

Abstract
The interaction of water molecules and hydroxyl groups with titanium dioxide (TiO2) surfaces is ubiquitous and very important in anatase nanoparticle photocatalytic processes. Infrared spectroscopy, assisted by ab initio calculations of vibrational frequencies, can be a powerful tool to elucidate the mechanisms behind water adsorption. However, a straightforward comparison between measurements and calculations remains a challenging task because of the complexity of the physical phenomena occurring on nanoparticle surfaces. Consequently, severe computational approximations, such as harmonic vibrational ones, are usually employed. In the present work we partially address this complexity issue by overcoming some of the standard approximations used in theoretical simulations and employ the Divide and Conquer Semiclassical Initial Value Representation (DC-SCIVR) molecular dynamics. This method allows to perform simulations of vibrational spectra of large dimensional systems accounting not only for anharmonicities, but also for nuclear quantum effects. We apply this computational method to water and deuterated water adsorbed on the ideal TiO2 anatase(101) surface, contemplating both the molecular and the dissociated adsorption processes. The results highlight not only the presence of an anharmonic shift of the frequencies in agreement with the experiments, but also complex quantum mechanical spectral signatures induced by the coupling of molecular vibrational modes with the surface ones, which are different in the hydrogenated case from the deuterated one. These couplings are further analyzed by exploiting the mode subdivision performed during the divide and conquer procedure.
Introduction
Understanding photocatalytic processes is important for their numerous potential applications in many fields, such as environmental remediation (e.g., mineralization of pollutants),1 and energy conversion (e.g., facilitating the process of water splitting).2,3 Among the several materials, titanium dioxide (TiO2) is one of the most promising, thanks to its abundance, high stability, and nontoxicity. Even if the rutile polymorph is the thermodynamically more stable one, it is the anatase polymorph that is more interesting for photocatalytic applications. However, anatase is available mainly in the form of nanoparticles, where the size is nanoscopic and quite complicated. Given the nanostructures involved in the photocatalytic processes, an atomistic comprehension of the mechanisms of molecular adsorption on TiO2 surfaces, both from an experimental and theoretical point of view, becomes crucial.4−11
Water adsorption is of tantamount importance in photocatalysis because it is ubiquitous and unavoidable, given water vapor in the air. During the synthesis of TiO2 and other oxide nanoparticles, water is an omnipresent component, despite the several treatments that have been proposed to dehydrate the nanoparticles. It is then realistic to consider that some amount of water or hydroxyl groups are present on the nanoparticle surfaces. This motivated a wide effort to understand the process of water adsorption on oxide surfaces and nanoparticles.7,12,13
Among the variety of experimental approaches adopted to investigate the behavior of water and hydroxyl groups on titanium nanoparticles, infrared spectroscopy can provide access to the vibrational frequencies of the adsorbed species and possibly allow an understanding of how water vibrational frequencies are modified by the interaction with the nanoparticle or other coadsorbed molecules.6,14 Unfortunately, the complexity and the variety of processes at nanoparticle surfaces cause the experimental spectra to be rather difficult to interpret. Even if an adsorption peak around 1615–1640 cm–1 can be assigned to the bending mode of the adsorbed molecular water, the IR spectra presents a broad adsorption band with several secondary structures in the frequency region, which is typical of OH stretchings (2500–3800 cm–1) and that are difficult to interpret.15−25 Roughly speaking, the stretching modes of an adsorbed molecular water are generally attributed to the lower frequency part of this absorption band, while the hydroxyl stretching has been assumed to be responsible for the high energy part of the band. This is clearly a simplified picture since, for example, the hydroxyl stretching can have a lower frequency when ascribed either to the formation of hydrogen bonds or to surface oxygen coordination or even in a water monolayer adsorption structure. In addition, the complex morphology of the nanoparticles exhibits not only different surfaces (mainly the (101) and (001) in the case of anatase TiO2), but also complex morphologies originated, for example, from high index facets, edges or corners, and defects.26 Additionally, side residuals from the synthesis and experimental limitations in controlling the environment during the measurements can further complicate the spectra interpretation.
In this paper, we focus on the relevance of anharmonic and quantum contributions to the vibrational spectra of water adsorption on anatase(101) surfaces and, more generally, in the field of molecular adsorption. We try to reach this goal by simulating the vibrational frequencies of the system composed by a water molecule and titania. Theoretical calculations can help in providing not only an interpretation of spectroscopy measurements, but also new atomistic physical insights. A standard harmonic estimation of the vibrational frequencies, based on Density Functional Theory (DFT) Potential Energy Surfaces (PES), can be inaccurate enough to deal with such a complex system, where intermolecular gas–surface interactions are important.27,28 While, from one side, it is difficult to improve the accuracy of the description of the electron–electron interaction, as provided by standard DFT functionals for these large periodic systems, also overcoming the harmonic approximations can still be a challenge.29 The inclusion of anharmonic contributions can be achieved, for example, by a scaled harmonic approximation23 or by means of the estimation of the velocity or dipole moment autocorrelation function on top of a molecular dynamic trajectory.30,31 Quantum mechanical effects can also be included, relying on approximate approaches, as applied to TiO2 surfaces,32 or can be addressed with complex theoretical approaches, as tested in metallic surfaces.33 These approaches, when applied to water molecules adsorbed on the TiO2 surfaces, can suffer from inaccuracies originating from the arbitrary choices of the scaling factors or from the reduced dimensionality of the calculation, as well as from the model employed to describe the nanoparticle surfaces. Consequently, when interpreting a complex experimental spectra, as in this case, it can become difficult to distinguish if the mismatch between theory and measurement can be ascribed to a limitation of the adopted approximation at the level of theory or to the complex morphology of the nanoparticles, which can hardly be addressed in an exhaustive way.
For these reasons, we tackle this spectroscopic problem with a quantum vibrational mechanic method that we have recently introduced,34 which can be an alternative with respect to the quantum mechanical approaches presented by Manzhos and Ihara.29 The Divide and Conquer Semiclassical Initial Value Representation (DC-SCIVR) approach34,35 to the semiclassical molecular dynamics is a useful tool that relies on classical Born–Oppenheimer trajectories and is able to address this issue since it has the capability of describing quantum nuclear effects, like zero point energy motion, overtones, and combination bands, which are supposed to be important in describing the water molecule adsorption. The basic idea that permits the application of such a computationally expensive approach to a large dimensional system as a titania slab with adsorbed molecules is to split the full problem in a set of vibrational degrees of freedom subspaces. In addition, the application of this approach to the problem of adsorption can be further facilitated from the fact that one is generally interested only in the high energy modes originating from the adsorbed molecule and a few phonon ones. Consequently, the simulation can be limited to the subspace, including the adsorbate modes and a few interacting ones.36 Moreover, a side result of the DC-SCIVR, which can be achieved by an inspection of the modes coupled in the same subspace, is the possibility to discuss the mechanism of interaction between the adsorbed molecule and the surface motion.
Specifically, in this paper, we apply DC-SCIVR to calculate the power spectra of water adsorbed on the stoichiometric TiO2 Anatase (101) surface, which is the most stable among the anatase surfaces. We restrict our analysis on a single molecule adsorbed on the surface thus neglecting the effect of the coverage and of the interaction between several water molecules at the level of vibrational properties. In addition, we will consider for adsorption the most energetically favorable configuration, which corresponds to the molecule on the 5-fold coordinated titanium atoms (Ti5c). There is also the possibility that the adsorption process yields the dissociation of the adsorbed molecule, thus leaving a hydroxyl group on the Ti5c site and the remaining hydrogen bonds with a two coordinate surface oxygen (O2c). We also contemplate these cases and we investigate the difference and similarities in the vibrational behavior of these different adsorption mechanisms in conjunction with the molecular hydrogen deuteration isotopic effect by performing similar DC-SCIVR calculations for the adsorption of deuterated water. A comparison of the results performed at different levels of theory (harmonic vs classical anharmonic vs semiclassical approximate quantum mechanical) will provide an estimate of the effect of some adopted approximations and, hopefully, help in unraveling some of the open questions in the interpretation of the experiments.
First, we briefly summarize the basics of the DC-SCIVR approach and the computational parameters used in this work and then we present our results. These address, in order, the adsorption geometry, the adsorbate vibrational frequencies, and the description of the vibrational couplings. A general discussion and comparison with the available measurements follows.
Computational Methods
Ab Initio Calculation Computational Details
All ab initio calculations are performed with the open source Quantum-Espresso (Q-E)37,38 suite of codes at the level of Density Functional Theory (DFT) with a Perdew–Burke–Ernzerhof (PBE) parametrization for the exchange and correlation functional. The PBE functional is known to provide a good description of hydrogen bonds, although the ionic and covalent bonds are usually slightly too long, which leads to underestimate the harmonic frequencies. In addition we adoptultrasoft pseudopotentials for the description of the core electrons. The plane wave energy cutoff was set to 40 Ry for the wave functions and 400 Ry for the density, and the Brillouin zone (BZ) was sampled using the Γ point only. This choice is justified by the size of the supercell, which is 7.58 Å along the [010] direction and 10.33 Å along [1̅01]. This limited k-point sampling does not affect the values of the harmonic frequencies (see Table S4). The surface has been modeled through a supercell generated by starting from the TiO2 anatase bulk ab initio equilibrium geometry and lattice constants. The supercell has been cut along the plane defined by the (101) Miller indexes. The choice of the (101) surface is motivated by the fact that it is the thermodynamic most stable one. Then, the adsorption geometries were determined by a further geometry optimization of the surface atoms and the adsorbants, by adopting a threshold of 10–7 Ry on the total energy and 10–7 Ry/a0 on the forces. The slab thickness consists of four Ti layers, with the two deepest ones frozen at the bulk geometry, while the other coordinates are free to relax to their equilibrium values. A separation between periodic replica of the slabs has been achieved by inserting ∼10 Å layer of vacuum. The most stable site for adsorption on the TiO2 anatase(101) surface is on top of the Ti5c atoms, and our choice of supercell makes four Ti5c sites available for adsorption. Thus, the adsorption of a single molecule per supercell corresponds to a coverage Θ = 0.25. The relative stability of the different adsorption geometries has been estimated though the evaluation of the Binding Energy (BE)
| 1 |
where Esys is the energy of the interacting surface–adsorbant system, Eslab is the energy of the bare slab, Eads is the energy of the isolated adsorbed molecule in its equilibrium configuration, and n is the number of the adsorbed molecules (n = 1 in our case).
The vibrational frequencies have been estimated at different level of accuracy with the purpose of highlighting the anharmonic effects. As a reference, the harmonic estimation of vibrational frequencies have been determined through the Density-Functional Perturbation Theory (DFPT)39 implementation available in Q-E. In this way, we compute the equilibrium Hessian matrix and, after its diagonalization, the matrix for coordinate transformation from Cartesian to normal modes. The nonanalytic term which corrects the long wavevector limit, that is, q → 0 limit, of the dynamical matrix for polar crystals has been neglected in the evaluation of the Hessian matrix, since it presents a limited effect on the vibrational frequencies of the adsorbate, as we have verified. We also checked that the choice of limiting the BZ sampling to the Γ point, the choice of a limited slab thickness, and of neglecting the Hubbard U parameter in the Hamiltonian or Empirical Dispersions yield to a limited effect on the geometries and harmonic frequency estimates (see Tables S1–S5 in the Supporting Information). In order to decouple the motion of the relaxed atoms from that of the atoms that are frozen in their bulk positions, we set the Hessian matrix elements of the latter ones to zero.
Classical Power Spectrum Calculation
We adopt a molecular dynamics approach to improve the accuracy with respect to the harmonic frequency estimates, including anharmonic effects. A classical estimation of anharmonic frequencies can be obtained from the Fourier transform of the velocity autocorrelation function. We run an ab initio molecular dynamics trajectory to calculate the velocity autocorrelation function. Specifically, we employ the time-averaging filtering technique40 to improve the numerical convergence with respect to the number of time-steps according to the following formula:
| 2 |
where pj(t) is the j-esime component of the normal mode p(t) momentum vector. A similar formula can be employed in atomic Cartesian coordinates. This kind of simulation will reproduce the fundamental frequency of each mode, including the anharmonic contributions. The Born–Oppenheimer molecular dynamics (BOMD) has been performed with an in-house adaptation of Plane-Wave Self-Consistent Field (PWScf) module to integrate the equation of motion using the symplectic velocity-Verlet algorithm. The BOMD equation of motion have been solved in the NVE ensemble for 2500 iterations with a time-step of 10 au (0.242 fs), thus yielding 0.6 ps of dynamics. The kinetic energy is initialized at the harmonic zero-point energy (ZPE), and for this reason, the method is also called “quasi-classical”.
Semiclassical Power Spectrum Calculation
Given a representation of the underlying PES, the semiclassical power spectrum reproduces all the vibrational eigenvalues on an absolute scale, including overtones and resonances. In contrast, the quasi-classical approximation is composed of a series of fundamental frequencies only. In the present context, the semiclassical approach can reveal the combination bands arising from the coupling between the modes of the adsorbate and the surface phonons.
Our quantum mechanical vibrational levels are determined though the Divide-and-Conquer Semiclassical Initial Value Representation (DC-SCIVR) method that we developed recently.34,35,41−51 Here we report only the working equations of the DC-SCIVR approach for the calculation of vibrational spectra. The reader can find further details in ref (52), and further details can be found in regard to the DC-SCIVR application to adsorption studies in ref (36). We present the method starting from the Multiple Coherent states Semiclassical Initial Value Representation (MC-SCIVR) version of the semiclassical molecular dynamics,53−58 on which the DC-SCIVR approach is based. The MC-SCIVR vibrational power spectra I(E) are calculated with the following sum over Ntraj classical trajectories:
| 3 |
where T is the total trajectory simulation time, St(p(0),q(0)) is the classical action, ϕt is the phase of the Herman–Kluk pre-exponential factor Ct(p(0), q(0))
| 4 |
where Mqq = ∂q(t)/∂q(0) and so on are the monodromy matrix blocks.
In eq 3, |p(t),q(t)⟩ is a coherent state of the following type:59−64
| 5 |
In eqs 4 and 5, Γ is approximated to be a diagonal width matrix for bound state calculations, with coefficients usually equal to the square root of the harmonic vibrational frequencies, and the time evolution of Ct(p(0),q(0)) is computed through the solution of the equation of motion for the monodromy matrix M, for which the knowledge of the Hessian matrix along the MD trajectory is required. The advantage of the MC-SCIVR method with respect to other quantum methods is that a suitable choice of initial conditions for the MD trajectories and of the reference coherent states |χ⟩ can enhance the spectral features corresponding to molecular vibrational excitations and avoid the burden of the phase space integration required by other semiclassical molecular dynamics approaches. Specifically, the trajectory initial conditions should be set to provide the system an energy close to a given quantum vibrational eigenvalue, whose condition can be satisfied by relying on its harmonic estimate, that is, peq2/2m = ℏω(n + 1/2). Additionally, the corresponding excitation can be selected by a suitable choice of the reference superposition of coherent states of the type:
| 6 |
where γ is a vector such that when all its elements are equal to 1, the ZPE peak and all even overtone intensities are enhanced,56 while when one of its component is equal to −1, the fundamental and the odd overtones of that mode component are enhanced. Unfortunately the MC-SCIVR is limited to a few tens of degrees of freedom calculations. For this reason, we have introduced the DC-SCIVR approach,34,35,41 whose strategy is to split the full dimensional Nvib problem into one at reduced dimensions (M < Nvib). This partition is 2-fold because it allows for faster numerical convergence with respect to the full dimensional calculation and also for the identification of the different spectroscopic signals. Therefore, the power spectrum I(E) of eq 3 is obtained as the composition of partial spectra Ĩ(E), computed in an M-dimensional subspace of the full Nvib-dimensional space. The DC-SCIVR power spectrum formula is
| 7 |
where all the quantities with the tilde superscript are evaluated by projecting the full dimensional ones onto the M-subspace. In this way, we can drastically reduce the dimensionality of the semiclassical calculation. For most of the quantities in eq 7, the projection onto each subspace starting from the full dimensional one consists of taking the related vector components, except for the classical action S̃t, due to the nonseparability of the potential energy term. We obviate to this issue by assuming the following expression for the reduced dimensional potential
| 8 |
where 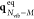 is the equilibrium values of the positions
for the degrees of freedom that do not belong to the M-dimensional
subspace S. The subspace subdivision should pair the stronger coupled
modes in the same subspace. Among the different approaches developed
in our group,65,66 in the present work we will employ
the “Hessian” method52,65 for the vibrational
space subdivision. Specifically, the magnitude of the coupling between
different modes is estimated by calculating the off-diagonal elements
of the average Hessian H̅ij matrix (in absolute value and normal mode coordinates) over
some representative trajectory steps, that is,
is the equilibrium values of the positions
for the degrees of freedom that do not belong to the M-dimensional
subspace S. The subspace subdivision should pair the stronger coupled
modes in the same subspace. Among the different approaches developed
in our group,65,66 in the present work we will employ
the “Hessian” method52,65 for the vibrational
space subdivision. Specifically, the magnitude of the coupling between
different modes is estimated by calculating the off-diagonal elements
of the average Hessian H̅ij matrix (in absolute value and normal mode coordinates) over
some representative trajectory steps, that is, 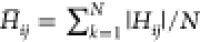 . Then, the subspace subdivision is obtained
by fixing a threshold value ϵ under which the H̅ij is set to zero. In this way the averaged
Hessian can be divided into sub-blocks such that all normal modes
within the same matrix sub-block are also in the same subspace. The
Hessian subblocks of interest are calculated at each time-step.
. Then, the subspace subdivision is obtained
by fixing a threshold value ϵ under which the H̅ij is set to zero. In this way the averaged
Hessian can be divided into sub-blocks such that all normal modes
within the same matrix sub-block are also in the same subspace. The
Hessian subblocks of interest are calculated at each time-step.
The DC-SCIVR implementation for the case of the adsorbed molecules
on solid surfaces is described in ref (36). Here, we adopt the same computational approach.
Specifically, we calculate the averaged Hessian over 20 full-dimensional
Hessian matrices computed within DFPT at time-steps uniformly distributed
along the trajectory. We think that this number of Hessian matrices
are representative enough of the trajectory geometries, as already
shown elsewhere.36 To identify the preferable
choice for each subspace dimension, we performed a DC-SCIVR power
spectrum calculation of increasing subspace dimensionality until the
quality of the spectrum was satisfactory. The quality has been evaluated
by considering mainly that there should not be any signal at energy
values lower than the ZPE value and that the position and the intensity
of each peak remains unaffected by increasing the subspace dimensionality.
Once we have identified how the full dimensional vibrational subspace
is divided into subspaces, we compute the ingredients in eq 7 that are necessary for the determination
of DC-SCIVR spectra. These are the projected potential, as in eq 8, and the Hessian at each
time-step. To reduce the computational cost, we calculate only the
Hessian elements of the subblocks that correspond to the vibrational
subspace of the adsorbate modes and the mostly coupled phonons. Most
of the phonon mode is weakly coupled with the adsorbant modes, and
we are not interested in their spectroscopic signal. For the Hessian
evaluation, we adopt a finite difference approach starting from the
forces obtained from the ab initio code and an atomic displacement 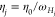 with η0 equal to 5 ×
10–4
with η0 equal to 5 ×
10–4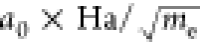 and
and 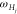 being the harmonic frequency of the mode.
being the harmonic frequency of the mode.
Results and Discussion
Geometry Considerations
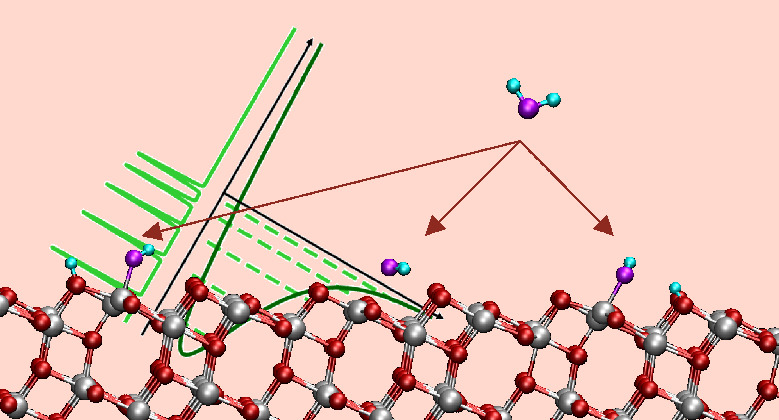
We start by determining which are the most stable water adsorption sites on the (101) anatase TiO2 surface. These sites correspond to the Ti5c atoms. Our geometry optimization includes the adsorbant and part of the slab, and it yields the conformations reported in Figure 1 (or in Figure S1 in the Supporting Information where the slab periodicity is more evident).We have identified three main different adsorption geometries, in agreement with the literature.67−75 Two geometries involve the molecule dissociation where a hydrogen atom is bonded to a surface O2c site, while the remaining one is the molecular adsorption. In this last case, the water oxygen Ow atom is bonded to a Ti5c site at a distance with ∼2.3 Å bond length and the two hydrogen atoms point toward two surface O2c sites forming hydrogen bonds. The geometry parameters for the molecular adsorption are reported in Table 1. The comparison with the available literature values shows a quantitative agreement.27,71−73
Figure 1.

Optimized geometries of molecular and dissociative H2O adsorption on the TiO2 anatase(101) surface. Titanium atoms are in gray, surface oxygen in red, molecular oxygen in violet, and hydrogen in cyan. Binding energies are reported in the boxes.
Table 1. Relevant Geometry Parameters and Binding Energies (BE) for Molecular H2O Adsorption on the TiO2 Anatase(101) Surface.
| layers | DFT Vxc | θ | r(Ti5c–Ow) (Å) | r(Ow–H) (Å) | d(O2c–H) (Å) | α(H–Ow–H) (°) | BEa (eV) | |
|---|---|---|---|---|---|---|---|---|
| Q-E present | 4L | PBE | 0.25 | 2.289 | 0.984 | 2.284 | 103.89 | 0.76 |
| CASTEP68 | 8L | PBE | 2.236 | 0.995 | 2.101 | 102.585 | 0.948 | |
| CASTEP67 | 4L | PBE | 2.245 | 0.993 | 2.122 | 0.916 | ||
| Q-E70 | PBE | 0.17 | 2.30 | 2.26 | 0.71 | |||
| Q-E69 | PBE | 0.17 | 2.300 | 2.257/2.263 | 0.720 | |||
| Q-E71 | 4L | PBE | 0.17 | 2.30 | 0.730 | |||
| Crystal1472 | 4L | PBE | 0.25 | 2.25 | 0.71 | |||
| Crystal1472 | 4L | HSE06 | 0.25 | 2.29 | 0.84 | |||
| Q-E73 | PBE | 2.32 | 2.35 | 0.73 | ||||
| Q-E76 | PBE | 2.31 | 2.34 | 0.67 | ||||
| CPMD74 | PW91 | 2.28 | 1.02 | 1.88/1.96 | 0.74 | |||
| Crystal1775 | 8L | PBE0 | 0.25 | 2.28/2.43 | 0.78 |
Experimental value 0.5–0.7 eV.77
Table 2 reports the geometry parameters for the dissociative adsorption. We distinguish two configurations, depending on the mutual positions of the hydroxyl groups that arise from the molecule dissociation: in the “intrapair” configuration (Figure 1b), O2c–H and Ow–H bind the same Ti5c. In the “interpair” configuration, reported in Figure 1, they bind different titanium atoms. The relevant interatomic distances and the binding energies are detailed in Table 2. In the interpair conformation a weak hydrogen bond is formed between the hydrogen adsorbed on the surface O2c site and the dissociated hydroxyl group (d(Ow–Hdiss) = 2.487 Å), while in the intrapair configuration, the hydrogen bond is absent or very weak (d(Ow–Hdiss) = 2.680 Å).
Table 2. Relevant Geometrical Parameters and Binding Energies (BE) for Dissociative H2O Adsorption on the TiO2 Anatase(101) Surface.
| layers | DFT Vxc | θ | r(Ti5c–Ow) (Å) | r(Ow–H) (Å) | d(O2c–H) (Å) | d(Ow–Hdiss) (Å) | α(Ow–Hdiss–O2c) (°) | BE (eV) | |
|---|---|---|---|---|---|---|---|---|---|
| Intrapair Dissociative Adsorption | |||||||||
| Q-E present | 4L | PBE | 0.25 | 1.824 | 0.976 | 0.973 | 2.680 | 79.578 | 0.41 |
| CASTEP68 | 8L | PBE | 1.816 | 0.983 | 1.039 | 0.658 | |||
| CASTEP67 | 4L | PBE | 1.819 | 0.981 | 0.978 | 2.549 | 0.622 | ||
| Crystal1472 | 4L | PBE | 0.25 | 2.71 | 0.38 | ||||
| Crystal1472 | 4L | HSE06 | 0.25 | 2.65 | 0.54 | ||||
| CPMD74 | PW91 | 1.83 | 0.99 | 0.30 | |||||
| Crystal1775 | 8L | PBE0 | 0.25 | 2.58 | 0.47 | ||||
| Interpair Dissociative Adsorption | |||||||||
| Q-E present | 4L | PBE | 0.25 | 1.854 | 0.975 | 0.982 | 2.487 | 138.282 | 0.29 |
| CASTEP67 | 4L | PBE | 1.835 | 0.977 | 0.985 | 2.598 | 0.5 | ||
| Crystal1472 | 4L | PBE | 0.25 | 2.75 | 0.34 | ||||
| Crystal1472 | 4L | HSE06 | 0.25 | 2.83 | 0.49 | ||||
| Q-E76 | PBE | 1.83 | 2.66 | 0.32 | |||||
| CPMD74 | PW91 | 1.85 | 1.00 | 1.00 | 2.39 | 0.23 | |||
| Crystal1775 | 8L | PBE0 | 0.25 | 2.74 | 0.42 | ||||
By comparing the value of the BE of the three type of adsorptions, we find that the molecular adsorption is the most stable one, followed by the intrapair dissociative configuration, which is slightly more stable than the interpair one. The larger binding energy of the molecular adsorption configuration is consistent with the formation of two quite strong hydrogen bonds between Hw and two surface O2c, which is not the case upon water dissociation.
Harmonic Frequencies Calculations
The calculations of harmonic vibrational frequencies is the first step toward the understanding of the relevance of the anharmonic contributions to the vibrational spectra. As a reference, the harmonic frequencies are reported in Table 3. When comparing with the literature ones, which are also calculated only at harmonic level, we find some differences, as well as there are significant differences between the literature values. We think that these discrepancies at the harmonic level of theory are mainly due to the difference computational setup, such as the slab dimension. However, for each type of calculation, we observe for the molecular adsorption the normal mode displacements of the adsorbate to be of the type of symmetric/asymmetric stretches and bending, as in the gas phase. While the bending frequency remains almost unchanged upon adsorption, the two stretching frequencies are smaller both in the hydrogenated and deuterated case. This red-shift can be ascribed to the formation of the hydrogen bonds between the water hydrogen atoms with the surface oxygen atoms, as described above. Differently from the molecular adsorption, the dissociative one is characterized by two hydroxyl stretching modes in the high frequency region without any bending mode signal. Thus, the experiments distinguish the molecular from the dissociative adsorption by the presence of the bending band. In the dissociated configurations, the two types of stretching modes correspond to the Ow–H and O2c–H stretch. In the case of the intrapair dissociative adsorption, the Ow–H and the O2c–H stretching frequencies are quite similar and greater than the symmetric and asymmetric frequencies of the molecular adsorption. This is equally valid when comparing the deuterated forms. Instead, in the case of the interpair dissociative adsorption, the O2c–H stretching frequency is comparable with the molecular adsorption, while the Ow–H stretching frequency is much greater. This observation suggests a significant difference between the two types of dissociative interactions, which we think is due to the presence of a weak hydrogen bond between the O2c–H group and the oxygen atom in the Ow–H adsorbate in the case of the interpair dissociative adsorption, as noted above from the distances values (Table 2). Same considerations can be applied to the deuterated case.
Table 3. Comparison of Harmonic Frequencies for H2O and D2O Adsorption on the TiO2 Anatase(101) Surface against Literature Dataa.
| layers | DFT Vxc | θ | H2O | D2O | |||||
|---|---|---|---|---|---|---|---|---|---|
| Gas Phase | |||||||||
| Q-E present | 1580 | 3731 | 3843 | 1156 | 2689 | 2815 | |||
| Molecular Adsorption | |||||||||
| Q-E present | 4L | PBE | 0.25 | 1586 | 3580 | 3647 | 1163 | 2581 | 2673 |
| Q-E78 | PBE+U | 1573 | 3702 | 3807 | 1153 | 2669 | 2791 | ||
| Q-E27 | PBE | 1579 | 3611 | 3686 | |||||
| DMol328 | PBE | 3516 | 3620 | ||||||
| Intrapair Dissociative Adsorption | |||||||||
| (Ow–H) | (O2c–H) | (Ow–D) | (O2c–D) | ||||||
| bond distance (Å) | 0.976 | 0.973 | 0.976 | 0.973 | |||||
| Q-E present | 4L | PBE | 0.25 | 3760 | 3797 | 2738 | 2765 | ||
| Q-E78 | PBE+U | 3750 | 3807 | 2726 | 2766 | ||||
| DMol328 | PBE | 3645 | 3689 | ||||||
| VASP23 | PBE+U | 3734/3722 | |||||||
| Interpair Dissociative Adsorption | |||||||||
| (Ow–H) | (O2c–H) | (Ow–D) | (O2c–D) | ||||||
| bond distance (Å) | 0.975 | 0.982 | 0.975 | 0.982 | |||||
| Q-E present | 4L | PBE | 0.25 | 3775 | 3644 | 2748 | 2654 | ||
All the frequencies are in cm–1.
Classical versus Semiclassical Power Spectra
The harmonic approximation may not be accurate enough to predict experimental frequencies. In particular, the zero-point energy of the OH (OD) stretching mode is significant, so that the atomic motion is sensitive to the PES much above the minimum, in sharp contrast to the harmonic picture. We now employ the methods described above, that is, the quasi-classical and the semiclassical molecular dynamics ones, to gain a complete spectroscopic picture of the water molecule adsorption on the (101) anatase surface.
As a first improvement, we include classical anharmonic effects by calculating the quasi-classical power spectra with the time-averaging filtering technique of the velocity autocorrelation function, as described in eq 2. As a further improvement, the quantum mechanical power spectra are calculated using the DC-SCIVR method of eq 7. We recall that with the semiclassical method, we account for not only the classical and quantum anharmonicities, but also the combination and overtone vibrational states.
Figure 2 shows the molecular adsorption power spectra, both in the gas phase (before adsorption) and after adsorption. Frequencies are red-shifted after the inclusion of anharmonic effects in a greater amount for the hydrogenated system respect to the deuterated one, as expected. In the gas phase, the fundamental frequencies are equally reproduced by both methods. Instead, when moving to the adsorbed water molecule there is a significant reduction of the frequency splitting between the symmetric and asymmetric modes which is larger for H2O than D2O. In addition, the semiclassical power spectra of the molecular adsorbed water show numerous combination bands with the low-frequency phonons of the titania substrate. This information provided by DC-SCIVR clearly proves the coupling between molecular modes and phonons. Specifically, DC-SCIVR indicates that lower frequency phonons are more strongly coupled to adsorbed H2O than D2O.
Figure 2.

Comparison between quasi-classical and semiclassical spectra for H2O in gas phase and adsorbed on anatase(101). Left panel: H2O adsorption. Right panel: D2O adsorption. DC-SCIVR semiclassical calculations refer to a 14-dimensional subspace in the case of H2O and 12-dimensional subspace for D2O.
Table 4 compares the results of Figure 2 with the available experimental results. Our Q-E computational setup is not so accurate for the gas phase as expected, since the PBE functional yields longer bonds and lower frequencies with respect to the experiments83 To check if this discrepancy is due to the plane-wave basis set, we perform a Gaussian basis sets harmonic, classical, and DC-SCIVR frequency calculation, and we find that this is consistent with our Q-E set up when adopting a PBE Vxc. However, frequencies are slightly more accurate with a hybrid PBE0 Vxc and almost exact with a classical and DC SCIVR B3LYP Vxc calculation (see Tables S6 and S7 in the Supporting Information) when compared with the gas-phase experimental values. This shows that the discrepancy is mainly due to the PBE functional and not to the DC SCIVR method. As far as the adsorbate frequencies are concerned, our quasi-classical and semiclassical fundamentals are anyway consistent with the experimental literature (see Table 4), apart from the bending case. Nevertheless, upon adsorption, the amount of downshift from the harmonic estimate to the semiclassical values ranges from 200 to 250 cm–1, thus much larger than for the isolated molecule (130 cm–1). This shows that the anharmonic contributions are much larger upon adsorption than in the gas phase. We believe that this is a genuine characteristic that is largely independent of the actual functional that is employed, as we observed the same amount of anharmonic contributions for different functionals in the gas phase (see Tables S6 and S7 in the Supporting Information). In the deuterated system, the gas-phase frequencies are about 50 cm–1 off the experimental values, while no terms of experimental comparison are available for the adsorbed system. In this case, the anharmonic correction for the stretching is ∼70 cm–1 both in gas and condensed phases.
Table 4. Comparison of Vibrational Frequencies of H2O in the Gas Phase and after Adsorption on the TiO2 Anatase(101) Surfacea.
| molecular
H2O |
molecular
D2O |
|||||
|---|---|---|---|---|---|---|
| bending | symmetric stretching | asymmetric stretching | bending | symmetric stretching | asymmetric stretching | |
| Gas Phase | Gas Phase | |||||
| experiment79−82 | 1595 | 3657 | 3756 | 1178 | 2671 | 2788 |
| harmonic | 1580 | 3731 | 3843 | 1156 | 2689 | 2815 |
| quasi-classical | 1545 | 3602 | 3683 | 1138 | 2617 | 2733 |
| DC SCIVR | 1546 | 3602 | 3682 | 1138 | 2618 | 2732 |
| Adsorbed | Adsorbed | |||||
| experiment | 1615,21 163519,20 | from 3300 to 370019,20 | 1205–121018,21 | |||
| harmonic | 1586 | 3580 | 3647 | 1163 | 2581 | 2673 |
| quasi-classical | 1567 | 3373 | 3376 | 1162 | 2517 | 2607 |
| DC SCIVR | 1572 | 3392 | 3392 | 1164 | 2518 | 2600 |
All the frequencies are in cm–1.
Figures 3 and 4 report the classical and semiclassical power spectra of the intra- and interpair adsorbates, respectively. In the case of the intrapair adsorption (Figure 3), the two Ow–H and O2c–H stretching modes are characterized by almost equal frequencies, both for the hydrogenated and deuterated systems at the classical level of theory. The semiclassical simulation (bottom panels of Figure 3) shows a slightly lower frequency for the Ow–H stretch than for to the O2c–H one, while such splitting is tiny for the deuterated case (Table 5). In addition, the semiclassical simulation yields a significant coupling between the water stretches and the phonon modes which is larger for the protonated than the deuterated surface. In particular, the O2c–H stretch is coupled to higher frequency phonons than the Ow–H one.
Figure 3.

Comparison of quasi-classical and semiclassical spectra for H2O dissociative adsorption on anatase(101) surface in the intrapair configuration. Left: H2O adsorption; right: D2O adsorption. Semiclassical calculations refer to a 6-dimensional subspace for H2O and 5-dimensional subspace for D2O.
Figure 4.

Same as in Figure 3 but for the interpair configuration. Semiclassical calculations refer to a 22-dimensional subspace for H2O and 14-dimensional subspace for D2O.
Table 5. Comparison of Vibrational Frequencies of H2O Adsorption on the TiO2 Anatase(101) Surface in the Dissociative Geometrya.
| dissociated
H2O |
dissociated
D2O |
|||
|---|---|---|---|---|
| Ow–H stretching | O2c–H stretching | Ow–D stretching | O2c–D stretching | |
| Intrapair Dissociative Adsorption | ||||
| bond distance (Å) | 0.976 | 0.973 | 0.976 | 0.973 |
| harmonic | 3760 | 3797 | 2738 | 2765 |
| quasi-classical | 3657 | 3683 | 2684 | 2707 |
| DC-SCIVR | 3572 | 3674 | 2682 | 2708 |
| Interpair Dissociative Adsorption | ||||
| bond distance (Å) | 0.975 | 0.982 | 0.975 | 0.982 |
| harmonic | 3775 | 3644 | 2748 | 2654 |
| quasi-classical | 3678 | 3555 | 2691 | 2573 |
| DC-SCIVR (6-dim.) | 3680 | 3550 | ||
| DC-SCIVR (14-dim.) | 2690 | 2474–2558–2688 | ||
| DC-SCIVR (22-dim.) | 3678 | 3560 | ||
All the frequencies are in cm–1. The two DC SCIVR calculations for H2O in the interpair dissociative adsorption are performed respectively with a 6-dimensional (upper line) and a 22-dimensional (lower line) vibrational subspace for comparison.
Figure 4 shows the analogous spectra of Figure 3 but for the interpair dissociative adsorption. In this case, both at quasi-classical and semiclassical levels, we find a significant difference in frequency between the Ow–H and the O2c–H stretching frequency. Also, by comparing the semiclassical results on the bottom panels of Figures 3 and 4, we can observe a significant involvement of the phonon modes in the interpair case respect to the intrapair one (as will be discussed in Adsorbate–Surface Interactions and shown in Figure 5). Specifically, beside the numerous combination peaks, the original streching peaks split both in classical and semiclassical simulations, suggesting the occurrence of Fermi-like resonances84 between the adsorbate and the titania.
Figure 5.
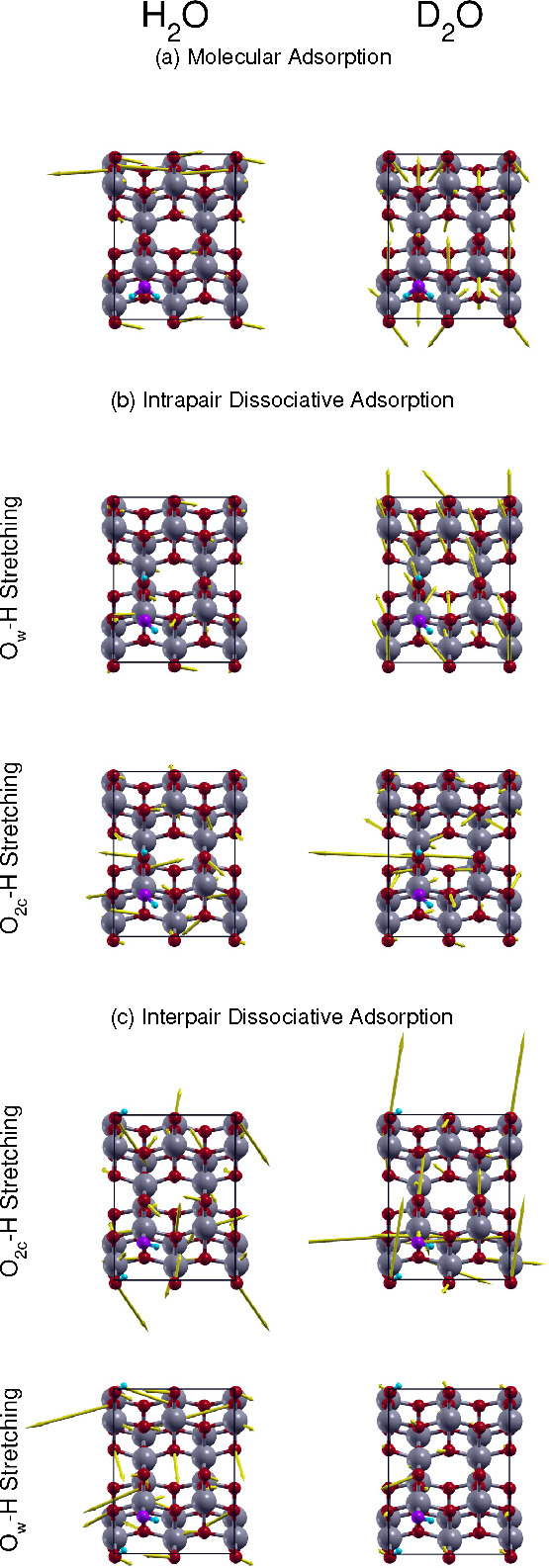
Displacement arrow plot for harmonic eigendisplacements of the phonon modes, which are coupled to the adsorbant modes, as obtained by inspection of the vibrational subspace grouping. The mass-scaled atomic displacement is larger the longer the arrow is of the corresponding atom.
A more detailed view on the frequency values is reported in Table 5 for the intra- and interpair dissociative adsorption of the hydrogenated and deuterated water molecule. No experimental values can be reported in this cases because the frequencies of the dissociative forms can not be assigned unequivocally, at variance with the molecular case, where the bending mode proves the presence of the ad-molecule.
As in the harmonic case, the main difference between the intra- and the interpair adsorption is that the fundamental Ow–H and O2c–H stretching frequencies are very similar in the intrapair case, while in the interpair configuration the O2c–H one is lower in frequency. This is due to the H-bonding interaction. The amount of anharmonicity, which is comparable for classical and semiclassical calculations, is quite consistent and of the order of hundred of wavenumbers. In Table 5 we also consider the effect of the DC-SCIVR vibrational subspace dimensionality in the case of the interpair dissociative adsorption and show that, for a 6-dimensional subspace, results are almost the same as taking an almost 4× larger (22-dimensional) vibrational subspace.
In the case of the dissociative deuterated water adsorption, the computed frequencies are reported in Table 5. As anticipated from Figures 3 and 4, in this case the D–O stretching modes are strongly coupled with substrate phonons. In Table 5 we report the related frequency values. In this case the anharmonicity amount is about 50 cm–1 as in the case of the harmonic estimates, that is, reduced by the larger mass of the deuterion. In the case of the DC-SCIVR interpair calculations, we also checked that the choice of the subspace dimension does not affect our results. This consideration confirms the resonant interaction between D–O modes and the surface phonons.
Adsorbate–Surface Interactions
An additional advantage of our DC-SCIVR methodology is the possibility of investigating the mechanism underlying the surface–adsorbate interactions by inspecting the vibrational subspace groups and in this way the coupling of surface phonons with the adsorbate normal modes. During the DC-SCIVR vibrational mode subspace determination, different normal mode groups are obtained for different averaged Hessian H̅ij course-graining thresholds. By starting from high enough threshold values, where all subspaces are monodimensional, and then gradually decreasing the threshold value, we can detect which phonon modes share in primis the same subspace with the adsorbant modes. Figure 5 reports these modes for each adsorption scenario. The phonon modes are represented with a displacement arrow plot corresponding to the harmonic eigendisplacements. The mass-scaled atomic displacement is larger the longer is the arrow of the corresponding atom. Other examples are reported in the Supporting Information (Figures S3–S7). The atoms without any arrow either correspond to the adsorbated molecule or have not been relaxed during the optimization and are not part of the dynamics. These last atoms belong to the layers deeper than the second one.
All the phonon modes of Figure 5 are of the type of frustrated rotations respect to the adsorbant. In other words, these modes involve an almost rigid motion of the molecule with respect to the surface with an additional vibration of the surface atoms. On the top panel of Figure 5, we report the phonon mode that is specific to the water molecular adsorption process. It involves the surface atoms to which the hydrogen are bonded, the four O2c surface sites, and for a smaller amount, the Ti5c atoms to which the surface O2c atoms are coordinated. We note that more many surface atoms are involved in the D2O molecular adsorption.
Both for the intrapair and the interpair dissociative adsorption, the surface modes that are coupled to the Ow–H stretching implicate the surface O2c and Ti5c atom. These surface phonons show a more pronounced delocalization for the dissociated configurations (in particular, for the intrapair one) than in the case of molecular adsorption.
Discussion
The results presented above are motivating us to provide new physical insight into the complex adsorption process of water molecules on titania anatase nanoparticles, mainly based on the comparison between experiments on one side and the classical and DC-SCIVR results on the other. The main open issue in the water adsorption is the identification of the hydroxyl stretching signals to be distinguished from the adsorbed molecular water one. The stretching of hydroxyl groups are generally attributed to higher frequencies compared to the molecular one and within the range 3600–3800 cm–1.15,16,19−23 However, the interpretation of the origin of the different bands is still controversial. There are two main interpretations that are based on the different explanation of the shift in the stretching frequency, which can be caused either by the oxygen coordination or by the differences in the OH bond length.15,17,23,32,85−87 Earlier work tried to distinguish between the two explanations relying on the spectral difference originated by the different oxygen coordination. Specifically, lower energy bands were assigned to the vibration of bridged hydroxyl groups (which in our case corresponds to the O2c–H stretching frequencies), while higher frequency modes were assigned to terminal hydroxyls (in our case, Ow–H stretchings). No criteria are provided for the molecular O–H stretching modes. However, alternative explanations correlate the aforementioned bands to the OH bond length, either adsorbed in the dissociated form or within the water ad-molecule. Actually, there are several factors that conspire to determine the OH bond length, among which the oxygen coordination number or the presence of hydrogen bonds (with other water ad-molecules or surface oxygen atoms).26,88
We start our analysis from the molecular adsorption that is experimentally detected through the presence of the bending peak in the range 1615–1640 cm–1.15−23 Our calculations provide for this type of adsorption a small underestimation of the bending frequency, which is evident already at the harmonic level for the isolated H2O molecule and in a smaller amount for D2O as well. The underestimation is typical of the PBE functional. Therefore, rather than discussing the values for the frequencies for the adsorbed species, we will focus on the shift (usually negative) existing between the frequencies that are obtained in the quasi-classical or semiclassical levels with respect to the harmonic calculations. Such a shift is indeed a reliable quantity that is largely independent of the employed functional and the algorithms for solving the Kohn–Sham equations (see Supporting Information, Tables S6 and S7). Actually, it is essential to take into account the anharmonic contributions to the experimental spectra since the zero-point energy contribution (hνs/2) of the OH stretching mode in the water molecule corresponds to more than 2000 K of kinetic energy. This implies that the vibrational motion is sensitive to the high part of the potential energy surface (rather than its minimum), which is highly anharmonic. Experimental spectra exhibit several spectroscopy features within the 2500–3800 cm–1 range, where signals can be attributed not only to water stretches, but also to hydroxyl stretching modes that originate from water dissociation or from the synthesis procedure. As a confirmation, in the case of the molecular adsorption where we can identify the bending signal, the stretching frequencies have been assigned in the range 2500–3600 cm–1,15−23 making it impossible to identify the hydroxyl stretching signal or exclude its presence. In the stretching case, our classical and semiclassical results are consistent with the experimental range, while the harmonic estimates fall outside the upper frequency limit.
Eventually, we obtain the following comprehensive picture of the water adsorption process. The symmetric and the asymmetric stretches of the molecular adsorption and the O2c–H in the interpair dissociative adsorption configurations share similar frequencies because the H atoms are hydrogen-bonded to the nearby oxygen atoms. These frequencies are red-shifted with respect to the O2c–H of the intrapair and the Ow–H of both the intra- and interpair adsorptions. Noteworthy, both classical and semicalssical dynamics results show degenerate frequencies for the adsorbed molecular symmetric and asymmetric stretching. This is due to the lower molecular symmetry after adsorption and to the strong coupling of the OH stretching to the surface modes, mainly frustrated rotations and translations, as obtained from the analysis of the vibrational eigenfunctions. In the intrapair dissociative configuration, the O2c–H stretching frequencies (both at the quasi- and semiclassical levels) are slightly higher with respect to their counterparts for the other adsorption configurations, which we attribute to the lack of hydrogen bonding. Generally, same considerations apply to D2O adsorption with the main difference between the deuterated water adsorption process and the hydrogenated one is unveiled by the DC-SCIVR spectra, where the Fermi resonances with the phonon bath is more evident in the deuterated case and in the interpair adsorption than in the other geometries, consistently with a strong coupling with the surface phonons for this case. Additionally, the eigenmode displacements suggest that in the molecular case the interactions are more local than in the dissociative one.
Finally, the previous analysis applies to the isolated water molecules. In the case of higher water coverage, we guess that more complex interactions, including hydrogen bonding between adsorbed molecules, or between ad-molecules and surface oxygen ions, would likely enhance anharmonic effects, making it even harder to disentangle all the individual contributions to the experimental spectra.
Conclusions
In this work we aim at gaining further insights into water adsorption on the TiO2 anatase(101) surface. The systems is extremely complex. First, the nanoparticle morphology is far from the stoichiometric surface period slab model usually adopted in theoretical simulations. Second, nanoparticles are usually composed by two titania polymorphs, that is, rutile and anatase. Third, nanoparticles expose different type of surfaces and most of the time these surfaces are defective ones. Clearly, defects can play an important role in the kinetic and thermodynamics of water adsorption. Similarly, the presence of nanoparticles edges and corners can modify the interaction with the adsorbate. Fourth, one should include the intramolecular interaction between several adsorbed molecules which can be significant in case of high coverage. All these considerations do not allow to straightforwardly compare experimental measurements with simulations, where usually the adsorption process is described not considering all the possible experimental scenarios and more often limited to a single-molecule adsorption. However, if one sticks on the experimental evidence, it is not possible to reach a definitive conclusion about the water atomic configuration for the adsorption on anatase(101) surface. Experimentalists tried to assign the water adsorption structure by comparing IR spectra of nanoparticles with different surface ratios21 or by inspecting IR spectra outgassed at different temperatures.15,17,23
For all these reasons, we decided to tackle this system with a novel computational approach, which is based on quantum anharmonic vibrational calculations. Eventually, we think that we have been able to provide clear spectroscopic information that can allow experimentalists to devise new experimental setups and disentagle the different types of water adsorption. We believe that the information hereby provided will be very helpful to determine which is the most stable water adsorption configuration if new experimental conditions are devised, such as ultravacuum ones.
Acknowledgments
The authors thank Dr. R. Conte, Dr. D. Ceresoli, Prof. L. Lo Presti, and Prof. I. Alessandri for useful discussions. We acknowledge financial support from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement No. [647107]-SEMICOMPLEX ERC-2014-CoG) and from the Italian Ministery of Education, University, and Research (MIUR; FARE Programme R16KN7XBRB-Project QURE). We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support (Grant Nos. HP10CV2CBQ and HP10CGH9YR).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcc.2c02137.
Functional dependency of the geometrical and vibrational properties in gas phase and upon adsorption, convergence of vibrational frequencies against the number of k-points and slab thickness, figures of adsorption geometries with periodic boundary conditions, and eigendisplacement plots for additional coupled modes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chen H.; Nanayakkara C. E.; Grassian V. H. Titanium Dioxide Photocatalysis in Atmospheric Chemistry. Chem. Rev. 2012, 112, 5919–5948. 10.1021/cr3002092. [DOI] [PubMed] [Google Scholar]
- Kudo A.; Miseki Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. 10.1039/B800489G. [DOI] [PubMed] [Google Scholar]
- Osterloh F. E. Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting. Chem. Soc. Rev. 2013, 42, 2294–2320. 10.1039/C2CS35266D. [DOI] [PubMed] [Google Scholar]
- Diebold U. The surface science of titanium dioxide. Surf. Sci. Rep. 2003, 48, 53–229. 10.1016/S0167-5729(02)00100-0. [DOI] [Google Scholar]
- Selloni A. In Handbook of Materials Modeling: Applications: Current and Emerging Materials; Andreoni W., Yip S., Eds.; Springer International Publishing, 2018; pp 1–23. [Google Scholar]
- Luo C.; Ren X.; Dai Z.; Zhang Y.; Qi X.; Pan C. Present Perspectives of Advanced Characterization Techniques in TiO2-Based Photocatalysts. ACS Appl. Mater. Interfaces 2017, 9, 23265–23286. 10.1021/acsami.7b00496. [DOI] [PubMed] [Google Scholar]
- Sun C.; Liu L.-M.; Selloni A.; Lu G. Q. M.; Smith S. C. Titania-water interactions: a review of theoretical studies. J. Mater. Chem. 2010, 20, 10319–10334. 10.1039/c0jm01491e. [DOI] [Google Scholar]
- De Angelis F.; Di Valentin C.; Fantacci S.; Vittadini A.; Selloni A. Theoretical Studies on Anatase and Less Common TiO2 Phases: Bulk, Surfaces, and Nanomaterials. Chem. Rev. 2014, 114, 9708–9753. 10.1021/cr500055q. [DOI] [PubMed] [Google Scholar]
- Guo Q.; Zhou C.; Ma Z.; Ren Z.; Fan H.; Yang X. Elementary photocatalytic chemistry on TiO2 surfaces. Chem. Soc. Rev. 2016, 45, 3701–3730. 10.1039/C5CS00448A. [DOI] [PubMed] [Google Scholar]
- Bourikas K.; Kordulis C.; Lycourghiotis A. Titanium Dioxide (Anatase and Rutile): Surface Chemistry, Liquid-Solid Interface Chemistry, and Scientific Synthesis of Supported Catalysts. Chem. Rev. 2014, 114, 9754–9823. 10.1021/cr300230q. [DOI] [PubMed] [Google Scholar]
- Lo Presti L.; Pifferi V.; Di Liberto G.; Cappelletti G.; Falciola L.; Cerrato G.; Ceotto M. Direct measurement and modeling of spontaneous charge migration across anatase–brookite nanoheterojunctions. J. Mater. Chem. A 2021, 9, 7782–7790. 10.1039/D1TA01040A. [DOI] [Google Scholar]
- Mu R.; Zhao Z.-J.; Dohnálek Z.; Gong J. Structural motifs of water on metal oxide surfaces. Chem. Soc. Rev. 2017, 46, 1785–1806. 10.1039/C6CS00864J. [DOI] [PubMed] [Google Scholar]
- Björneholm O.; Hansen M. H.; Hodgson A.; Liu L.-M.; Limmer D. T.; Michaelides A.; Pedevilla P.; Rossmeisl J.; Shen H.; Tocci G.; et al. Water at Interfaces. Chem. Rev. 2016, 116, 7698–7726. 10.1021/acs.chemrev.6b00045. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wöll C. IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: bridging the materials gap. Chem. Soc. Rev. 2017, 46, 1875–1932. 10.1039/C6CS00914J. [DOI] [PubMed] [Google Scholar]
- Finnie K. S.; Cassidy D. J.; Bartlett J. R.; Woolfrey J. L. IR Spectroscopy of Surface Water and Hydroxyl Species on Nanocrystalline TiO2 Films. Langmuir 2001, 17, 816–820. 10.1021/la0009240. [DOI] [Google Scholar]
- Maira A.; Coronado J.; Augugliaro V.; Yeung K.; Conesa J.; Soria J. Fourier Transform Infrared Study of the Performance of Nanostructured TiO2 Particles for the Photocatalytic Oxidation of Gaseous Toluene. J. Catal. 2001, 202, 413–420. 10.1006/jcat.2001.3301. [DOI] [Google Scholar]
- Soria J.; Sanz J.; Sobrados I.; Coronado J. M.; Maira A. J.; Hernandez-Alonso M. D.; Fresno F. FTIR and NMR Study of the Adsorbed Water on Nanocrystalline Anatase. J. Phys. Chem. C 2007, 111, 10590–10596. 10.1021/jp071440g. [DOI] [Google Scholar]
- Belhadj H.; Hakki A.; Robertson P. K. J.; Bahnemann D. W. In situ ATR-FTIR study of H2O and D2O adsorption on TiO2 under UV irradiation. Phys. Chem. Chem. Phys. 2015, 17, 22940–22946. 10.1039/C5CP03947A. [DOI] [PubMed] [Google Scholar]
- Sheng H.; Zhang H.; Song W.; Ji H.; Ma W.; Chen C.; Zhao J. Activation of Water in Titanium Dioxide Photocatalysis by Formation of Surface Hydrogen Bonds: An In Situ IR Spectroscopy Study. Angew. Chem., Int. Ed. 2015, 54, 5905–5909. 10.1002/anie.201412035. [DOI] [PubMed] [Google Scholar]
- Mino L.; Pellegrino F.; Rades S.; Radnik J.; Hodoroaba V.-D.; Spoto G.; Maurino V.; Martra G. Beyond Shape Engineering of TiO2 Nanoparticles: Post-Synthesis Treatment Dependence of Surface Hydration, Hydroxylation, Lewis Acidity and Photocatalytic Activity of TiO2 Anatase Nanoparticles with Dominant 001 or 101 Facets. ACS Appl. Nano Mater. 2018, 1, 5355–5365. 10.1021/acsanm.8b01477. [DOI] [Google Scholar]
- Zhang H.; Zhou P.; Chen Z.; Song W.; Ji H.; Ma W.; Chen C.; Zhao J. Hydrogen-Bond Bridged Water Oxidation on 001 Surfaces of Anatase TiO2. J. Phys. Chem. C 2017, 121, 2251–2257. 10.1021/acs.jpcc.6b11900. [DOI] [Google Scholar]
- Litke A.; Su Y.; Tranca I.; Weber T.; Hensen E. J. M.; Hofmann J. P. Role of Adsorbed Water on Charge Carrier Dynamics in Photoexcited TiO2. J. Phys. Chem. C 2017, 121, 7514–7524. 10.1021/acs.jpcc.7b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdavi-Shakib A.; Arce-Ramos J. M.; Austin R. N.; Schwartz T. J.; Grabow L. C.; Frederick B. G. Frequencies and Thermal Stability of Isolated Surface Hydroxyls on Pyrogenic TiO2 Nanoparticles. J. Phys. Chem. C 2019, 123, 24533–24548. 10.1021/acs.jpcc.9b05699. [DOI] [Google Scholar]
- Mino L.; Morales-García A.; Bromley S. T.; Illas F. Understanding the nature and location of hydroxyl groups on hydrated titania nanoparticles. Nanoscale 2021, 13, 6577–6585. 10.1039/D1NR00610J. [DOI] [PubMed] [Google Scholar]
- Chi M.; Sun X.; Lozano-Blanco G.; Tatarchuk B. J. XPS and FTIR investigations of the transient photocatalytic decomposition of surface carbon contaminants from anatase TiO2 in UHV starved water/oxygen environments. Appl. Surf. Sci. 2021, 570, 151147. 10.1016/j.apsusc.2021.151147. [DOI] [Google Scholar]
- Haque F.; Finocchi F.; Chenot S.; Jupille J.; Stankic S. Interplay between Single and Cooperative H2 Adsorption in the Saturation of Defect Sites at MgO Nanocubes. J. Phys. Chem. C 2018, 122, 17738–17747. 10.1021/acs.jpcc.8b03192. [DOI] [Google Scholar]
- Gala F.; Agosta L.; Zollo G. Water Kinetics and Clustering on the (101) TiO2 Anatase Surface. J. Phys. Chem. C 2016, 120, 450–456. 10.1021/acs.jpcc.5b10934. [DOI] [Google Scholar]
- Posternak M.; Baldereschi A.; Delley B. Dissociation of Water on Anatase TiO2 Nanoparticles: the Role of Undercoordinated Ti Atoms at Edges. J. Phys. Chem. C 2009, 113, 15862–15867. 10.1021/jp9032113. [DOI] [Google Scholar]
- Manzhos S.; Ihara M. Computational vibrational spectroscopy of molecule-surface interactions: what is still difficult and what can be done about it. Phys. Chem. Chem. Phys. 2022, na. 10.1039/d2cp01389d. [DOI] [PubMed] [Google Scholar]
- Jug K.; Nair N. N.; Bredow T. Molecular dynamics investigation of water adsorption on rutile surfaces. Surf. Sci. 2005, 590, 9–20. 10.1016/j.susc.2005.05.055. [DOI] [Google Scholar]
- Kavathekar R. S.; Dev P.; English N. J.; MacElroy J. Molecular dynamics study of water in contact with the TiO2 rutile-110, 100, 101, 001 and anatase-101, 001 surface. Mol. Phys. 2011, 109, 1649–1656. 10.1080/00268976.2011.582051. [DOI] [Google Scholar]
- Arrouvel C.; Digne M.; Breysse M.; Toulhoat H.; Raybaud P. Effects of morphology on surface hydroxyl concentration: a DFT comparison of anatase-TiO2 and gamma-alumina catalytic supports. J. Catal. 2004, 222, 152–166. 10.1016/j.jcat.2003.10.016. [DOI] [Google Scholar]
- Benoit D. M. Vibrational Signature of a Single Water Molecule Adsorbed on Pt(111): Toward a Reliable Anharmonic Description. J. Phys. Chem. A 2015, 119, 11583–11590. 10.1021/acs.jpca.5b08543. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Di Liberto G.; Conte R. Semiclassical ”Divide-and-Conquer” Method for Spectroscopic Calculations of High Dimensional Molecular Systems. Phys. Rev. Lett. 2017, 119, 010401. 10.1103/PhysRevLett.119.010401. [DOI] [PubMed] [Google Scholar]
- Di Liberto G.; Conte R.; Ceotto M. Divide and conquer” semiclassical molecular dynamics: A practical method for spectroscopic calculations of high dimensional molecular systems. J. Chem. Phys. 2018, 148, 014307. 10.1063/1.5010388. [DOI] [PubMed] [Google Scholar]
- Cazzaniga M.; Micciarelli M.; Moriggi F.; Mahmoud A.; Gabas F.; Ceotto M. Anharmonic calculations of vibrational spectra for molecular adsorbates: A divide-and-conquer semiclassical molecular dynamics approach. J. Chem. Phys. 2020, 152, 104104. 10.1063/1.5142682. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Baroni S.; Bonini N.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Chiarotti G. L.; Cococcioni M.; Dabo I.; et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.-Conddens. Mater. 2009, 21, 395502. 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- Giannozzi P.; Andreussi O.; Brumme T.; Bunau O.; Nardelli M. B.; Calandra M.; Car R.; Cavazzoni C.; Ceresoli D.; Cococcioni M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys.: Condens. Mater. 2017, 29, 465901. 10.1088/1361-648X/aa8f79. [DOI] [PubMed] [Google Scholar]
- Baroni S.; de Gironcoli S.; Dal Corso A.; Giannozzi P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515–562. 10.1103/RevModPhys.73.515. [DOI] [Google Scholar]
- Rognoni A.; Conte R.; Ceotto M. Caldeira-Leggett model vs ab initio potential: A vibrational spectroscopy test of water solvation. J. Chem. Phys. 2021, 154, 094106. 10.1063/5.0040494. [DOI] [PubMed] [Google Scholar]
- Di Liberto G.; Conte R.; Ceotto M. Divide-and-conquer” semiclassical molecular dynamics: An application to water clusters. J. Chem. Phys. 2018, 148, 104302. 10.1063/1.5023155. [DOI] [PubMed] [Google Scholar]
- Micciarelli M.; Conte R.; Suarez J.; Ceotto M. Anharmonic vibrational eigenfunctions and infrared spectra from semiclassical molecular dynamics. J. Chem. Phys. 2018, 149, 064115. 10.1063/1.5041911. [DOI] [PubMed] [Google Scholar]
- Micciarelli M.; Gabas F.; Conte R.; Ceotto M. An Effective Semiclassical Approach to IR Spectroscopy. J. Chem. Phys. 2019, 150, 184113. 10.1063/1.5096968. [DOI] [PubMed] [Google Scholar]
- Gabas F.; Di Liberto G.; Conte R.; Ceotto M. Protonated glycine supramolecular systems: the need for quantum dynamics. Chem. Sci. 2018, 9, 7894–7901. 10.1039/C8SC03041C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabas F.; Di Liberto G.; Ceotto M. Vibrational investigation of nucleobases by means of divide and conquer semiclassical dynamics. J. Chem. Phys. 2019, 150, 224107. 10.1063/1.5100503. [DOI] [PubMed] [Google Scholar]
- Conte R.; Parma L.; Aieta C.; Rognoni A.; Ceotto M. Improved semiclassical dynamics through adiabatic switching trajectory sampling. J. Chem. Phys. 2019, 151, 214107. 10.1063/1.5133144. [DOI] [PubMed] [Google Scholar]
- Aieta C.; Micciarelli M.; Bertaina G.; Ceotto M. Anharmonic quantum nuclear densities from full dimensional vibrational eigenfunctions with application to protonated glycine. Nat. Commun. 2020, 11, 1–9. 10.1038/s41467-020-18211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaina G.; Di Liberto G.; Ceotto M. Reduced rovibrational coupling Cartesian dynamics for semiclassical calculations: Application to the spectrum of the Zundel cation. J. Chem. Phys. 2019, 151, 114307. 10.1063/1.5114616. [DOI] [PubMed] [Google Scholar]
- Aieta C.; Bertaina G.; Micciarelli M.; Ceotto M. Representing molecular ground and excited vibrational eigenstates with nuclear densities obtained from semiclassical initial value representation molecular dynamics. J. Chem. Phys. 2020, 153, 214117. 10.1063/5.0031391. [DOI] [PubMed] [Google Scholar]
- Botti G.; Ceotto M.; Conte R. On-the-fly adiabatically switched semiclassical initial value representation molecular dynamics for vibrational spectroscopy of biomolecules. J. Chem. Phys. 2021, 155, 234102. 10.1063/5.0075220. [DOI] [PubMed] [Google Scholar]
- Gabas F.; Conte R.; Ceotto M. Quantum Vibrational Spectroscopy of Explicitly Solvated Thymidine in Semiclassical Approximation. J. Phys. Chem. Lett. 2022, 13, 1350–1355. 10.1021/acs.jpclett.1c04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte R.; Ceotto M.. Quantum Chemistry and Dynamics of Excited States: Methods and Applications; John Wiley & Sons, Ltd., 2020; p 595. [Google Scholar]
- Ceotto M.; Atahan S.; Shim S.; Tantardini G. F.; Aspuru-Guzik A. First-principles semiclassical initial value representation molecular dynamics. Phys. Chem. Chem. Phys. 2009, 11, 3861–3867. 10.1039/b820785b. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Atahan S.; Tantardini G. F.; Aspuru-Guzik A. Multiple coherent states for first-principles semiclassical initial value representation molecular dynamics. J. Chem. Phys. 2009, 130, 234113. 10.1063/1.3155062. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Dell’Angelo D.; Tantardini G. F. Multiple coherent states semiclassical initial value representation spectra calculations of lateral interactions for CO on Cu (100). J. Chem. Phys. 2010, 133, 054701. 10.1063/1.3462242. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Tantardini G. F.; Aspuru-Guzik A. Fighting the curse of dimensionality in first-principles semiclassical calculations: Non-local reference states for large number of dimensions. J. Chem. Phys. 2011, 135, 214108. 10.1063/1.3664731. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Valleau S.; Tantardini G. F.; Aspuru-Guzik A. First principles semiclassical calculations of vibrational eigenfunctions. J. Chem. Phys. 2011, 134, 234103. 10.1063/1.3599469. [DOI] [PubMed] [Google Scholar]
- Conte R.; Aspuru-Guzik A.; Ceotto M. Reproducing Deep Tunneling Splittings, Resonances, and Quantum Frequencies in Vibrational Spectra From a Handful of Direct Ab Initio Semiclassical Trajectories. J. Phys. Chem. Lett. 2013, 4, 3407–3412. 10.1021/jz401603f. [DOI] [PubMed] [Google Scholar]
- Heller E. J. The semiclassical way to molecular spectroscopy. Acc. Chem. Res. 1981, 14, 368–375. 10.1021/ar00072a002. [DOI] [Google Scholar]
- Heller E. J. Frozen Gaussians: A very simple semiclassical approximation. J. Chem. Phys. 1981, 75, 2923–2931. 10.1063/1.442382. [DOI] [Google Scholar]
- Heller E. J. Cellular dynamics: A new semiclassical approach to time-dependent quantum mechanics. J. Chem. Phys. 1991, 94, 2723–2729. 10.1063/1.459848. [DOI] [Google Scholar]
- Shalashilin D. V.; Child M. S. Multidimensional quantum propagation with the help of coupled coherent states. J. Chem. Phys. 2001, 115, 5367–5375. 10.1063/1.1394939. [DOI] [Google Scholar]
- Herman M. F.; Kluk E. A semiclassical justification for the use of non-spreading wavepackets in dynamics calculations. Chem. Phys. 1984, 91, 27–34. 10.1016/0301-0104(84)80039-7. [DOI] [Google Scholar]
- Kluk E.; Herman M. F.; Davis H. L. Comparison of the propagation of semiclassical frozen Gaussian wave functions with quantum propagation for a highly excited anharmonic oscillator. J. Chem. Phys. 1986, 84, 326–334. 10.1063/1.450142. [DOI] [Google Scholar]
- Di Liberto G.; Conte R.; Ceotto M. Divide and conquer” semiclassical molecular dynamics: A practical method for spectroscopic calculations of high dimensional molecular systems. J. Chem. Phys. 2018, 148, 014307. 10.1063/1.5010388. [DOI] [PubMed] [Google Scholar]
- Gandolfi M.; Rognoni A.; Aieta C.; Conte R.; Ceotto M. Machine learning for vibrational spectroscopy via divide-and-conquer semiclassical initial value representation molecular dynamics with application to N-methylacetamide. J. Chem. Phys. 2020, 153, 204104. 10.1063/5.0031892. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Li Z.; Zou Z. Understanding the interaction of water with anatase TiO2 (101) surface from density functional theory calculations. Phys. Lett. A 2011, 375, 2939–2945. 10.1016/j.physleta.2011.06.022. [DOI] [Google Scholar]
- Zhao Z.; Li Z.; Zou Z. A. Theoretical Study of Water Adsorption and Decomposition on the Low-Index Stoichiometric Anatase TiO2 Surfaces. J. Phys. Chem. C 2012, 116, 7430–7441. 10.1021/jp212407s. [DOI] [Google Scholar]
- Aschauer U.; He Y.; Cheng H.; Li S.-C.; Diebold U.; Selloni A. Influence of Subsurface Defects on the Surface Reactivity of TiO2: Water on Anatase (101). J. Phys. Chem. C 2010, 114, 1278–1284. 10.1021/jp910492b. [DOI] [Google Scholar]
- Aschauer U. J.; Tilocca A.; Selloni A. Ab initio simulations of the structure of thin water layers on defective anatase TiO2(101) surfaces. Int. J. Quantum Chem. 2015, 115, 1250–1257. 10.1002/qua.24918. [DOI] [Google Scholar]
- He Y.; Tilocca A.; Dulub O.; Selloni A.; Diebold U. Local ordering and electronic signatures of submonolayer water on anatase TiO2(101). Nat. Mater. 2009, 8, 585. 10.1038/nmat2466. [DOI] [PubMed] [Google Scholar]
- Martinez-Casado R.; Mallia G.; Harrison N. M.; Pérez R. First-Principles Study of the Water Adsorption on Anatase(101) as a Function of the Coverage. J. Phys. Chem. C 2018, 122, 20736–20744. 10.1021/acs.jpcc.8b05081. [DOI] [Google Scholar]
- Agosta L.; Gala F.; Zollo G. Water diffusion on TiO2 anatase surface. AIP Conf. Proc. 2015, 1667, 020006. 10.1063/1.4922562. [DOI] [Google Scholar]
- Vittadini A.; Selloni A.; Rotzinger F.; Grätzel M. Structure and energetics of water adsorbed at TiO2 anatase (101) and (001) surfaces. Phys. Rev. Lett. 1998, 81, 2954. 10.1103/PhysRevLett.81.2954. [DOI] [Google Scholar]
- Petersen T.; Klüner T. Water Adsorption on Ideal Anatase-TiO2(101): An Embedded Cluster Model for Accurate Adsorption Energetics and Excited State Properties. Z. Phys. Chem. 2020, 234, 813–834. 10.1515/zpch-2019-1425. [DOI] [Google Scholar]
- Selli D.; Fazio G.; Seifert G.; Di Valentin C. Water Multilayers on TiO2 (101) Anatase Surface: Assessment of a DFTB-Based Method. J. Chem. Theory Comput. 2017, 13, 3862–3873. 10.1021/acs.jctc.7b00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egashira M.; Kawasumi S.; Kagawa S.; Seiyama T. Temperature Programmed Desorption Study of Water Adsorbed on Metal Oxides. I. Anatase and Rutile. B. Chem. Soc. Jpn. 1978, 51, 3144–3149. 10.1246/bcsj.51.3144. [DOI] [Google Scholar]
- Dette C.; Pérez-Osorio M. A.; Mangel S.; Giustino F.; Jung S. J.; Kern K. Single-Molecule Vibrational Spectroscopy of H2O on Anatase TiO2(101). J. Phys. Chem. C 2017, 121, 1182–1187. 10.1021/acs.jpcc.6b10379. [DOI] [Google Scholar]
- NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101, Release 19, April, 2018, Johnson R. D., III, Ed.; http://cccbdb.nist.gov/.
- Shimanouchi T.Tables of Molecular Vibrational Frequencies Consolidated; National Bureau of Standard, 1972; Vol. I. [Google Scholar]
- Huber G. K. P.; Herzberg Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules; Springer: New York, NY, 1979. [Google Scholar]
- Varetti E. A far infrared and theoretical ab initio vibrational study of fluorosulfonic acid as monomer and cyclic dimer. J. Mol. Struc.-THEOCHEM 1998, 429, 121–130. 10.1016/S0166-1280(97)00347-3. [DOI] [Google Scholar]
- Xu X.; Goddard W. A. Bonding Properties of the Water Dimer: A Comparative Study of Density Functional Theories. J. Phys. Chem. A 2004, 108, 2305–2313. 10.1021/jp035869t. [DOI] [Google Scholar]
- Gygi F. m. c. Ab initio molecular dynamics in adaptive coordinates. Phys. Rev. B 1995, 51, 11190–11193. 10.1103/PhysRevB.51.11190. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Liu L.; Xiao W. Doping Effects on the Adsorption of a Nitric Oxide Molecule on an Anatase (101) Surface. ChemPhysChem 2017, 18, 653–661. 10.1002/cphc.201601164. [DOI] [PubMed] [Google Scholar]
- Chizallet C.; Digne M.; Arrouvel C.; Raybaud P.; Delbecq F.; Costentin G.; Che M.; Sautet P.; Toulhoat H. Insights into the Geometry, Stability and Vibrational Properties of OH Groups on c-Al2O3, TiO 2-Anatase and MgO from DFT Calculations. Top. Catal. 2009, 52, 1005–1016. 10.1007/s11244-009-9262-9. [DOI] [Google Scholar]
- Dzwigaj S.; Arrouvel C.; Breysse M.; Geantet C.; Inoue S.; Toulhoat H.; Raybaud P. DFT makes the morphologies of anatase-TiO2 nanoparticles visible to IR spectroscopy. J. Catal. 2005, 236, 245–250. 10.1016/j.jcat.2005.09.034. [DOI] [Google Scholar]
- Finocchi F.; Haque F.; Chenot S.; Jupille J.; Stankic S. Water dissociation on the low-coordinated sites of MgO nanopowders. J. Mater. Res. 2019, 34, 408–415. 10.1557/jmr.2018.461. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.