

Abstract
Chronic kidney disease (CKD) is a common complex condition associated with high morbidity and mortality. Polygenic prediction could enhance CKD screening and prevention, but this approach has not been optimized for ancestrally diverse populations. By combining APOL1 risk genotypes with GWAS for kidney function, we designed, optimized, and validated a genome-wide polygenic score (GPS) for CKD. The new GPS was tested in 15 independent cohorts, including 3 cohorts of European ancestry (total N=97,050), 6 cohorts of African ancestry (total N=14,544), 4 cohorts of Asian ancestry (total N=8,625), and 2 admixed Latinx cohorts (total N=3,625). We demonstrated score transferability with reproducible performance across all tested cohorts. The top 2% of the GPS was associated with nearly 3-fold increased risk of CKD across ancestries. In African-ancestry cohorts, APOL1 risk genotype and the polygenic component of the GPS had additive effects on the risk of CKD.
Keywords: chronic kidney disease (CKD), polygenic risk score (PRS), GWAS, APOL1
Editor summary:
A new study generated and optimized a polygenic score for chronic kidney disease with reproducible performance across 15 cohorts of different ancestries, and identified potentially clinically-relevant thresholds with predicted effects comparable to having family history of the disease
INTRODUCTION:
Chronic kidney disease (CKD) affects 10-16% of general population and has high morbidity and mortality1,2. In the US, CKD disproportionally affects African Americans (16.3%) when compared to European Americans (12.7%), Asian Americans (12.9%), or Hispanic Americans (13.6%)3. CKD stage 3 or greater is defined by a chronic loss of glomerular filtration rate (GFR) to below 60 mL/min/1.73m2. Because this definition is based on estimated kidney function rather than markers of specific kidney injury, it captures an etiologically heterogeneous set of primary and secondary kidney disorders. As expected for a highly heterogeneous trait, CKD has a complex determination with both genetic and environmental contributions. The observational heritability of CKD in the largest analysis of medical records ranged from 25% to 44%4. These estimates were generally consistent with smaller family-based studies of CKD and glomerular filtration rate5–7.
High heritability of CKD is attributed to both monogenic8,9 and polygenic causes10,11. Moreover, in individuals of African ancestry, two common risk alleles (G1 and G2) in APOL1 gene convey a large effect on the risk of kidney disease12,13. While heterozygotes for G1 or G2 alleles appear to be protected from trypanosomal sleeping sickness, kidney disease risk is conveyed under a recessive model in carriers of two risk alleles (G1G1, G2G2, or G1G2). Because of the selective pressure exerted by endemic trypanosomal species in certain parts of eastern and western Africa, G1 and G2 alleles are observed almost exclusively in individuals whose ancestry can be linked to those areas14,15. In the US population, frequency of APOL1 risk genotypes is estimated at approximately 15% in African Americans, 0.5-2% in Hispanic Americans, and <0.01% in Europeans16. These differences may be contributing to the higher prevalence of CKD in African Americans, but additional non-APOL1 genetic risk factors have not yet been elucidated.
Genome-wide polygenic scores (GPS) have emerged as promising tools for genetic risk stratification that can enhance traditional risk models for complex diseases. This approach has been applied to a variety of traits, including heart disease17,18, diabetes18,19, hypertension20,21, obesity22, schizophrenia23–25, and cancers26–31. One of the major limitations of the GPS approach is that existing GWAS are based predominantly on European cohorts and, as a result, most GPS do not perform well in more diverse cohorts, or in individuals with admixed ancestry32. Similar to other complex traits, GWAS for kidney function involved predominantly European cohorts. The latest study involved 765,348 participants, 75% of which were European, 23% East Asian, 2% African American, and <1% Hispanic11. Notably, this study did not capture the effects of APOL1 risk variants because of their recessive inheritance and very low frequencies in non-African populations.
The objective of the present study is to test if the existing knowledge on polygenic contributions to kidney function is sufficient to build a clinical risk predictor for moderate-to-advanced CKD with adequate performance across diverse ancestral groups. We specifically aimed to design, optimize, and test a new GPS for clinical risk prediction of kidney disease that maximizes the performance across ancestries. We combined information on APOL1 risk genotypes with the latest GWAS for kidney function to formulate a GPS that can reliably discriminate moderate to advanced CKD (stage 3 or greater) from population controls. In our approach, we took advantage of the power of the existing GWAS for a quantitative biomarker of kidney function (serum creatinine-based eGFR) to predict a disease state. To demonstrate transferability across different ancestral groups, we performed rigorous testing of our GPS in 15 independent and ancestrally diverse case-control cohorts following ClinGen standards33.
RESULTS:
GPS Optimization
The flowchart summary of our overall strategy is provided in Figure 1. In the GPS optimization step, a total of 19 candidate scores were generated using 1000G (all populations) linkage disequilibrium (LD) reference and summary statistics from GWAS for eGFR11. We then used a large optimization dataset comprised of 70% of European UK Biobank participants to select the best performing model (Table 1, Supplemental Table 1). The best model was based on the P-value thresholding (P+T) method and involved 41,426 markers with non-zero weights selected based on r2=0.2 and P=0.03. The score was standardized to zero-mean and unit-variance based on ancestry-matched population controls. In the optimization dataset, the polygenic component of the score explained 4% of variance (R2), with one standard deviation of the score increasing CKD risk by 86% (OR=1.86, 95%CI 1.83-1.89, P<1.00E-300) after controlling for age, sex, diabetes, center, genotyping/imputation batch, and genetic ancestry (Supplemental Table 1).
Figure 1: Overview of the study design.
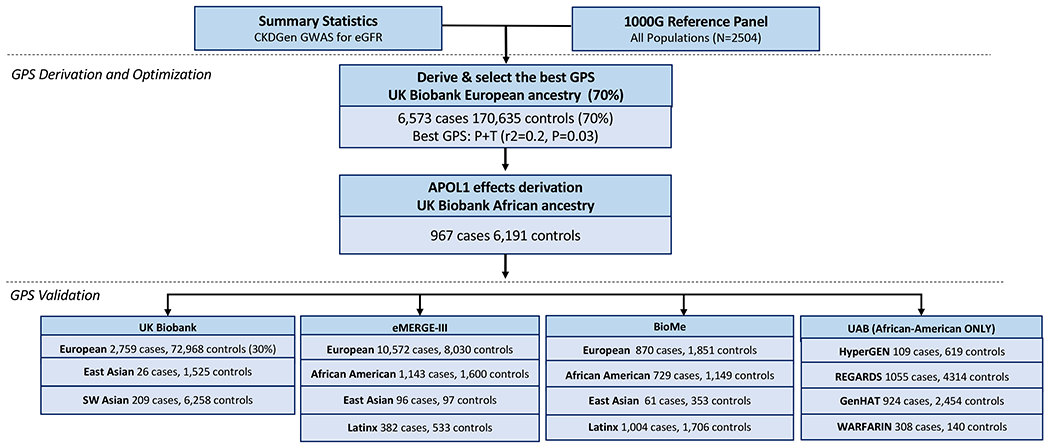
The CKD GPS was designed based on CKDGen GWAS summary statistics for eGFR and a cosmopolitan LD reference panel of 1000 Genomes (all populations); optimization was performed in two stages using UKBB participants of European (optimization 1) and African (optimization 2) ancestries; GPS performance validation was conducted in 15 additional independent testing cohorts of diverse ancestries.
Table 1:
Summary of study cohorts used for GPS optimization and testing.
| Study | Sub-Cohort | CKD Cases N=27,787 | Controls N=280,423 | Female (%) | Diabetes (%) | Mean Age (Years) |
|---|---|---|---|---|---|---|
| UKBB | Optimization 1: European Ancestry (70%) | 6,573 | 170,635 | 54 | 5 | 56.65 |
| Optimization 2: African Ancestry | 967 | 6,191 | 58 | 12 | 51.77 | |
|
| ||||||
| Testing 1: European Ancestry (30%) | 2,759 | 72,968 | 54 | 5 | 56.64 | |
| Testing 2: East Asian Ancestry | 26 | 1,525 | 68 | 5 | 52.37 | |
| Testing 3: South Asian Ancestry | 209 | 6,258 | 46 | 18 | 53.32 | |
|
| ||||||
| eMERGE | Testing 4: European Ancestry | 10,572 | 8,030 | 52 | 35 | 71.23 |
| Testing 5: African Ancestry | 1,143 | 1,600 | 70 | 40 | 66.76 | |
| Testing 6: Latinx Admixed Ancestry | 382 | 533 | 64 | 38 | 66.77 | |
| Testing 7: East Asian Ancestry | 96 | 97 | 59 | 27 | 72.81 | |
|
| ||||||
| UAB | Testing 8: Warfarin: African Ancestry | 308 | 140 | 58 | 50 | 61.48 |
| Testing 9: REGARDS: African Ancestry | 1,055 | 4,314 | 63 | 31 | 62.30 | |
| Testing 10: GenHAT: African Ancestry | 924 | 2,454 | 58 | 45 | 65.74 | |
| Testing 11: HyperGEN: African Ancestry | 109 | 619 | 62 | 31 | 52.23 | |
|
| ||||||
| BioMe | Testing 12: European Ancestry | 870 | 1,851 | 38 | 14 | 61.87 |
| Testing 13: African Ancestry | 729 | 1,149 | 56 | 32 | 61.65 | |
| Testing 14: Latinx Admixed Ancestry | 1,004 | 1,706 | 38 | 33 | 62.04 | |
| Testing 15: East Asian Ancestry | 61 | 353 | 41 | 15 | 55.75 | |
The second optimization step involved testing for independent contributions of APOL1 risk genotypes and included 7,158 UKBB participants of genetically defined African ancestry (967 cases and 6,191 controls). In the model adjusted for age, sex, diabetes, batch, and PCs of ancestry, we observed statistically significant independent effects of the polygenic component (OR per SD =1.16, 95%CI: 1.09-1.25, P=1.00E-04) and the recessive APOL1 risk genotype (OR=1.19, 95%CI: 1.01-1.38, P=4.00E-02), but no significant multiplicative interactions between the two predictors (P interaction=0.29) (Supplemental Table 2). Given these findings, we subsequently modeled APOL1 risk as additive to the polygenic component, assuming the APOL1 risk genotype effects approximately equivalent to one standard deviation of the standard-normalized polygenic score (the weight of one was used because the β per standard deviation of the polygenic score and the β for APOL1 risk genotype were comparable in magnitude).
Population differences in GPS distributions
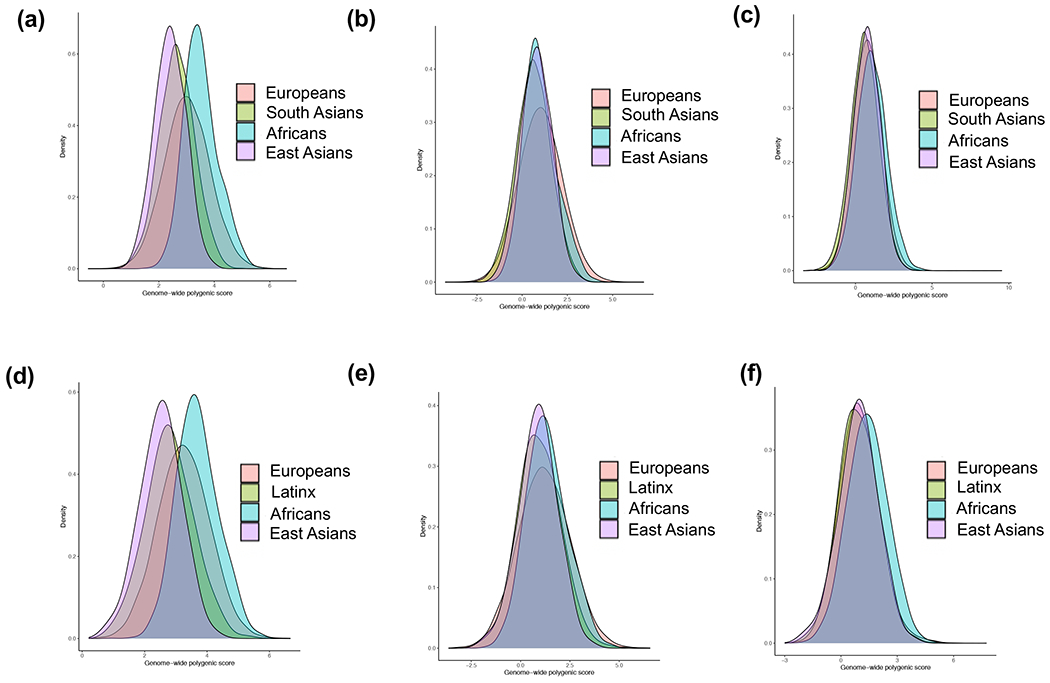
We next examined the distributions of the polygenic risk component (without APOL1), as well as the final combined GPS (with APOL1) in the reference populations of 1000G. We detected significant differences in the mean polygenic risk across populations (Figure 2, ANOVA P=3.40E-154), with a notable shift towards higher average risk in African ancestry compared to all other populations (P=4.92E-163). This shift became even more pronounced after including APOL1 risk genotype information in the combined GPS (P=1.58E-168). These results suggest that the polygenic risk score for CKD is considerably higher in African compared to non-African populations independent of APOL1.
Figure 2: Risk score distributions in five 1000 Genomes populations:
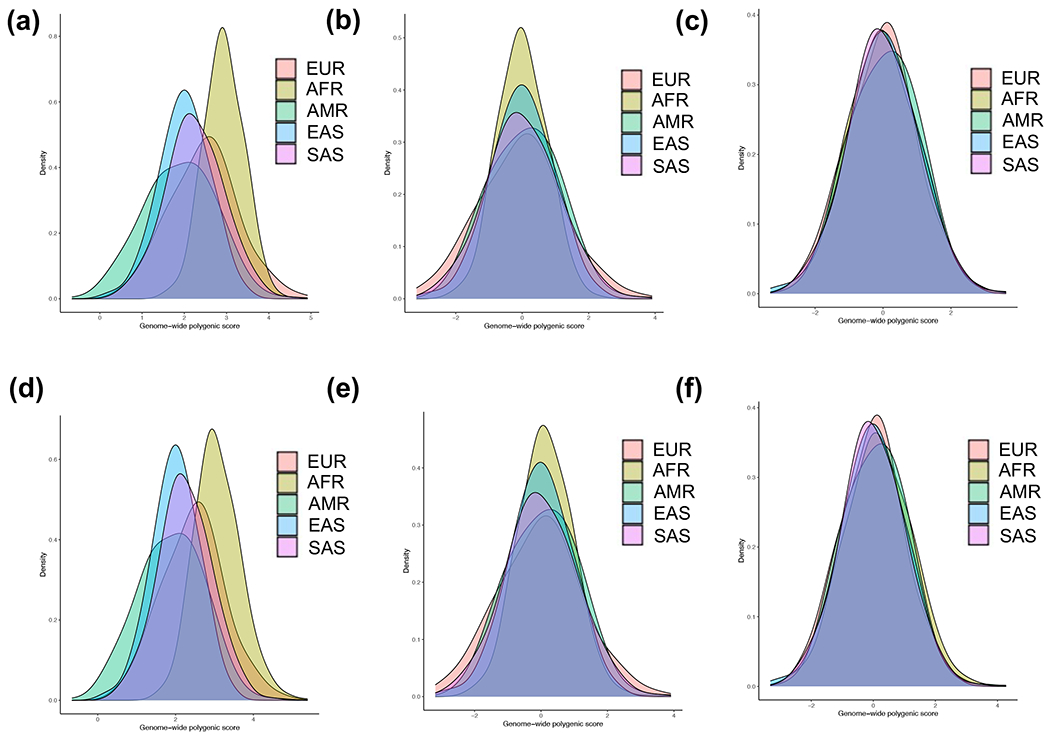
(a) raw polygenic score without APOL1; (b) ancestry-adjusted polygenic score without APOL1 (method 1: mean only); (c) ancestry-adjusted polygenic score without APOL1 (method 2: mean and variance); (d) raw combined GPS with APOL1; (e) ancestry-adjusted combined GPS with APOL1 (method 1) and (f) ancestry-adjusted combined GPS with APOL1 (method 2). AFR: African, AMR: Native American, EAS: East Asian, EUR: European, and SAS: South Asian.
Given that the weights of the score equation are fixed, we hypothesized that the observed distributional differences were driven by a higher frequency of CKD risk alleles in African genomes. Therefore, we examined the overall frequency spectrum of CKD risk alleles included in the GPS between European and African reference populations (Extended Data Figure 1). First, we observed a greater number of risk alleles in the African compared to European populations of 1000G at the extremes of the frequency spectrum (risk allele frequencies (RAF) < 0.01 or > 0.99). This observation is expected due to the MAF filter of 1% used in the European GWAS discovery cohorts. Second, across all variants included in the score, the mean difference in RAF between African and European populations was positive (i.e. greater than the expected mean of 0), indicating higher average frequency of risk alleles in African genomes. We further observed that the risk alleles with largest weights (effect sizes in GWAS) had a significantly higher frequency in African genomes compared to those with low effect sizes (P=0.025), or intermediate effect sizes (P=0.018) (Extended Data Figure 1d). Thus, it appears that the observed GPS distributional shifts between European and African populations are driven predominantly by frequency differences of large effect risk alleles.
GPS testing in cohorts of European ancestry
We next tested the final GPS in three European cohorts, including the remaining 30% of the UKBB (2,759 cases and 72,968 controls) and two large US-based European ancestry cohorts, eMERGE-III (10,572 cases and 8,030 controls) and BioMe (870 cases and 1,851 controls). In the combined meta-analysis, the GPS exhibited highly reproducible performance, with pooled OR per SD = 1.46, 95%CI: 1.43-1.48, P<1.00E-300 (Supplemental Table 3). While the UKBB testing cohort had nearly identical performance metrics to the optimization cohort, the effect sizes were attenuated slightly in the US-based cohorts. The frequency of APOL1 risk genotype was extremely low (Supplemental Table 4), thus its effect was negligible in the European cohorts.
GPS testing in cohorts of African ancestry
The GPS was tested in six independent African ancestry cohorts, including eMERGE-III (1,143 cases and 1,600 controls), BioMe (729 cases and 1,149 controls), HyperGEN (109 cases and 619 controls), REGARDS (1,055 cases and 4,314 controls), GenHat (924 cases and 2,454 controls) and Warfarin (308 cases and 140 controls). In the combined meta-analysis, the GPS had pooled OR per SD of 1.32, 95%CI: 1.26-1.38, P =1.78E-33 (Table 2 and Supplemental Table 5). The inclusion of APOL1 risk genotype considerably enhanced CKD risk prediction across all African ancestry cohorts, substantially improving tail cut-off discrimination i.e., the risk for the top 2% of individual was approximately 1.80-fold higher in the model without APOL1 and 2.70-fold higher in the model with APOL1 compared to the remaining 98% of individuals (Table 3). The effects of the GPS stratified by APOL1 risk genotype across all 6 African ancestry cohorts are depicted in Figure 3a.
Table 2:
The performance metrics of the GPS in the testing cohorts meta-analyzed by ancestry. For performance testing in individual cohorts, please refer to Supplemental Tables 4–7.
| Meta-analysis | Case/control | OR per SD (95% CI), P-value | AUC (Crude) | PRS Threshold | Odds ratio (95% CI), P-value |
|---|---|---|---|---|---|
| European (3 cohorts) | 14,201/82,849 | 1.46 (1.43-1.48), P<1.00E-300 | 0.81 (0.62) | Top 20% vs. other 80% | 2.30 (2.17-2.44), P=1.65E-174 |
| Top 10% vs. other 90% | 2.59 (2.40-2.78), P=1.27E-142 | ||||
| Top 5% vs. other 95% | 2.92 (2.65-3.21), P=2.64E-104 | ||||
| Top 2% vs. other 98% | 3.60 (3.11-4.17), P=4.26E-66 | ||||
| Top 1% vs. other 99% | 4.46 (3.66-5.44), P=7.82E-50 | ||||
|
| |||||
| African (6 cohorts) | 4,268/10,276 | 1.32 (1.26-1.38), P=1.78E-33 | 0.78 (0.57) | Top 20% vs. other 80% | 1.65 (1.49-1.82), P=1.17E-22 |
| Top 10% vs. other 90% | 1.84 (1.61-2.09), P=9.26E-20 | ||||
| Top 5% vs. other 95% | 2.06 (1.72-2.47), P=2.11E-15 | ||||
| Top 2% vs. other 98% | 2.66 (2.01-3.51), P= 4.93E-12 | ||||
| Top 1% vs. other 99% | 3.51 (2.37-5.22), P=4.21E-10 | ||||
|
| |||||
| Latinx (2 cohorts) | 1,386/2,239 | 1.42 (1.29-1.57), P=4.56E-12 | 0.88 (0.62) | Top 20% vs. other 80% | 1.88 (1.50-2.37), P=5.46E-08 |
| Top 10% vs. other 90% | 2.26 (1.66-3.06), P=1.56E-07 | ||||
| Top 5% vs. other 95% | 2.67 (1.75-4.07), P=4.96E-06 | ||||
| Top 2% vs. other 98% | 4.93 (2.46-9.89), P=6.69E-06 | ||||
| Top 1% vs. other 99% | 6.61 (2.46-17.75), P=1.77E-04 | ||||
|
| |||||
| Asian (4 cohorts) | 392/8,233 | 1.68 (1.45-2.06), P=7.11E-13 | 0.91 (0.61) | Top 20% vs. other 80% | 2.42 (1.81-2.27), P=4.39E-09 |
| Top 10% vs. other 90% | 2.95 (2.06-4.20), P=2.43E-09 | ||||
| Top 5% vs. other 95% | 3.56 (2.26-5.60), P=4.09E-08 | ||||
| Top 2% vs. other 98% | 3.81 (1.91-7.59), P=1.35E-04 | ||||
| Top 1% vs. other 99% | 8.46 (3.70-19.3), P=4.00E-07 | ||||
|
| |||||
| All 15 cohorts | 20,247/103,597 | 1.44 (1.42-1.47), P<1.00E-300 | 0.81 (0.61) | Top 20% vs. other 80% | 2.23 (2.11-2.35), P=8.02E-195 |
| Top 10% vs. other 90% | 2.31 (2.17-2.45), P=2.75E-168 | ||||
| Top 5% vs. other 95% | 2.58 (2.39-2.79), P=2.02E-123 | ||||
| Top 2% vs. other 98% | 3.26 (2.89-3.67), P=3.37E-84 | ||||
| Top 1% vs. other 99% | 4.61 (3.84-5.53), P=1.14E-60 | ||||
OR: Odds ratio for the model adjusted for age, sex, diabetes, principal components of ancestry and genotyping array or clinical site; SD: standard deviation of the GPS distribution in controls; AUC: area under the receiver-operator curve for the model adjusted for age, sex, diabetes, principal components of ancestry and genotyping array or clinical site (crude: AUC for GPS alone without any covariates).
Table 3:
Added value APOL1 risk genotype to polygenic risk components in predicting CKD using extreme tail (98th percentile) of the risk score distribution in African-American (4,268 cases and 10,276 controls) and admixed Latinx (1,386 cases and 2,239 controls) cohorts. All effect estimates are adjusted for age, sex, diabetes, and principal components of ancestry.
| Cohorts | APOL1 Risk Genotype OR (95%CI), P-value | Top 2% PRS without APOL1 OR (95%CI), P-value | Top 2% PRS with APOL1 OR (95%CI), P-value |
|---|---|---|---|
| African Ancestry: | |||
| eMERGE | 1.64 (1.42-1.86), P=2.00E-05 | 2.10 (1.46-2.74), P=2.00E-02 | 2.60 (1.38-4.90), P=3.10E-03 |
| BioMe | 1.38 (1.28-1.48), P=3.30E-10 | 2.70 (1.93-3.47), P=1.00E-02 | 5.75 (4.96-6.54), P=1.00E-05 |
| UAB HyperGen | 1.71 (0.93-3.12), P=8.20E-02 | 2.22 (0.59-8.44), P=2.40E-01 | 1.64 (0.43-6.20), P=4.65E-01 |
| UAB REGARDS | 1.35 (1.08-1.77), P=6.90E-03 | 1.26 (0.76-2.07), P=3.60E-01 | 1.56 (0.97-2.59), P=6.52E-02 |
| UAB GenHAT | 1.43 (1.12-1.81), P=3.20E-03 | 2.80 (1.64-4.77), P=1.50E-04 | 4.38 (2.56-7.50), P=6.80E-08 |
| UAB Warfarin | 1.93 (1.07-3.49), P=2.90E-02 | -- | 1.59 (0.28-8.73), P=5.96E-01 |
| Meta-analysis | 1.46 (1.38-1.54), P=2.70E-19 | 1.76 (1.41-2.20), P=5.90E-07 | 2.66 (2.01-3.51), P=4.93E-12 |
|
| |||
| Latinx Admixed Ancestry: | |||
| eMERGE | 16.5 (5.70-48.1), P=7.10E-04 | 1.41 (0.47-4.24), P=5.40E-01 | 6.89 (1.60-29.07), P=9.78E-03 |
| BioMe | 1.17 (1.10-1.24), P=2.40E-06 | 2.72 (1.97-3.47), P=1.00E-02 | 4.48 (3.69-5.27), P=2.10E-04 |
| Meta-analysis | 1.18 (1.09-1.27), P=4.40E-05 | 2.21 (1.19-4.10), P=1.10E-02 | 4.93 (2.46-9.89), P=6.69E-06 |
Figure 3. Effects of the genome-wide polygenic score (GPS) for chronic kidney disease (CKD):
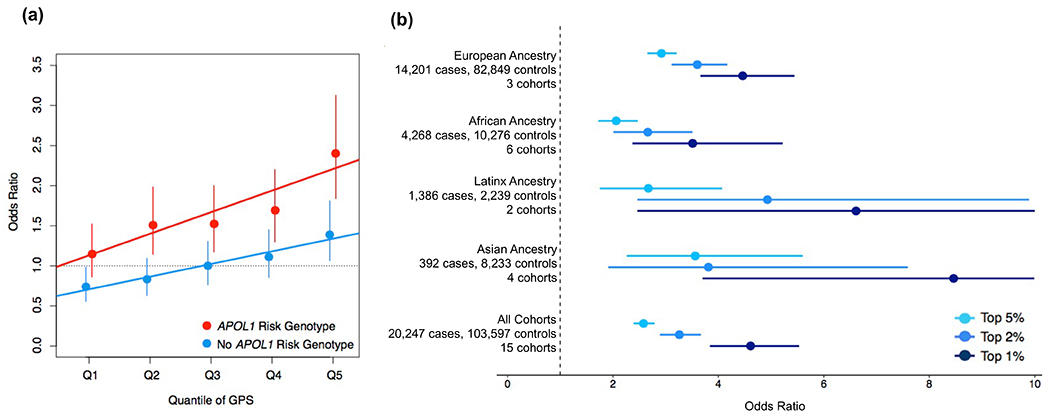
(a) GPS quantile effects stratified by the APOL1 risk genotype (a total N=2,020 with and N=12,526 without the APOL1 risk genotype, in red and blue, respectively). The X-axis depicts each quantile of the GPS ordered from the first (Q1) to the last (Q5) quantile. The Y-axis depicts odds ratios of CKD for each of the quantile-defined sub-groups in reference to the middle quantile (Q3) of those without the APOL1 risk genotype. The effect estimates (dots) and 95% confidence intervals (vertical bars) were derived based on a fixed-effects meta-analysis across all 6 African ancestry testing cohorts and adjusted for age, sex, diabetes, and principal components of ancestry. Regression lines were fitted for each group defined by the presence of APOL1 risk genotype. (b) GPS tail effects by ancestry. The X-axis depicts odds ratio of CKD, the Y-axis depicts testing cohort meta-analysis by ancestry (with numbers of cases, controls, and cohorts). The effect estimates (dots) and 95% confidence intervals (vertical bars) are provided for the top 5% versus bottom 95% (sky blue), top 2% versus bottom 98% (cobalt blue), and top 1% versus bottom 99% (navy blue). All effect estimates are adjusted for age, sex, diabetes, and principal components of ancestry.
GPS testing in admixed ancestry Latinx cohorts
The GPS was also tested in two admixed ancestry Latinx cohorts from eMERGE-III (382 cases and 533 controls) and BioMe (1,004 cases and 1,706 controls). The combined meta-analysis of these cohorts resulted in pooled OR per SD = 1.42, 95%CI: 1.29-1.57, P=4.56E-12 (Supplemental Table 6). Similar to African ancestry cohorts, the inclusion of APOL1 risk genotypes in the GPS improved risk prediction in these admixed cohorts (Table 3).
GPS testing in cohorts of Asian ancestry
We tested GPS in four diverse Asian cohorts including UKBB South-West Asian (209 cases and 6,258 controls), UKBB East Asian (26 cases and 1,525 controls), eMERGE-III East Asian (96 cases and 97 controls) and BioMe East Asian (61 cases and 353 controls) cohorts. The combined meta-analysis resulted in pooled OR per SD = 1.68, 95%CI: 1.45-2.06, P =7.11E-13 (Supplementary Table 7). APOL1 risk genotypes were absent in the Asian cohorts, thus the modelled risk was entirely attributable to the polygenic component.
Tail discrimination performance by ancestry
For each individual testing cohort, we derived risk estimates comparing extreme tails of the risk score distribution to all other cohort members, and estimated sensitivity and specificity for a range of tail cutoffs (20%, 10%, 5%, 2% and 1%). These metrics were meta-analyzed by ancestry and summarized in Table 2, Figure 3b, and Supplemental Tables 3, 5–7. Depending on ancestry, the top 2% tail of the risk score distribution was associated with 2.66-4.93 fold higher risk of CKD than for the remaining 98% of individuals, including in European (OR=3.60, 95%CI: 3.11-4.17, P=4.26E-66), African (OR=2.66, 95%CI: 2.01-3.51, P=4.93E-12), admixed Latinx (OR=4.93, 95%CI: 2.46-9.89, P=6.69E-06) and Asian ancestry (OR=3.81, 95%CI: 1.91-7.59, P=1.35E-04) cohorts. We consider this cut-off as clinically meaningful, since this degree of risk is approximately equivalent to the risk reported for a family history of kidney disease34. In Supplementary Table 8, we summarize various metrics of diagnostic performance for this cut-off by ancestry, including sensitivity, specificity, as well as prevalence-adjusted positive and negative predictive values. For comparison, we provide similar metrics for the top 5% of the risk score distribution.
Ancestry adjustments and calibration
We next compared the effect of two different ancestry adjustment methods on the GPS distributions in 1000G, eMERGE-III, and UKBB testing cohorts (Figure 2 and Extended Data Figure 2). Adjusting for mean only (method 1, see Methods) eliminated major distributional shifts by ancestry, but did not fully resolve the observed tail differences. The ancestry adjustment method 2 (adjusting for both mean and variance) resulted in comparable shapes of the GPS distributions by ancestry and facilitated the selection of a single trans-ancestry tail cut-off. Both methods result in comparably good risk score calibration when applied to the combined multiethnic eMERGE-III dataset (Extended Data Figure 3). As a trade-off, however, the ancestry adjustments reduced tail discrimination at extreme cut-offs as summarized in Supplementary Table 9. This trade-off appeared most pronounced for method 2, as well as for more admixed cohorts (African-American and Latinx).
Sensitivity analyses
The use of race in clinical predictive models has been scrutinized, and a new CKD-EPI 2021 equation without a race variable has recently been proposed35. We therefore performed sensitivity analyses to examine the effect of the new equation on the GPS performance within eMERGE-III, our largest and most diverse testing dataset. The CKD-EPI 2021 equation without race35 was applied to re-define cases and controls across among all eMERGE-III participants. As expected, the new equation reduced the number of CKD cases (and increased the numbers of controls) in the European, Latinx and East Asian cohorts, while the opposite effect was observed in the African ancestry cohort. Importantly, across all ancestral groups, the GPS had comparable performance between the new and old phenotype definitions, with a notable trend for improved performance in the African ancestry cohort (Supplemental Table 10a).
We additionally tested the effect of using self-reported race/ethnicity vs. genetic (principal component-based) ancestry to define our testing cohorts. Despite smaller sample sizes of the cohorts defined by self-report, we observed no major performance differences between self-report and genetic ancestry-defined cohorts. Similarly, the use of the 2021 CKD-EPI eGFR equation to define CKD in self-report-based cohorts resulted in comparable performance to the 2009 equation, with a similar trend for improved performance in African ancestry cohorts (Supplemental Table 10b).
Lastly, using our multiethnic eMERGE-III cohorts, we compared the performance of our GPS to an alternative score recently proposed by Yu et al36 (Supplementary Table 11). This score was based on a GWAS with higher proportion of Europeans, did not include APOL1 risk genotype, and was not optimized for transethnic performance. Given these differences, the score by Yu et al. performed better in cohorts of European ancestry but was less predictive in cohorts of African or Asian ancestry. Notably, the African ancestry distribution of the GPS by Yu et al. was also shifted towards higher values compared to other ancestries, confirming that the observed shift is independent of a specific method used to design the score (Extended Data Figure 4).
DISCUSSION:
We developed a GPS for CKD that captures polygenic determinants of kidney function emerging from recent GWAS studies and predicts CKD across four ancestry groups. Our score was designed and validated for individual level risk prediction following ClinGen guidelines33. The score had consistent performance despite heterogenous genotyping platforms and imputation methods employed in our testing studies. We also developed continuous ancestry adjustment methods to allow for cross-ancestry standardization of the score. Importantly, our testing studies demonstrated that extreme tails of the risk score distribution (top 2%) conveyed approximately 3-fold increase in the disease risk across all ancestries. This magnitude of risk is equivalent to a positive family history of kidney disease34.
In this study, we were unable to assess if the GPS conveys kidney disease risk independently of positive family history, mainly because family history is poorly captured in electronic health records, and thus it is not routinely available for large EHR-linked biobanks37. However, prior cohort studies in cardiovascular disease38–41 and breast cancer42 clearly demonstrate that polygenic scores for these conditions capture independent information from family history or traditional risk factors. For CKD, this specific question will be addressed by a prospective eMERGE-IV study testing the performance of our score against family history collected using MeTree software43. Additional studies are also needed to test if our GPS modifies penetrance of monogenic kidney disorders, similar to the effects reported for tier 1 genetic disorders44.
Beyond enhanced disease screening of asymptomatic individuals, other potential applications of the GPS may include improved risk stratification of potential living kidney donors, or enhanced quality assessment of deceased donor organs in the setting of kidney transplant45. The hypothesis that a donor polygenic risk is relevant to kidney allograft outcomes remains to be tested but is supported by the fact that most candidate causal genes from GWAS for kidney function map to kidney cell types46,47. The clinical practice and local guidelines for genetic screening of living donors continue to evolve rapidly, but presently only monogenic causes of kidney disease and APOL1 risk genotypes are being considered48. Our results stress an urgent need to test the utility donor GPS in this setting to better assess its impact on living donation risks as well as allograft outcomes.
Although our score is based on a multiethnic GWAS for eGFR, the allelic effect estimates remain heavily biased by the predominant Euro-Asian composition of the discovery GWAS that included 75% European, 23% East Asian, 2% African, and only <1% admixed ancestry Latinx participants. Because there are currently no studies of similar size performed in African and admixed ancestry participants, we were unable to improve the accuracy of effect estimates for these populations, and our model assumed fixed effects across ancestries. We used a diverse linkage disequilibrium reference in order to improve the trans-ethnic performance of the score, and we further enhanced the model by including African ancestry-specific recessive APOL1 risk genotypes known to have large effects on the risk of kidney disease. We demonstrated that APOL1 risk genotypes (coded under a recessive model) have an additive effect with the polygenic component, and significantly enhance case-control discrimination in African and admixed ancestry Latinx cohorts.
Several important limitations of this work need to be discussed. First, we are most limited by the lack of large-scale GWAS for kidney function in non-European populations, as well as small sizes of existing cohorts that could be used for performance optimization in non-Europeans49. As a result, the largest cohorts presently available for robust risk score optimization are also of predominantly European ancestry. The assumption of fixed allelic effects across different ancestral groups is likely inaccurate, because many disease-related lifestyle factors and environmental exposures correlated with ancestry could modify allelic effects. Accordingly, the overall tail discrimination of the score was lower in African than in European or Asian ancestry cohorts with notably lower sensitivity for the top 2% GPS cut-off. Although it is not possible to overcome this limitation in the present study, our GPS approach could be refined by inclusion of larger non-European GWAS studies for eGFR or CKD once available in the future.
Second, the performance comparisons between different ancestral groups could be biased by differences in genotyping platforms and ascertainment methods employed by various biobanks. For example, the UK Biobank represents a population-based cohort recruiting European participants in the 40-60 age group, while the eMERGE and BioMe case-control cohorts are ascertained among older patients with more comorbidities receiving care at tertiary U.S. medical centers. The inclusion of older participants in our testing cohorts might lead to some case misclassification due to age-related kidney function decline. To mitigate this issue, we excluded CKD stage 2 from all performance tests in our study. However, stage 3a may also be prone to residual misclassification50, resulting in risk underestimation for cohorts comprised of older participants.
Third, the ancestry definitions varied in our testing cohorts. While in eMERGE and UKBB the ancestry was defined agnostically using genetic approaches, all other testing cohorts relied on self-report. Despite these differences, the risk score performance was similar across all cohorts, and our sensitivity analyses demonstrated no major change in performance when cohorts were defined by genetics vs. self-report. Notably, the African ancestry cohorts included in this study were predominantly of West African descent. Due to the lack of relevant genetic cohorts, we were unable to test the GPS performance in other African populations, such as from East or South Africa. These populations have relatively low frequency of APOL1 risk alleles, which could potentially dampen the score effects, but follow-up studies are needed for these populations. Additional GPS validation is also needed in Native American and Pacific Islander populations that were not represented here.
Fourth, by design, our score models polygenic effects from GWAS for kidney function as approximated by estimated GFR from serum Cr (filtration marker) rather than CKD itself. We recognize multiple limitations to the use of estimated GFR as a phenotype in GWAS, including the fact that serum Cr level is influenced by the rate of Cr production and metabolism in addition to kidney clearance. Accordingly, to capture a clinically meaningful disease state, we designed the score to predict moderately advanced CKD (stage 3 and above) rather than a mild degree of kidney dysfunction. Notably, our risk score does not incorporate available information on the polygenic determination of albuminuria51 or primary kidney diseases52,53. However, current GWAS for these traits remain several orders of magnitude smaller in sample size compared to the GWAS for eGFR and thus incorporation of such data must await more powerful studies.
Fifth, we observed significant differences in the mean and variance of the GPS distributions by ancestry. The observed shift in the mean GPS towards higher values in individuals of African ancestry is independent of APOL1 and is driven by a higher average RAF in the African genomes. The inter-population RAF differences are greatest for the risk alleles with largest effects. This pattern may be consistent with polygenic adaptation, but the effects of uncorrected population stratification in the discovery GWAS may also potentially explain this phenomenon54. Therefore, based on this observation alone we are unable to determine if the observed shift contributes to the higher prevalence of CKD among individuals of African ancestry.
We note that the observed differences in the GPS distributions by ancestry represent a significant challenge for the clinical implementation of polygenic scores. The key problem is that it is not possible to select a single GPS threshold for all ancestries that results in the similar magnitude of risk. Therefore, we have explored several approaches that could be used to overcome this issue. One approach involves classifying individuals undergoing GPS testing into one of the four ancestry groups based on self-report, then using ancestry-specific cut-offs to interpret the results. However, because of a potential for racial bias, the use of race in clinical algorithms has been discouraged.55 One could also classify a tested individual based on genetic ancestry inferred from SNP data with subsequent application of ancestry-specific cut-offs. This approach still categorizes individuals into distinct groups, and can be inaccurate especially for those individuals with admixed genomes. We have therefore tested two different regression-based ancestry correction methods that model a continuous spectrum of genetic ancestry based on the diverse reference panel of 1000 Genomes. We demonstrate that the reference population-based correction for both mean and variance can best align distribution tails for selection of a single trans-ancestry GPS cut-off. This, however, results in some performance trade-offs, especially in admixed populations. Although still imperfect, this ancestry adjustment may be helpful in improving risk score standardization for clinical use in diverse populations.
Lastly, we used the 2009 CKD-EPI equation (with a race coefficient), since no alternatives were available at the time of our analyses. We do not expect this equation to affect the GPS performance, since our analyses were stratified by genetic ancestry, and the race coefficient was uniformly applied to all African ancestry cohorts. Importantly, our sensitivity analyses confirmed a comparable performance of the GPS when the case-control status was re-defined using the newly proposed 2021 CKD-EPI equation without a race variable35,56.
In summary, we derived, optimized, and tested a new GPS for CKD across major ancestries and proposed new methods for its trans-ethnic GPS standardization. We demonstrated that the polygenic component and APOL1 risk genotypes had additive effects on the risk of CKD. Our study showed that individuals in the highest 2% of the risk score distribution had nearly 3-fold increase in the disease risk, the degree of risk equivalent to a positive family history. The key advantage of the GPS over traditional screening is that it can identify at risk individuals before the onset of any disease manifestations. Timely communication of high-risk status may lead to adoption of protective lifestyle changes and improved adoption of the recommended screening guidelines. Because the cost of SNP arrays is no longer prohibitive, and multiple polygenic scores can be determined using a single array, a population-based genetic screening approach for common diseases (e.g. individuals >40 years old) may prove to represent a cost-effective public health strategy. While our study represents only the first step in this direction, prospective studies are needed to test the clinical utility and cost effectiveness of this approach. The prospective eMERGE-IV study is specifically designed to test this strategy in a newly recruited population-based cohort of over 20,000 volunteers.
METHODS:
Ethics Statement:
The study was approved by the Columbia University Institutional Review Board (IRB numbers IRB-AAAQ9205, IRB-AAAT8208, and IRB-AAAS3500). All participating studies were approved by their local institutional review boards, including all sites contributing human genetic and clinical data to the Electronic Medical Records and Genomics phase 3 (eMERGE-III) consortium. Of note, BioVU operated on an opt-out basis until January 2015 and on an opt-in basis since. The phenotypic data in BioVU are all deidentified, and the study was designated “nonhuman subjects” research by the Vanderbilt Institutional Review Board. All other participants provided written informed consent to participate in genetic studies.
Study cohorts:
Electronic Medical Records and Genomics (eMERGE):
The eMERGE network provides access to electronic health record (EHR) information linked to GWAS data for 102,138 individuals recruited in three phases (eMERGE-I, II, and III) across 12 participating medical centers in years 2007-2019 (54% female, mean age 69 years, 76% European, 15% African American, 6% Latinx, 1% East or Southeast Asian by self-report). All individuals were genotyped genome-wide, and details on genotyping and quality control analyses have been described previously4,57. Briefly, all GWAS datasets were imputed using the multiethnic Haplotype Reference Consortium (HRC) panel on Michigan Imputation Server58. The imputation was performed in 81 batches. Post-imputation, we included only markers with minor allele frequency (MAF) ≥ 0.01 and R2 ≥ 0.8 in ≥ 75% of batches. A total of 7,529,684 variants were retained for the GPS analysis. For principal component analysis (PCA), we used FlashPCA59 on a set of 48,509 common (MAF ≥ 0.01) and independent variants (pruned in PLINK with --indep-pairwise 500 50 0.05 command). The G1 and G2 alleles of APOL1 were imputed separately using the TOPMed imputation server60. The allelic frequencies of G1 and G2 alleles were comparable to previous studies61, as summarized in Supplementary Table 4. The analyses were performed using a combination of VCFtools62 and PLINK v.1.963.
UK Biobank (UKBB):
UKBB is a prospective cohort based in the United Kingdom that enrolled individuals ages 40-69 across the UK in years 2006-201064. This cohort comprised of 488,377 individuals (54% female, mean age 57 years, 94% European, 2% East or Southeast Asian, and 2% African ancestry by self-report), genotyped with high-density SNP arrays, and linked to EHR data. All individuals underwent genotyping with UK Biobank Axiom array from Affymetrix and UK BiLEVE Axiom arrays (~825,000 markers). Genotype imputation was carried out using a 1000 Genomes reference panel with IMPUTE4 software, as previously described64. We then applied QC filters similar to eMERGE-III, retaining 9,233,643 common (MAF ≥ 0.01) variants imputed with high confidence (R2 ≥ 0.8). For PCA by FlashPCA59, we used a set of 35,226 variants that were common (MAF ≥ 0.01) and pruned using --indep-pairwise 500 50 0.05 command in PLINK v.1.963. APOL1 G1 and G2 alleles were imputed separately using the TOPMed imputation server60.
BioMe Biobank:
The BioMe Biobank is an EHR-linked biorepository that has been enrolling participants non-selectively from across the Mount Sinai Health System in New York between 2007-2021. A total of 32,595 BioMe participants were genotyped on the Illumina Global Screening Array (GSA) through a collaboration with Regeneron Genetics Center and 11,953 on the Illumina Global Diversity Array (GDA) through a collaboration with Sema4. Population groups were determined by self-reported race/ethnicity as published previously, with 32% Hispanic/Latinx, 27% Europeans, 22% African Americans, and 2.6% East and Southeast Asian participants65. We removed participants under 40 years of age and those included in the CKDGen GWAS dataset11. We applied standard GWAS quality control analyses, removing participants with genotype missing rates >5% and variants with missing rates >5%, or Hardy-Weinberg Equilibrium violation per each ancestral group (P<1.00E-5 and P<1.00E-6 for GSA and GDA arrays respectively). We additionally removed individuals who were cryptically related (2nd degree or above), or whose genotype-inferred sex did not match the self-reported gender. Imputation (including G1 and G2 variants in APOL1) was performed using the TOPMed Imputation Server with the TOPMed Freeze 8 reference. Post-imputation, variants with quality scores <0.7 were removed. After QC, there were 9,154 BioMe participants of European ancestry, 7,318 African ancestry, 11,606 admixed ancestry Latinx cohorts, and 843 East Asian ancestry included in the analysis.
Reasons for Geographic and Racial Differences in Stroke Study (REGARDS):
REGARDS is a population-based, longitudinal study of incident stroke and associated risk factors in over 30,000 adults aged 45 years or older between 2003 and 2007 from all 48 contiguous US states and the District of Columbia66. By design, participants were oversampled if they were African American. Genotyping was performed on 8,916 self-identified African American participants using Illumina MEGA-EX arrays and imputed using the NHLBI TOPMed reference panel (Freeze 8). Participants were excluded if they had call rates less than 95%, if they were internal duplicates, had sex mismatches, or were outliers on PCA (outside of 6 standard deviations). After QC, there were 8,669 participants available for analysis (63% females, average age 62 years, 100% African-American by self-report). Over 99% of the variants with MAF>1% had an imputation quality of 0.6 or higher, and for GPS calculation we retained genotypes with genotypic probability of 0.9 or higher. APOL1 G1 and G2 alleles were genotyped directly using TaqMan SNP Genotyping Assays (Applied Biosystems/ThermoFisher Scientific).
The Hypertension Genetic Epidemiology Network (HyperGEN):
HyperGEN is a cross-sectional, population-based study and a component of the NHLBI Family Blood Pressure Program that was designed to identify genetic risk factors for hypertension and its complications67. The cohort was recruited in years 1995–2000 and comprised of sibships with at least two siblings diagnosed with hypertension before age 60, their adult offspring, and age-matched controls. The study was subsequently expanded to include additional siblings and offspring to a total N=5,000. African American participants (62% females, average age 52 years) underwent whole genome sequencing (WGS) through the NHLBI WGS program. To harmonize WGS data with the array-based studies, we compiled a set of non-monomorphic and non-multi-allelic SNPs with MAF >1% that were overlapping with our array-typed African American cohorts. This yielded a total of 2,204,415 SNPs that were used as fence post markers for imputation using the same TOPMed release 2 reference panel as for the REGARDS, GenHAT and WPC studies. Over 99% of the variants with MAF>1% had an imputation quality of 0.6 or higher. Only genotypes with genotypic probability over 0.9 were retained for the risk score calculation. APOL1 genotypes were called directly from the WGS data. Individuals younger than 40 years of age were excluded, and a total of 1,898 participants self-identified as African American were retained in the testing cohort.
Warfarin Pharmacogenomics Cohort (WPC):
WPC is a prospective cohort of first-time warfarin users aged 19 years or older starting anticoagulation for venous thromboembolism, stroke/transient ischemic attacks, atrial fibrillation, myocardial infarction, and/or peripheral arterial disease68. The genotype data was generated using Illumina’s MEGA-EX and 1M duo arrays for 599 and 297 participants, respectively (58% females, average age 61 years, 100% African American by self-report). Imputation was performed using the TOPMed r2 reference panel (Freeze 8). More than 99% of the imputed variants with MAF>1% had R2 of 0.6 or higher, and genotypes with genotypic probability of 0.9 or higher were retained for PRS calculation. PCA was performed using EIGENSOFT version 6.1.4 based on 44,137 high quality directly genotyped (missingness <5%), common (MAF ≥ 5%), and independent (r2<0.05) SNPs. APOL1 information was obtained from genotypic array data; rs143830837 was used as a proxy for rs71785313 since these SNPs represent the same G2 variant and were recently merged in dbSNP. For this analysis, only participants aged 40 years or older were included, leaving a total of 448 self-identified African-American participants.
The Genetics of Hypertension Associated Treatments (GenHAT) Study:
GenHAT is an ancillary study to the Antihypertensive and Lipid Lowering Treatment to Prevent Heart Attack Trial (ALLHAT)69. ALLHAT was a randomized, double blind, multicenter clinical trial with over 42,000 high-risk individuals with hypertension, aged 55 years or older, and with at least one additional risk factor for cardiovascular disease. ALLHAT was the largest antihypertensive treatment trial to date and was ethnically diverse, enrolling >15,000 African American participants70. Participants were randomized into four groups defined by the class of assigned antihypertensive medication including chlorthalidone, lisinopril, amlodipine, and doxazosin. The original GenHAT study (N=39,114) evaluated the effect of the interaction between candidate genetic variants and different antihypertensive treatments on the risk of cardiovascular outcomes69. In an ancillary study to the original study, genotyping using Illumina MEGA-EX arrays was performed on 7,546 African American participants. Samples with low call rate (<95%), sex mismatches, and outliers in the PCA (>6 standard deviations) were excluded, which resulted in 6,919 participants with genotypes available for analysis (58% females, average age 66 years, 100% African American by self-report). Genotype imputation was carried out using the NHLBI TOPMed r2 reference panel (Freeze 8), and only genotypes with genotypic probability of 0.9 or higher were retained. APOL1 information was extracted from the genotypic array data, similar to the WPC cohort.
Ancestry definitions
In UKBB and eMERGE-III datasets, the ancestry sub-cohorts were defined based on PCA-based clustering. We grouped all individuals into four major continental ancestry clusters by projecting each sample onto the reference principal components calculated from the 1000G reference panel71. Briefly, we merged our UKBB and eMERGE genotypes with 1000G data and kept only SNPs in common with 1000G. The markers were then pruned using PLINK --indep-pairwise 500 50 0.05. The numbers of pruned variants for UKBB and eMERGE were 35,091 and 43,080 respectively. We then calculated principal components for 1000G using FlashPCA and projected each of our samples onto those PCs. Ancestry assignments were then performed by co-clustering of the reference populations. Ancestry memberships were verified by visual inspection of PCA plots and projections of self-reported race and ethnicity labels on the genetically defined ancestral clusters (Extended Data Figure 5). Ancestry in BioMe, REGARDS, HyperGEN, WPC, and GenHAT was determined by self-reported race/ethnicity, and PCA was subsequently performed for ancestry verification and to exclude outliers.
CKD phenotyping and case-control definitions
For phenotyping, we used the computable CKD phenotype extensively validated by the eMERGE-III network4. We defined cases as having CKD stage 3 or above based on 2009 CKD-EPI72 eGFR <60 mL/min/1.73m2 on at least two serum Cr measurements 3 months apart, or patients on chronic dialysis, or after a kidney transplant. The controls were defined by eGFR > 90 mL/min/1.73m2 based on the latest serum Cr in the absence of CKD-related ICD codes4. The exclusion of individuals with CKD stage 2 (eGFR 60-90 mL/min/1.73m2) from case-control cohorts aimed to minimize potential case-control misclassification due to age-related decline in eGFR. The CKD definition of eGFR <60 mL/min/1.73m2 is thought to reflect <50% of the kidney function in adults, has been associated with increased morbidity and mortality, and has been endorsed as a clinically meaningful threshold by the Kidney Disease: Improving Global Outcomes (KDIGO) 2012 clinical practice guideline for the evaluation and management of CKD73. Only individuals 40 years of age or older were included across all datasets for consistency with the UKBB ascertainment strategy. Additional covariates used in the predictive models included age, sex, diabetes, and principal components of ancestry. The diagnosis of diabetes, an established risk factor for kidney failure, was defined based on ICD codes as previously published18. The diagnosis of hypertension was not added as a covariate to avoid over-adjustment, since cause-effect relationship of hypertension to CKD is difficult to establish based on EHR data, and CKD itself represents the most common cause of secondary hypertension.
Polygenic score design and optimization
We used 70% Europeans of UKBB (6,573 cases and 170,635 controls) to optimize the polygenic component of the GPS. The optimization was performed by selecting the best model between two commonly used methods and a range of input parameters (Table 1, Supplemental Table 1). We used the summary statistics for 8.2 million SNPs from the CKDGen consortium GWAS for eGFR11 in combination with the LD reference panel from phase 3 1000G project (all populations, N=2,504)71. We first computed 7 candidate GPSes using the LDPred algorithm74 across the following range of rho (fraction of casual variants): 1.00E+00, 1.00E-01, 1.00E-02, 1.00E-03, 3.00E-01, 3.00E-02 and 3.00E-03. We also generated 12 pruning and thresholding (P+T) scores with r2=0.2 and P-value thresholds of 1.0, 1.00E-02, 1.00E-03, 1.00E-04, 1.00E-05, 1.00E-06, 1.00E-07, 1.00E-08, 3.00E-02, 3.00E-03, 3.00E-04 and 3.00E-05. Based on the above parameters, each GPS was expressed as a weighted sum of alleles with weights based on the GWAS for the eGFR study:
where M is number of variants in the model and βj is the weight based on GWAS summary statistics and the negative sign reflects an inverse relationship between eGFR and CKD.
Each of the 19 scores derived above was subsequently assessed for discrimination of CKD cases from controls in the first UKBB optimization dataset after adjustment for age, sex, diabetes status and four principal components of ancestry. The score with the best performance was defined by the maximal area under the receiver operator curve and the largest fraction of variance explained. The best performing score was based on P+T method (r2=0.2, P=0.03) and comprised of 471,316 variants, 41,426 of which had non-zero weights (Supplemental Table 1). This score was normal-standardized (by subtracting control mean and dividing by control standard deviation) and advanced for testing in the second UKBB optimization cohort of African ancestry.
Modeling the effects of APOL1 risk genotypes
To optimize trans-ethnic performance, our final score was further optimized using the second UKBB optimization dataset of African ancestry (967 cases and 6,191 controls). We aimed to assess if adding APOL1 risk genotype (under a recessive model) enhanced CKD risk prediction. For this purpose, we first removed any variants in the APOL1 region from the GPS equation to avoid duplicate scoring of this region. Next, we tested the GPS and APOL1 risk genotype jointly for association with CKD in this dataset. The GPS (without APOL1 region) and recessive APOL1 risk genotypes both represented independently significant predictors of CKD before and after adjustment for age, sex, diabetes, and 4 principal components of ancestry. The risk effects of APOL1 and GPS were additive, with one standard deviation unit of the standard-normalized GPS conveying the risk that was approximately equivalent to APOL1 risk genotype (Supplemental Table 2). We also tested for effect modification of APOL1 risk genotype by the polygenic component in CKD prediction, but found no significant interactive effects (P interaction = 0.29). To account for the additive effect of APOL1 risk genotypes, we therefore updated the GPS for each subject using the following equation:
Predictive performance in independent testing datasets:
The predictive performance of the final GPS formulation was assessed in 15 ancestrally diverse testing datasets, including 3 cohorts of European ancestry (14,201 cases and 82,849 controls in total), 6 cohorts of African ancestry (4,268 cases and 10,276 controls), 4 cohorts of Asian (East and South-West) ancestry (392 cases and 8,233 controls), and 2 admixed ancestry Latinx cohorts (1,386 cases and 2,239 controls). We calculated a full set of standardized performance metrics following ClinGen guidelines33. Logistic regression models were used for predicting case-control status with adjustment for age, sex, diabetes, center and genotype/imputation batch (if relevant), and four principal components of ancestry using glm function in R version 3.6.3.
We used pROC R package to calculate the receiver operating characteristic area under curve (AUC). We calculated variance explained using the Nagelkerke’s pseudo-R2, including for the full model (GPS plus covariates), for the covariates-only model, and for the GPS component alone expressed as the R2 difference between the full and the covariates-only model. We also expressed the effect of standardized risk score as odds ratios (with 95% confidence intervals) per standard deviation unit of the control standard normalized risk score distribution in each of the validation cohorts. We examined the risk score discrimination at tail cut-offs corresponding to the top 20%, 10%, 5%, 2%, 1% of the GPS distribution by deriving odds ratios of disease for each tail of the distribution compared to all other individuals in each cohort. We also calculated sensitivities and specificities for each cut-off point in each cohort.
The performance metrics were meta-analyzed across the testing cohorts using an inverse variance weighed fixed-effects method to derive pooled performance metrics for each ancestral grouping75. Finally, we calculated prevalence-adjusted positive and negative predictive values for each GPS cut-off based on pooled estimates of sensitivity and specificity and known CKD prevalence in US population by ancestry. Statistical analyses were conducted using R version 3.6.3 software.
Comparing GPS distributions in the 1000G reference populations
To assess differences in the distributions of GPS by ancestry, we computed risk scores for the multiethnic reference of all 1000G phase 3 participants using our final equation:
where M is the total number of variants included the model, βj is the optimized weight based on GWAS summary statistics for each marker included in the score, and dosagej refers to effect allele dosage (0, 1, 2) for each variant j in 1000G samples. The distributions were examined visually in the form of histograms, and distributional differences by ancestry were tested using ANOVA.
Post-hoc ancestry adjustment
In order to express GPS effects on the same scale across ancestrally diverse individuals and to facilitate selection of a single cut-off for clinical implementation, we adjusted for differences in the first two moments of the GPS distributions by ancestry. Using multiethnic eMERGE cohorts, we tested two different regression-based ancestry adjustment strategies that utilize 1000G (all populations) reference: method 1 which adjusts for differences in mean, and method 2 which adjusts for both differences in mean and variance.
For method 1, we first regressed the GPS of 1000G participants against the first five PCs as proposed previously76:
Fitting the model to 1000G reference panel allows us to find α’s and generate residuals. Next, we used the estimated α’s to calculate the adjusted score for any individual projected onto the same PCA space:
where is the raw GPS, is the predicted (ancestry-adjusted) mean, and δ is the residual standard deviation from the 1000G model (all populations).
To adjust for ancestral differences in both mean and variance (method 2), we used the same method as above, but we also modeled residual variance (δ2) as a function of PCs of ancestry:
Next, we used the estimated α’s and β’s to calculate the adjusted Z-score:
Where is the predicted (ancestry-adjusted) residual variance.
The distributional transformations achieved by these methods were examined visually. We then compared the effects of these adjustments for the top percentile cut-offs in eMERGE-III cohorts.
Extended Data
Extended Data Fig. 1. Distribution of risk allele frequencies (RAF) and their effect sizes for the variants included in the GPS.
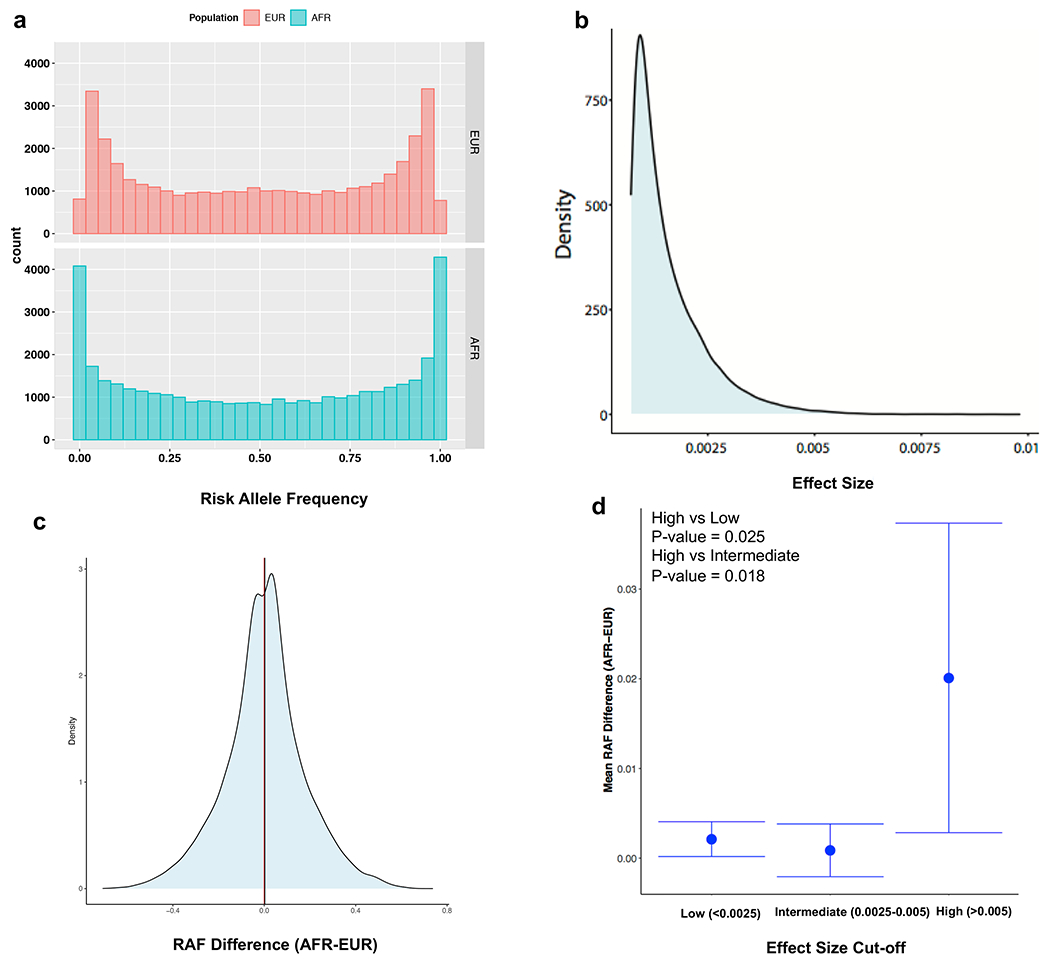
(a) comparison of RAF distributions for the risk variants included in the CKD GPS demonstrates higher frequency of rare (RAF<0.01) and common (RAF>0.99) risk alleles in African compared to European genomes (based on 1000G reference populations); this may be explained by the exclusion of variants with MAF<0.01 in European discovery GWAS; (b) highly skewed effect size (weight) distribution for the variants included in the GPS for CKD; (c) Distribution of RAF difference (AFR-EUR) demonstrating higher average frequency of risk alleles in African genomes (mean RAF difference = 0.002) and a slight rightward shift of the RAF difference distribution from the expected mean of 0; (d) Mean RAF difference (AFR-EUR) as a function of effect size binned into three categories (high, intermediate, and low) based on the observed distribution of effects sizes in panel b, demonstrating that the risk alleles with larger effect size have higher average frequency in African compared to European genomes. EUR: European (N=503) and AFR: African (N=661). The bars represent 95% confidence intervals around the mean RAF difference estimate for each bin; two-sided P-values were calculated using t-test.
Extended Data Fig. 2. Risk score distributions in eMERGE-III (N=22,453) and UKBB (N=77,584) validation datasets.
(a) the distribution of raw polygenic score without APOL1 in UKBB by ancestry; (b) the distribution of ancestry-adjusted polygenic score (method 1: mean-adjusted) in UKBB by ancestry; (c) the distribution of ancestry-adjusted polygenic score (method 2: mean and variance-adjusted) in UKBB by ancestry. Panels (d), (e) and (f) show the same analyses for the eMERGE-III dataset, respectively.
Extended Data Fig. 3. Final GPS calibration analysis in eMERGE-III cohorts combined (N=22,453).
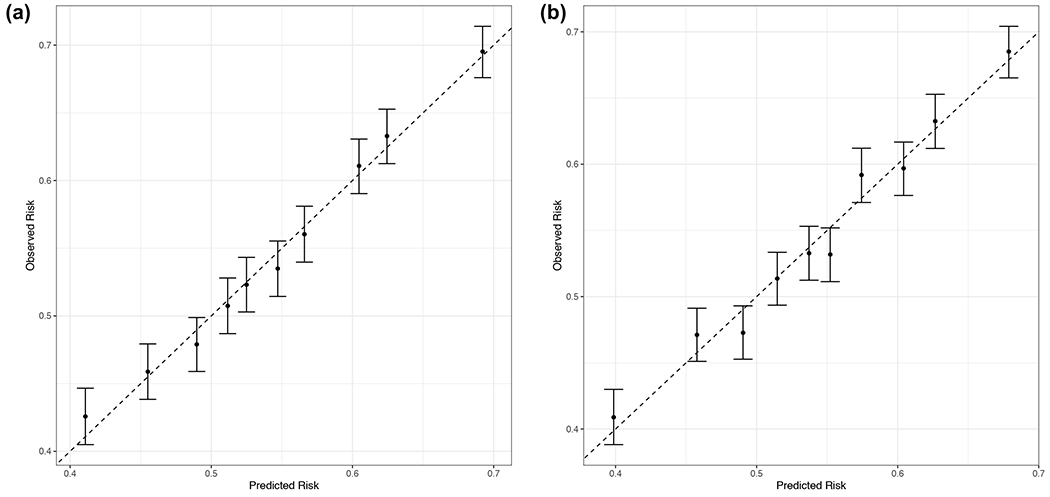
predicted risk (X-axis) as a function of the observed risk (Y-axis) in the multiethnic eMERGE-III dataset after ancestry adjustment with (a) method 1 and (b) method 2. The bars represent 95% confidence intervals.
Extended Data Fig. 4.
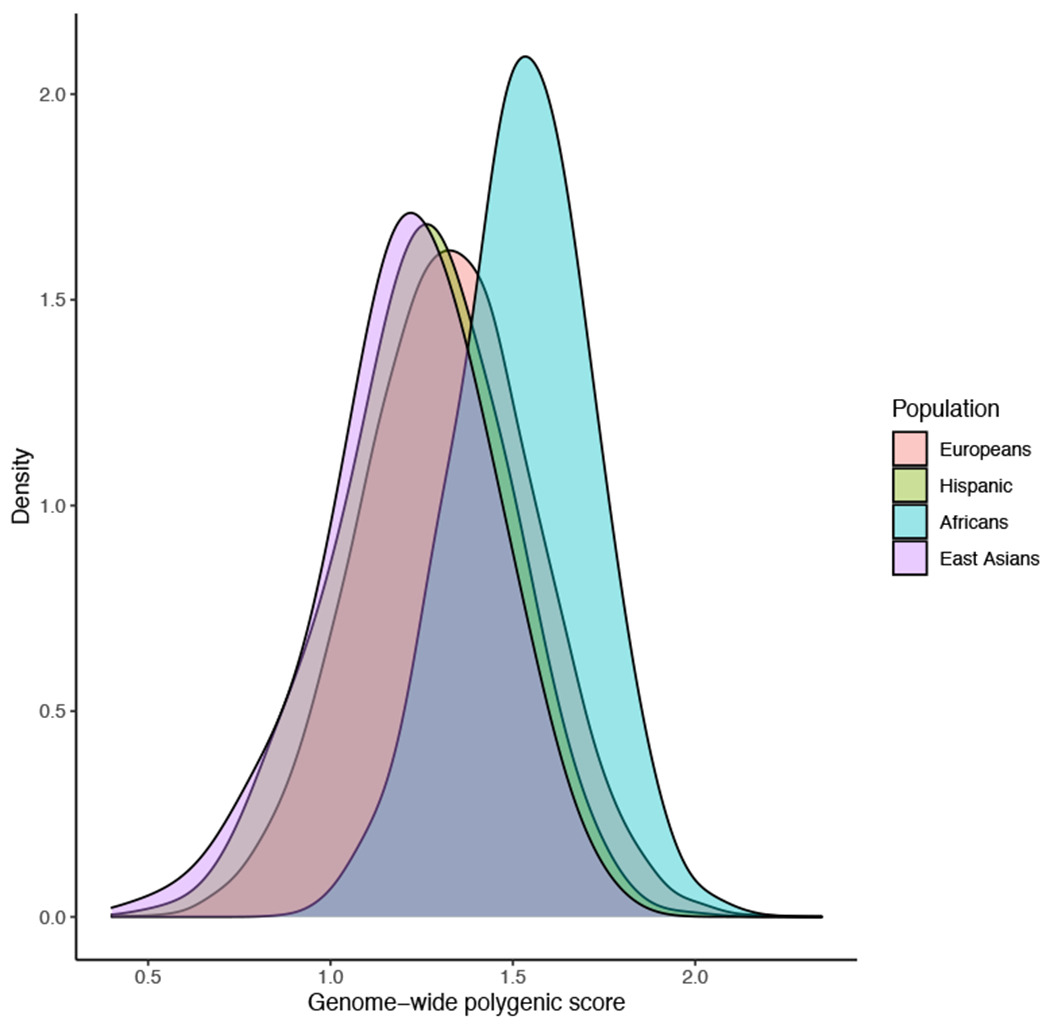
Distributions of the raw (non-standardized) genome-wide polygenic score (GPS) by Yu et al. in the eMERGE-III validation datasets by ancestry.
Extended Data Fig. 5. PCA projections of the study participants from the UKBB (top) and eMERGE-III (bottom) against the 1000G reference populations.
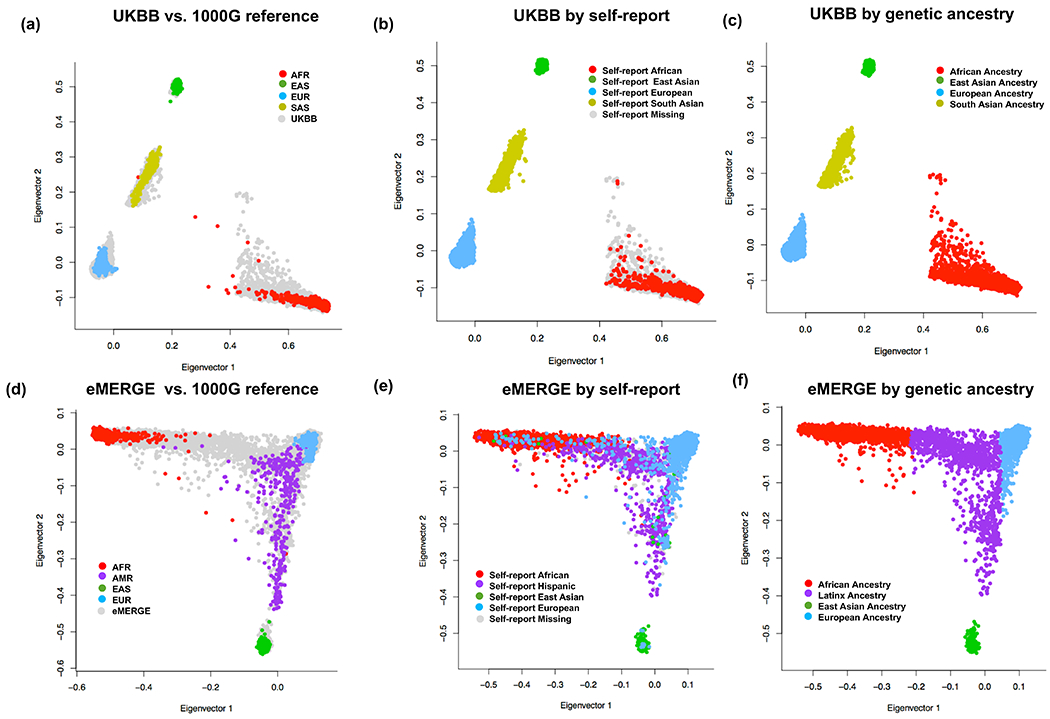
(a) UKBB (N=77,584) and (b) eMERGE-III (N=22,453) participants plotted against the reference 1000G populations (N=2,504); (b, e) plotted by self-reported race/ethnicity; and (c, f) plotted by final ancestry group assignment. X-axis: PC1; Y-axis: PC2; AFR: African; AMR: Native American; EAS: East Asian; EUR: European; and SAS: South Asian.
Supplementary Material
Acknowledgements
This work was funded by the National Human Genome Research Institute (NHGRI) Electronic Medical Records and Genomics-IV (eMERGE-IV grants 2U01HG008680-05, 1U01HG011167-01, 1U01HG011176-01). Additional sources of funding included UG3DK114926 (KK), RC2DK116690 (KK), R01LM013061 (CW, KK), K25DK128563 (AK) and UL1TR001873 (AK, KK), R01HL151855 (JBM), UM1DK078616 (JBM). The parent REGARDS study was supported by cooperative agreement U01NS041588 co-funded by the NINDS and the NIA, the NIH, and the Department of Health and Human Service. The HyperGEN (R01HL055673), GenHAT (R01HL123782), and WPC (R01HL092173, K24HL133373) studies were all supported by the National Heart, Lung, and Blood Institute (NHLBI). Parts of this study have been conducted using the UKBB Resource under UKBB project ID number 41849. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Code Availability Statement:
Electronic CKD Phenotype software is available from the Phenotype Knowledge (PheKB) Database at https://phekb.org/phenotype/chronic-kidney-disease. The CKD GPS score equation is available through GPS catalogue at https://www.pgscatalog.org/publication/PGP000269/ and through our lab website at http://www.columbiamedicine.org/divisions/kiryluk/study_GPS_CKD.php.
Competing Interests Statement
The authors declare no existing competing interest.
Data Availability Statement:
The final formulation of the GPS for CKD along with the standardized metrics of performance were deposited in the GPS catalogue: https://www.pgscatalog.org/publication/PGP000269/. The UK Biobank genotype and phenotype data are available through the UK Biobank web portal at https://www.ukbiobank.ac.uk/. The Electronic Medical Records and Genomics-III imputed genotype and phenotype data are available through dbGAP, accession number phs001584.v2.p2. The BioMe genotype datasets used in this study were generated by Regeneron and are not publicly available. However, the data will be made available for purposes of replicating the results by contacting the corresponding author and appropriate collaboration and/or data sharing agreements. The Warfarin and REGARDS imputed genotype and phenotype data are available through dbGAP, respective accession numbers phs000708.v1.p1 and phs002719.v1.p1. The GenHAT cohort is also available on dbGAP under accession number phs002716.v1.p1 (www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002716.v1.p1). The HyperGEN cohort has been sequenced by the TopMED consortium and WGS data along with the phenotype data are available through dbGAP, accession number phs001293.v3.p1. Minimum testing datasets with the GPS, CKD outcome, and a set of essential clinical covariates for each cohort are also available when consistent with the consent given by the participants and can be requested directly from the corresponding author (kk473@columbia.edu) with a 2–4-week response timeframe; because these datasets contain clinical data, access to these datasets may require a data use agreement.
References:
- 1.Coresh J, et al. Prevalence of chronic kidney disease in the United States. Jama-J Am Med Assoc 298, 2038–2047 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Collaborators GBDC o.D. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1151–1210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chronic Kidney Disease in the United States, (n.d.). https://www.cdc.gov/kidneydisease/publications-resources/ckd-national-facts.html (accessed February 21, 2022).
- 4.Shang N, et al. Medical records-based chronic kidney disease phenotype for clinical care and “big data” observational and genetic studies. Npj Digit Med 4(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox CS, et al. Genomewide linkage analysis to serum creatinine, GFR, and creatinine clearance in a community-based population: the Framingham Heart Study. J Am Soc Nephrol 15, 2457–2461 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Langefeld CD, et al. Heritability of GFR and albuminuria in Caucasians with type 2 diabetes mellitus. Am J Kidney Dis 43, 796–800 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Satko SG & Freedman BI The familial clustering of renal disease and related phenotypes. Med Clin N Am 89, 447-+ (2005). [DOI] [PubMed] [Google Scholar]
- 8.Groopman EE, et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. New Engl J Med 380, 142–151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lata S, et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease: A Pilot Study (vol 168, pg 100, 2018). Annals of Internal Medicine 168, 308-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kottgen A, et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat Genet 41, 712–717 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wuttke M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet 51, 957–972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsa A, et al. APOL1 risk variants, race, and progression of chronic kidney disease. New Engl J Med 369, 2183–2196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson R, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A 111, E2130–2139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko WY, et al. Identifying Darwinian Selection Acting on Different Human APOL1 Variants among Diverse African Populations (vol 93, pg 54, 2013). American Journal of Human Genetics 93, 191–191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nadkarni GN, et al. Worldwide Frequencies of APOL1 Renal Risk Variants. New Engl J Med 379, 2571-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gladding PA, Legget M, Fatkin D, Larsen P & Doughty R Polygenic Risk Scores in Coronary Artery Disease and Atrial Fibrillation. Heart Lung Circ (2019). [DOI] [PubMed] [Google Scholar]
- 18.Khera AV, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lall K, Magi R, Morris A, Metspalu A & Fischer K Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet Med 19, 322–329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffmann TJ, et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nat Genet 49, 54–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehret GB, et al. The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals. Nat Genet 48, 1171–1184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khera AV, et al. Polygenic Prediction of Weight and Obesity Trajectories from Birth to Adulthood. Cell 177, 587–596 e589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberger DR Polygenic Risk Scores in Clinical Schizophrenia Research. Am J Psychiatry 176, 3–4 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Reginsson GW, et al. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addict Biol 23, 485–492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Power RA, et al. Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat Neurosci 18, 953–955 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Aly M, et al. Polygenic risk score improves prostate cancer risk prediction: results from the Stockholm-1 cohort study. Eur Urol 60, 21–28 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fritsche LG, et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. American journal of human genetics 102, 1048–1061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeon J, et al. Determining Risk of Colorectal Cancer and Starting Age of Screening Based on Lifestyle, Environmental, and Genetic Factors. Gastroenterology 154, 2152–2164.e2119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huyghe JR, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nature Genetics 51, 76–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mavaddat N, et al. Polygenic Risk Scores for Prediction of Breast Cancer and Breast Cancer Subtypes. American journal of human genetics 104, 21–34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seibert TM, et al. Polygenic hazard score to guide screening for aggressive prostate cancer: development and validation in large scale cohorts. BMJ 360, j5757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin AR, et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wand H, et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591, 211–219 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, Thio CHL, Gansevoort RT & Snieder H Familial Aggregation of CKD and Heritability of Kidney Biomarkers in the General Population: The Lifelines Cohort Study. Am J Kidney Dis 77, 869–878 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Inker LA, et al. New Creatinine- and Cystatin C-Based Equations to Estimate GFR without Race. New Engl J Med 385, 1737–1749 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Z, et al. Polygenic Risk Scores for Kidney Function and Their Associations with Circulating Proteome, and Incident Kidney Diseases. J Am Soc Nephrol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polubriaginof F, Tatonetti NP & Vawdrey DK An Assessment of Family History Information Captured in an Electronic Health Record. AMIA Annu Symp Proc 2015, 2035–2042 (2015). [PMC free article] [PubMed] [Google Scholar]
- 38.Tada H, et al. Risk prediction by genetic risk scores for coronary heart disease is independent of self-reported family history. Eur Heart J 37, 561–567 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Timmerman N, et al. Family history and polygenic risk of cardiovascular disease: Independent factors associated with secondary cardiovascular events in patients undergoing carotid endarterectomy. Atherosclerosis 307, 121–129 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Hindy G, et al. Genome-Wide Polygenic Score, Clinical Risk Factors, and Long-Term Trajectories of Coronary Artery Disease. Arterioscl Throm Vas 40, 2738–2746 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inouye M, et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults Implications for Primary Prevention. J Am Coll Cardiol 72, 1883–1893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee A, et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med 21, 1708–1718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orlando LA, et al. Development and validation of a primary care-based family health history and decision support program (MeTree). N C Med J 74, 287–296 (2013). [PMC free article] [PubMed] [Google Scholar]
- 44.Fahed AC, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun 11, 3635 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zanoni F & Kiryluk K Genetic background and transplantation outcomes: insights from genome-wide association studies. Curr Opin Organ Transplant 25, 35–41 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellwege JN, et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat Commun 10, 3842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sheng X, et al. Mapping the genetic architecture of human traits to cell types in the kidney identifies mechanisms of disease and potential treatments. Nat Genet 53, 1322–1333 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neugut YD, Mohan S, Gharavi AG & Kiryluk K Cases in Precision Medicine: APOL1 and Genetic Testing in the Evaluation of Chronic Kidney Disease and Potential Transplant. Ann Intern Med 171, 659–664 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sirugo G, Williams SM & Tishkoff SA The Missing Diversity in Human Genetic Studies (vol 177, pg 26, 2019). Cell 177, 1080–1080 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Delanaye P, et al. CKD: A Call for an Age-Adapted Definition. J Am Soc Nephrol 30, 1785–1805 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teumer A, et al. Genome-wide association meta-analyses and fine-mapping elucidate pathways influencing albuminuria. Nat Commun 10, 4130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kiryluk K, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet 46, 1187–1196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie J, et al. The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun 11, 1600 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sohail M, et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vyas DA, Eisenstein LG & Jones DS Hidden in Plain Sight - Reconsidering the Use of Race Correction in Clinical Algorithms. N Engl J Med 383, 874–882 (2020). [DOI] [PubMed] [Google Scholar]
- 56.Delgado C, et al. A Unifying Approach for GFR Estimation: Recommendations of the NKF-ASN Task Force on Reassessing the Inclusion of Race in Diagnosing Kidney Disease. J Am Soc Nephrol 32, 2994–3015 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References:
- 57.Khan A, et al. Medical Records-Based Genetic Studies of the Complement System. J Am Soc Nephrol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abraham G & Inouye M Fast principal component analysis of large-scale genome-wide data. PLoS One 9, e93766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das S, et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nadkarni GN, et al. Worldwide Frequencies of APOL1 Renal Risk Variants. N Engl J Med 379, 2571–2572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Danecek P, et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203-+ (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Belbin GM, et al. Toward a fine-scale population health monitoring system. Cell 184, 2068–2083 e2011 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Howard VJ, et al. The reasons for geographic and racial differences in stroke study: objectives and design. Neuroepidemiology 25, 135–143 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Williams RR, et al. NHLBI family blood pressure program: methodology and recruitment in the HyperGEN network. Hypertension genetic epidemiology network. Ann Epidemiol 10, 389–400 (2000). [DOI] [PubMed] [Google Scholar]
- 68.Limdi NA, et al. Influence of kidney function on risk of supratherapeutic international normalized ratio-related hemorrhage in warfarin users: a prospective cohort study. Am J Kidney Dis 65, 701–709 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arnett DK, et al. Pharmacogenetic approaches to hypertension therapy: design and rationale for the Genetics of Hypertension Associated Treatment (GenHAT) study. Pharmacogenomics J 2, 309–317 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Major cardiovascular events in hypertensive patients randomized to doxazosin vs chlorthalidone: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). ALLHAT Collaborative Research Group. JAMA 283, 1967–1975 (2000). [PubMed] [Google Scholar]
- 71.Altshuler DM, et al. A global reference for human genetic variation. Nature 526, 68-+ (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Levey AS & Stevens LA Estimating GFR using the CKD Epidemiology Collaboration (CKD-EPI) creatinine equation: more accurate GFR estimates, lower CKD prevalence estimates, and better risk predictions. Am J Kidney Dis 55, 622–627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kidney Disease: Improving Global Outcomes (KDIGO) Chronic Kidney Disease Work Group: KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. . Kidney Int Suppl 3: 1–150(2013). [DOI] [PubMed] [Google Scholar]
- 74.Vilhjalmsson BJ, et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. American journal of human genetics 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khera AV, et al. Whole-Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized With Early-Onset Myocardial Infarction. Circulation 139, 1593–1602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final formulation of the GPS for CKD along with the standardized metrics of performance were deposited in the GPS catalogue: https://www.pgscatalog.org/publication/PGP000269/. The UK Biobank genotype and phenotype data are available through the UK Biobank web portal at https://www.ukbiobank.ac.uk/. The Electronic Medical Records and Genomics-III imputed genotype and phenotype data are available through dbGAP, accession number phs001584.v2.p2. The BioMe genotype datasets used in this study were generated by Regeneron and are not publicly available. However, the data will be made available for purposes of replicating the results by contacting the corresponding author and appropriate collaboration and/or data sharing agreements. The Warfarin and REGARDS imputed genotype and phenotype data are available through dbGAP, respective accession numbers phs000708.v1.p1 and phs002719.v1.p1. The GenHAT cohort is also available on dbGAP under accession number phs002716.v1.p1 (www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002716.v1.p1). The HyperGEN cohort has been sequenced by the TopMED consortium and WGS data along with the phenotype data are available through dbGAP, accession number phs001293.v3.p1. Minimum testing datasets with the GPS, CKD outcome, and a set of essential clinical covariates for each cohort are also available when consistent with the consent given by the participants and can be requested directly from the corresponding author (kk473@columbia.edu) with a 2–4-week response timeframe; because these datasets contain clinical data, access to these datasets may require a data use agreement.