

Abstract
Profiling of circulating tumor DNA (ctDNA) in the bloodstream shows promise for noninvasive cancer detection. Chromatin fragmentation features have previously been explored to infer gene expression profiles from cell-free DNA (cfDNA), but current fragmentomic methods require high concentrations of tumor-derived DNA and provide limited resolution. Here we describe promoter fragmentation entropy as an epigenomic cfDNA feature that predicts RNA expression levels at individual genes. We developed ‘epigenetic expression inference from cell-free DNA-sequencing’ (EPIC-seq), a method that uses targeted sequencing of promoters of genes of interest. Profiling 329 blood samples from 201 patients with cancer and 87 healthy adults, we demonstrate classification of subtypes of lung carcinoma and diffuse large B cell lymphoma. Applying EPIC-seq to serial blood samples from patients treated with PD-(L)1 immune-checkpoint inhibitors, we show that gene expression profiles inferred by EPIC-seq are correlated with clinical response. Our results indicate that EPIC-seq could enable noninvasive, high-throughput tissue-of-origin characterization with diagnostic, prognostic and therapeutic potential.
Cell-free DNA (cfDNA) molecules circulating in blood plasma largely arise from chromatin fragmentation accompanying cell death during homeostasis of diverse tissues throughout the body1–3. Accordingly, cfDNA profiling has been established for use in detection of tissue rejection after solid organ transplantation, noninvasive prenatal testing of fetal aneuploidy during pregnancy and noninvasive tumor genotyping, along with early evidence for detection of diverse cancer types4–12. For each application, current liquid biopsy testing approaches have largely relied on germline or somatic genetic variations to diagnose pathology in tissues of interest. Indeed, such variations in genetic sequences can be highly informative for biopsy-free tumor genotyping of circulating tumor DNA (ctDNA) and for monitoring of disease burden, with potential use for early cancer detection9,13–15.
Despite the many applications of cfDNA profiling for noninvasive detection of mutations, even in cancers with a high tumor mutation loads and even in patients with high disease burden, most cancer-derived fragments are unmutated. Accordingly, interrogating these cfDNA fragments using epigenetic features could have broad use for example to inform tissue-of-origin of unmutated molecules. Separately, such approaches could be useful for detection of tissue injury without associated genetic lesions16–21, and for cancer classification. Since circulating cfDNA molecules are primarily nucleosome-associated fragments, they reflect distinctive chromatin configurations of the nuclear genome of cells from which they derive22–24. Specifically, genomic regions densely associated with nucleosomal complexes are generally protected against the action of various endonucleases, while open chromatin regions are prone to such degradation25.
Several studies have identified specific chromatin fragmentation features as potentially useful for classification of tissue-of-origin by cfDNA profiling. These ‘fragmentomic’ features include lower depth of sequencing coverage26–29 and disruption of nucleosome positioning25 near transcription start sites (TSSs). Separately, several studies have shown that the length of cfDNA fragments can also inform tissue-of-origin, including tumor derivation, even when considered agnostic to genomic location or relation to gene promoters. For example, tumor-derived molecules bearing somatic variants tend to be shorter than wildtype counterparts29–32 as useful for distinguishing somatic variants that are tumor-derived from those arising from circulating leukocytes during clonal hematopoiesis9. Despite these advances, current fragmentomic methods, including those relying on shallow whole-genome sequencing (WGS) do not fully harness contributions of various tissues to the circulating DNA pool. Separately, current techniques provide inadequate genomic depth and breadth to enable gene-level resolution. Indeed, even when considering gene groups, such methods perform well for inferring expression only at high ctDNA levels. Accordingly, fragmentomic methods for inferring expression are limited to patients with very high tumor burden generally observed in advanced disease.
We addressed these limitations by evaluating additional cfDNA fragmentation features for predicting gene expression. We reasoned that by profiling cfDNA fragmentation in critical regions at high resolution, key fragmentomic features could capture gene-level associations with expression levels across the genome and could inform accurate models for predicting transcriptional output. If this hypothesis is indeed correct, then deep sequencing of informative genomic regions could overcome the limitations of previous approaches and allow profiling cfDNA fragmentation at high resolution and facilitate gene-level analyses. Here we describe a cfDNA fragmentation feature enabling prediction of expression for individual genes. We use this observation to develop epigenetic expression inference from cfDNA-sequencing (EPIC-seq), a method for analyzing expression based on cfDNA fragmentomics. We then applied EPIC-seq to classify histology of nonsmall-cell lung cancer (NSCLC), to distinguish molecular subtypes in diffuse large B cell lymphoma (DLBCL), to assess immunotherapy responses and to evaluate the prognostic value of individual genes.
Results
cfDNA features correlated with gene expression.
We hypothesized that cfDNA fragments from active promoters (which are less protected by nucleosomes) will exhibit more random cleavage patterns than fragments from inactive promoters (which are more protected by nucleosomes). If correct, this should allow inferences about expression of individual genes from cfDNA, reflecting contributions from various cell types in diverse tissues, including solid tumors (Fig. 1a). To explore this hypothesis, we profiled cfDNA by deep WGS (roughly 250×) from a patient with carcinoma of unknown primary (CUP) with very low ctDNA levels as quantified by cancer personalized profiling by deep sequencing (CAPP-seq)9 (<0.05%, Methods and Supplementary Table 1). Since most cfDNA molecules were of hematopoietic origin33, we correlated specific cfDNA fragmentomic features to expression levels of blood leukocytes determined by RNA-sequencing (RNA-seq). We then ranked genes by their expression levels and characterized the distribution of cfDNA fragments at promoters (Fig. 1b). Supporting our hypothesis, cfDNA molecules mapping to the roughly 2-kb region flanking the TSSs of highly expressed genes exhibit substantially more fragment length diversity than fragments mapping to TSSs of poorly expressed genes. This phenomenon is especially prominent in subnucleosomal fragments (<150 basepairs (bp) and 210–300 bp, Fig. 1b and Extended Data Fig. 1a,b).
Fig. 1 |. Correlation of gene expression and cfDNA molecular features.
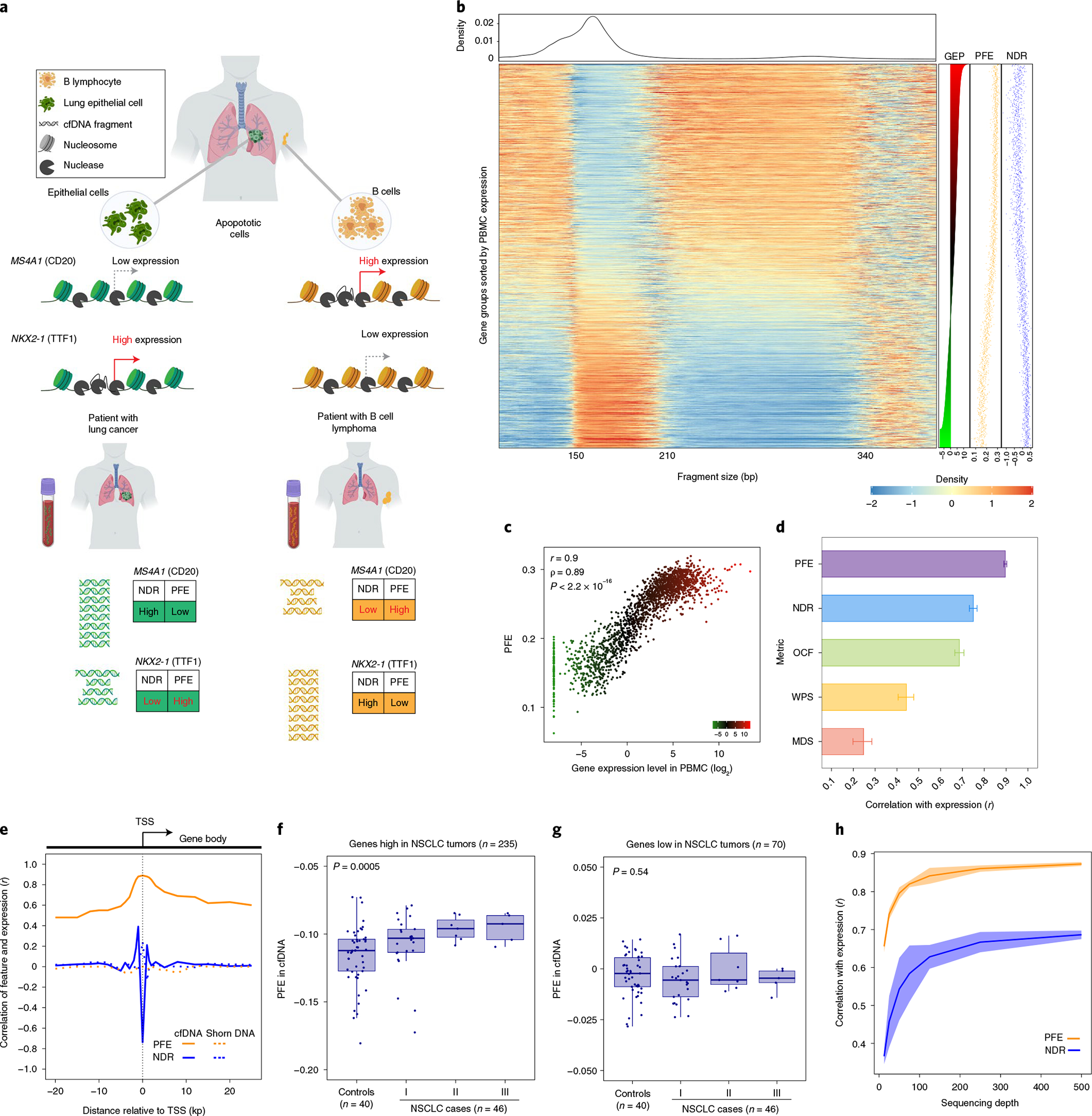
a, Chromatin accessibility footprints can be traced back to the tissue-of-origin. Open chromatin is subject to nuclease digestion resulting in decreased sequencing coverage depth, measured by NDR, and fragment length diversity, measured by PFE. In this cartoon, lung epithelial cells exhibit very low expression of MS4A1 (CD20) but high expression of NKX2-1 (TTF1). The cfDNA fragments of a patient with lung cancer consist of normal primarily hematopoietic cfDNA fragments mixed with fragments derived from LUAD cells undergoing apoptosis. Because the lung epithelial cell compartment has a lower NDR and higher PFE for NKX2-1 fragments, the resulting mixture shows similar changes with the net effect dependent on the total amount of circulating tumor-derived fragments. B cells, on the other hand, highly express MS4A1 with a very low expression level of NKX2-1. Accordingly, the cfDNA fragments of a patient with B cell lymphoma consist of normal cfDNA fragments admixed with B cell-derived ctDNA with overrepresentation of MS4A1 resulting in lower coverage and higher diversity of cfDNA fragment length values at the TSS. b, Heatmap depicting cfDNA fragment size densities at TSSs across the genome in an exemplar plasma sample profiled by high-depth WGS (roughly 250×). The x axis depicts cfDNA fragment size, while the rows of the heatmap capture fragment density as ordered by gene expression profile (GEP) in blood leukocytes assessed by RNA-seq using TPM (right). Each row corresponds to one meta-gene encompassing the TSSs of ten genes when ranked by a reference PBMC expression vector. The data are normalized column-wise for each cfDNA fragment size bin. Corresponding PFE, NDR and TPM levels are depicted for each bin in dot plots on the right. c, A scatterplot depicts the relationship between plasma cfDNA PFE versus leukocyte RNA expression levels (TPM), as in b. Both Pearson (r) and Spearman (ρ) correlation coefficients are reported. In both, P < 2.2 × 10−16. d, Pearson correlations between individual cfDNA fragment features and leukocyte gene expression levels. The error bars depict the 95% CIs resulted from 500 bootstrap replicates (resampling with replacement of gene groups). This analysis is performed by using the deep WGS profile used in b and c. e, The correlation between leukocyte gene expression and each of two leading cfDNA features as a function of distance to the TSS center. The dotted lines correspond to the concordance measure when evaluated on the shorn leukocyte DNA from a matched blood PBMC sample. f, Relationship between PFE of a NSCLC signature and cfDNA sample status and across stages. The PFE monotonically increases from noncancer to later stages patients with NSCLC (Jonckheere’s trend test P = 0.0005). g, Relationship between PFE of a gene set with low expression in NSCLC (and high in PBMC) and cfDNA sample status and across stages. The PFE of this set is not associated with disease status or disease stage (Jonckheere’s trend test P = 0.54). In box-and-whisker plots in f and g, the median is horizontally marked with a line in each box and whiskers span the 1.5 IQRs in each patient cohort. h, Effect of sequencing depth (x axis) on the correlation of cfDNA PFE and NDR with gene expression (y axis). For each down-sampled depth, three replicates were generated, and the shaded area illustrates three standard deviations above and below the mean.
We reasoned that nucleosome displacement or depletion at the TSS of active genes could result in more diverse digested fragments34, and that estimating this diversity could inform the corresponding expression level at individual gene TSS regions. We therefore captured this diversity in cfDNA fragment lengths as an entropy measure, calculating a modified Shannon index for fragments where both ends fell within the 2 kb flanking each gene’s TSS (1 kb on each side). After adjusting this cfDNA entropy measure using a Dirichlet-multinomial model for normalization, we refer to this metric as promoter fragmentation entropy (PFE) (Methods). We observed high transcriptome-wide correlation between PFE measured in cfDNA by WGS and expression levels measured by RNA-seq of peripheral blood mononuclear cells (PBMCs) (R = 0.89, P < 1 × 10−16; Fig. 1b,c and Supplementary Table 2). While sequencing depth at nucleosome-depleted regions (NDRs) flanking the TSS (NDR depth)27 was also significantly correlated with expression of corresponding genes, it showed substantially lower correlation than did PFE (Fig. 1b; r = −0.78, P < 1 × 10−16). The significant correlations between RNA expression levels and fragmentomic features were only observed in cfDNA and not in acoustically shorn high molecular-weight genomic DNA from matched leukocytes (PFE r = 0.003, NDR r = 0.24). Accordingly, expression inferences from cfDNA fragmentation profiles appear to reflect functional nucleosomal associations of DNA in vivo and not predictable from a primary DNA sequence. Furthermore, TSS regions were distinguished from exonic and intronic by having the highest representation of subnucleosomal fragments (P < 0.0001, Extended Data Fig. 1c). We also examined subnucleosomal cfDNA fragments in the context of mutant versus wildtype molecules in patients with NSCLC (Extended Data Fig. 1d and Supplementary Notes).
We next compared several other cfDNA fragmentation features for correlation with expression levels of blood leukocytes (Fig. 1d and Supplementary Table 2). While previous cfDNA profiling studies have reported lower sequencing coverage depth at NDRs within promoters of actively expressed genes27, the correlation between PFE and expression was stronger than between normalized NDR depth and expression (Fig. 1b,d). PFE also outperformed other previously defined fragmentomic metrics including windowed protection score (WPS)25, motif diversity score (MDS)35 and orientation-aware cfDNA fragmentation (OCF)20. We also examined whether the distance from the TSS affects correlations between cfDNA fragmentomic features and expression and observed that in comparison to NDR correlation of PFE had broader dispersion (Fig. 1e and Supplementary Notes).
We further confirmed our observations from deep WGS profiling of cfDNA by considering fragmentomic profiles of patients with lung cancer previously profiled at lower WGS depth (20–40×)15. We compared lung cancer cases and healthy control participants when inferring expression levels of two lung cancer gene signatures defined in primary tumor tissues, corresponding to genes variably expressed in NSCLC. We observed a substantial increase in inferred expression levels of the NSCLC-high signature as distinguishing lung cancer from healthy noncancer control participants, correlated with lung cancer stage (Fig. 1f and Methods). This increasing trend was not observed in the NSCLC-low expression signature (Fig. 1g), indicating the effect to be gene- and tissue-specific. Indeed, the NSCLC signature also showed modest performance in distinguishing lung cancer cases from control participants when cfDNA was profiled by WGS (area under curve (AUC), 0.76; Extended Data Fig. 1f). We also investigated the impact of sequencing depth on correlations between cfDNA fragmentomic signals and transcriptome-wide RNA expression. Correlations plateaued around 500× sequencing depth (Fig. 1h). Overall, these results indicated that cfDNA fragmentation features are strongly correlated with RNA expression, and that PFE better captures this correlation than previously described metrics.
To better resolve the association between cfDNA fragmentation entropy and expression levels, we next studied their relationship across individual gene bodies, when considering distance from the TSS and exon/intron organization. We found peak cfDNA fragmentation entropy to be centered at the TSS, with this effect being most prominent for highly expressed genes (Fig. 2a). When summarizing results across genes as a function of distance from the TSS, we observed a bimodal distribution of entropy values in a roughly 2.5-kb window flanking each TSS (Fig. 2b). When considering gene bodies, we found that while first exons display similar entropy signals as the TSS, this signal precipitously declines for subsequent introns and exons that are farther from the TSS (Fig. 2c). Therefore, cfDNA fragmentation features flanking TSS regions are highly correlated with expression levels across the transcriptome, with normalized entropy of cfDNA fragments overlapping first exons capturing much of this effect.
Fig. 2 |. Fragment size entropy in relation to gene structure informs expression inferences from whole-exome cfDNA profiling.
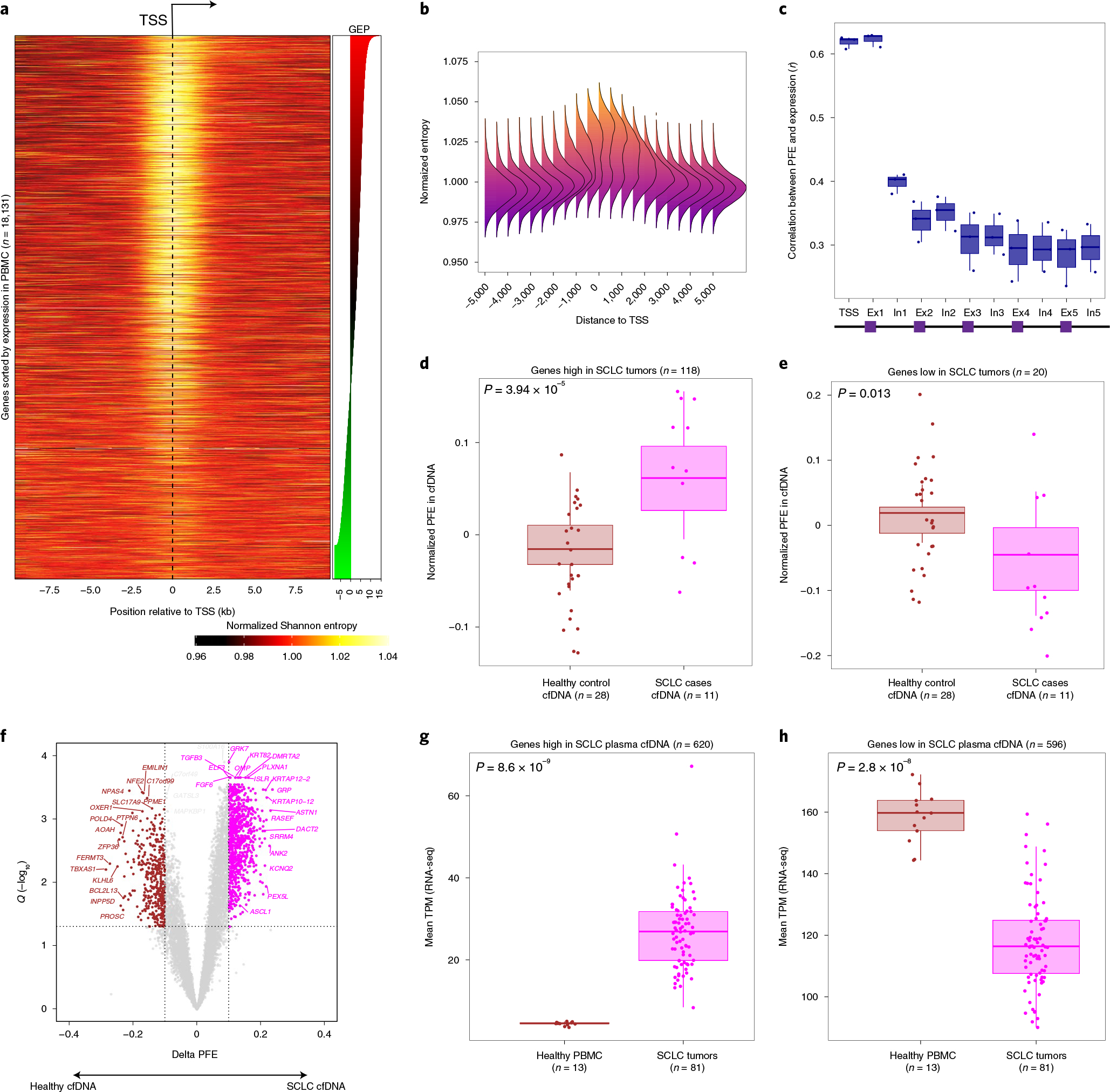
a, Heatmap depicting the mean normalized Shannon entropy of cfDNA fragment size distributions for 18,131 individual protein-coding genes when sorted by their expression in blood PBMC leukocytes, across a 20-kb region flanking each TSS. The heat illustrates the normalized entropy (normalization to the average entropy over the start to end of this 20-kb region). The underlying data are the deep whole-genome cfDNA profile from Fig. 1b. b, A summary representation of the heatmap in a. Each column reflects a window position across the TSS and is summarized by a histogram depicting the deviation of Shannon from the window centered at the TSS (position 0). c, Concordance analysis using a Pearson correlation between individual gene expression and PFEs when calculated in TSS, exon 1, intron 1 and so on. Each dot corresponds to one cfDNA sample profiled deeply by WGS (n = 3, Methods). d, Genes known to be highly expressed in SCLC tumors by RNA-seq (n = 118 genes from 81 tumors) exhibit significantly higher PFE in cfDNA samples from patients with SCLC (n = 11, pink dots) than healthy adult control participants (n = 28, brown dots; P = 3.94 × 10−5), as profiled by deep (roughly 2,000×) WES (Methods and Supplementary Fig. 1g). e, As in d, but showing significantly lower average PFE in cfDNA of patients with SCLC, when considering 20 genes known to be lowly expressed in SCLC tumors but highly expressed in PBMCs by RNA-seq (P = 0.02). f, DEGs associated with SCLC, identified directly from cfDNA using PFE analysis. Volcano plot depicts genes inferred to be more highly expressed in 11 cfDNA samples from SCLC cases (pink dots, n = 620), or in 28 cfDNA samples from healthy adult control participants (brown dots, n = 596). DEGs were determined by considering the magnitude of mean PFE difference between groups (x axis; |0.1|) and the false discovery rate (Q < 0.05) from t-tests between groups. These two sets of genes discovered noninvasively from cfDNA as differentially expressed in SCLC, were then assessed for expression in primary SCLC tumors in g and h. The box-and-whisker plots depict the median and IQR of the mean RNA expression levels (y axis, TPM) observed for the SCLC high (g) and SCLC low (h) gene sets, when comparing RNA-seq in SCLC tumors (n = 81, pink dots) versus healthy PBMCs (n = 13, brown dots). In all the box-and-whisker plots, the median is horizontally marked with a line in each box, and whiskers span the 1.5 IQRs in each patient cohort.
Validation of expression inferences from cfDNA in solid tumors.
Having observed that fragmentation entropy of cfDNA molecules overlapping first exons correlates with expression, we next asked whether whole-exome sequencing (WES) could further validate inferred expression estimates from cfDNA. Specifically, we sequenced plasma cfDNA of small-cell lung cancer cases (SCLC) (n = 11) and healthy control participants (n = 28) by ultradeep WES (median unique depth roughly 2,000×) to infer expression levels using PFE. We then compared these inferred results with expression levels observed in transcriptome profiling of solid tumor tissues (Extended Data Fig. 1g). When considering genes highly expressed in primary SCLC tumors compared with PBMCs by RNA-seq genes or vice versa (Methods), we found strong concordance in corresponding signatures in plasma cfDNA (Fig. 2d,e). Specifically, ‘SCLC high’ tumor genes had significantly higher normalized PFE levels in plasma cfDNA of patients with SCLC than healthy control participants (P = 3.9 × 10−5; Fig. 2d), and conversely, ‘SCLC low’ genes demonstrated the expected reciprocal pattern (P = 0.02; Fig. 2e). When combining these two signatures into a single ‘SCLC score’, we observed strong classification performance for distinguishing SCLC cases from control participants (AUC = 0.98, 95% CI: 0.94–1.00; Extended Data Fig. 1h).
Separately, we asked whether de novo discovery of SCLC-specific expression markers might be feasible noninvasively, when considering exome-wide cfDNA profiling and PFE overlapping first exons (Fig. 2f). Among such candidate differentially expressed genes (DEGs) distinguishing patients with SCLC from healthy adult control participants, we identified several well-known SCLC markers including ASCL1, ANK1 and ASTN1 (Fig. 2f). Indeed, genes whose differential expression was inferred from cfDNA exhibited highly significant and concordant differential expression in primary SCLC tumors and PBMCs when profiled by RNA-seq (Fig. 2g,h and Methods). SCLC-specific genes inferred from plasma cfDNA by WES were highly enriched for genes highly expressed in primary SCLC tumors by RNA-seq (P = 0.014, Extended Data Fig. 1i). Therefore, expression inference from cfDNA is feasible and can faithfully capture tumor-specific expression from solid lung cancer tissues at gene-level resolution.
Inferring gene expression from cfDNA fragmentation profiles.
We next attempted to predict expression from cfDNA fragmentomic features derived by WGS. When considering diverse metrics, we identified PFE and normalized NDR depth as complementary features predicting RNA expression in a generalized linear model (Methods). Specifically, PFE demonstrated better dynamic range for lowly expressed genes, while highly expressed genes appeared better captured by normalized NDR depth (Extended Data Fig. 1d). We then validated this model by applying it to a fragmentomic ‘meta-profile’ assembled by WGS profiling of plasma cfDNA from 27 healthy adults (Methods). Here again we observed high correlation between model-predicted expression levels and observed measurements by RNA-seq of PBMCs when considering groups of ten genes (r = 0.9, Extended Data Fig. 2a). Consistent with our previous observations (Fig. 1h), these correlations deteriorated at lower sequencing depth in a manner hampering single-gene resolution (Extended Data Fig. 2a,b). While cfDNA PFE outperformed NDR in correlations with expression, our composite model combining both PFE and NDR had higher correlations than either alone (Extended Data Fig. 2b). We also examined our expression inference model by considering its performance in several other validation studies, again observing high correlations (Extended Data Fig. 2c–h and Supplementary Notes).
EPIC-seq.
Having observed that higher sequencing depths improve correlations (Fig. 1h), we next set out to develop a method allowing predicting expression of individual genes by deeper profiling of TSS regions. While normalized entropy of cfDNA fragments overlapping first exons allowed expression inferences when using deep WES (as described above for SCLC), the nontranscribed 5′ flanking regions of most genes are uncaptured by typical commercially available exome bait sets, thereby precluding corresponding NDR estimates from these TSS regions. Therefore, we devised a new approach, EPIC-seq, that combines hybrid capture-based targeted deep sequencing of TSS flanking regions in cfDNA with machine learning for predicting RNA expression (Fig. 3a). The TSS regions targeted in an EPIC-seq experiment are tailored to include genes expected to be differentially expressed in the conditions of interest.
Fig. 3 |. EPIC-seq design and workflow.
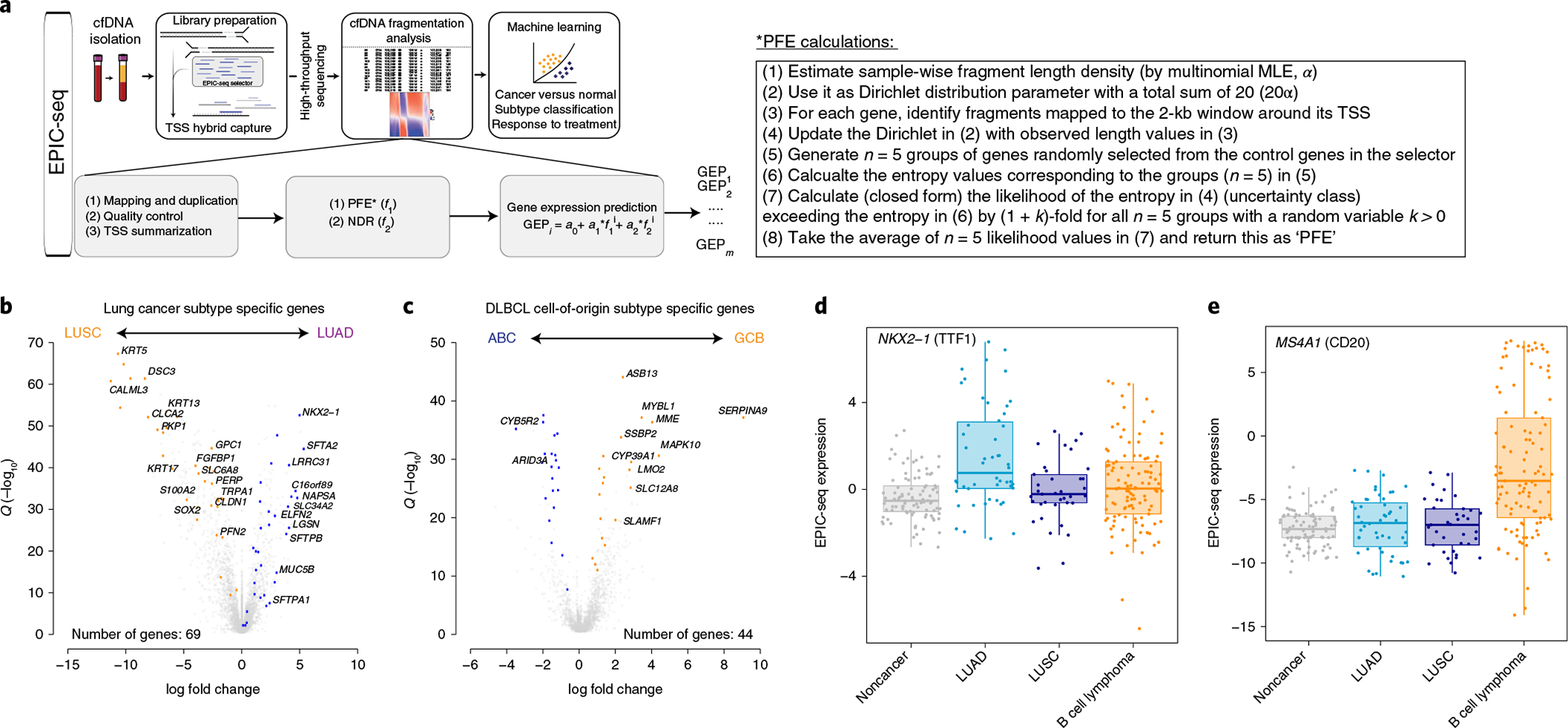
a, The schema depicts the general workflow of EPIC-seq, starting with cfDNA extraction from plasma, library preparation and capture of TSS of genes of interest, high-throughput sequencing of enriched regions and, finally, cfDNA fragmentation analysis followed by machine learning models for prediction of expression at each TSS and classification of the specimen. b,c, The volcano plots depict DEGs, as informative for histological classification in NSCLC subtypes (b) (LUAD versus LUSC from TCGA36,37) and in COO classification of DLBCL (c) (ABC versus GCB from Schmitz et al.101). Genes highlighted in colors other than gray were selected for TSS capture in EPIC-seq, after censoring genes with high expression in blood leukocytes (Methods). d, NKX2-1, encoding TTF1, known to be highly expressed in NSCLC-LUAD tumors, exhibits significantly higher predicted expression in cfDNA of patients with LUAD by EPIC-seq (LUAD versus others Wilcoxon test P = 5.7 × 10−6). e, MS4A1, encoding CD20, known to be a marker of DLBCL tumors, exhibits significantly higher predicted expression in cfDNA of patients with DLBCL by EPIC-seq (DLBCL versus others Wilcoxon test P = 5.44 × 10−9). Box-and-whisker plots depict predicted expression levels in individual samples profiled by EPIC-seq (dots), with boxes spanning the IQR; the median is horizontally marked with a line in each box, and whiskers span the 1.5 IQRs in each patient cohort. In d and e, individual patients are shown as dots (noncancer, n = 91; LUAD, n = 50; LUSC, n = 37; B cell lymphoma, n = 114).
As a proof-of-concept, we tested this framework by applying EPIC-seq to two cancer classification problems using cfDNA: (1) noninvasively distinguishing histological subtypes of the most common solid tumor (NSCLC), and (2) resolving molecular subtypes of the most common hematological malignancy (DLBCL). For each malignancy, we first identified genes highly expressed in tumor tissues, but with relatively low expression in whole blood (Methods). We then identified subtype-specific genes by evaluating those differentially expressed in NSCLC adenocarcinoma (lung adenocarcinoma, LUAD) versus lung squamous cell carcinoma (LUSC) and DLBCL germinal center B (GCB) versus activated B cell (ABC)-like subtypes36,37,39,101 (Supplementary Table 3, Fig. 3b,c and Supplementary Notes).
Using this workflow, we then profiled 373 plasma cfDNA samples, of which 329 were used for testing EPIC-seq in different applications (median roughly 2,000× unique coverage depth, Extended Data Fig. 3a and Supplementary Notes). This final set comprises 288 adults (Extended Data Fig. 3a,b and Supplementary Table 4), including 87 patients with NSCLC (n = 109 samples), 114 patients with DLBCL (n = 126 samples) and 87 otherwise healthy participants (n = 94 samples). Using a custom EPIC-seq analytical pipeline (Methods), we computed cfDNA fragmentomic features for each gene of interest and then estimated its predicted RNA expression level (Fig. 3a). To explore the ability of EPIC-seq to infer expression of individual genes, we next evaluated expression of NKX2-1 (TTF1), a gene highly expressed in LUAD and useful in histopathological diagnosis, and MS4A1 (CD20), a gene highly expressed in DLBCL and useful for immunophenotyping and classification of lymphomas38,39. The predicted expression level for NKX2-1 was significantly higher in plasma from patients with NSCLC-LUAD (Wilcoxon test P = 4.2 × 10−6; Fig. 3d). Conversely, the predicted expression level for MS4A1 was significantly higher in plasma from patients with DLBCL (Wilcoxon test P = 4.2 × 10−14, Fig. 3e). Collectively, these results illustrate that inference of expression is feasible by targeted deep cfDNA-seq using EPIC-seq, and that this framework can recover expected differences in tissue-derived expression at single-gene resolution.
EPIC-seq for lung cancer detection.
We next evaluated whether EPIC-seq might have use for cancer classification problems, starting with lung cancer, the leading cause of cancer-related death40,41. We asked whether noninvasive classification of NSCLC cases versus healthy control participants was feasible from cfDNA using EPIC-seq. The cohort was split into training (n = 138) and validation (n = 43). A classifier trained on EPIC-seq data to distinguish patients with NSCLC (n = 67: stage II (n = 7), stage III (n = 30) and stage IV (n = 30)) from noncancer control participants (n = 71) revealed robust performance (EPIC-lung AUC = 0.91, 95% confidence interval (CI), 0.86–0.96) when considering 141 TSS sites from 117 genes (Fig. 4a and Methods). When we applied this trained classifier to the validation subset of patients with NSCLC (n = 20) and noncancer control participants (n = 23), we again observed high classification accuracy, with only a modest decrease in performance (AUC = 0.83, 95% CI, 0.71–0.96; Fig. 4a).
Fig. 4 |. Application of EPIC-seq for lung cancer detection and histological classification.
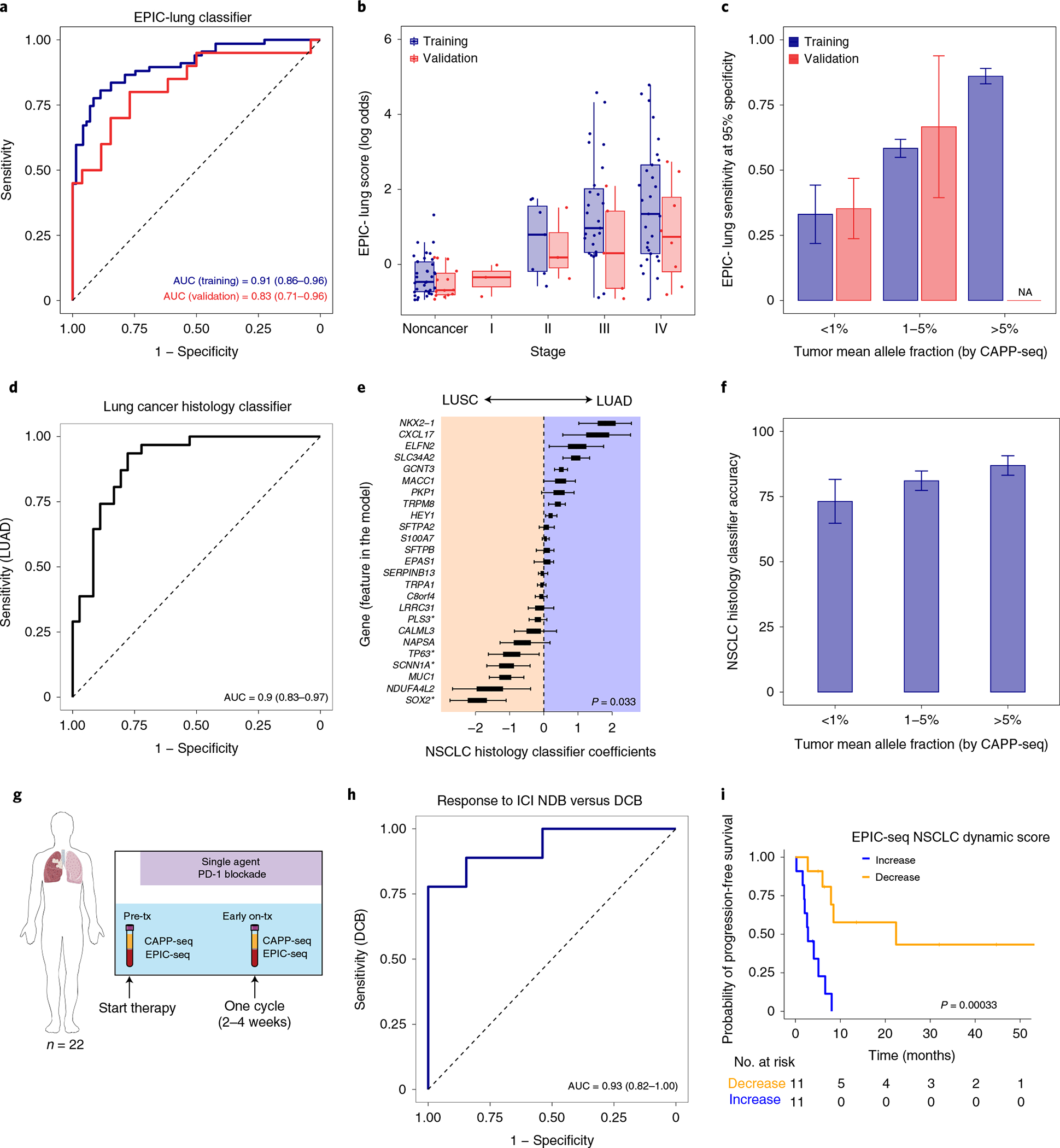
a, ROC capturing performance of the EPIC-lung classifier for distinguishing lung cancers from others in leave-one-batch-out analyses (AUC = 0.91). The 95% CI of the AUC is calculated using 2,000 bootstrap replicates. b, Relationship between EPIC-lung scores and NSCLC disease stage, measured by Jonckheere’s trend test (P = 0.08). Box-and-whisker plots depict the EPIC-lung classifier score in individual samples profiled by EPIC-seq (dots), with boxes spanning the IQR; the median is horizontally marked with a line in each box, and whiskers span the 1.5 IQRs in each disease stage group. Sample sizes are as follows: noncancer (n = 71 training; n = 23 validation), stage I (n = 0 training; n = 3 validation), stage II (n = 7 training; n = 4 validation), stage III (n = 30 training; n = 5 validation) and stage IV (n = 30 training, n = 8 validation). c, Sensitivity analysis of the EPIC-lung classifier at 95% specificity. Patients are grouped based on bins of mean circulating tumor AF (<1% (n = 8 training; n = 17 validation), 1–5% (n = 25 training; n = 3 validation) and >5% (n = 34 training)), estimated by CAPP-seq on the same samples. Sensitivity improves as ctDNA AF increases with roughly 33% of patients detectable when AF < 1%. The error bars for the training set depict the 95% CI of the sensitivity values resulted from 500 bootstrap replicates. The error bars for the validation set depict the sensitivity in the set ±s.e.m. taking sample size into account. d, ROC curve of the LUAD versus LUSC classifier when tested in a leave-one-out framework (AUC = 0.90, 95% CI (0.83–0.97)). e, Coefficients of the NSCLC histology classifier, with positive and negative coefficients favoring LUAD and LUSC, respectively. The coefficients are significantly associated with previous knowledge when comparing their magnitude and polarity by t-test (P = 0.033). Box-and-whisker plots are defined as in b and are resulted from 67 coefficient sets from classifiers trained in the leave-one-out cross-validation step. f, Accuracy of the histology classifier as a function of tumor ctDNA fraction as measured by CAPP-seq. The error bars are defined as in a. g, Application of EPIC-seq in predicting response to ICI within 4 weeks of treatment initiation. h, ROC curve of the EPIC-seq lung dynamics score calculated in g distinguishes patients with DCB versus those with NDB within 6 months (AUC = 0.93, 95% CI (0.78–1.00)). i, Prognostic value of EPIC-seq lung dynamics scores in Kaplan–Meier analysis of progression-free survival in the patients treated with immune-checkpoint inhibitors (log-rank P = 0.0003; hazard ratio 11.86). Patients are stratified by the median dynamics score.
Epigenetic signals in cfDNA captured by our EPIC-seq lung cancer classifier were significantly correlated with both total metabolic tumor volumes (MTV) and tumor-derived mutation signal measured by CAPP-seq42 (Extended Data Fig. 4a,b and Supplementary Notes). While most patients profiled had advanced NSCLC, our classifier showed a statistical trend for stage III–IV cases having higher scores compared to stage II cases (P = 0.08, Fig. 4b). We also assessed the importance of ctDNA concentration for the classifier’s performance. When binning cases by ctDNA concentrations determined using mutations (CAPP-seq), the EPIC-seq lung classifier achieved roughly 34% sensitivity at 95% specificity when allelic levels were below 1% and of roughly 86% sensitivity when ctDNA concentration exceeded 5% mean allele fractions (AF) (Fig. 4c). We observed similar sensitivity as a function of ctDNA fraction in the validation cohort (Fig. 4c). These results collectively demonstrate that RNA expression from lung tumors inferred by EPIC-seq can distinguish lung cancer cases from noncancer individuals and correlate with tumor burden.
Noninvasive classification of NSCLC subtypes.
Differentiating between the two most common histological subtypes of NSCLC43, LUAD and LUSC, is an important step in determining the optimal treatment for patients44,45. Currently, the morphologic and immunophenotypic criteria used for this classification are determined using tissue specimens43, but invasive evaluation can be fraught by diagnostic challenges and procedural risks46–48. Currently available mutation-based liquid biopsy methods are unable to reliably distinguish between LUAD and LUSC.
We therefore asked whether such classification could be performed noninvasively using EPIC-seq. In a cohort of 67 patients with NSCLC, a classifier for distinguishing histological subtypes (LUAD n = 36; LUSC n = 31) was trained on EPIC-seq data and demonstrated robust performance in cross-validation (AUC = 0.90, 95% CI: 0.83–0.97; Fig. 4d and Methods). The genes with largest coefficients and therefore strongest impact on the classification included canonical markers for LUAD (SLC34A2, NKX2-1 [TTF1]) and LUSC (SOX2), thus confirming biological plausibility of the classifier (Methods and Fig. 4e).
We evaluated the histology classifier’s accuracy as a function of ctDNA levels as determined by CAPP-seq (Methods) and as expected observed performance to be correlated with ctDNA concentration (Fig. 4f). Specifically, accuracy was highest at mean AFs above 5% (87%), with slight deterioration at levels between 1 and 5% (81%) and below 1% (73%) (Fig. 4f). These results demonstrate that inference of lung cancer expression differences by EPIC-seq allows for the noninvasive histological classification of NSCLC and that this framework appears robust across a range of ctDNA concentrations.
Predicting response to programmed death-ligand 1 (PD-(L)1) immune-checkpoint inhibition (ICI).
For patients with advanced NSCLC, therapeutic blockade of programmed death 1 and PD-(L)1 signaling using monoclonal antibodies has shown remarkable promise49,50. Trials combining PD-(L)1 blockade with cytotoxic therapy or with other ICI strategies have demonstrated improved response rates at the risk of higher toxicity51,52. Since only a minority of patients with NSCLC achieve durable benefit from ICI, there is a critical unmet need for reliable biomarkers that can accurately identify these patients before or early during ICI therapy53.
We therefore performed an exploratory analysis to test whether early, noninvasive assessment of response to ICI might be feasible using EPIC-seq. To do so, we analyzed 44 longitudinal blood specimens from 22 patients with NSCLC treated with PD-(L)1 blockade using EPIC-seq. Samples were collected immediately before PD-(L)1 therapy and within the first 4 weeks of therapy initiation (Fig. 4g). We developed a ‘lung dynamics index’ from EPIC-seq predicted gene expression as a function of therapeutic benefit (Methods). This index demonstrated a significant correlation to mutation-based response assessment using CAPP-seq54 (r = 0.526, P = 0.012, Extended Data Fig. 4c). This epigenetic metric was also able to distinguish patients achieving durable clinical benefit (DCB) (defined as no progression for at least 6 months after start of ICI) from those with no durable clinical benefit (NDB) achieving an AUC of 0.93, 95% CI: 0.78–1.00 (Fig. 4h). Moreover, when stratified by the median index score in Kaplan–Meier analysis, patients with higher scores had significantly better outcomes (log-rank P = 0.0003, Fig. 4i). Of note, within the limitations of this small cohort, we also observed a significant and continuous association of this index with progression-free survival (hazard ratio 11.38; Wald P = 0.006). Therefore, this proof-of-concept suggests that EPIC-seq can reliably detect tissue-specific signals in NSCLC and can faithfully monitor response to ICI in predicting durability of associated clinical benefit.
Noninvasive DLBCL quantitation using EPIC-seq.
DLBCL is the most common non-Hodgkin’s lymphoma and displays remarkable clinical and biological heterogeneity55. While aspects of this heterogeneity can be captured by clinical risk indices such as the International Prognostic Index (IPI)56, expression profiling57 or genotyping of primary tumor biopsies58,59, it remains unclear whether such stratification might also be feasible using less invasive approaches.
We therefore analyzed pretreatment blood samples from patients with DLBCL using EPIC-seq and tested whether epigenetic signals in cfDNA allow noninvasive detection of DLBCL cases, distinguishing patients with cancer from healthy control participants. Here again, a classifier trained on EPIC-seq data to distinguish patients with DLBCL (n = 91) from noncancer control participants (n = 71) revealed robust performance (AUC = 0.92, 95% CI 0.88–0.97; Fig. 5a and Methods). When we applied this classifier to a validation cohort of patients with DLBCL (n = 23) and noncancer control participants (n = 23), we observed similar performance in distinguishing cancer from noncancer (AUC = 0.96, 95% CI 0.9–1.00; Fig. 5a). We also observed a significant graded relationship between scores from this epigenetic classifier and the revised IPI (R-IPI) (P = 0.004, Fig. 5b). Separately, we observed the expected trend for scores from the epigenetic classifier with MTV and mean AFs of mutations measured by CAPP-seq59,60,61 (Extended Data Fig. 5a–c and Supplementary Notes). We also evaluated classifier performance at various ctDNA levels by calculating sensitivity at 95% specificity. While EPIC-seq’s sensitivity was strongly related to mean AF and showed most robust performance at ctDNA levels above 1%, we observed roughly 40% detection of DLBCL cases where mean AF was <1% (Fig. 5c).
Fig. 5 |. Application of EPIC-seq for DLBCL detection.
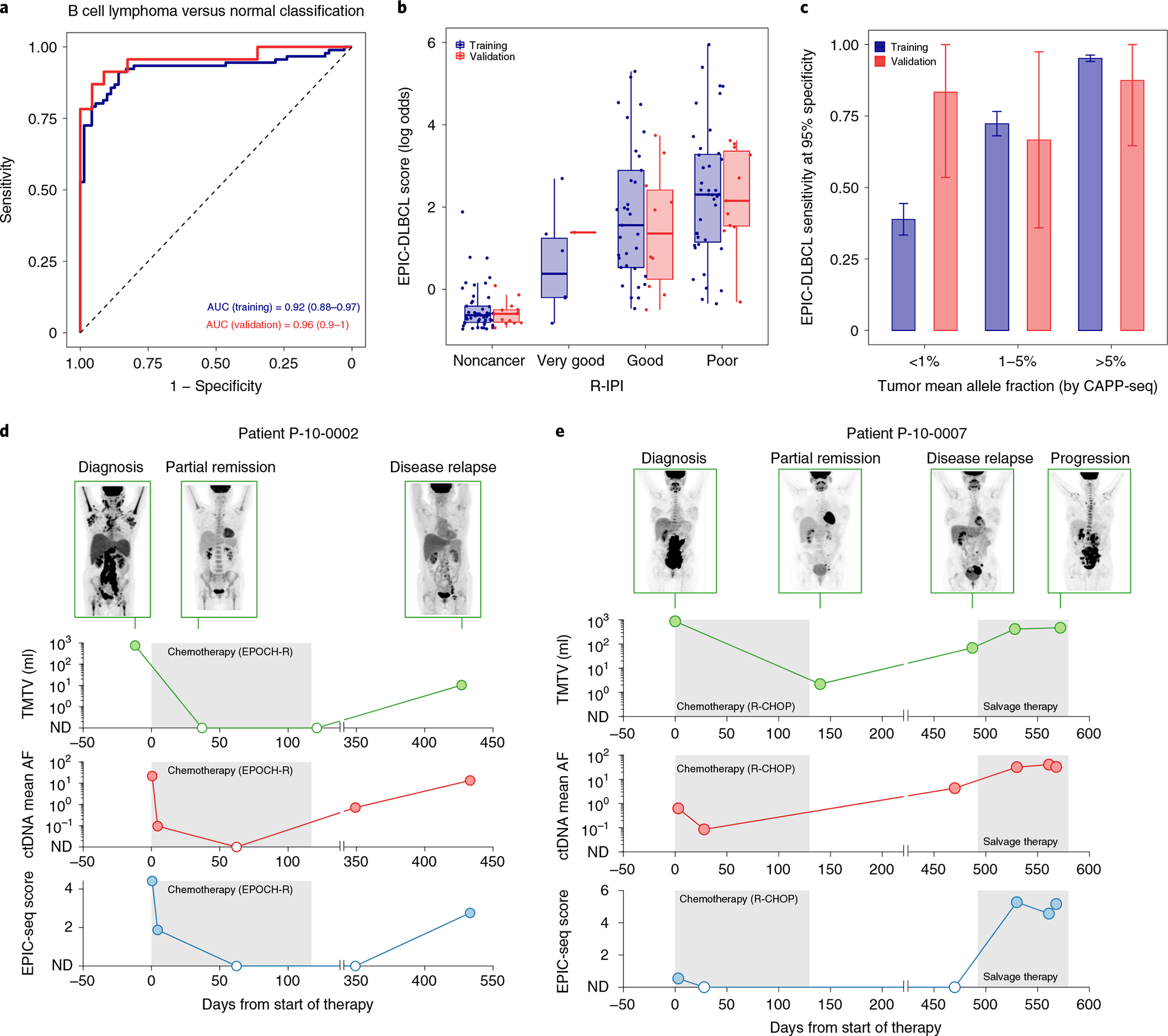
a, ROC analyses capture performance of the EPIC-DLBCL classifier for distinguishing lymphomas from others. Red and blue curves depict performance in the validation cohort (AUC = 0.96), versus leave-one-batch-out cross-validation analyses of the training cohort (AUC = 0.92), respectively. b, Relationship between EPIC-seq DLBCL classifier scores and clinical prognostic scores as measured by the R-IPI (Jonckheere’s trend test P = 4 × 10−4). Box-and-whisker plots depict the EPIC-DLBCL score in individual samples profiled by EPIC-seq (dots), with boxes spanning the IQR; the median is horizontally marked with a line in each box and whiskers span the 1.5 IQRs. Sample sizes are as follows: noncancer (n = 71 training; n = 23 validation); ‘very good’ (n = 7 training; n = 1 validation); ‘good’ (n = 38 training; n = 11 validation) and ‘poor’ (n = 46 training; n = 11 validation). c, Sensitivity analysis at 95% specificity for EPIC-DLBCL classifier. Similar to the EPIC-lung cancer classifier, sensitivity significantly improves as a function of ctDNA level (<1% (n = 16 training; n = 6 validation), 1–5% (n = 34 training; n = 9 validation) and >5% (n = 41 training; n = 8 validation). The error bars in the training set depict the 95% CI of the sensitivity values resulted from 500 bootstrap replicates. The error bars for the validation set depict the sensitivity in the set ±s.e.m. taking the sample size into account. d,e, Change of ctDNA disease burden in response to treatment and during clinical progression in two patients with DLBCL with GCB (d) and ABC (e) COO. Shown is the radiographic response as measured by PET/CT MTV (first row y axis), ctDNA mean AF measured by CAPP-seq (second row y axis) and the EPIC-seq lymphoma score (third row y axis) over serial, pre- and post-therapy time points (x axis).
To assess the relationship between epigenetic signals and somatic mutations during DLBCL therapy and their stability over time, we next profiled serial blood samples from two patients shortly after induction therapy with curative intent using both EPIC-seq and CAPP-seq (n = 12, Fig. 5d,e). Again, we observed strong and significant correlations between DLBCL EPIC-seq scores and ctDNA concentrations over time in both patients (ρ = 0.79, P = 0.004, Extended Data Fig. 5d), despite the administration of combined chemoimmunotherapy and the substantial attendant changes in leukocyte blood counts. Collectively, these results illustrate that expression inferences by EPIC-seq can noninvasively detect tissue-derived DLBCL signals and faithfully reflect disease burden before and after DLBCL therapy.
DLBCL cell-of-origin (COO) classification.
Most DLBCL tumors can be classified into two transcriptionally distinct molecular subtypes, each derived from a specific B cell differentiation state (COO): GCB and ABC57,62,63. These subtypes are prognostic with significantly better outcomes observed in patients with GCB tumors, and may also predict sensitivity to emerging targeted therapies64–70. While this classification of DLBCL is among the strongest prognostic factors and a potential biomarker for future personalized therapies, accurate subtyping remains challenging in clinical settings71.
We therefore used EPIC-seq profiling to develop a noninvasive probabilistic COO classifier from pretreatment plasma (Methods). When we benchmarked this classifier’s performance in our DLBCL cohort, we observed epigenetic scores to be significantly correlated with previously described mutation-based GCB scores60 (n = 90 with genetic scores available; ρ = 0.75, P = 1 × 10−5, Fig. 6a). When we examined this epigenetic COO classifier in the validation set, we observed a significant correlation between EPIC-seq scores and the mutation-based GCB scores (ρ = 0.64, P = 0.01, Extended Data Fig. 5e). When comparing patients classified by the more commonly clinically used immunohistochemical Hans algorithm72, we observed a significantly higher COO score for GCB cases compared with non-GCB (training n = 66, Wilcoxon P = 0.001, Fig. 6b; validation n = 18, P = 0.014, Extended Data Fig. 5f).
Fig. 6 |. Application of EPIC-seq for DLBCL Coo classification.
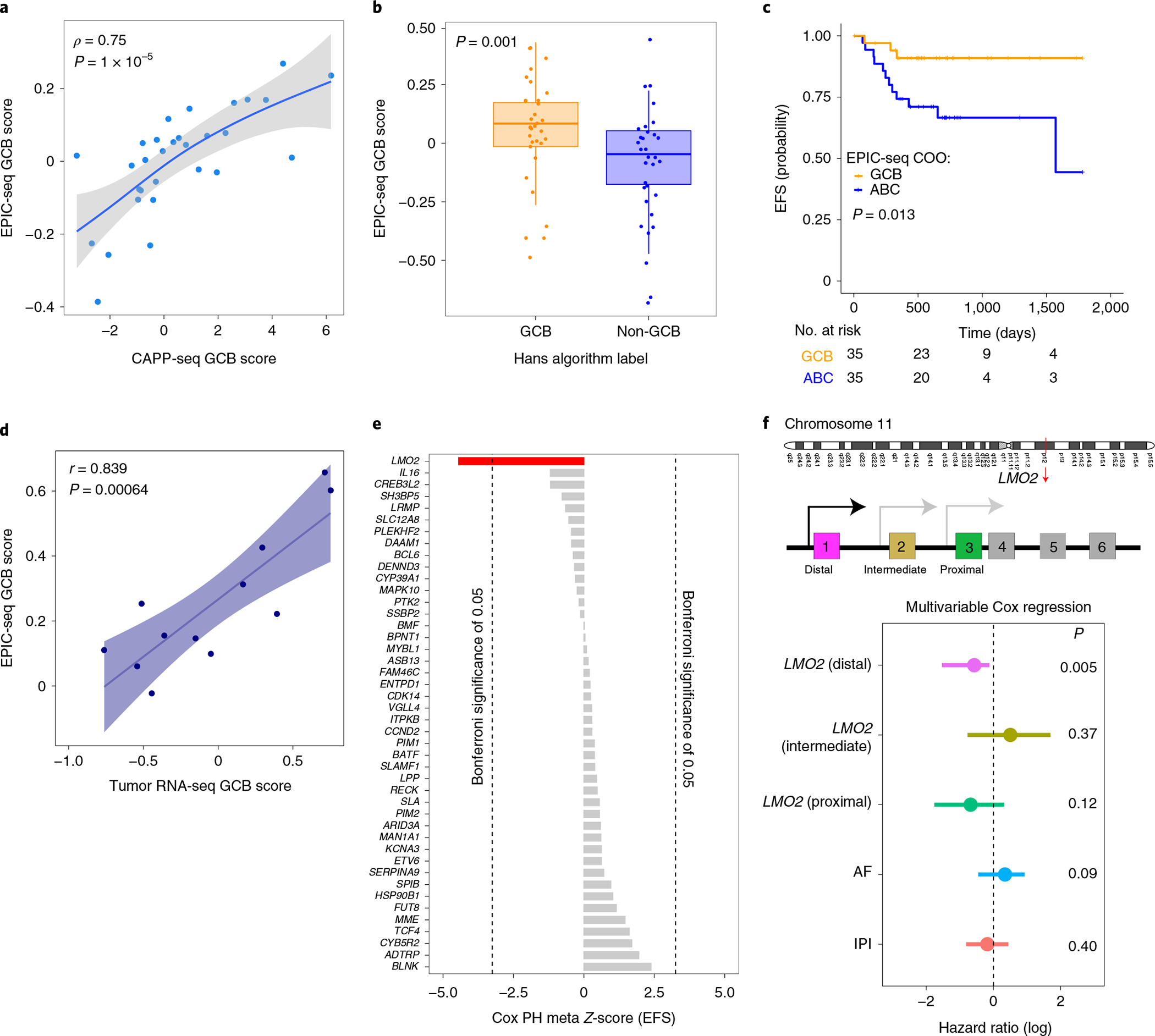
a, Relationship between DLBCL COO EPIC-seq GCB scores and mutation-based GCB scores as measured by CAPP-seq (Spearman ρ = 0.75, P = 1 × 10−5). Data were smoothed by three patient (nonoverlapping) bins after sorting by CAPP-seq scores before correlation analysis, and therefore there are 30 dots in the scatterplot. The grey region depicts the 95% CI around the smoothed line shown in blue. b, Relationship between EPIC-seq GCB scores from cfDNA and tumor tissue clinical classification by Hans immunohistochemical algorithm (GCB n = 33, non-GCB n = 33, Wilcoxon P = 0.001). Box-and-whisker plots depict the EPIC-seq GCB score in individual samples profiled by EPIC-seq (dots), with boxes spanning the IQR; the median is horizontally marked with a line in each box and whiskers span the 1.5 IQRs. c, Prognostic value of EPIC-seq COO scores in Kaplan–Meier analysis of EFS in DLBCL (log-rank P = 0.013). Patients are stratified by the median EPIC-COO score, with higher scores associated with GCB and lower levels with ABC subtype. d, Concordance analysis between EPIC-seq COO score and RNA-based scores (from matched tumor biopsy) for a cohort of 12 patients with DLBCL. Each dot represents one patient, with the x axis showing the GCB score from RNA-seq and y axis showing the EPIC-seq GCB score. The two scores exhibit reasonably strong correlation (r = 0.84, P = 0.0006). e, Prognostic value of individual genes profiled by EPIC-seq and EFS, as measured by Z-scores from univariate Cox proportional hazard models. For genes with multiple TSS regions, Z-scores were combined using Stouffer’s method102. After correcting for multiple hypothesis testing, only LMO2 (red) remains significantly associated with favorable DLBCL outcome. Dotted lines represent the significance threshold for Bonferroni-corrected P values of 0.05. f, Forest plot depicts multivariable Cox proportional hazard model results for EFS. After adjusting for IPI and ctDNA AF, only the distal TSS for LMO2 remains significantly prognostic for EFS (P = 0.005).
Comparing the expected prognostic power of epigenetic and mutation-based COO scores using univariate Cox regressions, we observed a stronger association between EPIC-seq GCB scores and favorable outcomes (n = 70, EPIC-seq: hazard ratio 0.13, P = 0.033 versus CAPP-seq: hazard ratio 0.95, P = 0.62). Indeed, when stratified by the median GCB score, patients with higher GCB scores had significantly better outcomes (log-rank P = 0.013, Fig. 6c). Among patients analyzed by both immunohistochemistry and DNA genotyping, the Hans algorithm failed to stratify patient clinical outcomes, suggesting more accurate classification by our approach (Extended Data Fig. 5h). To further characterize the fidelity of our plasma cfDNA classification results, we next profiled tumor biopsies of a subset of our DLBCL validation cases (n = 12) by RNA-seq. When assessing the concordance between EPIC-seq scores obtained from plasma cfDNA and COO scores from tumor tissues, we found a significantly high correlation between these two orthogonal approaches (r = 0.84, Fig. 6d and Extended Data Fig. 5g). Moreover, among patients analyzed by both immunohistochemistry and DNA genotyping, the Hans algorithm failed to stratify patient clinical outcomes, suggesting more accurate classification by our approach (Extended Data Fig. 5h). Overall, these results indicate that EPIC-seq has use for noninvasive classification of DLBCL COO and can stratify patients better than both the genetic classifier and the Hans algorithm.
Determining prognostic power of individual genes with EPIC-seq.
Expression profiling studies for a variety of tumor types have identified the prognostic power of individual genes for both risk stratification and therapeutic management. In DLBCL, previous studies have validated the prognostic use of several key genes in relatively large patient populations that were homogenously treated with modern combination immune-chemotherapy using R-CHOP (a combination of the drugs rituximab, cyclophosphamide, hydroxydaunorubicin hydrochloride (doxorubicin hydrochloride), vincristine (Oncovin) and prednisone)73–78. These studies have relied on expression profiling from tumor biopsy specimens, which can be hampered by limitations of RNA sample quality and quantity. Therefore, we evaluated the use of EPIC-seq for noninvasively measuring expression of genes with prognostic associations in DLBCL and observed significant concordance between our study using plasma cfDNA with previous studies using tumor RNA77,78 (Extended Data Fig. 5i and Supplementary Notes).
Within our cohort, only LMO2 emerged as significantly associated with progression-free survival after correction for multiple hypothesis testing (nominal P = 7.5 × 10−6, corrected P = 0.0055; Fig. 6e). This is consistent with previous data on its robust prognostic effect in DLBCL79. LMO2 is an oncogene consisting of six exons, of which three nearest the 3′ end are protein coding80. Inclusion of the three noncoding 5′ LMO2 exons is governed by alternative proximal81, intermediate82 and distal promoters83. When comparing predicted expression from each of these alternative promoters for prognostic strength in DLBCL using EPIC-seq, only the distal TSS (GRCh37/hg19-chr11:33,913,836) showed a significant association with outcome (Fig. 6f). Higher predicted expression from the distal LMO2 TSS remained prognostic of more favorable outcomes in multivariable Cox regression after adjusting for IPI and ctDNA level (Fig. 6f). This is consistent with the known importance of the distal promoter in driving LMO2 expression in tumors, as evidenced by retroviral insertional mutagenic events observed in gene therapy trials and chromosomal rearrangements mediating lymphomagenesis80. Collectively, these observations indicate that EPIC-seq has use for noninvasively measuring the expression and prognostic value of individual genes and for resolving individual TSS regions.
Discussion
In this study, we introduce EPIC-seq, a method using cfDNA fragmentation patterns to allow noninvasive inference of gene expression and which can be used for a wide variety of clinically relevant applications including tumor detection, subtype classification, response assessment and analysis of genes with prognostic implications. Compared to EPIC-seq, the sensitivity of previously described cfDNA fragmentomic techniques and features has been insufficient to resolve expression of individual genes with high fidelity19,20,24,26,81. The approach described here achieves substantially improved performance by using PFE as a metric, as well as higher sequencing depth achieved through targeted capture of promoter regions of genes of interest.
To allow inference of RNA expression levels from cfDNA fragmentomic features by EPIC-seq, we focused on capturing features at transcription sites that reflect epigenetically encoded signals from nucleosomal accessibility and positioning since these are key factors for determining transcriptional output84–101. These fragmentomic signals appeared strongest at promoters of actively expressed genes when profiling cfDNA by WGS, motivating our TSS capture approach. We also observed substantial signal at exonic regions of actively expressed genes in WES, suggesting opportunities to extend EPIC-seq more broadly to study expression of genes of interest.
As demonstrated above, EPIC-seq has potential use for a wide variety of clinically relevant cancer classification problems. While our study focused on tumor histological classification as a proof-of-concept, the approach described here will be likely be broadly generalizable to other tumor types. We demonstrate the biological plausibility of inferred expression levels from EPIC-seq using multiple independent lines of evidence. Specifically, we describe prominent correlations of EPIC-seq signals not only with expectations from tissue transcriptome profiles, but also with disease burden as measured by MTV and mutation-based ctDNA analysis. Furthermore, we observed strong correlation of EPIC-seq signals with anticancer therapeutic responses, as well as its ability to identify prognostically informative genes.
In our initial application of EPIC-seq, we focused on noninvasive histological classification of lung cancers and the molecular classification of aggressive B cell lymphomas, two common cancer types where such classification is clinically routine but at times fraught by diagnostic challenges. We did not observe a significant impact of several preanalytical factors on cfDNA fragmentation entropy measurements (Extended Data Fig. 6 and Supplementary Notes). Finally, we developed a mechanistic framework for how cfDNA fragmentation mirrors activity level of expressed genes in human tissues (Extended Data Fig. 7a–c). Using this model framework, we used simulations to explore the parameters influencing the likelihood of detection of expression of a given gene of interest within cfDNA as a function of tumor burden(Extended Data Fig. 7d). The robust performance we observed for accurate classification of each of these tumor subtypes is promising and suggests opportunities for extending this approach more broadly to other cancer types and other pathologies. For example, despite the many diagnostic tools already available in the USA, CUP cases continue to represent some 2–5% of incident cancers. EPIC-seq provides a promising avenue for the potential reclassification of such carcinomas using noninvasive methods. Separately, the methods we describe could have applications beyond cancer for the noninvasive detection of signals from cell types, tissues and pathways and pathologies of interest. These include noninvasive strategies to detect tissue injury and ischemia, as well as pharmacodynamic effects on specific therapeutically targeted pathways and toxicity profiles for diverse human tissues that are otherwise difficult to monitor noninvasively (for example, the brain and gastrointestinal tract), before symptomatic tissue damage occurs.
Despite the value of EPIC-seq for the applications shown here, further refinements are warranted to maximize potential use of such fragmentomic techniques. Considerable molecular heterogeneity exists within patients with localized tumors, with some small tumors shedding more cfDNA than others. Accordingly, low ctDNA levels could hamper immediate use for early cancer detection. As more genes and more fragmentomic features are studied using this approach, stronger signals could be gleaned to enable noninvasive detection of small tumor deposits, to interrogate the activity of specific biological pathways and to study other pathologies in target tissues of interest. Nevertheless, the method and applications described here hold imminent promise in personalized profiling efforts, enabling noninvasive, high-throughput tissue-of-origin characterization with diagnostic, prognostic and therapeutic potential.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-022-01222-4.
Methods
Human participants and cohorts.
Study overview.
All samples analyzed in this study were collected with informed consent from participants enrolled on Institutional Review Board-approved protocols complying with ethical regulations at their respective centers, Stanford University, MD Anderson Cancer Center, Memorial Sloan Kettering Cancer Center (MSKCC), Vanderbilt University, Mayo Clinic, the National Cancer Institute, CHU Dijon and Essen University Hospital, as detailed below. Fragmentomic features ultimately used for EPIC-seq were established and initially tested by profiling cfDNA through WGS and WES, as tabulated in Supplementary Table 1. These WGS and WES cfDNA profiling data derived from 150 participants that were either generated for this study (n = 64) or came from publicly available datasets (n = 86).
For initial model development and cfDNA fragmentomic feature selection, we profiled cfDNA from a patient with CUP by deep WGS to learn the relationships between cfDNA fragmentomic features and expression levels at whole-genome scale. After our initial cfDNA profiling of this patient by deep WGS with CUP to build expression predictors from cfDNA, we also profiled two healthy adult participants by WGS profiling of cfDNA (roughly 200×) and assessed robustness (Supplementary Table 1). For initial validation analyses using WGS cfDNA fragmentomics, we also reanalyzed samples from 40 healthy control participants and 46 patients with lung cancer previously described15.
We then extended our observations from WGS to WES of cfDNA, by deeply profiling 28 plasma specimens from healthy control participants, and 11 plasma specimens from six patients with extensive stage SCLC (deep WES). After identification and initial validation by WGS/WES of the key cfDNA fragmentomic signals informative for predicting gene expression in the participants described above, EPIC-seq was then applied to 329 blood samples from 201 patients with cancer and 87 healthy adults, as detailed below and as depicted in Supplementary Fig. 3. To select genes for the EPIC-seq capture panel focused on subclassification of lung cancers and lymphomas, we analyzed publicly available gene expression datasets for 1,156 lung cancers from The Cancer Genome Atlas and for 381 lymphomas from Schmitz et al., as described below60.
Healthy participants and noncancer control participants.
To identify and validate cfDNA fragmentomic features informing gene expression prediction, WGS was performed in 30 healthy participants. These participants were profiled at varying prespecified coverage depths (roughly 300×, n = 3; roughly 1–5×, n = 24; roughly 18–25×, n = 3), thereby allowing construction of meta-profiles for expression inferences, as described below (‘A gene expression model for predicting RNA output from TSS cfDNA fragmentomic features’). We separately profiled 94 peripheral blood samples from 87 participants without cancer using EPIC-seq. Among these participants, 35 (40%) qualified for lung cancer screening using low-dose CT (LDCT) due to a history of heavy smoking (≥30 pack years) and age (55–80 years).
EPIC-seq cancer cohorts.
Lung cancer cohort.
EPIC-seq was applied to 109 blood samples from 87 patients diagnosed with NSCLC (some with serial samples). Among these patients, 37 (43%) had a histological diagnosis of LUSC, while 50 (57%) patients had LUAD histology. Samples were collected at Stanford University, The University of Texas MD Anderson Cancer Center or MSKCC, with patient characteristics outlined in Supplementary Fig. 3b. A subset of patients with advanced NSCLC (n = 22) was treated with PD-(L)1 blockade-based ICI and had serial pre- and on-treatment samples available. These patients had stage IV disease and were treated with PD-(L)1 blockade-based ICI.
DLBCL cohort.
EPIC-seq was also applied to 126 samples from 114 patients diagnosed with large B cell lymphoma. Samples were collected at Stanford Cancer Center, CA, USA; MD Anderson Cancer Center, TX, USA; Dijon, France and Novara, Italy, and within the Phase III multicenter PETAL trial61, with baseline characteristics tabulated in Supplementary Fig. 3b.
Patient with CUP.
To assess with high resolution the relationship between fragmentomic features and gene expression, we compared deep WGS data and RNA-seq data from a patient with extremely low tumor burden. Tumor fraction was estimated using a tumor-informed plasma variant detection strategy. First, the patient’s tumor and germline DNA were prepared for exome capture using the Illumina Nextera Rapid Capture Exome Kit and sequenced on an Illumina NextSeq 500 machine using paired-end sequencing and 75-bp read lengths. Single nucleotide variant (SNV) calling was performed using Mutect and annotated by Annovar. A personalized targeted sequencing panel was generated using 120-bp IDT oligos overlapping SNVs detected in the tumor and applied to the tumor and germline sample. The variant set selected for monitoring consisted of 36 SNVs that both passed tumor/germline quality control filters and were present in at least 10% allele frequency in the tumor. The patient’s plasma sample was sequenced on an Illumina NovaSeq machine, achieving a deduplicated depth of 4,000×. The time point used in this study had a monitoring mean allele frequency of 0.056%, which is significantly lower than the lower limit of detection of disease at 250× coverage. Results from deep WGS cfDNA profiling of this patient with CUP were then reproduced by the independent WGS profiling of cfDNA (roughly 200×), and RNA-seq profiling of matched PBMCs from two healthy adult participants.
Clinical variables.
Histopathology.
Histological subtypes of each tumor type (SCLC, NSCLC, DLBCL) profiled in this study were established according to clinical guidelines using microscopy and immunohistochemistry and served as ground truths for assessing classification performance by trained pathologists. COO subtypes of DLBCL were assessed based on the Hans classifier per World Health Organization guidelines55. For NSCLC and DLBCL subtypes profiled in previous studies by RNA-seq, we relied on subtype labels from the Cancer Genome Atlas (TCGA) (for LUAD versus LUSC subtypes of NSCLC) or from Schmitz et al.101 (for GCB versus ABC subtypes of DLBCL).
MTV measurement.
Pretreatment tumor MTV was measured from 18FDG PET/CT scans, using semiautomated software tools: for NSCLC, it was done as previously described103 via MIM software by using PETedge. For DLBCL, three different software tools were used (Beth Israel Fiji, PETRA ACCURATE tool and Metavol) as previously described104. Regional volumes were automatically identified by the software and confirmed by visual assessment of the expert to confirm inclusion of only pathological lesions.
Clinical outcomes.
Event-free survival (EFS) and overall survival were calculated from time of treatment initiation. Overall survival events were death from any cause; EFS events were progression or relapse, unplanned retreatment of lymphoma and death resulting from any cause. Patients with NSCLC receiving PD(L)1 directed therapy were labeled as NDB or DCB for ‘experiencing progression or death’ and DCB within 6 months, respectively.
Specimen collection and molecular profiling.
Plasma collection and processing.
Peripheral blood samples were collected in K2EDTA or Streck Cell-Free DNA blood collection tubes and processed according to local standards to isolate plasma before freezing. Following centrifugation, plasma was stored at −80 °C until cfDNA isolation. The cfDNA was extracted from 2 to 16 ml of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen) according to the manufacturer’s instructions. After isolation, cfDNA was quantified using the Qubit double-stranded DNA High Sensitivity Kit (Thermo Fisher Scientific) and High Sensitivity NGS Fragment Analyzer (Agilent).
cfDNA-seq library preparation.
A median of 32 ng was input into library preparation. DNA input was scaled to control for high molecular-weight DNA contamination. End repair, A-tailing and custom adapter ligation containing molecular barcodes were performed following the KAPA Hyper Prep Kit manufacturer’s instructions with ligation performed overnight at 4 °C as previously described9,61. Shotgun cfDNA libraries were either subjected to WGS and/or subjected to hybrid capture of regions of interest as described below.
Hybrid capture and sequencing.
Exome capture.
For WES, shotgun genomic DNA libraries were captured with the xGen Exome Research Panel v.2 (IDT) per the manufacturer’s instructions with minor modifications. Hybridization was performed with 500 ng of each library in a single-plex capture for 16 h at 65 °C. After streptavidin bead washes and PCR amplification, postcapture PCR fragments were purified using the QIAquick PCR Purification Kit per the manufacturer’s instructions. Eluates were then further purified using a 1.5× AMPure XP bead cleanup.
Custom capture panels.
We used CAPP-seq to establish ctDNA levels, by genotyping of somatic variants including single nucleotide mutations42. We used entity-specific CAPP-seq capture panels for DLBCL or NSCLC (SeqCap EZ Choice, Roche NimbleGen)9,61 or personalized CAPP-seq selectors for CUP (IDT), as previously described9. Similarly, for EPIC-seq, we used the SeqCap EZ Choice platform (Roche NimbleGen) to target TSS regions of genes of interest, as described below. Enrichment for WES, CAPP-seq and EPIC-seq was done according to the manufacturers’ protocols. Hybridization captures were then pooled, and multiplexed samples were sequenced on Illumina HiSeq4000 instruments as 2 × 150 bp reads.
RNA-seq.
RNA-seq of PBMCs.
The Illumina TruSeq RNA Exome kit was used for RNA-seq library preparation starting from 20 ng of input RNA, per manufacturer’s instructions. When using peripheral blood as a source of leukocyte RNA, we used either plasma-depleted whole blood with globin depletion or enriched PBMCs without globin depletion. In brief, total RNA was fragmented and stranded complementary DNA libraries were created per the manufacturer’s protocol. The RNA libraries were then enriched for the coding transcriptome by exon capture using biotinylated oligonucleotide baits. Hybridization captures were then pooled, and samples were sequenced on an Illumina HiSeq4000 as 2 × 150 bp lanes of 16–20 multiplexed samples per lane, yielding roughly 20 million paired-end reads per case. After demultiplexing, the data were aligned and expression levels summarized using Salmon to transcript models annotated by GENCODE version 27105. We separately studied tumor RNA-seq data to identify DEGs of interest for EPIC-seq panel design, as described in detail below.
RNA-seq of lymphoma specimens.
Tumor-derived RNA was isolated from 2–4, 10-μm thick, formalin-fixed paraffin embedded (FFPE) scrolls of tumor tissue using the RNA Storm/DNA Storm Combination Kit (Cell Data Sciences), according to the manufacturer’s protocol. An off-column DNA digestion step was performed using Qiagen’s RNase-Free DNase Set followed by column purification using Zymo’s RNA Clean&Concentrator kit. RNA concentration was quantified using NanoDrop. The SMARTer Stranded Total RNA-seq Kit v2 (TaKaRa) was used for RNA-seq library preparation using 50 ng input RNA, according to the manufacturer’s protocol. Fragmentation steps were omitted as recommended for RNA isolated from FFPE specimens. Yield and fragment size of libraries were assessed using Qubit (dsDNA HS assay kit) and TapeStation. Libraries were sequenced on an Illumina HiSeq4000 or NovaSeq6000, respectively, with 2 × 150-bp paired-end reads.
Data analysis methods.
Mapping, deduplication and quality control of TSS sites and samples.
FASTQ files were demultiplexed using a custom pipeline wherein read pairs were considered only if both 8-bp sample barcodes and 6-bp unique identifiers matched expected the sequences after error correction9. After demultiplexing, barcodes were removed and adaptor read-through was trimmed from the 3′ end of the reads using fastp104 to preserve short fragments. Fragments were aligned to human genome (hg19) using BWA; we disabled the automated distribution inference in BWA ALN to allow inclusion of shorter and longer cfDNA fragments that would otherwise be anomalously flagged as improperly paired. We removed PCR duplicates using a customized barcoding approach, which combines endogenous and exogenous unique molecular identifiers, including cfDNA fragment start and end positions, as well as prespecified unique molecular identifiers within ligated adapters into account. To allow coverage uniformity for comparisons, we down-sampled data to a 2,000× depth using ‘samtools view -s’. Since in silico simulations showed >500× sequencing depth to be required for achieving reasonable correlations between entropy and expression, we considered any samples not meeting this depth threshold (median depth) as failing quality control. Any samples whose cfDNA fragment length density mode was below 140 or above 185 were also removed, since the expected fragment length density mode is 167 (corresponding to the chromatosomal DNA length). Together, these two criteria removed 21 samples as not meeting quality control. To identify and censor noisy sites among the 236 TSS regions profiled by our EPIC-seq panel, we profiled 23 control participants (Supplementary Table 3), allowing us to identify and remove stereotyped regions with reproducibly low TSS coverage (that is, any site with counts per million of less than one third of uniformly distributed coverage across the TSSs in the selector, that is, in more than 75% of control participants). This removed two TSS sites in FOXO1 and SFTA2 as not meeting quality control.
To guarantee adequate quality of fragments entering analysis, we required mapping quality (MAPQ, k) of >30 or >10 in the WGS and EPIC-seq data, respectively (using ‘samtools view -q k -F3084’). The more lenient EPIC-seq MAPQ threshold was qualified by more stringent mappability and uniqueness requirements already imposed on the TSS regions selected during EPIC-seq selector design. We also limited the analysis to reads with the following BAM FLAG set: 81, 93, 97, 99, 145, 147, 161 and 163. To ensure removal of nonunique fragments, reads with duplicate names were censored.
Fragmentomic feature extraction and summary.
We considered five cfDNA fragmentomic features at TSS regions and then compared each of these features to gene expression, including WPS25, OCF20, MDS35, NDR score27 and PFE (introduced here). MDS, NDR, OCF and WPS were each computed as per the conventions of the originally describing studies with minor modifications, as detailed below.
MDS.
We performed end-motif sequence analysis of individual cfDNA fragments to assess the distribution of nucleotides among the first few positions for the reads of each read pair, as previously described35. This was performed by computationally extracting the first four 5′ nucleotides of the genomic reference sequence for each sequence read, resulting in a 4-mer sequence motif. MDS was then computed as the Shannon index of the distribution across 256 motifs (4-mers) at each TSS site, when considering fragments overlapping the 2-kb window flanking each TSS. Of note, the first four 3′ nucleotides were not used as these may be altered by end repair during library preparation and may not reflect the native genomic sequence.
NDR.
To guard against variations in depth across the genome, including from GC-content variation or somatic copy number changes, depth was normalized within each 2-kilobase (kb) window flanking each TSS (−1,000 to +1,000 bp) in counts per million space. We denote this normalized measure as NDR score for each TSS.
PFE.
Shannon entropy was used to summarize the diversity in cfDNA fragment size values in the vicinity of each TSS site (−1 kb (5′; upstream) to +1 kb (3′, downstream)). We defined 201 size bins (from b1 = 100 bp to b201 = 300 bp) and estimated the density by the maximum likelihood, that is, with where ni and n denote the number of fragments with length bi and total number of fragments at the TSS, respectively. Shannon’s entropy was calculated as .
To account for variations in sequencing depth between samples as well as other hidden factors affecting overall cfDNA fragment length distributions as potential confounders, we performed normalization steps using a Bayesian approach through a Dirichlet-multinomial model.
For a given sample, we first built a sample-wide fragment length distribution using the multinomial maximum likelihood estimation. To minimize the impact of gene expression on this background fragment length distribution, we focused on the two 250-bp regions within the 2-kb window with the longest distance from the center of the TSS: (1) −1 kbp to −750 bp (upstream) and (2) from +750 bp to +1 kbp (downstream). Fragment length densities across the 201 size bins were then used as parameter vector α of a Dirichlet distribution with α0 = 20. For each TSS, we then updated the sample-wide background distribution to calculate the sample adjusted and gene-specific posterior of the Dirichlet distribution based on fragment counts in the 201 size bins within the 2-kb region around the TSS:
From Dir(α*), we sampled 2,000 fragment length distributions and calculated the corresponding Shannon’s entropies. Each value was then compared to the Shannon entropies of five randomly selected background gene sets, denoted as e1, e2, e3, e4 and e5. PFE was defined as the likelihood of the gene-specific entropy (uncertainty class) exceeding the control gene entropies by (1 + k) fold for all n = 5 groups with a random variable k. Here, we used a Gamma distribution for k ~ Γ(s = 0.5, r = 1), where Γ is the Gamma distribution with shape s and rate r. PFE therefore is a measure for the excess diversity in the fragment length distribution at a given TSS of interest compared to control genes, and is formally defined as
where Ek [.] denotes the expected value with respect to the excess parameter k, and P* is the probability with respect to the Dirichlet distribution Dir(α*) approximated by the 2,000 draws.
Preanalytical factors.
We examined robustness of PFE against preanalytical biases such as blood collection tubes, processing time and number of PCR cycles. To confirm that the type of collection tube does not confound the PFE, we collected blood from three healthy donors in K2EDTA and Streck blood collection tubes and compared PFE in the TSSs in the EPIC-seq selector, and measured concordance between the two using Pearson correlation. To evaluate the robustness against processing time, we used a cohort of patients with DLBCL captured by a CAPP-seq lymphoma panel106 and calculated PFE for the regions in the panel and summarized each patient by the median PFE across these regions. We compared samples grouped by the number of interval days before processing, and measured the correlation between median PFE and time at room temperature. We also tested effect of number of PCR cycles on PFE, by performing additional PCR cycles on cfDNA libraries from four healthy donors. Here, we compared the PFEs of regions in our NSCLC panel and measured Pearson correlation in PFEs with or without the additional PCR cycles.
cfDNA fragmentomic analysis by WES profiling.
Whole-exome PFE analysis.
For whole-exome analyses (Figs. 1g,h and 2d–h and Supplementary Fig. 2g–i), we used the raw Shannon entropy (as described in ‘PFE’ under section ‘Fragmentomic feature extraction and summary’) at any given gene, after transforming it into a z-score, using a cohort of 39 cfDNA WES profiles (each with 200–400× depth), including 28 plasma samples from healthy adult control participants and 11 plasma samples from patients with SCLC. To account for differences in depth in the cohort for normalization, we considered meta-profiles of five samples to achieve comparable depths as those initially used to relate PFE and gene expression levels when relying on WGS (roughly 2,000×).
SCLC gene signatures.
A SCLC-specific gene signature was generated using a previously described RNA-seq dataset of 81 surgically resected primary tumors107. To identify genes highly expressed in SCLC tumors but not circulating leukocytes (that is, ‘SCLC High Genes’, n = 118), we selected genes with mean transcripts per million (TPM) > 50 in these SCLC tumors and mean TPM < 0.5 in PBMCs from GSE107011 (n = 13). We limited our analyses to protein-coding genes, and renormalized expression levels to 1 × 106 after removal of individual genes with TPM > 100,000 (n = 16,865 genes). Conversely, we selected ‘SCLC Low Genes’ (n = 20) with TPM < 0.5 in SCLC tumors and >50 in PBMC. Using the deep whole-exome cfDNA profiles described above, we then calculated the mean Shannon entropy of first exons (that is, as an estimate for the residual PFE captured by exon 1 fragments) for each of the two SCLC signature sets, after subtracting the mean PFE of a set of control genes used throughout the whole-genome analyses. These two gene sets, which were originally defined in tumors and PBMCs by RNA-seq, were then compared for their mean PFE in cfDNA of a set of patients with SCLC and control participants that we profiled by deep WES. Next, we defined a ‘SCLC signature score’ as the difference between the ‘high’ and ‘low’ sets.
This allowed us to compare cfDNA profiles of SCLC cases versus healthy control participants for the discriminating power of the ‘SCLC score’ through calculation of the AUC of a receiver-operating curve (ROC). We separately identified DEGs directly from cfDNA, by comparing PFEs of SCLC cases versus healthy adult control participants in a volcano plot analysis. Specifically, we used t-tests with false discovery rate threshold of 0.05 and mean PFE difference of at least 0.1. Here again, we compared the mean expression level in TPM for these DEGs in RNA-seq data from SCLC tumors and PBMC samples described above. Overlap between two SCLC high gene sets identified by either tumor RNA-seq or by cfDNA WES profiling was performed using the hypergeometric test.
Genotyping of somatic copy number variants (CNVs).
Genomic copy number alterations in healthy and SCLC cfDNA samples profiled by deep WES were identified using CNVKit v.0.9.8 (ref. 108). Raw genomic coverage was calculated from deduplicated ‘bam’ files for each sample considering on-target (IDT xGen Exome Research Panel v.2) as well as off-target regions. To correct for potential biases in capture efficiency and GC content, a pooled per-region reference was generated from five healthy cfDNA samples that were held out. The remaining healthy and SCLC samples were then normalized using this pooled reference, with discrete copy number segments inferred using the default circular binary segmentation algorithm109. Corresponding log2 copy number values for each segment were then used in further analyses. We considered whether CNVs might disproportionately affect PFE estimates in two ways. First, we considered whether the PFE difference in genes falling within amplifications versus deletions was significantly different in SCLC compared to healthy control participants profiled by deep cfDNA WES. Second, using Fisher’s exact test, we tested whether genes inferred to be highly expressed in SCLC cfDNA were significantly more likely to fall in amplifications and, conversely, whether those inferred as lowly expressed were more likely to be deleted.
Consideration of GC correction.
Two healthy control cfDNA samples were profiled by deep WGS. For these two participants, we also profiled the matched PBMC by RNA-seq. We then compared the predicted values from cfDNA against observed values from RNA-seq for each of the different GC-correction scenarios and tested concordance. We tested the impact of correcting for GC content of TSS regions on gene expression model accuracy in several ways. We considered four scenarios were studied when correcting features using the GC values for NDR and PFE: PFE alone corrected, NDR alone corrected, both corrected and neither corrected. The correction was performed using a locally estimated scatterplot smoothing function with a span of 0.5. The concordance was evaluated using three metrics: Pearson correlation, Spearman correlation and root-mean-square error (r.m.s.e.). Since none of these GC-correction approaches significantly improved correlations or reduced associated error profiles when considering reproducibility across cfDNA samples, we opted not to correct for variability in GC content across the TSS regions of different genes.
A gene expression model for predicting RNA output from TSS cfDNA fragmentomic features.
To infer RNA expression levels from cfDNA fragmentation profiles at TSS regions of genes across the transcriptome, we built a prediction model using two features, PFE and NDR. Of note, among the five fragmentomic features considered, these indices demonstrate highest individual correlations as well as complementarity. For training, we used one cfDNA sample sequenced to high coverage depth by WGS. We performed RNA-seq on the PBMC of five healthy participants and used the average across three of these individuals as the ‘reference expression vector’. Next, to achieve a higher resolution at the core promoters, we grouped every ten genes, based on their expression in our reference RNA-seq vector. After removing genes used as background for calculating PFE, a total of 1,748 groups (of ten genes each) remained. We then pooled all the fragments at the extended core promoters (−1/+1 kb around the TSSs) of the genes within each group and extracted the two features: NDR and PFE. We then normalized the two features by 95% quantile over the background genes, where for PFE the normalization factor is and , where Q(.,k) denotes the kth quantile. By bootstrap resampling, we then built an ensemble of 600 models: 200 univariable PFE-alone models mPFE,1, mPFE,2, …, mPFE,200, 200 univariable NDR alone-models mNDR,1, mNDR,2, …, mNDR,200 and 200 NDR-PFE integrated models mInt,1, mInt,2, …, mInt,200.
To transfer this expression prediction model, which was originally derived from WGS, to the targeted TSS space (EPIC-seq), we evaluated each of the 600 models above, by measuring its r.m.s.e. on two held-out healthy participants. For each of these two healthy participants, we compared the cfDNA profile by EPIC-seq to the corresponding PBMC transcriptome profile by RNA-seq from the same blood specimen and computed the r.m.s.e. for each of the 600 models. The weight of each model was then proportionally scaled by the inverse r.m.s.e. of that model, with the final score then calculated as the linear sum of 600 models, weighted as described above.
EPIC-seq panel design.
Identification of cancer type-specific genes.
We downloaded TCGA and DLBCL gene expression data in the form of RNA-seq FPKM-UQ for all individuals using the GDC API. After removing samples from individuals with a history of more than one type of malignancy, we divided the remaining samples into two separate cohorts for training and validation (70 and 30% of each cancer type, respectively). In the training set for each cancer type, median gene expression (FPKM-UQ) was calculated and protein-coding genes in the upper 15th quantile were considered as highly expressed genes. To remove potentially confounding effects in cfDNA from variation in blood cells, we excluded genes within the upper fifth quantile of expression in peripheral blood, when considering whole-blood transcriptome profiles from GTEx (https://www.gtexportal.org/home/datasets).
Gene selection for EPIC-seq targeted sequencing panel design.
We considered NSCLC and DLBCL, with known molecular subtypes exhibiting distinct gene expression profiles. Cancer-specific genes for LUAD, LUSC and DLBCL were included. To find subtype-specific genes in NSCLC, we performed differential expression analysis using the DESeq2 package in R Bioconductor to distinguish LUAD and LUSC tumor transcriptomes from TCGA. For the lymphoma analysis, a list of genes previously shown as differentially expressed between ABC and GCB subtypes according to RNA-seq gene expression data was used101,110. In addition to these DLBCL and NSCLC specific genes, we included 50 genes from the LM22 gene set capturing variation in peripheral blood leukocyte counts111. Together, these and other control genes contributed to a total of 179 unique genes, with each gene contributing one or more TSS regions to EPIC-seq totaling 236 targeted TSS regions.
EPIC-seq classification analyses and machine learning.
Distinguishing lung cancer (EPIC-lung classifier).
The EPIC-lung classifier was trained to distinguish participants with lung cancer from those who were cancer free. All the TSSs for immune cell type and NSCLC histology classification were used in this classifier. For genes with multiple TSS regions, in each iteration of cross-validation, we first combined TSS regions with intragene correlation exceeding 0.95 and capturing the mean. For those with correlations less than 0.95, we preserved individual TSS regions as independent reporters. This resulted in 139 features in the model and 143 samples (67 lung cancer cases and 71 control participants). We then trained an ℓ1 − ℓ2–regularized logistic regression model (‘elastic net’) with α = 0.9, and an optimal λ obtained by cross-validation. The full model was evaluated through a leave-one-batch-out model. Here, every batch contained at least one sample, and representing a set of samples that were captured and/or sequenced together in one next-generation sequencing lane.
Subclassification of NSCLC (EPIC-NSCLC subtype).
A NSCLC histology subtype classifier was designed to distinguish the two major subtypes of NSCLC, that is, LUAD and LUSC. As in the model in ‘EPIC-lung classifier’, the classification model uses elastic net with α = 0.9, with multiple TSS sites corresponding to one gene being merged. The performance of this classifier was evaluated via leave-one-out analysis. The classifier was trained using 80 features with 67 samples (36 LUADs and 31 LUSCs). To evaluate performance, classification accuracy with equal weights was calculated.
Biological plausibility of classifier coefficients.
We assessed the significance of the model coefficients in the NSCLC histology classifier from plasma cfDNA using EPIC-seq and their concordance with previous design from tumor transcriptomes using RNA-seq. Specifically, we compared nonzero coefficients from the elastic net model from cfDNA profiling, and then performed a t-test for the LUAD gene coefficients versus LUSC gene coefficients.
EPIC-seq lung dynamics score for the ICI treated patients.
To predict the benefit from immune-checkpoint inhibitors, we first identified the differentially expressed TSSs in a discovery pretreatment cohort (non-ICI, lung cancer versus normal). We then nominated the following TSS regions from genes with Bonferroni-corrected P < 0.25 with a one-sided t-test: (FOLR1 TSS no. 3, ITGA3 TSS no. 1, LRRC31 TSS no. 1, MACC1 TSS no. 1, NKX2-1 TSS no. 2, SCNN1A TSS no. 2, SFTPB TSS no. 1, WFDC2 TSS no. 1, CLDN1 TSS no. 1, FSCN1 TSS no. 1, GPC1 TSS no. 1, KRT17 TSS no. 1, PFN2 TSS no. 1, PKP1 TSS no. 1, S100A2 TSS no. 1, SFN TSS no. 1, SOX2 TSS no. 2, TP63 TSS no. 2). Denoting the expression levels of these genes by and for time points t0 and t1, respectively, we defined (fold change) statistics where is used to denote averaging the vector elements. For each patient, we then empirically derived a null distribution for the s statistics by randomly selecting k sites from the EPIC-seq selector. An empirical left-sided P value was then calculated to measure response to therapy. The EPIC-seq dynamics score was then defined as the logarithm (base 10) of these empirical P values.
Distinguishing lymphoma (EPIC-DLBCL classifier).
This classifier was trained to distinguish participants with DLBCL from those who did not have cancer using elastic net, with regularization parameters being set as in the EPIC-lung classifier. The dataset used for leave-one-batch-out cross-validation comprised 129 features and 167 samples (91 DLBCL cases and 71 control participants).
Subclassification of DLBCL COO (EPIC-DLBCL COO).
For the classification of DLBCL COO, we defined a GCB score as follows: (1) within a leave-one-out cross-validation framework, we first standardized each gene expression (that is, the Z-score) and converted the Z-scores into probabilities, and then (2) defined a COO score as . Gene sets for each subtype were defined as originally selected in the EPIC-seq selector design for DLBCL classification. To evaluate performance, we measured the concordance between EPIC-seq scores and (1) genetic COO classification scores obtained from CAPP-seq62, as well as (2) labels from the Hans immunohistochemical algorithm. Finally, (3) for a subset of patients with available matched FFPE tumor specimens, we compared EPIC-seq COO scores for the GCB-signature from cfDNA against the corresponding GCB scores from RNA-seq profiling of tumor biopsies.
Mechanistic modeling of nucleosome accessibility.
In relying on several assumptions from the structural studies and the chromatin literature, we developed a mechanistic model linking nucleosome accessibility at TSS regions to corresponding cfDNA fragmentation profiles, and associated expression levels (depicted in Supplementary Fig. 7). Specifically, we assumed (1) starting with N = 5,000 genome templates of 2 kb in size centered at the TSS, wherein each has the ‘+1 nucleosome’ is well-positioned at the TSS (that is, position zero). Then, (2) within this 2,000-bp region with, we assumed that a total of 11 nucleosomes can be present and spaced at internucleosome distances of roughly 180 bp. When constrained by the well-positioned nucleosome at the TSS, (3) the position of other nucleosomes is determined by 147 (the length of DNA in the core octamer particle) plus a variably sized linker DNA segment. Here, we modeled the linker DNA size flanking each nucleosome as a random variable, defining as . Within this approach, the position of the 3′ nucleosomes downstream of +1 nucleosome is determined as for i ≥ 2 with pos1 = 0. The position of 5′ nucleosomes upstream of +1 nucleosome is determined as . We assumed that (4) the cut site positions are located either: within the linker segments (that is, within the interval (posi + 147, posi+1) with a cutting probability, pcut (in our simulations pcut=0.7), or anywhere within the interval (posi−2 + 147, posi) with pcut=0.7 for accessible sites (for example, at i=1 for active genes). A cfDNA fragment length was then generated by cutting the initial template at the cut sites. Finally, we assumed (5) a secondary ‘degradation event’ to generate smaller, subnucleosomal fragments as a Bernoulli process, and occurring with a probability, pdegrade, (set to 0.2 here). Within this secondary degradation process, in considering the roughly ten bases per helical DNA turn, we thus shortened the cfDNA fragment according to the random variable d = round(10 ×(1 + M)), where M ~ Γ(1, 3), that is, ldigested = l − d. We then plotted the associated size distributions for these pseudo-cfDNA molecules generated in silico, allowing us to compare profiles for genes with high versus low expression, when assuming their promoters to have variably accessible TSS with 1–3 nucleosome-free regions.
Simulating mixtures using the mechanistic model.
To simulate the effect of variable quantities of tumor-derived molecules in plasma cfDNA, we created mixtures controlled by a mixing factor τ ≥ 0. For each τ, we begin with N genomes, equivalent to coverage depth here. We then randomly selected a subset of genomes (Ntumor), using a Binomial distribution with probability of τand assigned them to one of three scenarios where the TSS is variably accessible, assuming that each of the three scenarios are equally probable (as depicted in Supplementary Fig. 7a). The remaining cfDNA molecules (that is, N − Ntumor) are then assumed to be fully nucleosome protected, and thus not accessible (as depicted in Supplementary Fig. 7a). We then mix the resulting fragments to calculate the associated entropy (PFE) at these modeled TSS. For the simulations, we varied the variable τ from zero to 0.15 and generated 100 simulated mixtures. To summarize the results, for each nonzero τ, we compared the entropy values with the inactive scenario (that is, τ = 0) and via ROC analysis, allowing us to determine the sensitivity corresponding to 85% specificity. To assess the effect of sequencing depth, the entire analysis was reperformed for three different unique cfDNA coverage depths, N ∈ {500, 2500, 5000}. Within each of these three sequencing depths, we identified ctDNA mixture (%) levels that were significantly discernable by Kolmogorov–Smirnov test, when comparing the simulated PFE of the cfDNA mixture including variable levels of ctDNA against a pure mixture devoid of ctDNA.
Statistical and patient survival analysis.
Throughout the study, associations between known and predicted variables were measured by Pearson correlation (r) or Spearman correlation (ρ) depending on data type. Whenever data were depicted as box-and-whisker plots, boxes spanned the interquartile range (IQR), while the median was horizontally marked with a line in each box and whiskers spanned the 1.5 IQRs. When data were normally distributed, group comparisons were determined using t-test with unequal variance or a paired t-test, as appropriate; otherwise, a two-sided Wilcoxon test was applied. To test for trend in continuous variables across categorical groups, Jonckheere’s trend test was used as implemented in the clinfun R package. Correction for multiple hypothesis testing was performed using the Bonferroni method. Results with two-sided P < 0.05 were considered significant. Statistical analyses were performed with R v.4.0.1. CIs were calculated by resampling with replacement (that is, bootstrapping). ROC curve analyses were performed using the R package, pROC. Survival analyses were performed using the R package, survival. When dichotomized, Kaplan–Meier estimates were used to plot stratified survival curves and statistical significance was evaluated by log-rank test. Otherwise, Cox proportional-hazards models were fitted to the data to determine the significance of each covariate using Wald log-likelihood testing to assess significance. The differential expression (or PFE) analyses are summarized by statistical significance (via false discovery rate adjustment by p.adjust R function) and change magnitude, which are visualized by volcano plots.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
For each sample profiled in this study, we also provide anonymized fragmentomic data for fragments meeting minimal MAPQ and read FLAGs, which are available at https://epicseq.stanford.edu. These data are summarized across TSS regions by fragment size distributions (as in Fig. 1b). Moreover, the anonymized sequencing reads of samples profiled whole-genome (n = 3 deep whole-genome; n = 3 whole-genome and n = 24 shallow whole-genome) and whole-exome (n = 39 deep WES) are deposited at SRA PRJNA795275.
Code availability
The custom EPIC-seq software code for fragmentomic featurization and gene expression inference from cfDNA BAM files can be accessed at https://epicseq.stanford.edu/.
Extended Data
Extended Data Fig. 1 |. Fragment length density at the transcription start sites varies with gene expression.
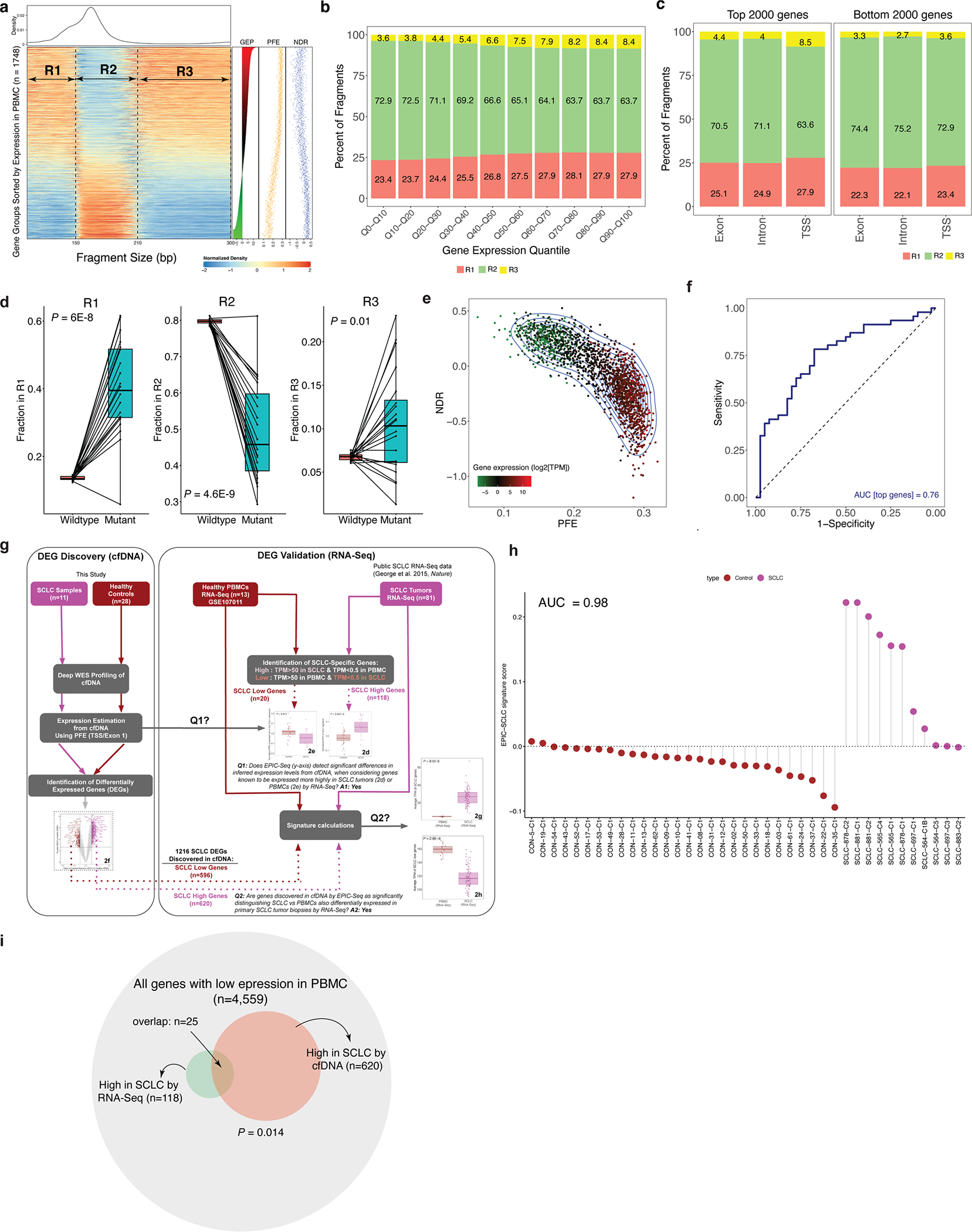
Fragment length density at the transcription start sites varies with gene expression. (a) A heatmap of fragment length densities across 1,748 groups of genes (similar to Fig. 1a). Three regions R1 (100–150 bps), R2 (151–210 bps), and R3 (211–300 bps) show enrichment in either high or low expression gene groups. (b) The percent of fragments within each region defined in panel (a) in the deep whole-genome sample across deciles of the reference PBMC gene expression vector, that is, 10 groups of genes when sorted by their expression values in PBMC. Highly expressed genes include fewer monosome fragments, indicating a wider distribution and thereby a higher PFE. (c) Fraction of fragments within the three regions, R1-R3, for exons vs introns vs TSS sites for the top (and bottom) 2000 genes as ranked by expression. The fraction of monosomal fragments within TSS regions is substantially lower than within intronic and exonic regions (63.5% at TSS vs ~71% at non-TSS). Pearson’s Chi-Squared goodness-of-fit tests resulted in the following test statistics (TSS vs Exon: G = 62,133 [P < 2.2E-16]; TSS vs Intron: G = 84,110 [P < 2.2E-16]). (d) Fraction of fragments falling within each region (R1, R2, and R3) for mutant cfDNA fragments and their wildtype counterparts. Each dot represents one tuple (variant-patient) and the connecting lines indicate the paired mutant-wildtype status. These results show that the mutant cfDNA fragments are enriched for R1 and R3 while wildtype fragments are enriched in R2. (e) A contour plot capturing the relationship between expression level (depicted by heat) as a function of two cfDNA fragmentomic features used in the gene inference model: PFE and NDR. (f) ROC analysis of a ‘NSCLC Score’ for noninvasively distinguishing patients with NSCLC from healthy controls (AUC = 0.76). The genes comprising this score were first defined from external RNA-Seq profiling data of primary NSCLC tumor tissues and blood samples, allowing subsequent calculation of their corresponding PFE in cfDNA samples profiled by WGS for independent NSCLC cases and healthy controls. (g) A schematic for the analyses performed for Fig. 2d–h. (h) Sample-level ‘SCLC Score’ from deep whole exome analysis of cfDNA and associated diagnostic performance. As in the exercise for NSCLC depicted in panel f, the genes comprising this SCLC score were first defined from external RNA-Seq profiling data of primary SCLC tumor tissues and blood samples. The corresponding PFEs (as the difference between the overall PFE level of top and bottom gene signatures) were subsequently calculated in cfDNA samples we profiled by deep WES for independent SCLC cases and healthy controls. Using these scores, an AUC of 0.9 was achieved in distinguishing cases from controls. (i) The Venn diagram of SCLC high genes identified in cfDNA (whole exome profiling) and tumor biopsy (RNA-Seq transcriptome profiling), with significance of overlap assessed by hypergeometric test.
Extended Data Fig. 2 |. Ensemble model accurately predicts gene expression in validation samples.
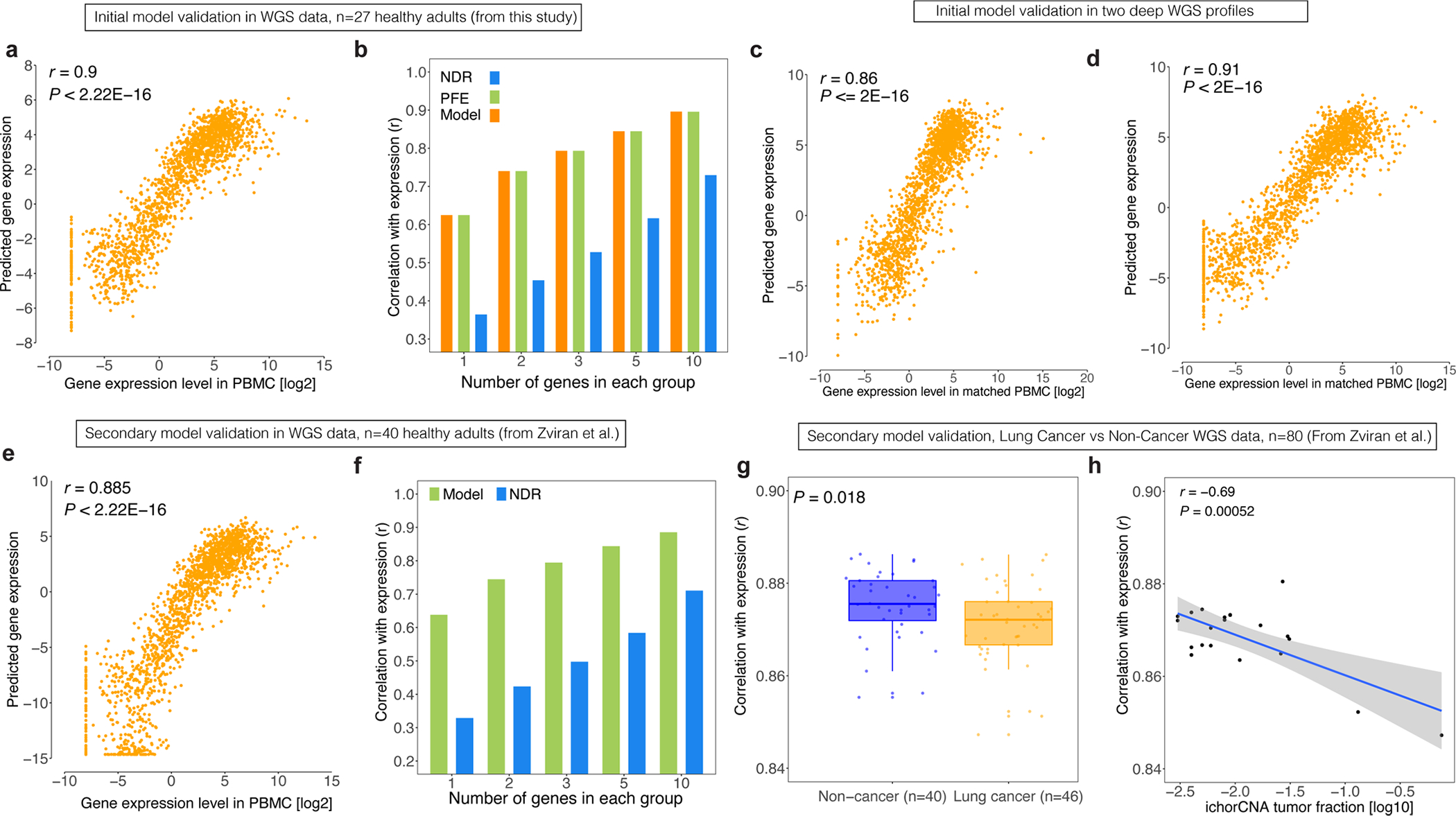
Ensemble model accurately predicts gene expression in validation samples. (a) The scatterplot of the predicted vs a population-averaged gene expression across 1,748 groups of genes. The underlying data are from a merged cfDNA ‘meta-sample’ (pooled from merger of 27 healthy subjects profiled by relatively shallow WGS), achieving a correlation of 0.9 in initial validation. (b) The meta sample from panel (a) was used to assess model performance, when considering TSS-level expression values without gene grouping (n = 1), as well as scenarios with 2, 3, 5 and 10 genes per group. The Pearson correlation between observed expression in PBMC versus predicted expression from our model (combining PFE and NDR) is shown in green bars. This correlation substantially improves as number of genes per group increases. The Pearson correlation values between observed gene expression and those predicted by NDR or PFE expression are shown in blue and green bars, respectively. (c) Scatterplot depicts predicted versus observed gene expression measurements across 1,748 groups of genes (dots), when comparing expression measurements by RNA-Seq on matched PBMC (x-axis) against plasma cfDNA inferences (y-axis), for a validation sample from a healthy adult that we also profiled by deep WGS (~200x). This achieved a Pearson correlation of 0.86. (d) Similar to panel c, but for a second healthy adult control subject also profiled for validation, by deep WGS of cfDNA and matched RNA-Seq of PBMC (Pearson r = 0.91). (e-f) The same analysis as in panels (a-b) for a meta whole-genome sample generated from healthy subjects from Zviran et al. (g) The whole genome samples (depth ~20–40x) from Zviran et al. were used with every ten genes grouped and the concordance between model-predicted expression and PBMC expression are evaluated using Pearson correlation (that is, each dot is one subject). The non-cancer samples show a significantly higher correlation with normal PBMC than lung cancer cases (Wilcoxon P = 0.018). (h) The ichorCNA tumor fraction estimates of the lung cancer cases in panel f are used to compare with the correlations in panel f. As shown in a scatterplot, as tumor fraction increases, the correlation decreases (r = −0.69, P = 0.00052).
Extended Data Fig. 3 |. Case-level information of samples profiled by EPIC-Seq.
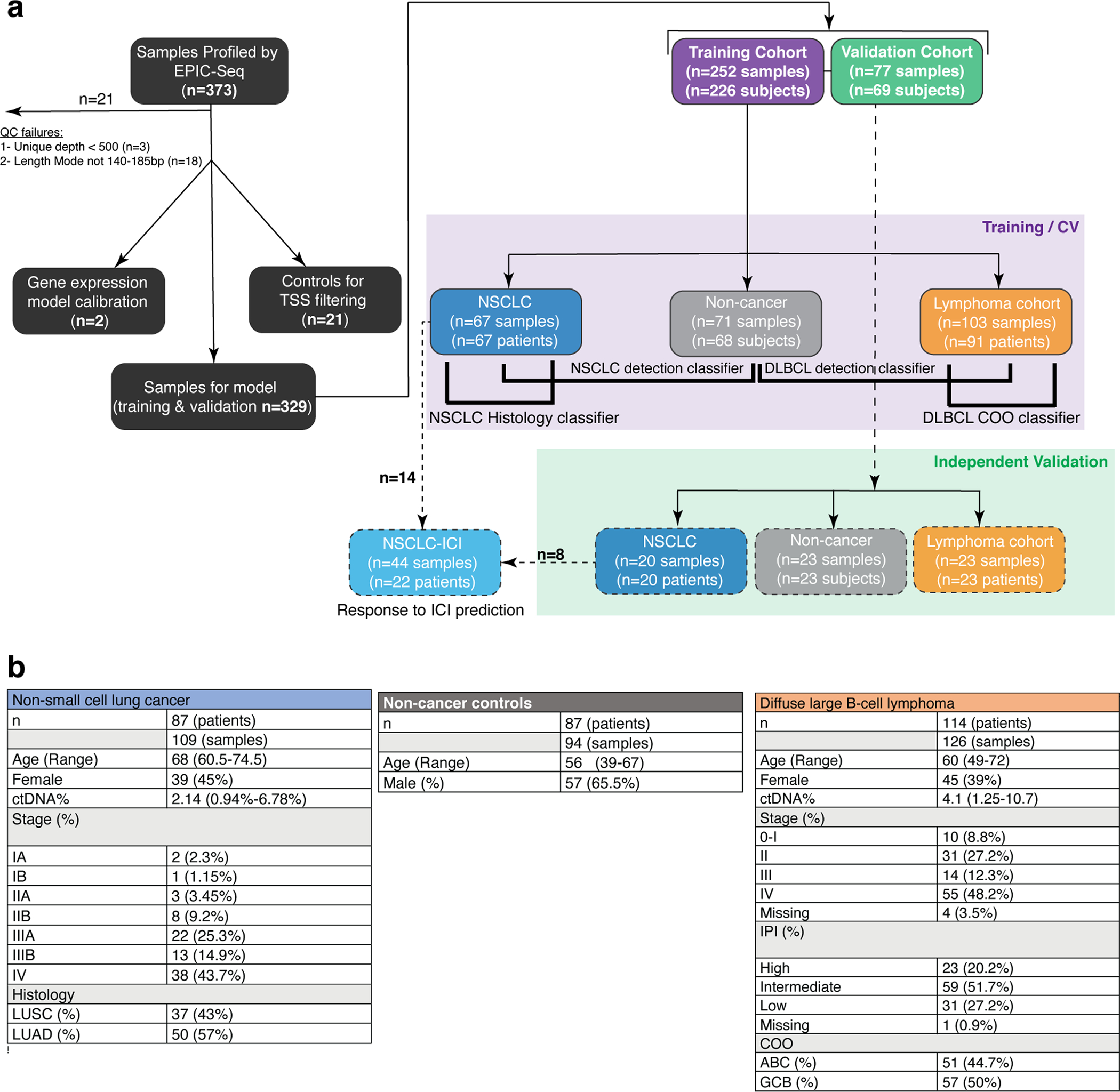
Cohorts and cell-free DNA samples profiled by EPIC-seq in this study, including Cancer Cases and Control Subjects. (a) Schema depicts the full set of specimens profiled by EPIC-Seq (n = 373), including those meeting Quality Control (QC) criteria (n = 352, 95%). A subset of samples were used for the initial gene expression model tuning (n = 2) and TSS filtering (n = 21). The remaining 329 samples were profiled by EPIC-Seq to address disease-specific questions, including utility for cancer detection, classification of histology and cell-of-origin, and response monitoring. These included 252 samples (76.6%) from 226 subjects that comprised our Discovery/Training cohort (large light purple rectangle), as well as subsequent profiling of a Validation Cohort of 77 samples (23.4%) from 69 subjects, after models were ‘locked down’ (large light green rectangle). A subset of 22 NSCLC patients where a pair of serial blood samples were monitored for ICI response (to allow comparisons of both EPIC-Seq and CAPP-Seq and assess biological plausibility), but this exercise was not subject to any model training. No samples were shared between Training and Validation exercises, with all models locked down before independent validations. Four healthy subjects (4.5%) provided more than one cfDNA specimen with one used for Training and the second for Validation. (b) Distribution of demographic, clinical, anatomic, and pathological variables for subjects profiled by EPIC-Seq. Tabulated are the relevant indices for cancer cases (235 blood samples 201 patients), including NSCLC patients (light blue; 109 blood samples from 87 patients), DLBCL patients (light orange; 126 blood samples from 104 patients), and non-cancer control subjects (gray; 94 blood samples from 87 adults).
Extended Data Fig. 4 |. Correlation between EPIC-lung score and clinical factors.
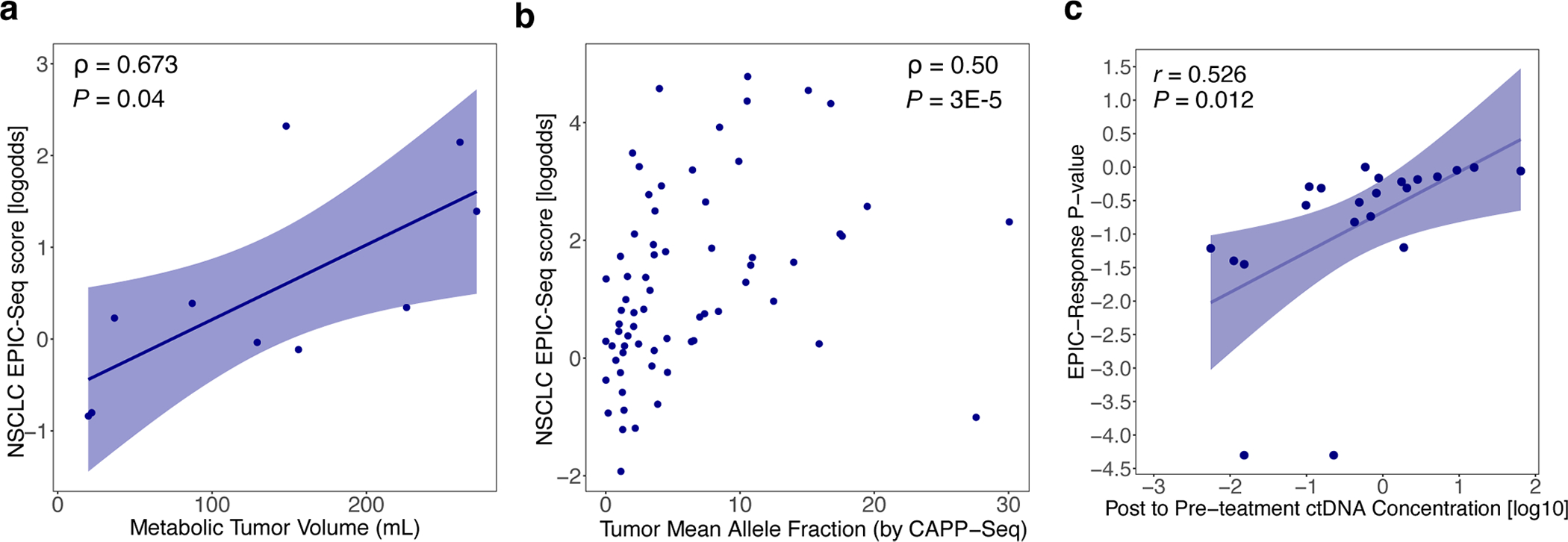
Concordance between EPIC-Seq measurements and established NSCLC risk factors including metabolic tumor burden, ctDNA level, and ctDNA response. (a) Concordance between EPIC-lung score and metabolic tumor volume (MTV), as measured by Spearman correlation (ρ = 0.67; P = 0.04). (b) Concordance between EPIC-lung score and the ctDNA mean allele fractions as measured by CAPP-Seq, evaluated using Spearman correlation (ρ = 0.5; P = 3E-5). (c) Relationships between genetic versus epigenetic molecular responses to Immune Checkpoint Inhibitor (ICI) therapy in advanced NSCLC. Scatterplot compares molecular responses measured noninvasively by CAPP-Seq (x-axis; fold change, Log10) and EPIC-Seq (lung dynamics score; y-axis) using serial plasma profiling before and after ICI therapy. The two orthogonal measures show moderate but significant correlation (r = 0.53, P = 0.012).
Extended Data Fig. 5 |. Correlation between EPIC-lymphoma score and clinical factors, results of the validation set and prognostic value of the LMO2 distal promoter.
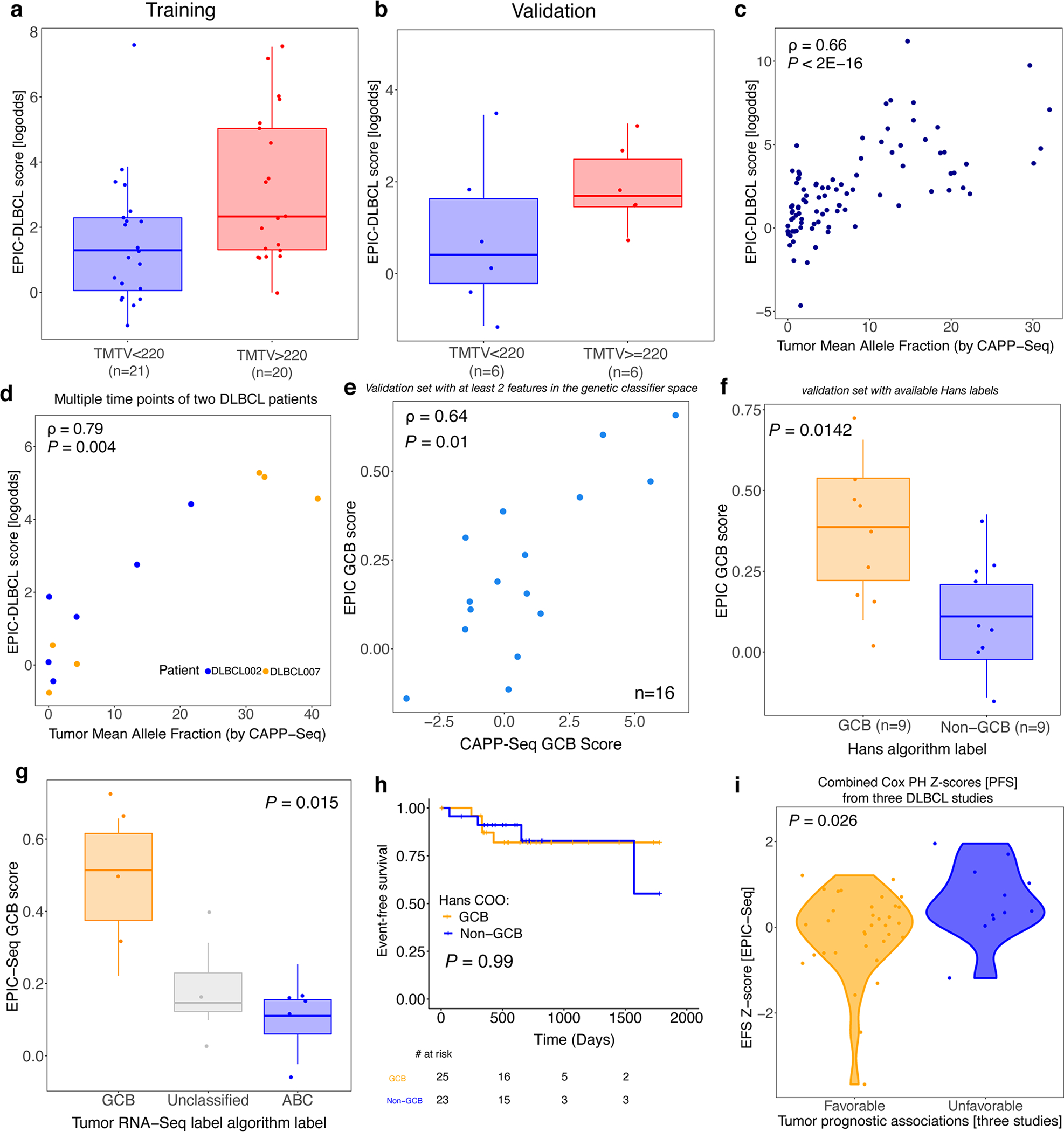
Concordance between EPIC-Seq measurements and established DLBCL risk factors impacting outcome, including metabolic tumor volume, ctDNA level, and Cell-of-Origin. (a) The boxplots illustrate the two groups of patients stratified by their metabolic tumor volumes (>220 vs <220 mL; Wilcoxon P = 0.015). (b) Similar to panel a, but for the DLBCL Validation Cohort. (c) Concordance between EPIC-DLBCL scores and ctDNA mean allele fractions (from CAPP-Seq), evaluated using Spearman correlation (ρ = 0.66; P < 2E-16). (d) The EPIC-DLBCL model is applied to the cfDNA profiles of 13 samples from two DLBCL patients (DLBCL002 [ABC] and DLBCL007 [GCB]). The concordance between the resulting scores and the ctDNA mean allele fractions is evaluated by Spearman correlation (ρ = 0.79; P = 0.004). (e) Relationship between DLBCL cell-of-origin EPIC-Seq GCB scores and mutation-based GCB scores as measured by CAPP-Seq in the validation set (Spearman ρ = 0.64, P = 0.01). Each dot represents one sample (related to Fig. 6a). (f) Relationship between EPIC-Seq GCB scores from cfDNA and matched tumor tissue classification by routine Hans immunohistochemical algorithm in the validation set (Wilcoxon P = 0.001; related to Fig. 6b). (g) Relationship between EPIC-Seq GCB scores from cfDNA and tumor classification by RNA-seq of paired tumor tissue (Jonckheere’s trend test, P = 0.015). Box-and-whisker plots depict the EPIC-Seq GCB score in individual samples profiled by EPIC-Seq (dots), with boxes spanning the inter-quartile range; the median is horizontally marked with a line in each box, and whiskers span the 1.5 IQRs. (h) The Kaplan-Meier curves of EFS of the patients when labeled by the Hans algorithm. The non-GCB group contains both Non-GCB and Unknown. (i) The violin plot shows the distributions of Cox Proportional Hazard Model Z-scores when genes are grouped according to their effects on outcome (measured as EFS) in three prior tumor studies.
Extended Data Fig. 6 |. Pre-analytical factors and TSS GC-content correction effect on PFE.
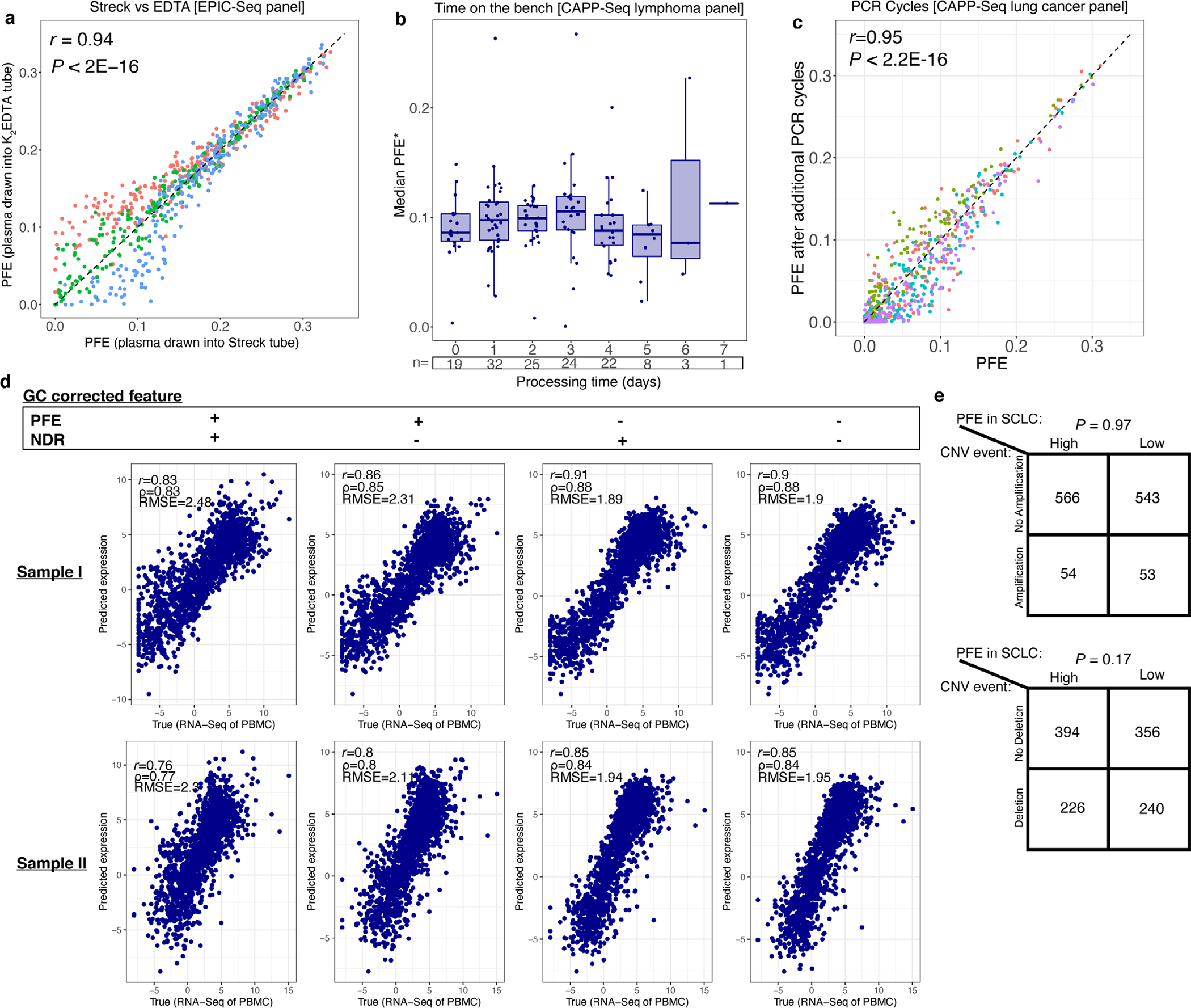
Effect of preanalytical factors on fragment size entropy and effect of GC-content correction on expression model performance. (a) The concordance between PFE values for three healthy controls profiled by EPIC-Seq using paired Streck BCT and K2EDTA tubes. A Pearson correlation of 0.94 was observed between tube types. (b) Effect of time on the bench (that is, in days) on the PFEs in a cohort of plasma cfDNA samples. (c) Effect of additional PCR cycles on PFE. Here we profiled 4 healthy control cfDNA samples by the CAPP-Seq lung cancer selector when 3 additional PCR cycles were included to study their effect. A Pearson correlation of 0.95 was observed between standard conditions versus those incorporating additional PCR cycles. (d) Effect of correction for GC-content of TSS regions on gene expression model accuracy. Four scenarios were studied when correcting features using the GC values for NDR and PFE: PFE alone corrected, NDR alone corrected, both corrected, and neither corrected. The correction was performed using a LOESS function with a span of 0.5. Two healthy control cfDNA samples were profiled by deep whole genome sequencing. For these two subjects, we also profiled the matched PBMC by RNA-Sequencing. We then compared the predicted values from cfDNA against observed values from RNA-Seq for each of the different GC-correction scenarios and tested concordance. The concordance was evaluated using three metrics: Pearson correlation, Spearman correlation, and root-mean-square error (RMSE). When considering both cfDNA samples, none of the four GC-correction approaches seemed to consistently improve correlations or reduce associated error profiles. (e) Whole exome profiling of small-cell lung cancer samples in Fig. 2 are used to investigate association between PFEs and copy number aberrations. We first determined genes with PFE significantly higher in SCLC cfDNA samples (n = 11) compared with healthy control cfDNA samples (n = 28) (‘High’ PFE). Similarly, we determined genes with significantly lower PFEs in SCLC cfDNA samples (‘Low’ PFE). Then, the copy number states (CNS) corresponding to all genes were identified by overlapping copy number profiles from CNVkit with the genomic coordinates of the first exons. The CNS values were then dichotomized into (i) amplification vs no-amplification and (ii) deletion vs no-deletion. Next, we summarized these by contingency tables for (i) vs PFE levels (top table) and (ii) vs PFE levels (bottom table). Finally, the association between the two was examined via Fisher’s exact test, which showed insignificant associations in both tests (P = 0.97 and P = 0.17; for amplifications and deletions, respectively).
Extended Data Fig. 7 |.
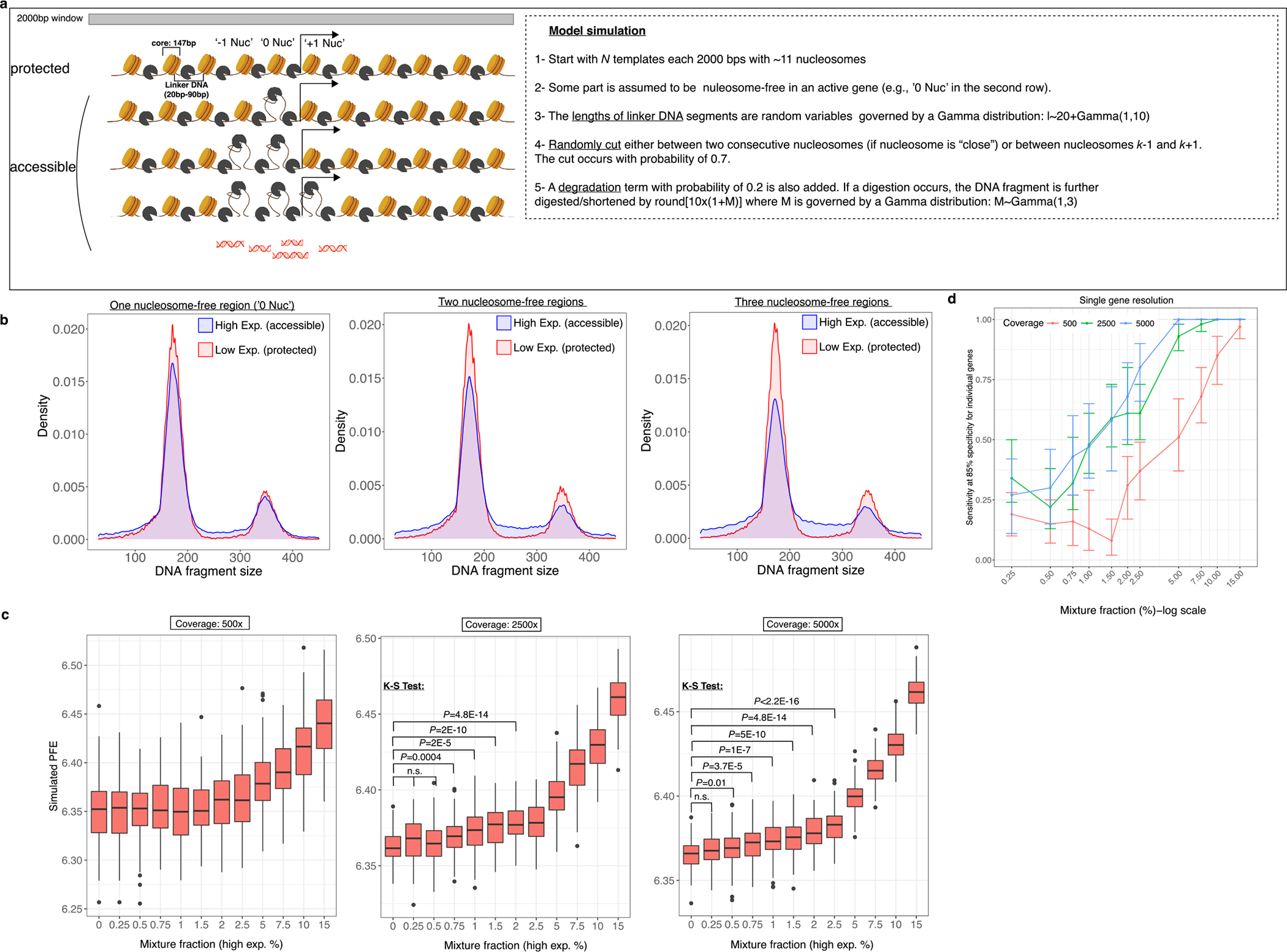
Mechanistic model and gene detection sensitivity with various parameters. (a) The cartoon shows four scenarios considered in our simulations: (i) protected, meaning that nucleosomes are well-positioned and are all present, (ii) one nucleosome-free position is present, (iii) two nucleosome-free positions are present and (iv) three nucleosome-free positions are present. (b) The density plots show the results of generating fragment lengths via the model described in panel a. Three panels correspond to scenarios (ii-iv) vs (i) in a. (c) A varying mixture parameters is considered and its effect on the entropy for three different coverages: 500x, 2500x and 5000x. (d) A summary of panel c for active gene detection sensitivity while achieving a specificity of 85%. The error bars are from the sensitivities calculated using the ‘ci.se’ function in R pROC package. The colors correspond to three different coverages in panel c.
Supplementary Material
Acknowledgements
We thank the patients and families who participated in this study, and we are grateful for all the insightful discussions with the members of the Alizadeh and Diehn laboratories. Funding sources: this work was supported by the National Cancer Institute (M.S.E., grant no. R25CA180993; M.S.E., grant no. T32CA009302; M.M., grant no. F99CA212457, M.D. and A.A.A., grant nos. R01CA188298, R01CA254179, R01CA257655, R01CA244526, R01CA233975, and R01CA229766), the US National Institutes of Health Director’s New Innovator Award Program (M.D.; grant no. 1-DP2-CA186569), the Virginia and D.K. Ludwig Fund for Cancer Research (M.D. and A.A.A.), and philanthropic support from the Bakewell Foundation (to M.D. and A.A.A.), the CRK Faculty Scholar Fund (M.D.), the Troper-Wojcicki Family Gift, The Shanahan Family Lymphoma Fund, Arzang Family Lymphoma Fund, The Skeff Family Lymphoma Fund, The Cane-Nowak Family Foundation, The Marc Benioff Fund, Jewish Communal Fund for Lymphoma Research, The Sara Schottenstein Memorial Fund, and The Moghadam Family Endowed Professorship (to A.A.A.). A.A.A. is a scholar of the Leukemia and Lymphoma Society. J.M.I., C.M.R. and B.T.L. are supported by the MSKCC Support grant no. P30 CA008748 from the NIH. B.Y.N. is a Stanford Cancer Systems Biology Scholar and supported by the NIH (grant no. 5R25CA180993) and by the Postdoctoral Research Fellowship (grant no. 134031-PF-19-164-01-TBG) from the American Cancer Society. Present address for M.M. is the Department of Biotechnology, College of Science, University of Tehran, Iran, and for D.A.K. is the Northwell Health Cancer Institute and Feinstein Institute of Research, Lake Success, NY, USA.
Footnotes
Competing interests
A.A.A. reports research funding from Celgene, Pfizer, ownership interests in FortySeven, CiberMed, ForeSight and paid consultancy from Roche, Genentech, Janssen, Pharmacyclics, Gilead, Celgene and Chugai. M.D. reports research funding from Varian Medical Systems and Illumina, ownership interest in CiberMed, ForeSight and paid consultancy from Roche, AstraZeneca, Illumina, RefleXion and BioNTech. J.J.C. reports paid consultancy from Lexent Bio Inc. and ownership interests in ForeSight. A.M.N. has patent filings related to expression deconvolution and cancer biomarkers and has served as a consultant for Roche, Merck and CiberMed. D.M.K. reports paid consultancy from Roche. B.T.L. has served as an uncompensated advisor and consultant to Amgen, Genentech, Boehringer Ingelheim, Lilly, AstraZeneca and Daiichi Sankyo. B.T.L. reports receiving research grants to his institution from Amgen, Genentech, AstraZeneca, Daiichi Sankyo, Lilly, Illumina, GRAIL, Guardant Health, Hengrui Therapeutics, MORE Health and Bolt Biotherapeutics. B.T.L. has received academic travel support from MORE Health and Jiangsu Hengrui Medicine. B.T.L. reports to be inventor on two institutional patents at MSKCC (US62/685,057, US62/514,661) and has intellectual property rights as a book author at Karger Publishers and Shanghai Jiao Tong University Press. J.M.I. reports serving as an unpaid consultant to Amgen and Roche-Genentech, institutional research support from Guardant Health and GRAIL, and ownership interest in LumaCyte. A.A.A., M.D., M.S.E., D.M.K., J.J.C., and B.Y.N. report patent filings related to cancer biomarkers. M.S.E., M.M., A.A.A. and M.D. have patent filing related to this paper. B.Y.N. is currently an employee and holds stock from Roche/Genentech. The remaining authors declare no competing interests.
Extended data are available for this paper at https://doi.org/10.1038/s41587-022-01222-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01222-4.
Peer review information Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Jahr S et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 61, 1659–1665 (2001). [PubMed] [Google Scholar]
- 2.Lo YM et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci. Transl. Med. 2, 61ra91 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Heitzer E, Auinger L & Speicher MR Cell-free DNA and apoptosis: how dead cells inform about the living. Trends Mol. Med. 26, 519–528 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Newman AM et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phallen J et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 9, eaan2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JD et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cristiano S et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 570, 385–389 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heitzer E, Haque IS, Roberts CES & Speicher MR Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Chabon JJ et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 580, 245–251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Opstal D et al. Origin and clinical relevance of chromosomal aberrations other than the common trisomies detected by genome-wide NIPS: results of the TRIDENT study. Genet. Med. 20, 480–485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan HC et al. Non-invasive prenatal measurement of the fetal genome. Nature 487, 320–324 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knight SR, Thorne A & Lo Faro ML Donor-specific cell-free DNA as a biomarker in solid organ transplantation. a systematic review. Transplantation 103, 273–283 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Chaudhuri AA et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 7, 1394–1403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lennon AM et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 369, eabb9601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zviran A et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 26, 1114–1124 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo YM et al. Presence of donor-specific DNA in plasma of kidney and liver-transplant recipients. Lancet 351, 1329–1330 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Snyder TM, Khush KK, Valantine HA & Quake SR Universal noninvasive detection of solid organ transplant rejection. Proc. Natl Acad. Sci. USA 108, 6229–6234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehmann-Werman R et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl Acad. Sci. USA 113, E1826–1834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang P et al. Preferred end coordinates and somatic variants as signatures of circulating tumor DNA associated with hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 115, E10925–E10933 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun K et al. Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 29, 418–427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadeh R et al. ChIP–seq of plasma cell-free nucleosomes identifies gene expression programs of the cells of origin. Nat. Biotechnol. 39, 586–598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lui YY et al. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin. Chem. 48, 421–427 (2002). [PubMed] [Google Scholar]
- 23.Fleischhacker M & Schmidt B Circulating nucleic acids (CNAs) and cancer—a survey. Biochim. Biophys. Acta 1775, 181–232 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Ramachandran S, Ahmad K & Henikoff S Transcription and remodeling produce asymmetrically unwrapped nucleosomal intermediates. Mol. Cell 68, 1038–1053 e1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snyder MW, Kircher M, Hill AJ, Daza RM & Shendure J Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164, 57–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanov M, Baranova A, Butler T, Spellman P & Mileyko V Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 16, S1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulz P et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 48, 1273–1278 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Wu J et al. Decoding genetic and epigenetic information embedded in cell free DNA with adapted SALP-seq. Int. J. Cancer 145, 2395–2406 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Jiang P et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl Acad. Sci. USA 112, E1317–1325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Underhill HR et al. Fragment length of circulating tumor DNA. PLoS Genet. 12, e1006162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mouliere F et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 10, eaat4921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulz P et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 10, 4666 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss J et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 9, 5068 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weintraub H & Groudine M Chromosomal subunits in active genes have an altered conformation. Science 193, 848–856 (1976). [DOI] [PubMed] [Google Scholar]
- 35.Jiang P et al. Plasma DNA end-motif profiling as a fragmentomic marker in cancer, pregnancy, and transplantation. Cancer Discov. 10, 664–673 (2020). [DOI] [PubMed] [Google Scholar]
- 36.The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puglisi F et al. Prognostic value of thyroid transcription factor-1 in primary, resected, non-small cell lung carcinoma. Mod. Pathol. 12, 318–324 (1999). [PubMed] [Google Scholar]
- 39.Maloney DG et al. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood 84, 2457–2466 (1994). [PubMed] [Google Scholar]
- 40.Ferlay J et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–386 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Torre LA, Siegel RL & Jemal A Lung cancer statistics. Adv. Exp. Med. Biol. 893, 1–19 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Newman AM et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 34, 547–555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Travis WD et al. The 2015 World Health Organization Classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Reck M & Rabe KF Precision diagnosis and treatment for advanced non-small-cell lung cancer. N. Engl. J. Med. 377, 849–861 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Ettinger DS et al. NCCN guidelines insights: non-small cell lung cancer, version 1.2020. J. Natl Compr. Cancer Netw. 17, 1464–1472 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Wiener RS, Schwartz LM, Woloshin S & Welch HG Population-based risk for complications after transthoracic needle lung biopsy of a pulmonary nodule: an analysis of discharge records. Ann. Intern. Med. 155, 137–144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bubendorf L, Lantuejoul S, de Langen AJ & Thunnissen E Nonsmall cell lung carcinoma: diagnostic difficulties in small biopsies and cytological specimens: number 2 in the series ‘Pathology for the clinician’ edited by Peter Dorfmuller and Alberto Cavazza. Eur. Respir. Rev. 26, 170007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McLean AEB, Barnes DJ & Troy LK Diagnosing lung cancer: the complexities of obtaining a tissue diagnosis in the era of minimally invasive and personalised medicine. J. Clin. Med. 7, 163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reck M et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Socinski MA et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Gandhi L et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Hellmann MD et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Camidge DR, Doebele RC & Kerr KM Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat. Rev. Clin. Oncol. 16, 341–355 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Nabet BY et al. Noninvasive early identification of therapeutic benefit from immune checkpoint inhibition. Cell 183, 363–376 e313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Menon MP, Pittaluga S & Jaffe ES The histological and biological spectrum of diffuse large B-cell lymphoma in the World Health Organization classification. Cancer J. 18, 411–420 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sehn LH et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood 109, 1857–1861 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Alizadeh AA et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Pasqualucci L et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 43, 830–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cottereau AS et al. Molecular profile and FDG-PET/CT total metabolic tumor volume improve risk classification at diagnosis for patients with diffuse large B-cell lymphoma. Clin. Cancer Res. 22, 3801–3809 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Scherer F et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 8, 364ra155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurtz DM et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. J. Clin. Oncol. 36, 2845–2853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosenwald A et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 1937–1947 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Basso K & Dalla-Favera R Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 15, 172–184 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Dunleavy K et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 113, 6069–6076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thieblemont C et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J. Clin. Oncol. 29, 4079–4087 (2011). [DOI] [PubMed] [Google Scholar]
- 66.Scott DW et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 123, 1214–1217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nowakowski GS et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-cell lymphoma: a phase II study. J. Clin. Oncol. 33, 251–257 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Wilson WH et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 21, 922–926 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Young RM & Staudt LM Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 12, 229–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lenz G et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 359, 2313–2323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zelenetz AD et al. NCCN guidelines insights: B-cell lymphomas, version 3.2019. J. Natl Compr. Cancer Netw. 17, 650–661 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Hans CP et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103, 275–282 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Lossos IS et al. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. N. Engl. J. Med. 350, 1828–1837 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Malumbres R et al. Paraffin-based 6-gene model predicts outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Blood 111, 5509–5514 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alizadeh AA, Gentles AJ, Lossos IS & Levy R Molecular outcome prediction in diffuse large-B-cell lymphoma. N. Engl. J. Med. 360, 2794–2795 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Alizadeh AA et al. Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood 118, 1350–1358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chapuy B et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 24, 679–690 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ennishi D et al. Double-Hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 190–201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gentles AJ & Alizadeh AA A few good genes: simple, biologically motivated signatures for cancer prognosis. Cell Cycle 10, 3615–3616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chambers J & Rabbitts TH LMO2 at 25 years: a paradigm of chromosomal translocation proteins. Open Biol. 5, 150062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Royer-Pokora B et al. The TTG-2/RBTN2 T cell oncogene encodes two alternative transcripts from two promoters: the distal promoter is removed by most 11p13 translocations in acute T cell leukaemia’s (T-ALL). Oncogene 10, 1353–1360 (1995). [PubMed] [Google Scholar]
- 82.Oram SH et al. A previously unrecognized promoter of LMO2 forms part of a transcriptional regulatory circuit mediating LMO2 expression in a subset of T-acute lymphoblastic leukaemia patients. Oncogene 29, 5796–5808 (2010). [DOI] [PubMed] [Google Scholar]
- 83.Boehm T et al. An unusual structure of a putative T cell oncogene which allows production of similar proteins from distinct mRNAs. EMBO J. 9, 857–868 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smale ST & Kadonaga JT The RNA polymerase II core promoter. Annu. Rev. Biochem. 72, 449–479 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Bernstein BE et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181 (2005). [DOI] [PubMed] [Google Scholar]
- 86.Wong IH et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 59, 71–73 (1999). [PubMed] [Google Scholar]
- 87.Chim SS et al. Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc. Natl Acad. Sci. USA 102, 14753–14758 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernandez AF et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 22, 407–419 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Houseman EA et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chan KC et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl Acad. Sci. USA 110, 18761–18768 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lun FM et al. Noninvasive prenatal methylomic analysis by genomewide bisulfite sequencing of maternal plasma DNA. Clin. Chem. 59, 1583–1594 (2013). [DOI] [PubMed] [Google Scholar]
- 92.Ou X et al. Epigenome-wide DNA methylation assay reveals placental epigenetic markers for noninvasive fetal single-nucleotide polymorphism genotyping in maternal plasma. Transfusion 54, 2523–2533 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Jensen TJ et al. Whole genome bisulfite sequencing of cell-free DNA and its cellular contributors uncovers placenta hypomethylated domains. Genome Biol. 16, 78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Visel A et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koh W et al. Noninvasive in vivo monitoring of tissue-specific global gene expression in humans. Proc. Natl Acad. Sci. USA 111, 7361–7366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Srinivasan S et al. Small RNA sequencing across diverse biofluids identifies optimal methods for exRNA isolation. Cell 177, 446–462 e416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ibarra A et al. Non-invasive characterization of human bone marrow stimulation and reconstitution by cell-free messenger RNA sequencing. Nat. Commun. 11, 400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou Z et al. Extracellular RNA in a single droplet of human serum reflects physiologic and disease states. Proc. Natl Acad. Sci. USA 116, 19200–19208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Verwilt J et al. When DNA gets in the way: a cautionary note for DNA contamination in extracellular RNA-seq studies. Proc. Natl Acad. Sci. USA 117, 18934–18936 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schmitz R et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 378, 1396–1407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 103.Binkley MS et al. KEAP1/NFE2L2 mutations predict lung cancer radiation resistance that can be targeted by glutaminase inhibition. Cancer Discov. 10, 1826–1841 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen S, Zhou Y, Chen Y & Gu J fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Patro R, Duggal G, Love MI, Irizarry RA & Kingsford C Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alig S et al. Short diagnosis-to-treatment interval is associated with increased tumor burden measured by circulating tumor DNA and metabolic tumor volume in diffuse large B-cell lymphoma. J. Clin. Oncol. 39, 2605–2616 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.George J et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.U, M.Talevich E, Katiyar S, Rasheed K & Kannan N Prediction and prioritization of rare oncogenic mutations in the cancer Kinome using novel features and multiple classifiers. PLoS Comput. Biol. 10, e1003545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Venkatraman ES & Olshen AB A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23, 657–663 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Newman AM et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For each sample profiled in this study, we also provide anonymized fragmentomic data for fragments meeting minimal MAPQ and read FLAGs, which are available at https://epicseq.stanford.edu. These data are summarized across TSS regions by fragment size distributions (as in Fig. 1b). Moreover, the anonymized sequencing reads of samples profiled whole-genome (n = 3 deep whole-genome; n = 3 whole-genome and n = 24 shallow whole-genome) and whole-exome (n = 39 deep WES) are deposited at SRA PRJNA795275.