions that lost chromatin accessibility in MZnps embryos and were therefore dependent on the pioneering activity of NPS (Group 1) (Figure 4B and Table S1).
Figure 4. Identification of TF motifs enriched in accessible regions and their dependence on NPS.

(A) Heatmaps of Omni-ATAC signal at differentially affected accessible regions in wild-type (WT) and MZnps embryos. Heatmaps are centered at Omni-ATAC peak summits and ranked by the average Omni-ATAC signal intensity in WT. (B) Enrichment of representative TF motifs at promoters, active enhancers, and other accessible regions within the three groups of accessible regions from (A). (C) Heatmaps of Eomesa and Nfya ChIP-seq signal within the three groups of accessible regions from (A) in WT and MZnps embryos. Heatmaps are centered at the Omni-ATAC peak summits and ranked by average ChIP-seq signal in WT embryos. (D) Heatmaps showing Eomesa and Nfya levels at active enhancers and promoters of differentially affected zygotic genes. Heatmaps are centered at the TSS (promoters) and the summit of the Omni-ATAC peak (active enhancers) and ranked by average Pol II signal within 1kb of the TSS of the associated zygotic genes in WT embryos. (E) Representative genome tracks of TF binding and Omni-ATAC show that Eomesa binding depends on NPS pioneering activity, while Nfya binding is largely independent of NPS. (F) Diagram illustrating TF motifs located at the accessible regions near klf17. See also Figure S4.
To further investigate the dependency of these TFs on NPS pioneering activity, we selected one representative from Group 3 (Nfya) and Group 1 (Eomesa) and used ChIP-seq to examine their binding profiles in wild-type and MZnps embryos. The majority (73%) of the Nfya-binding regions remain bound in MZnps embryos (Figures 4C–4E, and S4A-C), indicating that Nfya establishes chromatin opening independently of NPS. In contrast, most (87%) of the Eomesa-binding sites lost Eomesa occupancy and chromatin accessibility in MZnps embryos (Figures 4C–4E and S4). Together, these results provide a set of TFs that potentially regulate chromatin opening during ZGA (Figure 4F) and establish the relative dependency of these factors on NPS pioneering function.
Independent and cooperative NPS pioneering functions during ZGA
Our results indicate that NPS co-bind many genomic regions (Veil et al., 2019) (Figures 1A and S1A); however, it remains unclear how these factors work together (i.e. redundantly or additively) to initiate chromatin opening during ZGA (Cirillo et al., 2002; Pálfy et al., 2020; Veil et al., 2019; Zaret and Carroll, 2011). These are challenging questions that cannot be easily addressed using single mutants (Pálfy et al., 2020; Veil et al., 2019). To address this, we assessed chromatin accessibility (i) after rescuing N, P, or S function separately or in combination (NP, NS, PS, NPS) by injecting mRNAs into MZnps embryos or (ii) in single, double, and triple mutant backgrounds where the maternal contribution of N, P or S is lost (Figure 5A). To identify the physiological levels of mRNA required to restore the function of each factor, we titrated mRNA levels to rescue gastrulation and survival to adulthood without causing an overexpression phenotype. We found that each individual factor has pioneering activity, however Nanog is required to open chromatin at more regions during ZGA than Pou5f3 and Sox19b are, and many regions were only rescued by a combination of factors (Figures 5B, 5C, S5A-S5C). Among the 5,854 N/P/S-bound regions that lost accessibility in MZnps embryos (Group 1), the rescue of chromatin accessibility was different for each condition (2,318 regions were opened by N; 250 by P; 131 by S; 3,559 by NP; 2,796 by NS; 770 by PS; 4,468 by NPS). 2,303 regions only became accessible when simultaneously rescued by two or three factors (Figure S5C), suggesting that NPS work additively to pioneer chromatin opening. Conversely, NPS can also work redundantly, as two or more factors can function interchangeably to initiate chromatin accessibility in 130 regions (Figure S5D). Our results also show that N, P, and S have a concentration-dependent effect on chromatin accessibility; a higher concentration of each factor increases the number of regions in which accessibility is rescued (Figure 5D), suggesting that the dynamic increase in chromatin accessibility over development (Liu et al., 2018; Pálfy et al., 2020; Veil et al., 2019) could be caused by the accumulation of pioneer factors (Bazzini et al., 2012).
Figure 5. Nanog, Pou5f3, and Sox19b function independently and cooperatively to remodel chromatin.

(A) Schematic illustrating the approach to test the pioneering activity of N, P, and S – alone and in combination - by either TF restoration (N, P, and S denote the injected mRNAs) in MZnps embryos, or different combinations of mutants deprived of maternal contribution of the corresponding TFs. (B) Venn diagrams showing significant overlap of Group 1 regions that are rescued in different TF conditions (top left of each diagram) and corresponding genotypes (top right of each diagram). For example, most regions where accessibility was rescued by N (restoring Nanog in Maternal-zygotic-nanog−/−;pou5f3−/−;sox19b−/−) remain accessible in the N+/−;P−/−;S−/− (Maternal-nanog+/−;pou5f3−/−;sox19b−/−) mutant embryos. (N (P < 10−100); NP (P < 10−100); NPS (P = 1.1 × 10−93) Fisher’s exact test). (C) Representative genome tracks showing accessibility in different rescue conditions and corresponding genotypes at regions that require one, two, and three NPS factors to rescue accessibility. (D) Bar plot comparing the number of regions in Group 1 that were bound and can be rescued by N, P, and S at low and high dosage. See also Figure S5.
Of all regions co-bound by all three factors (1,613 regions; Group 1) we identified 582 regions that were opened exclusively by only one factor (8, 60, and 514 regions opened by S, P, and N respectively) (Figure S5E). These results are consistent with the rescue observed in different double mutants N+/−;P−/−;S−/− 299 (rescued by nanog+/− but not pou5f3+/−) and N−/−;P+/−;S−/− 37 (rescued by pou5f3+/− but not nanog+/−) (Figure S5F). Hence, in these 582 co-bound regions, one factor functions as the pioneer factor for the other two. Together these results demonstrate that N, P, and S can initiate chromatin opening either alone, interchangeably, or additively and indicate that their pioneering activity is modulated by sequence context and co-factors in vivo.
Intrinsic nucleosome occupancy primes chromatin opening in early embryos
N, P, and S can initiate chromatin opening either alone or in combination at different genomic sites, yet how the genomic context regulates their pioneering function in vivo is poorly understood. It is generally believed that nucleosomes serve as barriers that pioneer factors must overcome to facilitate the binding of other TFs (Joseph et al., 2017; Pálfy et al., 2020; Zaret and Carroll, 2011). While pioneer factors can bind nucleosomal DNA (Zaret, 2020; Zaret and Carroll, 2011), several studies suggest that regions with high affinity for nucleosomes require more pioneering factors for opening (Soufi et al., 2015; Veil et al., 2019). These studies were focused on pioneer factor binding, yet pioneer factors only open a fraction of their bound regions, indicating that binding is not equivalent to pioneering activity (Larson et al., 2021; Zaret, 2020) (Figure S1A). To define features that facilitate pioneering function beyond motif frequency (Grant et al., 2011) (Figures S5G and S5H), we determined whether genomic regions with different nucleosome occupancy respond differently to N/P/S. Several lines of evidence indicate that high nucleosome occupancy facilitates pioneering activity. i) We used a computational model to predict the intrinsic ability of different DNA sequences to bind nucleosomes (nucleosome occupancy score) (Kaplan et al., 2009). We found that regions with higher intrinsic nucleosome occupancy require fewer factors for opening (Figure 6A). Conversely, sites that require multiple pioneer factors for opening have lower nucleosome occupancy scores (Figures 6B and S6A). ii) Nucleosome occupancy also affected how different regions respond to TF levels, since regions that were opened by lower concentrations of N, P, or S had significantly higher nucleosome occupancy scores than those requiring higher concentrations of the transcription factors (Figure 6C). These results indicate that nucleosome occupancy regulates NPS pioneering activity in vivo, where sites with lower nucleosome occupancy are more likely to be regulated by the combined action of multiple pioneer factors or by a higher concentration of pioneer factors. iii) To validate the nucleosome occupancy score, we performed ChIP-seq for H3 as a proxy for nucleosome position in wild-type and MZnps mutant embryos. H3 signal showed a strong overlap with high intrinsic nucleosome occupancy across closed regions (Figures S6B and S6C). iv) H3-ChIP-seq signal was higher at the center of regions that lost accessibility in MZnps compared with wild-type embryos (Figures 6D and 6E), suggesting that nucleosomes occupy those sites that will be preferentially opened by NPS. In the absence of NPS, we observe the native preference for nucleosomes in the genome. Together these data indicate that nucleosomes facilitate, rather than impede, NPS pioneering activity during ZGA (Figure 6F; two-sample t-test, P < 1×10−100). Our analyses found this to be true for other TFs identified in early embryos beyond NPS, since TF motifs with the highest nucleosome occupancy or H3 ChIP-seq signal (top quartile), were significantly enriched in accessible sites across multiple TFs (Figure 6G; Fisher’s exact test, P < 0.01). These results suggest that differential nucleosome occupancy may be a general feature that facilitates pioneer factor activity in early development (Figures 6H and S6D).
Figure 6. Nucleosome occupancy facilitates pioneering activity of Nanog, Pou5f3, and Sox19b.

(A) Stacked bar plot showing the percentage of N/P/S-bound Group 1 regions that require one, two, or three NPS factors to rescue accessibility. All regions were ranked by average nucleosome occupancy score and then divided into quartiles. (B) Violin plot comparing the nucleosome occupancy scores between Nanog-bound regions that are rescued by single, double, or triple factor combination (two-sample t-test, *** P <0.001). On the x-axis, † denotes regions that require two or three factors for rescue. (C) Violin plot comparing nucleosome occupancy between low and high dosage groups in Figure 5D (two-sample t-test, *** P <0.001, ** P <0.01). (D) Heatmaps of Omni-ATAC, H3, and NPS binding signal, and nucleosome occupancy score (nuc score) at N/P/S-bound Group 1 regions that can be rescued by any single/double/triple NPS factors. Heatmaps are centered at Omni-ATAC peak summits and ranked by average intensity of Omni-ATAC signal in wild-type (WT) embryos. (E) Line plot of nucleosome occupancy score, H3 signal in MZnps embryos, and H3 signal (top 25% accessible) in WT embryos at the regions in (D). (F) Violin plot comparing the nucleosome occupancy between N/P/S-bound accessible regions and inaccessible regions (two-sample t-test, *** P <0.001). (G) Heatmaps showing accessibility enrichment for TF motifs (Figure 4B) in different quartiles ranked by nucleosome occupancy score, and H3 ChIP-seq signal in MZnps and WT. (H) Schematic illustrating how nucleosome occupancy facilitates pioneering activity. See also Figure S6.
Discussion
Here, we define how genomic context modulates the pioneering function of NPS in vivo; identify the network of TFs that, together with NPS, regulate chromatin accessibility during activation of the zygotic genome; and reveal that binding of these factors activates transcription through histone acetylation. These results provide the sequence of events and their causal relationships required for genome activation. We propose a model where nucleosome occupancy facilitates NPS pioneering function; NPS open half of the early enhancers, and activate the zygotic genome by recruiting histone acetyltransferases, such as p300, which acetylate the surrounding histone tails at enhancers and promoters; Brd4 then binds to acetylated histones and promotes Pol II recruitment and transcriptional activation, possibly via the Mediator complex (Cho et al., 2018; Gibson et al., 2019; Itzen et al., 2014; Sabari et al., 2018; Yang et al., 2008). Consistent with this model, Med13 is required for ZGA in mice (Miao et al., 2018), Zelda is required to promote H3K18ac in flies (Li et al., 2014), and chemical inhibition of p300 and Brd4 block ZGA in zebrafish (Chan et al., 2019). H4K16ac has been shown to regulate chromatin accessibility in flies (Samata et al., 2020). However, most promoters at NPS target genes remain open despite the loss of histone acetylation across canonical core histones in MZnps embryos, indicating that histone acetylation is dispensable to either initiate or maintain chromatin accessibility during ZGA. We identified an extensive network of TFs associated with accessible regions during ZGA, including SP1, NFY, FOXK1, BBX, and KLF9, which have the potential to regulate ZGA independently of NPS pioneering activity. Indeed, NFY and FoxH1 regulate early zygotic gene expression in mice during preimplantation development and in Xenopus during mesendoderm formation, respectively (Charney et al., 2017; Lu et al., 2016), suggesting a conserved regulatory mechanism for ZGA across vertebrates. Future studies will be needed to investigate the combinatorial role of these TFs during genome activation.
Pioneer factors play critical roles in embryo development, cellular reprogramming, and cancer (Zaret, 2020), however, the genomic features that define pioneering activity in vivo remain poorly understood. In our model, nucleosome occupancy provides a flexible landscape across the genome to determine where NPS function as pioneer factors or regular TFs. We show that each factor (N, P, or S) displays pioneering activity in vivo (Pálfy et al., 2020; Veil et al., 2019) (Figures 5C, S5B, and S5D), can initiate chromatin opening in the absence of the other two factors, and preferentially open regions of high nucleosome occupancy, while in other regions they function as canonical TFs that require the activity of other pioneer factors. Regions with lower nucleosome occupancy require a higher concentration of pioneer factors or a combination of NPS (Figures 6A-C and S6A), where these factors work cooperatively or additively to open the chromatin. We observe that high nucleosome occupancy is associated with chromatin opening across multiple TFs (Figure 6G), although we cannot exclude that this effect is secondary due to the association of some motifs with other genomic features correlated with high nucleosome occupancy. Our results are consistent with the role of nucleosomes in motif recognition in vitro (Soufi et al., 2015; Zhu et al., 2018) and the pioneering function for other TFs such as the progesterone receptor (Ballaré et al., 2013). High nucleosome affinity is associated with slow nucleosome conformational fluctuations (Tims et al., 2011). Thus, we speculate that nucleosomes at regions with high intrinsic nucleosome occupancy hold the DNA motif in a more favorable conformation for the pioneer factors to open the chromatin (Ballaré et al., 2013; Donaghey et al., 2018; Zhu et al., 2018). Alternatively, interactions between pioneer factors and core histones may facilitate pioneering function (Iwafuchi et al., 2020). Furthermore, the rearrangement of nucleosomes by the initial pioneer factors can then change the sites such that they have preferential affinity for a subsequent pioneer TFs (Figure S6D), providing differential chromatin accessibility across development. This might explain how the same pioneer TF can regulate different sites across different cell types (Donaghey et al., 2018). Altogether, our results provide insights into the molecular mechanisms underlying genome activation and reprogramming during the MZT and suggest that nucleosome occupancy is a general determinant for pioneering specificity, which provides a flexible platform to interpret the genome and integrate the input of different TFs across different cell types and developmental transitions.
Limitations of the Study
Our study reveals that NPS are required to establish chromatin opening, H3K4me1, and histone acetylation across core histones during ZGA. We show H3K27ac is dispensable for chromatin opening at 4 hpf. Because the fertilized egg shows an initial presence of H3K27ac that is quickly lost after fertilization (Zhang et al., 2018), we cannot exclude that the initial H3K27ac is required to initiate chromatin opening. Because of the technical challenges present at early-stage embryos due to the low cell number, we focused our analysis at 4 hpf when the major wave of zygotic transcription is established, and the number of cells facilitate genomic analysis. Future studies will be needed to investigate the relevance of these early acetylation events in chromatin remodeling and genome activation. While we focus on the early zygotic genes, further studies will be required to analyze the global structure of the genome, later zygotic gene expression, pluripotency, and differentiation. NPS are required to open more than half of active enchanters in the genome, however, only a fraction of zygotic genes is downregulated in MZnps, future studies will be needed to understand potential redundant roles of other TFs during ZGA.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Antonio J. Giraldez (antonio.giraldez@yale.edu).
Materials Availability
All mutant fish lines and reagents generated in this study are available upon request from the lead contact.
Data and Code Availability
Raw reads generated in this study are publicly available in the Sequence Read Archive (SRP355652). Processed data are available at https://data.giraldezlab.org/. All high-throughput sequencing datasets used in this paper are listed in Table S3. Original western blot images and microscopy data have been deposited at Mendeley and will be shared by the lead contact upon request.
This paper does not report original code. Software used are listed in the Key resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP | Thermo Fisher Scientific | Cat#A-11120; RRID:AB_221568 |
| Goat anti-mouse secondary antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat#A32723; RRID:AB_2633275 |
| Goat Anti-Rabbit IgG H&L (HRP) | Abcam | Cat#ab6721 RRID:AB_955447 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat#ab4729; RRID:AB_2118291 |
| Rabbit polyclonal anti-H2A.Z | Abcam | Cat#ab4174; RRID:AB_304345 |
| Rabbit polyclonal anti-H2BK15ac | Diagenode | Cat#C15410220 |
| Rabbit polyclonal anti-H2AK5ac | Diagenode | Cat#C15410215; |
| Rabbit monoclonal anti-BRD4 | Cell Signaling Technology | Cat#13440; RRID:AB_2687578 |
| Rabbit monoclonal anti-H2A.Z(Lys4/Lys7)ac | Cell Signaling Technology | Cat#75336, RRID:AB_2799867 |
| Rabbit monoclonal anti-H2BK5ac | Cell Signaling Technology | Cat#12799; RRID:AB_2636805 |
| Rabbit monoclonal anti-H3K14ac | Cell Signaling Technology | Cat#7627; RRID:AB_10839410 |
| Rabbit monoclonal anti-H3K36ac | Cell Signaling Technology | Cat#27683; RRID:AB_2798946 |
| Rabbit monoclonal anti-H4K16ac | Cell Signaling Technology | Cat#13534; RRID:AB_2687581 |
| Rabbit polyclonal anti-H3K9ac | Active Motif | Cat#39137; RRID:AB_2561017 |
| Rabbit polyclonal anti-H3K18ac | Abcam | Cat#ab1191; AB_298692 |
| Rabbit polyclonal anti-H4K8ac | Abcam | Cat#ab15823; RRID:AB_880455 |
| Rabbit polyclonal anti-H3K4me3 | Abcam | Cat#ab8580 RRID:AB_306649 |
| Rabbit polyclonal anti-H3K4me1 | Diagenode | Cat#C15310037 |
| Mouse monoclonal anti-Pol II [8WG16] | Abcam | Cat#ab817; RRID:AB_306327 |
| Mouse monoclonal anti-Pol II [8WG16] | BioLegend | Cat#664912; RRID:AB_2650945 |
| Goat polyclonal anti-Myc tag | Abcam | Cat#ab9132; RRID:AB_307033 |
| Goat anti-rabbit-IgG-Atto647N | Sigma-Aldrich | Cat#40839; RRID:AB_1137669 |
| Rabbit polyclonal anti-Actin | Sigma-Aldrich | Cat#A5060; RRID:AB_476738 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F3165; RRID:AB_259529 |
| Rabbit polyclonal anti-p300a | This paper | N/A |
| Rabbit polyclonal anti-Nfya | Charles Sagerström lab | N/A |
| Rabbit polyclonal anti-Eomesa | Fiona Wardle lab | N/A |
| Rabbit polyclonal anti-Brd4 | Dawid lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-amanitin | Sigma-Aldrich | Cat#A2263 |
| Triptolide | Sigma-Aldrich | Cat#T3652 |
| Flavopiridol hydrochloride hydrate | Sigma-Aldrich | Cat# F3055 |
| Critical Commercial Assays | ||
| ChIP DNA Clean & Concentrator Kits | ZYMO RESEARCH | Cat#D5205 |
| Nextera DNA Library Prep Kit | Illumina | Cat#FC-121–1030 |
| Click-iT™ RNA Alexa Fluor™ 594 Imaging Kit | Thermo Fisher Scientific | Cat#C10330 |
| Click-iT™ Nascent RNA Capture Kit | Thermo Fisher Scientific | Cat#C10365 |
| SuperScript™ VILO™ cDNA Synthesis Kit | Thermo Fisher Scientific | Cat#11754–050 |
| MAXIscript™ SP6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1308 |
| mMESSAGE mMACHINE™ T3 Transcription Kit | Thermo Fisher Scientific | Cat#AM1348 |
| mMESSAGE mMACHINE™ SP6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1340 |
| AmpliScribe™ T7-Flash™ Transcription Kit | Lucigen | Cat#ASF3257 |
| Deposited Data | ||
| Datasets for Omni-ATAC seq, ChIP-seq, Click-iT-seq, RNA-seq, CUT&TAG in WT and MZnps embryos | This paper | SRP355652 |
| Nanog ChIP-seq datasets | Xu et al., 2012 | GSE34683 |
| ATAC-seq datasets at oblong and sphere stages | Pálfy et al., 2020 | GSE130944 |
| ATAC-seq datasets at oblong and dome stages | Liu et al., 2018 | GSE101779 |
| CAGE datasets at high/oblong/sphere/shield stages | Nepal et al., 2013 | SRA055273 |
| Ribosome profiling datasets at 2 hpf | Bazzini et al., 2014 | GSE53693 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: MZnps, triple maternal-zygotic mutant for nanog−/−;pou5f3−/−;sox19b−/− | This paper | N/A |
| Zebrafish: TU-AB, and TL strains | N/A | N/A |
| Oligonucleotides (See Table S1 for oligonucleotides used) | ||
| Recombinant DNA | ||
| pT3TS-zdCas9–3XGFP | Chan et al., 2019 | N/A |
| pcDNA-dCas9-p300 Core | Hilton et al., 2015 | Addgene Plasmid #61357; RRID:Addgene_61357 |
| pT3TS-dCas9-HAT | This paper | N/A |
| pT3TS-dCas9-dHAT | This paper | N/A |
| pCS2+nanog | Lee et al., 2013 | N/A |
| pCS2+pou5f3 | Lee et al., 2013 | N/A |
| pCS2+pou5f3–6Xmyc | This paper | N/A |
| pCS2+sox19b-6Xmyc | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| Skewer | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| STAR | Dobin et al., 2012 | https://github.com/alexdobin/STAR |
| SAMtools | Li et al., 2009 | http://www.htslib.org |
| BEDTools | Quinlan et al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/ht ml/DESeq2.html |
| MEME suite | Bailey et al., 2009 | http://meme-suite.org |
| Nucleosome occupancy prediction model | Kaplan et al. 2009 | https://github.com/KaplanLab/NucleosomeModel |
| Python | Python 3.8 | https://www.python.org |
| R | R 3.6.2 | https://www.r-project.org |
| GeneAbacus | Charles E. Vejnar | https://github.com/vejnar/geneabacus |
| FONtools | Charles E. Vejnar | https://github.com/vejnar/fontools |
| LabxDB seq | Vejnar et al., 2020 | https://gitlab.com/vejnar/labxdb |
Experiment Model and Subject Details
Zebrafish maintenance, embryo treatment, and mutant generation
Fish lines were maintained in accordance with the AAALAC research guidelines following a protocol approved by the Yale University IACUC. Zebrafish husbandry and manipulation were performed as described (Howe et al., 2016; Westerfield, 2000).
Drug treatment: 1) α-amanitin, 1-cell stage embryos were injected with 0.2 ng of α-amanitin; 2) triptolide, embryos were bathed in 5.8 μM triptolide from the 1-cell stage; 3) triptolide + flavopiridol, embryos were bathed in 5.8 μM triptolide from the 1-cell stage, and 11.4 uM of flavopiridol was added starting from the 128-cell stage; 4) JQ1, embryos were bathed in 43.8 μM JQ1 (stock: 4.38 mM in DMSO) from 1-cell stage; 5) SGC-CBP30, embryos were bathed in 20 mg/ml SGC-CBP30 (stock: 10mg/ml in DMSO) from 1-cell stage.
nanog and sox19b mutants were generated using CRISPR/Cas9 (Deltcheva et al., 2011; Haurwitz et al., 2010; Moreno-Mateos et al., 2015) targeting the following sequences: “AGCCCGGGTCTTGCGGGGCAGGG” and “GCAAACGGCTCGGCGCGGAGTGG,” respectively. A 4-nucleotide deletion mutant for nanog and a 5-nucleotide deletion mutant for sox19b were identified and maintained. See Table S2 for all oligos used to generate the mutants. MZnanog embryos are not viable, as previously described (Gagnon et al., 2018; Veil et al., 2018). The nanog homozygous mutant line was maintained by injecting MZnanog embryos with 25 pg of nanog mRNA (Lee et al., 2013) at the 1-cell stage. Consistent with published work (Pálfy et al., 2020), the sox19b homozygous mutants are viable.
The nanog homozygous mutant was crossed with the sox19b homozygous mutant to obtain nanog+/−;sox19b+/− double heterozygotes, which were then crossed with pou5f3hi349Tg/hi349Tg mutants (Burgess et al., 2002) to obtain nanog+/−;pou5f3+/−;sox19b+/− triple heterozygotes. The nanog+/−;pou5f3+/−;sox19b+/− triple heterozygotes were intercrossed and the resulting embryos were injected with 30 pg of pou5f3 mRNA at the 1-cell stage. These embryos were raised and genotyped to obtain the nanog−/−;pou5f3−/−;sox19b−/− triple homozygous mutant. To maintain the MZnps mutant line, the nanog−/−;pou5f3−/−;sox19b−/− triple mutants were intercrossed and the embryos were injected with 25 pg of nanog mRNA and 30 pg of pou5f3 mRNA at the 1-cell stage. sox19b mRNA was not injected because the sox19b homozygous mutants are viable (Pálfy et al., 2020).
sgRNA synthesis and injection
sgRNAs were designed and synthesized as previously described (Vejnar et al., 2016). Briefly, sgRNAs were designed using CRISPRscan (Moreno-Mateos et al., 2015). sgRNA templates were amplified using the oligos listed in Table S2 together with the following universal reverse primer: 5′-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′ (Vejnar et al., 2016). sgRNAs were then synthesized using the AmpliScribe T7-Flash Transcription Kit (Lucigen) and purified by ethanol precipitation or with the RNeasy MinElute Cleanup Kit (QIAGEN). To generate mutants, 100 pg of Cas9 mRNA and 20 pg of sgRNA were injected into 1-cell stage embryos.
Histone acetylation restoration in MZnps embryos with the dCas9-HAT system
The histone acetyltransferase domain from human p300 was subcloned from a published construct (Addgene, 61357) (Hilton et al., 2015) into the pT3TS-dCas9 plasmid to obtain pT3TS-dCas9-HAT. The HAT domain was inactivated by introducing a point mutation (D1399Y) in pT3TS-dCas9-HAT with the primers listed in Table S2 to produce pT3TS-dCas9dHAT. dCas9-HAT mRNA and dCas9-dHAT mRNA were synthesized using the mMESSAGE mMACHINE T3 Transcription Kit (Thermo Fisher Scientific) according to manufacturer’s instructions.
For qPCR and RNA in situ hybridization following histone acetylation by dCas9-HAT, asb11 and her5 were targeted separately. Specifically, 200 pg of dCas9-HAT mRNA or dCas9-dHAT mRNA and 30 pg of each sgRNA targeting either asb11 or her5 (sgRNAs 1–3 for asb11 or sgRNAs 1–4 for her5) were injected into 1-cell stage embryos. When measuring transcript level by qPCR, asb11 and her5 sgRNAs were used as control sgRNAs for each other. For high-throughput sequencing experiments, asb11 and her5 were targeted simultaneously. 200 pg of dCas9-HAT mRNA or dCas9-dHAT mRNA together with 15 pg of each sgRNA (sgRNAs 1–3 for asb11 and sgRNAs 1–4 for her5) were injected into 1-cell stage embryos. For the NPS-bound enhancer targeting experiments, the following mix (see Table S2 for details) was injected into 1-cell stage embryos: single sgRNA, 26 sgRNAs (targeting 15 strongly downregulated genes) were mixed (5 pg each, one sgRNA per enhancer) with dCas9-HAT/dHAT; multiple sgRNAs, 10 sgRNAs (targeting asb11, her5, and apela) were mixed (15 pg each, two to three sgRNAs per enhancer) with dCas9-HAT/dHAT. Embryos were collected at 4 hpf for subsequent assays.
Method details
RNA in situ hybridization
Template DNAs for antisense RNA probes were amplified from 6 hpf cDNA using primers containing the T3-promoter sequence in the forward primer as listed in Table S2. Digoxigenin (DIG) labeled RNA probes were synthesized using T3 RNA Polymerase (Roche) and purified by ethanol precipitation. RNA in situ hybridization was performed as described (Giraldez et al., 2005). Briefly, MZnps embryos were injected at the 1-cell stage for targeted histone acetylation. Uninjected wild-type, uninjected MZnps embryos, and injected MZnps embryos were fixed at 4 hpf with 4% paraformaldehyde (PFA) overnight at 4°C. Fixed embryos were washed three times with phosphate-buffered saline (PBS) and dehydrated with a methanol series (25%, 50%, 75%, and 100% methanol). Dehydrated embryos were stored at −20°C for at least 24 hours and then rehydrated with a reverse methanol series. Pre-hybridization and hybridization were performed at 65°C for 3 hours and overnight, respectively. Embryos were washed extensively and blocked for 3 hours at room temperature before anti-DIG antibody incubation overnight at 4°C. After antibody incubation, embryos were stained with BCIP/NBT. Staining was stopped by incubating the embryos with 4% PFA overnight at 4°C. Embryos were washed briefly, mounted with a glycerol series (30%, 50%, 70%, and 86%), and imaged in 86% glycerol with a Zeiss stereo Discovery.V12 microscope.
RNA-seq and real-time quantitative PCR (qPCR)
Total RNA was extracted from embryos at 4 hpf using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer’s instructions and was treated with DNase I to remove any DNA contamination. Poly(A)-selected RNA-seq libraries were prepared and sequenced with an Illumina HiSeq system at the Yale Center for Genome Analysis. For qPCR, the extracted RNA was reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). qPCR was performed using Power Sybr Green PCR Master Mix (Thermo Fisher Scientific). The efficiency of all primer-sets used, listed in Table S2, was determined to be between 90%−110%. asb11 and her5 levels were normalized to actb1 (beta-actin1) (Yuan et al., 2010). Data were collected from three biological replicates. The two-tailed unpaired t-test was used to evaluate whether the transcription level was significantly different between two groups.
Western blotting
Zebrafish embryos from wild-type and MZnps embryos were dechorionated at the one-cell stage and were deyolked at 4 hpf following the batch deyolking method previously published (Link et al., 2006). Briefly, embryos were first shaken in deyolking buffer (55 mM NaCl, 1.8 mM KCl, 1.25 mM NaHCO3) before pelleting the cells at 300 g for 30 sec, and the cells were washed in deyolking wash buffer (110 mM NaCl, 3.5 mM KCl, 2.7 mM CaCl2, 10 mM Tris/Cl, pH 8.5). For western blotting, the cell pellets were resuspended in sample buffer (1x NuPAGE LDS Sample Buffer supplemented with DTT) and heated for 10 min at 95°C. Protein samples representing 20–40 deyolked embryos were resolved on a NuPAGE gel (3–8% Tris-Acetate gel with NuPAGE Tris-Acetate SDS Running Buffer for Brd4 and p300, 4–12% Bis-Tris gel with NuPAGE MOPS Running Buffer for all other proteins; Thermo Fisher Scientific) and transferred to a nitrocellulose membrane using the iBlot 2 Gel Transfer Device (Thermo Fisher Scientific). The membranes were blocked in 3–5% milk / PBS with 0.1% Tween-20 (PBST), incubated with primary antibody solution prepared in block solution, and then incubated with a peroxidase-conjugated secondary antibody solution prepared in block solution. Proteins were detected using SuperSignal West Pico PLUS Chemiluminescent Substrate or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
For detection of histone modifications, primary antibodies were used at 1:1000 dilution. Other primary antibodies were used at 1:2000 (actin), 1:500 (p300), 1:20,000 (Brd4) (Toyama et al., 2008), 1:600 (Eomesa), and 1:500 (Nfya). Secondary antibodies were used at 1:5000–1:10000 dilution.
Click-iT-seq
Click-iT-seq was performed as previously described (Chan et al., 2019). Briefly, 50 pmol of EU (5-ethynyl uridine), from the Click-iT™ Nascent RNA Capture Kit (C10365), was injected into 1-cell stage embryos. Total RNA was extracted from 20 embryos at 4 hpf using TRIzol reagent (Thermo Fisher Scientific). Nascent RNAs were captured, and cDNA was synthesized with the SuperScript VILO cDNA synthesis kit (11754–050) following the manufacturer’s instructions (C10365). Libraries were prepared from cDNA following the dUTP protocol and Illumina TruSeq protocol and were sequenced with an Illumina HiSeq system at the Yale Center for Genome Analysis.
Immunofluorescence and Click-iT RNA imaging
As previously described (Chan et al., 2019), 25 pg of dCas9–3xGFP mRNA together with two sgRNAs targeting miR-430 (100 pg each, see Table S2 for the amplification primers) were injected into 1-cell stage embryos. Embryos were fixed with 4% PFA at 2.5 hpf (the 256-cell stage) to perform immunofluorescence against GFP to label the miR-430 locus. To ensure that the loss of transcription at the miR-430 locus in the MZnps embryos can be detected by imaging, Click-iT RNA imaging was performed together with miR-430 locus labeling. Specifically, wildtype and MZnps embryos were injected with 25 pg of dCas9–3xGFP mRNA, 100 pg each of the two miR-430 targeting sgRNAs, and 50 pmol of EU at the 1-cell stage and fixed with 4% PFA at 2.5 hpf. Fixed embryos were washed three times with 1X PBST (phosphate-buffered saline + 0.5% Triton X-100), dehydrated with a methanol series (25%, 50%, 75%, and 100% methanol), stored at −20°C for at least 2 hours, and then rehydrated with a reverse methanol series.
Rehydrated embryos were blocked with 10% BSA (in PBST) for 2–3 hours and incubated with anti-GFP antibody (1:1000; Thermo Fisher Scientific, A-11120) overnight at 4°C. After three washes with 1X PBST, nascent transcripts were labeled using the Click-iT RNA Alexa Fluor 594 Imaging Kit (C10330) following the manufacturer’s instructions. Then, the embryos were incubated with Alexa Fluor Plus 488 anti-Mouse secondary antibody (1:1,000; Thermo Fisher Scientific, A32723). For H3K27ac staining, primary antibody (ab4729) and secondary antibody (40839, goat anti-rabbit-IgG-Atto647N) were diluted to 1:1000. Embryos were stained with DAPI, mounted in ProLong™ Diamond Antifade Mountant (Thermo Fisher Scientific, P36965) or 1% low-melt agarose, and imaged using confocal fluorescence microscopy (ZEISS LSM 880, Airyscan, ZEISS LSM 980 Airyscan2, or Leica TCS SP8). Images were processed using Image-J software (Schneider et al., 2012) and are displayed as maximum intensity Z-projections unless otherwise noted.
Image analysis
To measure the enrichment of Click-iT or H3K27ac at the miR-430 locus, nuclei were segmented based on DAPI and the mean nuclear intensity of Click-iT or H3K27ac was measured using FIJI (Schindelin et al., 2012). Then, individual miR-430 loci were manually identified based on dCas9–3xGFP staining and a circular region of interest was used to measure the intensity of Click-iT or H3K27ac at the loci in a single z-slice. Enrichment was determined by subtracting the mean nuclear intensity from the intensity measured at the miR-430 loci for each nucleus.
To quantify H3K27ac levels after SGC-CBP30 treatment, nuclei were initially segmented based on DAPI using the pixel classification and object classification workflows in ilastik (Berg et al., 2019). Object predictions were then imported to FIJI and a series of binary operations were performed to fill any holes in the nuclei. Connected components labelling was performed to label individual nuclei and then the intensity of H3K27ac was measured in each nuclei using MorphoLibJ (Legland et al., 2016).
Omni-ATAC
To reduce contamination by mitochondrial DNA, Omni-ATAC (Corces et al., 2017) was adapted to zebrafish based on published methods (Buenrostro et al., 2013; Corces et al., 2017; Liu et al., 2018). Briefly, at 4 hpf, five embryos were manually deyolked in PBS and resuspended in cold lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% NP40, 0.1% Tween-20, and 0.01% digitonin). The lysate was incubated on ice for 5 minutes, then 1 ml of dilution buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20) was mixed with the lysate by inverting several times and samples were centrifuged at 500 g for 10 minutes at 4°C. The supernatant was removed, and the purified nuclei were resuspended in the transposition reaction mixture (25 μl 2×TD Buffer, 2.5 μl Tn5 transposase, 22.5 μl Nuclease-Free water) and incubated for 30 minutes at 37°C. DNA was then purified with the Qiagen MinElute Kit (Qiagen, 28004). Libraries were prepared using NEBNext High-Fidelity 2X PCR Master Mix (NEB, M0541) with the following conditions: 72°C, 5 minutes; 98°C, 30 seconds; 15 cycles of 98°C, 10 seconds; 63°C, 30 seconds; and 72°C, 1 minute. Libraries were purified with Agencourt AMPureXP beads (Beckman Coulter Genomics, A63881) and sequenced with the Illumina NovaSeq 6000 System at the Yale Center for Genome Analysis.
To investigate how Nanog, Pou5f3, and Sox19b work separately and combinatorially to initiate chromatin opening, 25 pg of nanog mRNA, 30 pg of pou5f3 mRNA, and 5 pg of sox19b mRNA were injected separately or in combinations at the 1-cell stage. To test concentration dependency, a high dosage (25 pg of nanog mRNA, 30 pg of pou5f3 mRNA and 20 pg of sox19b mRNA) and a low dosage (2.5 pg of nanog mRNA, 5 pg of pou5f3 mRNA and 5 pg of sox19b mRNA) were injected at 1-cell stage. Genetically relevant females (nanog+/−;pou5f3−/−;sox19b−/−, nanog−/−;pou5f3+/−;sox19b−/−, nanog+/−;pou5f3+/−;sox19b−/−, nanog−/−;pou5f3+/−;sox19b+/−, nanog+/−;pou5f3+/;sox19b+/−) were used to confirm the requirement of different factors. Omni-ATAC was performed at 4 hpf to assess chromatin accessibility.
ChIP-seq
Chromatin immunoprecipitation (ChIP) was performed as described (Bogdanovic et al., 2013; Leichsenring et al., 2013; Nelson et al., 2014; Zhang et al., 2014). Briefly, embryos were dechorionated at the 1-cell stage. For each assay, 1000 embryos were fixed at 4 hpf with 1.9% PFA for 15 minutes, quenched with 0.125 M glycine, washed with cold PBS, and then frozen with liquid nitrogen and stored at −80°C until use. Nuclei were purified and lysed in 100 μl nuclei lysis buffer (50 mM Tris–HCl pH 7.5, 10 mM EDTA, 1% SDS) and diluted with 200 μl IP dilution buffer (16.7 mM Tris–HCl pH 7.5, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS). Lysates were sonicated with a Bioruptor Pico sonication device (Diagenode) using two rounds of 15 cycles of 30 seconds ON and 30 seconds OFF, with 15 minutes on ice in between rounds. Then, 27.6 μl of 10% Triton X-100 was added to the sonicated chromatin before centrifugation at 14,000 rpm for 10 minutes at 4°C and 5% of the supernatant was taken as input. Protein G Dynabeads (Invitrogen, 10003D) were pre-bound to antibodies overnight at 4°C and washed three times with 0.5% BSA in 1X PBS. 25 μl of antibody-bound beads was added to the remaining supernatant and incubated overnight at 4°C. Samples were washed five times with RIPA wash buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 0.7% DOC, 1% Igepal, 0.5 M LiCl) and two times with 1X TBS (Tris-buffered saline; 50 mM Tris pH 7.5, 150 mM NaCl). DNA– protein complexes were eluted with 660 μl elution buffer (50 mM NaHCO3, 1% SDS) at 65°C for 15 minutes with occasional vortexing. Input samples were thawed, and three volumes of elution buffer was added. NaCl was added to samples to a final concentration of 0.2 M and then the samples were reverse-crosslinked overnight at 65°C. Samples were treated with RNase A (final concentration: 0.33 μg/μl) for 2 hours and Proteinase K (final concentration: 0.2 μg/μl) for 2 hours before purification. Libraries were prepared following the Illumina TruSeq protocol and sequenced with the Illumina NovaSeq 6000 System at the Yale Center for Genome Analysis.
Antibodies against H3K27ac, H2A.Z, H3K4me1, H3K4me3, Eomesa, NFYa, and RNA Polymerase II were ChIP-validated in zebrafish embryos (Bogdanovic et al., 2013; Murphy et al., 2018; Nelson et al., 2014; Stanney et al., 2020; Vastenhouw et al., 2010; Zhang et al., 2014). Antibodies against H3K18ac and H4K8ac were ChIP-validated in Drosophila embryos (Li et al., 2014). For ChIP-seq against H3K27ac (ab4729), H3K18ac (ab1191), H4K8ac (ab15823), H2A.Z (ab4174), H2BK15ac (C15410220), H2AK5ac (C15410215), and p300 (using a rabbit polyclonal custom antibody generated by YenZyme Antibodies against the “CQGTNDMLTNNQDMGSNINH” peptide of zebrafish p300a; available upon request), 4 μg of antibody was used for each sample. 40 μl of anti-Eomesa antibody (Nelson et al., 2014), 10 μl of anti-Nfya antibody (Stanney et al., 2020), 20 μl of anti-BRD4 antibody (13440S), and 6 μl of acetylation antibodies (CST, [H2A.Z acetyl-Histone H2A.Z (Lys4/Lys7), 75336S; H2BK5ac, 12799S; H3K14ac, 7627S; H3K36ac, 27683S; and H4K16ac, 13534S], Active Motif, [H3K9ac, 39137]) were used for each sample. For ChIP-seq against RNA Polymerase II, 4 μg of anti-RNA polymerase II CTD repeat YSPTSPS antibody [8WG16] from Abcam (ab817) and 4 μg from BioLegend (664912) were used for each sample.
Because there are no antibodies available to perform ChIP-seq against zebrafish Pou5f3 or Sox19b, we overexpressed Myc-tagged Pou5f3 and Myc-tagged Sox19b and performed ChIP-seq using 4 μg of anti-Myc-tag antibody (ab9132) for each sample. In detail, a 6x Myc tag was cloned into to the N-terminus of Pou5f3 and C-terminus of Sox19b in the pCS2 backbone. The plasmids were linearized with NotI (NEB, R0189) and mRNA (myc-pou5f3 and sox19b-myc) was synthesized with the mMESSAGE mMACHINE SP6 Transcription Kit according to the manufacturer’s instructions. Wild-type embryos were injected with 20 pg of sox19b-myc mRNA or 30 pg of myc-pou5f3 mRNA at the 1-cell stage. To ensure that the enrichment of precipitated DNA was not due to non-specific binding of the anti-Myc antibody, ChIP-seq using the anti-Myc antibody was also performed on uninjected wild-type embryos.
CUT&TAG against H3K27ac
CUT&TAG was adapted from the published protocol (Kaya-Okur et al., 2019). Briefly, at 4 hpf, five embryos were manually deyolked in PBS and then dissociated by pipetting up and down in 400 μl Dig-wash Buffer (20 mM HEPES, pH 7.5; 150 mM NaCl; 0.5 mM Spermidine; 1X Protease inhibitor cocktail; 0.015% Digitonin). Cells were spun down (600 g for 3 minutes at 4°C), washed briefly with Dig-wash Buffer, and then resuspended in 50 μl of H3K27ac antibody buffer (2 mM EDTA and a 1:50 dilution of the H3K27ac antibody in Dig-wash Buffer). Cells were incubated with primary antibody at room temperature for 2 hours with gentle rotation. Cells were spun down and washed with Dig-wash Buffer (3 × 5 minutes) to remove unbound antibody. Then, cells were resuspended in 50 μl of Dig-med Buffer (20 mM HEPES, pH 7.5; 300 mM NaCl; 0.5 mM Spermidine; 1X Protease inhibitor cocktail; 0.015% Digitonin) containing a 1:200 dilution of the pA-Tn5 adapter complex (~0.04 μM) and incubated at room temperature for 1 hour with gentle rotation. After incubation, cells were spun down and washed with Dig-med Buffer (4 × 5 minutes) to remove unbound pA-Tn5 complexes. Then, cells were resuspended in 100 μl of Tagmentation buffer (10 mM MgCl2 in Dig-med Buffer) and incubated at 37°C for 1 hour. Tagmentation was stopped by adding 2.25 μl of 0.5 M EDTA, 5.5 μl of 10% SDS, and 1 μl of 20 mg/mL Proteinase K to the reaction. Next, DNA was extracted with 244 μl of AMPureXP beads (Beckman Coulter Genomics, A63881). Libraries were prepared with the NEBNext High-Fidelity 2X PCR Master Mix as in Omni-ATAC, purified with Agencourt AMPureXP beads, and sequenced with the Illumina NovaSeq 6000 System at the Yale Center for Genome Analysis.
High-throughput sequencing data management
LabxDB seq (Vejnar and Giraldez, 2020) was used to manage our high-throughput sequencing data and configure our analysis pipeline. Export to the Sequence Read Archive was achieved using the “export_sra.py” script from LabxDB Python.
Omni-ATAC data processing and analysis
Raw paired-end Omni-ATAC reads were adapter trimmed using Skewer (Jiang et al., 2014) and mapped to the zebrafish GRCz11 genome sequence (Yates et al., 2019) using Bowtie2 (v2.3.4.1) (Langmead and Salzberg, 2012) with parameters ‘-X 2000, --no-unal’. Unpaired and discordant reads were discarded. The alignments were deduplicated using samtools markdup (Li et al., 2009). Reads mapped to the + strand were offset by +4 bp and reads mapped to the – strand were offset by −5 bp (Buenrostro et al., 2013). Only fragments with insert size <=100 bp (effective fragments) were used to determine accessible regions. Genome tracks were created using BEDTools (Quinlan and Hall, 2010) and utilities from the UCSC genome browser (Lee et al., 2020). For all the genome tracks in the paper, signal intensity was in RPM (reads per million). Fragment coverage on each nucleotide was normalized to the total number of effective fragments in each sample per million fragments. For genome-wide analysis, reads mapped to mitochondrial DNA and contigs were filtered out and only uniquely mapped reads (with alignment quality ≥ 30) were used.
ChIP-seq data processing and analysis
Raw ChIP-seq reads were adapter trimmed, mapped, and deduplicated using the same method described in the previous section but using the default parameters for Bowtie2 for read mapping. GeneAbacus (https://github.com/vejnar/geneabacus) was used to create genomic profiles for creating tracks. -end samples, fragment coverage on each nucleotide was normalized to the total fragments in each sample per million fragments. For single-read samples, reads were extended to the predicted fragment size from MACS2 (see Peak calling below) (Zhang et al., 2008), using the GeneAbacus “--profile_type all-extension” and“--profile_extension_length” options, and read coverage on each nucleotide was calculated based on the extended reads and was normalized to the total number of reads in each sample per million reads. For genome-wide analysis, reads mapped to mitochondrial DNA and contigs were filtered out and only uniquely mapped reads (with alignment quality ≥ 30) were used.
CUT&TAG data processing and analysis
Raw CUT&TAG reads were adapter trimmed, mapped, deduplicated, and tracks were created using the same method as the Omni-ATAC reads. Reads mapped to the + strand were offset by +4 bp and reads mapped to the – strand were offset by −5 bp. Fragment coverage on each nucleotide was normalized to the total fragments in each sample per million fragments.
Peak calling
Peaks were called using MACS2 (Zhang et al., 2008) for Omni-ATAC and ChIP-seq data. For Omni-ATAC, peaks were called with the additional parameters ‘-f BEDPE --nomodel --keep-dup all’ with significance cutoff at P = 10-8. To determine high-confidence accessible regions, the three wild-type Omni-ATAC replicates were merged, and then peaks were called from the merged data. Only those peaks called from merged data that overlap with peaks called from all three biological replicates individually were defined as accessible regions. To compare the pioneering activity between Nanog, Pou5f3, and Sox19b, an accessible region was defined as being rescued if a peak was called from Omni-ATAC data in that rescue condition or genotype (at the significance level at P = 10−8) and overlap with the accessible region in wildtype.
For ChIP-seq of transcription factors, narrow peaks were called using MACS2 with the additional parameters ‘-f BEDPE --nomodel --keep-dup all’ for paired-end samples and ‘-f BAM --keep-dup all’ for single-read samples. Peaks from ChIP-seq of Nanog, Pou5f3, and Sox19b were called with the default significance cut-off (q = 0.05).
For ChIP-seq of histone modifications, broad peaks were called with the additional parameters ‘-f BEDPE --nomodel --keep-dup all -q 0.05 --broad --broad-cutoff 0.05’ to identify peaks for broad regions.
Differential accessibility analysis
To identify regions where accessibility was significantly changed in MZnps embryos, the fragment coverage of each accessible region of the three wild-type replicates was compared with that of the three MZnps replicates using DESeq2 (Love et al., 2014) with false discovery rate (FDR) < 0.01. Accessible regions with significantly lower Omni-ATAC signal in MZnps embryos were further subdivided into two categories: regions with no overlap with peaks called at a low significance cutoff (P = 10−3) from any MZnps Omni-ATAC replicate were classified as Group 1 (accessibility completely lost); and the other regions were classified as Group 2 (accessibility decreased but still accessible). Additionally, accessible regions where accessibility was not significantly changed and the fragment coverage difference between wild-type embryos and MZnps embryos was less than 30% were classified as Group 3 (accessibility unaffected).
Definition of promoters and active enhancers
Promoters were defined as the region within 500 bp of a transcription start site (TSS) of annotated transcripts from Ensembl v92 (Yates et al., 2019) and Cap Analysis of Gene Expression (CAGE) datasets (Nepal et al., 2013) at 3.3 hpf, 3.7 hpf, 4 hpf, and 6 hpf. The promoter of the most abundant transcript isoform, as determined by the CAGE data, was used to define the promoter of a gene. If no isoform had a maximum CAGE signal ≥ 0.5 RPM, the promoter was defined using a gene model that was constructed by merging all transcript isoforms annotated from Ensembl.
Active enhancers were defined as accessible regions outside of the promoter regions (TSS ± 500 bp) and overlapping with H3K27ac region (H3K27ac peak region with 200 bp extension on both sides). Active enhancers within 4.5 kb of the promoter of a zygotic gene (see below) were considered to be associated with the gene. If an active enhancer was within 4.5 kb of the promoters of multiple genes, the active enhancer was considered to be associated with the gene closest to it. The histone modification, H3 and H2AZ signal of each active enhancer was defined as the average signal across all nucleotides within the flanking 500 bp region from the Omni-ATAC peak summit. The histone modification, H3 and H2AZ signal of promoters was defined as the average signal within the promoter region.
RNA-seq and Click-iT-seq analysis
Raw reads were aligned to the zebrafish GRCz11 genome sequence using STAR 2.7.5b (Dobin et al., 2012) with the parameters ‘--alignEndsType Local –sjdbScore 2’. Genomic sequence index for STAR was built including exon-junction coordinates from Ensembl v92. Gene models were constructed using the “import_ensembl” tool from the FONtools (https://github.com/vejnar/fontools), which concatenates the isoforms of each gene together using the “--method union” option of the “fon_transform” tool. GeneAbacus (https://github.com/vejnar/geneabacus) was used to obtain read counts per gene with the following rules. Reads overlapping at least 10 nucleotides of the gene annotation were considered to be mapped to the gene. For read counts of the genes, each locus where a read was mapped was assigned a weight equal to 1 divided by the total number of loci to which the read was mapped. Read counts per gene were sum of the weights assigned to the gene. For miR-430, reads overlapping the region from coordinate 28,693,371 to 28,709,534 on chromosome 4 were counted as miR-430 cluster reads. Read counts from regular RNA-seq experiments were normalized to the total number of reads mapped to the zebrafish genes per million.
For Click-iT-seq experiments, read counts were normalized by the total number of mitochondrial reads mapped to mitochondrial protein coding genes. Zygotic genes were determined using a previously described method (Chan et al., 2019). Briefly, a zygotic gene was defined as a gene identified by (Heyn et al., 2014; Lee et al., 2013) as a zygotic gene, or a gene with a nascent transcription level ≥ 10 RPKM in exon regions and at least 4-fold more nascent transcription in wild-type than α-amanitin-treated embryos in exon or intron regions. Zygotic genes were classified into four categories based on the transcriptional change in the MZnps embryos compared to wild-type embryos: Strongly down: zygotic genes with nascent transcription down-regulated ≥ 8-fold. Mildly down: zygotic genes with nascent transcription down-regulated between 8- and 2-fold. Unaffected: zygotic genes with transcription not substantially changed (< 2-fold). Up: zygotic genes with nascent transcription up-regulated ≥ 2-fold.
Motif enrichment analysis
Motifs for Nanog, Pou5f3, Sox19b in zebrafish were identified using DREME in MEME suite (Bailey, 2011) based on all peaks called from the zebrafish ChIP-seq datasets at a significance cutoff at P = 10−5. Due to the high co-occupancy of Nanog and Pou5f3 and the similarity of their binding motifs, a second Nanog binding motif (Nanog motif 2) was identified from regions bound by Nanog that do not overlap with the binding regions of Pou5f3 and Sox19b. Pou5f3 motif, Sox19b motif, and both Nanog motifs were used to determine the contribution of the sequence context to the pioneering activities of different transcription factors. Motif enrichment was performed using AME in MEME suite (McLeay and Bailey, 2010) with default parameters on all known transcription factor binding motifs from the Motif database on the MEME suite website (http://meme-suite.org/doc/download.html) and HOMER website (http://homer.ucsd.edu/homer/custom.motifs). Motifs for human and mouse transcription factors were used as the motifs for their homologous transcription factors in zebrafish. Homologs between zebrafish and human and mouse were identified using BioMart on Ensembl genome browser (Yates et al., 2019). Only transcription factors (or transcription factor homologs in zebrafish) with mRNA translation rate ≥ 5 RPKM at 2 hpf in zebrafish embryos (Chan et al., 2019) were included in this analysis. The same sample size (either 1000 or the minimum sample size among the groups) was used for motif enrichment across different conditions to generate comparable significance levels between conditions.
Motif frequency and nucleosome occupancy
The motif frequency within each region was determined by the number of sequences matching the binding motif of the specific transcription factors using FIMO (Grant et al., 2011). Motif co-occurrence was determined if a region contains sequences that match the binding motifs of both transcription factors.
Nucleosome occupancy scores at nucleotide resolution were predicted using the model from (Kaplan et al., 2009). Nucleosome occupancy score of each accessible region was defined as the average nucleosome occupancy scores on all the nucleotides within the region. A nucleosome occupancy peak was defined as a region ≥ 100 bp, and each nucleotide has a nucleosome occupancy score ≥ 0.5 within that region. To assess the correlation between nucleosome occupancy and the number of factors required for rescue on N/P/S-bound Group 1 regions, all regions were ranked according to nucleosome occupancy score of the regions and were then divided into quartiles, and percentage of regions that require one, two and three factors to rescue accessibility within each quartile was then calculated. To calculate the enrichment of accessible motif within each quartile, the average nucleosome occupancy score and H3 ChIP-seq signal in MZnps and wild-type embryos were calculated at all motif locations and ranked from high to low, into quartiles. locations respectively. For each quartile, the enrichment of accessible motif was calculated by the percentage of open motifs within the quartile divided by the percentage of open motifs at all motif locations.
Heatmaps, plots, and networks
Heatmaps based on Omni-ATAC and ChIP-seq data were created using R 3.6 and the pheatmap package (https://github.com/raivokolde/pheatmap). Motif enrichment and histone acetylation/transcription correlation heatmaps were created using the R package gplots (https://github.com/talgalili/gplots). Box plots, bar plots, and violin plots were created using the R package ggplot2 (Wickham, 2016). Biplots and line plots were created using Python 3.8 and the Matplotlib library (Hunter, 2007).
Quantification and statistical analysis
For qPCR, statistical tests were performed on the data from three biological replicates using a two-tailed unpaired t-test. For imaging analysis, two-tailed unpaired t-test was performed on data from at least three embryos. Fisher’s exact test was performed to calculate the significance of co-binding of different factors in Venn diagrams and the difference in percentage between different groups. The two-sample t-test was performed to calculate the significance between different groups for the rest of this paper. Wilcoxon sign-ranked test was performed to calculate the significance between signals from the heatmaps in WT and MZnps. Pearson correlation was used to represent the correlation between two variables.
Supplementary Material
Supplemental Table 1 Maternally deposited sequence specific TFs and their motif enrichment in differentially affected accessible regions in MZnps, related to Figure 4
Supplemental Table 2 Oligonucleotides used, related to the STAR Methods
Supplemental Table 3 Summary of high-throughput sequencing experiments, related to the STAR Methods
Highlights.
NPS pioneer chromatin opening at more than half of active enhancers
NPS regulate histone acetylation across core histones at enhancers and promoters
Recruiting histone acetylation can bypass the function of NPS in gene activation
High nucleosome occupancy facilitates NPS pioneering activity
Acknowledgments
We thank Steven Henikoff, Karen Adelman, Fiona Wardle, Charles Sagerström, Igor Dawid, Jose Luis Gomez-Skarmeta, Juan Ramon Martinez Morales, Yong Zhang and Guifen Liu for sharing reagents and protocols; Kaya Bilguvar, Christopher Castaldi, Irina Tikhonova, and Emma Sykes from the Yale Center for Genome Analysis for sequencing support; Hiba Codore, Nitya Khatri, Eliakim de Guzman, Sarah Dube, Valeria Schmidt, and Timothy Gerson for technical help; Bluma Lesch, Zachary Smith, Andrew Xiao, Melissa Harrison, Ethan Strayer, Maria Benitez, Shawna Hiley, and Guy Riddihough for feedback on the manuscript.
Funding:
We thank the Surdna Foundation and the Yale Genetics Venture Fund for supporting L.M.; the Jane Coffin Childs Memorial Fund for supporting M.L.K.; the 2T32HD007149-41A1 NIH training grant and the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number F31HD104443 for supporting M.E.P. This work is supported by NIH grants R01 HD100035, HD074078, GM103789, GM102251, GM101108, and GM081602, R35 GM122580 and the 4DNucleome program.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballaré C, Castellano G, Gaveglia L, Althammer S, González-Vallinas J, Eyras E, Le Dily F, Zaurin R, Soronellas D, Vicent GP, and Beato M. (2013). Nucleosome-driven transcription factor binding and gene regulation. Mol Cell 49, 67–79. 10.1016/j.molcel.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Bazzini AA, Lee MT, and Giraldez AJ (2012). Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. 10.1126/science.1215704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nature Methods 16, 1226–1232. 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- Blythe SA, and Wieschaus EF (2016). Establishment and maintenance of heritable chromatin structure during early Drosophila embryogenesis. Elife 5. 10.7554/eLife.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic O, Fernandez-Minan A, Tena JJ, de la Calle-Mustienes E, and Gomez-Skarmeta JL (2013). The developmental epigenomics toolbox: ChIP-seq and MethylCap-seq profiling of early zebrafish embryos. Methods 62, 207–215. 10.1016/j.ymeth.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956. 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Reim G, Chen W, Hopkins N, and Brand M. (2002). The zebrafish spiel-ohne-grenzen (spg) gene encodes the POU domain protein Pou2 related to mammalian Oct4 and is essential for formation of the midbrain and hindbrain, and for pre-gastrula morphogenesis. Development (Cambridge, England) 129, 905–916. [DOI] [PubMed] [Google Scholar]
- Calo E, and Wysocka J. (2013). Modification of enhancer chromatin: what, how, and why? Mol Cell 49, 825–837. 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary PD, Crane-Robinson C, Bradbury EM, and Dixon GH (1982). Effect of acetylation on the binding of N-terminal peptides of histone H4 to DNA. Eur J Biochem 127, 137–143. 10.1111/j.14321033.1982.tb06847.x. [DOI] [PubMed] [Google Scholar]
- Chan SH, Tang Y, Miao L, Darwich-Codore H, Vejnar CE, Beaudoin JD, Musaev D, Fernandez JP, Benitez MDJ, Bazzini AA, et al. (2019). Brd4 and P300 Confer Transcriptional Competency during Zygotic Genome Activation. Dev Cell 49, 867–881 e868. 10.1016/j.devcel.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney RM, Forouzmand E, Cho JS, Cheung J, Paraiso KD, Yasuoka Y, Takahashi S, Taira M, Blitz IL, Xie X, and Cho KW (2017). Foxh1 Occupies cis-Regulatory Modules Prior to Dynamic Transcription Factor Interactions Controlling the Mesendoderm Gene Program. Dev Cell 40, 595–607 e594. 10.1016/j.devcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, and Zaret KS (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell 9, 279–289. 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY, et al. (2016). Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537, 548–552. 10.1038/nature19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghey J, Thakurela S, Charlton J, Chen JS, Smith ZD, Gu H, Pop R, Clement K, Stamenova EK, Karnik R, et al. (2018). Genetic determinants and epigenetic effects of pioneer-factor occupancy. Nat Genet 50, 250–258. 10.1038/s41588-017-0034-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckersley-Maslin MA, Alda-Catalinas C, and Reik W. (2018). Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat Rev Mol Cell Biol 19, 436–450. 10.1038/s41580-018-0008-z. [DOI] [PubMed] [Google Scholar]
- Fang F, Xu Y, Chew KK, Chen X, Ng HH, and Matsudaira P. (2014). Coactivators p300 and CBP maintain the identity of mouse embryonic stem cells by mediating long-range chromatin structure. Stem Cells 32, 1805–1816. 10.1002/stem.1705. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Obbad K, and Schier AF (2018). The primary role of zebrafish nanog is in extra-embryonic tissue. Development (Cambridge, England) 145. 10.1242/dev.147793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Wu K, Liu Z, Yao X, Yuan S, Tao W, Yi L, Yu G, Hou Z, Fan D, et al. (2018). Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell 173, 248–259 e215. 10.1016/j.cell.2018.02.028. [DOI] [PubMed] [Google Scholar]
- Garcia-Ramirez M, Rocchini C, and Ausio J. (1995). Modulation of chromatin folding by histone acetylation. J Biol Chem 270, 17923–17928. 10.1074/jbc.270.30.17923. [DOI] [PubMed] [Google Scholar]
- Ge SX, Jung D, and Yao R. (2019). ShinyGO: a graphical enrichment tool for animals and plants. Bioinformatics. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421. 10.1016/j.cell.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ (2010). microRNAs, the cell’s Nepenthe: clearing the past during the maternal-to-zygotic transition and cellular reprogramming. Curr Opin Genet Dev 20, 369–375. 10.1016/j.gde.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Cinalli RM, Glasner ME, Enright AJ, Thomson JM, Baskerville S, Hammond SM, Bartel DP, and Schier AF (2005). MicroRNAs regulate brain morphogenesis in zebrafish. Science 308, 833–838. 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, and Doudna JA (2010). Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355–1358. 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DA, Fedorov O, Martin S, Singleton DC, Tallant C, Wells C, Picaud S, Philpott M, Monteiro OP, Rogers CM, et al. (2014). Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J Am Chem Soc 136, 9308–9319. 10.1021/ja412434f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbes TR, Clayton AL, Thorne AW, and Crane-Robinson C. (1994). Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J 13, 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, and Neugebauer KM (2014). The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep 6, 285–292. 10.1016/j.celrep.2013.12.030. [DOI] [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517. 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe DG, Bradford YM, Eagle A, Fashena D, Frazer K, Kalita P, Mani P, Martin R, Moxon ST, Paddock H, et al. (2016). The Zebrafish Model Organism Database: new support for human disease models, mutation details, gene expression phenotypes and searching. Nucleic Acids Research 45, D758–D768. 10.1093/nar/gkw1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- Itzen F, Greifenberg AK, Bösken CA, and Geyer M. (2014). Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res 42, 7577–7590. 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M. (2019). The mechanistic basis for chromatin regulation by pioneer transcription factors. Wiley Interdiscip Rev Syst Biol Med 11, e1427. 10.1002/wsbm.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, and Zaret KS (2014). Pioneer transcription factors in cell reprogramming. Genes Dev 28, 2679–2692. 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, and Zaret KS (2016). Cell fate control by pioneer transcription factors. Development (Cambridge, England) 143, 1833–1837. 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi M, Cuesta I, Donahue G, Takenaka N, Osipovich AB, Magnuson MA, Roder H, Seeholzer SH, Santisteban P, and Zaret KS (2020). Gene network transitions in embryos depend upon interactions between a pioneer transcription factor and core histones. Nature Genetics 52, 418–427. 10.1038/s41588-020-0591-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lei R, Ding S-W, and Zhu S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182. 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SR, Pálfy M, Hilbert L, Kumar M, Karschau J, Zaburdaev V, Shevchenko A, and Vastenhouw NL (2017). Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife 6, e23326. 10.7554/eLife.23326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane DA, Hammerschmidt M, Mullins MC, Maischein HM, Brand M, van Eeden FJ, FurutaniSeiki M, Granato M, Haffter P, Heisenberg CP, et al. (1996). The zebrafish epiboly mutants. Development (Cambridge, England) 123, 47–55. [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, and Segal E. (2009). The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458, 362–366. 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications 10, 1930. 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski WA, and Becker PB (1998). Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc Natl Acad Sci U S A 95, 1540–1545. 10.1073/pnas.95.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladstätter S, and Tachibana K. (2019). Genomic insights into chromatin reprogramming to totipotency in embryos. J Cell Biol 218, 70–82. 10.1083/jcb.201807044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson ED, Marsh AJ, and Harrison MM (2021). Pioneering the developmental frontier. Mol Cell. 10.1016/j.molcel.2021.02.020. [DOI] [PMC free article] [PubMed]
- Lee CM, Barber GP, Casper J, Clawson H, Diekhans M, Gonzalez JN, Hinrichs AS, Lee BT, Nassar LR, Powell CC, et al. (2020). UCSC Genome Browser enters 20th year. Nucleic Acids Res 48, D756–D761. 10.1093/nar/gkz1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MT, Bonneau AR, Takacs CM, Bazzini AA, DiVito KR, Fleming ES, and Giraldez AJ (2013). Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature 503, 360–364. 10.1038/nature12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, and Andrey P. (2016). MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32, 3532–3534. 10.1093/bioinformatics/btw413. [DOI] [PubMed] [Google Scholar]
- Leichsenring M, Maes J, Mossner R, Driever W, and Onichtchouk D. (2013). Pou5f1 transcription factor controls zygotic gene activation in vertebrates. Science 341, 1005–1009. 10.1126/science.1242527. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Subgroup GPDP (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Guo F, Gao Y, Ren Y, Yuan P, Yan L, Li R, Lian Y, Li J, Hu B, et al. (2018). Single-cell multiomics sequencing of human early embryos. Nature Cell Biology 20, 847–858. 10.1038/s41556-018-0123-2. [DOI] [PubMed] [Google Scholar]
- Li XY, Harrison MM, Villalta JE, Kaplan T, and Eisen MB (2014). Establishment of regions of genomic activity during the Drosophila maternal to zygotic transition. Elife 3. 10.7554/eLife.03737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link V, Shevchenko A, and Heisenberg C-P (2006). Proteomics of early zebrafish embryos. BMC Developmental Biology 6, 1. 10.1186/1471-213X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Xu Q, Wang Q, Feng S, Lai F, Wang P, Zheng F, Xiang Y, Wu J, Nie J, et al. (2020). The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 587, 139–144. 10.1038/s41586-020-2847-y. [DOI] [PubMed] [Google Scholar]
- Liu G, Wang W, Hu S, Wang X, and Zhang Y. (2018). Inherited DNA methylation primes the establishment of accessible chromatin during genome activation. Genome Res 28, 998–1007. 10.1101/gr.228833.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. (2006). The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38, 431–440. 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Inoue A, Suzuki T, Zhao K, and Zhang Y. (2016). Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell 165, 1375–1388. 10.1016/j.cell.2016.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. 10.1186/1471-2105-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019a). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47, D419–D426. 10.1093/nar/gky1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019b). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14, 703–721. 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao YL, Gambini A, Zhang Y, Padilla-Banks E, Jefferson WN, Bernhardt ML, Huang W, Li L, and Williams CJ (2018). Mediator complex component MED13 regulates zygotic genome activation and is required for postimplantation development in the mouse. Biol Reprod 98, 449–464. 10.1093/biolre/ioy004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, and Giraldez AJ (2015). CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12, 982–988. 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PJ, Wu SF, James CR, Wike CL, and Cairns BR (2018). Placeholder Nucleosomes Underlie Germline-to-Embryo DNA Methylation Reprogramming. Cell 172, 993–1006 e1013. 10.1016/j.cell.2018.01.022. [DOI] [PubMed] [Google Scholar]
- Nelson AC, Cutty SJ, Niini M, Stemple DL, Flicek P, Houart C, Bruce AE, and Wardle FC (2014). Global identification of Smad2 and Eomesodermin targets in zebrafish identifies a conserved transcriptional network in mesendoderm and a novel role for Eomesodermin in repression of ectodermal gene expression. BMC Biol 12, 81. 10.1186/s12915-014-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal C, Hadzhiev Y, Previti C, Haberle V, Li N, Takahashi H, Suzuki AM, Sheng Y, Abdelhamid RF, Anand S, et al. (2013). Dynamic regulation of the transcription initiation landscape at single nucleotide resolution during vertebrate embryogenesis. Genome Res 23, 1938–1950. 10.1101/gr.153692.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pálfy M, Schulze G, Valen E, and Vastenhouw NL (2020). Chromatin accessibility established by Pou5f3, Sox19b and Nanog primes genes for activity during zebrafish genome activation. PLoS Genet 16, e1008546. 10.1371/journal.pgen.1008546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Coactivator condensation at super-enhancers links phase separation and gene control. Science 361. 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samata M, Alexiadis A, Richard G, Georgiev P, Nuebler J, Kulkarni T, Renschler G, Basilicata MF, Zenk FL, Shvedunova M, et al. (2020). Intergenerationally Maintained Histone H4 Lysine 16 Acetylation Is Instructive for Future Gene Activation. Cell 182, 127–144 e123. 10.1016/j.cell.2020.05.026. [DOI] [PubMed] [Google Scholar]
- Sato Y, Hilbert L, Oda H, Wan Y, Heddleston JM, Chew TL, Zaburdaev V, Keller P, Lionnet T, Vastenhouw N, and Kimura H. (2019). Histone H3K27 acetylation precedes active transcription during zebrafish zygotic genome activation as revealed by live-cell analysis. Development (Cambridge, England) 146. 10.1242/dev.179127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KN, and Harrison MM (2019). Mechanisms regulating zygotic genome activation. Nature Reviews Genetics 20, 221–234. 10.1038/s41576-018-0087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, and Vakoc CR (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54, 728–736. 10.1016/j.molcel.2014.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555568. 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanney W 3rd, Ladam F, Donaldson IJ, Parsons TJ, Maehr R, Bobola N, and Sagerström CG (2020). Combinatorial action of NF-Y and TALE at embryonic enhancers defines distinct gene expression programs during zygotic genome activation in zebrafish. Dev Biol 459, 161–180. 10.1016/j.ydbio.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, and Lipshitz HD (2009). The maternal-to-zygotic transition: a play in two acts. Development (Cambridge, England) 136, 3033–3042. 10.1242/dev.033183. [DOI] [PubMed] [Google Scholar]
- Tims HS, Gurunathan K, Levitus M, and Widom J. (2011). Dynamics of nucleosome invasion by DNA binding proteins. J Mol Biol 411, 430–448. 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama R, Rebbert ML, Dey A, Ozato K, and Dawid IB (2008). Brd4 associates with mitotic chromosomes throughout early zebrafish embryogenesis. Dev Dyn 237, 1636–1644. 10.1002/dvdy.21576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Sera T, Wolffe AP, and Hansen JC (1998). Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol 18, 4629–4638. 10.1128/mcb.18.8.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vastenhouw NL, Cao WX, and Lipshitz HD (2019). The maternal-to-zygotic transition revisited. Development (Cambridge, England) 146. 10.1242/dev.161471. [DOI] [PubMed] [Google Scholar]
- Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, and Schier AF (2010). Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464, 922–926. 10.1038/nature08866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veil M, Schaechtle MA, Gao M, Kirner V, Buryanova L, Grethen R, and Onichtchouk D. (2018). Maternal Nanog is required for zebrafish embryo architecture and for cell viability during gastrulation. Development (Cambridge, England) 145. 10.1242/dev.155366. [DOI] [PubMed] [Google Scholar]
- Veil M, Yampolsky LY, Grüning B, and Onichtchouk D. (2019). Pou5f3, SoxB1, and Nanog remodel chromatin on high nucleosome affinity regions at zygotic genome activation. Genome Res 29, 383–395. 10.1101/gr.240572.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vejnar CE, and Giraldez AJ (2020). LabxDB: Versatile databases for genomic sequencing and lab management. Bioinformatics. 10.1093/bioinformatics/btaa557. [DOI] [PMC free article] [PubMed]
- Vejnar CE, Moreno-Mateos MA, Cifuentes D, Bazzini AA, and Giraldez AJ (2016). Optimized CRISPR-Cas9 System for Genome Editing in Zebrafish. Cold Spring Harb Protoc; 2016. 10.1101/pdb.prot086850. [DOI] [PubMed] [Google Scholar]
- Wang Z, Oron E, Nelson B, Razis S, and Ivanova N. (2012). Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 10, 440–454. 10.1016/j.stem.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, and Zhao K. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40, 897–903. 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. (2000). The zebrafish book: a guide for the laboratory use of zebrafish (Danio rerio). Eugene OR, University of Oregon Press. [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2016). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 978-3-319-24277-4. [Google Scholar]
- Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W, et al. (2016). The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 534, 652–657. 10.1038/nature18606. [DOI] [PubMed] [Google Scholar]
- Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, et al. (2018). Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557, 256–260. 10.1038/s41586-018-0080-8. [DOI] [PubMed] [Google Scholar]
- Xia W, Xu J, Yu G, Yao G, Xu K, Ma X, Zhang N, Liu B, Li T, Lin Z, et al. (2019). Resetting histone modifications during human parental-to-zygotic transition. Science, eaaw5118. 10.1126/science.aaw5118. [DOI] [PubMed]
- Yang Z, He N, and Zhou Q. (2008). Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol Cell Biol 28, 967–976. 10.1128/mcb.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yartseva V, and Giraldez AJ (2015). The Maternal-to-Zygotic Transition During Vertebrate Development: A Model for Reprogramming. Curr Top Dev Biol 113, 191–232. 10.1016/bs.ctdb.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al. (2019). Ensembl 2020. Nucleic Acids Research 48, D682–D688. 10.1093/nar/gkz966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Zhou J, Deng M, Liu X, Le Bras M, de The H, Chen SJ, Chen Z, Liu TX, and Zhu J. (2010). Small ubiquitin-related modifier paralogs are indispensable but functionally redundant during early development of zebrafish. Cell Res 20, 185–196. 10.1038/cr.2009.101. [DOI] [PubMed] [Google Scholar]
- Zaret KS (2020). Pioneer Transcription Factors Initiating Gene Network Changes. Annu Rev Genet. 10.1146/annurev-genet-030220-015007. [DOI] [PMC free article] [PubMed]
- Zaret KS, and Carroll JS (2011). Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25, 2227–2241. 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Wu X, Zhang W, Shen W, Sun Q, Liu K, Zhang Y, Wang Q, Li Y, Meng A, and Xie W. (2018). Widespread Enhancer Dememorization and Promoter Priming during Parental-to-Zygotic Transition. Mol Cell 72, 673–686 e676. 10.1016/j.molcel.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Vastenhouw NL, Feng J, Fu K, Wang C, Ge Y, Pauli A, van Hummelen P, Schier AF, and Liu XS (2014). Canonical nucleosome organization at promoters forms during genome activation. Genome Res 24, 260–266. 10.1101/gr.157750.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. 10.1038/s41586-018-0549-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Maternally deposited sequence specific TFs and their motif enrichment in differentially affected accessible regions in MZnps, related to Figure 4
Supplemental Table 2 Oligonucleotides used, related to the STAR Methods
Supplemental Table 3 Summary of high-throughput sequencing experiments, related to the STAR Methods
Data Availability Statement
Raw reads generated in this study are publicly available in the Sequence Read Archive (SRP355652). Processed data are available at https://data.giraldezlab.org/. All high-throughput sequencing datasets used in this paper are listed in Table S3. Original western blot images and microscopy data have been deposited at Mendeley and will be shared by the lead contact upon request.
This paper does not report original code. Software used are listed in the Key resource table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GFP | Thermo Fisher Scientific | Cat#A-11120; RRID:AB_221568 |
| Goat anti-mouse secondary antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat#A32723; RRID:AB_2633275 |
| Goat Anti-Rabbit IgG H&L (HRP) | Abcam | Cat#ab6721 RRID:AB_955447 |
| Rabbit polyclonal anti-H3K27ac | Abcam | Cat#ab4729; RRID:AB_2118291 |
| Rabbit polyclonal anti-H2A.Z | Abcam | Cat#ab4174; RRID:AB_304345 |
| Rabbit polyclonal anti-H2BK15ac | Diagenode | Cat#C15410220 |
| Rabbit polyclonal anti-H2AK5ac | Diagenode | Cat#C15410215; |
| Rabbit monoclonal anti-BRD4 | Cell Signaling Technology | Cat#13440; RRID:AB_2687578 |
| Rabbit monoclonal anti-H2A.Z(Lys4/Lys7)ac | Cell Signaling Technology | Cat#75336, RRID:AB_2799867 |
| Rabbit monoclonal anti-H2BK5ac | Cell Signaling Technology | Cat#12799; RRID:AB_2636805 |
| Rabbit monoclonal anti-H3K14ac | Cell Signaling Technology | Cat#7627; RRID:AB_10839410 |
| Rabbit monoclonal anti-H3K36ac | Cell Signaling Technology | Cat#27683; RRID:AB_2798946 |
| Rabbit monoclonal anti-H4K16ac | Cell Signaling Technology | Cat#13534; RRID:AB_2687581 |
| Rabbit polyclonal anti-H3K9ac | Active Motif | Cat#39137; RRID:AB_2561017 |
| Rabbit polyclonal anti-H3K18ac | Abcam | Cat#ab1191; AB_298692 |
| Rabbit polyclonal anti-H4K8ac | Abcam | Cat#ab15823; RRID:AB_880455 |
| Rabbit polyclonal anti-H3K4me3 | Abcam | Cat#ab8580 RRID:AB_306649 |
| Rabbit polyclonal anti-H3K4me1 | Diagenode | Cat#C15310037 |
| Mouse monoclonal anti-Pol II [8WG16] | Abcam | Cat#ab817; RRID:AB_306327 |
| Mouse monoclonal anti-Pol II [8WG16] | BioLegend | Cat#664912; RRID:AB_2650945 |
| Goat polyclonal anti-Myc tag | Abcam | Cat#ab9132; RRID:AB_307033 |
| Goat anti-rabbit-IgG-Atto647N | Sigma-Aldrich | Cat#40839; RRID:AB_1137669 |
| Rabbit polyclonal anti-Actin | Sigma-Aldrich | Cat#A5060; RRID:AB_476738 |
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F3165; RRID:AB_259529 |
| Rabbit polyclonal anti-p300a | This paper | N/A |
| Rabbit polyclonal anti-Nfya | Charles Sagerström lab | N/A |
| Rabbit polyclonal anti-Eomesa | Fiona Wardle lab | N/A |
| Rabbit polyclonal anti-Brd4 | Dawid lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-amanitin | Sigma-Aldrich | Cat#A2263 |
| Triptolide | Sigma-Aldrich | Cat#T3652 |
| Flavopiridol hydrochloride hydrate | Sigma-Aldrich | Cat# F3055 |
| Critical Commercial Assays | ||
| ChIP DNA Clean & Concentrator Kits | ZYMO RESEARCH | Cat#D5205 |
| Nextera DNA Library Prep Kit | Illumina | Cat#FC-121–1030 |
| Click-iT™ RNA Alexa Fluor™ 594 Imaging Kit | Thermo Fisher Scientific | Cat#C10330 |
| Click-iT™ Nascent RNA Capture Kit | Thermo Fisher Scientific | Cat#C10365 |
| SuperScript™ VILO™ cDNA Synthesis Kit | Thermo Fisher Scientific | Cat#11754–050 |
| MAXIscript™ SP6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1308 |
| mMESSAGE mMACHINE™ T3 Transcription Kit | Thermo Fisher Scientific | Cat#AM1348 |
| mMESSAGE mMACHINE™ SP6 Transcription Kit | Thermo Fisher Scientific | Cat#AM1340 |
| AmpliScribe™ T7-Flash™ Transcription Kit | Lucigen | Cat#ASF3257 |
| Deposited Data | ||
| Datasets for Omni-ATAC seq, ChIP-seq, Click-iT-seq, RNA-seq, CUT&TAG in WT and MZnps embryos | This paper | SRP355652 |
| Nanog ChIP-seq datasets | Xu et al., 2012 | GSE34683 |
| ATAC-seq datasets at oblong and sphere stages | Pálfy et al., 2020 | GSE130944 |
| ATAC-seq datasets at oblong and dome stages | Liu et al., 2018 | GSE101779 |
| CAGE datasets at high/oblong/sphere/shield stages | Nepal et al., 2013 | SRA055273 |
| Ribosome profiling datasets at 2 hpf | Bazzini et al., 2014 | GSE53693 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: MZnps, triple maternal-zygotic mutant for nanog−/−;pou5f3−/−;sox19b−/− | This paper | N/A |
| Zebrafish: TU-AB, and TL strains | N/A | N/A |
| Oligonucleotides (See Table S1 for oligonucleotides used) | ||
| Recombinant DNA | ||
| pT3TS-zdCas9–3XGFP | Chan et al., 2019 | N/A |
| pcDNA-dCas9-p300 Core | Hilton et al., 2015 | Addgene Plasmid #61357; RRID:Addgene_61357 |
| pT3TS-dCas9-HAT | This paper | N/A |
| pT3TS-dCas9-dHAT | This paper | N/A |
| pCS2+nanog | Lee et al., 2013 | N/A |
| pCS2+pou5f3 | Lee et al., 2013 | N/A |
| pCS2+pou5f3–6Xmyc | This paper | N/A |
| pCS2+sox19b-6Xmyc | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| Skewer | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| STAR | Dobin et al., 2012 | https://github.com/alexdobin/STAR |
| SAMtools | Li et al., 2009 | http://www.htslib.org |
| BEDTools | Quinlan et al., 2010 | https://bedtools.readthedocs.io/en/latest/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/ht ml/DESeq2.html |
| MEME suite | Bailey et al., 2009 | http://meme-suite.org |
| Nucleosome occupancy prediction model | Kaplan et al. 2009 | https://github.com/KaplanLab/NucleosomeModel |
| Python | Python 3.8 | https://www.python.org |
| R | R 3.6.2 | https://www.r-project.org |
| GeneAbacus | Charles E. Vejnar | https://github.com/vejnar/geneabacus |
| FONtools | Charles E. Vejnar | https://github.com/vejnar/fontools |
| LabxDB seq | Vejnar et al., 2020 | https://gitlab.com/vejnar/labxdb |