

Abstract
Nuclear pore complexes (NPCs) mediate communication between the nucleus and the cytoplasm, and regulate gene expression by interacting with transcription and mRNA export factors. Lysine acetyltransferases (KATs) promote transcription through acetylation of chromatin‐associated proteins. We find that Esa1, the KAT subunit of the yeast NuA4 complex, also acetylates the nuclear pore basket component Nup60 to promote mRNA export. Acetylation of Nup60 recruits the mRNA export factor Sac3, the scaffolding subunit of the Transcription and Export 2 (TREX‐2) complex, to the nuclear basket. The Esa1‐mediated nuclear export of mRNAs in turn promotes entry into S phase, which is inhibited by the Hos3 deacetylase in G1 daughter cells to restrain their premature commitment to a new cell division cycle. This mechanism is not only limited to G1/S‐expressed genes but also inhibits the expression of the nutrient‐regulated GAL1 gene specifically in daughter cells. Overall, these results reveal how acetylation can contribute to the functional plasticity of NPCs in mother and daughter yeast cells. In addition, our work demonstrates dual gene expression regulation by the evolutionarily conserved NuA4 complex, at the level of transcription and at the stage of mRNA export by modifying the nucleoplasmic entrance to nuclear pores.
Keywords: G1‐S transition, Hos3, mRNA export, NuA4, nuclear pore complex
Subject Categories: Cell Cycle, Post-translational Modifications & Proteolysis, RNA Biology
The Esa1/NuA4 acetyltransferase complex regulates gene expression not just at the level of transcription, but also by controlling nuclear mRNA export via the basket protein Nup60.
Introduction
Nuclear pores are macromolecular assemblies composed of approximately 30 different nucleoporins that form a channel across the nuclear envelope (Knockenhauer & Schwartz, 2016; Hampoelz et al, 2019; Raices & D'Angelo, 2021). The central channel mediates communication between the nucleus and cytoplasm. Other NPC substructures include the cytoplasmic filaments and the nuclear basket, associated with the cytoplasmic and nuclear sides of the central channel, respectively. The nuclear basket regulates gene expression through interactions with active genes (Casolari et al, 2004; Cabal et al, 2006; Light et al, 2010; Brickner et al, 2019) and with regulators of transcription (Texari et al, 2013; Schneider et al, 2015) and mRNA export (Fischer et al, 2002; Dieppois et al, 2006). These and other studies have suggested that NPCs can act as regulatory sites for the coordination of transcript elongation, processing and export (Sood & Brickner, 2014; Ibarra & Hetzer, 2015).
The nuclear basket also recruits lysine acetyltransferases (KATs) and deacetylases (KDACs). Acetylation of histones is tightly associated with transcription, and KATs and KDACs, often residing in large multiprotein complexes, are thought to target nucleosomes to regulate transcriptional activity (Sterner & Berger, 2000; Lee & Workman, 2007). In addition, acetylation of non‐histone proteins is common in eukaryotes and has been implicated in a variety of biological processes in addition to transcription, such as DNA damage repair, cell division and signal transduction (Kaluarachchi Duffy et al, 2012; Narita et al, 2019). Among the best‐characterised yeast KATs are Esa1 and Gcn5, contained in the NuA4 and SAGA complexes, respectively. Esa1 (known as Kat5 or Tip60 in mammals) is the only essential KAT in budding yeast and is involved in DNA transcription and repair (Allard et al, 1999; Clarke et al, 1999; Doyon & Côté, 2004; Bruzzone et al, 2018). Gcn5 (also known as KAT2A) is not essential but plays a role in the transcription of most yeast genes (Baptista et al, 2017; Bruzzone et al, 2018). KATs and KDACs known to associate with NPCs include Gcn5 and the type II deacetylases Hos3 (in yeast) and HDAC4 (in mammals) (Cabal et al, 2006; Kurshakova et al, 2007; Kehat et al, 2011; Kumar et al, 2018). Despite their presence at NPCs, how KATs and KDACs act to regulate gene expression at these sites is poorly understood.
Regulatory principles of the G1/S transition (known as Start in yeast) are evolutionarily conserved: activation of cyclin‐dependent kinase (CDK) drives the transcription of hundreds of genes involved in the start of S phase (Bertoli et al, 2013). Indeed, defects in G1/S control are tightly associated with oncogenesis. For example, pRB is a repressor of the G1/S transition thought to be functionally inactivated in most tumour cells (Frolov & Dyson, 2004). In both yeast and animal cells, inhibition of premature G1/S transition involves the targeting of KDACs to chromatin, generating an environment that is unfavourable for transcription (Frolov & Dyson, 2004; Huang et al, 2009; Takahata et al, 2009; Wang et al, 2009). We recently discovered that in budding yeast, NPC acetylation regulates the G1/S transition (Kumar et al, 2018; Gomar‐Alba & Mendoza, 2019). The KDAC Hos3 is cytoplasmic during interphase, but associates with the yeast division site (the mother‐bud neck) in mitosis and then binds to daughter cell NPCs as they traverse the bud neck during anaphase, leading to Hos3 association with the nuclear basket specifically in daughter cells (Fig 1A). Hos3‐dependent deacetylation of central pore channel nucleoporins in daughter cells enhances nuclear accumulation of the main Start inhibitor (the transcriptional repressor Whi5, functional analog of pRB). In addition, deacetylation of the nuclear basket nucleoporin Nup60 has relatively minor effects on Whi5 nuclear accumulation but is associated with the perinuclear tethering of a key cell cycle control gene (encoding the G1/S cyclin Cln2, homologue of mammalian Cyclin E) (Kumar et al, 2018). Thus, acetylation of specific nucleoporins promotes different aspects of nuclear pore function necessary for S‐phase entry, and their deacetylation in daughter cells reinforces cell size control mechanisms that prevent premature S phase in small daughters (Turner et al, 2012). However, the identity of the KAT(s) targeting the NPC for acetylation is unknown, and the molecular mechanism by which NPC acetylation status affects S‐phase entry remains unclear.
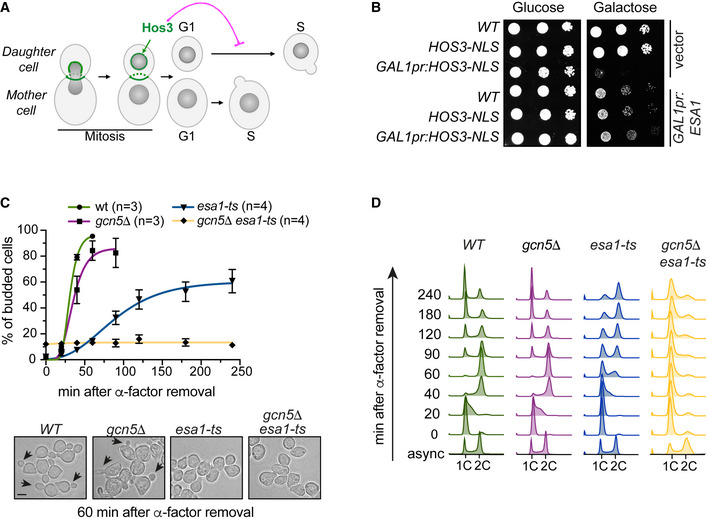
Figure 1. KATs Esa1 and Gcn5 counteract the KDAC Hos3 to promote the G1/S transition.
-
ALocalisation and function of the Hos3 deacetylase during mitotic division. Hos3 (in green) associates with the bud neck and with daughter cell NPCs during nuclear migration into the bud. Hos3 delays the G1/S transition specifically in daughter cells through deacetylation of NPCs (Kumar et al, 2018).
-
BGrowth inhibition upon overexpression of HOS3‐NLS driven by the GAL1 promoter is suppressed by overexpression of the KAT Esa1. 10‐fold serial dilutions of the indicated strains transformed with an empty vector or with the indicated plasmids were spotted onto SC‐Glu and SC‐Gal medium (to activate the GAL1 promoter) and incubated at 25°C for 3 days. Note that HOS3‐NLS (under the control of the native HOS3 promoter) does not affect cell growth.
-
Cesa1‐ts and gcn5Δ esa1‐ts mutants have bud emergence defects. (Top) Cells of the indicated strains were arrested in G1 by treatment with ɑ‐factor for 2.5 h at 25°C, shifted to 37°C for 1 h and released from the G1 arrest at 37°C. Cells were fixed at the indicated times, and the presence of buds was assessed by microscopy. Mean and SEM are derived from n = number of independent experiments. At least 200 cells were scored for each strain and time point. (Bottom) Bright‐field images of the indicated strains 60 min after the ɑ‐factor washout. Arrowheads point to cell buds. Scale bar, 4 μm.
-
DInactivation of ESA1 and GCN5 delays DNA replication. Cells of the indicated genotypes were synchronised as in panel C, and DNA content was evaluated by flow cytometry. Numbers indicate time in minutes after the release. This experiment was repeated three times with similar results; one experiment is shown.
Source data are available online for this figure.
Here, we show that the KAT Esa1 acetylates the nuclear basket component Nup60 to promote mRNA export and the G1/S transition. Furthermore, we demonstrate that Hos3‐dependent deacetylation of Nup60 displaces mRNA export complexes from daughter cell NPCs to inhibit Start. We propose that, in addition to modulating cell cycle entry and preventing premature division of daughter cells, this pathway regulates general mRNA export. Thus, the evolutionarily conserved NuA4 complex drives gene expression and cell cycle progression not only by acetylating chromatin and promoting transcription but also by acetylating the nucleoplasmic entrance to NPCs to facilitate export of nuclear mRNA, thereby dually controlling the gene expression state of the cell.
Results
Esa1 is the main lysine acetyltransferase promoting cell cycle entry
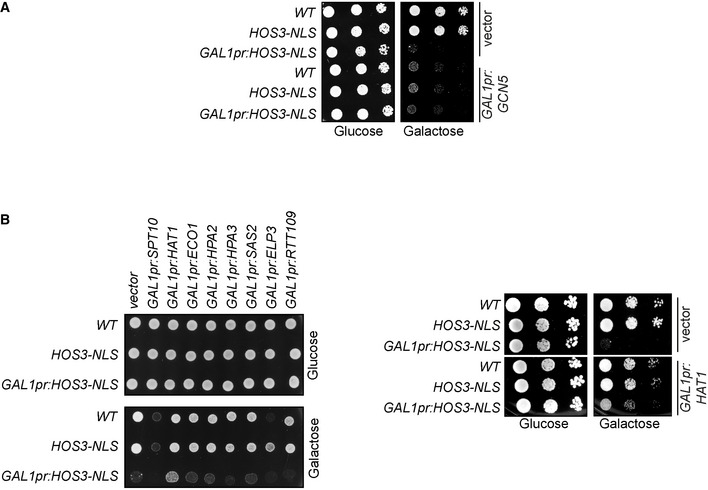
To understand how NPC acetylation regulates the G1/S transition (Start), we sought to identify the lysine acetyltransferases (KATs) counteracting the activity of the Hos3 deacetylase. Hos3 displays asymmetric distribution between mother and daughter cells in wild‐type Saccharomyces cerevisiae. Overexpression of a version of Hos3 fused to a nuclear localisation signal (GAL1pr‐HOS3‐NLS) leads to targeting of Hos3 to mother and daughter cell nuclei, deacetylation of nucleoporins and inhibition of cell proliferation (Kumar et al, 2018). We tested whether this inhibition could be relieved by overexpression of yeast KATs, including Eco1, Elp3, Esa1, Gcn5, Hat1, Hpa2, Hpa3, Rtt109, Sas2 and Spt10. Overexpression of Elp3, Gcn5 and Spt10 was toxic in wild‐type cells, and therefore, their potential role in opposing Hos3 could not be established using this assay (Fig EV1). However, we found that of the remaining KATs, only Esa1 and Hat1 overexpression suppressed Hos3‐NLS lethality (Figs 1B and EV1B).
Figure EV1. Effect of KAT overexpression in Hos3‐NLS‐dependent growth inhibition.
-
AOverexpression of the KAT Gcn5 is toxic. 10‐fold serial dilutions of wild‐type (WT), HOS3‐NLS‐GFP and GAL1pr:HOS3‐NLS‐GFP (single copy at the endogenous locus) transformed with an empty vector or the GAL1pr:GCN5‐HA plasmid were spotted onto SC‐Glu and SC‐Gal medium and incubated at 25°C for 3 days.
-
BRole of KAT overexpression in cell viability and ability to rescue growth in the presence of overexpressed HOS3‐NLS. (Left) Exponential cultures of the indicated strains transformed with an empty vector or plasmids overexpressing the KATs Spt10, Eco1, Hpa2, Hpa3, Sas2, Elp3 or Rtt109 were spotted onto SC‐Glu and SC‐Gal medium and incubated at 25°C for 3 days. (Right) The effect of HAT1 overexpression was also assessed in 10‐fold serial dilutions as in (A).
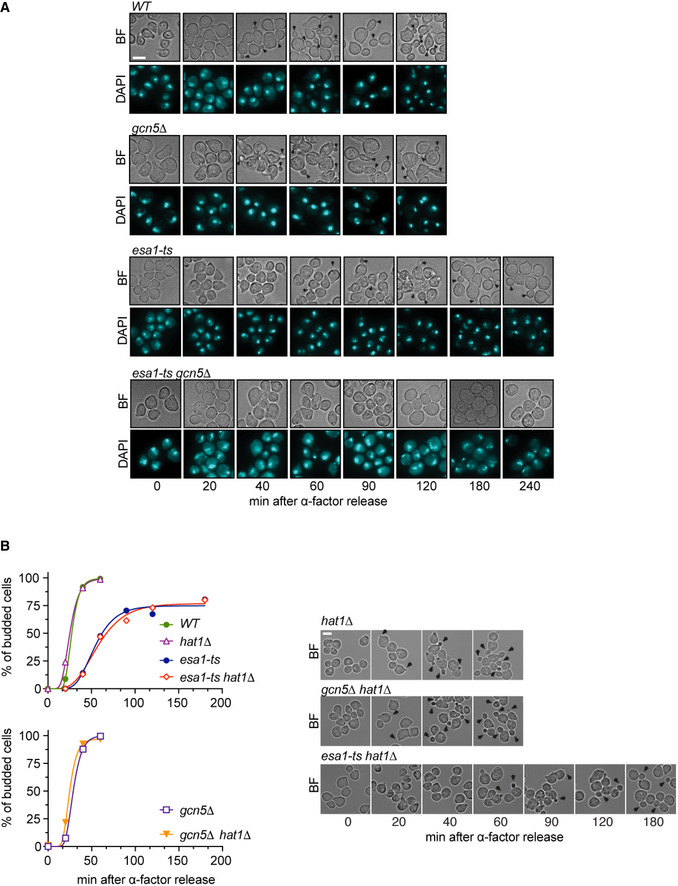
Inactivation of Hos3 leads to premature onset of S phase in daughter cells (Kumar et al, 2018). We thus tested whether inactivation of Esa1 or Hat1 (alone or in combination) inhibits the G1/S transition, as would be expected of KATs counteracting the KDAC Hos3. We also tested the role of Gcn5, since its loss was previously reported to cause a mild delay in the G1/S transition (Kishkevich et al, 2019). The non‐essential genes HAT1 and GCN5 were deleted, whereas Esa1 was inactivated using the well‐characterised thermosensitive (ts) mutation esa1‐L254P (hereafter called esa1‐ts) (Clarke et al, 1999). Wild‐type and mutant cells were arrested in G1 at 25°C by addition of alpha factor, shifted to the restrictive temperature for esa1‐ts (37°C), and released from the cell cycle arrest by alpha‐factor removal. The fraction of S‐phase cells at different times after alpha‐factor washout was determined by monitoring bud emergence. More than 95% of wild‐type cells budded within 60 min of alpha‐factor removal, and gcn5∆ cells exhibited a 15‐min delay in budding as previously reported (Kishkevich et al, 2019). In contrast, budding was strongly delayed in esa1‐ts cells: on average, only 10% of these cells had formed a bud after 60 min, and approximately 40% lacked a bud after 4 h. Moreover, cells lacking both Esa1 and Gcn5 had stronger defects in budding than either single mutant: esa1‐ts gcn5∆ cells remained unbudded after 4 h of alpha‐factor washout (Figs 1C and EV2A). Deletion of HAT1 did not delay budding of either wild‐type, gcn5∆ or esa1‐ts cells (Fig EV2B). Thus, HAT1 does not play a role in Start and was not characterised further. DNA replication, assayed by flow cytometry, was also delayed in Esa1‐deficient cells. Whereas most wild‐type cells replicated their DNA 40 min after alpha‐factor removal, replication was still incomplete after 4 h in esa1‐ts cells, and was undetectable in esa1‐ts gcn5∆ (Fig 1D). In summary, Esa1 promotes budding and DNA replication, which are hallmarks of the G1/S transition. In the absence of Esa1, these functions can be partially compensated by Gcn5.
Figure EV2. Role of ESA1, GCN5 and HAT1 in Start.
-
Aesa1‐ts and gcn5Δ esa1‐ts mutants have bud emergence defects. Cells of the indicated strains were arrested in G1 by treatment with ɑ‐factor for 2.5 h at 25°C, shifted to 37°C for 1 h and released from the G1 arrest at 37°C. Cells were fixed at the indicated times, and the presence of buds (arrowheads) was assessed by microscopy.
-
BHAT1 is not involved in Start. Bright‐field images of cells of the indicated strains, treated as in (A) and scored at the indicated times after ɑ‐factor washout. The left subpanel shows the fraction of budded cells in one of two independent experiments with similar results. At least 200 cells were scored for each strain and time point. Arrowheads point to cell buds. Scale bars in (A and B), 5 μm.
Source data are available online for this figure.
Esa1 acts through Nup60 acetylation to promote Start
These data raised the possibility that Esa1, Gcn5 and Hos3 regulate the G1/S transition, at least in part, by modulating the acetylation level of shared target proteins. Proteomic studies have indicated that budding yeast nucleoporins are targeted by multiple KATs, including Esa1 and Gcn5, although the role of these modifications remained unclear (Henriksen et al, 2012; Downey et al, 2015). Furthermore, Hos3‐dependent deacetylation of the nuclear basket component Nup60 lysine 467 is important for inhibition of Start in daughter cells (Kumar et al, 2018). Therefore, we investigated whether Esa1 and Gcn5 promote Start through Nup60 acetylation. Nup60‐GFP was immunoprecipitated from cells expressing Esa1 or Gcn5 under the control of the inducible GAL1 promoter, and its acetylation state was assayed with an anti‐acetyl‐lysine (AcLys) antibody. This revealed increased Nup60 acetylation after addition of galactose in GAL1pr‐ESA1 and GAL1pr‐GCN5 cells (Fig 2A). Thus, Esa1 and Gcn5 can acetylate Nup60.
Figure 2. Acetyl‐mimic Nup60 partially rescues the Start defects of esa1‐ts cells.
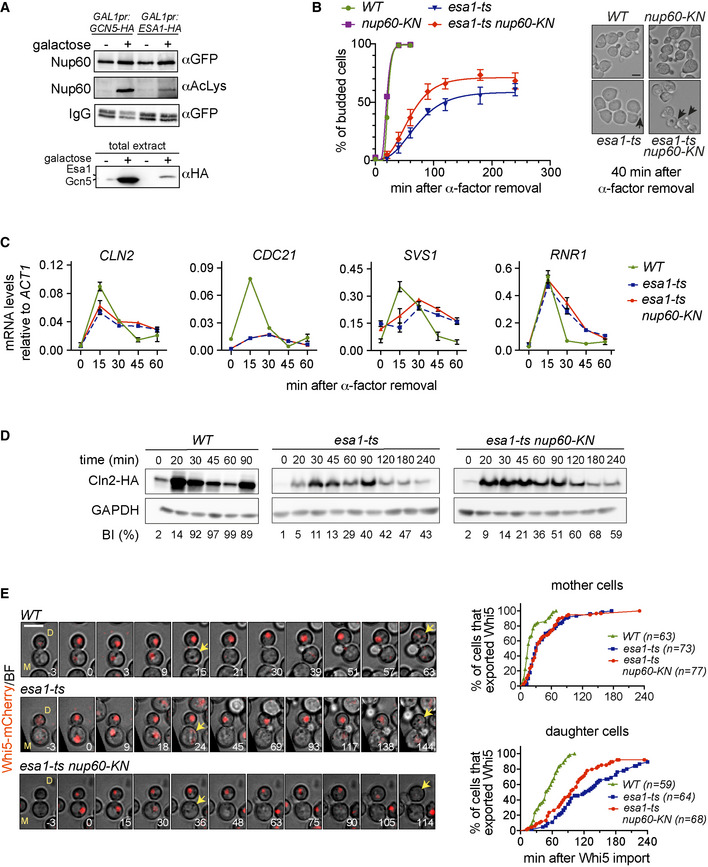
-
AOverexpression of Esa1 and Gcn5 KATs leads to increased acetylation levels of the nuclear basket nucleoporin Nup60. (Top) Nup60‐GFP was immunoprecipitated from extracts of the indicated strains, and its acetylation state probed with anti‐AcLys antibodies. (Bottom) Total extracts probed with anti‐HA antibodies to verify KAT overexpression.
-
Bnup60‐KN partially rescues the budding defect of esa1‐ts cells. (Left) Budding of cells of the indicated strains was determined as in Fig 1C. At least 200 cells were scored for each strain and time point. Data from three independent experiments are represented as mean and SEM (esa1‐ts, esa1‐ts nup60‐KN). (Right) Bright‐field images of the indicated strains 40 min after the ɑ‐factor washout. Arrowheads point to cell buds. Scale bar, 4 μm.
-
CmRNA levels of CLN2, CDC21, SVS1 and RNR1 were determined for cells of the indicated strains after G1 arrest and release at restrictive temperature, with samples collected at indicated times. Data from three independent experiments are represented as mean and SEM.
-
Dnup60‐KN mutation partially rescues the delay in synthesis of the G1/S cyclin Cln2 in esa1‐ts cells. Cells of the indicated strains were processed as in (B), and the amount of Cln2‐HA protein at the indicated times was assessed by Western blot. % of budded cells (budding index, BI) is indicated below for each corresponding strain and time point. Note the slow, inefficient budding in esa1‐ts mutants. In WT, the reduction in Cln2 at 60 min and its increase at 90 min reflect the start of a second cycle, which is absent in esa1‐ts cells.
-
Enup60‐KN partially rescues the Whi5 export defect of esa1‐ts daughter cells. Composite of bright field and Whi5‐mCherry (left) and quantification of Whi5 nuclear export (right) in mother (M) and daughter (D) cells of the indicated strains. Whi5 export (arrows) is delayed in esa1‐ts mothers and daughters compared to WT (P < 0.001, log‐rank Mantel–Cox test); esa1‐ts nup60‐KN advances Whi5 export relative to esa1‐ts in daughters (P = 0.0105), but not in mothers (P > 0.05). 3 z‐confocal slices spaced 0.5 μm were acquired every 3 min; maximum projections of selected timepoints are shown. Time is indicated in minutes; t = 0 marks Whi5 nuclear import. Scale bar, 5 μm. n = number of cells, pooled from two independent experiments with similar results.
Source data are available online for this figure.
We next tested whether acetylation of Nup60 can mediate the G1/S function of Esa1 and Gcn5. Lysine (K) 467 of Nup60 was replaced with an asparagine (N) residue, whose biophysical properties resemble those of acetylated lysine, to generate the acetyl‐mimic Nup60‐KN. Cells were released from a G1 block, and their budding efficiency was determined as previously. Expression of Nup60‐KN partially rescued the budding efficiency of the single mutant esa1‐ts (Fig 2B), although it was not sufficient to restore budding in cells lacking both Esa1 and Gcn5 (Fig EV3A). Furthermore, the replacement of Nup60 K467 with arginine (R) to mimic the lack of acetylation (Nup60‐KR) led to the opposite phenotype of Nup60‐KN, further delaying the budding of esa1‐ts (Fig EV3B). These results suggest that Esa1 promotes the G1/S transition in part by acetylation of Nup60.
Figure EV3. G1/S transition in KAT and Nup60 KN/KR mutants.
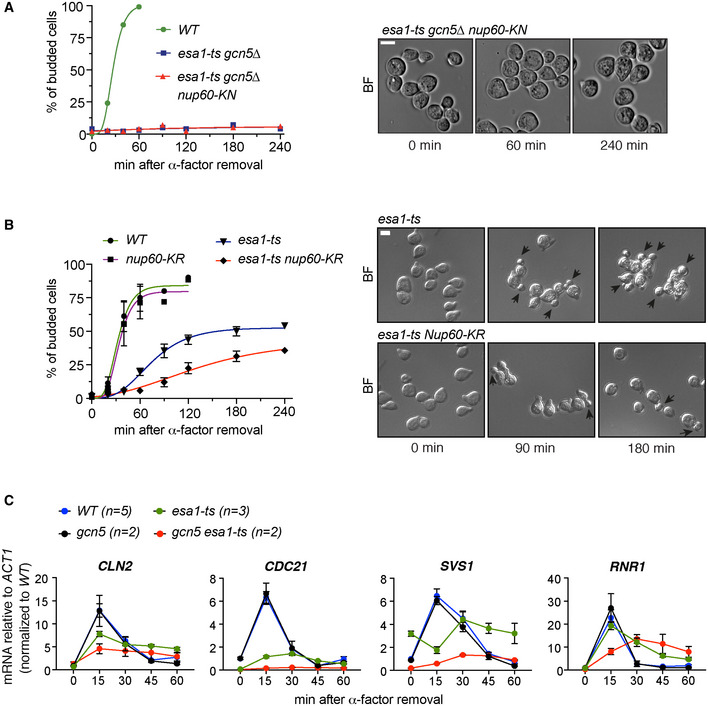
-
AAcetyl‐mimic Nup60‐KN does not rescue the budding defect of the double mutant esa1‐ts gcn5∆. Cells were arrested in G1 by treatment with ɑ‐factor for 2.5 h at 25°C, shifted to 37°C for 1 h and released from the G1 arrest at 37°C. Cells were fixed at the indicated times, and the presence of buds was assessed by microscopy. The left subpanel shows the fraction of budded cells in one of two independent experiments with similar results.
-
BNon‐acetylatable Nup60‐KR increases the budding defect of esa1‐ts cells. In (A and B), at least 200 cells were scored for each strain and time point. In (B), data from three independent experiments are represented as mean and SEM. In bright‐field images of cells at the indicated times after ɑ‐factor washout, arrowheads point to cell buds.
-
CmRNA levels of CLN2, CDC21, SVS1 and RNR1 were determined for cells of the indicated strains after G1 arrest and release at restrictive temperature, with samples collected at indicated times. Data from n > 2 independent experiments are represented as mean and SEM (WT, esa1‐ts) and data from n = 2 independent experiments (gcn5∆, gcn5∆ esa1‐ts) as mean and range. The 2ΔCt values were then normalised relative to the wild‐type value at 0 min.
Data information: (A, B) Scale bar, 4 μm.
Source data are available online for this figure.
Start is marked by the transcription of hundreds of genes of the G1/S regulon, which are required for budding and DNA replication. To gain insight into how Nup60 acetylation promotes the G1/S transition, we first determined the mRNA levels of four representative regulon genes (CLN2, CDC21, SVS1 and RNR1) during the G1/S transition using quantitative reverse transcription PCR (RT–qPCR) in KAT‐deficient cells. In wild‐type synchronous cultures, CLN2, CDC21, SVS1 and RNR1 are induced 15 min after alpha‐factor removal, and their mRNA levels decrease as cells enter S phase (Fig 2C, WT). In agreement with the observed budding and DNA replication defects, transcription of G1/S genes was impaired in esa1‐ts cells. CLN2 was induced at lower levels and with slower kinetics than in wild type, whereas CDC21 and SVS1 mRNA levels did not oscillate during the experiment, and RNR1 was induced at normal levels but its mRNA remained unusually stable (Fig 2C, esa1‐ts). As previously reported (Kishkevich et al, 2019), deletion of GCN5 did not affect the mRNA levels of any of the tested genes. However, the double mutant gcn5∆ esa1‐ts showed stronger transcriptional defects than the esa1‐ts single mutant (Fig EV3C). Together, these results indicate that Esa1 drives the coordinated induction of G1/S genes and that Gcn5 can partially compensate for the absence of Esa1. Notably, the acetyl‐mimic version of Nup60, which partially rescued budding efficiency of the esa1‐ts mutant (Fig 2B), did not improve its transcriptional defects (Fig 2C, esa1‐ts nup60‐KN). To understand how Nup60‐KN promotes budding of Esa1‐deficient cells, western blotting was used to determine the protein levels of the G1/S cyclin Cln2 in esa1‐ts and esa1‐ts nup60‐KN. Cln2 plays a critical role in driving activation of CDK in late G1 and robust, irreversible G1/S transition via positive feedback (Skotheim et al, 2008; Charvin et al, 2010). As expected, Cln2 protein synthesis occurred later and at lower levels in esa1‐ts than in wild‐type cells released from a G1 block. Importantly, the delay in Cln2 protein synthesis was alleviated in esa1‐ts nup60‐KN (Fig 2D and Appendix Fig S1). This suggests that Nup60 acetylation promotes Cln2 expression at the post‐transcriptional level.
Finally, we examined the requirement for Esa1 and Nup60 acetylation in the G1/S transition of mother (M) and daughter (D) cells, using time‐lapse microscopy of freely cycling cells. To determine the time of the G1/S transition in single cells, we monitored the nuclear localisation changes of the Whi5 transcriptional repressor, a G1 marker. Whi5 is imported into the nucleus of M and D cells in late anaphase, and its export in G1, driven by CDK phosphorylation, marks the irreversible commitment to S phase (Costanzo et al, 2004; de Bruin et al, 2004; Charvin et al, 2010). Cells were incubated at 37°C and imaged at 3‐min intervals. In wild‐type cells, nuclear export of Whi5‐mCherry occurred first in mothers and later in daughters relative to Whi5 import (median times, 15 min [M cells] and 59 min [D cells]; note that the duration of G1 phase in cells synchronised with alpha factor is not directly comparable with that of freely cycling cells) (Fig 2E, WT and Appendix Fig S2). This dichotomy is due to both cell size control in small daughters and size‐independent mechanisms that delay Start specifically in daughters, including NPC deacetylation (Di Talia et al, 2007; Kumar et al, 2018). In esa1‐ts cells, Whi5 export was markedly delayed in both M and D cells (median times: 30 min [M] and 123 min [D]; Fig 2E, esa1‐ts). Furthermore, the presence of Nup60‐KN partially restored the delay in Whi5 export caused by Esa1 inactivation, specifically in daughter cells (median times: 33 min [M] and 96 min [D]; Fig 2E, esa1‐ts nup60‐KN). Thus, Esa1 promotes Start in both mother and daughter cells, but constitutive Nup60 acetylation advances Start specifically in Esa1‐deficient daughters. This is consistent with daughter‐specific Nup60 deacetylation caused by asymmetric inheritance of the Hos3 KDAC (Kumar et al, 2018), and further supports the hypothesis that acetylation of Nup60 in mothers is reversed by Hos3 in daughter cells.
Esa1 and Nup60‐KN promote the export of CLN2 mRNA
Next, we used time‐lapse microscopy to determine CLN2 mRNA localisation, by inserting PP7 stem loops in its 3’ UTR. PP7 stem loops bind to the bacteriophage coat protein fused to a nuclear localisation signal and GFP (PCP‐GFP‐NLS), allowing visualisation of CLN2 mRNA foci (Neurohr et al, 2018). We used Whi5‐tdTomato to monitor the G1/S transition in the same cells. Freely cycling cells (wild type, esa1‐ts and esa1‐ts nup60‐KN) were incubated at 37°C and imaged at 5‐min intervals for up to 6 h. We restricted our analysis to daughter cells, which are most affected by inactivation of Esa1. In wild type, a single bright perinuclear focus indicative of CLN2 transcription was detected for 1–2 consecutive frames at the time of Whi5 nuclear export. This occurred approximately 30 to 50 min after Whi5 was imported into the nucleus (Fig 3A–C). CLN2 transcription was followed by the appearance of 5–20 mRNA foci in the cytoplasm (Fig 3A and D). In esa1‐ts, nuclear mRNA foci appeared later (relative to Whi5 import) than in wild‐type cells (Fig 3B); short‐lived nuclear foci could be detected during the extended G1 phase in these cells (see example in Fig 3A). Importantly, the fraction of cells with nuclear CLN2 mRNA foci peaked in both wild‐type and esa1‐ts at the time of Whi5 export, indicating that Whi5 export is associated with CLN2 transcription even in the absence of Esa1 (Fig 3C). However, the fluorescence intensity of nuclear foci was lower in esa1‐ts than in wild type (Fig 3E). Thus, Esa1 promotes the synthesis of CLN2 mRNA. Inactivation of Esa1 was also associated with changes in the localisation of CLN2 transcripts; nuclear mRNA foci were associated with the nuclear periphery in 95% of wild‐type cells, compared to 80% in esa1‐ts (Fig 3F). In addition, the number of cytoplasmic foci at the time of Whi5 export was lower in esa1‐ts than in wild‐type cells (Fig 3A and E). Strikingly, Nup60‐KN did not alter the intensity of CLN2 nuclear foci in esa1‐ts cells, but rescued the fraction of cells with perinuclear mRNA, and increased the number of CLN2 cytoplasmic foci (Fig 3D and E). These results raised the possibility that Nup60 acetylation is specifically required for the nuclear export of CLN2 mRNA.
Figure 3. Esa1 and Nup60‐KN promote the nuclear export of CLN2 mRNA.
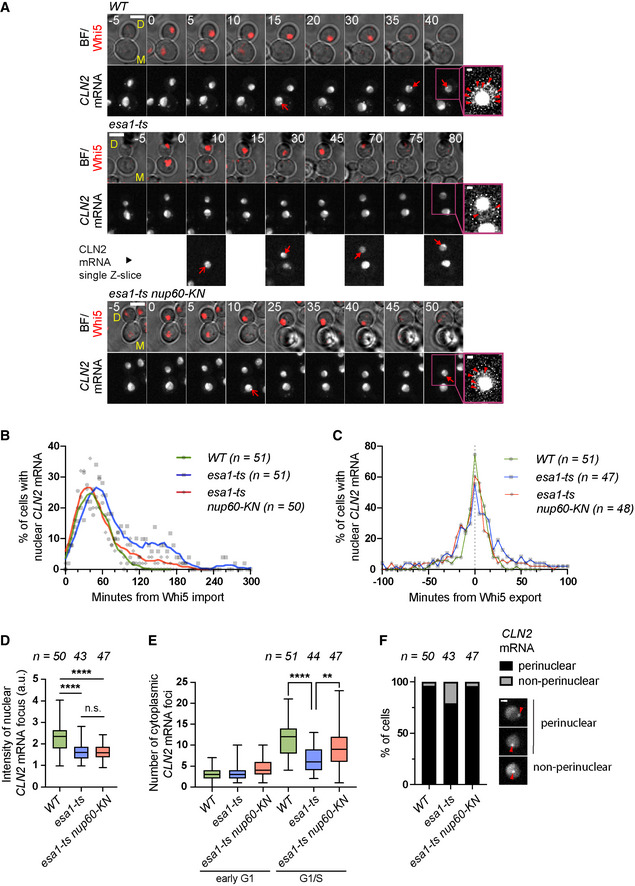
-
AComposite of bright field and Whi5‐tdTomato (BF/Whi5) and CLN2‐PP7 mRNA labelled with PCP‐NLS‐GFP (CLN2 mRNA) in mother (M) and daughter (D) cells of the indicated strains. Arrows indicate nuclear foci. Brighter GFP nucleoplasmic areas may correspond to the nucleolus. Insets show enhanced‐contrast images to visualise cytoplasmic mRNA particles (arrowheads). Numbers indicate minutes relative to Whi5 nuclear import. Maximum projections of whole‐cell Z‐stacks are shown for Whi5 and CLN2 mRNA except in the inset and in selected esa1‐ts images, where single Z‐slices are shown for clarity. Scale bar, 4 μm (inset: 1 μm).
-
BFraction of cells of the indicated strains with nuclear CLN2‐PP7 mRNA foci, aligned relative to Whi5 import. Symbols represent individual values; lines were generated by smoothing of the nearest three neighbouring values (0th order polynomial).
-
CFraction of cells of the indicated strains with nuclear CLN2‐PP7 mRNA foci, aligned relative to Whi5 export.
-
DMean fluorescence intensity of nuclear CLN2 mRNA foci at the time of Whi5 export, normalised relative to the nuclear background.
-
ENumber of cytoplasmic CLN2‐PP7 mRNA foci in early G1 (5 min after Whi5 import) and at the G1/S transition (5 min after Whi5 export).
-
FThe position of nuclear CLN2‐PP7 foci (marked with arrowheads in the examples) was scored relatively to the nuclear periphery (visualised with PCP‐NLS‐GFP) in the indicated strains. The fraction of non‐perinuclear CLN2‐PP7 foci is significantly increased in esa1‐ts cells compared to the WT and rescued by nup60‐KN mutation (two‐sided Fisher's exact test, P = 0.0209 and P = 0.0227). Scale bar, 1 μm.
Data information: In (D and E), boxes include 50% of data points, lines represent the median, and whiskers extend to maximum and minimum values. ****, P ≤ 0.0001; **, P ≤ 0.01; and n.s., P > 0.05, ordinary one‐way ANOVA with Tukey's multiple comparisons test. Adjusted P‐values in (D): WT vs. esa1‐ts, P < 0.0001; WT vs. esa1‐ts nup60‐KN, P < 0.0001; and esa1‐ts vs. esa1‐ts nup60‐KN, P = 0.9845; and in (E): WT vs. esa1‐ts, P < 0.0001; and esa1‐ts vs. esa1‐ts nup60‐KN, P = 0.002. Foci were scored in individual Z‐slices spanning the entire cell volume. n = number of cells, pooled from two independent experiments with similar results.
Source data are available online for this figure.
Together, these findings suggest two distinct roles for Esa1 in driving cell cycle commitment. First, a major function of Esa1 in promoting the G1/S transition is to drive the timely transcription of genes required for cell cycle entry. This is in keeping with an established role of Esa1 in promoting transcription of most yeast genes (Bruzzone et al, 2018). Second, Esa1 may play additional positive roles in Start, independently of transcription, that are mediated by Nup60 acetylation and may include mRNA export. To further test the hypothesis that Nup60 acetylation plays mostly a post‐transcriptional role in gene regulation, we determined genome‐wide mRNA levels in wild‐type, nup60‐KN and nup60‐KR cells by RNA sequencing. With the exception of minor changes in a handful of subtelomeric genes, this analysis found no significant changes in transcription among these strains (Fig EV4). A non‐transcriptional role of Nup60 acetylation in gene expression could also explain why Nup60‐KN rescues the Start defect of esa1‐ts but not those of the double mutant esa1‐ts gcn5∆. Indeed, levels of G1/S mRNAs in this mutant may be too low to promote S phase, even in the presence of increased mRNA export efficiency promoted by Nup60‐KN.
Figure EV4. Nup60‐KN and Nup60‐KR alleles do not alter mRNA levels.
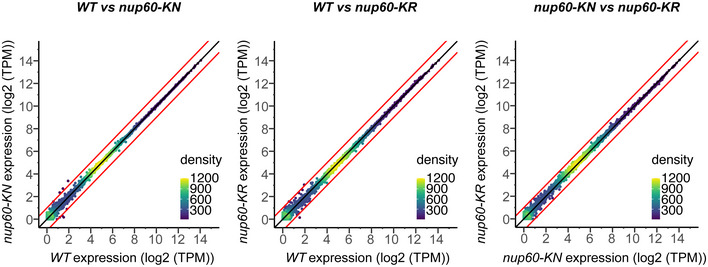
Expression levels in nup60‐KN and nup60‐KR strains compared to expression in the wild‐type strain (WT) and to each other. Red lines show a fold change of 1. Genes with FC > 1 are YCR107W, YCR106W and YGL263W (subtelomeric); genes with FC < −1 are YFL065C (subtelomeric), YLR124W and tF(GAA)F (phenylalanine tRNA).
Esa1 and Nup60 acetylation promote bulk mRNA export
Nuclear basket components are required for the efficient export of nuclear mRNA through their association with the Transcription and Export 2 (TREX‐2) complex (Fischer et al, 2002). Our previous findings raised the possibility that Nup60 acetylation promotes mRNA export. We therefore tested whether the cell proliferation defect of GAL1pr‐HOS3‐NLS cells, in which Nup60 and other nucleoporins are deacetylated, can be alleviated by increased levels of mRNA export factors (Fig 4A). Indeed, we found that the growth of GAL1pr‐HOS3‐NLS cells was restored by overexpression of the mRNA export receptors Mtr2 and Mex67, which escort mRNA molecules through the NPC (Strässer et al, 2000; Strawn et al, 2001) (Fig 4B). Likewise, overexpression of the scaffolding subunit of the TREX‐2 complex, Sac3 (Fischer et al, 2002; Jani et al, 2014), also restored growth of GAL1pr‐HOS3‐NLS cells (Fig 4C). Note that a truncated Sac3 version was used, since overexpression of full‐length Sac3 is toxic (Appendix Fig S3A–C). This further suggests that Hos3, possibly via Nup60 deacetylation, prevents cell proliferation by inhibiting export of nuclear mRNA. Overexpression of Mtr2 and Mex67 did not rescue the growth of Esa1‐deficient cells, confirming that mRNA export is not the only essential function of this KAT (Appendix Fig S3D).
Figure 4. Nuclear export of mRNA is inhibited by the KDAC Hos3 and promoted by the KAT Esa1 and Nup60 acetylation.
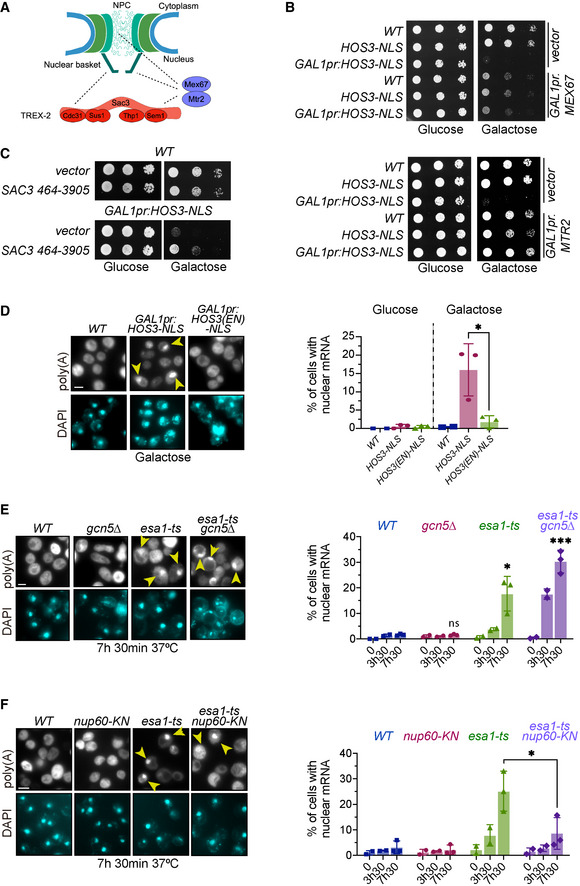
-
AIllustration of the physical interactions (dashed lines) of the NPC with mRNA export factors.
-
BOverexpression of MEX67 and MTR2 rescues the lethality of Hos3‐NLS overexpression. 10‐fold serial dilutions of the indicated strains transformed with the indicated plasmids spotted onto SC‐Glu and SC‐Gal medium and incubated at 25°C for 3 days.
-
CA high‐copy plasmid containing SAC3 rescues the lethality of Hos3‐NLS overexpression. Strains carrying a high‐copy plasmid containing the SAC3 ORF (nucleotides 467–3,905), or the empty vector, were grown as in (B).
-
DOverexpression of Hos3‐NLS promotes nuclear accumulation of mRNA. (Left) Cultures of the indicated strains were treated with galactose overnight to induce HOS3‐NLS expression, cells were fixed, and FISH was performed using Cy3‐Oligo(dT). (Right) The fraction of cells with nuclear mRNA accumulation was determined for the indicated strains and conditions.
-
EInactivation of Esa1 impairs export of poly(A) RNA. (Left) Cultures of the indicated strains were incubated at 37°C. (Right) The fraction of cells with nuclear mRNA accumulation was determined for the indicated strains and conditions as in (D).
-
Fnup60‐KN mutation partially rescues the mRNA export defects of esa1‐ts. Cells of the indicated strains were processed as in (E).
Data information: In (D–F), arrows point to polyadenylated RNA in the nucleus, which was visualised by DAPI staining. Data from three independent biological replicates (7h30) are represented as mean and s.d., and data from two independent biological replicates (0 and 3h30) as mean and range. *, P ≤ 0.05; ***, P ≤ 0.001; and ns, P > 0.05, two‐tailed unpaired t‐test. At least 200 cells were scored for each time point and condition. Scale bar, 4 μm.
Source data are available online for this figure.
To directly test whether Nup60 deacetylation inhibits bulk mRNA export, we imaged polyadenylated mRNA by fluorescence in situ hybridisation (FISH) using a poly‐dT probe. This was done in wild type and in cells in which Nup60 acetylation was reduced by overexpression of nuclear Hos3 (GAL1pr:HOS3‐NLS) or by inactivation of Esa1 and Gcn5 (esa1‐ts, gcn5∆). As expected, all wild‐type cells showed mRNA localisation diffusely in both nucleus and cytoplasm. In contrast, approximately 15% of cells exhibited nuclear mRNA accumulation upon induction of GAL1pr:HOS3‐NLS (Fig 4D and Appendix Fig S4A). Nuclear accumulation of mRNA was dependent on Hos3 KDAC activity, as it was not observed upon overexpression of a catalytically inactive mutant (Hos3EN‐NLS) (Fig 4D). Furthermore, inactivation of Esa1 (esa1‐ts) led to accumulation of nuclear mRNA in up to 20% of cells, and this fraction rose to 30% in the double mutant esa1‐ts gcn5∆ (Fig 4E and Appendix Fig S4B). Notably, deletion of GCN5 alone did not cause nuclear mRNA accumulation, suggesting that this KAT does not play an important role in promoting mRNA export. The Esa1 function in RNA export appeared to be specific for mRNA, since depletion of Esa1 (alone or in combination with Gcn5) did not affect export of ribosomal RNA (Appendix Fig S5A and B). Importantly, the fraction of Esa1‐deficient cells with nuclear mRNA accumulation was significantly reduced in esa1‐ts cells carrying the acetyl‐mimic Nup60 mutation (Fig 4F). These results suggest that acetylation and deacetylation of Nup60, mediated by Esa1 and Hos3, respectively, regulate mRNA export.
Nup60 acetylation recruits the TREX‐2 complex to the nuclear basket to promote Start
We previously found that constitutive nuclear localisation of Hos3 (Hos3‐NLS) reduces the amount of NPC‐associated mRNA export factors, such as Sac3, that localise to the nuclear periphery (Kumar et al, 2018). This suggests that in wild‐type cells, Nup60 acetylation facilitates the recruitment of Sac3 to NPCs and this is inhibited in G1 daughter cells via Hos3‐dependent Nup60 deacetylation. To test this prediction, we measured the nuclear intensity of GFP‐tagged Sac3 relative to that of the structural NPC component Nup49‐mCherry in mother (M) and daughter (D) nuclei immediately after cytokinesis. Interestingly, loss of Hos3 (hos3∆) lead to an increase in Sac3 nuclear localisation in D cells, and expression of acetyl‐mimic Nup60 (nup60‐KN) caused an increase in nuclear Sac3 in both M and D cells (Fig 5A, nucleus). Importantly, cellular Sac3 levels were not affected by mutations in HOS3 or NUP60 (Fig 5A, whole cell; see also Appendix Fig S6 for analysis of Sac3 levels by Western blotting). As wild‐type D cells entered S phase (and Nup60 was acetylated), their nuclear Sac3 levels increased; in contrast, Sac3 levels remained constant from G1 to S phase in hos3 and nup60‐KN daughter cells (Fig EV5). We conclude that Hos3 and deacetylation of Nup60 reduce the enrichment of Sac3 in G1 daughter cell nuclei. We then tested whether Esa1 promotes the localisation of Sac3 to the nuclear basket. To avoid potential confounding effects due to Esa1 inactivation during S phase, cells were treated with the microtubule polymerisation inhibitor nocodazole at 25°C, to arrest esa1‐ts cells in mitosis in the presence of Esa1 function. Nocodazole was then removed, cells were shifted to 37°C to inactivate Esa1, and the nuclear intensity of Sac3‐GFP, normalised to that of Nup49‐mCherry, was determined in the following G1. This revealed that nuclear enrichment of Sac3 is significantly reduced in both M and D esa1‐ts cells, while total cellular levels of Sac3 were not affected (Fig 5B and Appendix Fig S6). Together, these data indicate that Nup60 acetylation, driven by Esa1 and inhibited by Hos3 in G1 daughter cells, is important for perinuclear recruitment of the mRNA export factor Sac3.
Figure 5. Nup60 deacetylation in daughter cells displaces Sac3 from NPCs and delays Start.
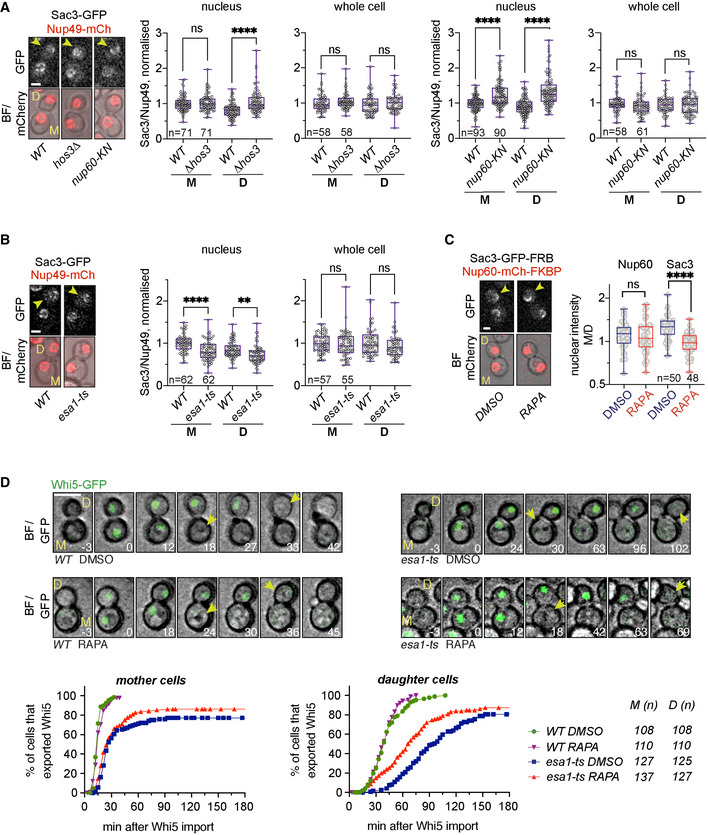
-
ADepletion of Hos3 or expression of acetyl‐mimic Nup60 (nup60‐KN) increases the nuclear localisation of Sac3 in G1. Cells of the indicated strains were imaged by time‐lapse microscopy, and the fluorescence levels of the indicated proteins were determined in G1 (after cytokinesis). The NPC component Nup49 was used as a control for nuclear pore complex protein levels. Fluorescence intensity was measured in sum projections of whole‐cell Z‐stacks, by segmentation of either the nuclear area in the mCherry channel or the whole cell in the bright‐field channel. The ratio of Sac3 to Nup49 intensities was then normalised relative to the mean intensity of wild‐type mothers.
-
BInactivation of Esa1 decreases Sac3 nuclear levels. Wild‐type (WT) and esa1‐ts cells were arrested in mitosis by treatment with nocodazole at 25°C, shifted to 37°C, released from the mitosis block in fresh medium at 37°C and imaged by time‐lapse microscopy. Fluorescence levels were quantified in G1 as in (A).
-
CRapamycin‐dependent dimerisation abolishes Sac3 mother/daughter asymmetries. NUP60‐mCherry‐FKBP SAC3‐GFP‐FRB cells were incubated with rapamycin (RAPA) to trigger FRB‐FKBP heterodimerisation, or with DMSO as control. Fluorescence levels were quantified in G1 cells as in (A), 15 to 30 min after addition of the drug.
-
DSac3 anchoring to the nuclear basket advances Start in esa1‐ts daughter cells. Composite of bright field and Whi5‐mGFP (top) and quantification of Whi5 nuclear export timing (bottom) in wild‐type (WT) and esa1‐ts mother (M) and daughter (D) cells treated with either rapamycin (RAPA) or DMSO and expressing Nup60‐FRB and Sac3‐mCherry‐FKBP. Sac3 anchoring to Nup60 does not alter Whi5 export timing in WT mother or daughter cells (DMSO vs. RAPA, P > 0.05, log‐rank Mantel–Cox test), but it advances Whi5 export in esa1‐ts daughters (P = 0.0001). Whi5 export efficiency was slightly improved also in mother cells (P = 0.0374). 8 z‐confocal slices spaced 0.4 μm were acquired every 3 min; maximum projections are shown. Time is indicated in minutes; t = 0 marks Whi5 nuclear import. Scale bar, 5 μm.
Data information: In (A–C), arrowheads point to daughter cells, and in (D), to Whi5 export. In (A–C), boxes include 50% of data points, the line represents the median, and whiskers extend to maximum and minimum values. ****, P ≤ 0.0001; **, P ≤ 0.01; and ns, P > 0.05, two‐tailed unpaired t‐test. Scale bar, 2 μm. n = number of cells, pooled from three independent experiments with similar results.
Source data are available online for this figure.
Figure EV5. Sac3 recruitment to daughter cell nuclei in G1 and S phase.
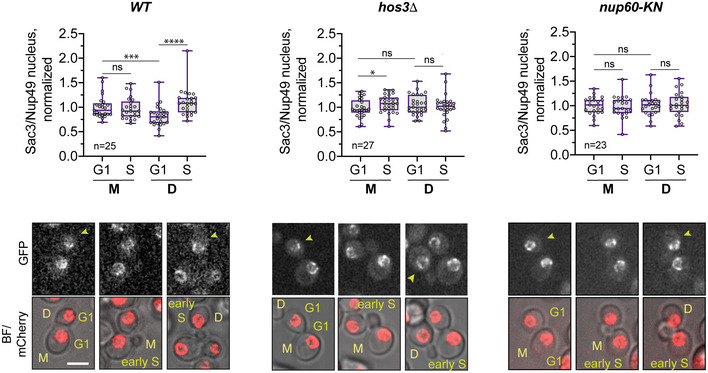
Cells of the indicated strains were imaged by time‐lapse microscopy, and the fluorescence levels of Sac3‐GFP and Nup49‐mCherry were determined in G1 (unbudded cells after cytokinesis) and S phase (cells with small buds). The NPC component Nup49 was used as a control for nuclear pore complex protein levels. Fluorescence intensity was measured in sum projections of whole‐cell Z‐stacks, by segmentation of the nuclear area in the mCherry channel. The ratio of Sac3 to Nup49 intensities was then normalised relative to the mean intensity of wild‐type mothers. Boxes include 50% of data points, the line represents the median, and whiskers extend to maximum and minimum values. ****, P ≤ 0.0001; ***, P < 0.001, *, P ≤ 0.05; and ns, P > 0.05, two‐tailed paired t‐test. Scale bar, 4 μm. n = number of cells, pooled from three independent experiments with similar results. Arrows point to daughter cells.
Source data are available online for this figure.
Sac3 is tethered to the nuclear pore basket, and this localisation is required for its mRNA export function (Fischer et al, 2002). To test whether Esa1 promotes Start by targeting Sac3 to NPCs, we asked whether artificially anchoring Sac3 to the nuclear basket is sufficient to rescue their Start delay in Esa1‐deficient cells. We used the anchor‐away system, which triggers dimerisation of FK506‐binding protein (FKBP) and FKBP–rapamycin binding (FRB) in the presence of rapamycin (Gallego et al, 2013) to anchor Sac3‐FKBP and Nup60‐FRB protein fusions. Consistent with results in Fig 5A, the distribution of fluorescently labelled Sac3‐FRB and Nup60‐FKBP was biased towards mother cells, with a higher bias for Sac3 than for Nup60 (P < 0.005, unpaired t‐test) (Fig 5C). The addition of rapamycin did not alter the accumulation of Nup60‐FKBP in mother cells. In contrast, rapamycin altered the asymmetric localisation of Sac3‐FRB: whereas Sac3 accumulated preferentially in mother cell nuclei in DMSO‐treated cells, it was partitioned equally to M and D nuclei in the presence of rapamycin (Fig 5C). Thus, rapamycin increases the localisation of Sac3‐FRB in daughter cells. G1 duration was then determined by time‐lapse microscopy of esa1‐ts cells expressing Whi5‐GFP, as in Fig 2E, in the presence of either rapamycin or DMSO. Rapamycin slightly increased Whi5 export efficiency in esa1‐ts NUP60‐FRB SAC3‐FKBP mother cells, and specifically advanced Whi5 export in daughter cells (Fig 5D). This is consistent with Hos3‐dependent Nup60 deacetylation in daughter cells (Kumar et al, 2018). We conclude that acetylation of Esa1 and Nup60 promotes Start, at least in part, by targeting Sac3 to the nuclear basket, where it mediates mRNA export. Consistent with the requirement of mRNA export to trigger Start, we find that inactivation of the essential mRNA export factor Mex67 is sufficient to prevent entry into S phase (Appendix Fig S7).
Nup60 acetylation regulates expression of the inducible GAL1 gene
Our results indicate that Nup60 deacetylation inhibits mRNA export and reduces the NPC recruitment of Sac3 to delay Start in daughter cells, presumably by inhibiting the export of mRNAs required for S phase. Next, we asked whether Nup60 acetylation can also affect the expression of genes that are not required for the G1/S transition, such as the inducible galactokinase (GAL1) gene. To measure GAL1 expression, fast‐folding, destabilised GFP (sfGFP) was placed under the control of the GAL1‐10 promoter (GAL1pr) and inserted next to the endogenous GAL1 locus. Thus, measuring GFP fluorescence with time‐lapse microscopy allows tracking of GAL1 expression in single cells. Wild‐type and Nup60 acetyl‐mimic (nup60‐KN) were placed in a microscope chamber, and GAL1 expression was induced with galactose (Fig 6A). GFP fluorescence appeared earlier and increased to higher levels in nup60‐KN than in wild‐type cells, indicating that Nup60 acetylation promotes GAL1 expression (Fig 6B and Appendix Fig S8). Notably, mRNA levels of GAL1 (measured by RT–qPCR) were comparable in wild type and nup60‐KN. This indicates that Nup60 acetylation promotes GAL1 expression at the post‐transcriptional level (Fig 6C). Nup60 levels were equivalent throughout all experiments, suggesting that Nup60 acetylation is unlikely to affect gene expression through changes in Nup60 stability (Appendix Fig S9A and B).
Figure 6. Daughter cell‐specific Nup60 deacetylation inhibits GAL1 expression.
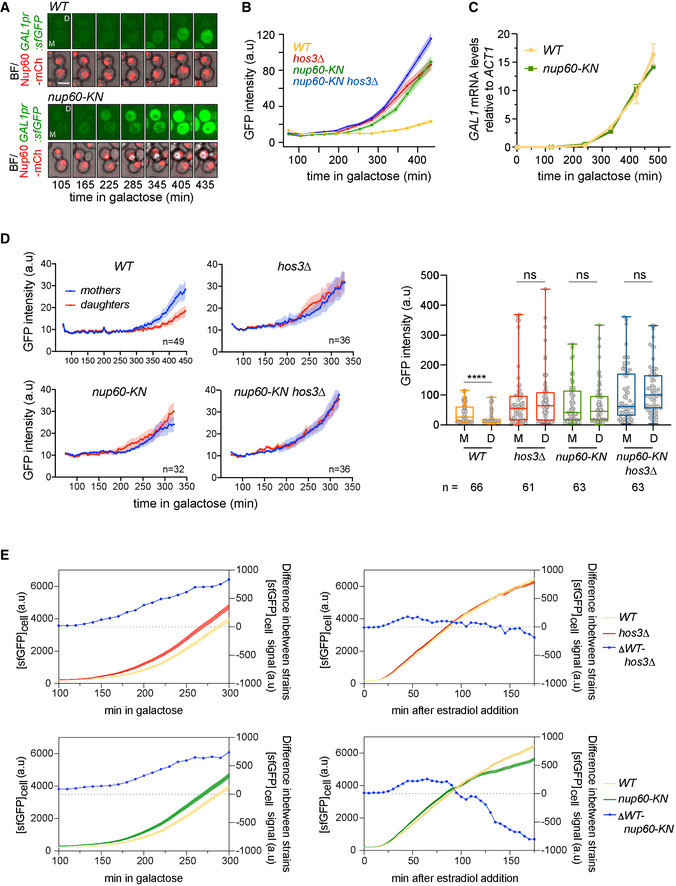
-
ATime‐lapse microscopy of WT and nup60‐KN cells expressing GAL1pr:sfGFP and Nup60‐mCherry at the indicated times of galactose induction. Scale bar, 4 μm.
-
BDepletion of Hos3, and expression of acetyl‐mimic Nup60 (nup60‐KN) enhance GAL1 expression. WT, hos3Δ, nup60‐KN and nup60‐KN hos3Δ cells were shifted to galactose and imaged by time‐lapse microscopy to monitor GAL1pr:sfGFP expression during 7 h. Nuclear fluorescence was scored by segmentation of the nuclear area in the mCherry channel, and mean fluorescence of nuclear GFP and Nup60‐mCherry was quantified from sum projections of whole‐cell Z‐stacks at the indicated times. At least 200 cells were scored for each strain and time point. Shaded areas indicate the SEM.
-
CmRNA levels of GAL1 were determined for wild‐type (WT) and nup60‐KN cells at the indicated times after galactose addition. One of two independent experiments with similar results is shown (mean and SEM from three technical replicates).
-
DThe GFP intensity of mother/daughter pairs for cells in (B), at 5‐min intervals after galactose addition (left) and 425 min after galactose addition (right). Boxes include 50% of data points, the line represents the median, and whiskers extend to maximum and minimum values. ****, P ≤ 0.001; and ns, P > 0.05, two‐tailed paired t‐test. N = number of cells. One of two independent experiments with similar results is shown.
-
EExpression of GAL1pr:sfGFP induced with galactose (left) or ß‐oestradiol (right), in the presence of the ß‐oestradiol‐dependent transactivator Gal4‐ER‐VP16. Smooth lines show GFP fluorescence intensity (left x axis); lines with circles show the difference in GFP intensity between the indicated strains (right x axis). The difference between wild type and hos3∆, and between wild type and nup60‐KN, increases continuously over time in response to galactose, but not to ß‐oestradiol.
Source data are available online for this figure.
Because Nup60 acetylation is inhibited in daughter cells, we tested whether GAL1 expression occurs with different strengths in mothers and daughters. Indeed, GAL1 expression levels were higher in wild‐type mother cells than in their daughters, and these differences were absent in cells lacking Hos3 or expressing Nup60‐KN (Fig 6D). We conclude that acetylation of Nup60 in mother cells promotes GAL1 expression, whereas its deacetylation in daughter cells inhibits expression. Expression of GAL1 was slightly increased in the double mutant hos3∆ nup60‐KN relative to either hos3∆ and nup60‐KN single mutants (Fig 6D). This suggests that Hos3 inhibits GAL1 expression in daughters largely, but not entirely through deacetylation of Nup60 at Lys 467.
Induction of GAL1 by galactose is associated with tethering of its gene locus to the NPC (Cabal et al, 2006). To test whether NPC association is required for GAL1 regulation by acetylated Nup60, we induced the GAL1 gene in wild‐type and nup60‐KN cells with an oestradiol‐dependent hybrid transactivator (the DNA binding domain of Gal4 fused to the hormone‐binding domain of the human oestrogen receptor and the activation domain of the viral transcriptional activator VP16; GEV) that does not induce gene tethering (Appendix Fig S10). In cells expressing the GEV hybrid protein, addition of oestradiol led to similar GAL1 induction profiles in wild‐type, hos3∆ and nup60‐KN cells. This was in contrast to GAL1 expression upon galactose addition, in which hos3∆ and nup60‐KN induced GAL1 more strongly than wild type (Fig 6E and Appendix Fig S11A and B). Note that the effect of hos3 and nup60‐KN on GAL1 expression in response to galactose was milder in GEV than in non‐GEV strains, probably due to differences in genetic background. GEV strains are derived from W303, which carries a functional copy of the galactose permease GAL2. Non‐GEV strains in Fig 6A–D are derived from S288c, which is gal2‐ and relies on lower‐affinity permeases to import galactose (Mortimer & Johnston, 1986; Donnini et al, 1992). Thus, Nup60 acetylation may be particularly important when galactose availability is limited. These data suggest that for optimal regulation of GAL1 expression by Nup60 acetylation, the GAL1 gene must be associated with the NPC. Furthermore, forcing the symmetric distribution of the mRNA export factor Sac3 to the nuclear basket of mother and daughter nuclei using the FRB‐FKBP system (as in Fig 5C) increased GAL1 expression specifically in daughter cells (Fig 7A and B and Appendix Fig S12). These data further support the notion that Hos3‐dependent deacetylation of Nup60 inhibits general mRNA export in daughter cells.
Figure 7. Sac3 anchoring to Nup60 promotes GAL1 expression.
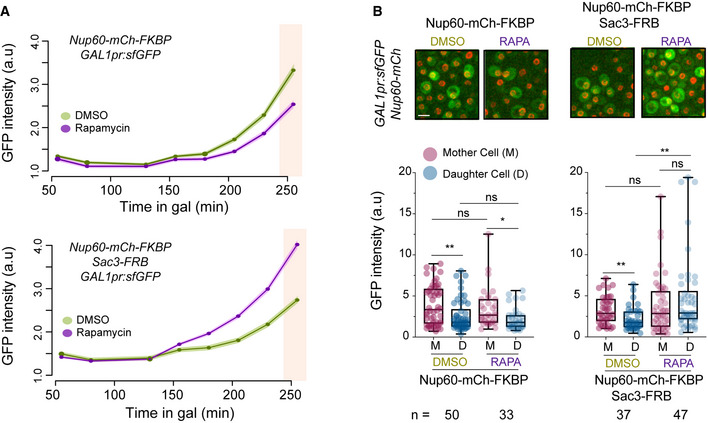
-
ACells of the indicated strains were incubated with galactose in the presence of either rapamycin (RAPA) to induce FRB‐FKBP heterodimerisation, or DMSO as control. GAL1pr:sfGFP expression was monitored over time as in Fig 6B.
-
B(Top) Representative images of the indicated cells in rapamycin or DMSO, 250 min after galactose addition. Scale bar, 4 μm. (Bottom) Mother/daughter pairs were quantified as in Fig 6D at 250 min after galactose addition (pink shaded area in A). Boxes include 50% of data points, the line represents the median, and whiskers extend to maximum and minimum values. **, P ≤ 0.01; *, P ≤ 0.05; and ns, P > 0.05, two‐tailed paired t‐test for M‐D comparisons, unpaired for comparisons between strains. n = number of cells. One of two independent experiments with similar results is shown.
Source data are available online for this figure.
Discussion
Our data indicate that in budding yeast, the lysine acetyltransferase subunit of the NuA4 complex (Esa1) promotes cell cycle entry. In the absence of Esa1, the KAT subunit of the SAGA complex (Gcn5) can partially fulfil this function. Given that the G1/S transition depends on the coordinated expression of hundreds of genes, the involvement of NuA4 and SAGA components in this cell cycle transition is in line with their common role as transcriptional coactivators with known functional overlaps. Indeed, NuA4 and SAGA complexes share the targeting subunit Tra1 (Helmlinger & Tora, 2017) and are thought to drive gene expression mainly through acetylation of histone H3 (for SAGA) and H4 (for NuA4) and subsequent chromatin decompaction (Sterner & Berger, 2000; Lee & Workman, 2007). However, we demonstrate that Esa1 is required not only for gene transcription but also for mRNA export. Also in this case, Gcn5 can partially compensate for the absence of Esa1. Importantly, expression of an acetyl‐mimic Esa1/Gcn5 substrate, the nuclear pore component Nup60, partially alleviates the G1/S transition and mRNA export defects of Esa1‐defective cells. Thus, we propose that Esa1 is the main KAT driving transcription of G1/S genes (most likely through acetylation of chromatin proteins) and that it also promotes mRNA export by acetylation of Nup60. To our knowledge, this is the first time that a KAT is linked to post‐transcriptional regulation of gene expression by modification of NPCs. Esa1 may acetylate other NPC components in addition to Nup60 to promote mRNA export (Henriksen et al, 2012; Kumar et al, 2018). Importantly, we show that Esa1 activity and Nup60 acetylation facilitate the nuclear enrichment of the TREX‐2 complex scaffolding subunit Sac3, which promotes mRNA export at the nucleoplasmic side of the nuclear pores, thereby driving mRNA export and cell cycle entry (Fig 8). Our finding that Nup60 acetylation promotes the Sac3‐dependent expression of GAL1, which is not required for cell cycle entry, further suggests that Esa1 and acetylation of Nup60 are part of a pathway promoting general mRNA export during the entire cell cycle.
Figure 8. Esa1 coordinates mRNA synthesis and export during the G1/S transition through Nup60 acetylation.
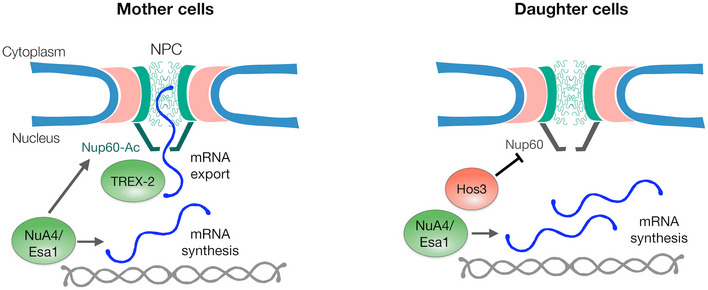
In mother cells, Esa1 promotes both mRNA synthesis and export. Mechanistically, mRNA export is promoted by acetylation of Nup60, which increases the association between the mRNA export factor TREX‐2 and the nuclear pore basket. In daughter cells, Hos3 deacetylates Nup60, which reduces TREX‐2 association with the NPC, and thus mRNA export. Inhibition of Nup60 acetylation in daughter cells contributes to their longer G1 phase, possibly by delaying the export of mRNAs required for entry into S phase such as CLN2, and inhibits the expression of the GAL1 gene in response to galactose.
Our results also indicate that Esa1‐dependent mRNA export, which may be constitutively active in mother cells, is specifically inhibited in daughter cells to restrain their G1/S transition. This inhibition is caused by the KDAC Hos3, which deacetylates Nup60 in G1 daughters, contributing to their prolonged G1 phase and enforcing cell size control. Nup60 acetylation in daughter cells is restored in S phase, presumably due to removal of Hos3 from NPCs in G1 (Kumar et al, 2018), thus allowing resumption of normal mRNA export in daughter cells later in the cell cycle. These observations reveal an additional level of control of the G1/S transition, which is also regulated by the differential scaling of Start inhibitors and activators with cell size (Schmoller et al, 2015; Chen et al, 2020) and by the daughter‐specific inheritance of transcriptional regulators (Di Talia et al, 2009; Kumar et al, 2018). Notably, our data suggest that gene expression at the G1/S transition is controlled not only through transcription but also at the level of mRNA export. We speculate that the Hos3‐Nup60 pathway downregulates mRNA export during G1, because Nup60 deacetylation is largely restricted to this cell cycle phase. Moreover, it is possible that Nup60 deacetylation does not specifically inhibit expression of the G1/S regulon, because Hos3 also inhibits expression of the nutrient‐responsive GAL1 gene.
Esa1‐deficient yeast cells arrest in late S phase or early mitosis in a manner dependent on the DNA damage checkpoint (Clarke et al, 1999). This cell cycle arrest probably masked the role of Esa1 in earlier cell cycle stages, which we reveal here through analysis of both synchronised populations and freely cycling cells. Moreover, our results raise the possibility that DNA damage in esa1‐ts cells may stem (at least in part) from replicative stress during S phase caused by inefficient synthesis and/or export of G1/S mRNAs. Supporting this hypothesis, DNA replication (although delayed) seems to proceed in many unbudded esa1‐ts cells (Fig 1C and D). In addition, lack of Esa1 activity is associated with aberrant nucleolar fragmentation (Clarke et al, 1999). Although the molecular basis of the nucleolar defect in Esa1‐defective cells is unclear, it is interesting that nucleolar fragmentation is also observed after inactivation of mRNA export factors, provided that mRNA synthesis is ongoing (Kadowaki et al, 1994; Schneiter et al, 1995). Thus, nucleolar fragmentation after inactivation of Esa1 may be caused by abnormal accumulation of mRNA in the cell nucleus.
mRNA export factors, including Mex67 and Sac3, contribute to NPC tethering of active yeast genes and may contribute to their optimal expression (Cabal et al, 2006; Dieppois et al, 2006; Brickner et al, 2019). Our data indicate that Esa1 promotes the perinuclear enrichment of Sac3, but whether this localisation affects the interaction of chromosomal loci with NPCs is not known. Interestingly, the CLN2 locus interacts with NPCs specifically in G1, and this interaction is stabilised by Nup60 deacetylation in daughter cells (Kumar et al, 2018). It will be of interest to establish how NPC tethering of G1/S genes such as CLN2 is related to the elongation and export state of their respective mRNAs, and whether stabilisation of gene tethering by Nup60 deacetylation corresponds to primed or active transcriptional conditions.
Whether the role of NuA4 in NPC acetylation and mRNA export is evolutionarily conserved is not known. As in yeast, the TREX‐2 complex is associated with the nuclear pore basket in human cells (Umlauf et al, 2013) and promotes export of a subset of mRNAs together with the basket component TPR (Wickramasinghe et al, 2010, 2014; Aksenova et al, 2020). Furthermore, human nucleoporins are acetylated, including TPR and the Nup60 homologue Nup153 (Choudhary et al, 2009), and TPR physically interacts with Tip60/KAT5, the mammalian homologue of Esa1 (Chen et al, 2013); however, the physiological relevance of these modifications and interactions has not been determined. Our findings raise the possibility that mammalian nucleoporins represent a novel category of substrates for KATs and for the multiprotein complexes in which these enzymes reside, with important roles in gene expression. Testing this possibility, and identifying the molecular mechanisms by which KATs such as NuA4 and SAGA regulate mRNA export in mammalian cells (e.g. by acetylation of non‐histone proteins such as nucleoporins) remain therefore a key subject for future studies.
In summary, these data reveal a novel role in mRNA export for the evolutionarily conserved KAT‐containing coactivator complex, NuA4. They also demonstrate that differences in Nup60 acetylation determined by the interplay between a KAT (in mother cells) and a KDAC (in daughter cells) allow the modulation of mRNA export capabilities of NPCs in different cell types, shaping their gene expression and cell proliferation profiles. Furthermore, our findings on the regulation of GAL1 expression by Nup60 acetylation indicate that differences in NPC acetylation between mother and daughter cells contribute to the development of heterogeneous gene expression responses among a population of genetically identical cells. This type of phenotypic variability (often interpreted as a bet‐hedging strategy) could provide a growth advantage for a clonal population upon sudden changes in environmental conditions (Veening et al, 2008). Thus, while specifically assessing the role of NPC acetylation in cell cycle entry, our study raises the possibility that acetylation of nuclear pores and regulation of mRNA export define an important regulatory step in cell identity establishment. Analogous mechanisms may also contribute to cell differentiation during the development of multicellular organisms.
Materials and Methods
Strains, plasmids and cell growth
Saccharomyces cerevisiae strains are derivatives of S288c or BY4741 except when indicated (Appendix Table Table S1). Gene deletions and insertions of C‐terminal tags were generated by standard one‐step PCR‐based methods (Longtine et al, 1998; Janke et al, 2004). The acetyl‐mimic nup60‐KN mutant was generated using CRISPR/Cas9 to replace the acetylated lysine 467 by asparagine, as described (Kumar et al, 2018). The deacetyl‐mimic nup60‐KR mutant was obtained by homologous recombination with a PCR product generated from a nup60‐KR strain, a gift from Dr. Yves Barral. The esa1‐ts thermosensitive strain carrying the L254P mutation (Clarke et al, 1999) linked to the kanamycin‐resistance cassette KanMX (Li et al, 2011) was used to obtain the esa1‐ts‐kanMX cassette and integrate the ts allele in the corresponding S288c‐derived strains. The mex67‐ts strain is a gift from Dr. M. del Olmo (Estruch et al, 2009).
pBG1805 2μ multicopy plasmids expressing KATs, Mtr2, Mex67 or Sac3 under the control of the GAL1 promoter and carrying a HA C‐terminal tag (Gelperin et al, 2005) were obtained from the Yeast ORF collection (Thermo Scientific Open Biosystems, YSC3868). pGP565 2μ multicopy plasmids (Jones et al, 2008) containing Sac3, Sus1, Cdc31, Sem1 and Thp1 are a gift from Dr. S. Leon (Yeast Genomic Tiling Collection, Open Biosystems). The pUC57 vector containing the superfolder (sf) GFP fused to the CLN2‐PEST degron under the control of GAL1pr (AP2) is described in Goulev et al (2019), and the Rpl25‐GFP plasmid is a gift from Dr. H. Schmidt.
Cells were grown in exponential conditions (below OD600 = 1) at 25°C in standard yeast extract–peptone–dextrose medium supplemented with adenine 70 μg/ml (YPDA) or synthetic complete (SC) medium with 2% glucose, 2% raffinose (SC‐Raf) or 2% galactose (SC‐Gal). Where indicated, cells were incubated in the presence of 15 μg/ml nocodazole and 15 μg/ml α‐factor, or transferred to 37°C. For growth assays in solid media, 10‐fold serial dilutions of exponential cultures were spotted onto SC‐Glu and SC‐Gal medium and incubated at 25°C for 3 days.
For G1 arrest, exponential cells growing in YPDA medium were synchronised with 15 μg/ml α‐factor (GenScript, Cat. No: RP01002) for 2 h at 25°C, supplemented with additional α‐factor (5 μg/ml) and incubated 30 min more at 25°C. Then, cells were shifted to 37°C during 1 h, washed three times with pre‐warmed YPDA and released in fresh pre‐warmed YPDA medium at 37°C. For the nocodazole arrest, cells were incubated during 2 h with nocodazole 15 μg/ml, washed three times and then released in fresh pre‐warmed YPDA medium at 37°C.
For the analyses of GAL1pr‐driven expression of the sfGFP reporter, the GAL1pr:sfGFP‐CLN2‐PEST cassette was integrated between the GAL1 and FUR4 loci using the following oligonucleotides:
GALsfGFP‐Fw (5’‐AAAGTCATTTGCGAAGTTCTTGGCAAGTTGCCAACTGACGtACGGATTAGAAGCCGCCGA‐3′) and GALsfGFP‐Rv (5’‐AGGACAAAAAGTTTCAAGACGGCAATCTCTTTTTACTGCAATGGttagaaaaactcatcgag‐3′) and the AP2 plasmid as a PCR template. For induction of the GAL1pr:sfGFP reporter (Fig 6), exponential cells were grown in glucose, washed three times with SC‐Gal and resuspended in SC‐Gal (2% galactose, 0.1% glucose) for time‐lapse imaging. For Nup60‐GFP IP assays (Fig 2A), cells were grown in glucose until exponential phase, diluted and incubated in SC‐Raf (2% raffinose, 0.1% glucose) overnight until exponential phase, and 2% galactose was added to induce GAL1pr:GCN5‐HA and GAL1pr:ESA1‐HA expression for 2 h.
For analysis of Gal1‐LacO gene tethering, cells were grown in glucose, washed three times with SC‐Gal and resuspended in SC‐Glu, SC‐Glu + 90 μM oestradiol or SC‐Gal for time‐lapse imaging.
For tethering of Sac3 to Nup60, we used inducible dimerisation of FK506‐binding protein (FKBP) and FKBP–rapamycin binding (FRB) domain, as described in Gallego et al (2013). Sac3 and Nup60 were tagged at the C‐terminus. The background of the anchoring strains harbours the tor1‐1 mutation and lacks the endogenous FPR1 gene rendering growth insensitive to rapamycin. Exponential cells were incubated with 20 μM rapamycin (Sigma‐Aldrich, Cat. No R8781) throughout the imaging experiment. Association between Sac3‐GFP‐FRB and Nup60‐mCherry‐FKBP was checked by measuring the disruption of Sac3 asymmetries within 15–30 min of rapamycin addition (Fig 5C). Similar results were obtained for Sac3‐mCherry‐FKBP and Nup60‐FRB. For the induction of GAL1pr:sfGFP, cells were grown in glucose, washed three times with SC‐Gal and shifted to SC‐Gal (2% galactose, 0.1% glucose) with rapamycin or DMSO for the time‐lapse imaging (Fig 7). For inactivation of esa1‐ts, cells were imaged at 37°C at the moment of rapamycin or DMSO addition (Fig 5D).
Fluorescence microscopy
For time‐lapse microscopy, cells were grown overnight in 50‐ml flasks containing 10 ml of SC medium at 25°C, then diluted to OD600 = 0.1–0.3 in fresh medium, grown at least for 4 h to mid‐log phase and plated in minimal synthetic medium on concanavalin A‐coated (Sigma‐Aldrich) Lab‐Tek chambers (Thermo Fisher Scientific). Images were acquired on a Nikon TiE inverted microscope (or on a Leica DMi8 for Fig 3) equipped with a X1 Yokogawa Spinning disk confocal head using a Leica HC PL APO 100× NA1.4 objective. The excitation/emission parameters were as follows: for GFP laser excitation at 488 nm/bandpass emission filter 525/50 nm and for tdTomato laser excitation at 561 nm/bandpass emission filter 605/64 nm. Images were captured with a Photometrics Prime 95B sCMOS camera. Laser power and camera exposure times were modulated depending on the samples. A Tokai Hit Environmental Chamber maintained the sample temperature at 37°C. Time‐lapse series of 4‐μm stacks spaced 0.2–0.3 μm were acquired every 2–5 min. For Sac3‐GFP, a maximum of 13 z‐stacks were taken; for Whi5‐GFP, 8 z‐stacks spaced by 0.4 μm were taken; for Whi5‐tdTomato/CLN2‐PP7, 16 z‐stacks spaced by 0.25 μm were taken; and in case of Whi5‐mCherry due to low protein abundance and poor fluorophore stability, 3 z‐stacks spaced by 0.5 μm were used. Red channel images (Whi5‐mCherry, Whi5‐tdTomato) were subject to Gaussian blur (radius = 2 px), and Whi5‐GFP images were subject to median filtering (radius = 1 px) to remove noise. The images were processed and analysed on 2D maximum or sum projections (unless mentioned otherwise) using Fiji (https://imagej.net/software/fiji/). In Figs 2E and 5D, only cells that imported Whi5 30 min after the start of imaging were included in the analysis. Maximum projections are shown throughout, except in Figs 5A–C and EV5, where sum projections are shown.
Nuclear CLN2‐PP7 focus intensity (Fig 3D) was calculated by determining the mean fluorescence of nuclear foci during Whi5 export in a single confocal Z‐slice, normalised by the mean fluorescence of the whole nucleus in the same slice. Fluorescent levels of Sac3‐GFP, and Nup49‐ or Nup60‐mCherry were determined in background‐subtracted 2D sum projections of whole‐cell Z‐stacks, with the nuclear area defined by Nup49‐mCherry or by Nup60‐mCherry. For Sac3 (Fig 5A), G1 mother and daughter cells were quantified in G1, defined as the first 30–45 min after completion of anaphase and the absence of bud. For the GAL1pr:sfGFP reporter, the GFP mean fluorescence was determined (Fig 6). For mother/daughter measurements (Figs 6D right and 7B), mother/daughter pairs with individual nuclei at the moment of galactose shift were tracked and their fluorescence was measured at the indicated times. For the quantification of GAL1pr:sfGFP activation over time (Figs 6B and 7A), a custom Fiji macros segmented the nuclear signal (Nup60‐mCherry), and after manual correction of ROI, total fluorescence and mean fluorescence of the mCherry and GFP channels were automatically determined for all the individual cells. For measuring GAL1pr‐GFP fluorescence in mother and daughter cells over time (Fig 6D left), sequences of bright‐field / fluorescence confocal images were processed using DetecDiv (preprint: Aspert et al, 2021) as follows: cells were segmented using a pre‐trained deep learning‐based pixel classification model, after training it using 14 manually annotated images. Then, the model was deployed on all (roughly ~1,000) images and slight post‐processing (i.e. small object removal and watershed) was applied to refine cell segmentation. We then used a tracking routine based on the Hungarian method for assignment to map individual cell trajectories over time, which allowed us to quantify the dynamics of their mean cytoplasmic fluorescence.
Conventional epifluorescence microscopy was carried out with a Leica DM4000B widefield microscope equipped with HCX PL APO 100X/1.40 OIL PH3 CS objective, and image acquisition was performed with Hamamatsu ORCA‐Flash4.0 LT digital CMOS camera with the help of Leica Application Suite X (LAS X) software. For DNA staining, cells were fixed for 5 min by addition of 70% ethanol and resuspended in 1 μg/ml DAPI (4′,6‐diamidino‐2‐phenylindole). Budding index was scored manually using the transmitted light images and the cell‐counter plug‐in of Fiji.
Microfluidics
Microfluidics devices (Fig 6E and Appendix Fig S11) were fabricated and handled as in Jacquel et al (2021). Cells were imaged with a Zeiss Axio Observer Z1 microscope. Before image acquisition, cells were washed for 1 h with SC‐Glu media, and afterwards, the media flow was changed either to SC‐Glu + 90 μM oestradiol or to SC‐Gal. Raw images were processed using MATLAB‐based software PhyloCell (Goulev et al, 2017). Imaged cells of all time points were segmented by the software, and the mean intensity of each segment was calculated.
Western blotting
Approximately 10 ml of exponential growing cells (OD600 = 0.3–0.6) was collected, resuspended in 200 μl of 0.1 M NaOH and incubated for 5 min at room temperature. Cells were collected by centrifugation, resuspended in 50 μl of Laemmli buffer and incubated for 5 min at 95°C. Extracts were clarified by centrifugation, and equivalent amounts of protein were resolved in an SDS–PAGE and transferred onto a nitrocellulose membrane. Membranes were blocked with milk powder 5% in TBS–Tween 0.01% or FBS 10% in TBS–Tween 0.1% (anti‐AcLys) and incubated overnight with primary antibodies. Primary antibodies were anti‐HA peroxidase 3F10 (Roche Diagnostics, Cat. No: 12013819001) diluted 1:5,000, anti‐GFP (Roche Diagnostics, Cat. No: 11814460001) diluted 1:5,000, anti‐GAPDH (Thermo Fisher Scientific Cat. No: MA5‐15738) diluted 1:2,000, anti‐G‐6‐PDH diluted 1:20,000 (Sigma, Cat. No: A9521), anti‐AcLys diluted 1:1,000 (Cell Signalling, Cat. No: 9681), and anti‐Cdc28 (anti‐Cdk1/cdc2 (PSTAIR)) (Merck, Cat. No: 06–923) diluted 1:2,000. Blots were developed with anti‐mouse IgG and anti‐rabbit IgG horseradish peroxidase conjugate (Thermo Fisher Scientific, Cat. No: 170–6,516 or 31,460 respectively) diluted 1:20,000 using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Bands were acquired with ImageQuant™ LAS 4000 mini biomolecular imager (GE Healthcare) and quantified in ImageJ.
Protein Immunoprecipitation assays
Approximately 100 ml of exponential growing cells (OD600 = 0.5–0.8) expressing Nup60‐GFP were collected and resuspended in 300 μl of Lysis Buffer (50 mM Tris–HCl, pH 8, 250 mM NaCl, 5 mM EDTA, 1% Triton X‐100, 1 mM PMSF, 2.5 mM benzamidine, and 0.1% (w/v) sodium deoxycholate) with Complete Mini Protease Inhibitor (Roche Diagnostics) and histone deacetylase inhibitors (iHDAC) 5 μM trichostatin A (Sigma‐Aldrich), 10 mM nicotinamide (Sigma‐Aldrich) and 10 mM sodium butyrate (Sigma‐Aldrich). Cells were broken with vigorous shaking in the presence of glass beads, the cellular debris was removed, and the supernatant was clarified by centrifugation at 13,400 g for 5 min. 50 μl Dynabeads Protein G magnetic beads (Invitrogen) were incubated with anti‐GFP antibody (Roche Diagnostics) for 20 min at room temperature and after washing with phosphate‐buffered saline (PBS) containing Tween 0.02%, incubated with the cell extract for 2 h at 4°C. After washing the beads four times with PBS Tween 0.02%, the immunoprecipitated Nup60 was eluted by boiling the beads for 5 min with 100 μl of Laemmli buffer 2× and analysed by SDS–PAGE followed by Western blot analysis.
PolyA fluorescence in situ hybridisation
Cells were grown in SC to exponential phase at 25°C, and the culture was divided into two to incubate half of the culture at 25°C or 37°C or in SC or SC‐gal during the indicated times. Approximately 10 ml of exponential cultures (OD600 = 0.5–1) was fixed with 4% (v/v) formaldehyde (Sigma‐Aldrich) during 15 min at room temperature or 37°C. Cells were collected and resuspended in 0.1 M potassium phosphate (KPi), pH = 6.4/4% formaldehyde and fixated for 1 h in agitation at room temperature. The fixation agent was removed by washing two times with 0.1 M KPi, pH 6.4. Cells were washed one time with ice‐cold washing buffer (0.1 M KPi (pH = 6.4)/1.2 M sorbitol) and resuspended in 200 μl of ice‐cold washing buffer, and subsequently, cell wall was digested by incubating 100 μl of cells with 0.4 mg/ml of Zymolyase 100T (SEIKAGAKU CORPORATION) at 30°C for 5–15 min. Partially spheroplasted cells were recovered by centrifugation (3,000 g for 5 min), washed one time with washing buffer and resuspended in 30 μl 0.1 M KPi (pH = 6.4)/1.2 M sorbitol. Samples were then applied on Teflon slides with wells previously coated with poly‐L‐lysine. Non‐adhering cells were removed by aspiration, cells were rehydrated with 2X SSC (0.15 M NaCl and 0.015 M sodium citrate) and incubated with prehybridisation buffer (formamide 50%, dextran sulphate 10%, 125 μg/ml of Escherichia coli tRNA, 4× SSC, 1× Denhardt's solution and 500 μg/ml herring sperm DNA) for 1 h at 37°C in a humid chamber. The hybridisation was incubated overnight at 37°C in a humid chamber with 20 μl of prehybridisation buffer supplemented with 1 μM of Cy3‐end‐labelled oligo(dT), 1 mM DTT and RNasin (Promega) 4 U/ml. After hybridisation, slides were washed with 2× SSC and 1× SSC at room temperature for 5 min, and subsequently, cells were incubated for 1 min with 2 μl of DAPI 2.5 mg/ml. Cells were washed twice with 1× SSC, washed with 0.5× SSC, air‐dried and mounted with 50% glycerol. Detection of Cy3‐oligo(dT) and DAPI was performed using a Leica DM4000B fluorescence microscope.
Flow cytometry
DNA content analysis was performed according to the protocol from Rosebrock (2017). Briefly, cells were fixed in two volumes of 100% ethanol (overnight −20°C), rehydrated with PBS and digested with proteinase K (20 μg/ml, Thermo Scientific) and RNase A (10 μg/ml) for 2 h at 50°C in the presence of SYTOX Green (0.5 μM, Invitrogen). Flow cytometry was performed on a LSRII (BD) instrument, and the data were analysed with the FlowJo software. Gating strategies are shown in Appendix Fig S13.
RT–qPCR
Samples for gene expression analysis containing approximately 4 × 108 cells were frozen in liquid nitrogen and stored at −80°C. RiboPure‐Yeast (Invitrogen) kit was used to isolate total RNA according to the manufacturer's protocol. Briefly, cells were broken in a Disruptor Genie (Scientific Industries) with Zirconia Beads in Lysis Buffer with one volume of phenol:chloroform. The lysate was centrifuged at 16,100 g for 10 min, and the aqueous phase was separated and mixed with corresponding amounts of 100% ethanol and binding buffer. The resulting solution was drawn through the column filter, which was further washed with wash solutions. RNA was eluted from the filter with 2 × 50 μl of elution buffer and 1 × 50 μl with DEPC‐treated Molecular Biology Grade Water (Merck). RNA samples were quantified with NanoDrop 2000 Spectrophotometer (Thermo Scientific). 2.5 μg of RNA was incubated with ezDNase, and cDNA was obtained with SuperScript IV VILO Master Mix (both Invitrogen) following the manufacturer's instructions. cDNA was analysed in triplicate by quantitative RT–PCR with LightCycler 480 SYBR Green I Master Mix (Roche) using the Roche LightCycler 480 II instrument. mRNA levels of genes of interest were quantified relative to ACT1 mRNA by the ΔCt method.
RNA‐seq
Cells were harvested by centrifugation, and RNA was extracted from snap‐frozen pellets using the RiboPure‐Yeast Kit (Ambion). RNA concentrations were determined using NanoDrop 1000 (Thermo Scientific), while quality and integrity were checked using a Bioanalyzer 2100 (Agilent Technologies). RNA sequencing (RNA‐seq) was performed on a HiSeq 4000 (Illumina). Single‐end reads were preprocessed using cutadapt (Martin, 2011) to remove adapters, polyA and low‐quality sequences. Reads longer than 40 bp were aligned to the reference S. cerevisiae genome (R64) using STAR version 2.5.3a (Dobin et al, 2013). Gene expression was quantified from uniquely aligned reads using HTSeq‐count (Anders et al, 2014), v.0.6.1p1, with annotations from Ensembl release 94 and union mode. TPM was calculated from raw read counts, and average TPM in each condition was performed and transformed using log2.
Statistical methods and reproducibility
Graphs and statistical analysis (two‐tailed unpaired/paired t‐test, one‐sample t‐test, ANOVA with Tukey's multiple comparisons test and log‐rank Mantel–Cox test) were performed with GraphPad Prism software and R. For all the strains used in this work, at least two independent clones were tested with similar results. All data were obtained from at least two independent biological replicates. Each experiment was repeated on at least two different days. For most experiments, at least 100 cells were counted for each replicate (50 cells for time‐lapse microscopy). Given two populations of cells, and two behaviours (0 and 1), a sample size of 100 cells can detect a difference between the two populations with P < 0.05 of more than five cells (more than 5%) and with P < 0.001 of more than 10 cells (more than 10%) (Fisher's exact test).
Author contributions
Mercè Gomar‐Alba: Conceptualization; validation; investigation; visualization; methodology. Vasilisa Pozharskaia: Conceptualization; validation; investigation; visualization; methodology. Bogdan Cichocki: Investigation. Celia Schaal: Investigation. Arun Kumar: Conceptualization. Basile Jacquel: Investigation. Gilles Charvin: Conceptualization; supervision; investigation; methodology. J Carlos Igual: Conceptualization; supervision. Manuel Mendoza: Conceptualization; supervision.
In addition to the CRediT author contributions listed above, the contributions in detail are:
MG‐A, VP, AK, GC, JCI and MM conceptualised the study. MG‐A, VP, BC, CS, BJ and GC investigated the study. MGA, VP, BC, CS, GC and MM performed the formal analysis. MGA, VP and MM wrote the original draft. MGA, VP, GC, JCI and MM wrote, reviewed and edited the manuscript. JCI and MM acquired funding. GC, JCI and MM supervised the study.
Disclosure and competing interest statement
The authors declared that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are grateful to the IGBMC Imaging Centre and Flow Cytometry Facility; Kenny Schumacher for assistance with RT–qPCR; Damien Plassard for analysis of RNA‐seq data; Didier Devys, Gabriel Neurohr, Snezhana Oliferenko, Laszlo Tora and all members of the Mendoza Lab for helpful comments; and Life Science Editors for editorial assistance. This study was supported by Ministerio de Ciencia e Innovación, Spain, and co‐financed by the European Regional Development Fund from the European Union, grant numbers BFU2017‐88692 and PID2020‐119793GB‐I00 to JCI; by the Fondation ARC pour la recherche sur le cancer www.fondation‐arc.org and “Équipe FRM” EQU202003010561 to MM; and by the grant ANR‐10‐LABX‐0030‐INRT, which is a French state fund managed by the Agence Nationale de la Recherche under the frame programme Investissements d'Avenir ANR‐10‐IDEX‐0002‐02 to the IGBMC. MGA was a Postdoctoral Fellow from the Generalitat Valenciana (APOSTD/2017/094) and is a recipient of a Juan de la Cierva Incorporación Fellowship (IJC2018‐036206‐I) from the Ministerio de Ciencia e Innovación, Spain. VP was supported by “Fin de thèse” FRM fellowship FDT202106012921.
The EMBO Journal (2022) 41: e110271.
Data availability
All strains, plasmids and data are available from the authors upon request. RNA‐seq raw data are available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199740
References
- Aksenova V, Smith A, Lee H, Bhat P, Esnault C, Chen S, Iben J, Kaufhold R, Yau KC, Echeverria C, et al (2020) Nucleoporin TPR is an integral component of the TREX‐2 mRNA export pathway. Nat Commun 11: 4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Côté J (1999) NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM‐related cofactor Tra1p. EMBO J 18: 5108–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2014) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspert T, Hentsch D & Charvin G (2021) DetecDiv, a deep‐learning platform for automated cell division tracking and replicative lifespan analysis. bioRxiv 10.1101/2021.10.05.463175 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Baptista T, Grünberg S, Minoungou N, Koster MJE, Timmers HTM, Hahn S, Devys D, Tora L (2017) SAGA is a general cofactor for RNA polymerase II transcription. Mol Cell 68: 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RAM (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner DG, Randise‐Hinchliff C, Lebrun Corbin M, Liang JM, Kim S, Sump B, D'Urso A, Kim SH, Satomura A, Schmit H, et al (2019) The role of transcription factors and nuclear pore proteins in controlling the spatial organization of the yeast genome. Dev Cell 49: 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin RAM, Mcdonald WH, Kalashnikova TI, Yates J, Wittenberg C (2004) Cln3 activates G1‐specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117: 887–898 [DOI] [PubMed] [Google Scholar]
- Bruzzone MJ, Grünberg S, Kubik S, Zentner GE, Shore D (2018) Distinct patterns of histone acetyltransferase and Mediator deployment at yeast protein‐coding genes. Genes Dev 32: 1252–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabal GG, Genovesio A, Rodriguez‐Navarro S, Zimmer C, Gadal O, Lesne A, Buc H, Feuerbach‐Fournier F, Olivo‐Marin J‐C, Hurt EC, et al (2006) SAGA interacting factors confine sub‐diffusion of transcribed genes to the nuclear envelope. Nature 441: 770–773 [DOI] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, Silver PA (2004) Genome‐wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117: 427–439 [DOI] [PubMed] [Google Scholar]
- Charvin G, Oikonomou C, Siggia ED, Cross FR (2010) Origin of irreversibility of cell cycle start in budding yeast. PLoS Biol 8: e1000284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Hung J‐H, Hickman TL, Coles AH, Carey JF, Weng Z, Chu F, Fazzio TG (2013) Hdac6 regulates Tip60‐p400 function in stem cells. elife 2: e01557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhao G, Zahumensky J, Honey S, Futcher B (2020) Differential scaling of gene expression with cell size may explain size control in budding yeast. Mol Cell 78: 359–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M (2009) Lysine acetylation targets protein complexes and co‐regulates major cellular functions. Science 325: 834–840 [DOI] [PubMed] [Google Scholar]
- Clarke AS, Lowell JE, Jacobson SJ, Pillus L (1999) Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol Cell Biol 19: 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M (2004) CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117: 899–913 [DOI] [PubMed] [Google Scholar]
- Di Talia S, Skotheim JM, Bean JM, Siggia ED, Cross FR (2007) The effects of molecular noise and size control on variability in the budding yeast cell cycle. Nature 448: 947–951 [DOI] [PubMed] [Google Scholar]
- Di Talia S, Wang H, Skotheim JM, Rosebrock AP, Futcher B, Cross FR (2009) Daughter‐specific transcription factors regulate cell size control in budding yeast. PLoS Biol 7: e1000221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieppois G, Iglesias N, Stutz F (2006) Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genes. Mol Cell Biol 26: 7858–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnini C, Lodi T, Ferrero I, Algeri A, Puglisi PP (1992) Allelism of IMP1 and GAL2 genes of Saccharomyces cerevisiae . J Bacteriol 174: 3411–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey M, Johnson JR, Davey NE, Newton BW, Johnson TL, Galaang S, Seller CA, Krogan N, Toczyski DP (2015) Acetylome profiling reveals overlap in the regulation of diverse processes by sirtuins, gcn5, and esa1. Mol Cell Proteomics 14: 162–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Côté J (2004) The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev 14: 147–154 [DOI] [PubMed] [Google Scholar]
- Estruch F, Peiró‐Chova L, Gómez‐Navarro N, Durbán J, Hodge C, Del Olmo M, Cole CN (2009) A genetic screen in Saccharomyces cerevisiae identifies new genes that interact with mex67‐5, a temperature‐sensitive allele of the gene encoding the mRNA export receptor. Mol Gen Genomics 281: 125–134 [DOI] [PubMed] [Google Scholar]
- Fischer T, Strässer K, Rácz A, Rodriguez‐Navarro S, Oppizzi M, Ihrig P, Lechner J, Hurt E (2002) The mRNA export machinery requires the novel Sac3p‐Thp1p complex to dock at the nucleoplasmic entrance of the nuclear pores. EMBO J 21: 5843–5852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov MV, Dyson NJ (2004) Molecular mechanisms of E2F‐dependent activation and pRB‐mediated repression. J Cell Sci 117: 2173–2181 [DOI] [PubMed] [Google Scholar]
- Gallego O, Specht T, Brach T, Kumar A, Gavin A‐C, Kaksonen M (2013) Detection and characterization of protein interactions in vivo by a simple live‐cell imaging method. PLoS One 8: e62195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin DM, White MA, Wilkinson ML, Kon Y, Kung LA, Wise KJ, Lopez‐Hoyo N, Jiang L, Piccirillo S, Yu H, et al (2005) Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes Dev 19: 2816–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomar‐Alba M, Mendoza M (2019) Modulation of cell identity by modification of nuclear pore complexes. Front Genet 10: 1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulev Y, Matifas A, Heyer V, Reina‐San‐Martin B, Charvin G (2019) COSPLAY: an expandable toolbox for combinatorial and swift generation of expression plasmids in yeast. PLoS One 14: e0220694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulev Y, Morlot S, Matifas A, Huang B, Molin M, Toledano MB, Charvin G (2017) Nonlinear feedback drives homeostatic plasticity in H2O2 stress response. elife 6: e23971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B, Andres‐Pons A, Kastritis P, Beck M (2019) Structure and assembly of the nuclear pore complex. Annu Rev Biophys 48: 515–536 [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Tora L (2017) Sharing the SAGA. Trends Biochem Sci 42: 850–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen P, Wagner SA, Weinert BT, Sharma S, Bacinskaja G, Rehman M, Juffer AH, Walther TC, Lisby M, Choudhary C (2012) Proteome‐wide analysis of lysine acetylation suggests its broad regulatory scope in Saccharomyces cerevisiae . Mol Cell Proteomics 11: 1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Kaluarachchi S, van Dyk D, Friesen H, Sopko R, Ye W, Bastajian N, Moffat J, Sassi H, Costanzo M, et al (2009) Dual regulation by pairs of cyclin‐dependent protein kinases and histone deacetylases controls G1 transcription in budding yeast. PLoS Biol 7: e1000188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra A, Hetzer MW (2015) Nuclear pore proteins and the control of genome functions. Genes Dev 29: 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquel B, Aspert T, Laporte D, Sagot I, Charvin G (2021) Monitoring single‐cell dynamics of entry into quiescence during an unperturbed life cycle. elife 10: e73186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani D, Valkov E, Stewart M (2014) Structural basis for binding the TREX2 complex to nuclear pores, GAL1 localisation and mRNA export. Nucleic Acids Res 42: 6686–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno‐Borchart A, Doenges G, Schwob E, Schiebel E, et al (2004) A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962 [DOI] [PubMed] [Google Scholar]
- Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G (2008) A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae . Nat Methods 5: 239–241 [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Hitomi M, Chen S, Tartakoff AM (1994) Nuclear mRNA accumulation causes nucleolar fragmentation in yeast mtr2 mutant. Mol Biol Cell 5: 1253–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluarachchi Duffy S, Friesen H, Baryshnikova A, Lambert J‐P, Chong YT, Figeys D, Andrews B (2012) Exploring the yeast acetylome using functional genomics. Cell 149: 936–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehat I, Accornero F, Aronow BJ, Molkentin JD (2011) Modulation of chromatin position and gene expression by HDAC4 interaction with nucleoporins. J Cell Biol 193: 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishkevich A, Cooke SL, Harris MRA, de Bruin RAM (2019) Gcn5 and Rpd3 have a limited role in the regulation of cell cycle transcripts during the G1 and S phases in Saccharomyces cerevisiae . Sci Rep 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knockenhauer KE, Schwartz TU (2016) The nuclear pore complex as a flexible and dynamic gate. Cell 164: 1162–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Sharma P, Gomar‐Alba M, Shcheprova Z, Daulny A, Sanmartín T, Matucci I, Funaya C, Beato M, Mendoza M (2018) Daughter‐cell‐specific modulation of nuclear pore complexes controls cell cycle entry during asymmetric division. Nat Cell Biol 20: 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshakova MM, Krasnov AN, Kopytova DV, Shidlovskii YV, Nikolenko JV, Nabirochkina EN, Spehner D, Schultz P, Tora L, Georgieva SG (2007) SAGA and a novel Drosophila export complex anchor efficient transcription and mRNA export to NPC. EMBO J 26: 4956–4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KK, Workman JL (2007) Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol 8: 284–295 [DOI] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, VanderSluis B, Bellay J, Devit M, et al (2011) Systematic exploration of essential yeast gene function with temperature‐sensitive mutants. Nat Biotechnol 29: 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light WH, Brickner DG, Brand VR, Brickner JH (2010) Interaction of a DNA zip code with the nuclear pore complex promotes H2A.Z incorporation and INO1 transcriptional memory. Mol Cell 40: 112–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnetjournal 17: 10–12 [Google Scholar]
- Mortimer RK, Johnston JR (1986) Genealogy of principal strains of the yeast genetic stock center. Genetics 113: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T, Weinert BT, Choudhary C (2019) Functions and mechanisms of non‐histone protein acetylation. Nat Rev Mol Cell Biol 20: 156–174 [DOI] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Sandikci A, Zou K, Li H, Amon A (2018) Deregulation of the G1/S‐phase transition is the proximal cause of mortality in old yeast mother cells. Genes Dev 32: 1075–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M, D'Angelo MA (2021) Structure, maintenance, and regulation of nuclear pore complexes: the gatekeepers of the eukaryotic genome. Cold Spring Harb Perspect Biol 14: a040691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosebrock AP (2017) Analysis of the budding yeast cell cycle by flow cytometry. Cold Spring Harb Protoc 2017 10.1101/pdb.prot088740 [DOI] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Kõivomägi M, Skotheim JM (2015) Dilution of the cell cycle inhibitor Whi5 controls budding‐yeast cell size. Nature 526: 268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M, Hellerschmied D, Schubert T, Amlacher S, Vinayachandran V, Reja R, Pugh BF, Clausen T, Köhler A (2015) The nuclear pore‐associated TREX‐2 complex employs mediator to regulate gene expression. Cell 162: 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter R, Kadowaki T, Tartakoff AM (1995) mRNA transport in yeast: time to reinvestigate the functions of the nucleolus. Mol Biol Cell 6: 357–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR (2008) Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454: 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood V, Brickner JH (2014) Nuclear pore interactions with the genome. Curr Opin Genet Dev 25: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL (2000) Acetylation of histones and transcription‐related factors. Microbiol Mol Biol Rev 64: 435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strässer K, Bassler J, Hurt E (2000) Binding of the Mex67p/Mtr2p heterodimer to FXFG, GLFG, and FG repeat nucleoporins is essential for nuclear mRNA export. J Cell Biol 150: 695–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn LA, Shen T, Wente SR (2001) The GLFG regions of Nup116p and Nup100p serve as binding sites for both Kap95p and Mex67p at the nuclear pore complex. J Biol Chem 276: 6445–6452 [DOI] [PubMed] [Google Scholar]
- Takahata S, Yu Y, Stillman DJ (2009) The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. EMBO J 28: 3378–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texari L, Dieppois G, Vinciguerra P, Contreras MP, Groner A, Letourneau A, Stutz F (2013) The nuclear pore regulates GAL1 gene transcription by controlling the localization of the SUMO protease Ulp1. Mol Cell 51: 807–818 [DOI] [PubMed] [Google Scholar]
- Turner JJ, Ewald JC, Skotheim JM (2012) Cell size control in yeast. Curr Biol 22: R350–R359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf D, Bonnet J, Waharte F, Fournier M, Stierle M, Fischer B, Brino L, Devys D, Tora L (2013) The human TREX‐2 complex is stably associated with the nuclear pore basket. J Cell Sci 126: 2656–2667 [DOI] [PubMed] [Google Scholar]
- Veening J‐W, Smits WK, Kuipers OP (2008) Bistability, epigenetics, and bet‐hedging in bacteria. Annu Rev Microbiol 62: 193–210 [DOI] [PubMed] [Google Scholar]
- Wang H, Carey LB, Cai Y, Wijnen H, Futcher B (2009) Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol 7: e1000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, Andrews R, Ellis P, Langford C, Gurdon JB, Stewart M, Venkitaraman AR, Laskey RA (2014) Selective nuclear export of specific classes of mRNA from mammalian nuclei is promoted by GANP. Nucleic Acids Res 42: 5059–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, McMurtrie PIA, Mills AD, Takei Y, Penrhyn‐Lowe S, Amagase Y, Main S, Marr J, Stewart M, Laskey RA (2010) mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol 20: 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]