

Abstract
While epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have changed the treatment landscape for EGFR mutant (L858R and ex19del)-driven non-small-cell lung cancer (NSCLC), most patients will eventually develop resistance to TKIs. In the case of first- and second-generation TKIs, up to 60% of patients will develop an EGFR T790M mutation, while third-generation irreversible TKIs, like osimertinib, lead to C797S as the primary on-target resistance mutation. The development of reversible inhibitors of these resistance mutants is often hampered by poor selectivity against wild-type EGFR, resulting in potentially dose-limiting toxicities and a sub-optimal profile for use in combinations. BLU-945 (compound 30) is a potent, reversible, wild-type-sparing inhibitor of EGFR+/T790M and EGFR+/T790M/C797S resistance mutants that maintains activity against the sensitizing mutations, especially L858R. Pre-clinical efficacy and safety studies supported progression of BLU-945 into clinical studies, and it is currently in phase 1/2 clinical trials for treatment-resistant EGFR-driven NSCLC.
Introduction
In 2020, lung cancer accounted for 18% of deaths caused by cancer, making it the leading cause of cancer mortality globally.1 Non-small-cell lung cancer (NSCLC) accounts for the majority of these cases (80–85%), and mutations in the kinase domain of the epidermal growth factor receptor (EGFR) are oncogenic drivers in a subset of this disease, adenocarcinoma.2−4 The most common EGFR-activating mutations (EGFR+) in NSCLC are deletions in exon 19 (ex19del) of the EGFR gene and a single point mutation in exon 21 (L858R).5,6 Tyrosine kinase inhibitors (TKIs) developed over the past two decades that target mutated EGFR have shown superiority in treating patients with EGFR-positive NSCLC over chemotherapy and are now considered the standard of care in this area.7−9
The first-generation EGFR TKIs developed for NSCLC were reversible ATP-competitive inhibitors such as gefitinib (Figure 1).10−13 Although targeted therapies such as these show improved durations of survival, after 1–2 years patients experience a recurrence of disease due to acquired resistance to these inhibitors.14 The most common acquired resistance is the formation of a secondary mutation to the gatekeeper residue (T790M), which accounts for 50–70% of resistance cases.15−18 This gatekeeper mutation is also the prevailing acquired resistance mechanism to afatinib, an irreversible second-generation EGFR inhibitor.19 In addition to acquired resistance, first- and second-generation EGFR inhibitors suffer from limited therapeutic windows, which can be attributed to toxicity caused by wild-type (WT) EGFR inhibition.20−22
Figure 1.

Selected FDA-approved first-, second-, and third-generation EGFR tyrosine kinase inhibitors.
Since the identification of the T790M resistance mutation and WT-EGFR-driven toxicities, several newer EGFR TKIs have been developed to address these issues.23−25 Osimertinib, an irreversible inhibitor that targets C797 in the EGFR active site, is currently the only widely used third-generation EGFR TKI and has inhibitory activity against the T790M resistance mutation as well as the primary, sensitizing mutations (Figure 1).26,27 While osimertinib shows clear patient benefit in first- and second-line settings, acquired resistance also emerges over time.28−30 The most commonly occurring EGFR-dependent resistance mutation resulting from treatment with osimertinib is the C797S mutation, which disrupts covalent binding of the inhibitor, resulting in progression of the disease.31−35
Currently, when patients progress on osimertinib in second-line settings, the resulting EGFR+/T790M/C797S mutant is undruggable with currently available EGFR TKIs.36 Since EGFR is an extensively studied oncogenic target for kinase inhibitors and the NSCLC resistance mutations are well known, there have been several recent publications describing a variety of inhibitors with activity against EGFR+/T790M and EGFR+/T790M/C797S mutant EGFR that do not rely on covalent inhibition (selected examples shown in Figure 2).37−49
Figure 2.

Selected examples of published mutant-selective EGFR inhibitors.
Genentech has documented the optimization of several reversible WT-sparing EGFR inhibitors targeting EGFR+/T790M, shown in Figure 2, based on aminopyrimidine (1, 2) and pyrazole hinge binders (3).50,51 An additional example to address EGFR resistance is the combination of brigatinib (Figure 2) with cetuximab (anti-EGFR antibody), which has shown significant suppression of tumor growth in an EGFR-ex19del/T790M/C797S mouse xenograft model derived from the PC9 cell line.52 Pre-clinically this combination demonstrated superior activity when compared to brigatinib monotherapy and was well tolerated in both treatment groups. There has also been some progress in the use of allosteric inhibitors to overcome EGFR resistance and WT selectivity issues faced by ATP-competitive inhibitors. EAI045 (Figure 2) shows high WT selectivity and potency against the EGFR L858R and EGFR L858R/T790M mutant but unfortunately is not active against EGFR ex19del.43 Most recently, Engelhardt et al. detailed the discovery of the conformationally restricted macrocycle to target EGFR+/T790M/C797S mutant EGFR, BI-4020 (Figure 2), which shows WT selectivity similar to that of osimertinib.53 Despite several publications detailing next-generation EGFR TKIs, there are currently no approved agents that target EGFR+/T790M and EGFR+/T790M/C797S.
Given the high unmet medical need for an effective next-generation EGFR inhibitor, we set out to develop a best-in-class, WT-sparing, potent, EGFR TKI with activity against the EGFR+/T790M and EGFR+/T790M/C797S mutants. Since inhibition of WT-EGFR with approved TKIs is known to cause adverse events, including skin rashes and gastroenterological side effects,20−22 discovery efforts began with the aim of identifying EGFR+/T790M and EGFR+/T790M/C797S inhibitors with increased selectivity over WT-EGFR.
Results and Discussion
Blueprint Medicines’ proprietary compound library contains >25,000 agnostically designed small-molecule kinase inhibitors spanning more than 100 diverse scaffolds. The majority of these molecules have been screened up-front against a large portion of the human kinome (>400 kinases, using the KINOMEscan screening platform).54 This compound library and the associated data allow us to quickly identify chemical starting points for new programs from multiple scaffolds, circumventing traditional high-throughput screening. From this data set we identified 4, which showed moderate potency against EGFR+/T790M and EGFR+/T790M/C797S paired with good metabolic stability and excellent selectivity over WT-EGFR in biochemical and cellular assays.
With a selective library hit in hand, we started to explore various vectors with the aim of improving EGFR+/T790M and EGFR+/T790M/C797S mutant potency. Although most modifications to the piperidinol ring led to reduced activity, we found that installation of a fluorine next to the alcohol led to a 2-fold improvement in potency without diminishing WT selectivity (5, Table 1), similar to findings from Genentech.50 Unfortunately, exploration of substituents off of the amide or the amine of the pyridine to improve potency was mostly unsuccessful. Meanwhile, we proposed that cyclization through the amide carbonyl and aminopyridine NH to yield a 2,7-napthyridine would mimic the proposed intramolecular hydrogen bond of 5 and generate a structurally similar scaffold for additional structure–activity relationship (SAR) exploration. We were pleased to see that cyclized compound 6 showed a significant increase in biochemical and cellular potency while, importantly, maintaining WT selectivity. Compound 6 was a promising early lead and, while achieving excellent selectivity over EGFR WT, displayed only moderate kinome selectivity, with S(10) at 3 μM = 0.14.55
Table 1. Properties of EGFR Inhibitors 4–6a.

| compound |
|||
|---|---|---|---|
| 4 | 5 | 6 | |
| Enz EGFR LR/TM IC50 (nM) | 290 | 99 | 50 |
| Enz EGFR LR/TM/CS IC50 (nM) | 266 | 105 | 46 |
| Enz EGFR WT IC50 (nM) | >10 000 | >10 000 | >10 000 |
| pEGFR H1975 LR/TM IC50 (nM) | 914 | 420 | 67 |
| pEGFR A431 WT (nM) | >25 000 | >25 000 | >25 000 |
| HLM Clint (μL/min/mg) | 4.0 | 9.1 | 19.0 |
Biochemical assays using different EGFR variants measure inhibition in the presence of 1 mM ATP, and compounds were incubated with enzymes for 10 min before ATP and peptide substrate were added (for more details see Experimental Section). EGFR LR/TM means EGFR L858R/T790M, and EGFR LR/TM/CS means L858R/T790M/C790S. HLM Clint is the measurement of intrinsic clearance obtained from isolated human liver microsomes. H1975 is a human lung cancer cell line harboring the EGFR L858R/T790M mutation. A431 is a cell line in which EGFR is amplified.
In addition to improving kinome selectivity, further optimization around 6 was focused on improving potency and metabolic stability. In order to aid in optimization, we obtained an X-ray structure of 7, a closely related analog, in EGFR L858R/T790M protein. The X-ray structure of compound 7 shows several key interactions (Figure 3): (1) an expected two-point interaction with the hinge, the amino-naphthyridine NH making a hydrogen bond to the backbone carbonyl of Gln791 and naphthyridine N2 forming a hydrogen bond with the backbone NH of Met793; (2) a hydrogen-bonding interaction in the back-pocket between the piperidinol and Lys745; and (3) a weak interaction (3.5 Å) between the NH of the methyl amine in the front-pocket and the backbone carbonyl of Met793. This interaction is of note, since typically the Met793 backbone carbonyl is involved in an intramolecular hydrogen bond with the NH of Gly796. Here that hydrogen bond is broken, and the carbonyl is rotated into the binding pocket and closer to the inhibitor NH, providing a more polar environment. Additionally, similar to reports by Hanan et al., there is a favorable hydrophobic interaction between the piperidine group and M790, the gatekeeper residue, which is mutated from WT-EGFR.51,56
Figure 3.

Compound 7 (left) and X-ray crystal structure of 7 (right) in the ATP pocket of the EGFR L858R/T790M protein (PDB: 8D73).
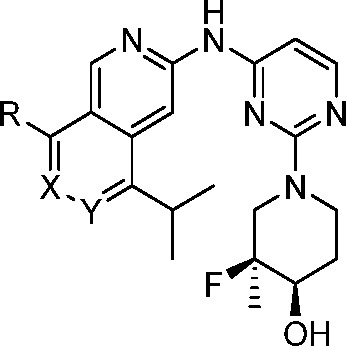
From initial lead 6, further expansion of the back-pocket piperidinol revealed that the addition of a methyl group at the fluorine-containing stereocenter provided an improvement in potency: pEGFR IC50 in the H1975 cell line = 20 nM (8, Table 2). The rest of the 2,7-naphthyridine SAR investigations were performed using this optimized back-pocket piece. Previous internal SAR efforts had demonstrated that removing the hydrogen bond donor from NH-alkyl R groups could significantly enhance kinome selectivity. Replacing the methylamine with azetidine 9 indeed led to improved kinome selectivity (S(10) at 3 μM = 0.037) while maintaining the favorable EGFR potency and WT selectivity of 8. Removal of the NH is thought to disrupt the hydrogen bond from the inhibitor to the carbonyl of Met793, restoring the carbonyl to its canonical position in hydrogen-bonding distance to the Gly796 NH. Meanwhile, a bulkier methylene is now directed toward the side chain of Leu792. Since approximately 70% of kinases have a larger residue at this position, commonly Phe or Tyr, the additional inhibitor bulk likely creates a steric clash with these kinases. Increasing the size of the ring to pyrrolidine 10 further increases kinome selectivity (S(10) at 3 μM = 0.022) but in turn leads to decreased potency on EGFR mutants of interest. Turning our focus back to modifications of the azetidine ring, we found the addition of a chiral methyl group in 11 resulted in a meaningful improvement in potency (pEGFR H1975 = 7 nM). While the removal of the NH and additional methyl group proved to be beneficial for kinome selectivity and potency, the additional lipophilicity led to higher clearance in human microsomes (Clint = 80 μL/min/mg).
Table 2. Structure–Activity Relationship of Selected Analogs of Compound 6.

DiscoverX’s KINOMEscan selectivity profiling at 3 μM, S(10) = (number of non-mutant kinases with %Ctrl < 10)/(number of non-mutant kinases tested).
Biochemical assays using different EGFR variants measure inhibition in the presence of 1 mM ATP, and compounds were incubated with enzymes for 10 min before ATP and peptide substrate were added (for more details see Experimental Section). EGFR LR/TM = EGFR L858R/T790M, EGFR LR/TM/CS = L858R/T790M/C790S, and ex19/TM/CS = ex19del(746–750)/L858R/C790S.
H1975 is a human lung cancer cell line harboring the EGFR L858R/T790M mutation.
A431 is a cell line in which EGFR is amplified.
Intrinsic clearance obtained from isolated human liver microsomes.
LipE = −log(LRTMCS Enz IC50) – log D (calculated from ACD labs @ pH = 7.4). ND: not determined.
We explored replacing the 2,7-naphthyridine with a more polar heterocyclic group to improve metabolic stability, but pyrido[3,4-d]pyridazine 13 led to a loss in potency and LipE compared to 11. We then identified the 3-position of the azetidine ring as a vector to explore the addition of polar substituents to tune properties. 3-Hydroxylazetidine 15 led to a 1.4-unit improvement in LipE but a 2-fold loss in potency. Re-introduction of the 2-methyl group to give 16 recovered the potency loss and maintained the improvement in LipE and metabolic stability (Clint = 25 μL/min/mg). While 16 was one of the most promising compounds to date, we surveyed several other polar substituents on the azetidine, targeting improved metabolic stability. While most modifications did not show improvements over the 3-hydroxyl (compounds 19–24, Table 2), sulfone-containing 24 was distinctly superior, with excellent stability and potency compared to azetidine 9. The significant potency against EGFR+/T790M and EGFR+/T790M/C797S and high WT-EGFR selectivity (335-fold selective, pEGFR WT/LRTM) prompted us to obtain a co-crystal of 24 in the kinase domain of EGFR L858R/T790M to understand this increase in potency (Figure 4). The X-ray crystal structure revealed this improvement could be attributed to the S(O) of the sulfone hydrogen-bonding to Lys716 and Lys728 in the front-pocket, stabilized by a nearby helix from the kinase domain carboxy terminus, which is not commonly observed in published EGFR structures.
Figure 4.
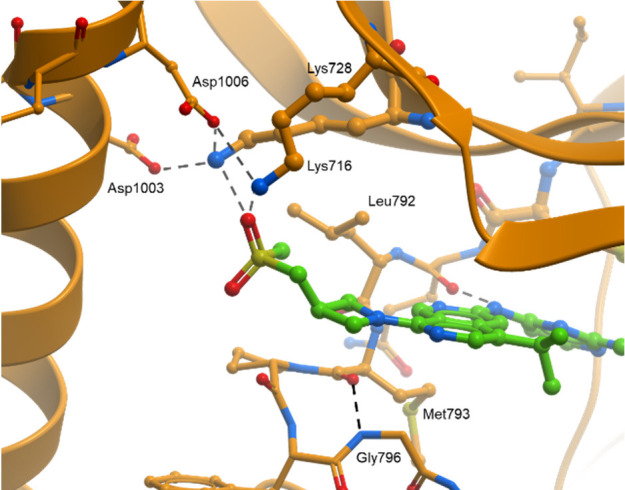
X-ray co-crystal structure of 24 in EGFR LR/TM, highlighting interactions of the sulfone substituent with Lys716 and Lys728 (PDB: 8D76).
To further assess the potential to progress compound 24, it was evaluated in vivo. Rat pharmacokinetics (PK) studies showed that the intravenous clearance was close to hepatic blood flow (∼70 mL min–1 kg–1), and the compound suffered from low oral bioavailability (Table 3). Assessment of 24 in the MDCK-MDR1 assay revealed that the compound has poor passive permeability and a high efflux ratio, indicating a risk of P-gp-mediated active efflux.57 In order to mitigate this potential risk, we added a methyl group on the azetidine ring, with the aim of increasing the steric bulk around the polar substituent to reduce efflux. The additional methyl group of 25 did reduce efflux but likely not enough to have a substantial impact on bioavailability, and therefore was not profiled further. We then attempted to reduce the TPSA of the core by replacing one of the nitrogen atoms with carbon to give isoquinoline 26, which resulted in a superior MDCK-MDR1 profile. The improvement in passive permeability and reduction in efflux resulted in an improved rat PK profile (F = 85%, Cl = 20 mL min–1 kg–1) while having little effect on cellular EGFR L858R/T790M potency. In the earlier SAR investigation, combination of a 2-methyl on the azetidine with the isoquinoline core resulted in a loss in potency (16 vs 18, Table 2). We were pleased to see that this was not the case with the sulfone azetidine, and the additional methyl group in 27 gave an improvement in potency and WT selectivity.
Table 3. Strategies to Improve Bioavailabilitya.

| compound |
||||
|---|---|---|---|---|
| 24 | 25 | 26 | 27 | |
| Enz EGFR LR/TM IC50 (nM) | 0.1 | 0.2 | 0.2 | 0.3 |
| Enz EGFR LR/TM/CS IC50 (nM) | 0.1 | 0.2 | 0.2 | 0.2 |
| Enz EGFR WT IC50 (nM) | 1050 | 270 | 385 | 505 |
| pEGFR H1975 LR/TM IC50(nM) | 4.8 | 1.7 | 2.7 | 1.0 |
| pEGFR A431 WT IC50 (nM) | 1608 | 781 | 1362 | 1780 |
| MDCK-MDR1 PA-B/efflux | 2/32 | 5/16 | 17/4 | 9/3 |
| rat IV PKb Cl (mL min–1 kg–1) (Clu)c, t1/2, F (%) | 67 (838), 1.6 h, 2% | – | 20 (833), 3.0 h, 85% | 25 (847), 1.3 h, 50% |
Biochemical assays using different EGFR variants measure inhibition in the presence of 1 mM ATP, and compounds were incubated with enzymes for 10 min before ATP and peptide substrate were added (for more details see Experimental Section). EGFR LR/TM means EGFR L858R/T790M, and EGFR LR/TM/CS means L858R/T790M/C790S. HLM Clint is the measurement of intrinsic clearance obtained from isolated human liver microsomes. H1975 is a gefitinib resistance human cancer cell line harboring the EGFR L858R/T790M mutation. A431 is a cell line in which EGFR is amplified.
Sprague–Dawley rats (n = 3) were dosed at 1 mg/kg IV and 5 mg/kg PO dose using the following formulations. For 24: IV, solution of 10% DMSO, 10% solutol, 80%–“20% HP-β-CD in water” PO; suspension of “20% solutol in “0.5% MC in water”. For 26 and 27: IV and PO solution of 10% DMSO, 10% solutol, 80% “20% HP-β-CD in saline”.
Clu: unbound in vivo clearance (in vivo rat clearance/free fraction in rat), free fraction calculated from plasma protein binding determined by ultracentrifugation method.
Cynomolgus monkey (cyno) PK was obtained for compound 27 and revealed a poor in vitro/in vivo correlation (determined using incubational and PPB corrections and the well-stirred model58) with respect to the moderate cyno microsomal intrinsic clearance (Clint = 55 μL/min/mg) observed in vitro but high in vivo clearance (Cl = 31.8 mL min–1 kg–1). In vitro profiling of 27 across species in hepatocytes revealed relatively high turnover in human and cyno (Table 4). This indicated that 27 was likely subject to phase II metabolism, which could be driving the high in vivo cyno clearance. Subsequent cross-species hepatocyte metabolite identification studies revealed that 27 was subject to UGT-mediated glucuronidation in human and cyno hepatocytes but not in rats or dogs, and suggested the site of glucuronidation as the hydroxyl of the piperidinol.
Table 4. In Vitro/In Vivo Profile of Compound 27a.
| 27: MW = 557, TPSA = 112, log D7.4 = 3.8 | |
|---|---|
| human LM Clint/Hep Clint | 7.2/41 |
| rat LM Clint/Hep Clint | 43/33 |
| dog LM Clint/Hep Clint | 20/21 |
| cyno LM Clint/Hep Clint | 55/80 |
| cyno IV PK Cl (mL min–1 kg–1)b (Clu)c | 31.8 (611) |
| cyno IV PK t1/2 (h) | 2.9 |
LM Clint represents intrinsic clearance obtained from isolated microsomes (units = μL/min/mg) with NADPH, and Hep Clint represents intrinsic clearance obtained from isolated hepatocyte cells (units = μL/min/million cells).
Cynomolgus monkey, 0.5 mg/kg IV and 5 mg/kg PO dose. For IV and PO dosing, 27 was formulated in 5% DMSO + 5% kolliphor HS 15 + 90% saline vehicle. Cl: plasma clearance after administration of single IV bolus dose.
Clu: unbound in vivo clearance (in vivo cyno clearance/free fraction in cyno), free fraction calculated from plasma protein binding determined by ultracentrifugation method.
The next focus of optimization was aimed at reducing UGT-mediated clearance by modifying the site of gluceronidation (Table 5). Since this clearance mechanism was not observed in rats, we used hepatocytes (human and cyno) and IV PK (cyno) to monitor for improved clearance. Increasing steric bulk adjacent to the site of glucuronidation via the ethylated analog 28 did not improve the hepatocyte stability and was not profiled further. Moving the steric bulk closer to the hydroxyl group, 29 resulted in lower turnover in cyno hepatocytes, which translated into reduced cyno PK clearance (Cl = 8.8 mL min–1 kg–1); however, this modification led to a decrease in EGFR+/T790M potency. Methylation of the hydroxyl to block the site of glucuronidation in 30 led to a compound with an improved in vivo profile while maintaining excellent potency and EGFR WT selectivity. The methylated analog was promising but had lower microsomal stability, a result of the increased lipophilicity. In order to build back in polarity and further improve clearance, 31 was made. The additional hydroxyl in this case was not subject to glucuronidation, but ultimately this compound led to elevated cyno clearance compared to 30, despite the improved in vitro profile. Based on the improved in vivo profile, compound 30 was chosen for further characterization to determine its potential as a development candidate.
Table 5. Lead Optimization to Mitigate UGT-Mediated Clearance.

Intrinsic clearance obtained from isolated human microsomes (units = μL/min/mg).
Intrinsic clearance obtained from isolated human hepatocyte cells (units = μL/min/million cells).
Intrinsic clearance obtained from isolated cyno microsomes (units = μL/min/mg).
Intrinsic clearance obtained from isolated cyno hepatocyte cells (units = μL/min/million cells).
Cynomolgus monkey, 0.5 mg/kg IV and 2.5 mg/kg PO dose. For IV and PO dosing, 29, 30, and 31 were formulated in 5% DMSO + 5% kolliphor HS 15 + 90% saline vehicle; Cl: plasma clearance after administration of single IV bolus dose.
Clu: unbound in vivo clearance (in vivo cyno clearance/free fraction in cyno), free fraction calculated from plasma protein binding determined by ultracentrifugation method.
In enzymatic assays, 30 displays sub-nanomolar activity across the EGFR+/T790M and EGFR+/T790M/C797S mutants and maintains activity against the EGFR-activating mutations (L858R, ex19del), especially EGFR L858R. Compound 30 showed excellent inhibitory activity against the pEGFR H1975 cell line (IC50 = 1.1 nM) with about 500-fold greater potency than in pEGFR A431, the EGFR-WT amplified cell line. Additionally, 30 potently inhibited EGFR phosphorylation in Ba/F3 engineered cell lines (L858R/T790M/C797S IC50= 3.2 nM, and ex19del/T790M/C797S IC50 = 4.0 nM).
Consistent with previous analogs, 30 was found to have a high level of kinome selectivity, S(10) = 0.010.55 Pre-clinical PK profiles for 30 in rat, dog, and cyno are also summarized in Table 6. Compound 30 showed low to moderate clearance, moderate volume of distribution, and good oral bioavailability across species. The human PK profile of 30 was predicted using a combination of in vitro/in vivo extrapolation from hepatocyte data and in vivo-scaling methodology based on the non-clinical PK.59,60 These different methodologies showed a high level of agreement and predicted 30 to have low clearance (<25% QH) and a terminal half-life of 6–7 h.
Table 6. In Vitro and In Vivo Profile of Compound 30a.
| 30: MW = 556/TPSA = 102/log D7.4 = 3.8 | |
|---|---|
| S(10) @ 3 μMb | 0.010 |
| Enz LR IC50 (nM) | 7.4 |
| Enz LR/TM IC50 (nM) | 0.4 |
| Enz LR/TM/CS IC50 (nM) | 0.5 |
| Enz ex19del IC50 (nM) | 25 |
| Enz ex19del/TM IC50 (nM) | 0.8 |
| Enz ex19del/TM/CS IC50 (nM) | 0.7 |
| Enz WT IC50 (nM) | 683 |
| pEGFR PC-9 ex19del IC50 (nM) | 130 |
| pEGFR H1975 LR/TM IC50 (nM) | 1.1 |
| pEGFR A431 WT IC50 (nM) | 544 |
| Ba/F3-EGFR-LR/TM/CS IC50 (nM) | 3.2 |
| Ba/F3-EGFR-ex19del/TM/CS IC50 (nM) | 4.0 |
| thermodynamic solubility at pH 1.5/6.5 (mg/mL) | 11/0.01 |
| plasma protein binding (%fu): human, rat, cyno, dogc | 1.1, 0.9, 3.5, 1.4 |
| rat: Clp (mL min–1 kg–1), t1/2 (h), Vss (L/kg), %Fd | 32.4, 1.3, 2.3, 48% |
| cyno: Clp (mL min–1 kg–1), t1/2 (h), Vss (L/kg), %Fe | 4.9, 3.5, 1.6, 33% |
| dog: Clp (mL min–1 kg–1), t1/2 (h), Vss (L/kg), %Ff | 13.7, 4.9, 3.9, 49% |
Biochemical assays using different EGFR variants measures inhibition in the presence of 1 mM ATP, and compounds were incubated with enzymes for 10 min before ATP and peptide substrate were added (for more details see Experimental Section). EGFR LR/TM = EGFR L858R/T790M, EGFR LR/TM/CS = L858R/T790M/C790S, ex19/TM = ex19del(746–750)/L858R, and ex19/TM/CS = ex19del(746–750)/L858R/C790S. PC-9 is a human lung cancer cell line harboring the EGFR ex19del(746–750) mutation. H1975 is a human lung cancer cell line harboring the EGFR L858R/T790M mutation. A431 is a cell line in which EGFR is amplified. Ba/F3 cells are transduced with lentiviral particles encoding for mutant EGFR.
DiscoverX’s KINOMEscan selectivity profiling at 3 μM, S(10) = (number of non-mutant kinases with %Ctrl < 10)/(number of non-mutant kinases tested).
Plasma–protein binding was determined by an ultracentrifugation method.
Sprague–Dawley rats (n = 3); IV dose = 1 mg/kg using 10% DMSO, 10% solutol, 80% “20% HP-β-CD in water” and PO dose = 2.5 mg/kg, solution of 10% DMSO, 10% solutol, 80% “20% HP-β-CD in water”.
Cynomolgus monkey (n = 3), IV dose = 0.5 and PO dose = 2.5 mpk using formulation vehicle: solution of 5% DMSO + 5% kolliphor HS 15 + 90% saline.
Beagle dogs, IV dose = 0.5 mg/kg using 5% DMSO + 5% kolliphor HS 15 + 90% saline and PO dose = 2.5 mg/kg using suspension of 0.5% (w/v) CMC-Na + 0.1% (v/v) Tween 80 in Milli-Q water.
The activity of 30 was next evaluated in a series of in vivo tumor models. Compound 30 was tested in PK/PD studies using the NCI-H1975 EGFR L858R/T790M-driven mouse tumor model, which confirmed it potently inhibits EGFR L858R/T790M phosphorylation, with an unbound IC50 of 0.6 nM (Figure 5A), in good agreement with the cellular IC50 (Table 5).
Figure 5.

(A) PK/PD measurements in plasma and PK/PD relationship of 30 in the NCI-H1975 tumor model. (B) Activity in the NCI-H1975 tumor model. (C) NCI-H1975 tolerability in female BALB/c nude mice.
The in vivo activity of 30 was evaluated in mice bearing NCI-H1975 xenografts (Figure 5B). Compound 30 at 30 mg/kg BID resulted in tumor stasis, while 100 mg/kg BID of 30 BID led to tumor regression over 14 days of dosing, on par with the anti-tumor activity of osimertinib (25 mg/kg QD) in this model. Both doses were well tolerated, without any significant weight loss in the animals (Figure 5C). The activity of 30 was subsequently assessed in engineered Ba/F3 EGFR L858R/T790M/C797S and Ba/F3 ex19del/T790M/C797S osimertinib-resistant tumor models (Figure 6A,B). In both models, the 100 mpk BID dose of 30 produced strong tumor regression while, as predicted, osimertinib did not show any anti-tumor effects.
Figure 6.

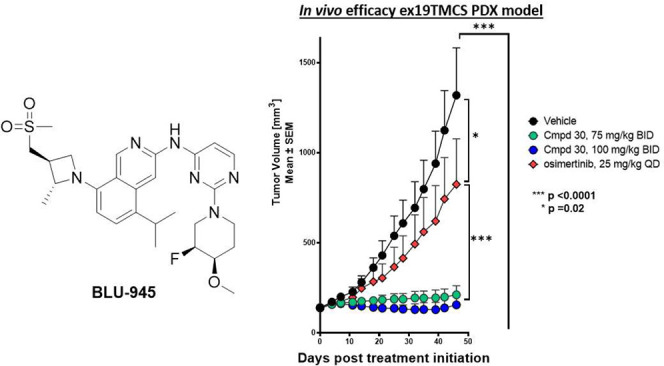
(A) Activity of 30 in NOD SCID mice bearing engineered Ba/F3 (EGFR L858R/T790M/C797S) tumors. (B) Activity of 30 in NOD SCID mice bearing engineered Ba/F3 (EGFR ex19del/T790M/C797S) tumors. (C) Activity of 30 and osimertinib in EGFR ex19del/T790M/C797S PDX model.
With promising in vivo activity in engineered tumor models expressing EGFR+/T790M or EGFR+/T790M/C797S, we were interested in further profiling the activity of 30 in a patient-derived cell-line xenograft (PDX) model. Samples from a patient with EGFR-driven NSCLC who progressed after seven lines of treatment including gefitinib and osimertinib were used to develop an osimertinib-resistant EGFR ex19del/T790M/C797S mouse model. In this model, after treating mice with 30 (75 and 100 mg/kg BID) for 56 days, we were pleased to see substantial tumor growth inhibition with compound treatment (Figure 6C). Supported by these encouraging in vivo activity results, 30 was selected as a development candidate (BLU-945) and advanced into key non-clinical safety studies.
In pre-clinical 28-day GLP toxicity studies in rats and non-human primates (NHP), BLU-945 achieved suitable safety margins to support advancement into human testing with a starting dose of 25 mg in the dose escalation phase.61 We elected to use a spray-dry dispersion formulation due to the pH-dependent solubility of BLU-945 and low aqueous solubility at higher pH (Table 6). A first-in-human, phase 1 dose escalation study is currently underway with BLU-945 in patients with EGFR-mutated NSCLC who have previously received at least one prior EGFR-targeted TKI (NCT04862780). BLU-945 was administered to patients orally once daily on a continuous schedule. The plasma concentrations versus time PK profile of the starting 25 mg dose in one patient is shown in Figure 7, indicating that BLU-945 has low clearance and a long plasma half-life (t1/2).
Figure 7.
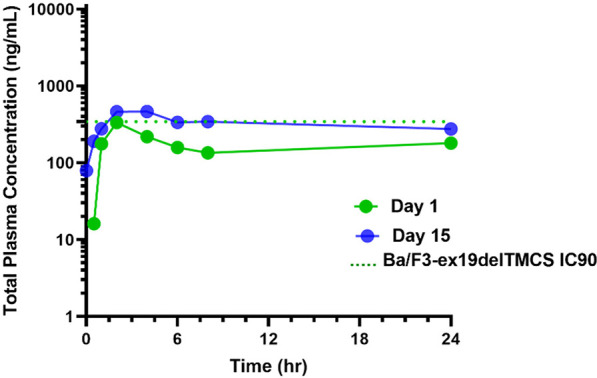
Concentration vs time profile of BLU-945 after 25 mg dose in one patient.
Synthesis
The synthesis of BLU-945 and related analogs began with the construction of a modular isoquinoline core (Scheme 1).62 Regioselective bromination of commercially available isoquinoline 32, followed by demethylation, provided 33 in 75% yield over two steps. Triflate 34 was prepared under typical conditions, followed by Suzuki–Miyaura coupling with isopropenyl boronic ester at 45 °C, resulting in selective coupling of the triflate to form 35. Treatment of 35 with platinum oxide under an atmosphere of hydrogen yielded isopropyl-containing 36. Intermediate 36 was then used in subsequent Buchwald–Hartwig couplings with the appropriate azetidine to furnish the penultimate intermediate to the desired analogs shown in Table 5.
Scheme 1. Synthesis of Isoquinoline Core 36.

Reagents and conditions: (a) Br2, AcOH, rt followed by BBr3, CH2Cl2, 0 °C–rt, 75%; (b) Tf2O, TEA, CH2Cl2, −60 °C, 85%; (c) 4,4,5,5-tetramethyl-2-(prop-1-en-2-yl)-l,3,2-dioxaborolane, K2CO3, Pd(dppf)Cl2·CH2Cl2, dioxane, H2O, 45 °C, 67%; (d) PtO2, H2, EtOAc, rt, 93%.
For BLU-945, the stereospecific synthesis of azetidine 42 is shown in Scheme 2. Commercially available enantiopure (2R,3S)-1-benzhydryl-2-methylazetidin-3-ol 37 was treated with methanesulfonyl chloride to form activated alcohol 38. Deprotonation of methyl 2-(methylsulfonyl)acetate with sodium hydride followed by reaction with 38 led to mesylate displacement with retention of stereochemistry, 39. This retention of configuration has been previously described in substitution reactions with 3-azetidinyl tosylates and mesylates, where retention is attributed to the participation of the azetidine nitrogen.63,64 The reaction is proposed to occur via formation of a bicyclobutonium ion, 40, followed by nucleophilic ring opening. Krapcho decarboxylation reaction with lithium chloride followed by removal of the benzhydryl protecting group afforded azetidine 42.
Scheme 2. Stereospecific Synthesis of Azetidine 42.

Reagents and conditions: (a) mesyl chloride, TEA; CH2Cl2, rt, 98%; (b) methyl 2-(methylsulfonyl)acetate, NaH; DMF, 80 °C, 80%; (c) LiCl; DMA, 150 °C, 93%; (d) Pd(OH)2, TFA; MeOH, rt, 75%.
Aminopyrimidine 46 was available in three steps from tert-butyl (3S,4R)-3-fluoro-4-hydroxypiperidine-1-carboxylate (Scheme 3). Alcohol 43 was methylated with iodomethane in the presence of sodium hydride to afford 44 in 94% yield. Boc deprotection followed by SNAr with 2-chloropyrimidin-4-amine yielded the desired aminopyrimidine 46.
Scheme 3. Synthesis of Aminopyrimidine 46.

Reagents and conditions: (a) NaH, iodomethane, THF, 0 °C, 94%; (b) TFA, DCM, rt; (c) 2-chloropyrimidin-4-amine, TEA, IPA, rt, 66%, over two steps.
The final synthetic sequence began with Buchwald–Hartwig coupling reaction between azetidine 42 and isoquinoline 36 using Pd-Xantphos G4 pre-catalyst to provide azetidinyl isoquinoline 47 (Scheme 4). The final product BLU-945 was prepared by a second Buchwald–Hartwig coupling between 47 and aminopyrimidine 46.
Scheme 4. Synthesis of BLU-945 via Subsequent Buchwald–Hartwig Couplings.

Reagents and conditions: (a) 42, XantphosPd G4, Cs2CO3, 1,4-dioxane, 100 °C, 63%; (b) 46, BrettPhosPd G4, Cs2CO3, 1,4-dioxane, 100 °C, 38%.
Conclusion
Herein we have reported drug discovery efforts resulting in the identification of BLU-945 (30), a potent and selective EGFR+/T790M and EGFR+/T790M/C797S inhibitor. Using Blueprint Medicines’ proprietary compound library, we identified compound 4 with moderate EGFR mutant potency but excellent selectivity over WT-EGFR. The initial phase of optimization focused on improving potency, and a scaffold-hop to a 2,7-naphthyridine improved EGFR+/T790M and EGFR+/T790M/C797S enzymatic and cellular potency without compromising WT-EGFR selectivity.
Subsequent optimization of kinome selectivity, metabolic stability, and cellular potency resulted in lead compound 27. Further analysis of 27 revealed a glucuronidation liability leading to high in vivo clearance in cyno. A strategy of mitigating glucuronidation by sterically encumbering the site of glucuronidation paired with analysis of new compounds in monkey IV PK studies for improved clearance enabled the identification compounds with a reduced glucuronidation liability. This effort ultimately led to the identification of BLU-945. Evaluation of BLU-945 in osimertinib-resistant mouse xenograft models showed robust tumor growth inhibition. In addition to excellent in vivo tumor activity, an acceptable non-clinical safety profile supported selection as a clinical candidate. BLU-945 is currently being evaluated in a phase 1/2 clinical trial (NCT 04862780).
Experimental Section
Compound Synthesis and Characterization
All solvents employed were commercially available anhydrous grade, and reagents were used as received unless otherwise noted. Compound purity of all compounds was assessed by HPLC to confirm >95% purity. The liquid chromatography–mass spectrometry (LC-MS) data were obtained with an Agilent model-1260 LC system using an Agilent model 6120 mass spectrometer utilizing ES-API ionization fitted with an Agilent Poroshel 120 (EC-C18, 2.7 μm particle size, 3.0 × 50 mm dimensions) reverse-phase column. The mobile phase consisted of a mixture of solvent 0.1% formic acid in water and 0.1% formic acid in acetonitrile. A constant gradient from 95% aqueous/5% organic to 5% aqueous/95% organic mobile phase over the course of 4 min was utilized. The flow rate was constant at 1 mL/min. Alternatively, the LC-MS data were obtained with a Shimadzu LC-MS system using a Shimadzu LC-MS mass spectrometer utilizing ESI fitted with an Agilent (Poroshel HPH-C18 2.7 μm particle size, 3.0 × 50 mm dimensions) reverse-phase column. The mobile phase consisted of a mixture of solvent 5 mM NH4HCO3 (or 0.05% TFA) in water and acetonitrile. A constant gradient from 90% aqueous/10% organic to 5% aqueous/95% organic mobile phase over the course of 2 min was utilized. The flow rate was constant at 1.5 mL/min. Preparative HPLC was performed on a Shimadzu Discovery VPR preparative system fitted with a Luna 5 μm C18(2) 100 Å, AXIA packed, 250 × 21.2 mm reverse-phase column. Alternatively, the preparative HPLC was performed on a Waters Preparative system fitted with an XBridge Shield RP18 OBD column, 30 × 150 mm, 5 μm; the mobile phase consisted of a mixture of solvent water (10 mmol/L NH4CO3 + 0.05% NH3·H2O) and acetonitrile. A constant gradient from 95% aqueous/5% organic to 5% aqueous/95% organic mobile phase over the course of 11 min was utilized. The flow rate was constant at 60 mL/min. Reactions carried out in a microwave were performed in a Biotage Initiator microwave unit. Silica gel chromatography was performed on a Teledyne Isco CombiFlash Rf unit, a BiotageR Isolera Four unit, or a BiotageR Isolera Prime unit. 1H NMR spectra were obtained with a Varian 400 MHz Unity Inova 400 MHz NMR instrument, an Avance 400 MHz Unity Inova 400 MHz NMR instrument, or an Avance 300 MHz Unity Inova 300 MHz NMR instrument. Unless otherwise indicated, all protons were reported in DMSO-d6 solvent as parts per million (ppm) with respect to residual DMSO (2.50 ppm). Chiral-HPLC was performed on an Agilent 1260 Preparative system. Chiral-SFC purification was performed with a Waters preparative system.
Synthesis of compounds 4–27 can be found in the Supporting Information.
Synthesis of BLU-945, N-(2-((3S,4R)-3-fluoro-4-methoxypiperidin-1-yl)pyrimidin-4-yl)-5-isopropyl-8-((2R,3S)-2-methyl-3-((methylsulfonyl)methyl)azetidin-1-yl)isoquinolin-3-amine
8-Bromo-3-chloroisoquinolin-5-yl Trifluoromethanesulfonate (34)
Trifluoromethanesulfonyl trifluoromethanesulfonate (45.7 g, 162 mmol) was added dropwise to 8-bromo-3-chloroisoquinolin-5-ol (14 g, 54.1 mmol) and TEA (21.8 g, 216 mmol) in DCM (400 mL) at −60 °C. The resulting mixture was warmed to room temperature naturally and stirred at rt for 1 h. The mixture was concentrated under vacuum. The residue was purified by a silica gel column with PE:EA = 5:1 to afford 18 g (85%) of the title compound as a white solid. LC-MS: (ES, m/z) = 392 [M+1]; 1H NMR (400 MHz, DMSO-d6) δ 9.46 (d, 1H, J = 0.8 Hz), 8.20 (d, 1H, J = 8.3 Hz), 8.02 (d, 1H, J = 8.4 Hz), 7.93 (d, 1H, J = 0.7 Hz).
8-Bromo-3-chloro-5-(prop-1-en-2-yl)isoquinoline (35)
The mixture of K2CO3 (6 g, 43.5 mmol), 8-bromo-3-chloroisoquinolin-5-yl trifluoromethanesulfonate (17 g, 43.5 mmol, 34), 4,4,5,5-tetramethyl-2-(prop-1-en-2-yl)-l,3,2-dioxaborolane (7.30 g, 43.5 mmol), and Pd(dppf)Cl2·CH2Cl2 (2.83 g, 3.48 mmol) in dioxane (200/20 mL) was stirred for 3 h at 45 °C. The mixture was diluted with 500 mL of EA and washed two times with brine (200 mL). The organic layer was dried with Na2SO4 and concentrated under vacuum. The residue was purified by a silica gel column with PE:EA = 20:1 to afford 8.0 g (67%) of the title compound as an off-white solid. LC-MS: (ES, m/z) = 282 [M+1].
8-Bromo-3-chloro-5-isopropylisoquinoline (36)
PtO2 (1.7 g 7.04 mmol) and 8-bromo-3-chloro-5-(prop-1-en-2-yl)isoquinoline (7.1 g, 25.1 mmol, 35) in EA (300 mL) were stirred under an atmosphere of H2 at rt for l h. The solid was filtered out. The mother solvent was concentrated under vacuum. The crude product was purified by a silica gel column with PE:EA = 10:1 to get 6.7 g (93%) of the title compound as a brown solid.
(2R,3S)-1-Benzhydryl-2-methylazetidin-3-yl Methanesulfonate (38)
(2R,3S)-1-Benzhydryl-2-methylazetidin-3-ol (Pharmablock, 20 g, 78.9 mmol) was dissolved in 300 mL of DCM, TEA (9.55 g, 94.6 mmol) was added, and the reaction mixture was cooled in an ice bath. Mesyl chloride (9.93 g, 86.7 mmol) was added dropwise and allowed to stir, warming slowly to rt and continuing stirring overnight. The mixture was diluted with DCM and washed with water, and the organic phase was dried over sodium sulfate, filtered, and evaporated to give 26 g (98%) of the title compound as a viscous yellow oil. LC-MS: (ES, m/z) = 332 [M+1].
(S)-Methyl 2-((2R,3S)-1-Benzhydryl-2-methylazetidin-3-yl)-2-(methylsulfonyl)acetate (39)
(2R,3S)-1-Benzhydryl-2-methylazetidin-3-yl methanesulfonate (26 g, 78.4 mmol, 38) and methyl 2-(methylsulfonyl)acetate (15.3 g, 101 mmol) were dissolved in 260 mL of DMF, and then NaH (3.75 g of 60% dispersion in mineral oil, 6.63 mmol) was added and the mixture was stirred for ∼15 min, until hydrogen evolution had ceased. The reaction mixture was heated to 80 °C overnight. The reaction was cooled, then diluted with 200 mL of water and extracted with EA. The combined organics were washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give the crude product. The residue was purified by chromatography (0 to 7% MeOH/DCM). Pure fractions were combined and evaporated to give 24 g (80%) of the title compound as a pale-yellow foam.
(2R,3S)-1-Benzhydryl-2-methyl-3-(methylsulfonylmethyl)azetidine (41)
(S)-Methyl 2-((2R,3S)-1-benzhydryl-2-methylazetidin-3-yl)-2-(methylsulfonyl)acetate (24 g, 61.9 mmol, 39) was dissolved in 240 mL of DMA, lithium chloride (20.9 g, 495 mmol) was added, and the flask was put into a preheated block that was kept at 150 °C. LC-MS indicated the starting material was consumed after 1.5 h. The mixture was cooled to rt, diluted with water, and extracted with EA, and the combined organics were washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give the crude product, which was further purified by chromatography (0 to 5% MeOH/DCM). Pure fractions were combined and evaporated to give 19 g (93%) of the title compound as a pale-yellow foam. LC-MS: (ES, m/z) = 330 [M+1].
(2R,3S)-2-Methyl-3-(methylsulfonylmethyl)azetidine (42)
To a solution of (2R,3S)-1-(diphenylmethyl)-3-(methanesulfonylmethyl)-2-methylazetidine (1 9 g, 57.3 mmol, 41) in MeOH (270 mL) was added TFA (9 mL) and Pd(OH)2 (5.7 g), the reaction was stirred overnight at rt under H2 atmosphere. The reaction mixture was filtered and evaporated to give the crude title compound (17 g) as a light-brown oil. LC-MS: (ES, m/z) = 164 [M+1].
(3S,4R)-tert-Butyl 3-Fluoro-4-methoxypiperidine-1-carboxylate (44)
Sodium hydride (218.90 mg, 9.122 mmol, 4 equiv) was added to tert-butyl (3S,4R)-3-fluoro-4-hydroxypiperidine-1-carboxylate (500 mg, 2.280 mmol, 1 equiv, 42) in THF (10 mL) at 0 °C. After the mixture was stirred for 20 min, methyl iodide (1294.73 mg, 9.122 mmol, 4 equiv) was added. The resulting solution was stirred for an additional 1 h at 0 °C. The reaction was then quenched by addition of 10 mL of water. The solids were filtered out. The resulting solution was extracted with EA and concentrated under vacuum. This resulted in 500 mg (94.1%) of the title compound as a light-yellow oil. LC-MS: (ES, m/z) = 178 [M+l–56].
(3S,4R)-3-Fluoro-4-methoxypiperidine (45)
The solution of tert-butyl (3S,4R)-3-fluoro-4-methoxypiperidine-1-carboxylate (500 mg, 2.143 mmol, 1 equiv, 42) in TFA/DCM (3/10 mL) was stirred for 1 h at rt. The resulting mixture was concentrated under vacuum to afford 500 mg (crude) of the title compound as a solid.
2-((3S,4R)-3-Fluoro-4-methoxypiperidin-1-yl)pyrimidin-4-amine (46)
The mixture of (3S,4R)-3-fluoro-4-methoxypiperidine (3 g, 22.528 mmol, 1 equiv, 45), 2-chloropyrimidin-4-amine (2.33 g, 0.018 mmol, 0.8 equiv), and TEA (6.84 g, 0.068 mmol, 3 equiv) in IPA (3 mL) was stirred for 12 h at 100 °C. The solvent was removed under vacuum, and THE residue was purified by FLASH (5% MeOH in DCM) to give 3.3 g (66%) of the title compound as a light-yellow solid. LC-MS: (ES, m/z) = 227 [M+1]. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.72 (d, 1H, J = 5.6 Hz), 6.39 (s, 2H), 5.71 (d, 1H, J = 5.6 Hz), 4.83 (d, 1H, J = 49.3 Hz), 4.60–4.49 (m, 1H), 4.29 (d, 1H, J = 13.3 Hz), 3.55–3.42 (m, 1H), 3.28 (d, 1H, J = 13.3 Hz), 3.20–3.04 (m, 1H), 1.76–1.48 (m, 2H).
3-Chloro-5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl)isoquinoline (47)
To a solution of 8-bromo-3-chloro-5-(propan-2-yl)isoquinoline (9 g, 31.6 mmol, 36) in 1,4-dioxane (130 mL) were added (2R,3S)-3-(methanesulfonylmethyl)-2-methylazetidine (5.15 g, 31.6 mmol, 42), Cs2CO3 (20.6 g, 63.2 mmol), and XantphosPd G4 (1.51 g, 1.58 mmol) under nitrogen. The mixture was stirred at 100 °C for 3 h under nitrogen. The reaction mixture was cooled to rt and diluted with 300 mL of water. The resulting solution was extracted with EA, washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The crude product was purified by silica gel chromatography (0–60% EA in PE) to give 7.2 g (62.6%) of 3-chloro-8-[(2R,3S)-3-(methanesulfonylmethyl)-2-methylazetidin-1-yl]-5-(propan-2-yl)isoquinoline as a yellow solid. LC-MS: (ES, m/z) = 367 [M+1].
N-(2-((3S,4R)-3-Fluoro-4-methoxypiperidin-1-yl)pyrimidin-4-yl)-5-isopropyl-8-((2R,3S)-2-methyl-3-((methylsulfonyl)methyl)azetidin-1-yl)isoquinolin-3-amine (BLU-945)
To a solution of 2-((3S,4R)-3-fluoro-4-methoxypiperidin-1-yl)pyrimidin-4-amine (18.50 mg, 0.082 mmol, 1 equiv, 46) 3-chloro-5-isopropyl-8-((2R,3S)-2-methyl-3-((methylsulfonyl)methyl)azetidin-1-yl)isoquinoline (30 mg, 0.082 mmol, 1 equiv, 47), and Cs2CO3 (53.3 mg, 0.164 mmol, 2 equiv) in 1,4-dioxane (0.82 mL) was added BrettPhos precatalyst (Gen IV) (3.76 mg, 4.09 pmol, 0.05 equiv) under nitrogen. The mixture was stirred at 90 °C for 16 h and then filtered and concentrated in vacuo. The crude mixture was purified by reverse-phase chromatography (0–60% acetonitrile/water containing 0.1% TFA). Pure fractions were combined and neutralized with saturated sodium bicarbonate solution and then extracted with 10% MeOH/DCM (5 mL × 3). The combined organic phases were dried over sodium sulfate, filtered, and evaporated to give 17.4 mg of the title compound (38%) as a yellow solid. LC-MS: (ES, m/z) = 557 [M+1]. 1H NMR (400 MHz, DMSO-d6) δ 9.90 (s, 1H), 9.06 (s, 1H), 8.63 (s, 1H), 8.00 (d, 1H, J = 5.6 Hz), 7.42 (d, 1H, J = 8.0 Hz), 6.56 (d, 1H, J = 8.1 Hz), 6.47 (d, 1H, J = 5.7 Hz), 4.94 (d, 1H, J = 49.3 Hz), 4.69 (dt, J = 25.9, 6.4 Hz, 2H), 4.47 (d, 1H, J = 13.2 Hz), 4.27–4.11 (m, 1H), 3.72–3.42 (m, 5H), 3.37 (s, 3H), 2.99 (s, 3H), 2.89 (q, 1H, J = 7.3 Hz), 1.86–1.65 (m, 2H), 1.42 (d, 3H, J = 6.0 Hz), 1.31 (dd, 6H, J = 6.8, 1.9 Hz).
General Procedure for the Synthesis of Compounds 27–29 and 31
3-Chloro-5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl)isoquinoline (47) was combined with aminopyrimidine derivatives (SI1–SI7, see Supporting Information), Cs2CO3, and BrettPhos precatalyst (Gen IV) (5 mol%) in 1,4-dioxane (0.1 M) and stirred under nitrogen at 90 °C for 2–16 h. Reactions were determined complete by LC-MS filtered and concentrated in vacuo. Crude mixtures were purified by reverse-phase chromatography, and pure fractions were neutralized with saturated sodium bicarbonate solution and then extracted with 10% MeOH/DCM (5 mL × 3). Combined organic phases were dried over sodium sulfate, filtered, and evaporated to give the title compounds.
(3S,4R)-3-Fluoro-1-(4-(5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl)isoquinolin-3-ylamino)pyrimidin-2-yl)-3-methylpiperidin-4-ol (27)
The general procedure using SI1 yielded the title compound as a yellow solid (4.6 g, 8.3 mmol, 61%). LC-MS: (ES, m/z) = 557 [M+1]. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.88 (s, 1H), 9.05 (s, 1H), 8.63 (s, 1H), 7.99 (d, 1H, J = 5.6 Hz), 7.42 (d, 1H, J = 8.0 Hz), 6.56 (d, 1H, J = 8.1 Hz), 6.52–6.43 (m, 1H), 5.32 (d, 1H, J = 4.4 Hz), 4.65 (t, 1H, J = 7.5 Hz), 4.19 (t, 1H, J = 6.3 Hz), 4.04–3.82 (m, 4H), 3.75 (s, 1H), 3.64 (t, 1H, J = 7.1 Hz), 3.53 (hept, 3H, J = 7.9, 7.3 Hz), 2.99 (s, 3H), 2.89 (q, 1H, J = 7.3 Hz), 1.92–1.88 (m, 1H), 1.57–1.51 (m, 1H), 1.42 (d, 3H, J = 6.1 Hz), 1.35–1.21 (m, 9H).
(3R,4S)-3-Ethyl-3-fluoro-1-(4-((5-isopropyl-8-((2R,3S)-2-methyl-3-((methylsulfonyl)methyl)azetidin-1-yl)isoquinolin-3-yl)amino)pyrimidin-2-yl)piperidin-4-ol (28)
The general procedure using SI3, followed by chiral separation (column: CHIRALPAK IG-3, 0.46 × 5 cm, 3 μm, mobile phase (Hex:DCM = 1:1) (0.1% DEA):EtOH = 50:50, flow rate: 1.0 mL/min), gave the title compound (peak 2, 29.8 mg, 0.05 mmol, 51%). LC-MS: (ES, m/z) = 571 [M+1]. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.03 (s, 1H), 9.07 (s, 1H), 8.60 (s, 1H), 7.99 (d, 1H, J = 5.8 Hz), 7.43 (d, 1H, J = 8.0 Hz), 6.57 (d, 1H, J = 8.1 Hz), 6.50 (d, 1H, J = 5.8 Hz), 5.00 (s, 1H), 4.71–4.54 (m, 3H), 4.20 (p, 1H, J = 6.1 Hz), 3.66 (dt, 2H, J = 20.7, 7.2 Hz), 3.61–3.44 (m, 3H), 3.31–3.19 (m, 2H), 2.99 (s, 3H), 2.96–2.84 (m, 1H), 1.86 (dt, 1H, J = 16.6, 7.8 Hz), 1.80–1.60 (m, 3H), 1.42 (d, 3H, J = 6.1 Hz), 1.34–1.26 (m, 6H), 0.93 (t, 3H, J = 7.5 Hz).
(3S,4R)-3-Fluoro-1-(4-(5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl)isoquinolin-3-ylamino)pyrimidin-2-yl)-4-methylpiperidin-4-ol (29)
The general procedure using SI4 yielded the title compound as a light-yellow solid (28.8 g, 51.1 mmol, 67%). LC-MS: (ES, m/z) = 557 [M+1] .1H NMR (400 MHz, DMSO-d6) δ ppm 9.95 (s, 1H), 9.06 (s, 1H), 8.67 (s, 1H), 8.01 (d, 1H, J = 5.6 Hz), 7.42 (d, 1H, J = 8.0 Hz), 6.57 (d, 1H, J = 8.1 Hz), 6.47 (d, 1H, J = 5.7 Hz), 4.86 (s, 1H), 4.66 (t, 1H, J = 7.5 Hz), 4.52–4.36 (m, 2H), 4.25–4.14 (m, 2H), 3.69–3.47 (m, 6H), 3.00 (s, 3H), 2.89 (q, 1H, J = 7.3 Hz), 1.73–1.67 (m, 1H), 1.62–1.50 (m, 1H), 1.43 (d, 3H, J = 6.0 Hz), 1.34–1.23 (m, 9H).
(3R,4R)-5,5-Difluoro-1-(4-(5-isopropyl-8-((2R,3S)-2-methyl-3-(methylsulfonylmethyl)azetidin-1-yl)isoquinolin-3-ylamino)pyrimidin-2-yl)-4-methoxypiperidin-3-ol (31)
The general procedure using SI6 gave the title compound as a yellow solid (50 mg, 0.08 μmol, 44%). LC-MS: (ES, m/z) = 591 [M+1]. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.97 (s, 1H), 9.05 (s, 1H), 8.52 (s, 1H), 8.02 (d, 1H, J = 5.7 Hz), 7.41 (d, 1H, J = 8.0 Hz), 6.57 (d, 1H, J = 5.5 Hz), 6.41 (d, 1H, J = 8.1 Hz), 5.31 (d, 1H, J = 5.8 Hz), 4.80 (s, 1H), 4.56–4.48 (m, 1H), 4.43–4.34 (m, 2H), 3.96 (td, 2H, J = 7.0, 2.7 Hz), 3.82–3.66 (m, 2H), 3.64–3.51 (m, 6H), 3.54–3.48 (m, 1H), 3.30–3.23 (m, 1H), 3.22–3.04 (m, 3H), 1.33–1.20 (m, 9H).
Inhibition of EGFR Mutant Biochemical Enzymatic Activity
Inhibitory effects of the compounds were determined by measuring the enzymatic activity of EGFR enzyme phosphorylates’ 2.5 μM fluorescent substrate (5-FAM-EEPLYWSFPAKKK-CONH2, ProfilerPro kinase peptide substrate 22, PerkinElmer) in the presence of 1 mM adenosine-5′-triphosphate (ATP) and varying concentrations of the test compound. The enzyme reaction buffer contains 10 mM MgCl2, 0.015% Brij-35, 1 mM dithiothreitol (DTT), 1.0% dimethyl sulfoxide (DMSO), and 100 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), pH 7.5, at 25 °C. The WT and mutant EGFR enzymes (SignalChem) were allowed to incubate with the inhibitor for 10 min. The kinase reaction was activated by the addition of ATP and the peptide substrate. Reactions proceeded until 10%–20% total peptides were phosphorylated and were terminated with 35 mM 2,2′,2″,2‴-(ethane-1,2-diyldinitrilo)tetraacetic acid (EDTA). Analysis of the proportion of phosphorylated substrate peptide was performed automatically on the Caliper EZReader 2 (PerkinElmer), where the phosphorylated peptide (product) and substrate were electrophoretically separated and measured. Percent activity was plotted against log concentration of compound to generate an apparent IC50 using a 4-parameter fit in CORE LIMS.
Measurement of EGFR Inhibition in Cells
Cell Lines
Ba/F3 cells were transduced with lentiviral particles encoding mutant EGFR. After 48 h, cells were placed in blasticidin-containing media for 14 days, followed by incubation in IL-3-free media for another 14 days to enable selection of mutant EGFR Ba/F3 cells that were IL-3-independent. These cells were then expanded and used in subsequent assays. A-431 and NCI-H1975 cells in 10% FBS DMEM + 1x Pen-Strep were purchased from ATCC, PC-9 cells in 10% FBS RPMI + 1x Pen-Strep were purchased from Millipore Sigma, Osimertinib was purchased from LC Laboratories, and Gefitinib was purchased from Selleckchem.
Assessing Inhibition of EGFR by AlphaLISA (A431, PC9 and NCI-H1975)
Ba/F3 cells were diluted with phenol-free DMEM with 10% FBS ,while A431 cells were diluted with DMEM lacking 10% FBS (for serum starvation) to 3.125 × 105 cells/mL. Next, 40 μL of cell suspension was added into each well of a 384-well microplate, which was then placed in an incubator containing 5% CO2 at 37 °C overnight for cells to adhere. The following day, experimental compounds were serial-diluted in DMSO, added to the cells, and then placed in a humidified 37 °C incubator for 4–5 h. After incubation with compounds, A431 cells were stimulated for 10 min with EGF (30 ng/mL final concentration). Media was then removed from all cell plates. All cells were then lysed and processed per the Phospho-EGFR (Tyr1068) AlphaLISA SureFire Ultra Detection Kit protocol, and the plate was read on an EnVision multilabel reader. All IC50 representative curves were plotted using GraphPad Prism (version 8.00 for Windows, GraphPad Software, San Diego, California, USA). All values quoted are the average of at least two independent experiments.
Assessing Inhibition of EGFR by AlphaLISA (Mutant EGFR Ba/F3 Lines)
Mutant EGFR-expressing Ba/F3 cells were resuspended in fresh 10% FBS RPMI and plated at 1.0 × 106 cells/mL. Cells were harvested the next day and then diluted in fresh media at 1.25 × 106 cells/mL and plated (40 μL of cells) to each well of a 384-well microplate. Experimental compounds were serial-diluted in DMSO, added to the cells, and then placed in a humidified 37 °C incubator for 4 h. The plate was then spun at 3000 rpm for 5 min to pellet the cells, and media was removed. All cells were then lysed and processed per the Phospho-EGFR (Tyr1068) AlphaLISA SureFire Ultra Detection Kit protocol, and the plate was read on an EnVision multilabel reader. All IC50 representative curves were plotted using GraphPad Prism (version 8.00 for Windows, GraphPad Software, San Diego, California, USA). All values quoted are the average of at least two independent experiments.
Crystallization and Structure Determination
Protein expression and purification of a triple-mutant EGFR protein (L858R, T790M, V948R) were performed as previously published.65 Mutant EGFR at 5–6 mg/mL (25 mM HEPES/NaOH, 300 mM NaCl, 10% glycerol, 4 mM TCEP, pH 8.0) was incubated with compound to a final concentration of 0.7 mM for 1 h on ice. The complex was crystallized in 0.10 M sodium acetate, pH 5.30, 0.20 M potassium acetate, and 3% (w/v) PEG 8000 at 12 °C over 3–7 days. Crystals were cryoprotected by addition of 20% glycerol prior to mounting, and diffraction data were collected at ESRF beamline ID30a1 and Diamond beamline i04-1. The structure was solved using molecular replacement followed by multiple rounds of refinement with REFMAC5 to produce the final models. Crystal structures have been deposited in the RCSB PDB with accession codes 8D73 and 8D76, and coordinates will be released upon publication.
In Vivo Pharmacokinetics and Pharmacodynamics Studies
All the procedures related to animal handling, care, and treatment in the study were performed according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) of WuXi AppTec following the guidance of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
BALB/c nude female mice (Zhejiang Vital River Laboratory Animal Technology Co., Ltd.) weighing 18–22 g were used for studies. NCI-H1975 cells (5 × 106) were inoculated subcutaneously at the right flank for tumor development. The treatments started when the average tumor size reached approximately 389 mm3. Treatment was given by oral gavage (PO doses). Blood samples were collected from all animals at 2, 6, and 12 h post dose. Plasma was separated from blood by centrifugation at 4 °C. Compound concentrations in both plasma and tumor were quantified using a liquid chromatography with tandem mass spectrometry (LC-MS/MS) method.
Cell-Line-Derived Xenograft (CDX) Efficacy Studies
The protocol and any amendment(s) or procedures involving the care and use of animals in this study were reviewed and approved by the IACUC of WuXi AppTec prior to conduct.
BALB/c nude female mice (Zhejiang Vital River Laboratory Animal Technology Co., Ltd.), 6–8 weeks old, were used for our studies. Each mouse was inoculated subcutaneously at the right flank with the tumor cells (5 × 106) in 0.2 mL of PBS supplemented with Matrigel (PBS:Matrigel = 1:1) for tumor development. Animal randomization and treatments started when the average tumor volume reached approximately 155 mm3. Animals were dosed twice (BID) or once daily (QD) by oral gavage. Tumor size and body weight were measured every second day. After the last dose, blood was collected at 2, 6 and 12 h for plasma preparation to assess compound concentration (LC-MS/MS). Tumor volume was calculated using the formula V = 0.5ab2, where a and b are the long and short diameters of the tumor in mm, respectively. Statistical analysis was performed by using a two-way RM ANOVA analysis followed by Dunnett’s multiple comparison test.
Patient-Derived Xenograft (PDX) Efficacy Studies
The protocol and any amendment(s) or procedures involving the care and use of animals in this study were reviewed and approved by the IACUC of Lide Biotech prior to conduct. During the study, the care and use of animals were conducted in accordance with the regulations of the AAALAC.
LUPF104 human tumor fragments, 15–30 mm3, were implanted in the right flanks of 5–7-week-old NU/NU female mice (Zhejiang Vital River Laboratory Animal Technology Co. Ltd.) under isoflurane anesthesia. Animal randomization and treatments started when the tumor average reached 200 mm3. Treatment, animal monitoring, end of study plasma collection, and statistical analysis were performed in the same way as the CDX efficacy studies.
Acknowledgments
The authors are grateful to Luz E. Tavera-Mendoza for assistance in preparing in vivo experiment figures and associated experimental details, Tyler Rouskin-Faust for help with cell biology experimental description, and Jagan Parepally for reviewing clinical pharmacology. The authors would also like to thank Percy Carter and Robert Meissner for critically reading the manuscript and providing helpful comments. We would like to acknowledge Yanhai Bai and the Pharmaron chemistry team for their synthetic efforts.
Glossary
Abbreviations Used
- Cl
in vivo clearance
- Clu
unbound in vivo clearance
- GLP
good laboratory practice
- HLM
human liver microsomes
- LipE
lipophilic efficiency
- t1/2
half-life
- UGT
uridine 5′-diphospho-glucuronosyltransferase
- Vss
volume of distribution measured at steady-state
- WT
wild-type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00704.
Accession Codes
Crystal structures have been deposited in the RCSB PDB with accession codes 8D73 and 8D76, and coordinates will be released upon publication.
Author Present Address
† Reverie Laboratories, Cambridge, Massachusetts 02139, United States
Author Present Address
‡ Kymera Therapeutics, Watertown, Massachusetts 02472, United States
Author Present Address
§ MOMA Therapeutics, Brighton, Massachusetts 02135, United States
Author Present Address
⊥ Tango Therapeutics, Cambridge, Massachusetts 02139, United States
Author Present Address
∥ Foghorn Therapeutics, Cambridge, Massachusetts 02139, United States
Author Present Address
∇ Scorpion Therapeutics, Boston, Massachusetts 02110, United States
Author Present Address
○ Nested Therapeutics, Cambridge, Massachusetts 02138, United States
Author Present Address
◆ Bristol Myers Squibb, Cambridge, Massachusetts 02142, United States
Author Present Address
¶ Constellation Pharmaceuticals, Cambridge, Massachusetts, 02142, United States
Author Present Address
# OnKure Therapeutics, Boulder, Colorado 80301, United States
Author Present Address
□ Forma Therapeutics, Watertown, Massachusetts 02472, United States
The authors declare the following competing financial interest(s): All authors are or were employees of Blueprint Medicines and may hold company stocks or stock options with Blueprint Medicines.
Supplementary Material
References
- International Agency for Research on Cancer . Lung. GLOBOCAN World Fact Sheet, 2020, https://gco.iarc.fr/today/data/factsheets/cancers/15-Lung-fact-sheet.pdf (accessed Oct 4, 2021).
- Zhang Y.-L.; Yuan J.-Q.; Wang K.-F.; Fu X.-H.; Han X.-R.; Threapleton D.; Yang Z.-Y.; Mao C.; Tang J.-L. The Prevalence of EGFR Mutation in Patients With Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Oncotarget 2016, 7 (48), 78985–78993. 10.18632/oncotarget.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midha A.; Dearden S.; McCormack R. EGFR Mutation Incidence in Non-Small-Cell Lung Cancer of Adenocarcinoma Histology: a systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [PMC free article] [PubMed] [Google Scholar]
- Hsu W.; Yang J.; Mok T.; Loong H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29 (suppl. 1), i3–i9. 10.1093/annonc/mdx702. [DOI] [PubMed] [Google Scholar]
- Rosell R.; Morán T.; Carcereny E; Quiroga V; Molina M.-A.; Costa C.; Benlloch S.; Taron M. Non-Small-Cell Lung Cancer Harbouring Mutations in the EGFR Kinase Domain. Clin. Transl. Oncol. 2010, 12, 75–80. 10.1007/S12094-010-0473-0. [DOI] [PubMed] [Google Scholar]
- Gazdar A. F. Activating and Resistance Mutations of EGFR in Non-Small-Cell Lung Cancer: Role in Clinical Response to EGFR Tyrosine Kinase Inhibitors. Oncogene 2009, 28, S24–S31. 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X.; Xie C.; Yu X.; Liu J. EGFR TKI as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer. Oncotarget 2017, 8 (43), 75712–75726. 10.18632/oncotarget.20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester J. F.; Shah R. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non–Small-Cell Lung Cancer: A Clash of the Generations. Clinical Lung Cancer 2019, 21 (3), E216–E228. 10.1016/j.cllc.2019.12.003. [DOI] [PubMed] [Google Scholar]
- Riely G. J.; Yu H. A. EGFR: The Paradigm of an Oncogene-Driven Lung Cancer. Clin. Cancer. Res. 2015, 21 (10), 2221–2226. 10.1158/1078-0432.CCR-14-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K.; Tan E.-H.; O’Byrne K.; Zhang Li.; Boyer M.; Mok T.; Hirsh V.; Yang J.-C.; Lee K.-H.; Lu S.; Shi Y.; Kim S. W.; Laskin J.; Kim D.-W.; Arvis C. D.; Kolbeck K.; Laurie S. A.; Tsai C.-M.; Shahidi M.; Kim M.; Massey D.; Zazulina V.; Paz-Ares L. Afatinib Versus Gefitinib as First-Line Treatment of Patients with EGFR Mutation-Positive Non-Small-Cell Lung Cancer (LUX-Lung 7): A Phase 2B, Open-Label, Randomised Controlled Trial. Lancet Oncol. 2016, 17 (5), 577–589. 10.1016/S1470-2045(16)30033-X. [DOI] [PubMed] [Google Scholar]
- Muhsin M.; Graham J.; Kirkpatrick P. Gefitinib. Nature Reviews Cancer. 2003, 3, 556–557. 10.1038/nrc1159. [DOI] [PubMed] [Google Scholar]
- Zhou C.; Wu Y. L.; Chen G.; Feng J.; Liu X.-Q.; Wang C.; Zhang S.; Wang J.; Zhou S.; Ren S.; Lu S.; Zhang L.; Hu C.; Hu C.; Luo Y.; Chen L.; Ye M.; Huang J.; Zhi X.; Zhang Y.; Xiu Q.; Ma J.; Zhang L.; You C. Erlotinib Versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011, 12 (8), 735–742. 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- Dowell J.; Minna J. D.; Kirkpatrick P. Erlotinib Hydrochloride. Nat. Rev. Drug Discov. 2005, 4, 13–14. 10.1038/nrd1612. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K.-A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib is Associated with a Second Mutation in the EGFR Kinase Domain. Plos Med. 2005, 2 (3), 225–235. 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belani C. P. The Role of Irreversible EGFR Inhibitors in the Treatment Of Non-Small Cell Lung Cancer: Overcoming Resistance to Reversible EGFR Inhibitors. Cancer Investigation 2010, 28 (4), 413–423. 10.1080/07357901003631072. [DOI] [PubMed] [Google Scholar]
- Blakely C. M.; Bivona T. G. Resiliency of Lung Cancers to EGFR Inhibitor Treatment Unveiled, Offering Opportunities to Divide and Conquer EGFR Inhibitor Resistance. Cancer Discov. 2012, 2 (10), 872–875. 10.1158/2159-8290.CD-12-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Janne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352 (8), 786–792. 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of Tumor Specimens at the Time of Acquired Resistance To EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19 (8), 2240–2247. 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmair M. J.; Buder A.; Schwab S.; Burghuber O. C.; Prosch H.; Hilbe W.; Cseh A.; Fritz R.; Filipits M. Liquid-Biopsy-Based Identification of EGFR T790M Mutation-Mediated Resistance to Afatinib Treatment in Patients with Advanced EGFR Mutation-Positive NSCLC, and Subsequent Response to Osimertinib. Target Oncol. 2019, 14 (1), 75–83. 10.1007/s11523-018-0612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsh V.; Melosky B. Management of Common Toxicities in Metastatic NSCLC Related to Anti-Lung Cancer Therapies with EGFR–Tkis. Front. Oncol. 2014, 4, 238. 10.3389/fonc.2014.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding P. N.; Lord S. J.; Gebski V.; Links M.; Bray V.; Gralla R. J.; Yang J. C.-H.; Lee C. K. Risk of Treatment-Related Toxicities from EGFR Tyrosine Kinase Inhibitors: A Meta-Analysis of Clinical Trials of Gefitinib, Erlotinib, And Afatinib in Advanced EGFR-Mutated Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12 (4), 633–643. 10.1016/j.jtho.2016.11.2236. [DOI] [PubMed] [Google Scholar]
- Takeda M.; Nakagawa K. Toxicity Profile of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Patients with Epidermal Growth Factor Receptor Gene Mutation-Positive Lung Cancer. Molecular And Clinical Oncology 2017, 6 (1), 3–6. 10.3892/mco.2016.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasaka M.; Zhu V. W.; Lim S. M.; Greco M.; Wu F.; Ou S.-H. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J. Thorac. Oncol. 2021, 16 (5), 740–763. 10.1016/j.jtho.2020.11.028. [DOI] [PubMed] [Google Scholar]
- Tan A. C.; Teh Y. L.; Lai G. G. Y.; Tan D. S. W. Third Generation EGFR TKI Landscape for Metastatic EGFR Mutant Non-Small Cell Lung Cancer (NSCLC). Expert Review Of Anticancer Therapy 2019, 19 (6), 431–435. 10.1080/14737140.2019.1604228. [DOI] [PubMed] [Google Scholar]
- Chen L.; Fu W.; Zheng L.; Liu Z.; Liang G. Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors Against C797S Resistance in Non-Small-Cell Lung Cancer. J. Med. Chem. 2018, 61 (10), 4290–4300. 10.1021/acs.jmedchem.7b01310. [DOI] [PubMed] [Google Scholar]
- Cross D. A.E.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R. V.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Brewer M. R.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H.P.; Cantarini M.; Kim D.-W.; Ranson M. R.; Pao W. AZD9291, An Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4 (9), 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay M. R. V.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D. A. E.; Currie G. S.; Grist M.; Hassall L.; Hill G. B.; James D.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamont S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. L. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations that Spares the Wild Type Form of the Receptor. J. Med. Chem. 2014, 57, 8249–8267. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Ramalingam S. S.; Vansteenkiste J.; Planchard D.; Cho B. C.; Gray J. E.; Ohe Y.; Zhou C.; Reungwetwattana T.; Cheng Y.; Chewaskulyong B.; Shah R.; Cobo M.; Lee K. H.; Cheema P.; Tiseo M.; John T.; Lin M.-C.; Imamura F.; Kurata T.; Todd A.; Hodge R.; Saggese M.; Rukazenkov Y.; Soria J.-C. Overall Survival with Osimertinib in untreated, EGFR-Mutated Advanced NSCLC. N. Engl. J. Med. 2020, 382, 41–50. 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- Mok T. S.; Wu Y.-L.; Ahn M.-J.; Garassino M. C.; Kim H. R.; Ramalingam S. S.; Shepherd F. A.; He Y.; Akamatsu H.; Theelen W. S.M.E.; Lee C. K.; Sebastian M.; Templeton A.; Mann H.; Marotti M.; Ghiorghiu S.; Papadimitrakopoulou V. A. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria J.; Ohe Y.; Vansteenkiste J.; Reungwetwattana T.; Chewaskulyong B.; Lee K.; Dechaphunkul A.; Imamura F.; Nogami N.; Kurata T.; Okamoto I.; Zhou C.; Cho B.; Cheng Y.; Cho E.; Voon P.; Planchard D.; Su W.; Gray J.; Lee S.; Hodge R.; Marotti M.; Rukazenkov Y.; Ramalingam S. Osimertinib in untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378 (2), 113–125. 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- Ortiz-Cuaran S.; Scheffler M.; Plenker D.; Dahmen I.; Scheel A. H.; Fernandez-Cuesta L.; Meder L.; Lovly C. M.; Persigehl T.; Merkelbach-Bruse S.; Bos M.; Michels S.; Fischer R.; Albus K.; Konig K.; Schildhaus H.-U.; Fassunke J.; Ihle M. A.; Pasternack H.; Heydt C.; Becker C.; Altmuller J.; Ji H.; Muller C.; Florin A.; Heuckmann J. M.; Nuernberg P.; Ansen S.; Heukamp L. C.; Berg J.; Pao J.; Peifer M.; Buettner R.; Wolf J.; Thomas R. K.; Sos M. L. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22 (19), 4837–4847. 10.1158/1078-0432.CCR-15-1915. [DOI] [PubMed] [Google Scholar]
- Schmid S.; Li J. J. N.; Leighl N. B. Mechanisms of Osimertinib Resistance and Emerging Treatment Options. Lung Cancer 2020, 147, 123–129. 10.1016/j.lungcan.2020.07.014. [DOI] [PubMed] [Google Scholar]
- Leonetti A.; Sharma S.; Minari R.; Perego P.; Giovannetti E.; Tiseo M. Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121 (9), 725–737. 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper-Vallillo A. J.; Sequist L. V.; Piotrowska Z. Emerging Treatment Paradigms for EGFR-Mutant Lung Cancers Progressing on Osimertinib: A Review. J. Clin. Oncol. 2020, 38 (25), 2926–2936. 10.1200/JCO.19.03123. [DOI] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Janne P. A.; Oxnard G. R. Acquired EGFR C797S Mutation Mediates Resistance to AZD9291 in Non–Small Cell Lung Cancer Harboring EGFR T790M. Nat. Medicine 2015, 21, 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh M.; Shinde Y.; Pawara R.; Noolvi M.; Surana S.; Ahmad I.; Patel H. Emerging Approaches to Overcome Acquired Drug Resistance Obstacles to Osimertinib in Non-Small-Cell Lung Cancer. J. Med. Chem. 2022, 65, 1008–1046. 10.1021/acs.jmedchem.1c00876. [DOI] [PubMed] [Google Scholar]
- Lu X.; Zhang T.; Zhu S. J.; Xun Q.; Tong L.; Hu X.; Li Y.; Chan S.; Su Y.; Sun Y.; Chen Y.; Ding J.; Yun C. H.; Xie H.; Ding K. Discovery of JND3229 as a New EGFR C797S Mutant Inhibitor with in Vivo Mono Drug Efficacy. ACS Med. Chem. Lett. 2018, 9 (11), 1123–1127. 10.1021/acsmedchemlett.8b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther M.; Juchum M.; Kelter G.; Fiebig H.; Laufer S. Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity Against a Therapy-Resistant L858R/T790M/C797S Mutant. Angew. Chem., Int. Ed. 2016, 55 (36), 10890–10894. 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Wang J.; Shen Y.; Wang H. Y.; Duan W. M.; Zhao H. Y.; Hei Y. Y.; Xin M.; Cao Y. X.; Zhang S. Q. Discovery of 2,4,6-Trisubstitued Pyrido[3,4-d]Pyrimidine Derivatives as New EGFR TKIs. Eur. J. Med. Chem. 2018, 148, 221–237. 10.1016/j.ejmech.2018.02.051. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Wang Y.; Wang J.; Liu Z.; Shi J.; Li M.; Zhu Y.; Wang S. Design, Synthesis and Biological Evaluation of the Quinazoline Derivatives as L858R/T790M/C797S Triple Mutant Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Chem. Pharm. Bull. 2020, 68 (10), 971–980. 10.1248/cpb.c20-00411. [DOI] [PubMed] [Google Scholar]
- Heald R.; Bowman K. K.; Bryan M. C.; Burdick D.; Chan B.; Chan E.; Chen Y.; Clausen S.; Dominguez-Fernandez B.; Eigenbrot C.; Elliott R.; Hanan E. J.; Jackson P.; Knight J.; La H.; Lainchbury M.; Malek S.; Mann S.; Merchant M.; Mortara K.; Purkey H.; Schaefer G.; Schmidt S.; Seward E.; Sideris S.; Shao L.; Wang S.; Yeap K.; Yen I.; Yu C.; Heffron T. P. Non-covalent Mutant Selective Epidermal Growth Factor Receptor Inhibitors: A Lead Optimization Case Study. J. Med. Chem. 2015, 58 (22), 8877–8895. 10.1021/acs.jmedchem.5b01412. [DOI] [PubMed] [Google Scholar]
- Heppner D. E.; Gunther M.; Wittlinger F.; Laufer S. A.; Eck M. J. Structural Basis for EGFR Mutant Inhibition by Trisubstituted Imidazole Inhibitors. J. Med. Chem. 2020, 63 (8), 4293–4305. 10.1021/acs.jmedchem.0c00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Yun C.-H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P.-Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K.-K.; Janne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) Resistance with Mutant-Selective Allosteric Inhibitors. Nature 2016, 534, 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellmon N.; Li C.; Yang Z. Allosterically Targeting EGFR Drug-Resistance Gatekeeper Mutations. J. Thorac. Dis. 2017, 9 (7), 1756–1758. 10.21037/jtd.2017.06.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel H. M.; Pawara F.; Ansari A.; Noolvi M.; Surana S. Design and Synthesis of Quinazolinones as EGFR Inhibitors to Overcome EGFR Resistance Obstacle. Bioorg. Med. Chem. 2017, 25 (10), 2713–2723. 10.1016/j.bmc.2017.03.039. [DOI] [PubMed] [Google Scholar]
- Park H.; Jung H.-Y.; Mah S.; Hong S. Discovery of EGF Receptor Inhibitors that are Selective for the D746–750/T790M/C797S Mutant Through Structure-Based De Novo Design. Angew. Chem., Int. Ed. 2017, 56 (26), 7634–7638. 10.1002/anie.201703389. [DOI] [PubMed] [Google Scholar]
- Hei Y.-Y.; Shen Y.; Wang J.; Zhang H.; Zhao H.-Y.; Xin M.; Cao Y.-X.; Li Y.; Zhang S.-Q. Synthesis and Evaluation o 2,9-Disubstituted 8-Phenylthio/Phenylsulfinyl-9H-Purine as New EGFR Inhibitors. Bioorg. Med. Chem. 2018, 26 (8), 2173–2185. 10.1016/j.bmc.2018.03.025. [DOI] [PubMed] [Google Scholar]
- To C.; Jang J.; Chen T.; Park E.; Mushajiang M.; De Clercq D. J. H.; Xu M.; Wang S.; Cameron M. D.; Heppner D. E.; Shin B. H.; Gero T. W.; Yang A.; Dahlberg S. E.; Wong K.-K.; Eck M. J.; Gray N. S.; Janne P. A. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov. 2019, 9 (7), 926–943. 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C.; Beyett T. S.; Jang J.; Feng W. W.; Bahcall M.; Haikala H. M.; Shin B. H.; Heppner D. E.; Rana J. K.; Leeper B. A.; Soroko K. M.; Poitras M. J.; Gokhale P. C.; Kobayashi Y.; Wahid K.; Kurppa K. J.; Gero T. W.; Cameron M. D.; Ogino A.; Mushajiang M.; Xu C.; Zhang Y.; Scott D. A.; Eck M. J.; Gray N. S.; Janne P. A. An Allosteric Inhibitor Against the Therapy-Resistant Mutant Forms of EGFR in Non-Small Cell Lung Cancer. Nature Cancer 2022, 3, 402–417. 10.1038/s43018-022-00351-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R.; Bowman K. K.; Bryan M. C.; Burdick D.; Chan B.; Chan E.; Chen Y.; Clausen S.; Dominguez-Fernandez B.; Eigenbrot C.; Elliott R.; Hanan E. J.; Jackson P.; Knight J.; La H.; Lainchbury M.; Malek S.; Mann S.; Merchant M.; Mortara K.; Purkey H.; Schaefer G.; Schmidt S.; Seward E.; Sideris S.; Shao L.; Wang S.; Yeap K.; Yen I.; Yu C.; Heffron T. P. Noncovalent Mutant Selective Epidermal Growth Factor Receptor Inhibitors: A Lead Optimization Case History. J. Med. Chem. 2015, 58, 8877–8895. 10.1021/acs.jmedchem.5b01412. [DOI] [PubMed] [Google Scholar]
- Chan B. K.; Hanan E. J.; Bowman K. K.; Bryan M. C.; Burdick D.; Chan E.; Chen Y.; Clausen S.; Dela Vega T.; Dotson J.; Eigenbrot C.; Elliott R. L.; Heald R. A.; Jackson P. S.; Knight J. D.; La H.; Lainchbury M. D.; Malek S.; Purkey H. E.; Schaefer G.; Schmidt S.; Seward E. M.; Sideris S.; Shao L.; Wang S.; Yeap S. K.; Yen I.; Yu C.; Heffron T. P. Discovery of a Noncovalent, Mutant-Selective Epidermal Growth Factor Receptor Inhibitor. J. Med. Chem. 2016, 59, 9080–9093. 10.1021/acs.jmedchem.6b00995. [DOI] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Araki M.; Kamada M.; Sato S.; Okuno Y.; Fujita N.; Katayama R. Brigatinib Combined with Anti-EGFR Antibody Overcomes Osimertinib Resistance in EGFR-Mutated Non-Small-Cell Lung Cancer. Nat. Commun. 2017, 8, 14768. 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt H.; Böse D.; Petronczki M.; Scharn D.; Bader G.; Baum A.; Bergner A.; Chong E.; Döbel S.; Egger G.; Engelhardt C.; Ettmayer P.; Fuchs J.; Gerstberger T.; Gonnella N.; Grimm A.; Grondal E.; Haddad N.; Hopfgartner B.; Kousek R.; Krawiec M.; Kriz M.; Lamarre L.; Leung J.; Mayer M.; Patel N.; Simov B.; Reeves J.; Schnitzer R.; Schrenk A.; Sharps B.; Solca F.; Stadtmüller H.; Tan Z.; Wunberg T.; Zoephel A.; Mcconnell D. Start Selective and Rigidify: The Discovery Path Toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62, 10272–10293. 10.1021/acs.jmedchem.9b01169. [DOI] [PubMed] [Google Scholar]
- Eurofins . KINOMEscan - World’s Largest Kinase Assay Panel (489 kinases), https://www.Discoverx.Com/Services/Drug-Discovery-Development-Services/Kinase-Profiling/Kinomescan (accessed Jan 17, 2022)
- The selectivity score is the percent of non-mutant kinases inhibited at 90% at 3 μM, screening against a panel of 489 kinases.
- Hanan E. J.; Eigenbrot C.; Bryan M. C.; Burdick D. J.; Chan B. K.; Chen Y.; Dotson J.; Heald R. A.; Jackson P. S.; La H.; Lainchbury M. D.; Malek S.; Purkey H. E.; Schaefer G.; Schmidt S.; Seward E. M.; Sideris S.; Tam C.; Wang S.; Yeap S. K.; Yen I.; Yin J.; Yu C.; Zilberleyb I.; Heffron T. P. Discovery of Selective snd Noncovalent Diaminopyrimidine-Based Inhibitors of Epidermal Growth Factor Receptor Containing the T790M Resistance Mutation. J. Med. Chem. 2014, 57, 10176–10191. 10.1021/jm501578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine J. D.; Takahashi L.; Lockhart K.; Cheong J.; Tolan J. W.; Selick H. E.; Grove J. R. MDCK (Madin–Darby Canine Kidney) Cells: A Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88, 28–33. 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Clearance in Drug Design. J. Med. Chem. 2019, 62 (5), 2245–2255. 10.1021/acs.jmedchem.8b01263. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Waters N. J.; Argikar U. A.; Dennehy M. K.; Zhan J.; Gunduz M.; Harriman S. P.; Berellini G.; Liric Rajlic I.; Obach R. S. Comprehensive Assessment of Human Pharmacokinetic Prediction Based on in Vivo Animal Pharmacokinetic Data, Part 2: Clearance. J. Clin. Pharm. 2013, 53 (2), 178–191. 10.1177/0091270012440282. [DOI] [PubMed] [Google Scholar]
- Jones R. D.; Jones H. M.; Rowland M.; Gibson C. R.; Yates J. W.T.; Chien J. Y.; Ring B. J.; Adkison K. K.; Ku M. S.; He H.; Vuppugalla R.; Marathe P.; Fischer V.; Dutta S.; Sinha V. K.; Bjornsson T.; Lave T.; Poulin P. PhRMA CPCDC Initiative on Predictive Models of Human Pharmacokinetics, Part 2: Comparative Assessment of Prediction Methods of Human Volume of Distribution. J. Pharm. Sci. 2011, 100 (10), 4074–4089. 10.1002/jps.22553. [DOI] [PubMed] [Google Scholar]
- Conducted in rodent (Sprague–Dawley) at 10, 30, and 100 mg/kg/day and NHP (cynomolgus monkeys) at 3, 10, and 30 mg/kg/day. These studies provided a safety margin of >5× above the anticipated exposure at the projected efficacious H1975 cellular IC90.
- Synthetic routes to compounds in Table 2 and 3 with alternate cores can be found in the Supporting Information.
- Higgins R. H.; Cromwell N. H. Stereochemistry, Kinetics, and Mechanism of the Hydrolysis of 3-Azetidinyl Tosylates. J. Am. Chem. Soc. 1973, 95 (1), 120–124. 10.1021/ja00782a020. [DOI] [Google Scholar]
- Frigola J.; Torrens A.; Castrillo J. A.; Mas J.; Vano D.; Berrocal J. M.; Calvet C.; Salgado L.; Redondo J. 7-Azetidinylquinolones as Antibacterial Agents. 2. Synthesis and Biological Activity of 7-(2,3-disubstituted-1-azetidinyl)-4-oxoquinoline- and -1,8-Naphthyridine-3-carboxylic acids. Properties and Structure-Activity Relationships of Quinolones with an Azetidine Moiety. J. Med. Chem. 1994, 37, 4195–4210. 10.1021/jm00050a016. [DOI] [PubMed] [Google Scholar]
- Gajiwala K. S.; Feng J.; Ferre R.; Ryan K.; Brodsky O.; Weinrich S.; Kath J. C.; Stewart A. Insights into the Aberrant Activity of Mutant EGFR Kinase Domain and Drug Recognition. Structure 2013, 21 (2), 209–219. 10.1016/j.str.2012.11.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.