

Abstract
Polymer–protein hybrids are intriguing materials that can bolster protein stability in non-native environments, thereby enhancing their utility in diverse medicinal, commercial, and industrial applications. One stabilization strategy involves designing synthetic random copolymers with compositions attuned to the protein surface, but rational design is complicated by the vast chemical and composition space. Here, a strategy is reported to design protein-stabilizing copolymers based on active machine learning, facilitated by automated material synthesis and characterization platforms. The versatility and robustness of the approach is demonstrated by the successful identification of copolymers that preserve, or even enhance, the activity of three chemically distinct enzymes following exposure to thermal denaturing conditions. Although systematic screening results in mixed success, active learning appropriately identifies unique and effective copolymer chemistries for the stabilization of each enzyme. Overall, this work broadens the capabilities to design fit-for-purpose synthetic copolymers that promote or otherwise manipulate protein activity, with extensions toward the design of robust polymer–protein hybrid materials.
Keywords: active learning, Bayesian optimization, combinatorial polymer design, machine learning, polymer–protein conjugates, protein formulations, single-enzyme nanoparticles
Graphical Abstract
Polymer-protein hybrids (PPHs) offer new opportunities for protein stabilization in industrial and medical applications. Utilizing a combination of automated polymer synthesis and active machine learning, we report a “Learn-Design-Test-Build” strategy for protein-tailored copolymer design and synthesis. Further, utilizing large quantities of experimental data, structure-function relationships within PPHs are probed and biophysical characterization is performed to elucidate potential mechanisms of stability within PPHs.
1. Introduction
Polymer–protein hybrids (PPHs) have emerged as attractive materials that leverage polymers to improve protein solubility and stability in often denaturing and abiological environments.[1–5] One strategy, which has resulted in remarkable hours-long enzyme activity in toluene,[6] tailors the composition of random copolymers based on protein surface chemistry. In principle, copolymers might be precisely designed to stabilize any given protein without compromising activity. However, identifying such copolymers, whether via rational design or screening, is challenging due to a large combinatorial design space (e.g., monomer chemistry, chain length, architecture).[7] Thus, fit-for-purpose PPHs could facilitate myriad applications—biofuel production,[8] plastics degradation,[9,10] pharmaceutical synthesis[11]—but a robust strategy for their design remains elusive.
Over the last decade, machine learning (ML) has dramatically accelerated materials discovery across disciplines,[12–14] enabling more efficient identification of materials with target properties.[12,15–20] Nonetheless, ML-guided copolymer design is limited by several factors, including the availability of quality data necessary to train models.[7,21–24] Most polymer databases predominantly feature homopolymers,[25] and the laborious nature of polymer synthesis and characterization severely limits the number of systems that can be examined “in-house”.[26] Several copolymer design efforts have thus relied on data generated in silico.[20,27,28] Meanwhile, recent experimental work has used flow reactors or parallel batch synthesizers to provide modest data (<500 samples).[17,29,30] More scalable approaches would substantially extend capabilities to design copolymers for PPHs and other materials applications.
Here, we use active ML to rapidly design copolymers to form thermostable PPHs with glucose oxidase (GOx), lipase (Lip), and horseradish peroxidase (HRP) (Figure 1). To efficiently acquire data, we use automated oxygen-tolerant radical polymerization for copolymer synthesis[31,32] and develop a facile, thermal-stability assay to characterize PPHs. With this platform and five iterations of a Learn–Design–Build–Test cycle for each enzyme, we successfully identify PPHs with significant enzyme activity; these PPHs generally outperform those derived from a systematic screen with over 500 unique copolymers. Notably, we demonstrate that our strategy, which utilizes active ML, appropriately adapts data acquisition to yield chemically distinct sets of top-performing copolymers for each enzyme. Post hoc analysis of our data and ML models reveals important relationships between specific copolymer chemistries and PPH stability, while biophysical characterization of our most efficacious PPHs provide mechanistic insight into how copolymers may preserve enzyme function under thermal stress. Overall, this framework will automate and accelerate the design of copolymers for stable PPHs across applications.
Figure 1.

Overview of study. a) Schematic illustration of the surface chemistry for horseradish peroxidase (HRP), glucose oxidase (GOx), and lipase (Lip). Amino acids are colored based on classification as ionic (blue), hydrophilic (green), and hydrophobic (magenta). Images for the protein are rendered using Visual Molecular Dynamics.[33] b) Monomers utilized for copolymer design. The colored boxes delineate rough classifications as ionic (blue), hydrophilic (green), and hydrophobic (magenta). c) Schematic representation of closed-loop Learn–Design–Build–Test discovery process used in this work. After initialization with a seed dataset, the process consists of: training an enzyme-specific Gaussian process regression (GPR) surrogate model to predict the retained enzyme activity (REA) of a polymer–protein hybrid (PPH) based on copolymer characteristics (learn); Bayesian optimization of copolymers to satisfy an expected improvement acquisition function and subsequent filtering to propose new copolymers (design) (ii); automated synthesis of proposed copolymers via photoinduced electron/energy transfer reversible addition–fragmentation chain transfer (PET-RAFT) polymerization (build) (iii); and mixing of synthesized copolymers with enzyme to form PPHs that are thermally stressed and assessed for REA (test) (iv). The newly acquired and existing data is then used to begin a new Learn–Design–Build–Test iteration.
2. Overview of Design Space and Strategy
2.1. Design Space and Initial Screen
To test our ML-based design paradigm, we consider three chemically distinct enzymes–HRP, GOx, and Lip–with the design goal to maximize retained enzyme activity (REA) following thermal stressing. For reference, a PPH exhibiting 100% REA provides the same level of activity as the enzyme prior to thermal stressing. Because these enzymes possess distinct surface chemistries and molecular weights (Figure 1a), we consider a copolymer design space with eight possible monomers (Figure 1b) copolymerized with target degree of polymerization (DP) between 50 and 200 in increments of 25. The chosen monomers are classified as hydrophobic (2-diethylamino ethyl methacrylate (DEAMA), hydroxypropyl methacrylate (HPMA), butyl methacrylate (BMA), methyl methacrylate (MMA)), hydrophilic (N-(3-(dimethylamino)propyl) methacrylamide (DMAPMA), poly(ethyleneglycol) (n) monomethyl ether monomethacrylate (PEGMA)), or ionic (3-sulfopropyl methacrylate potassium (SPMA), (2-(methacryloyloxy)ethyl) trimethylammonium chloride (TMAEMA)); this set enables various interactions (e.g., van der Waals, hydrogen-bonding, electrostatic) with the enzyme, while balancing aqueous solubility. To encourage reproducible synthesis and minimize latency, up to four distinct monomers are selected for copolymerization for any given copolymer design. These choices (i.e., fractions of incorporation of up to four monomers and the degree of polymerization) result in a design space of ≈545 622 synthetically unique copolymers.
Before evaluating an iterative Learn–Design–Build–Test approach, we sought to gain perspective on the viability of a systematic search, relying on high-throughput experimentation and polymer chemist intuition. We first performed an initial screen with synthetic limits on certain monomers to ensure copolymer solubility and conversion. In particular, hydrophobic monomer content was limited to ≤ 70% mol fraction and ionic monomer content was limited to ≤ 50%. Additionally, in this screen, no copolymers were allowed to include both ionic monomers. Then, systematic composition-based perturbations were made to design copolymers with unique combinations of hydrophilic, hydrophobic, and ionic properties at three degrees of polymerization (50, 100, 200). This resulted in 504 unique copolymers; the systematic nature can be readily identified by principal component analysis (Figures S1 and S2, Supporting Information). All copolymers constituting this seed dataset were tested with each of the three enzymes using enzyme-specific stability assays. To minimize wasted resources, the data obtained from the systematic screens are used to seed an active learning guided search.
2.2. Learn–Design–Build–Test Cycle
We iterate with a Learn–Design–Build–Test cycle (Figure 1) to identify high-performing PPHs. Each iteration consists of four steps: i) developing ML models to predict REA from copolymer characteristics; ii) identifying batches of 24 candidate copolymers for PPHs using active and unsupervised ML; iii) synthesizing candidate copolymers; and iv) performing thermal activity assays to determine REA for candidate PPHs. The results from step (iv) augment the dataset for a given enzyme before beginning the next iteration.
Our discovery process invoked five total iterations based on experimental resources and demonstrated feasibility of enhancements to REA. As such, copolymers proposed in step (ii) during the first four iterations are generated to simultaneously explore and exploit knowledge of the chemical space. In the final iteration, dubbed “exploit round” or iteration 5, we simply aim to maximize the REA of copolymers generated, subject to the constraint that they are unique (to within synthetic confidence) compared to other candidates. While our stopping criterion is principally exhaustion of a fixed budget for optimization, other reasonable criteria from active learning and optimization may be devised and deployed.[34,35]
Below we further describe other methodological aspects of our Learn–Design–Build–Test cycle:
Learn: To cheaply assess the prospective stability of new PPHs, we trained Gaussian process regression (GPR) models to make surrogate predictions of REA directly from representations of the copolymer chemistry[36] (see Section 5). These models provided instantaneous estimates of the REA for any given PPH based on data collected to that point.
Design: The GPR models were combined with Bayesian optimization (BO) in an active learning paradigm to identify candidate copolymers according to prescribed objectives. In each of the first four iterations, 200 initial copolymers were produced by maximizing a data-acquisition utility function that biased optimal designs to favor designs across the explore–exploit spectrum (see Section 5). Similar acquisition functions have been used in previous work related to polymer design.[37,38] To preserve the diversity of candidates and match experimental capabilities to minimize latency, unsupervised ML clustering algorithms were used to identify and select 24 distinct copolymer candidates (see Section 5) during iterations 1–4.
Build: Proposed copolymers from the Design step were synthesized by automated photoinduced electron/energy transfer reversible addition–fragmentation chain transfer (PET-RAFT) polymerization in 96 well plates as previously described.[31,32,39,40] Briefly, synthetic information regarding copolymer designs is converted to synthesis procedures, which are undertaken by a Hamilton MLSTARlet liquid-handling robot, enabling highly parallelized preparation (see Section 5).
Test: Once copolymerizations are complete, copolymers undergo a dilution series into DMSO and then an enzyme-specific assay buffer. Following this dilution, PPHs are formed through mixing copolymers with each enzyme (see Section 5). After PPH formation, REA is determined for each proposed PPH by measuring REA following enzyme-specific thermal stress assays, providing new data for the next iteration.
3. Results and Discussion
3.1. Inefficiency of Screening
The vast majority of copolymers in the seed dataset did not result in substantial REA, with the mean values of 15.7% ± 21.3% (HRP), 12.9% ± 10.3% (GOx), and 2.1% ± 7.6% (Lip). These poor results are partly explained by the limited chemical space surveyed during systematic screening (Figures S1 and S2, Supporting Information); copolymers in the seed dataset account for only ≈0.1% of the total design space. Additionally, the REA for PPHs with Lip, HRP, and GOx vary significantly for any given copolymer design in the seed dataset, suggesting that copolymers should be tuned to specific enzymes and that systematic screening is likely to have mixed success across different enzymes.
3.2. Active Learning in a Combinatorial Design Space
Figure 2a–c shows that active learning facilitated identification of numerous, diverse copolymers that enhanced retained activity for each of the three enzymes. The median REA of PPHs found in the intermediate and final iterations of active learning show progressive and significant increase over those in the seed dataset. In particular, there is a difference of 46.2%, 31.5%, and 87.6% between the median REA of seed PPHs and those found in the exploit round for HRP, GOx, and Lip, respectively. Even within the intermediate iterations (1–4), we typically find improvements in median REA iteration-over-iteration (Figure S4, Supporting Information), despite data acquisition sometimes foregoing potentially promising designs in favor of diversity or uncertainty. For Lip and GOx, the best PPHs are found within the exploit round and exhibit remarkable REA values of 107.9% and 67.4%, which significantly improve upon both the average and maximum values observed in the seed datasets. For HRP, the top-performing PPH is found during the initial screen with a measured REA of 93.1%; however, many of the top hybrids are still identified by active learning including one with an REA of 81.0%. More generally, we find that a large number of diverse copolymers offer reasonable stabilization of HRP, and active learning identifies some promising regions of the chemical space that are not exposed by our systematic search. Quantitatively, copolymers discovered using active learning are disproportionately represented as top performers, comprising 70.2%, 40.5%, and 42.5% of the top twentieth percentile of PPHs sorted by REA for Lip, GOx, and HRP, respectively. Interestingly, the exploit round also produces three PPHs for Lip that not only preserve but enhance its activity relative to the unstressed enzyme.
Figure 2.
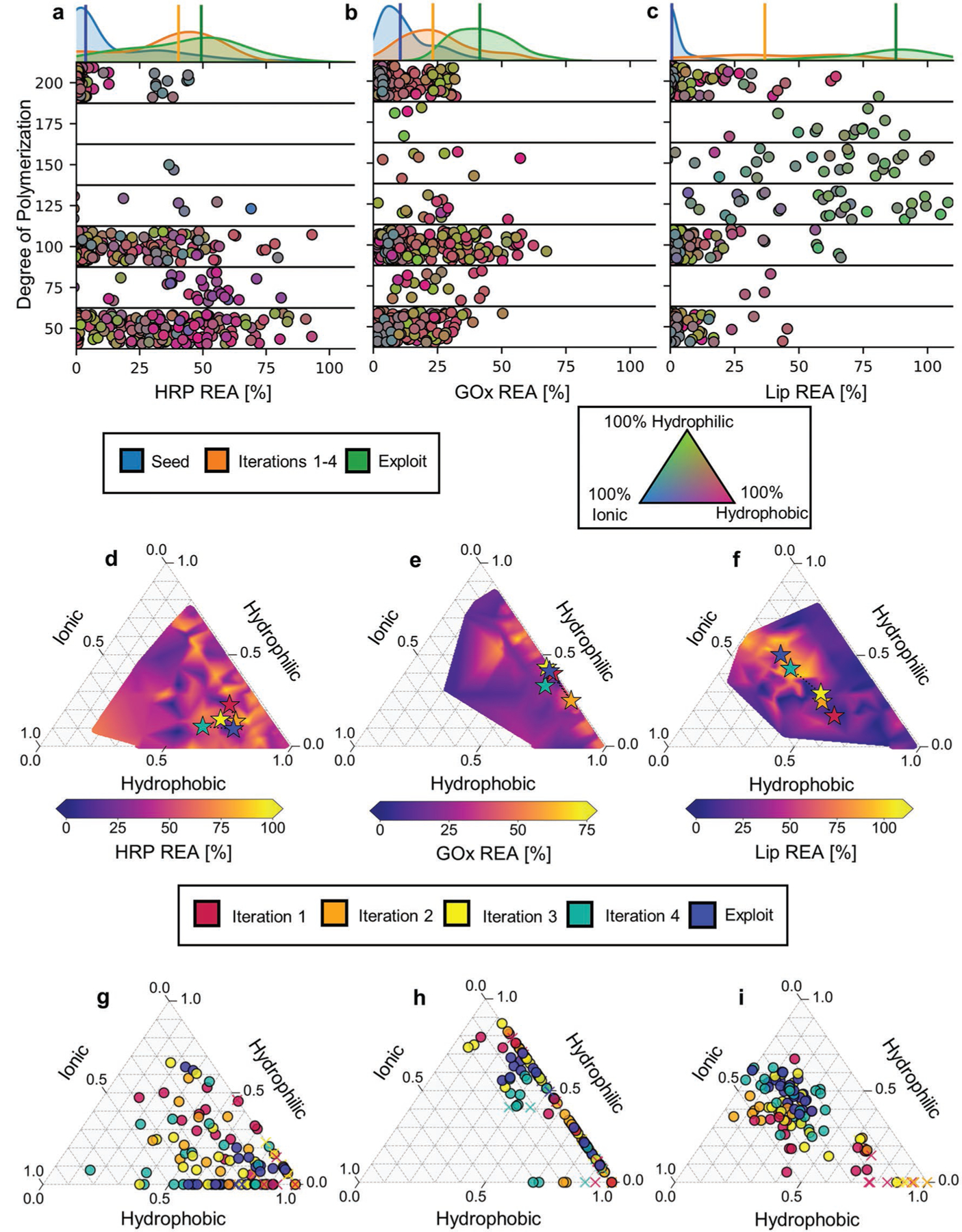
ML guides design of highly stable polymer–protein hybrids. a–c) Copolymer designs and their measured REAs for HRP, GOx, and Lip. Marginal axes at the top contain Gaussian kernel density estimate distributions of REA in the seed dataset (blue), Learn–Design–Build–Test iterations 1–4 (orange), and the final exploitation round (green). Medians of distributions are indicated by vertical lines. Main axes show the experimentally measured REA for all tested PPHs; individual markers are vertically located in bins according to their degree of polymerization with jitter added within bins to improve visual clarity. The marker color reflects the composition of the copolymer according to the ternary diagram (bottom right). d–f) Representation of active learning path traversed through copolymer chemical space for each enzymes. The chemical space is represented as a ternary diagram with coordinates providing the fraction of incorporation of hydrophobic, hydrophilic, and ionic monomers in copolymers. Colored stars indicate the mean composition of copolymers proposed during a given iteration. The ternary diagrams are additionally colored by maximum REA observed for a PPH in a given region of the chemical space spanned by the ternary axes. g–i) Individual chemical compositions of copolymers proposed during each stage of active learning. The centroid of all points at a given iteration yields the position of the stars (d–f). The crosses denote copolymers that showed undesirable gelation during synthesis (see Section 5).
Figure 2d–i examines both the progression of active learning and PPH performance as a function of the chemical constitution of copolymers. Based on the totality of measured REA values, we find that best-performing PPHs for each enzyme utilize entirely different copolymer chemistries, which justifies a tailored design strategy. In particular, optimal copolymers for HRP stabilization predominantly feature hydrophobic and ionic monomers and smaller DP (<100) (Figure 2a,d). While active-learning-generated candidates primarily focus on uncovering this region of the chemical space, there are also many effective PPHs that limit ionic content as identified by the seed dataset (Figure 2g and Figure S2c, Supporting Information). In this case, a wide range of diverse, high-performing PPHs are identified by active learning, despite outlier points in the HRP dataset (Table S1, Supporting Information). For GOx, optimal copolymers are either predominantly hydrophobic or hydrophilic with very little ionicity and have DP typically in the range of 100–150 (Figure 2b,e). Accordingly, active learning for GOx stabilization predominantly probed these regions of the chemical space and remained globally stagnant in its search (Figure 2e,h), fine-tuning relatively promising regions identified in the seed dataset (Figure S2a, Supporting Information). Conversely, optimal copolymers for Lip stabilization possess sizable incorporations of monomers from all three chemical groupings with generally larger DP (Figure 2c,f). Active learning-proposed candidates progress toward this promising region of the chemical space with each subsequent iteration (Figure 2f,i). Notably, this region of the chemical space is completely avoided in the seed dataset (Figure S2b, Supporting Information), suggesting that the Lip design campaign benefited from both exploration- and exploitation-based candidate proposals. Therefore, the active learning paradigm appropriately adapted optimization to identify high-performing PPHs for each enzyme across chemical space, accounting for less than 20% additional data beyond the initial systematic screen and ≈0.02% of the total design space.
3.3. Understanding Chemical Features Driving PPH Performance
Given the identification of highly stable PPHs for each enzyme, we sought to understand the specific chemical features of copolymers that gave rise to their performance. Figure 3a compares the features of copolymers underlying PPHs with the top ten highest REA for each enzyme. While top-performing PPHs for a given enzyme tend to have some chemical similarity across effective copolymers, there is substantial chemical diversity between PPHs for different enzymes. This suggests that copolymer pairing with HRP, GOx, and Lip may be highly enzyme-specific. To investigate this, we cross-evaluated the efficacy of the top-performing copolymers discovered for each enzyme to retain the activity of all three enzymes in our study. For example, the top ten copolymers identified as highly effective for stabilizing HRP were additionally formulated into GOx-PPHs and Lip-PPHs. Then, respective GOx and Lip stability assays (see Section 5) were performed to determine the efficacy of top performing HRP copolymers in stabilizing GOx and Lip, for which the copolymers were not designed. We then repeated this process for the top ten performing GOx and Lip copolymers to observe all combinations of top-performing copolymers with each enzyme in this study. Experimentally, we observe that the REA of PPHs designed for a specific enzyme are significantly higher than that of PPHs formed by non-specific copolymers (Figure 3b). Further, virtual cross-evaluation using enzyme-specific GPR models trained on all iterations of data similarly suggest that REA is significantly diminished when top-performing copolymers for one enzyme are paired with another. Together, these results not only suggest an intricate connection between copolymer chemistry and size with the stability of PPHs, but such correlations can be effectively learned from data.
Figure 3.
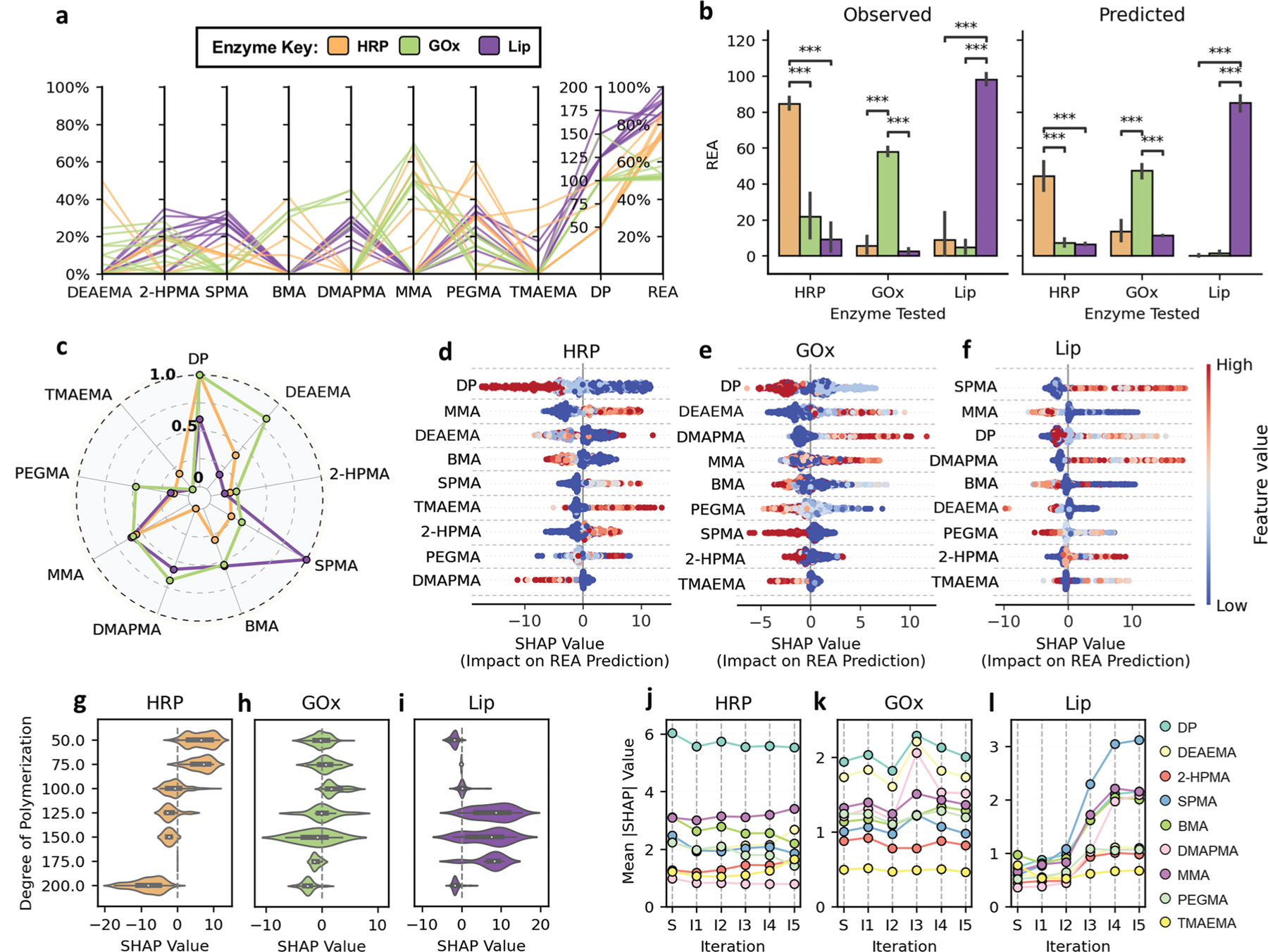
Analysis reveals distinct priorities in copolymer features for each protein. a) Copolymer compositions and degree of polymerization (DP) for the top ten performing PPHs for HRP (orange), GOx (green), and Lip (purple). b) Cross-evaluation of top-performing copolymers across enzymes showing mean observed and predicted REA for each copolymer–enzyme pairing. Statistical significance was determined by Mann–Whitney U test. *(p < 0.05), **(p < 0.005), ***(p < 0.0005), unlabeled pairs are not significantly different. Top ten performers for each enzyme demonstrate high specificity in agreement with predicted activity. c) Normalized mean absolute Shapley additive explanations (SHAP) values calculated for HRP, GOx, and Lip for each model to quantify relative feature importance. d–f) Summary of SHAP values for GPR models calculated from available data after all five Learn–Design–Build–Test iterations. Each point corresponds to a uniquely evaluated PPH, and the point’s position along the X-axis shows the impact of a feature on predicted REA. g–i) SHAP value distributions demonstrating the effect of degree of polymerization on REA predictions. Black candlesticks range from second to third quartiles of SHAP values and white dots represent the distribution mean. j–l) Mean absolute SHAP values calculated for all model features after model training on the seed dataset and after each iteration of active learning.
To further explore the relationship between copolymer features and PPH activity, we computed Shapley additive explanations (SHAP) values[41,42] to quantify how chemical features of the copolymers (fractions of incorporation and DP) contributes to REA predictions by our GPR models. Here, positive SHAP values indicate positive contributions REA (negative SHAP values suggest negative contributions), and we use the mean absolute SHAP value of a feature as a proxy for its overall importance to model prediction. Figure 3c shows that different copolymer features have distinct impact on REA predictions. To elucidate these differences, we compare SHAP values for the fractions of incorporation for each monomer (Figure 3d–f) and DP (Figure 3g–i) for each enzyme. Although we previously associated hydrophobic chemistry with high-performing PPHs for HRP (Figure 2f,i), Figure 3d reveals that the exclusion of BMA is favorable (higher REA), while the inclusion of MMA, a similar hydrophobic monomer, is associated with higher REA. Similar observations can be readily identified for Lip (Figure 3f), for which SPMA and TMAEMA monomers (both highly ionic) represent the most and least important features based on their mean absolute SHAP values. Such differences in SHAP values between monomers with the same chemical classifications underscore the intricacy of designing effective polymer–enzyme pairing.
Figure 3c–i also indicates that the relative importance of copolymer features varies across enzyme models. For example, we find that different chain length regimes favor high predictions on REA, depending on the enzyme-specific GPR model (Figure 3g–i). For HRP, smaller copolymers (DP = 50, 75) display the highest SHAP values, while the highest SHAP values for Lip are observed for DP = 125 or 150. DP = 200 is generally associated with lower REA, perhaps suggesting that shorter copolymer sequences enable more facile pairing with enzyme chemical domains to promote stabilization.
To understand the evolution of feature importance during discovery, we compared mean absolute SHAP values for all non-gelling copolymers derived from GPR models trained after each stage of data acquisition. Figure 3j–l shows that the importance of features can shift significantly, even with the addition of small amounts of data (typically 20 data points added per iteration or less than 4% increase in prior data available). This is most evident following for Lip, wherein mean absolute SHAP values for SPMA, MMA, DMAPMA, and DP all substantially increase after the third and fourth iteration. This behavior might be related to data acquisition over previously unexplored regions of chemical space, which is partly shown in Figure 2e. The effects for HRP and GOx are overall less dramatic; most rankings are unchanged between iterations, with occasional shifts of one or two ranks upon exposure to new data. Nonetheless, even if the rank-ordering of features is unchanged, mean improvement in measured REA for PPHs across iterations suggests that GPR models had sufficient fidelity to effectively optimize REA, at least within a local chemical space.
3.4. Revealing Mechanisms with Biophysical Characterization
Although mechanisms of stabilization for PPHs based on random copolymers have been hypothesized and studied in limited fashion using molecular dynamics simulation,[6] experimental examination of these biophysical interactions is nascent. Therefore, we characterized (Figure S5, Supporting Information) and investigated a particular PPH for HRP identified in the exploit round—dubbed HRP-Exploit Polymer 1 (HRP-EP1)—using circular dichroism (CD) spectroscopy, small-angle X-ray scattering (SAXS), dynamic light scattering (DLS), and quartz crystal microbalance with dissipation (QCM-D). HRP was selected due to its amenability to these characterization techniques, while detailed characterization of other enzyme systems proved challenging due to weak CD spectroscopy signal-to-noise and solubility limitations. We first investigated the impact of heating and cooling on the secondary structure of HRP by CD spectroscopy (Figure 4a). The corresponding measured α-helix, β-sheet, and random coil content is provided in Table S2, Supporting Information. We initially hypothesized that the addition of copolymer EP1 would reduce thermally induced unfolding of HRP; however, the CD data suggests only a slight retardation of unfolding. Upon heating, the α-helix content for HRP degrades from ≈34.8% to 17.4%, while the α-helix content for the HRP-EP1 system is 20.3% after heating. However, following cooling, HRP-EP1 exhibited 31.6% α-helix content compared to just 24.6% for HRP alone. This suggests that EP1 facilitates significant refolding of HRP in a chaperone-like manner.
Figure 4.
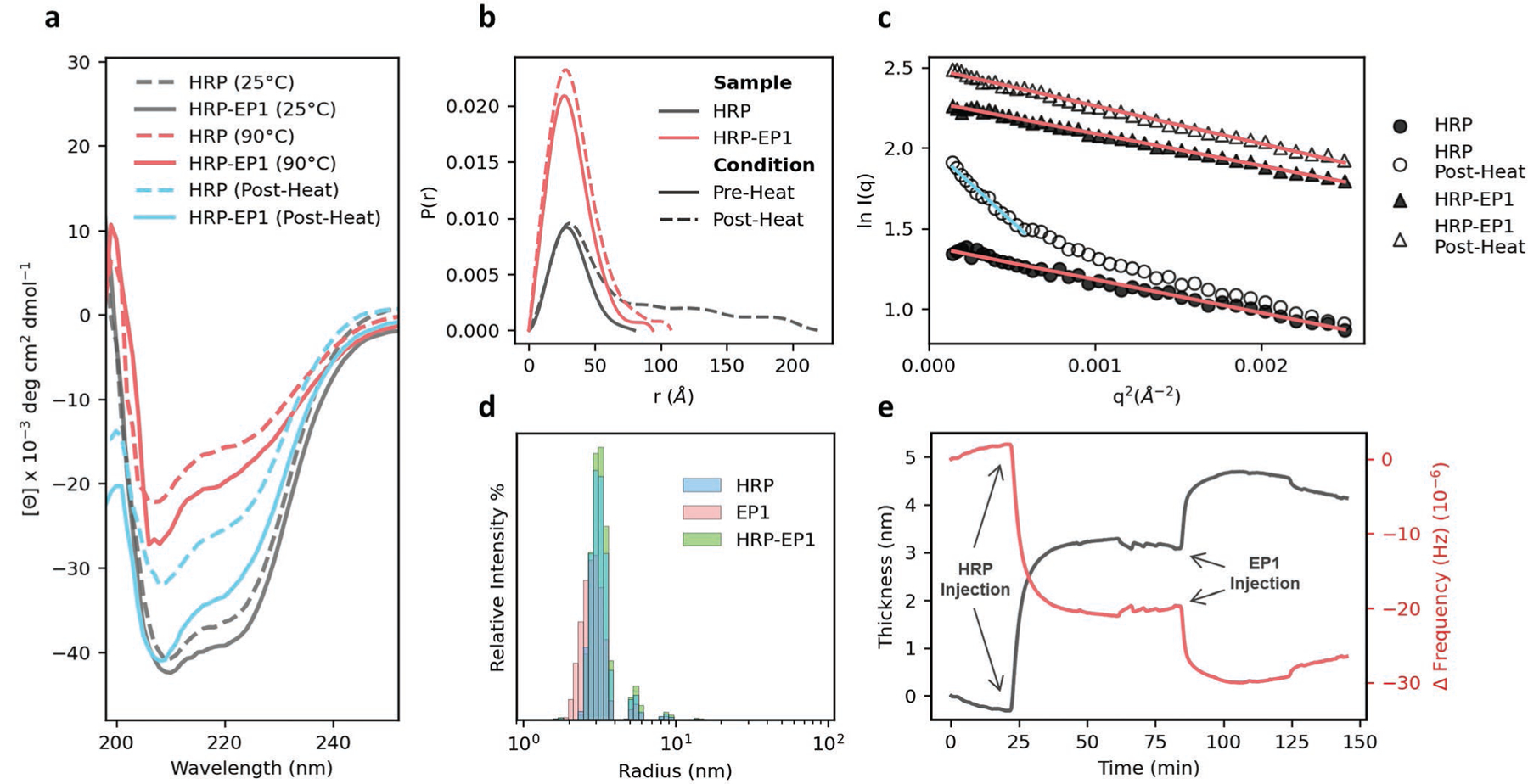
Biophysical characterization indicates copolymer-assisted refolding. a) Circular dichroism wavelength scans of HRP (dashed lines) and HRP-EP1 (solid lines) at room temperature (black), upon heating (red), and after cooling for 24hrs (blue), demonstrating that HRP-EP1 promotes retention of secondary structure in HRP during thermal stress and promotes significant protein refolding in comparison to HRP control. b) Pair-distance distribution function of HRP and HRP-EP1 by small-angle X-ray scattering demonstrating retained HRP-PPH morphology and size after exposure to thermal stress in comparison to native enzyme. c) Guinier analysis of HRP and HRP-EP1 before and after heating suggesting the development of a denatured or aggregated sub-population of HRP (blue line) in comparison to a single species observed in HRP, HRP-EP1, and HRP-EP1 after thermal stress (red lines). d) Dynamic light scattering size distributions of HRP with and without polymer EP1, demonstrating that no larger structures were observed after mixing. e) Surface thickness measured by Quartz crystal microbalance with dissipation after direct adsorption of HRP (t = 22 min) followed by injection of polymer EP1 (t = 82 min).
To further understand the nature of the HRP-EP1 interactions, we used SAXS to compare the physical dimensions of HRP and its complexes in pre- and post-stress states. Guinier analysis of the data (Table S3, Figure S6, Supporting Information) showed that both HRP and HRP-EP1 have the same radius of gyration (Rg, 24.6–25.0 Å) in the pre-stressed state. Similarly, in the pre-stressed state, the pair-distance distribution function P(r) remains highly similar upon complexation of HRP with EP1 (Figure 4b). Post-stress, the differences are dramatic in the pair-distance distribution function. While the maximum particle diameter (Dmax) of native HRP increases from 80 to 200 Å, that of HRP-EP1 increased only to 94 Å (Table S3, Supporting Information). Additionally, while the Rg of HRP-EP1 increases only slightly to 26.9 Å, a larger 51.9 Å component appears in the Guinier plots of HRP (Figure 4c, blue line), likely indicative of a denatured or aggregated sub-species of HRP created through thermal stress. Additionally, Kratky plots (Figure S7, Supporting Information) show peaks at q = 0.065 and 0.075 Å−1 in HRP and HRP-EP1, respectively, which indicates a compact structure similar to that of the native protein. This clearly suggests that the complex promotes a certain level of conformational integrity in HRP even if secondary structure is impacted.
Finally, DLS was performed to complement the SAXS results by providing the distribution of hydrodynamic radii (Rh) in the samples (Figure 4d). All samples show peak intensities between 3.0–3.3 nm with minimal signal intensity for Rh > 10 nm. Additionally, measured polydispersity index remained under 0.2 for all samples, suggesting relatively monodisperse solutions (Figure S8, Table S4, Supporting Information). These results indicate that stabilization of HRP in PPH-EP1 is indeed driven by the formation of a complex rather than via larger macromolecular assembly. Further support of complex formation by QCM-D showed significant differences in the Sauerbrey mass thickness following injection of EP1 onto surface immobilized HRP (Figure 4e and Figure S9, Supporting Information). While native HRP exhibited a thickness of 3.6 nm, HRP-EP1 increased to 5.1 nm post injection at 80 min.
4. Conclusion
Polymer–protein hybrids offer a powerful approach to stabilize sensitive proteins in a range of environments. Here, we developed a robust design framework integrating automated polymer chemistry and ML to efficiently discover polymer–protein hybrids with enhanced thermostability for three chemically distinct enzymes. Notably, the ML-guided acquisition of data was effectively tailored to each enzyme. In addition, by analysis of developed surrogate ML models, we determined particular chemical features of copolymers that drive increased retained activity for each enzyme. Furthermore, the biophysical characterization of a successful polymer–protein hybrid design reveals chaperone-like assistance in structural refolding as a possible mechanism of stabilization. Taken together, these results highlight the existence of a complex structure–function relationship underlying protein-polymer hybrid activity that can be learned and exploited for materials optimization.
This discovery platform for polymer–protein hybrids can be extended in numerous directions. First, it provides an exemplary approach that can be extended to other proteins, other copolymer chemistries, and/or alternative design objectives, such as other environmental stresses. Furthermore, enabled by the vast and flexible chemical space spanned by the copolymer chemistries, the platform can be expanded accommodate the simultaneous pursuit of multiple design objectives. Advancement in this area could significantly accelerate their use as functional, commercial materials in myriad applications. One intriguing possibility is also to generalize the surrogate models to incorporate chemical features of both proteins and their encapsulating copolymers. This would not only be a step toward constructing more physically informed surrogate models, but it would also open the door to using protein features as additional degrees of freedom for design. In a similar vein, the assay data collected in this study can be used in conjunction with simulation-based models to further elucidate and validate molecular-level mechanisms for stability. Such simulations might also aid in identifying and selecting key features for surrogate models or even provide in silico figures of merit that correlate with stability. Last, while our ML workflow appeared generally insensitive to the biased nature of the seed dataset, it is possible high-performing PPHs could have been discovered starting from a smaller, more targeted selection of experiments. Insights in this area could help reduce resources required for high-throughput materials discovery efforts.
5. Experimental Section
Materials:
Hydroxypropyl methacrylate (HPMA), 2-diethylamino ethyl methacrylate (DEAEMA), (2-(methacryloyloxy)ethyl) trimethylammonium chloride solution (TMAEMA), and N-(3-(dimethylamino)propyl) methacrylamide (DMAPMA) were purchased from Sigma-Aldrich; methyl methacrylate (MMA) and 3-sulfopropyl methacrylate potassium salt (SPMA) from VWR; butyl methacrylate (BMA) from Alfa Aesar; and poly(ethylene glycol) (n) monomethyl ether monomethacrylate (PEGMA, Mn ≈ 400 g mol−1) from Polysciences. PEGMA was deinhibited prior to use by passing over mono-methyl ether hydroxyquinone inhibitor removal resin. Ethyl 2-(phenylcarbonothioylthio)-2-phenylacetate, 4-nitrophenyl butyrate (PNB), hydrogen peroxide (H2O2), D-(+)-glucose, sodium acetate, lithium bromide were purchased from Sigma-Aldrich; zinc tetraphenyl porphyrin (ZnTPP), dimethyl sulfoxide (DMSO), 3,3ꞌ,5,5ꞌ-tetramethylbenzidine (TMB) from Fisher Scientific; and potassium phosphate (mono and dibasic) and sodium acetate anhydrous from VWR.
Automated PET-RAFT Synthesis:
Copolymers were prepared by automated photoinduced electron/energy transfer reversible addition–fragmentation chain transfer (PET-RAFT) polymerization in 96 well plates as previously described.[31,32,39,40] Briefly, the sequences and processes to be conducted by the Hamilton MLSTARlet liquid-handling robot were programmed in Python, indicating information on sample concentration, reagent volumes, and well position. Files containing reaction information were transferred to the Hamilton MLSTARlet to prime the robotic transfers. Stock solutions of monomer (2 m), ethyl 2-(phenylcarbonothioylthio)-2-phenylacetate (RAFT chain-transfer agent (CTA), 100 or 50 × 10−3 m) and ZnTPP (photocatalyst, 4 or 2 × 10−3 m) were prepared in DMSO as 1 mL aliquots. Aliquots were loaded into the Hamilton MLSTARlet liquid-handling robot and automatically pipetted into 96-wells clear flat-bottom well plates (Greiner Bio-One). Monomer/CTA ratio was varied from 50–200 to control degree of polymerization while ZnTPP/CTA remained at 0.01. Polymer mixtures were dispensed to a total volume of 200 µL and final monomer concentration of 1 m. The mixtures were then covered with well-plate sealing tape and radiated under 560 nm LED light (5 mW cm−2, TCP 12 Watt Yellow LED BR30 bulb) for 16 h.
HRP Thermal Stability Assay:
The activities of PPHs for HRP were evaluated by its ability to oxidize TMB in the presence of H2O2. Copolymers were synthesized and diluted in DMSO before further dilution into assay buffer (50 × 10−3 m sodium acetate, pH 5.0) to a final concentration of 22.7 × 10−6 m (<1% DMSO). From the 22.7 × 10−6 m polymer samples, 50 µL were mixed with 50 µL of 10 µg mL−1 HRP (0.11 × 10−6 m) in polystyrene 96 well plates. The solutions were thermally sealed with plate-sealing film and then thermally challenged in a water bath at 60 ºC for 30 min. This temperature was chosen as it reliably diminishes all HRP activity and is above HRP’s reported melting temperature of 55 ºC.[43] Substrate solution was prepared by diluting 40 × 10−3 m of TMB in DMSO to a final concentration of 0.4 × 10−3 m in 1% H2O2 assay buffer. 5 µL of polymer–enzyme mixtures were added to 245 µL of substrate solution. Absorbance was measured in kinetic mode for 5 min in 20 s intervals; measurements were made at 653 nm, which is the maximum of the absorption peak. The initial rate of change of absorbance (ΔOD) was used to calculate the activity of HRP. Native HRP activity without heating served as a positive control (PC), while HRP heated at 60 ºC for 30 min served as the negative control (NC). REA was calculated for each PPH by the following equation
| (1) |
GOx Thermal Stability Assay:
The activities of PPHs for GOx were evaluated using an assay buffer containing glucose, TMB, and HRP. Copolymers were diluted in DMSO and then in assay buffer (50 × 10−3 m sodium acetate, pH 5.0) to a final concentration of 12 × 10−6 m (<1% DMSO). Resulting solutions were mixed with equal volumes of stock GOx solution (5 µg mL−1, 30 × 10−9 m) in polystyrene 96 well plates. The solutions were thermally sealed with plate-sealing film and then thermally challenged in a water bath at 65 ºC for 30 min. This temperature was chosen as it reliably diminishes all GOx activity and is above GOx’s reported melting temperature of 60 ºC.[44] After heating, 20 µL of the PPH samples were added to 100 µL of substrate solution (5% glucose, 0.4 × 10−3 m TMB, 0.11 × 10−6 m HRP in assay buffer). Absorbance was measured in kinetic mode for 5 min in 20 s intervals; measurements were made at 653 nm, which is the maximum of the absorption peak. The initial rate of change of absorbance (ΔOD) was used to calculate the enzyme activity. Native GOx activity without heating served as a positive control (PC), while GOx heated at 65 ºC for 30 min served as the negative control (NC). REA for all GOx-PPHs was calculated as previously described.
Lip Thermal Stability Assay:
Activities of PPHs for Lip were evaluated using PNB as the substrate. Copolymers were diluted in DMSO and then in assay buffer (50 × 10−3 m K2HPO4, 16.66 × 10−3 m K2HPO4, pH 7.4) to a final concentration of 120 × 10−6 m (<1.5% DMSO). From the 120 × 10−6 m copolymer solutions, 50 µL were mixed with 50 µL of stock lipase solution (0.8 mg mL−1 24 × 10−6 m) in polystyrene 96 well plates. The solutions were thermally sealed with plate-sealing film and heated in a water bath at 70 ºC for 1 h. This temperature was chosen as it reliably diminishes all Lip activity and is above Lip’s reported melting temperature of 60 ºC.[45] Substrate solution was prepared by diluting stock PNB solution (5.4 m) first to 10 × 10−3 m in DMSO, followed by a final dilution to 0.5 × 10−3 m in assay buffer. Absorbance was measured in kinetic mode for 10 min in 20 s intervals; measurements were made at 410 nm to monitor the production of p-nitrophenol. The initial rate of change of absorbance (ΔOD) was used to calculate the enzyme activity. Native Lip activity without heating served as a positive control (PC), while Lip heated at 70 ºC for 1 h served as the negative control (NC). REA for all Lip-PPHs were calculated as previously described.
Circular Dichroism Spectroscopy:
CD wavelength and temperature scans of samples were collected using an AVIV Model 400 CD spectrometer (AVIV Biomedical Inc.). Wavelength scans consisted of measurements from 260 to 190 nm, collecting points every 0.5 nm with a 1 nm bandwidth for 5 s, at all required temperatures. Temperature scans were consisted of measuring mean residue ellipticity at 222 nm from 30 to 90 ºC with a 5 s averaging time and 1.5 nm bandwidth. The ramp rate was 2 ºC min−1, and samples were equilibrated for 5 min at each temperature before measurement. The fraction of protein unfolding at different temperatures were calculated by assuming fully folded state at 30 ºC and fully unfolded state at 90 ºC. The melting temperature Tm was determined by fitting the temperature scans to a Boltzmann sigmoidal equation. The fractions of α-helices and β-sheets in the protein samples were calculated using CD deconvolution algorithms for wavelength scans (Table S2, Supporting Information).
Dynamic Light Scattering:
DLS of copolymers and polymer–enzyme mixtures were performed on a DynaPro DLS Plate Reader III, Wyatt Technologies. Concentration of HRP for DLS experiments was maintained at 0.2 mg mL−1 while polymer concentration was at 1 mg mL−1. The data was collected using a wavelength of 830 nm and a scattering angle of 173º. Fifteen acquisitions were collected for each sample with an acquisition time of 5 s per acquisition using auto attenuation. Regularization analysis was performed using Rayleigh spheres model for hydrodynamic size measurement.
Small-Angle X-ray Scattering:
All scattering experiments were carried out at the Life Science X-ray Scattering (LiX) beamline 16-ID of the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Laboratory (Upton, NY, USA). HRP was prepared at a final concentration of 1 mg mL−1 in 50 × 10−3 m sodium acetate (pH 5.15) while lyophilized copolymers were reconstituted in sodium acetate buffer and mixed with HRP at a final concentration of 2.61 mg mL−1 (10:1 molar concentration of polymer:HRP). Samples were denatured by heating in a water bath at 65 ºC for 1 h. All solutions were loaded into 96-well PCR plates and mailed in for data collection. An X-ray energy of 15.14 keV was utilized for solution SAXS. Three Pilatus detectors were employed to provide a q range of 0.005–3.13 Å, while the range 0.005–0.25 Å was taken as the small-angle region. For background subtraction, sodium acetate buffer blanks were run for every three samples. The subtracted data were analyzed in BioXTAS RAW 2.1 with ATSAS 3.0.4–6. Guinier analysis was performed to quantify the radius of gyration Rg, whereas pair-distance distribution analysis by an indirect Fourier transform method was conducted to quantitatively assess Rg, maximum dimension, and macromolecular structure.[46–48]
Quartz Crystal Microbalance with Dissipation:
All quartz crystal microbalance experiments were carried out on the Q-Sense Omega Auto (Biolin Scientific) with 5 MHz sensitivity, less than 1 nm surface roughness, and theoretical mass sensitivity of 17.7 ng cm−2 Hz−1. HRP was dissolved in 50 × 10−3 m sodium acetate buffer (pH 5.15) at 0.2 mg mL−1 whereas the final concentration of lyophilized copolymers was set to 0.52 mg mL−1 (10:1 molar concentration of polymer:HRP). Sodium acetate buffer was flowed as an initial equilibration step at 20 µL min−1 for 25 min. HRP, polymer, and mixtures of HRP with polymer were flowed at 40 µL min−1 for 10 min. Sodium acetate was flowed after each step at 20 µL min−1 for 25 min to remove any loosely associated enzyme or polymer. Transformations using the Sauerbery equation[49,50] were completed on the fifth harmonic frequency and dissipation responses to obtain surface thickness.
Polymer Characterization:
The molecular weights (Mw and Mn) and dispersity (Đ) were measured by gel-permeation chromatography using an Agilent 1260 Infinity II. Polymer samples were eluted through a Phenomenex 5.0 µm guard column (50 × 7.5 mm) preceded by superose Phenogel 12 10/300 GL column (Cytiva 17–5173-01, column L × I.D. 30 cm × 10 mm, 11 µm avg. part. size) in 0.5× PBS (0.2% N3) using a flow rate of 0.5 mL min−1. GPC calibration was completed with Agilent PEG standards. Copolymers were prepared at 50:1 eluent/polymer ratio in 0.5× PBS (0.2% NaN3) and filtered with a 0.45 µm nylon filter. Polymer conversion was calculated by obtaining 1H NMR spectra using a Varian VNMRS 500 MHz spectrometer with mesitylene as an internal standard and processed using Mestrenova 11.0.4.
Machine-Learning Surrogate Models:
All copolymers were featurized as DP-explicit composition vectors with one-hot encoding vectors used as fingerprints for monomer units.[36] With eight possible monomers, the resulting feature vector possesses nine dimensions, with the first containing the DP of the copolymer divided by 200 and the remaining eight containing the fractions of incorporation for each monomer; the division in the first dimension represents DP on a similar scale as the remaining features. Gaussian process regression (GPR) models, trained to predict the Yeo–Johnson transformation[51] of the REA for a PPH, were preferred due to their superior predictive performance compared to other ML algorithms (Figure S3, Supporting Information). The Yeo–Johnson transformation is given by
| (2) |
and is used to transform REA measurements, which resemble random variables sampled from power-law distributions, to values that exhibit draws from a Gaussian distribution. The exponential parameter λ was found using maximum likelihood estimation, as implemented by python package scikit-learn. Use of this transformation was empirically found to improve predictive performance of models. In addition, preliminary comparisons amongst GPR models trained over the seed datasets revealed no evident advantage to using more advanced fingerprinting strategies over simple one-hot encoding (Figure S3, Supporting Information). Using available experimental data of various PPHs, enzyme-specific datasets were constructed wherein each datum is described by this feature vector and labeled by REA.
The relationship between the copolymer features and REA was modeled using GPR to both capture the nontrivial, nonlinear mapping and to facilitate active learning as GPR naturally provides uncertainty estimates on predicted labels. Covariances modeled by the Gaussian Process are calculated using the squared exponential kernel basis function
| (3) |
where is the feature vector of the copolymer, and l, σ, σn are kernel hyperparameters. Anisotropic kernels were explored but did not improve model performance. GPR models for each enzyme were constructed as follows: the dataset was first split into fivefolds. Four of five of the folds were then used to tune the GPR model hyperparameters, which were identified with 20-fold cross-validation and optimization by the Tree-structured Parzen Estimator (TPE) approach[52] to minimize the mean squared error of labels. The optimal hyperparameters, along with data from four of five folds, were used to train a GPR model that made predictions on the remaining fold of data. This process was repeated four more times, such that all five of the original folds served as test sets. The five sets of optimized hyperparameters were then averaged and used to define a final GPR model with the full set of data available for an enzyme at a given iteration. The five sets of held-out test performance metrics were also averaged to quantify and validate the predictive capabilities of the model.
Candidate Copolymer Generation:
Bayesian optimization (BO) was used in tandem with a GPR model to propose promising candidate copolymers. For the first four rounds of active learning, candidates that maximize the expected improvement (EI) acquisition function were selected and given by
| (4) |
| (5) |
where is the predicted mean REA from the GPR, fꞌ is the current largest mean REA observed by the model, is the standard deviation from the GPR, Φ and ϕ are the cumulative and probability density functions of the normal distribution, respectively, and ξ is a hyperparameter that controls the balance between exploring unobserved regions of the chemical space and exploiting known regions of it to obtain high performing copolymers.
To effectively sample copolymer designs across the exploit–explore spectrum, 200 copolymer candidates were sequentially generated for distinct ξ values that logarithmically vary from 0.001 to 30. To avoid proposing previously synthesized copolymers or those within the margin of synthetic experimental error previously synthesized or already proposed copolymers, an additional penalty function was added to the acquisition function based on (see also Supporting Information). In the final iteration or exploit round, copolymers that simply maximize REA predictions from the GPR model were proposed as candidates, although the penalty function was retained to avoid redundant proposals.
Candidate Copolymer Down-Selection:
While copolymer candidate generation is performed by maximizing acquisition functions that uniquely weight the balance between exploration and exploitation, it was found that weightings over a similar range yield similar optima, or designs. Unsupervised clustering methods were used to select 24 diverse candidates for synthesis from the larger set of 200 candidates generated by the BO procedure. In general, this strategy helped to ensure that final candidate proposals were optimal, mutually diverse, and could be synthesized and characterized with minimal latency. Related clustering methods have been deployed to enhance candidate diversity in other polymer design campaigns.[29]
In particular, the following protocol was used for candidate selection in the first four active learning iterations. First, a filter was applied to ensure that no copolymer featured fractions of incorporation of any given monomer that was less than 5%. This filter was imposed to establish reasonable margins of experimental control over the process of dispensing the monomer reagents with the robotic arm used to automatically synthesize the copolymers. Second, candidates were subsequently clustered using density-based spatial clustering of applications with noise (DBSCAN) using a distance threshold of and a minimum of three points per cluster. Following the formation of clusters, the copolymer with the shortest Euclidean distance to the centroid position of the cluster in the copolymer feature vector space was selected as a representative candidate for further consideration. All non-clustered candidates, or noise-points, were also considered in this fashion, the procedure produced a set of relatively diverse and representative copolymer candidates that fairly considers “outliers.” Third, in cases where DBSCAN produced more than 24 candidates (this always occurred), precisely 24 candidates were proposed by application of k-Means clustering. Again, representative candidates were chosen based on proximity to the cluster centroid. If a cluster consisted of only two points, then the candidate with the higher REA was used. A different downsampling procedure was used in the exploit round, since diversity was no longer a priority for selection. Specifically, after producing the 200 polymer designs with BO, candidates were ranked by their REA in descending order and iteratively chosen for the final set of 24 candidates, provided they had compositions that were unique (within synthetic precision) from any copolymers that constituted the growing list at that point.
Handling Polymer Gelation:
Upon construction of the seed dataset and throughout the active learning, a handful of copolymers were found to seemingly undergo gelation. While gelling copolymers recorded nonzero REA values, they were excluded from the dataset used to train the GPR models from iteration 1 onward due to the potential uncontrolled differences in copolymer–enzyme interaction environments that could obfuscate model training. However, the penalty function was used during the active learning procedure to avoid suggesting polymer candidates proximate to gelling copolymers across discovery campaigns across all three enzymes up to that iteration. While this strategy limited the number of gelled copolymers per iteration per enzyme to an average of six copolymers in the first two rounds of active learning, it ultimately proved ineffective for GOx as hydrophobic monomers were found to be effective for GOx stabilization but increased polymer gelation (Figure S11, Supporting Information). To combat this issue, a classifier that leveraged knowledge of prior polymer gelation across all enzymes and iterations up to that point was designed and integrated in the active learning scheme. The use of the classifier was limited to and ultimately facilitated the discovery of primarily soluble copolymers for iterations 4 and 5 of active learning for GOx. Further discussion on the development and integration of the classifier into the active learning scheme is supplied in the Supporting Information (Table S5, Figures S11 and S12, Supporting Information).
Supplementary Material
Acknowledgements
A.J.G. acknowledges support from the National Institutes of Health (NIH) under NIGMS MIRA Award R35GM138296, and the National Science Foundation under DMREF Award NSF-DMR-2118860 and CBET Award Number NSF-ENG-2009942. R.A.P., C.H.B., and M.A.W. acknowledge support from the National Science Foundation under DMREF Award Number NSF-DMR-2118861 as well as start-up funds from Princeton University. M.J.T. acknowledges additional support from the National Institutes of Health (GM135141). The training and optimization with ML models was performed with resources from Princeton Research Computing at Princeton University, which is a consortium led by the Princeton Institute for Computational Science and Engineering (PICSciE) and Office of Information Technology’s Research Computing. A.J.G. and N.S.M. acknowledge James Byrnes, beamline scientist at NSLS-II beamline 16-ID for Life Science X-ray Scattering (LiX), for his assistance with conducting experiments at Brookhaven National Laboratory. The LiX beamline is part of the Center for BioMolecular Structure (CBMS), which is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS) through a P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). LiX also received additional support from NIH Grant S10 OD012331. As part of NSLS-II, a national user facility at Brookhaven National Laboratory, work performed at the CBMS was supported in part by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Program under contract number DE-SC0012704.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
M.J.T. and A.J.G. have filed a PCT patent application and are co-founders of Plexymer, Inc.
Contributor Information
Matthew J. Tamasi, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
Roshan A. Patel, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA
Carlos H. Borca, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA
Shashank Kosuri, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Heloise Mugnier, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Rahul Upadhya, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
N. Sanjeeva Murthy, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
Michael A. Webb, Department of Chemical and Biological Engineering, Princeton University, Princeton, NJ 08544, USA
Adam J. Gormley, Department of Biomedical Engineering, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
Data Availability Statement
The data that support the findings of this study are openly available in DataSpace at https://doi.org/10.34770/h938-nn26, ref. [53]. Trained machine-learning models that support the findings of this study are openly available in Github at https://github.com/webbtheosim/PPH_ public, ref. [54].
References
- [1].Chapman R, Stenzel MH, J. Am. Chem. Soc 2019, 141, 2754. [DOI] [PubMed] [Google Scholar]
- [2].Lancaster L, Abdallah W, Banta S, Wheeldon I, Chem. Soc. Rev 2018, 47, 5177. [DOI] [PubMed] [Google Scholar]
- [3].Pelegri-O’Day EM, Lin E-W, Maynard HD, J. Am. Chem. Soc 2014, 136, 14323. [DOI] [PubMed] [Google Scholar]
- [4].Ko JH, Maynard HD, Chem. Soc. Rev 2018, 47, 8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kosuri S, Borca CH, Mugnier H, Tamasi M, Patel RA, Perez I, Kumar S, Finkel Z, Schloss R, Cai L, Yarmush ML, Webb MA, Gormley AJ, Adv. Healthcare Mater 2022, 11, 2102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Panganiban B, Qiao B, Jiang T, DelRe C, Obadia MM, Nguyen TD, Smith AAA, Hall A, Sit I, Crosby MG, Dennis PB, Drockenmuller E, de la Cruz MO, Xu T, Science 2018, 359, 1239. [DOI] [PubMed] [Google Scholar]
- [7].Gormley AJ, Webb MA, Nat. Rev. Mater 2021, 6, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Satari B, Karimi K, Kumar R, Sustainable Energy Fuels 2019, 3, 11. [Google Scholar]
- [9].DelRe C, Chang B, Jayapurna I, Hall A, Wang A, Zolkin K, Xu T, Adv. Mater 2021, 33, 2105707. [DOI] [PubMed] [Google Scholar]
- [10].DelRe C, Jiang Y, Kang P, Kwon J, Hall A, Jayapurna I, Ruan Z, Ma L, Zolkin K, Li T, Scown CD, Ritchie RO, Russell TP, Xu T, Nature 2021, 592, 558. [DOI] [PubMed] [Google Scholar]
- [11].Wu S, Snajdrova R, Moore JC, Baldenius K, Bornscheuer UT, Angew. Chem., Int. Ed 2021, 60, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ramprasad R, Batra R, Pilania G, Mannodi-Kanakkithodi A, Kim C, npj Comput. Mater 2017, 3, 54. [Google Scholar]
- [13].de Pablo JJ, Jackson NE, Webb MA, Chen L-Q, Moore JE, Morgan D, Jacobs R, Pollock T, Schlom DG, Toberer ES, Analytis J, Dabo I, DeLongchamp DM, Fiete GA, Grason GM, Hautier G, Mo Y, Rajan K, Reed EJ, Rodriguez E, Stevanovic V, Suntivich J, Thornton K, Zhao J-C, npj Comput. Mater 2019, 5, 41. [Google Scholar]
- [14].MacLeod BP, Parlane FG, Morrissey TD, Häse F, Roch LM, Dettelbach KE, Moreira R, Yunker LP, Rooney MB, Deeth JR, Lai V, Ng GJ, Situ H, Zhang RH, Elliott MS, Haley TH, Dvorak DJ, Aspuru-Guzik A, Hein JE, Berlinguette CP, Sci. Adv 2020, 6, eaaz8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gómez-Bombarelli R, Aguilera-Iparraguirre J, Hirzel TD, Duvenaud D, Maclaurin D, Blood-Forsythe MA, Chae HS, Einzinger M, Ha D-G, Wu T, Markopoulos G, Jeon S, Kang H, Miyazaki H, Numata M, Kim S, Huang W, Hong SI, Baldo M, Adams RP, Aspuru-Guzik A, Nat. Mater 2016, 15, 1120. [DOI] [PubMed] [Google Scholar]
- [16].Kumar JN, Li Q, Tang KYT, Buonassisi T, Gonzalez-Oyarce AL, Ye J, npj Comput. Mater 2019, 5, 73. [Google Scholar]
- [17].Kumar R, Le N, Tan Z, Brown ME, Jiang S, Reineke TM, ACS Nano 2020, 14, 17626. [DOI] [PubMed] [Google Scholar]
- [18].Wu Y, Guo J, Sun R, Min J, npj Comput. Mater 2020, 6, 120. [Google Scholar]
- [19].Barnett JW, Bilchak CR, Wang Y, Benicewicz BC, Murdock LA, Bereau T, Kumar SK, Sci. Adv 2020, 6, eaaz4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang Y, Xie T, France-Lanord A, Berkley A, Johnson JA, Shao-Horn Y, Grossman JC, Chem. Mater 2020, 32, 4144. [Google Scholar]
- [21].Audus DJ, de Pablo JJ, ACS Macro Lett 2017, 6, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Upadhya R, Kosuri S, Tamasi M, Meyer TA, Atta S, Webb MA, Gormley AJ, Adv. Drug Delivery Rev 2021, 171, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen L, Pilania G, Batra R, Huan TD, Kim C, Kuenneth C, Ramprasad R, Mater. Sci. Eng., R 2021, 144, 100595. [Google Scholar]
- [24].Lin T-S, Coley CW, Mochigase H, Beech HK, Wang W, Wang Z, Woods E, Craig SL, Johnson JA, Kalow JA, Jensen KF, Olsen BD, ACS Cent. Sci 2019, 5, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ma R, Luo T, J. Chem. Inf. Model 2020, 60, 4684. [DOI] [PubMed] [Google Scholar]
- [26].Knox ST, Warren NJ, React. Chem. Eng 2020, 5, 405. [Google Scholar]
- [27].Webb MA, Jackson NE, Gil PS, de Pablo JJ, Sci. Adv 2020, 6, eabc6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jablonka KM, Jothiappan GM, Wang S, Smit B, Yoo B, Nat. Commun 2021, 12, 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Reis M, Gusev F, Taylor NG, Chung SH, Verber MD, Lee YZ, Isayev O, Leibfarth FA, J. Am. Chem. Soc 2021, 143, 17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rubens M, Vrijsen JH, Laun J, Junkers T, Angew. Chem., Int. Ed 2019, 58, 3183. [DOI] [PubMed] [Google Scholar]
- [31].Tamasi M, Kosuri S, DiStefano J, Chapman R, Gormley AJ, Adv. Intell. Syst 2020, 2, 1900126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gormley AJ, Yeow J, Ng G, Conway Ó, Boyer C, Chapman R, Angew. Chem., Int. Ed 2018, 57, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Humphrey W, Dalke A, Schulten K, J. Mol. Graphics 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- [34].Nguyen V, Gupta S, Rana S, Li C, Venkatesh S, Proc. Ninth Asian Conf. on Machine Learning (Eds: Zhang M-L, Noh Y-K), Vol. 77, PMLR, Cambridge, MA, USA: 2017, pp. 279–294. [Google Scholar]
- [35].Zhu J, Wang H, Hovy E, in Proc. 22nd Int. Conf. on Computational Linguistics (Coling 2008), Association for Computational Linguistics, Stroudsburg, PA, USA: 2008, pp. 1129–1136. [Google Scholar]
- [36].Patel RA, Borca CH, Webb MA, Mol. Syst. Des. Eng 2022, 7, 661. [Google Scholar]
- [37].Kim C, Chandrasekaran A, Jha A, Ramprasad R, MRS Commun 2019, 9, 860. [Google Scholar]
- [38].Shmilovich K, Mansbach RA, Sidky H, Dunne OE, Panda SS, Tovar JD, Ferguson AL, J. Phys. Chem. B 2020, 124, 3873. [DOI] [PubMed] [Google Scholar]
- [39].Xu J, Jung K, Boyer C, Macromolecules 2014, 47, 4217. [Google Scholar]
- [40].Ng G, Yeow J, Chapman R, Isahak N, Wolvetang E, Cooper-White JJ, Boyer C, Macromolecules 2018, 51, 7600. [Google Scholar]
- [41].Lundberg SM, Lee S-I, in Proc. of the 31st Int. Conf. on Neural Information Processing Systems, Curran Associates Inc., Red Hook, NY, USA: 2017, pp. 4768–4777. [Google Scholar]
- [42].Lundberg SM, Erion G, Chen H, DeGrave A, Prutkin JM, Nair B, Katz R, Himmelfarb J, Bansal N, Lee S-I, Nat. Mach. Intell 2020, 2, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chattopadhyay K, Mazumdar S, Biochemistry 2000, 39, 263. [DOI] [PubMed] [Google Scholar]
- [44].Zoldák G, Zubrik A, Musatov A, Stupák M, Sedlák E, J. Biol. Chem 2004, 279, 47601. [DOI] [PubMed] [Google Scholar]
- [45].Sawada S.-i., Akiyoshi K, Macromol. Biosci 2010, 10, 353. [DOI] [PubMed] [Google Scholar]
- [46].Hopkins JB, Gillilan RE, Skou S, J. Appl. Crystallogr 2017, 50, 1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, Kikhney AG, Hajizadeh NR, Franklin JM, Jeffries CM, Svergun DI, J. Appl. Crystallogr 2017, 50, 1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Petoukhov MV, Franke D, Shkumatov AV, Tria G, Kikhney AG, Gajda M, Gorba C, Mertens HD, Konarev PV, Svergun DI, J. Appl. Crystallogr 2012, 45, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang X, Bai Q, Hu J, Hou D, Sensors 2017, 17, 1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Su X, Zong Y, Richter R, Knoll W, J. Colloid Interface Sci 2005, 287, 35. [DOI] [PubMed] [Google Scholar]
- [51].Yeo I-K, Johnson RA, Biometrika 2000, 87, 954. [Google Scholar]
- [52].Bergstra J, Bardenet R, Bengio Y, Kégl B, in Proc. of the 24th Int. Conf. on Neural Information Processing Systems, NIPS’11, Curran Associates Inc, Red Hook, NY, USA: 2011, pp. 2546–2554. [Google Scholar]
- [53].Webb MA, Patel R, Gormley A, Tamasi M, Borca C, Kosuri S, Mugnier H, Upadhya R, Murthy NS, DataSpace repository at Princeton University, 2022, 10.34770/h938-nn26. [DOI] [PMC free article] [PubMed]
- [54].Webb MA, Patel RA, PPH_public on GitHub, https://github.com/webbtheosim/PPH_public.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in DataSpace at https://doi.org/10.34770/h938-nn26, ref. [53]. Trained machine-learning models that support the findings of this study are openly available in Github at https://github.com/webbtheosim/PPH_ public, ref. [54].