

Abstract
Coronaviruses (CoVs) have complex genomes that encode a fixed array of structural and nonstructural components, as well as a variety of accessory proteins that differ even among closely related viruses. Accessory proteins often play a role in the suppression of immune responses and may represent virulence factors. Despite their relevance for CoV phenotypic variability, information on accessory proteins is fragmentary. We applied a systematic approach based on homology detection to create a comprehensive catalogue of accessory proteins encoded by CoVs. Our analyses grouped accessory proteins into 379 orthogroups and 12 super‐groups. No orthogroup was shared by the four CoV genera and very few were present in all or most viruses in the same genus, reflecting the dynamic evolution of CoV genomes. We observed differences in the distribution of accessory proteins in CoV genera. Alphacoronaviruses harboured the largest diversity of accessory open reading frames (ORFs), deltacoronaviruses the smallest. However, the average number of accessory proteins per genome was highest in betacoronaviruses. Analysis of the evolutionary history of some orthogroups indicated that the different CoV genera adopted similar evolutionary strategies. Thus, alphacoronaviruses and betacoronaviruses acquired phosphodiesterases and spike‐like accessory proteins independently, whereas horizontal gene transfer from reoviruses endowed betacoronaviruses and deltacoronaviruses with fusion‐associated small transmembrane (FAST) proteins. Finally, analysis of accessory ORFs in annotated CoV genomes indicated ambiguity in their naming. This complicates cross‐communication among researchers and hinders automated searches of large data sets (e.g., PubMed, GenBank). We suggest that orthogroup membership is used together with a naming system to provide information on protein function.
Keywords: accessory proteins, coronavirus, naming system, phosphodiesterase, remote homology
1. INTRODUCTION
Coronaviruses (CoVs, family Coronaviridae, order Nidovirales) are a large family of nonsegmented, positive‐sense RNA viruses that infect a wide range of animal hosts. As of 2022, seven human CoVs are known, all of them zoonotic in origin (Cui et al., 2019; Forni et al., 2017; Zhou et al., 2020). Three of such human CoVs are highly pathogenic (SARS‐CoV‐2, SARS‐CoV and MERS‐CoV), whereas the other four (HCoV‐OC43, HCoV‐NL63, HCoV‐229E and HCoV‐HKU1) usually cause mild symptoms (Cui et al., 2019; Forni et al., 2017; Zhou et al., 2020). All these viruses belong either to the genus Alphacoronavirus or genus Betacoronavirus, which are divided into several subgenera. For instance, SARS‐CoV and SARS‐CoV‐2 belong to the subgenus Sarbecovirus, whereas MERS‐CoV is classified in the subgenus Merbecovirus (https://talk.ictvonline.org/taxonomy/) (Coronaviridae Study Group of the International Committee on Taxonomy of Viruses, 2020). Two additional CoV genera, Gammacoronavirus and Deltacoronavirus, include viruses that mainly infect birds, but also cetaceans, pigs and other mammals (Wille & Holmes, 2020).
The advent of high‐throughput sequencing technologies has resulted in the identification of an outstanding number of novel viruses, and the emergence of SARS‐CoV‐2 has spurred efforts to characterize CoV genetic diversity, particularly in bats, which are considered the ultimate reservoir from which SARS‐CoV‐2 spilled over (Annan et al., 2013; Anthony et al., 2017; Corman et al., 2015; Corman, Ithete, et al., 2014; Hu et al., 2017; Lam et al., 2020; Latinne et al., 2020; Lau et al., 2015; Tao et al., 2017; Wang, Fu et al., 2017; Yang et al., 2014; Zhou et al., 2021). These studies have indicated that alphaCoVs and betaCoVs are highly diverse, especially in bats, rodents and other small mammals, and that they often switch among hosts (Latinne et al., 2020; Li et al., 2021; Tsoleridis et al., 2019; Wang et al., 2015, 2020; Wang, Lin et al., 2017; Zhou et al., 2021). Likewise, a large diversity of gammaCoVs and deltaCoVs is hosted by wild birds, and cross‐species transmission from birds to pigs is probably responsible for the emergence of porcine deltacoronavirus (Wille & Holmes, 2020).
The host‐switching ability of CoVs is thought to be at least partially due to their genome plasticity and rampant recombination (Forni et al., 2017). CoV genomes are unusually long and complex compared to those of other RNA viruses. The genomic organization and some encoded proteins are shared among all CoVs. Specifically, two‐thirds of the genome consists of two large open reading frames (ORF1a and ORF1b), which overlap by a few nucleotides at their termini. Both ORF1a and ORF1b are translated into polyproteins. The remaining portion of the genome includes ORFs for the structural proteins, namely spike (S), envelope (E), membrane (M) and nucleoprotein (N) (Forni et al., 2017). However, CoVs also encode a variable number of accessory proteins, which differ in sequence and number even among closely related viruses. Some of these accessory products such as phosphodiesterases (PDEs), lectin‐like molecules and immunoglobulin domain‐containing proteins were probably acquired from vertebrate hosts, whereas others originated by duplication or by the exchange of genetic material with other viruses (De Sabato et al., 2020; Forni et al., 2017; Tan et al., 2020; Wang et al., 2020; Zhang et al., 2013). Because they are usually dispensable for viral replication, these accessory ORFs are frequently lost through mutation or deletion, leading to a highly dynamic evolution of gene content. This ability to acquire and shuffle protein‐coding genes is thought to contribute to host adaptation (Forni et al., 2017). Importantly, CoV accessory proteins often play a role in the suppression of immune responses or in immune evasion and, for this reason, some of them represent virulence factors (Forni et al., 2017). This is the case, for instance, of the MHV (mouse hepatitis virus) NS2a protein, a PDE that blocks the oligoadenylate synthetase (OAS)‐RNase L pathway and leads to hepatitis development (Zhao et al., 2012). In general, the dispensability of accessory proteins for viral replication makes them an exceptional raw material for the evolution of new phenotypes.
Despite their potential relevance for CoV ecology and phenotypic variability, a comprehensive catalogue of accessory ORFs is lacking and the evolutionary history of most of these proteins remains unexplored. Moreover, the partial annotation of several CoV genomes and the absence of a standardized nomenclature system to classify these proteins makes it difficult to obtain a full picture of the overall distribution and representation of accessory ORFs in CoV genomes. To fill such gap in knowledge, we performed a systematic search and evolutionary analysis of accessory proteins in CoVs.
2. MATERIAL AND METHODS
2.1. Viral sequence selection
All complete or coding‐complete CoV genomes were retrieved from the NCBI virus database (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/) and viral genomes belonging to the same genus were aligned using mafft version 7.475 (Katoh & Standley, 2013). We then calculated nucleotide pairwise identity for each genus alignment and we retained strains showing less than 99% identity. Specifically, we calculated pairwise identity scores between all sequence combinations and, for each genome, we removed all those showing identity ≥99% (Table S1). Pairwise identity scores were calculated as 1 − (M/N), where M is the number of mismatching nucleotides and N is the total number of positions along the alignment at which neither sequence has a gap or an undetermined character. This procedure generated a list of 107 alphaCoVs, 136 betaCoVs, 293 gammaCoVs and 34 deltaCoVs.
2.2. Accessory protein identification and orthology inference
For each of these selected genomes, we downloaded all annotated accessory protein sequences from the NCBI database. Proteins annotated as “nonfunctional” were removed from the analyses.
To limit biases of annotation accuracy among different viral strains, all viral genomes (with the exclusion of the ORF1a/ORF1b regions) were also analysed with the orffinder tool version 0.4.3 (Sayers et al., 2011). This tool searches for ORFs in a given genome and identifies potential protein‐coding segments. Specifically, we searched the positive strand in all three frames for all possible ORFs with a length of at least 25 codons and with “ATG” as the start codon. Because of the large number of resulting ORFs, additional filtering procedures were adopted. Thus, out‐of‐frame overlapping genes (OLGs) were analysed using the CodScr+SeqComp method (Pavesi, 2021). This approach relies on one of five prediction criteria: one from the codon scrambling (CodScr) method (Pavesi, 2000) and four from the sequence composition (SeqComp) method (Pavesi, 2020). The former is based on codon usage, whereas the latter uses known differences in nucleotide and amino acid composition to predict OLGs. Only OLGs confirmed by the five criteria were included in downstream analyses. These ORFs and all non‐OLGs predicted by orffinder were then merged with NCBI annotations.
Annotated and predicted proteins were analysed to infer possible orthologies with orthofinder version 2.5.4 (Emms & Kelly, 2015, 2019). orthofinder identifies hierarchical OrthoGroups (OGs) as groups of genes that are descended from a common gene in the last common ancestor; an OG can be composed of orthologues and paralogues.
orthofinder was run using “multiple sequence alignment” as the method for gene tree inference, mafft as an aligner (Katoh & Standley, 2013) and fasttree (Price et al., 2010) for tree inferences. Coronavirus genera were analysed independently.
All non‐OLGs predicted by orffinder and longer than 50 codons were included. However, to limit the number of potentially spurious ORFs, we retained non‐OLGs shorter than 50 codons only if, based on orthofinder results, they clustered with one or more annotated proteins.
2.3. Remote homology detection
Protein sequences belonging to the same OG were then aligned with mafft and the generated multiple sequence alignment was used as a template to search for remote homology.
Sequence identity searches are not well suited for the inference of distant relationship among protein families. Conversely, profile hidden Markov models (HMMs) and 3D‐structure comparisons have proven to be more sensitive. We thus applied the hhpred and hhblits tools (Alva et al., 2016; Remmert et al., 2011) to characterize all OGs identified by orthofinder. Only hhpred/hhblits hits with a probability higher than 90% were considered significant (Gabler et al., 2020). The probability criterion is the most sensitive for both hhpred and hhblits (Gabler et al., 2020). For hhpred we used PDB_mmCIF70_12_Oct, SCOPe70_2.07, ECOD_F70_20200717, and UniProt‐SwissProt‐viral70_23_Aug_2020 as target databases; all other parameters were set as defaults, both for hhpred and hhblits.
2.4. Alignment and phylogenetic analyses
Phylogenetic maximum‐likelihood (ML) trees were generated by the iq‐tree software version 1.6.12 (Trifinopoulos et al., 2016). iq‐tree was run by selecting the best‐fitting evolutionary model selected according to the Bayesian Information Criterion (BIC), followed by reconstruction of an ML tree (MFP + MERGE option). For all trees, branch supports were calculated using the ultrafast bootstrap method (UFBoot) with 1000 bootstraps.
To infer phylogenetic relationships among distantly related proteins, two different approaches were applied. In the first approach, sequences were aligned using the “Expresso” mode (Armougom et al., 2006) from the t‐coffee multiple sequence alignment package (Notredame et al., 2000). Expresso uses blast to identify homologues within the PDB database and uses the 3D structure information as a template to build the multiple sequence alignment (Armougom et al., 2006). This alignment was then used as input for the iq‐tree software as described above.
In the second approach, phylogenetic relationships were reconstructed using bali‐phy (Redelings & Suchard, 2005; Suchard & Redelings, 2006). This method jointly infers the alignment and the tree by applying a Bayesian Markov chain Monte Carlo (MCMC) approach. Specifically, we ran three different analyses with default parameters and, after checking for the potential scale reduction factor to be <1.01, we combined them to find the consensus tree. Branch support was evaluated by posterior probabilities.
2.5. Recombination
Alignments of the RdRp domains were screened for the presence of recombination using the gard tool (Kosakovsky Pond et al., 2006) from the hyphy package (Pond et al., 2005). This tool uses phylogenetic incongruence among segments in the alignment to identify recombination breakpoints. Recombination events were considered significant when showing a p‐value <.01.
No breakpoint was detected for alphaCoVs, betaCoVs and gammaCoVs. gard detected two breakpoints in the deltaCoV RdRP alignment. Thus, the longest nonrecombinant region was selected (420 aa) and a phylogenetic tree were generated as described above.
2.6. Molecular modelling and protein analysis
Ab initio structural modelling was performed using rosettafold (Baek et al., 2021), an automated software tool that uses deep learning to accurately predict protein structures based on their sequence. The global confidence of each model is evaluated according to the mean of the per‐residue local distance difference test (lDDT), ranging from 0.0 (low quality) to 1.0 (high quality), by using the deep learning network DeepAccNect (Hiranuma et al., 2021). Low‐confidence portions characterized by a per‐residue error estimate >5 Å, probably corresponding to highly flexible/disordered regions, have been excluded both from model structures and estimation of lDDT scores.
Signal peptides were predicted using signalp‐5.0 (Almagro Armenteros et al., 2019), transmembrane helices using tmhmm and phobius (Kall et al., 2007; Krogh et al., 2001).
3D structures were analysed with the software pymol (Schrödinger, 2017), which was also used to create protein figures.
3. RESULTS
3.1. Accessory ORF identification and classification
Our first aim was to obtain a comprehensive catalogue of accessory proteins encoded by CoVs and to reconstruct their evolutionary relationships. CoV genome sequences available in public repositories are annotated with different levels of accuracy, and some of them display no accessory ORF annotation. We thus used a strategy based on both available annotations and ORF prediction, as summarized in Figure 1. Specifically, we analysed CoV genomes selected to have less than 99% identity in pairwise comparisons (Table S1). Annotated accessory protein sequences were downloaded; also, all genomes (with the exclusion of the ORF1a/ORF1b regions) were used as an input for orffinder, a program that identifies all ORFs of a selected minimum size (25 amino acids in this case) in a sequence (Sayers et al., 2011). OLGs (out‐of‐frame overlapping genes) were only included if they were also confirmed by the CodScr+SeqComp method (see Materials and Methods). We next combined annotated and predicted proteins in a common data set and we identified OGs with orthofinder, which defines an OG as a group of genes that are descended from a common gene in the last common ancestor (Emms & Kelly, 2015, 2019). To limit the number of false positive orffinder predictions, we discarded predicted non‐OLGs shorter than 50 codons that did not belong to an OG including at least one annotated protein. Using these criteria, orthofinder identified 188 OGs for alphaCoVs, 107 for betaCoVs, 50 for gammaCoVs and 34 for deltaCoVs. Such OGs included very different numbers of orthologues, from one to 288 (https://github.com/dforni5/CoVaccessory).
FIGURE 1.
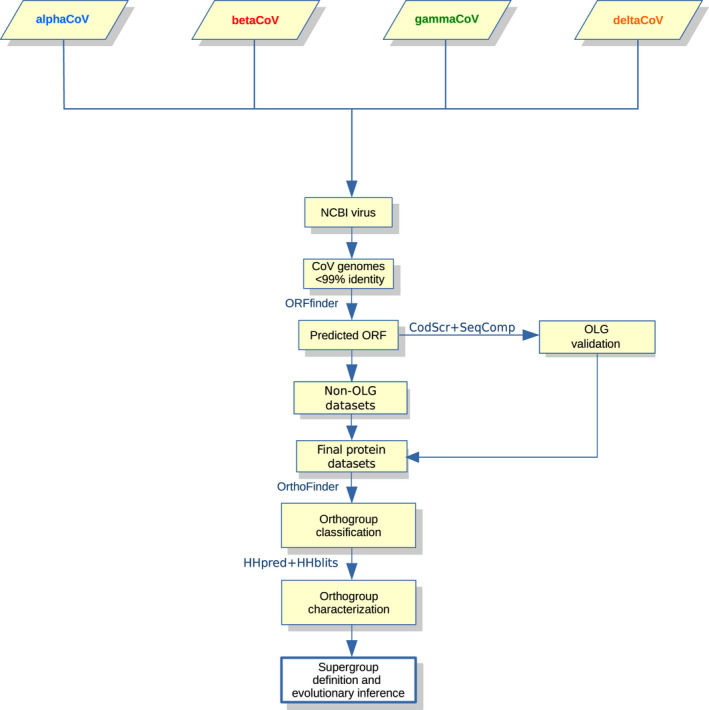
Overview of the applied workflow. Schematic representation of the workflow applied in this study for the characterization of CoV accessory proteins and for the identification of orthogroups/supergroups
We next characterized OGs and determined whether some of them might share distant sequence or structure homology. To this end, we ran hhpred and hhblits on alignments of orthologs in all OGs (Alva et al., 2016; Remmert et al., 2011). Both methods were developed to search for remote homologues and we only considered hits with a probability higher than 90%, which corresponds to a conservative cutoff (Gabler et al., 2020). These analyses revealed known and unknown homologies among OGs. For instance, in line with recent works (Tan et al., 2021), we found that orthologues in the large OG to which the SARS‐CoV‐2 ORF3a protein belongs form a super‐group with other coronavirus proteins (all denoted by the light green colour in Figure 2). These include ORF5 (also known as NS5) from MERS‐CoV, proteins in alphaOG01, which are encoded by all analysed alphaCoVs, as well as M proteins from CoVs and toroviruses (Table 1, Figure 2). Indeed, it has been suggested that accessory proteins in this OG emerged via duplication of a gene encoding an M protein followed by diversification (as opposed to conservation of M) (Ouzounis, 2020; Tan et al., 2021). We will refer to these OGs as the ORF3a‐like super‐group (Table 1). Likewise, orthofinder placed the sarbecovirus ORF8 and ORF7a proteins in the same OG, which showed homology to alphaOG18, as previously reported (Neches et al., 2021; Tan et al., 2020). These OGs are thus referred to as the ORF7a/ORF8‐like super‐group (denoted by the dark green colour in Figure 2) (Table 1). Similarly, homology was found between two OGs in deltaCoVs and gammaCoVs (Figure 3 and Table 1). By searching for among‐OG homologies we reconstructed a total of 12 super‐groups (Table 1). However, no OG was found to be common to more than two subgenera (Table 1).
FIGURE 2.
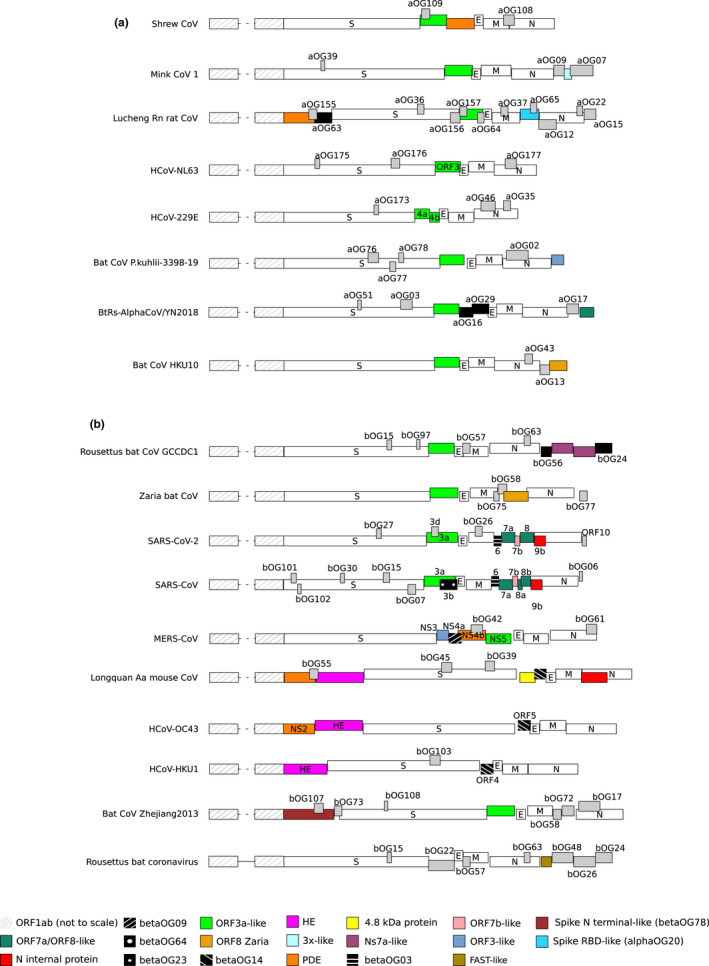
AlphaCoV and betaCoV genome organization. A schematic genome organization of representative alphaCoV (a) and betaCoV (b) genomes. Super‐groups and relevant orthogroups are coloured as shown in the key; known and unknown orthogroups are coloured in black and grey, respectively, and orthogroup names are reported. For all viruses, ORF1ab is not shown to scale and structural proteins are coloured in white. For human coronaviruses, accessory protein names are also reported. Viruses reported in the figure are as follows: Common shrew coronavirus Tibet‐2014: KY370053; mink coronavirus 1: MN535737; Lucheng Rn rat coronavirus: MT820627; HCoV‐NL63: NC_005831; HCoV‐229E: NC_002645; Alphacoronavirus bat‐CoV/P.kuhlii/Italy/3398–19/2015: NC_046964; coronavirus BtRs‐AlphaCoV/YN2018: MK211373; Rousettus bat coronavirus HKU10: NC_018871; Rousettus bat coronavirus GCCDC1: MT350598; Zaria bat coronavirus: HQ166910; SARS‐CoV‐2: NC_045512; SARS‐CoV: NC_004718; MERS‐CoV: NC_019843; Longquan aa mouse coronavirus: KF294357; HCoV‐OC43: NC_006213; HCoV‐HKU1: NC_006577; bat Hp‐betacoronavirus/Zhejiang2013: NC_025217; Rousettus bat coronavirus isolate GCCDC1 356: NC_030886
TABLE 1.
Super‐group classification and description
| Super‐group | OGs | Contributing viruses | hhpred/hhblits results |
|---|---|---|---|
| ORF3a‐like | alphaOG01 | All alphaCovs | Homology to a number of proteins encoded by betaCoVs (betaOG02 and betaOG22) |
| betaOG02 | Most sarbecoviruses; includes SARS‐CoV‐2 ORF3a | Homology to proteins encoded by alphaCoVs (alphaOG01) and betaCoVs (betaOG22), as well to the coronavirus/torovirus M protein | |
| betaOG08 | All merbecoviruses; includes MERS‐CoV ORF5/NS5 | Homology to HKU9 proteins in betaOG22 | |
| betaOG22 | Most hibecoviruses and nobecoviruses | Homology to a number of proteins encoded by alphaCoVs (alphaOG01) and beta CoVs (betaOG02), as well to the coronavirus/torovirus M protein | |
| ORF7a/ORF8‐like | alphaOG18 | Rodent coronaviruses (luchacoviruses) | Homology to SARS‐Cov‐2 ORF7a (betaOG01) |
| betaOG01 | All sarbecoviruses; includes SARS‐CoV‐2 ORF7a/ORF8 | Homology only to ORFs in the same OG | |
| PDE | alphaOG21 | Most rodent alphaCoVs (luchacoviruses) | Homology to coronavirus/torovirus/rotavirus PDEs; homology to cellular PDEs (AKAP7) |
| alphaOG107 | Only one alphaCoV (Shrew‐CoV) | Homology to coronavirus/torovirus/rotavirus PDEs; homology to cellular PDEs (AKAP7) | |
| betaOG10 | All merbecoviruses; includes MERS‐CoV NS4b | Homology to coronavirus/torovirus/rotavirus PDEs; homology to cellular PDEs (AKAP7) | |
| betaOG16 | Most embecoviruses | Homology to coronavirus/torovirus/rotavirus PDEs; homology to cellular PDEs (AKAP7) | |
| N internal protein | alphaOG02 | Several alphaCoVs | Homology to proteins encoded by betaCoVs (betaOG17) |
| betaOG04 | Subset of sarbecoviruses; includes SARS‐CoV‐2 ORF9b | Homology only to ORFs in the same OG | |
| betaOG12 | Most merbecoviruses | Homology to proteins encoded by betaCoVs (betaOG17). Also homology (84% probability) to sarbecovirus ORF9b protein (betaOG04) | |
| betaOG17 | Most embecoviruses | Homology to proteins encoded by betaCoVs (betaOG12) | |
| 4.8‐kDa protein | betaOG18 | Subset of embecoviruses | No homology (excluding embecovirus proteins) |
| betaOG66 | Only one embecovirus (Buffalo coronavirus B1‐28F) | Homology to the HS4 protein of MHV (betaOG18) | |
| Ns7a‐like | betaOG33 | Subset of nobecoviruses | Homology to HKU9 nonstructural protein 7a |
| betaOG40 | Subset of nobecoviruses | Homology to HKU9 nonstructural protein 7a | |
| betaOG48 | Subset of nobecoviruses; includes HKU9 | No homology | |
| ORF7b‐like | betaOG05 | Several sarbecoviruses; includes SARS‐CoV‐2 ORF7b | Homology (probability 92%) to uncharacterized baculovirus proteins; however, homology extends across a short low‐complexity region, suggesting a false positive result |
| betaOG65 | Subset of sarbecoviruses | Homology to SARS‐CoV and SARS‐CoV‐2 ORF7b | |
| ORF8 Zaria | alphaOG11 | Subset of decacoviruses; one duvinacovirus | Homology to Zaria bat coronavirus ORF8 (betaOG76) |
| betaOG76 | Only Zaria bat coronavirus | No homology | |
| ORF3‐like | alphaOG50 | Two Nyctacovirus; one unclassified alphaCoV | Homology to MERS‐CoV and HKU5 ORF3 (betaOG11) |
| betaOG11 | All merbecoviruses; includes MERS‐CoV ORF3/NS3 | No homology | |
| 3x‐like | alphaOG08 | Carnivore CoVs (minacoviruses) | No homology |
| alphaOG172 | FIPV | Homology to 3x‐like proteins of ferret/mink CoVs (alphaOG08) | |
| 4b‐like | gammaOG03 | Most gammaCoVs; includes IBV 4b | Homology to deltaCoV NS6 proteins (deltaOG1) |
| deltaOG01 | All deltaCoVs | Homology to IBV 4b protein (gammaOG03) | |
| 3b‐like | gammaOG05 | Most gammaCoVs; includes IBV 3b | Homology to deltaCoV 7b proteins (deltaOG11) |
| deltaOG11 | Subset of deltaCoVs | Homology to IBV 3b protein (gammaOG05) |
FIGURE 3.
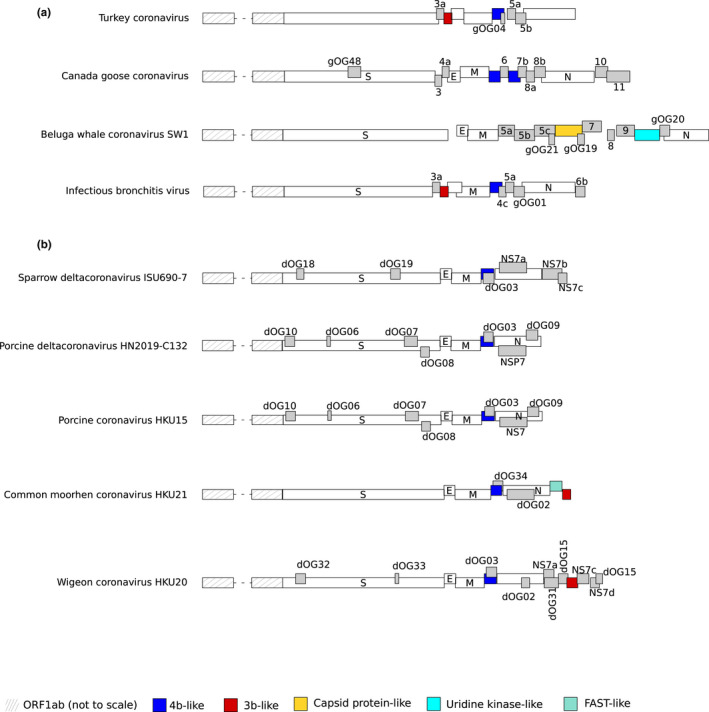
GammaCoV and deltaCoV genome organization. A schematic genome organization of representative gammaCoV (a) and deltaCoV (b) genomes. Super‐groups and relevant orthogroups are coloured as shown in the key; unknown orthogroups are coloured in grey and orthogroup names are reported. For all viruses, ORF1ab is not shown to scale and structural proteins are coloured in white. Viruses reported in the figure are as follows: Turkey coronavirus: NC_010800; Canada goose coronavirus: NC_046965; beluga whale coronavirus: NC_010646; infectious bronchitis virus: KP662631; sparrow deltacoronavirus: MG812376; porcine deltacoronavirus HN2019‐C132: MN520206; porcine coronavirus HKU15: NC_039208; common moorhen coronavirus: NC_016996; Wigeon coronavirus: NC_016995
Clearly, a few super‐groups simply reflect the fact that some accessory proteins encoded by viruses in the same CoV genus/subgenus diverged to such a degree that orthofinder assigned them to distinct OGs. Others unveiled the ancestral common origin of CoVs or the exchange of genetic material between genera. Thus, in addition to the ORF3a‐like and ORF7a/ORF8‐like super‐groups, we found that a small subset of alphaCoVs encode a putative accessory ORF (denoted by the light orange colour in Figure 2) showing homology to a protein encoded by Zaria bat CoV, a betaCoV sequenced from Nigerian bats (Figure 2; Table 1) (Quan et al., 2010). Likewise, three alphaCoV genomes harbour ORFs related to the ORF3 protein encoded by MERS‐CoV (light blue colour in Figure 2) (Table 1). Some other super‐groups suggest that CoVs independently acquired genetic material from their hosts or from other viruses, as exemplified by PDEs (Forni et al., 2017; Zhang et al., 2013). One super‐group included PDEs from alphaCoVs and betaCoVs, but we also identified a divergent, unannotated PDE in a shrew alphaCoV (all of them denoted in orange, Figure 2; Table 1) (see below).
Particular mention is also necessary for a super‐group of proteins internal to the N gene. AlphaOG02 included proteins predicted in several alphaCoV genomes, whereas betaOG12 and betaOG17 included internal proteins encoded by merbecoviruses (including MERS‐CoV) and embecoviruses (including HKU1) (red colour in Figure 2; Table 1). Proteins in these OGs were previously reported to display no mutual sequence homology, and also no similarity to the ORF9b proteins (betaOG04) encoded by sarbecoviruses (Wong et al., 2020). However, hhpred identified mutual structural homology between betaOG12 and betaOG17. hhpred also detected structural homology between proteins in betaCoV OG12 and SARS‐CoV‐2 ORF9b, although with only 84% probability.
Finally, homology was found between proteins in gammaOG05 (sometimes annotated as 3b) and proteins in deltaOG11 (referred to as 7b). Because these proteins show distinct locations in gammaCoV and deltaCoV genomes, they may have originated from intergenus recombination (Figure 3).
3.2. Distribution of accessory proteins and OGs in CoV genomes
We next analysed the distribution of OGs and of accessory proteins in CoV genomes. BetaCoV genomes displayed, on average, more accessory ORFs (median: eight ORFs per genome) than all other genera (medians: six, seven and seven for alphaCoVs, deltaCoVs and gammaCoVs, respectively) (Figures 2 and 3). In fact, although we identified a larger overall number of OGs in alphaCoVs, OGs tended to be specific to one or a few viruses in this genus (Figure 4a). This held true for the other genera as well, but an appreciable number of OGs were also shared by several viruses (Figure 4a). Thus, alphaCoVs encode, on average, fewer accessory proteins per genome than viruses in other genera, but accessory ORFs are more specific to subsets of alphaCoVs. With the exclusion of the ORF3a‐like proteins in alphaCoVs (Figure 5a) and of deltaOG1 (Table 1, Figure 6a), no OG was shared by all CoVs in the same genus, and very few were present in more than 50% of the analysed viruses (Figure 4a, https://github.com/dforni5/CoVaccessory), reflecting the high plasticity and dynamic evolution of CoV genomes. Also, as previously shown for SARS‐CoV ORF8 and HCoV‐229E ORF4, some CoVs carry split accessory ORFs (Figure 2) (Forni et al., 2017).
FIGURE 4.
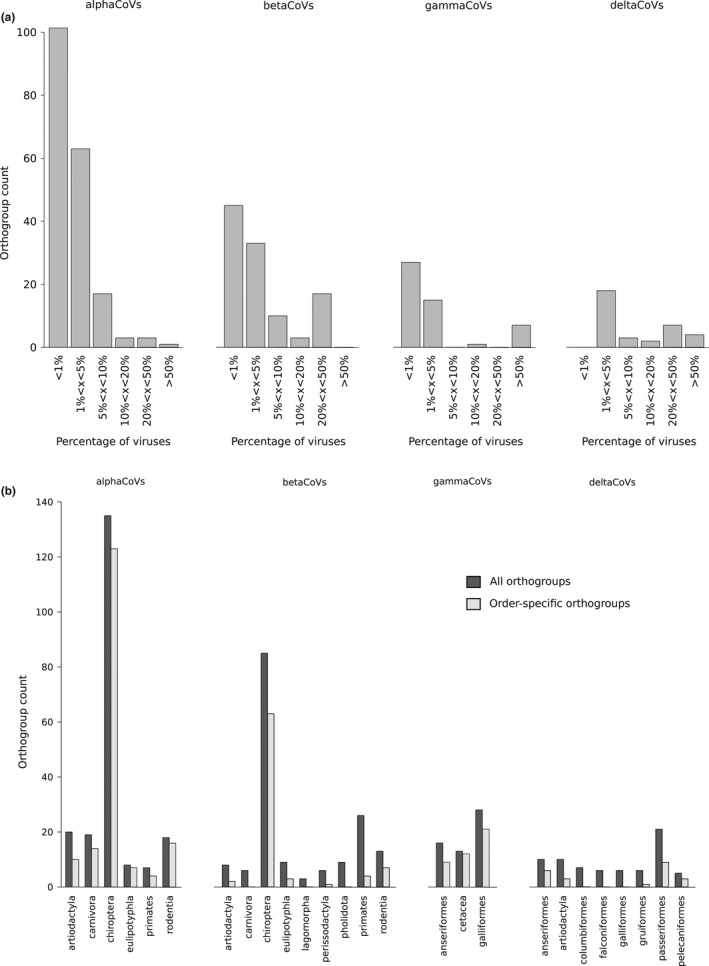
OG representation in coronaviruses. (a) Counts of OGs shared by different fractions of viruses (from less than 1% to more than 50%). (b) Distribution of OGs by host group. OG counts in different virus host groups are shown (dark bars). Light grey bars indicate the number of OGs that are host group‐specific
FIGURE 5.
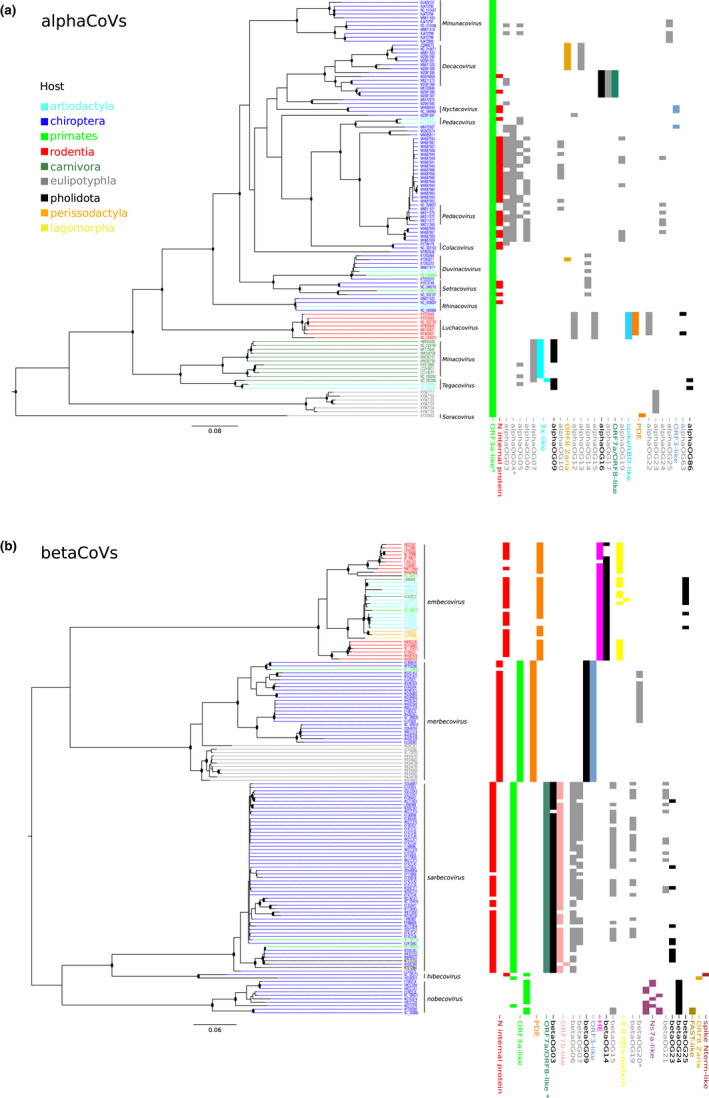
Alpha CoV and beta CoV phylogenies and orthogroup distribution. Maximum‐likelihood phylogenetic tree of the RdRp region of alphaCoVs (a) and betaCoVs (b) generated by iq‐tree using the LG + I + F + G4 and LG + R5 models. Internal nodes with bootstrap values >80% are shown as black dots and tips are coloured according to viral hosts (see key). The distribution of all orthogroups among viral species is also reported. Super‐groups and relevant orthogroups are shown with the same colours as in Figure 2; known and unknown orthogroups are coloured in black and grey, respectively. Asterisks indicate orthogroups with split/paralogous ORFs. Scale bars are expressed as substitutions per site
FIGURE 6.
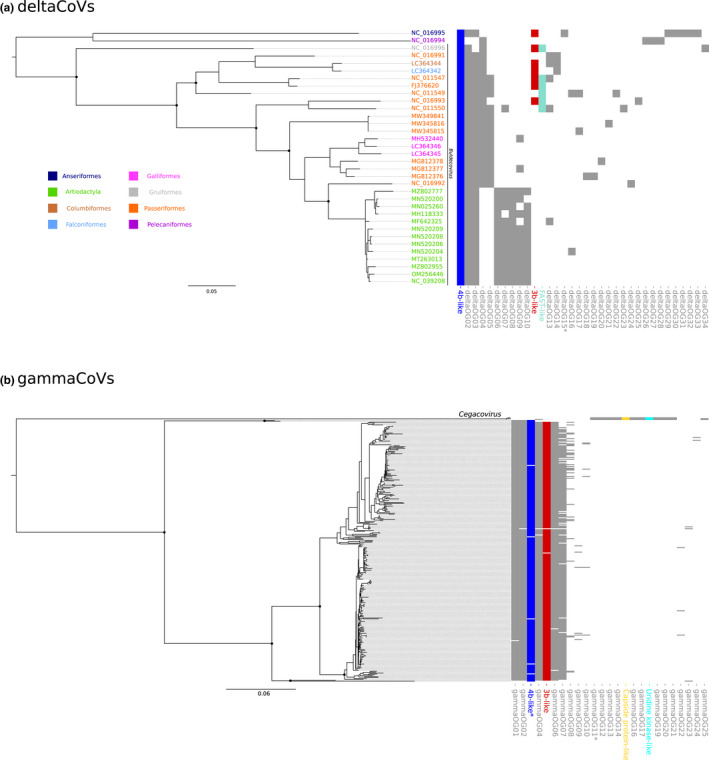
DeltaCoV and GammaCoV phylogeny and orthogroup distribution. Maximum‐likelihood phylogenetic tree of the RdRp region of deltaCoV (a) and gammaCoV (b) generated by iq‐tree using the LG + G4 and JTT + F + R6 models. Relevant internal nodes with bootstrap values >80% are shown as black dots. Super‐groups and relevant orthogroups are shown with the same colours as in Figure 3; unknown orthogroups are coloured in grey. Asterisks indicate orthogroups with split/paralogous ORFs. Scale bars are expressed as substitutions per site
To get a general picture of OG and super‐group representation in CoV genomes, we mapped their occurrences on phylogenetic trees (Figures 5 and 6). Specifically, we identified nonrecombining regions within the RdRp domains of the viral polymerases of all genera and we used them for ML phylogenetic reconstruction. As expected, viral genomes clustered by subgenera and, in the case of betaCoVs, largely also by OG presence/absence. Indeed, several OGs or super‐groups were shared by all or most viruses in the same betaCoV subgenus (Figure 5b). This was not the case for alphaCoVs, as only a minority of OG/super‐groups defined viral subgenera. However, some level of OG clustering by host order was observed, as viruses borne by rodents, carnivores and shrews tended to harbour specific OGs (Figure 5a). With respect to gammaCoVs, OGs clustered by subgenus and divided viruses hosted by cetaceans (cegacoviruses) from those hosted by birds (igacoviruses) (Figure 6b). Likewse, in deltaCoVs, OGs tended to separate CoVs hosted by birds and pigs (Figure 6a).
Analysis of OGs/supergroups by host order indicated that most (93%) OGs are host order‐specific in alphaCoVs and, to a lesser extent, in gammaCoVs (84%), whereas less than 74% are host order‐specific in betaCoVs and deltaCoVs (Figure 4b). As expected, chiroptera displayed the largest diversity, both in alphaCoVs and in betaCoVs. This is unsurprising because bats are the original reservoir of most alphaCoV and betaCoV subgenera and they account for 65% of genomes analysed herein. Nonetheless, for chiroptera, as well as for other orders (e.g., rodentia, carnivora and eulipotyphla), more OGs were specific in alphaCoVs than in betaCoVs.
With respect to gammaCoVs, the largest OG diversity was observed in galliformes, which account for 97% of the hosts. However, cegacoviruses, although represented by only three members, showed a remarkable diversity of accessory proteins (Figure 6a), most of them specific to this subgenus. Finally, deltaCoVs hosted by passeriformes showed the most diverse repertoire of accessory proteins (Figure 4b).
3.3. Evolutionary analysis of OGs
Previous analyses of specific CoV accessory ORFs indicated that these originate from either cellular or viral genes (Forni et al., 2017). We thus used hhpred and hhblits to search for remote homologies between OGs and known proteins (Alva et al., 2016; Remmert et al., 2011). Again, we set the cutoff for reliable hits at 90% probability (Gabler et al., 2020). No homology was detected for the large majority of OGs, reflecting the difficulty of defining relationships among distantly related homologues. However, for some OGs we detected known and unknown similarities suggestive of ORF origin and function.
We confirmed that most embecoviruses express haemagglutinin‐esterases that are probably of viral origin (Smits et al., 2005) and that two nobecoviruses harbour a protein derived from the ns1‐1 product (also known as p10) of bat orthoreovirus (Table 2). This protein belongs to the FAST (fusion‐associated small transmembrane) family of viral proteins (Huang et al., 2016). ORFs derived from other viruses were also detected in deltaCoVs. For instance, proteins in deltaOG12, which are harboured by a subset of buldecoviruses, showed homology to the ns1‐1 protein (a FAST protein) from human Rotavirus B. Because rotaviruses and orthoreoviruses belong to two distinct subfamilies in the family Reoviridae, we investigated whether the ns1‐1 ORFs were acquired independently in nobecoviruses and buldecoviruses. Thus, we generated a phylogenetic tree with ns1‐1 sequences from coronaviruses, orthoreoviruses and rotaviruses. The results indicated that the nobecovirus and buldecovirus proteins cluster with bat/avian orthoreoviruses and human/porcine rotaviruses, respectively (Figure 7). This suggests strongly that FAST proteins were independently acquired by viruses in the two genera.
TABLE 2.
List of relevant orthogroups
| OG | Viruses | Homology results |
|---|---|---|
| alphaOG16 | Seven bat viruses | Family of viral and host proteins containing Ig‐like domains (e.g., HCMV RL11 and adenovirus protein E3) |
| alphaOG20 | All rodent CoVs (luchacoviruses) | Spike protein of alphaCoVs (SADS‐CoV and HKU2) |
| alphaOG63 | Two rodent viruses | C‐type lectin domain |
| alphaOG86 | FIPV and TGEV | Phosphoribosylamine–glycine ligase domain of bacterial and eukaryotic organisms |
| betaOG09 | Most merbecoviruses; includes MERS‐CoV ORF4a/NS4a | RNA binding proteins, either from viruses (e.g., nonstructural protein 3 of Rotavirus C, Pernambuco mycovirus 1, Thika virus, NSP1 from Porcine rotavirus) or from cellular organisms (e.g., rat dsRNA‐specific editase 1, human interferon‐inducible double stranded RNA‐dependent protein kinase activator A, human TAR RNA‐binding protein 2) |
| betaOG13 | All embecoviruses | Influenza virus and torovirus haemagglutinin‐esterase |
| betaOG36 | Subset of merbecoviruses hosted by hedgehogs | Several viral and cellular proteins carrying Ig‐like domains. Most viral proteins with high similarity are encoded by human herpesviruses |
| betaOG56 | Two bat nobecoviruses | Rotavirus ns1‐1 proteins |
| betaOG78 | Only one hibecovirus | SARS‐CoV‐2 spike protein |
| gammaOG15 | Three cegacoviruses | Capsid protein (VP90) of mamastoviruses and human/porcine artroviruses |
| gammaOG18 | Three cegacoviruses | Uridine kinases from many different cellular organisms |
| deltaOG12 | Six Buldecoviruses | ns1‐1 product of Rotavirus B |
FIGURE 7.
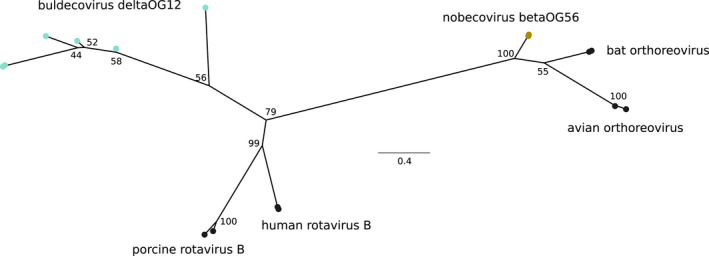
FAST proteins in nobecoviruses and buldecoviruses. Maximum‐likelihood phylogenetic tree showing the relationship among coronavirus and reovirus FAST proteins. The tree with bootstrap values was generated by iq‐tree using the best fitting substitution model (LG + G4). In addition to nobecovirus (betaOG56) and buldecovirus (deltaOG12) proteins, the FAST proteins of human rotavirus B (YP_008126848 and AAF69263), porcine rotavirus B (BAL04357 and AUG44809), bat orthoreovirus (JF811580 and NC_020448) and avian orthoreovirus (AIA57457 and DQ996607) were included. Orthogroups are shown with the same colours as in Figures 2 and 3. Scale bar is expressed as substitutions per site
An unexpected finding was that accessory proteins in alphaOG20 show sequence and structure similarity to the spike proteins of alphaCoVs (Table 2). ORFs belonging to this OG are only found in rodent alphaCoVs (luchacoviruses) and are located immediately 3′ of the M coding region (Figures 2a and 5a). This observation is reminiscent of a previous description of a spike‐related accessory protein in a hibecovirus (herein ascribed to betaOG78), but which is located upstream of the S ORF (Figures 2b and 5b) (Wu et al., 2016). We thus analysed these spike‐like proteins in further detail.
For the spike‐like proteins in alphaOG20, hhblits found homology to the spike protein of BtRf‐AlphaCoV/YN2012, a bat alphaCoV. hhpred confirmed structural similarity to the spike protein of swine acute diarrhoea syndrome CoV (SADS‐CoV) and HKU2. Alignment of the predicted proteins to the spike proteins of BtRf‐AlphaCoV/YN2012 and SADS‐CoV indicated that the homologous region overlaps with the receptor binding domain (RBD) (Figure 8a). Likewise, ab initio modelling using RoseTTAfold (Baek et al., 2021) showed very good superimposition with the RBD of the SADS‐CoV spike protein (Figure 8b) (Guan et al., 2020).
FIGURE 8.
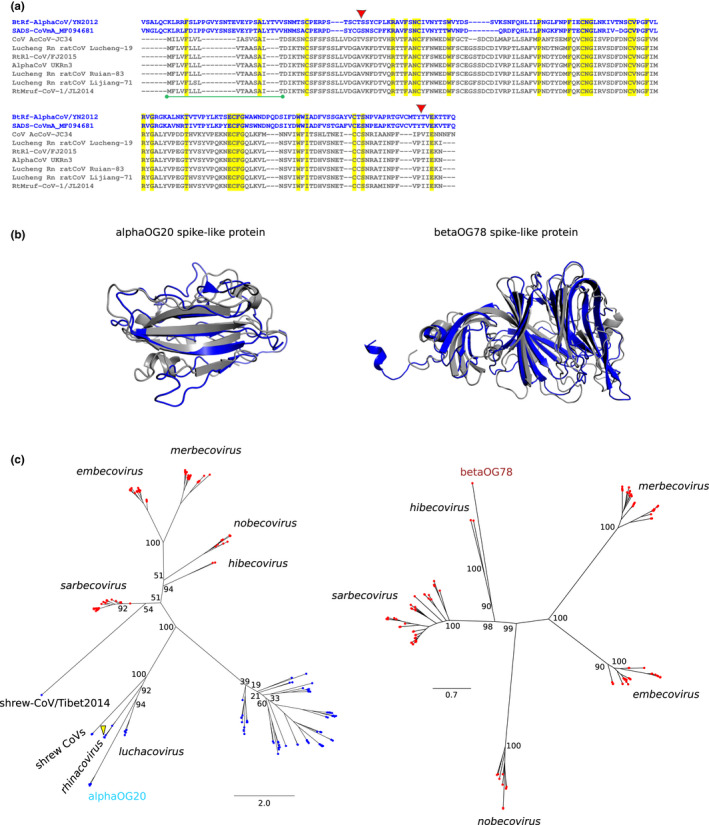
Spike‐like proteins in alphaCoVs and betaCovs. (a) Alignment of the spike‐like proteins of luchacoviruses (dark grey) with the spike proteins of SADS‐CoV and BtRf‐AlphaCoV/YN2012 (blue). Fully conserved residues are shaded in yellow. The red triangles denote the start and end of the predicted RBD of SADS‐CoV spike protein. The green bar indicates the predicted signal peptide in spike‐like proteins. (b) 3D structure superimposition of (left) the ab initio 3D model of alphaOG20 spike‐like protein (NC_032730) to the RBD of SADS‐CoV spike protein (PDB ID: 6 M39) and of (right) the ab initio 3D model of betaOG78 spike‐like protein (NC_025217) to the N‐terminal domain of SARS‐CoV2 spike protein (PDB ID: 7B62). The lDDT scores for alphaOG20 and betaOG78 spike‐like protein models were 0.75 and 0.73, respectively. Dark grey cartoons correspond to spike‐like ab initio models, and blue cartoons to spike protein structures. (c) Phylogenetic trees of spike proteins and spike‐like proteins identified in luchacoviruses (left, alphaOG20) and hibecoviruses (right, betaOG78). Tip colour indicates alpha (red) and beta (blue) CoVs. The position of SADS‐CoV is indicated by a yellow triangle, along with relevant CoV subgenera. The trees were generated with iq‐tree using the WAG+I + G4 model. Bootstrap values for relevant internal nodes are also reported. Scale bars are expressed as substitutions per site
With respect to the hibecovirus spike‐like protein, hhpred and hhblits found the S proteins of SARS‐CoV‐2 and SARS‐CoV as best hits. Sequence and structural alignment using a model built with RoseTTAfold indicated that the region of homology corresponds to the N‐terminal domain of the sarbecovirus spike protein (Figures 8b and S1).
Although they derive from coronavirus spike proteins, the accessory ORFs in alpha and betaCoVs probably represent independent acquisitions, either through duplication or recombination. We thus generated phylogenetic trees of these ORFs and the spike proteins of all analysed alpha and betaCoVs. Because the spike proteins of luchacoviruses and SADS‐CoV are thought to have originated through recombination with betaCoVs, analysis of proteins in alphaOG20 was performed by including both alphaCoVs and betaCoVs (Pan et al., 2017; Tsoleridis et al., 2019). The results showed both hibecovirus and luchacovirus spike‐like proteins to be more closely related to the S proteins of the respective viruses than to other CoV spikes (Figure 8c). This suggests that the spike‐like accessory proteins originated from independent duplications of the hibecovirus and luchacovirus spike genes (either entire or partial). The duplications were followed by divergence of the newly acquired ORFs.
Finally, we investigated the evolutionary history of coronavirus PDEs. Enzymes belonging to the 2H‐PDE family (characterized by the presence of two HxT/S motifs, where x in a hydrophobic residue) are encoded by embecoviruses and merbecoviruses, although their genomic locations differ (Figures 2b and 5b) (Thornbrough et al., 2016; Zhang et al., 2013). These coronavirus PDEs show homology to enzymes encoded by toroviruses and rotaviruses, and also to cellular proteins (AKAP7) (Forni et al., 2017; Zhang et al., 2013). We found that ORFs encoding PDEs are also present in the genomes of most luchacoviruses and in a shrew alphaCoV (Figure 5a, Table 1). These alphaCoV proteins also belong to the 2H‐PDE family and share the same homologies to viral and cellular genes as the betaCoV counterparts. To explore the relationships among the viral and the cellular PDEs, we selected the protein region covering the two HxT/S motifs and the intervening sequence. Because phylogenetic inference is challenging for highly diverged proteins, and alignment errors impact topological accuracy (Ogden & Rosenberg, 2006), we applied two different methodologies and compared the resulting trees. Thus, we aligned the PDE sequences with t‐coffee “Expresso” (Armougom et al., 2006), which uses structural data to inform the alignment, and generated a tree with iq‐tree (Trifinopoulos et al., 2016). In parallel, we used bali‐phy to jointly infer the alignment and the tree (Redelings & Suchard, 2005; Suchard & Redelings, 2006) (see Methods). The two approaches yielded very similar tree topologies, which only differed in the placement of the torovirus protein (Figure 9). Luchacovirus and embecovirus PDEs formed a clade that was more closely related to the cellular and torovirus/rotavirus than to merbecovirus PDEs (Figure 9). The PDE encoded by the shrew alphaCoV was closely related to the cellular AKAP7 proteins. These observations suggest that the PDEs of rodent alphaCoVs and embecoviruses were acquired by an ancestor of these viruses, as also suggested by the identical genome location of the ORFs (Figure 2). Indeed, the PDE might have been already present in an ancestor of nidoviruses, to which toroviruses also belong. Whereas it is impossible to determine with certainty whether this ancestral PDE was derived from a cellular or viral gene, it seems safe to assume that the shrew alphaCoV PDE derived from an independent gene transfer from a mammalian genome or from an unidentified virus (Figure 9). Regarding the merbecovirus proteins, they probably represent yet another independent gene gain event.
FIGURE 9.

Phylogenetic relationship among phosphodiesterases. Phylogenetic trees showing the relationship among viral and host phosphodiesterases. A maximum‐likelihood tree with bootstrap values (a) generated by iq‐tree using the JTT + I + G4 model and a Bayesian tree with posterior probabilities (b) generated by bali‐phy are reported. Values are shown for relevant internal nodes only. IDs for human AKAP7, mouse AKAP7, porcine torovirus PDE and rotavirus a VP3 are NP_057461.2, NP_001366167, YP_008798231 and YP_002302228, respectively. Scale bars are expressed as substitutions per site
3.4. Accessory protein nomenclature
As recently highlighted in the case of SARS‐CoV‐2, the nomenclature of CoV accessory proteins is ambiguous and confusing, both in annotations and in the scientific literature (Jungreis, Nelson, et al., 2021). More generally, the same holds true for alphaCoVs and betaCoVs. For instance, we retrieved the gene and/or product annotations for 181 proteins belonging to the ORF3a‐like super‐group and we counted at least 19 different names, which appear with different frequencies. Very similar results were obtained when we analysed 61 viral PDEs with 12 different names (Figure 10). The same babel of names can be observed for most CoV accessory ORFs (https://github.com/dforni5/CoVaccessory).
FIGURE 10.
Ambiguous nomenclature of accessory proteins. Word clouds for gene and/or product annotations for 181 proteins belonging to the ORF3a‐like super‐group (a) and 61 viral PDEs (b). Word clouds were generated with WordItOut (https://worditout.com/)
4. DISCUSSION
The emergence of three epidemic coronaviruses in the last two decades has kindled interest in zoonotic viruses. As a consequence, a number of studies have contributed to widen our knowledge of CoV distribution and genetic diversity. Whereas bats have remained the main focus of field surveys, and probably represent the major natural reservoir of alphaCoVs and betaCoVs, other small mammals such as rodents and shrews are increasingly recognized to host a large variety of coronaviruses (Annan et al., 2013; Anthony et al., 2017; Corman et al., 2015; Corman, Ithete, et al., 2014; Corman, Kallies, et al., 2014; De Sabato et al., 2020; Hu et al., 2017; Lam et al., 2020; Latinne et al., 2020; Lau et al., 2015; Li et al., 2021; Tao et al., 2017; Tsoleridis et al., 2016, 2019; Wang et al., 2015, 2020; Wang, Fu et al., 2017; Wang, Lin et al., 2017; Yang et al., 2014; Zhou et al., 2021). Also, wild birds are known to host a substantial diversity of deltaCoVs and gammaCoVs (Wille & Holmes, 2020). These sampling efforts generated a large number of CoV genomes, which were annotated with different levels of accuracy, depending on the main purpose for which they were initially sequenced. In general, information on accessory ORFs has remained fragmentary and scattered. This is unfortunate for at least two reasons. First, although dispensable for virus replication, accessory proteins often play a role at the host–virus interface and represent virulence factors (Forni et al., 2017). Thus, the complement of accessory proteins contributes to the pathogenetic potential of specific CoVs. Second, the characterization of accessory ORFs in terms of origin and diversity provides a window to understand CoV evolution. We thus applied a systematic approach to identify, categorize and characterize CoV accessory proteins.
Our analyses identified 379 OGs and 12 super‐groups. In line with previous analyses, we found that some super‐groups are shared by alphaCoVs and betaCoVs, some others by deltaCoVs and gammaCoVs. Whereas the relationships among members in the ORF3a‐like and ORF7a/ORF8‐like super‐groups were previously described (Neches et al., 2021; Tan et al., 2020, 2021), we also detected homology between an accessory ORF in the genomes of Asian HKU10‐related bat alphaCoVs and a putative protein (annotated as ORF8) encoded by Zaria bat CoV. The latter was sequenced from Commerson's leaf‐nosed bats in Nigeria and is one of the few representatives of the hibecovirus subgenus (Quan et al., 2010). Proteins in this super‐group show no homology to any other know protein, either of viral or of cellular origin, and they display distinct locations in alphaCoV and in Zaria bat CoV genomes. Another similar instance of shared accessory proteins in alphaCoVs and betaCoVs involves the ORF3 protein encoded by all merbecoviruses and by three bat alphaCoVs isolated in Italy and Australia. As in the case above, the locations of these accessory proteins differ in merbecoviruses (3′ of the S ORF) and alphaCoVs (3′ of N). These observations suggest that the sharing of these ORFs in alphaCoVs and betaCoVs is due to recombination rather than to common ancestry. Similarly, the genomic locations of 3b proteins in gammaCoVs and 7b proteins in deltaCoVs are distinct, again suggesting the action of recombination. Indeed, recombination events among viruses in different subgenera are thought to have generated the spike proteins of a subset of alphaCoVs, including SADS‐CoV and luchacoviruses (Pan et al., 2017; Tsoleridis et al., 2019). This indicates that recombination is not limited to closely related CoVs, but can also occasionally occur between members of different genera. A recent analysis of the four CoV genera, though, showed that recombination is ubiquitous among genetically similar viruses, but less common when genetic distances increase (Vakulenko et al., 2021). This pattern was observed for all genera and probably results from incompatibilities of genome fragment combinations (Vakulenko et al., 2021). Their study, however, did not analyse accessory ORFs, but only the essential genes. It is thus unclear how often recombination events can lead to the exchange of accessory ORFs between CoV genomes.
It is similarly uncertain whether recombination patterns play a role in explaining differences in the number and distribution of accessory proteins in coronaviruses from different genera. AlphaCoVs display, on average, fewer accessory ORFs per genome compared to the other genera, but the encoded proteins belong to many different OGs and tend to be virus‐specific. With the exclusion of the ORF3a‐like proteins, which are a hallmark of alphaCov genomes (https://talk.ictvonline.org/ictv‐reports/ictv_9th_report/positive‐sense‐rna‐viruses‐2011/w/posrna_viruses/222/coronaviridae), most OGs only appear in one or a few alphaCoVs. Conversely, in betaCoVs and deltaCoVs several accessory proteins are shared among viruses in the same subgenera, and their distribution mirrors phylogenetic relationships. This also holds true, to some extent, for gammaCoVs, as a large fraction of accessory protein diversity and suborder‐specific ORFs are accounted for by the presence of cegacoviruses, which have the largest genomes among CoVs and display a number of unique ORFs (Woo et al., 2014).
The reason(s) for the different distribution of accessory ORFs in CoVs genomes are presently unknown. Experimental and theoretical work on virus genome evolution has indicated that the stability of a new insert (in this case an accessory ORF) depends on the nature of the genome, the site, size and sequence of the insert, and the recombination rate (Willemsen et al., 2017; Willemsen & Zwart, 2019). However, other factors play a very important role, as well. These include host range, host‐switch frequency and demographic conditions, such as viral population and bottleneck sizes (Willemsen & Zwart, 2019). Unfortunately, detailed and systematic information on these parameters is not available for CoVs.
Despite these differences, it is worth noting that some evolutionary strategies seem to be shared by CoVs in different genera. Thus, both alphaCoVs and betaCoVs probably acquired PDEs and spike‐like accessory proteins independently. Although it is difficult to reliably infer the evolutionary history of distantly related proteins, the two methods we used showed good concordance in the topology of the PDE tree. If the genomic locations of these ORFs is also taken into account, the most likely scenario is that the PDEs encoded by rodent alphaCoVs and by embecoviruses were acquired by an ancestor of these viruses from a cellular or viral source. The clustering of the shrew alphaCoV PDE with human and rodent AKAP7 proteins suggests a recent, independent horizontal gene transfer from a mammalian genome, although transfer from a presently unidentified (corona)virus cannot be excluded. Concerning the merbecovirus proteins, these probably represent another independent acquisition, through either horizontal gene transfer or recombination. Interestingly, all these CoV PDEs, as well as those encoded by toroviruses and rotavirus A, belong to the 2H‐phosphoesterases superfamily and recent analyses indicated that they all have 2′,5′‐oligoadenilates as the preferred substrate (Asthana et al., 2021). This is consistent with data showing that the MHV and MERS‐CoV enzymes can block RNAse L activation and promote evasion from the host innate immune system (Thornbrough et al., 2016; Zhang et al., 2013). Clearly, the finding that these viral enzymes were acquired multiple times underlines their relevance in CoV evolution and adaptation.
Likewise, we found that the acquisition of ns1‐1‐related proteins occurred independently in nobecoviruses and buldecoviruses. Notably, ns1‐1 proteins from orthoreovirius and rotavirus B function as FAST proteins. These proteins are fusogenic and mediate the formation of syncytia in mammalian cells, thus enhancing cell‐to‐cell virus spread (Diller et al., 2019; Huang et al., 2016). Thus, the FAST proteins encoded by CoV genomes might favour viral dissemination in the host independently of receptor binding. Importantly, FAST proteins represent major pathogenicity determinants in orthoreoviruses (Kanai et al., 2019).
The spike‐like accessory proteins we identified in luchacoviruses and the one previously described in a hibecovirus (Wu et al., 2016) probably also represent independent acquisitions from different sources (i.e., viruses closely related to those carrying the respective spike‐like accessory ORFs). It is worth noting that proteins in the two OGs correspond to distinct regions of the spike protein, but in both cases they cover the S1 region, which is a major target of neutralizing antibodies (Du et al., 2009; Kubo et al., 1994; Sadarangani et al., 2021; Zhou et al., 2018). The spike‐like proteins of luchacoviruses are predicted to present a signal peptide, but no transmembrane region. These findings, as well as the sequence and structure similarity restricted to the RBD region, suggest that these proteins are secreted and may function as decoys for the host immune system. Such a strategy was suggested to be exploited by Ebola virus: a soluble form of the viral glycoproteins (sGP) is secreted during infection and functions as a modulator of the host immune response (Zhu et al., 2019). sGP absorbs antibodies against the full‐length protein and, at least in mouse models, induces cross‐reactivity with epitopes it shares with membrane‐bound GP (Mohan et al., 2012; Murin et al., 2014). Thus, the characterization of accessory proteins in terms of homology and sequence features can generate testable hypotheses about their function and possible relevance for CoV biology.
Of course, our study has limitations. The most relevant one is that the accessory protein sequences we analysed derive from annotations and/or ORF predictions. Thus, some of these proteins may not be translated during viral infection. The other side of the coin is that proteins that are not included in our catalogue may be produced and may have functional effects. Indeed, to limit false positive predictions, we restricted our analyses to OLGs longer than 25 codons that are predicted by the CodScr+SeqComp method. For instance, in the case of SARS‐CoV‐2, these criteria result in the exclusion of ORF3c (internal to ORF3a and not predicted by CodScr+SeqComp), which is likely to be functional, as assessed by sequence conservation and experimental data (Finkel et al., 2021; Jungreis, Nelson, et al., 2021; Jungreis, Sealfon, & Kellis, 2021; Nelson et al., 2020). Also, non‐OLGs shorter than 50 codons were discarded if they did not cluster in an OG with annotated proteins. Taking SARS‐CoV‐2 again as an example, failure to apply these criteria would result in the prediction of three short ORFs partially overlapping with S and ORF3a, ORF3a and M, and M and E. The resulting proteins have never been described and are unlikely to be produced. Although necessary, these criteria are arbitrary and are likely to introduce biases and inconsistencies, as ORFs from the best annotated genomes and the most investigated genera/subgenera are preferentially retained. In this respect our analysis is not truly comprehensive. Nonetheless, it is difficult to reliably estimate the coding potential of so many CoVs without extensive proteomic data or ribosome profiling experiments. Methods based on measuring the selective pressure acting on single ORFs (e.g., by estimating dN/dS, the ratio of divergence at nonsynonymous and synonymous sites) can provide valuable information and complement the approach we applied herein, as previously shown in SARS‐CoV‐2 (Cagliani et al., 2020; Jungreis, Nelson, et al., 2021; Jungreis, Sealfon, & Kellis, 2021; Nelson et al., 2020).
Another shortcoming is that OG and super‐group classification, as well as inference of ORF origin, are influenced by the ability of existing tools to detect remote homologies. Consequently, we cannot exclude that proteins in different OGs once shared a common ancestor (i.e., that they are orthologues). Likewise, we were unable to trace the evolutionary history of most OGs. Nevertheless, one of the main goals of this work was to provide an overview of accessory protein distribution and diversity, regardless of their ultimate origin, to facilitate future studies. Such an overview is necessarily limited to and possibly biased by the available sample of CoV genomes.
Finally, it is important to underline that an analysis of published studies indicated that conflicting names have been used for the accessory proteins of SARS‐CoV‐2 with, consequently, erroneous functional inferences (Jungreis, Nelson, et al., 2021). Our analysis of ORF names in several annotated CoV genomes confirms that substantial ambiguity exists. This complicates the cross‐communication among researches and hinders automated searches of large data sets (e.g., PubMed, GenBank). Jungreis and co‐workers, who included members of the Coronaviridae Study Group of the ICTV, suggested that accessory protein nomenclatures should be based on homology recognition (Jungreis, Nelson, et al., 2021). We suggest that OG membership might be used together with the name to provide information about protein function. We thus make sequences of all OGs available (https://github.com/dforni5/CoVaccessory), in the hope that they will facilitate the annotation of CoV genomes that will be sequenced in the future, as well as the analysis of those genomes that are already available.
AUTHOR CONTRIBUTIONS
Conceptualization, Diego Forni and Manuela Sironi; formal analysis, Diego Forni, Rachele Cagliani, Cristian Molteni, Federica Arrigoni, Alessandra Mozzi and Manuela Sironi; investigation, Diego Forni, Rachele Cagliani, Federica Arrigoni, Alessandra Mozzi and Manuela Sironi; visualization, Diego Forni, Rachele Cagliani, Cristian Molteni, Federica Arrigoni; writing—original draft, Manuela Sironi, Diego Forni; writing—review & editing, Manuela Sironi, Mario Clerici, Luca De Gioia; funding acquisition, Manuela Sironi and Diego Forni; supervision, Manuela Sironi, Mario Clerici and Luca De Gioia.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Figure S1
Table S1
ACKNOWLEDGEMENTS
This work was supported by the Italian Ministry of Health (“Ricerca Corrente 2021” to M.S.), by Fondazione Cariplo (grant CORONA, no. 2020‐1353), and by Regione Lombardia (Bando Progetti Ricerca Covid 19 – H44I20000470002).
Forni, D. , Cagliani, R. , Molteni, C. , Arrigoni, F. , Mozzi, A. , Clerici, M. , De Gioia, L. , & Sironi, M. (2022). Homology‐based classification of accessory proteins in coronavirus genomes uncovers extremely dynamic evolution of gene content. Molecular Ecology, 31, 3672–3692. 10.1111/mec.16531
Handling Editor: Camille Bonneaud
DATA AVAILABILITY STATEMENT
The list of NCBI IDs of the viral sequences analysed is provided in Table S1. ORF sequences for all orthogroups and tables with all OGs for each subgenus are available at https://github.com/dforni5/CoVaccessory.
REFERENCES
- Almagro Armenteros, J. J. , Tsirigos, K. D. , Sønderby, C. K. , Petersen, T. N. , Winther, O. , Brunak, S. , von Heijne, G. , & Nielsen, H. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 37, 420–423. 10.1038/s41587-019-0036-z [DOI] [PubMed] [Google Scholar]
- Alva, V. , Nam, S. Z. , Soding, J. , & Lupas, A. N. (2016). The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Research, 44, W410–W415. 10.1093/nar/gkw348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annan, A. , Baldwin, H. J. , Corman, V. M. , Klose, S. M. , Owusu, M. , Nkrumah, E. E. , Badu, E. K. , Anti, P. , Agbenyega, O. , Meyer, B. , Oppong, S. , Sarkodie, Y. A. , Kalko, E. K. , Lina, P. H. , Godlevska, E. V. , Reusken, C. , Seebens, A. , Gloza‐Rausch, F. , Vallo, P. , … Drexler, J. F. (2013). Human betacoronavirus 2c EMC/2012‐related viruses in bats, Ghana and Europe. Emerging Infectious Diseases, 19, 456–459. 10.3201/eid1903.121503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, S. J. , Johnson, C. K. , Greig, D. J. , Kramer, S. , Che, X. , Wells, H. , Hicks, A. L. , Joly, D. O. , Wolfe, N. D. , Daszak, P. , Karesh, W. , Lipkin, W. I. , Morse, S. S. , PREDICT Consortium , JAK, M. , & Goldstein, T. (2017). Global patterns in coronavirus diversity. Virus Evolution, 3, vex012. 10.1093/ve/vex012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armougom, F. , Moretti, S. , Poirot, O. , Audic, S. , Dumas, P. , Schaeli, B. , Keduas, V. , & Notredame, C. (2006). Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D‐coffee. Nucleic Acids Research, 34, W604–W608. 10.1093/nar/gkl092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana, A. , Gaughan, C. , Dong, B. , Weiss, S. R. , & Silverman, R. H. (2021). Specificity and mechanism of coronavirus, rotavirus, and mammalian two‐histidine Phosphoesterases that antagonize antiviral innate immunity. mBio, 12, e0178121‐21. 10.1128/mBio.01781-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, M. , DiMaio, F. , Anishchenko, I. , Dauparas, J. , Ovchinnikov, S. , Lee, G. R. , Wang, J. , Cong, Q. , Kinch, L. N. , Schaeffer, R. D. , Millán, C. , Park, H. , Adams, C. , Glassman, C. R. , DeGiovanni, A. , Pereira, J. H. , Rodrigues, A. V. , van Dijk, A. A. , Ebrecht, A. C. , … Baker, D. (2021). Accurate prediction of protein structures and interactions using a three‐track neural network. Science, 373, 871–876. 10.1126/science.abj8754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagliani, R. , Forni, D. , Clerici, M. , & Sironi, M. (2020). Coding potential and sequence conservation of SARS‐CoV‐2 and related animal viruses. Infection, Genetics and Evolution, 83, 104353. 10.1016/j.meegid.2020.104353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V. M. , Baldwin, H. J. , Tateno, A. F. , Zerbinati, R. M. , Annan, A. , Owusu, M. , Nkrumah, E. E. , Maganga, G. D. , Oppong, S. , Adu‐Sarkodie, Y. , Vallo, P. , da Silva Filho, L. V. , Leroy, E. M. , Thiel, V. , van der Hoek, L. , Poon, L. L. , Tschapka, M. , Drosten, C. , & Drexler, J. F. (2015). Evidence for an ancestral Association of Human Coronavirus 229E with bats. Journal of Virology, 89, 11858–11870. 10.1128/JVI.01755-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V. M. , Ithete, N. L. , Richards, L. R. , Schoeman, M. C. , Preiser, W. , Drosten, C. , & Drexler, J. F. (2014). Rooting the phylogenetic tree of middle east respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. Journal of Virology, 88, 11297–11303. 10.1128/JVI.01498-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman, V. M. , Kallies, R. , Philipps, H. , Gopner, G. , Muller, M. A. , Eckerle, I. , Brunink, S. , Drosten, C. , & Drexler, J. F. (2014). Characterization of a novel betacoronavirus related to middle east respiratory syndrome coronavirus in European hedgehogs. Journal of Virology, 88, 717–724. 10.1128/JVI.01600-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . (2020). The species severe acute respiratory syndrome‐related coronavirus: Classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nature Microbiology, 5, 536–544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, J. , Li, F. , & Shi, Z. L. (2019). Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology, 17, 181–192. 10.1038/s41579-018-0118-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sabato, L. , Di Bartolo, I. , De Marco, M. A. , Moreno, A. , Lelli, D. , Cotti, C. , Delogu, M. , & Vaccari, G. (2020). Can coronaviruses steal genes from the host as evidenced in Western European hedgehogs by EriCoV genetic characterization? Viruses, 12, 1471. 10.3390/v12121471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diller, J. R. , Parrington, H. M. , Patton, J. T. , & Ogden, K. M. (2019). Rotavirus species B encodes a functional fusion‐associated small transmembrane protein. Journal of Virology, 93, e00813‐19. 10.1128/JVI.00813-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, L. , He, Y. , Zhou, Y. , Liu, S. , Zheng, B. J. , & Jiang, S. (2009). The spike protein of SARS‐CoV—A target for vaccine and therapeutic development. Nature Reviews Microbiology, 7, 226–236. 10.1038/nrmicro2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D. M. , & Kelly, S. (2015). OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology, 16, 1–14. 10.1186/s13059-015-0721-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D. M. , & Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biology, 20, 1–14. 10.1186/s13059-019-1832-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel, Y. , Mizrahi, O. , Nachshon, A. , Weingarten‐Gabbay, S. , Morgenstern, D. , Yahalom‐Ronen, Y. , Tamir, H. , Achdout, H. , Stein, D. , Israeli, O. , Beth‐Din, A. , Melamed, S. , Weiss, S. , Israely, T. , Paran, N. , Schwartz, M. , & Stern‐Ginossar, N. (2021). The coding capacity of SARS‐CoV‐2. Nature, 589, 125–130. 10.1038/s41586-020-2739-1 [DOI] [PubMed] [Google Scholar]
- Forni, D. , Cagliani, R. , Clerici, M. , & Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends in Microbiology, 25, 35–48. 10.1016/j.tim.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabler, F. , Nam, S. Z. , Till, S. , Mirdita, M. , Steinegger, M. , Söding, J. , Lupas, A. N. , & Alva, V. (2020). Protein sequence analysis using the MPI bioinformatics toolkit. Current Protocols in Bioinformatics, 72, e108. 10.1002/cpbi.108 [DOI] [PubMed] [Google Scholar]
- Guan, H. , Wang, Y. , Perčulija, V. , Saeed, A. F. U. H. , Liu, Y. , Li, J. , Jan, S. S. , Li, Y. , Zhu, P. , & Ouyang, S. (2020). Cryo‐electron microscopy structure of the swine acute diarrhea syndrome coronavirus spike glycoprotein provides insights into evolution of unique coronavirus spike proteins. Journal of Virology, 94, e01301‐20. 10.1128/JVI.01301-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiranuma, N. , Park, H. , Baek, M. , Anishchenko, I. , Dauparas, J. , & Baker, D. (2021). Improved protein structure refinement guided by deep learning based accuracy estimation. Nature Communications, 12, 1–11. 10.1038/s41467-021-21511-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B. , Zeng, L. P. , Yang, X. L. , Ge, X. Y. , Zhang, W. , Li, B. , Xie, J. Z. , Shen, X. R. , Zhang, Y. Z. , Wang, N. , Luo, D. S. , Zheng, X. S. , Wang, M. N. , Daszak, P. , Wang, L. F. , Cui, J. , & Shi, Z. L. (2017). Discovery of a rich gene pool of bat SARS‐related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathogens, 13, e1006698. 10.1371/journal.ppat.1006698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Liu, W. J. , Xu, W. , Jin, T. , Zhao, Y. , Song, J. , Shi, Y. , Ji, W. , Jia, H. , Zhou, Y. , Wen, H. , Zhao, H. , Liu, H. , Li, H. , Wang, Q. , Wu, Y. , Wang, L. , Liu, D. , Liu, G. , … Gao, G. F. (2016). A bat‐derived putative cross‐family recombinant coronavirus with a Reovirus Gene. PLoS Pathogens, 12, e1005883. 10.1371/journal.ppat.1005883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungreis, I. , Nelson, C. W. , Ardern, Z. , Finkel, Y. , Krogan, N. J. , Sato, K. , Ziebuhr, J. , Stern‐Ginossar, N. , Pavesi, A. , Firth, A. E. , Gorbalenya, A. E. , & Kellis, M. (2021). Conflicting and ambiguous names of overlapping ORFs in the SARS‐CoV‐2 genome: A homology‐based resolution. Virology, 558, 145–151. 10.1016/j.virol.2021.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungreis, I. , Sealfon, R. , & Kellis, M. (2021). SARS‐CoV‐2 gene content and COVID‐19 mutation impact by comparing 44 Sarbecovirus genomes. Nature Communications, 12, 1–20. 10.1038/s41467-021-22905-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall, L. , Krogh, A. , & Sonnhammer, E. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Research, 35, W429–W432. 10.1093/nar/gkm256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai, Y. , Kawagishi, T. , Sakai, Y. , Nouda, R. , Shimojima, M. , Saijo, M. , Matsuura, Y. , & Kobayashi, T. (2019). Cell‐cell fusion induced by reovirus FAST proteins enhances replication and pathogenicity of non‐enveloped dsRNA viruses. PLoS Pathogens, 15, e1007675. 10.1371/journal.ppat.1007675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosakovsky Pond, S. L. , Posada, D. , Gravenor, M. B. , Woelk, C. H. , & Frost, S. D. (2006). Automated phylogenetic detection of recombination using a genetic algorithm. Molecular Biology and Evolution, 23, 1891–1901. 10.1093/molbev/msl051 [DOI] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , von Heijne, G. , & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology, 305, 567–580. 10.1006/jmbi.2000.4315 [DOI] [PubMed] [Google Scholar]
- Kubo, H. , Yamada, Y. K. , & Taguchi, F. (1994). Localization of neutralizing epitopes and the receptor‐binding site within the amino‐terminal 330 amino acids of the murine coronavirus spike protein. Journal of Virology, 68, 5403–5410. 10.1128/JVI.68.9.5403-5410.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam, T. T. , Shum, M. H. , Zhu, H. C. , Tong, Y. G. , Ni, X. B. , Liao, Y. S. , Wei, W. , Cheung, W. Y. , Li, W. J. , Li, L. F. , Leung, G. M. , Holmes, E. C. , Hu, Y. L. , & Guan, Y. (2020). Identifying SARS‐CoV‐2 related coronaviruses in Malayan pangolins. Nature, 583, 282–285. 10.1038/s41586-020-2169-0 [DOI] [PubMed] [Google Scholar]
- Latinne, A. , Hu, B. , Olival, K. J. , Zhu, G. , Zhang, L. , Li, H. , Chmura, A. A. , Field, H. E. , Zambrana‐Torrelio, C. , Epstein, J. H. , Li, B. , Zhang, W. , Wang, L. F. , Shi, Z. L. , & Daszak, P. (2020). Origin and cross‐species transmission of bat coronaviruses in China. Nature Communications, 11, 1–15. 10.1038/s41467-020-17687-3 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lau, S. K. , Woo, P. C. , Li, K. S. , Tsang, A. K. , Fan, R. Y. , Luk, H. K. , Cai, J. P. , Chan, K. H. , Zheng, B. J. , Wang, M. , & Yuen, K. Y. (2015). Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. Journal of Virology, 89, 3076–3092. 10.1128/JVI.02420-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Wang, L. , Liu, P. , Li, H. , Huo, S. , Zong, K. , Zhu, S. , Guo, Y. , Zhang, L. , Hu, B. , Lan, Y. , Chmura, A. , Wu, G. , Daszak, P. , Liu, W. J. , & Gao, G. F. (2021). A novel potentially recombinant rodent coronavirus with a polybasic cleavage site in the spike protein. Journal of Virology, 95, e0117321. 10.1128/JVI.01173-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan, G. S. , Li, W. , Ye, L. , Compans, R. W. , & Yang, C. (2012). Antigenic subversion: A novel mechanism of host immune evasion by Ebola virus. PLoS Pathogens, 8, e1003065. 10.1371/journal.ppat.1003065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murin, C. D. , Fusco, M. L. , Bornholdt, Z. A. , Qiu, X. , Olinger, G. G. , Zeitlin, L. , Kobinger, G. P. , Ward, A. B. , & Saphire, E. O. (2014). Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proceedings of the National Academy of Sciences of the United States of America, 111, 17182–17187. 10.1073/pnas.1414164111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neches, R. Y. , Kyrpides, N. C. , & Ouzounis, C. A. (2021). Atypical divergence of SARS‐CoV‐2 Orf8 from Orf7a within the coronavirus lineage suggests potential stealthy viral strategies in immune evasion. mBio, 12, e03014‐20. 10.1128/mBio.03014-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C. W. , Ardern, Z. , Goldberg, T. L. , Meng, C. , Kuo, C. H. , Ludwig, C. , Kolokotronis, S. O. , & Wei, X. (2020). Dynamically evolving novel overlapping gene as a factor in the SARS‐CoV‐2 pandemic. eLife, 9, e59633. 10.7554/eLife.59633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C. , Higgins, D. G. , & Heringa, J. (2000). T‐coffee: A novel method for fast and accurate multiple sequence alignment. Journal of Molecular Biology, 302, 205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- Ogden, T. H. , & Rosenberg, M. S. (2006). Multiple sequence alignment accuracy and phylogenetic inference. Systematic Biology, 55, 314–328. 10.1080/10635150500541730 [DOI] [PubMed] [Google Scholar]
- Ouzounis, C. A. (2020). A recent origin of Orf3a from M protein across the coronavirus lineage arising by sharp divergence. Computational and Structural Biotechnology Journal, 18, 4093–4102. 10.1016/j.csbj.2020.11.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y. , Tian, X. , Qin, P. , Wang, B. , Zhao, P. , Yang, Y. L. , Wang, L. , Wang, D. , Song, Y. , Zhang, X. , & Huang, Y. W. (2017). Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Veterinary Microbiology, 211, 15–21. 10.1016/j.vetmic.2017.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi, A. (2000). Detection of signature sequences in overlapping genes and prediction of a novel overlapping gene in hepatitis G virus. Journal of Molecular Evolution, 50, 284–295. 10.1007/s002399910033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi, A. (2020). New insights into the evolutionary features of viral overlapping genes by discriminant analysis. Virology, 546, 51–66. 10.1016/j.virol.2020.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavesi, A. (2021). Prediction of two novel overlapping ORFs in the genome of SARS‐CoV‐2. Virology, 562, 149–157. 10.1016/j.virol.2021.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond, S. L. , Frost, S. D. , & Muse, S. V. (2005). HyPhy: Hypothesis testing using phylogenies. Bioinformatics, 21, 676–679. 10.1093/bioinformatics/bti079 [DOI] [PubMed] [Google Scholar]
- Price, M. N. , Dehal, P. S. , & Arkin, A. P. (2010). FastTree 2—Approximately maximum‐likelihood trees for large alignments. PLoS One, 5, e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan, P. L. , Firth, C. , Street, C. , Henriquez, J. A. , Petrosov, A. , Tashmukhamedova, A. , Hutchison, S. K. , Egholm, M. , Osinubi, M. O. , Niezgoda, M. , Ogunkoya, A. B. , Briese, T. , Rupprecht, C. E. , & Lipkin, W. I. (2010). Identification of a severe acute respiratory syndrome coronavirus‐like virus in a leaf‐nosed bat in Nigeria. mBio, 1, e00208‐10. 10.1128/mBio.00208-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redelings, B. D. , & Suchard, M. A. (2005). Joint Bayesian estimation of alignment and phylogeny. Systematic Biology, 54, 401–418. 10.1080/10635150590947041 [DOI] [PubMed] [Google Scholar]
- Remmert, M. , Biegert, A. , Hauser, A. , & Soding, J. (2011). HHblits: Lightning‐fast iterative protein sequence searching by HMM‐HMM alignment. Nature Methods, 9, 173–175. 10.1038/nmeth.1818 [DOI] [PubMed] [Google Scholar]
- Sadarangani, M. , Marchant, A. , & Kollmann, T. R. (2021). Immunological mechanisms of vaccine‐induced protection against COVID‐19 in humans. Nature Reviews Immunology, 21, 475–484. 10.1038/s41577-021-00578-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers, E. W. , Barrett, T. , Benson, D. A. , Bolton, E. , Bryant, S. H. , Canese, K. , Chetvernin, V. , Church, D. M. , DiCuccio, M. , Federhen, S. , Feolo, M. , Fingerman, I. M. , Geer, L. Y. , Helmberg, W. , Kapustin, Y. , Landsman, D. , Lipman, D. J. , Lu, Z. , Madden, T. L. , … Ye, J. (2011). Database resources of the National Center for biotechnology information. Nucleic Acids Research, 39, D38–D51. 10.1093/nar/gkq1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger, L. (2017). The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC. [Google Scholar]
- Smits, S. L. , Gerwig, G. J. , van Vliet, A. L. , Lissenberg, A. , Briza, P. , Kamerling, J. P. , Vlasak, R. , & de Groot, R. J. (2005). Nidovirus sialate‐O‐acetylesterases: Evolution and substrate specificity of coronaviral and toroviral receptor‐destroying enzymes. The Journal of Biological Chemistry, 280, 6933–6941. 10.1074/jbc.M409683200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard, M. A. , & Redelings, B. D. (2006). BAli‐Phy: Simultaneous Bayesian inference of alignment and phylogeny. Bioinformatics, 22, 2047–2048. 10.1093/bioinformatics/btl175 [DOI] [PubMed] [Google Scholar]
- Tan, Y. , Schneider, T. , Leong, M. , Aravind, L. , & Zhang, D. (2020). Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS‐CoV‐2‐related viruses. mBio, 11, e00760‐20. 10.1128/mBio.00760-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, Y. , Schneider, T. , Shukla, P. K. , Chandrasekharan, M. B. , Aravind, L. , & Zhang, D. (2021). Unification and extensive diversification of M/Orf3‐related ion channel proteins in coronaviruses and other nidoviruses. Virus Evolution, 7, veab014. 10.1093/ve/veab014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, Y. , Shi, M. , Chommanard, C. , Queen, K. , Zhang, J. , Markotter, W. , Kuzmin, I. V. , Holmes, E. C. , & Tong, S. (2017). Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. Journal of Virology, 91, e01953‐16. 10.1128/JVI.01953-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornbrough, J. M. , Jha, B. K. , Yount, B. , Goldstein, S. A. , Li, Y. , Elliott, R. , Sims, A. C. , Baric, R. S. , Silverman, R. H. , & Weiss, S. R. (2016). Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. mBio, 7, e00258‐16. 10.1128/mBio.00258-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifinopoulos, J. , Nguyen, L. T. , von Haeseler, A. , & Minh, B. Q. (2016). W‐IQ‐TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Research, 44, W232–W235. 10.1093/nar/gkw256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoleridis, T. , Chappell, J. G. , Onianwa, O. , Marston, D. A. , Fooks, A. R. , Monchatre‐Leroy, E. , Umhang, G. , Müller, M. A. , Drexler, J. F. , Drosten, C. , Tarlinton, R. E. , McClure, C. P. , Holmes, E. C. , & Ball, J. K. (2019). Shared common ancestry of rodent Alphacoronaviruses sampled globally. Viruses, 11, 125. 10.3390/v11020125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoleridis, T. , Onianwa, O. , Horncastle, E. , Dayman, E. , Zhu, M. , Danjittrong, T. , Wachtl, M. , Behnke, J. M. , Chapman, S. , Strong, V. , Dobbs, P. , Ball, J. K. , Tarlinton, R. E. , & McClure, C. P. (2016). Discovery of novel Alphacoronaviruses in European rodents and shrews. Viruses, 8, 84. 10.3390/v8030084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulenko, Y. , Deviatkin, A. , Drexler, J. F. , & Lukashev, A. (2021). Modular evolution of coronavirus genomes. Viruses, 13, 1270. 10.3390/v13071270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , Fu, S. , Cao, Y. , Zhang, H. , Feng, Y. , Yang, W. , Nie, K. , Ma, X. , & Liang, G. (2017). Discovery and genetic analysis of novel coronaviruses in least horseshoe bats in southwestern China. Emerging Microbes & Infections, 6, e14. 10.1038/emi.2016.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Lin, X. D. , Guo, W. P. , Zhou, R. H. , Wang, M. R. , Wang, C. Q. , Ge, S. , Mei, S. H. , Li, M. H. , Shi, M. , Holmes, E. C. , & Zhang, Y. Z. (2015). Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China. Virology, 474, 19–27. 10.1016/j.virol.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Lin, X. D. , Liao, Y. , Guan, X. Q. , Guo, W. P. , Xing, J. G. , Holmes, E. C. , & Zhang, Y. Z. (2017). Discovery of a highly divergent coronavirus in the Asian house shrew from China illuminates the origin of the Alphacoronaviruses. Journal of Virology, 91, e00764‐17. 10.1128/JVI.00764-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Lin, X. D. , Zhang, H. L. , Wang, M. R. , Guan, X. Q. , Holmes, E. C. , & Zhang, Y. Z. (2020). Extensive genetic diversity and host range of rodent‐borne coronaviruses. Virus Evolution, 6, veaa078. 10.1093/ve/veaa078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille, M. , & Holmes, E. C. (2020). Wild birds as reservoirs for diverse and abundant gamma‐ and deltacoronaviruses. FEMS Microbiology Reviews, 44, 631–644. 10.1093/femsre/fuaa026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, A. , & Zwart, M. P. (2019). On the stability of sequences inserted into viral genomes. Virus Evolution, 5, vez045. 10.1093/ve/vez045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, A. , Zwart, M. P. , Ambrós, S. , Carrasco, J. L. , & Elena, S. F. (2017). 2b or not 2b: Experimental evolution of functional exogenous sequences in a plant RNA virus. Genome Biology and Evolution, 9, 297–310. 10.1093/gbe/evw300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, L. R. , Ye, Z. W. , Lui, P. Y. , Zheng, X. , Yuan, S. , Zhu, L. , Fung, S. Y. , Yuen, K. S. , Siu, K. L. , Yeung, M. L. , Cai, Z. , Woo, P. C. , Yuen, K. Y. , Chan, C. P. , & Jin, D. Y. (2020). Middle East respiratory syndrome coronavirus ORF8b accessory protein suppresses type I IFN expression by impeding HSP70‐dependent activation of IRF3 kinase IKKε. Journal of Immunology, 205, 1564–1579. 10.4049/jimmunol.1901489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo, P. C. , Lau, S. K. , Lam, C. S. , Tsang, A. K. , Hui, S. W. , Fan, R. Y. , Martelli, P. , & Yuen, K. Y. (2014). Discovery of a novel bottlenose dolphin coronavirus reveals a distinct species of marine mammal coronavirus in Gammacoronavirus. Journal of Virology, 88, 1318–1331. 10.1128/JVI.02351-13 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wu, Z. , Yang, L. , Ren, X. , He, G. , Zhang, J. , Yang, J. , Qian, Z. , Dong, J. , Sun, L. , Zhu, Y. , Du, J. , Yang, F. , Zhang, S. , & Jin, Q. (2016). Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. The ISME Journal, 10, 609–620. 10.1038/ismej.2015.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L. , Wu, Z. , Ren, X. , Yang, F. , Zhang, J. , He, G. , Dong, J. , Sun, L. , Zhu, Y. , Zhang, S. , & Jin, Q. (2014). MERS‐related betacoronavirus in Vespertilio superans bats, China. Emerging Infectious Diseases, 20, 1260–1262. 10.3201/eid2007.140318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, R. , Jha, B. K. , Ogden, K. M. , Dong, B. , Zhao, L. , Elliott, R. , Patton, J. T. , Silverman, R. H. , & Weiss, S. R. (2013). Homologous 2′,5′‐phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proceedings of the National Academy of Sciences of the United States of America, 110, 13114–13119. 10.1073/pnas.1306917110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Jha, B. K. , Wu, A. , Elliott, R. , Ziebuhr, J. , Gorbalenya, A. E. , Silverman, R. H. , & Weiss, S. R. (2012). Antagonism of the interferon‐induced OAS‐RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host & Microbe, 11, 607–616. 10.1016/j.chom.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Ji, J. , Chen, X. , Bi, Y. , Li, J. , Wang, Q. , Hu, T. , Song, H. , Zhao, R. , Chen, Y. , Cui, M. , Zhang, Y. , Hughes, A. C. , Holmes, E. C. , & Shi, W. (2021). Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS‐CoV‐2 and related viruses. Cell, 184, 4380–4391.e14. 10.1016/j.cell.2021.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X. L. , Wang, X. G. , Hu, B. , Zhang, L. , Zhang, W. , Si, H. R. , Zhu, Y. , Li, B. , Huang, C. L. , Chen, H. D. , Chen, J. , Luo, Y. , Guo, H. , Jiang, R. D. , Liu, M. Q. , Chen, Y. , Shen, X. R. , Wang, X. , … Shi, Z. L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579, 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Jiang, S. , & Du, L. (2018). Prospects for a MERS‐CoV spike vaccine. Expert Review of Vaccines, 17, 677–686. 10.1080/14760584.2018.1506702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W. , Banadyga, L. , Emeterio, K. , Wong, G. , & Qiu, X. (2019). The roles of Ebola virus soluble glycoprotein in replication, pathogenesis, and countermeasure development. Viruses, 11, 999. 10.3390/v11110999 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
The list of NCBI IDs of the viral sequences analysed is provided in Table S1. ORF sequences for all orthogroups and tables with all OGs for each subgenus are available at https://github.com/dforni5/CoVaccessory.