

Abstract
Ophthalmic neurodegenerative diseases encompass a wide array of molecular pathologies unified by calpain dysregulation. Calpains are calcium-dependent proteases that perpetuate cellular death and inflammation when hyperactivated. Calpain inhibition trials in other organs have faced pharmacological challenges, but the eye offers many advantages for the development and testing of targeted molecular therapeutics, including small-molecules, peptides, engineered proteins, drug implants, and gene-based therapies. This review highlights structural mechanisms underlying calpain activation, distinct cellular expression patterns, and in vivo models that link calpain hyperactivity to human retinal and developmental disease. Optimizing therapeutic approaches for calpain-mediated eye diseases can help accelerate clinically feasible strategies for treating calpain dysregulation in other diseased tissues.
Keywords: Calpain, retina, blindness, calcium dysregulation, therapeutics
Advances in molecular therapeutics for the eye
The human eye is at the frontier for testing novel molecular therapeutics. It is an immune privileged site, protected by the blood-retina-barrier (BRB), and advanced surgical techniques allow for highly precise therapeutic delivery that can target not just tissues but specific cells. The cellular components of the eye are easily and non-invasively monitored by well-established live imaging tools. Functional and physiological assessment modalities can track disease progression and treatment responses longitudinally, allowing for robust clinical trial design. The pharmacokinetic challenges of systemic drug delivery are greatly simplified in the eye because of its compartmentation. Following intraocular delivery, molecular therapeutics generally remain within the eye, reducing systemic toxicity. The small volume of the eye significantly reduces manufacturing requirements, which can otherwise be cost prohibitive in systemic delivery. Additionally, many ocular diseases exhibit some degree of symmetry such that the untreated fellow eye can serve as an internal control. These features, in combination with advancements in drug formulation, have paved the way for numerous state-of-the-art treatment methods for ophthalmic disorders, especially for retinal diseases.
Retinal diseases have become attractive targets for gene-based therapeutics, such as gene supplementation, clustered regularly interspaced short palindromic repeats (CRISPR) gene editing, and antisense oligonucleotide-based therapies. For example, voretigene neparvovec (AAV2.hRPE65v2; i.e., Luxturna) became the first FDA-approved gene therapy medication for the treatment of Leber congenital amaurosis (LCA; Figure 1) [1]. In acquired diseases like macular degeneration, gene therapy is being used to express therapeutic proteins. Other acquired eye diseases (e.g., diabetic retinopathy and ocular inflammatory disorders) have benefited from the therapeutic injection of monoclonal antibodies (mAbs), engineered protein inhibitors, or peptides targeting various cytokines (e.g., vascular endothelial growth factor [VEGF], complement, and interleukin-6), facilitating direct administration of high local drug concentrations while minimizing systemic dissemination (Figure 1).[2]. Further, development of sustained release intraocular implants (e.g., fluocinolone acetonide [Retisert™, Bausch & Lomb, Rochester, NY] and bimatoprost [Durysta™, Allergan, Irvine, CA] implants for non-infectious posterior uveitis and glaucoma, respectively) have allowed for continuous dosing of medications while simultaneously reducing the risks associated with repeated intraocular injections (Figure 1) [3,4].
Figure 1. Molecular therapeutics in the human eye.
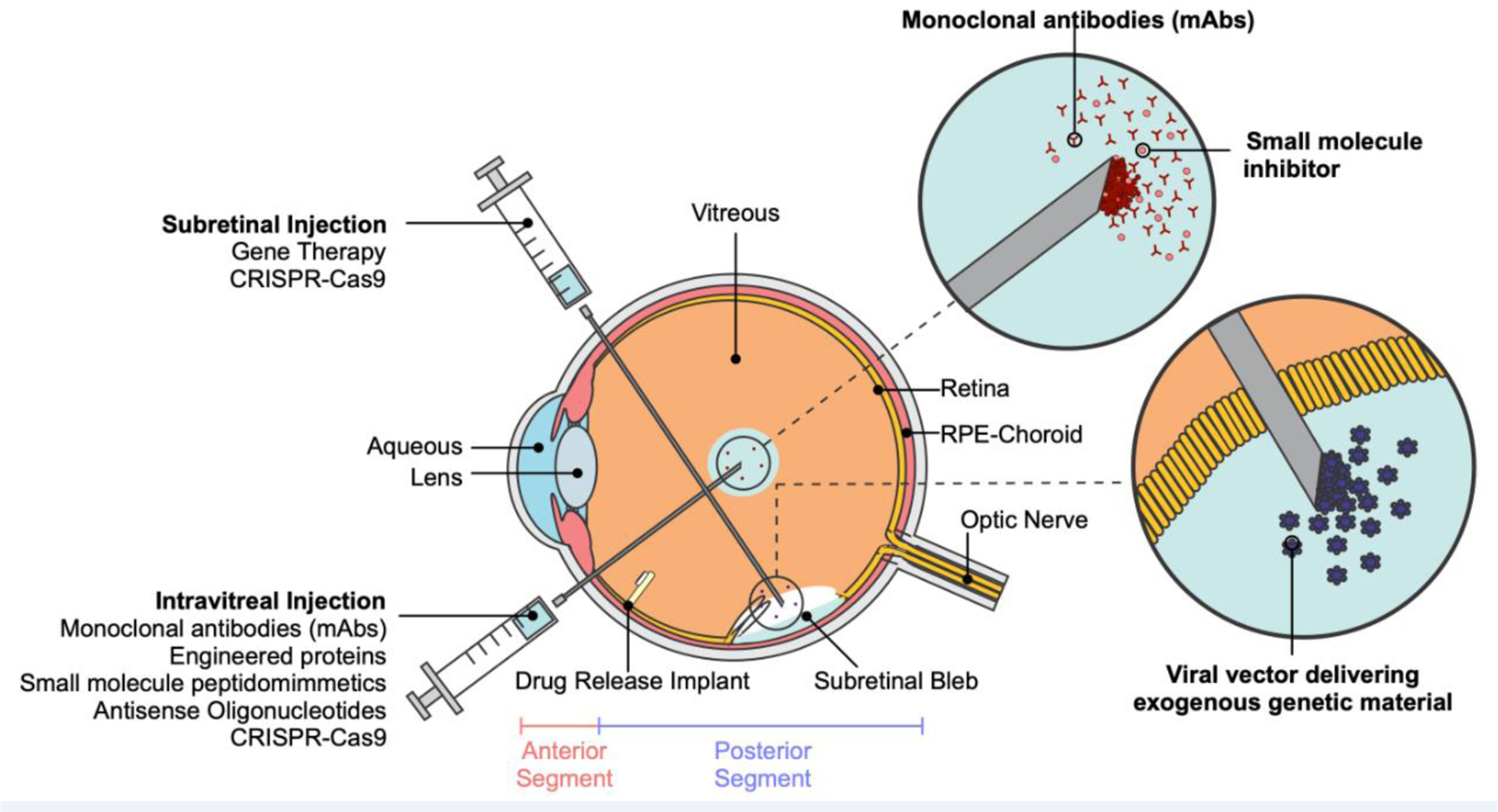
Cross-sectional anatomical illustration of the human eye. Molecular therapies can be administered via intravitreal injection (e.g., monoclonal antibodies, small molecule inhibitors, antisense oligonucleotides, CRISPR-Cas9, etc.), subretinal injection (e.g., gene therapy, CRISPR-Cas9, etc.), or intraocular drug release implants.
Calpains are a broad class of proteases amenable to molecular therapies and they are involved in a wide range of eye diseases. Previous clinical trials for calpain-related disorders have utilized systemic small-molecule inhibitors with limited success [5]. In light of the recent advances in ophthalmic molecular therapy, reconsideration of calpain inhibition strategies is warranted.
Calpains are implicated in a wide range of human diseases
Calpains (CAPNs) are a family of calcium-dependent non-lysosomal cysteine proteases (see Glossary) that mediate a range of physiological processes [6]. Aberrant elevation in intracellular calcium concentrations (e.g., by cellular stress or pathologic conditions) can result in the dysregulation of calpain-mediated proteolysis, triggering calpain overactivation [7]. Calpain hyperactivity is associated with ophthalmic diseases [8], neurodegenerative disorders (e.g., Alzheimer disease) [9], cardiovascular diseases [10–12], muscular dystrophies [13,14], gastropathies [15–17], pulmonary fibrosis [18], diabetes [19,20], and cancer [21–24]. In addition, inherited genetic defects can render calpains hypersensitive to physiological calcium levels resulting in aggravated inflammation and cell death.
Disruptions in calcium homeostasis in the eye result in calpain overactivation leading to prolonged proteolysis, necroptosis, apoptosis, and irreversible retinal damage [25]. Mounting evidence in human patients implicates calpain overactivity in a variety of retinal neurodegenerative diseases, including neovascular inflammatory vitreoretinopathy (NIV) [8], retinitis pigmentosa (RP), diabetic retinopathy (DR) [26], uveitis (i.e., intraocular inflammation) [8,27–29], glaucoma [30–37], and cataractogenesis [38,39]. However, the clinical utility of systemic calpain inhibitors, which have been aimed at treating fibrotic and neurological disorders, may be limited by their lack of target specificity [40]. Notably, two orally administered small molecule calpain inhibitors have reached Phase 1 clinical trials: one which evaluated the safety and pharmacokinetics of a peptidomimetic drug (i.e., ABT-957) for Alzheimer’s disease (NCT02220738) I [41] and another for an inhibitor (i.e., BLD-2660) to treat coronavirus disease-19 (COVID-19; NCT04334460) II [42].
In vivo models simulating diverse ocular diseases also confirm the link between calpains and disease progression such that calpain inhibition showed promise in attenuating damage and preserving vision [39,43–51]. Nevertheless, concern remains regarding off-target effects. Even with intraocular drug delivery, calpain inhibitors may need to be highly specific to avoid off-target inhibition of other proteases (e.g., cathepsins) [52–56]. Prior to clinical application, fine-tuning strategies to selectively target pathologic calpain activity while sparing normal calpain activity in healthy cells is possible. New insights into the structural variation and tissue-specific expression between calpains, for example, can be used to optimize therapeutic development.
Ocular calpains: distribution, classification, structure, and calcium-activation mechanisms
Calpain classification
Evolutionary gene duplication events have generated 15 mammalian genes that encode a conserved calpain-like protease core domain [57]. There are nine classical calpains, five non-classical calpains, and one demi-calpain classified based on domain topology (Figure 2A). The nomenclature for calpain domains highlights each domain’s structure-function relationship [57,58]. The signature feature of a calpain is the protease core (PC) domain, which is formed by two subdomains, PC1 and PC2 [59,60].
Figure 2. Expression, domain topology, and structural diversity of calpains.
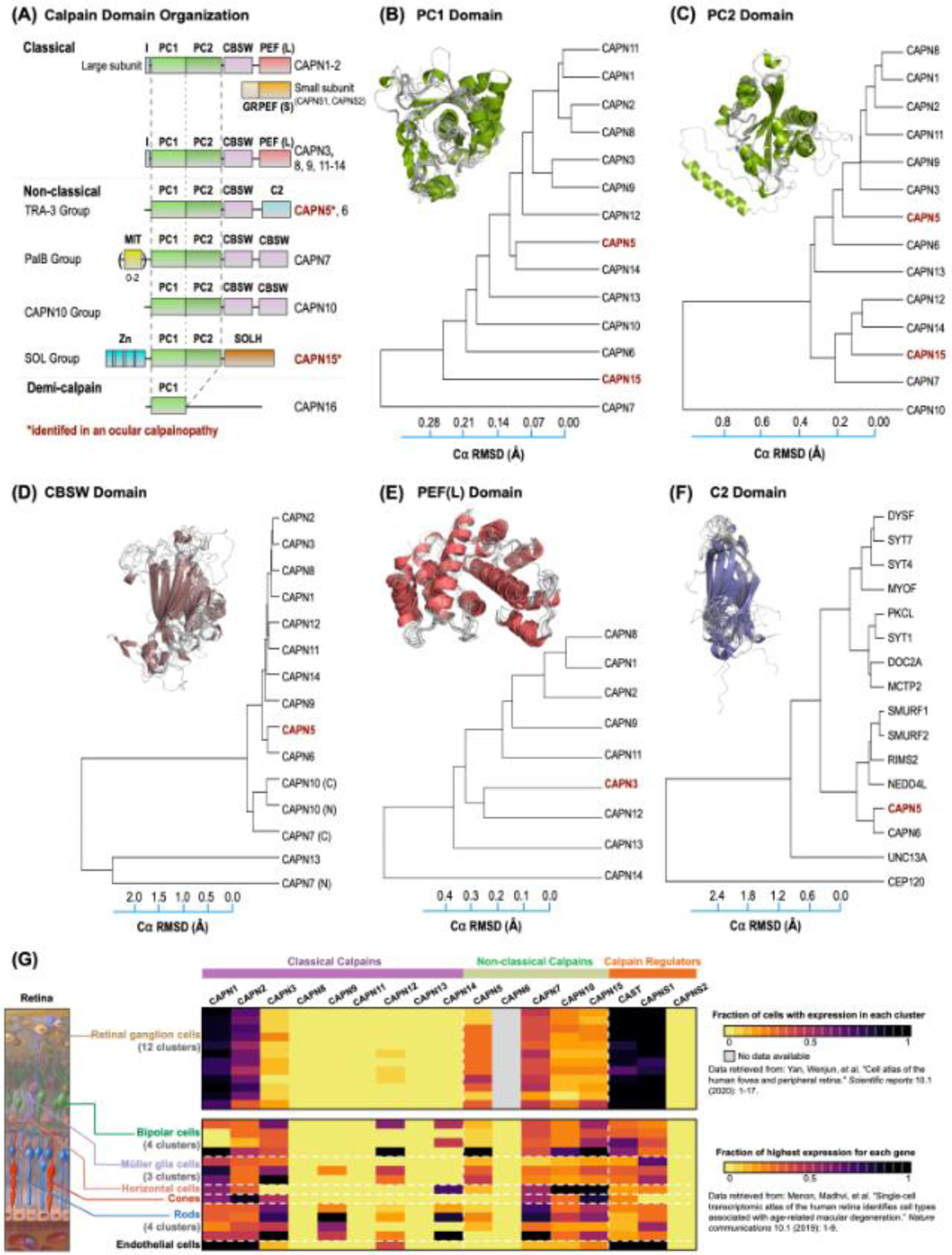
(A) Calpains are predominantly classified based on their domain topology. The protease core (PC) domain, made up of two sub-domains, is conserved among all classical and non-classical calpains. Structure-based phylogenetic tree of human calpain (B) PC1, (C) PC2, (D) CBSW, (E) PEF(L), and (F) related C2 domains (e.g., SYT1, RIMS2, etc.). The evolutionary distance was inferred using the UPGMA method. The tree is drawn to scale, with branch lengths corresponding to the Cα RMSD (Å) of the pairwise structural alignments used to infer the phylogenetic tree. Branches are labeled according to the calpain paralog structures. Superimposition of corresponding domain structures (from AlphaFold) were used to construct the phylogenetic tree. (G) Single-cell RNA sequencing (scRNA-seq) data of the human retina (Menon M et al., 2019) was clustered and normalized by The Human Protein Atlas (bottom panel). Fourteen distinct clusters were identified in the eye (including bipolar cells, Müller glia cells, horizontal cells, rods, and endothelial cells). However, retinal ganglion cells (RGCs) were excluded from this clustering. Therefore, another scRNA-seq dataset of the human retina (Yan W et al., 2020) that included RGC clusters determined by the Broad Institute’s Single Cell Portal was analyzed (top panel). The scRNA-seq data are represented as a heatmap and display gene expression based on the fraction of cells with expression (top panel) or on the fraction of highest expression determined by maximum norm constraints (bottom panel). Black indicates high expression, purple indicates mid-level expression, and yellow indicates low or no expression. Genes are separated based on their identification as classical calpains, non-classical calpains or calpain regulators.
Classical calpains (CAPN1-3, 8, 9, and 11–14) function as a large catalytic subunit (80 kDa) and heterodimerize with the small regulatory subunit (28 kDa; encoded by CAPNS1 and CAPNS2). The large subunit is comprised of PC1 and PC2 followed by a calpain-beta-sandwich (CBSW) domain and a penta-EF hand domain (PEF[L]; Figure 2A) [61–63]. The small subunit contains two domains: a glycine-rich region and a PEF small (PEF[S]) domain [5,6,64], the latter of which has been proposed to constrain the protease core in the absence of calcium to prevent spurious proteolytic activation. Classical calpain activation involves a two-step unique structural mechanism regulated by multiple steps which are impaired in calpain pathologies [61]. For CAPN1 and 2, binding of Ca2+ to the PEF-hand domains leads to disruption of the interactions between the small and large subunit [60]. Ca2+ then binds to PC1 and PC2 on their cognate sites leading to formation of the catalytic triad. While this mechanism has been extensively characterized in CAPN1, 2, and 5 [55,60,61], several calcium binding residues are not conserved in the CAPN10 and −15 PC domains, suggesting that these non-classical calpains may be active in the absence of calcium [60,65]. The EF hands also interact with the endogenous calpain inhibitor, calpastatin (encoded by the CAST gene) [5,60]. Calpastatin’s specificity is determined by the simultaneous binding of its three subdomains to both subunits of heterodimeric calpains. Nevertheless, the crystal structure of the calpain-calpastatin complex has guided rational design of peptidomimetic inhibitors targeting the active site [66,67].
CAPN5, 6, 7, 10 and 15 are considered non-classical because they do not have PEF domains, making them refractory to calpastatin inhibition [58]. Non-classical calpains display a highly modular domain organization and their specific functions can be attributed to their unique non-catalytic domains (Figure 2A) [59,63,68]. For example, CAPN5’s C2 domain is involved in membrane association and regulation of autoproteolytic processing [68]. Additionally, CAPN5 has been shown to exhibit non-proteolytic functions, such as facilitating hepatitis C virus (HCV) entry into liver cells through binding interactions with Casitas B-lineage lymphoma proto-oncogene B (CBLB) and CD81 [69]. Although all calpains possess the PC domain, several lack critical residues required to form the catalytic triad (i.e., CAPN6 and −15) or modules known to regulate proteolytic activity (e.g., anchor helix, PEF-hand, etc.; Figure 2A) [55]. This structural diversity suggests that the non-classical calpains have regulatory roles beyond proteolysis. In line with this reasoning, the non-catalytic CAPN6 has been shown to play a role in cytoskeletal stabilization and organization via its CBSW domain [70]. CAPN15 contains five N-terminal zinc fingers, suggesting that it may bind RNA or mediate protein-protein interactions (e.g., polyubiquitin; Figure 2A) [65,71,72]. These differences could be exploited in the development of isoform-specific inhibitors.
Primary sequence analysis has helped to characterize the evolutionary diversity of calpain domains and has been performed extensively. However, sequence-based predictions often miss key functional differences between protein structures [73–75]. Instead, a structure-based, comparative analysis pipeline (i.e., 3DPhyloFold) can point to functional similarities that can and have been used to identify small-molecule protease inhibitors and identify potential drug off-targeting [55,76]. 3DPhyloFold performs pairwise structural comparisons, which are independent from sequence analyses, and calculates structural dissimilarity matrices (SDM) based on the root mean square deviation (RMSD) between protein alpha carbon (Cα) atoms. Structure-based phylogenetic trees are then generated from these matrices using the UPGMA (Unweighted Pair Group Method with Arithmetic Mean) method and allow for direct structural comparisons between calpain family members (Figure 2B–F) [77]. 3DPhyloFold was previously utilized to examine the functional diversity among 20 calpain PC domain structures [55,76]. For instance, CAPN5-PC contain three elongated flexible loops (termed PC1L1, PC2L1, and PC2L2, respectively) and, consistent with previous sequence-based analysis and homology modeling, clusters close to CAPN9-PC [78]. Further, the presence of a NIV disease-causing mutation (p.G267S) in the CAPN5 PC2L2 loop suggested a role in the allosteric regulation of CAPN5’s activity. This analysis was limited to the comparison of only the PC domain and by the lack of available structures for many other calpain isoforms [55]. Since then, AlphaFold models for CAPN1–15 allow for more comprehensive structure-based comparative analyses (Figure 2 and Figure S1) [79].
The structural phylogenetic tree for the calpain-beta-sandwich (CBSW) domain demonstrated the largest degree of separation among calpain family members with an average Cα RMSD of 2.71 Å (Figure 2D), making this domain an attractive target for allosteric inhibition and drug specificity. Analysis of the PEF(L) domain demonstrated less variation among calpains (average Cα RMSD of 1.02) compared to the CBSW domain, highlighting its conserved role in classical calpain regulation (Figure 2E). To assess the suitability of these domains for targeted drug development, high-resolution structures of full-length calpains could experimentally confirm each calpain domain structure and their respective conformational states (i.e., Ca2+- and inhibitor-bound).
Ocular calpains identified by genomic and proteomic studies
The eye has a combination of anatomical features that make it ideally suited as a target organ for the study of many calpain-related diseases. It is a unique sensory organ with complex anatomy and physiology. It can be anatomically divided into two main components: the anterior and posterior segments (Figure 1). The anterior segment occupies approximately one-third of the eye and is comprised of tissues anterior to the vitreous: the cornea, aqueous humor, iris, ciliary body, and lens. The posterior segment is comprised of the vitreous humor, sclera, choroid, retinal pigment epithelium (RPE), retina, and optic nerve.
Bulk RNA-sequencing (RNA-seq) has revealed that calpain expression is similar between human and murine retinas [28]. CAPN1, 2, 3, 5, and 7 are most abundantly expressed in both species [80], while mouse retinas also express CAPN6 and 10. Subsequently, single-cell RNA sequencing (scRNA-seq) experiments of human eyes were consistent with bulk RNA-seq results, enabling localization of specific calpains to various retinal cell types (Figure 2G) [81,82]. Further, these cell type clusters were determined by previously established cell-type specific markers. The complex expression patterns of calpains are typified by CAPN3, which expresses twelve distinct splicing variants depending on tissue type, developmental stage, and species. These include Lp82, Cn94, and Rt88, which were first identified in the lens, cornea, and retina, respectively [83]. Such data highlight that cell type determines the precise transcription and splicing patterns of ocular calpains.
Proteomic studies also indicated that CAPN1, CAPN2, and CAPNS1 were the most abundant calpains in the human retina and vitreous [84,85]. However, a shotgun proteomic approach might not detect calpains expressed at lower levels, thus requiring more sensitive methods such as western blotting, immunohistochemistry, ELISA, or aptamer-based detection platforms. Taken together, the broad distribution of calpains reflects their pathophysiological and biochemical roles in a myriad of ocular diseases.
Calpains are central mediators of eye pathologies
Calpain-associated human eye diseases are induced by various factors including genetic mutations, chronic exposure to lifestyle-related factors (e.g., smoking, alcohol, diet, occupation, or climate), aging, and/or acute injury to the eye (Figure 3A). Regardless of etiology, many exhibit similar clinical features including ocular cell death, inflammation, and/or neovascularization. Mechanistically, the major underlying molecular pathology driving these diseases is calcium dysregulation leading to calpain overactivation. In the following sections, we will review the extent by which in vivo models phenocopy human disease and pathologies, highlight the advantages and limitations of calpain-targeting therapeutics tested in vivo, and explore opportunities for future translational research (see Clinician’s Corner).
Figure 3. Calcium-mediated calpain hyperactivity is the central molecular mediator of ocular diseases.
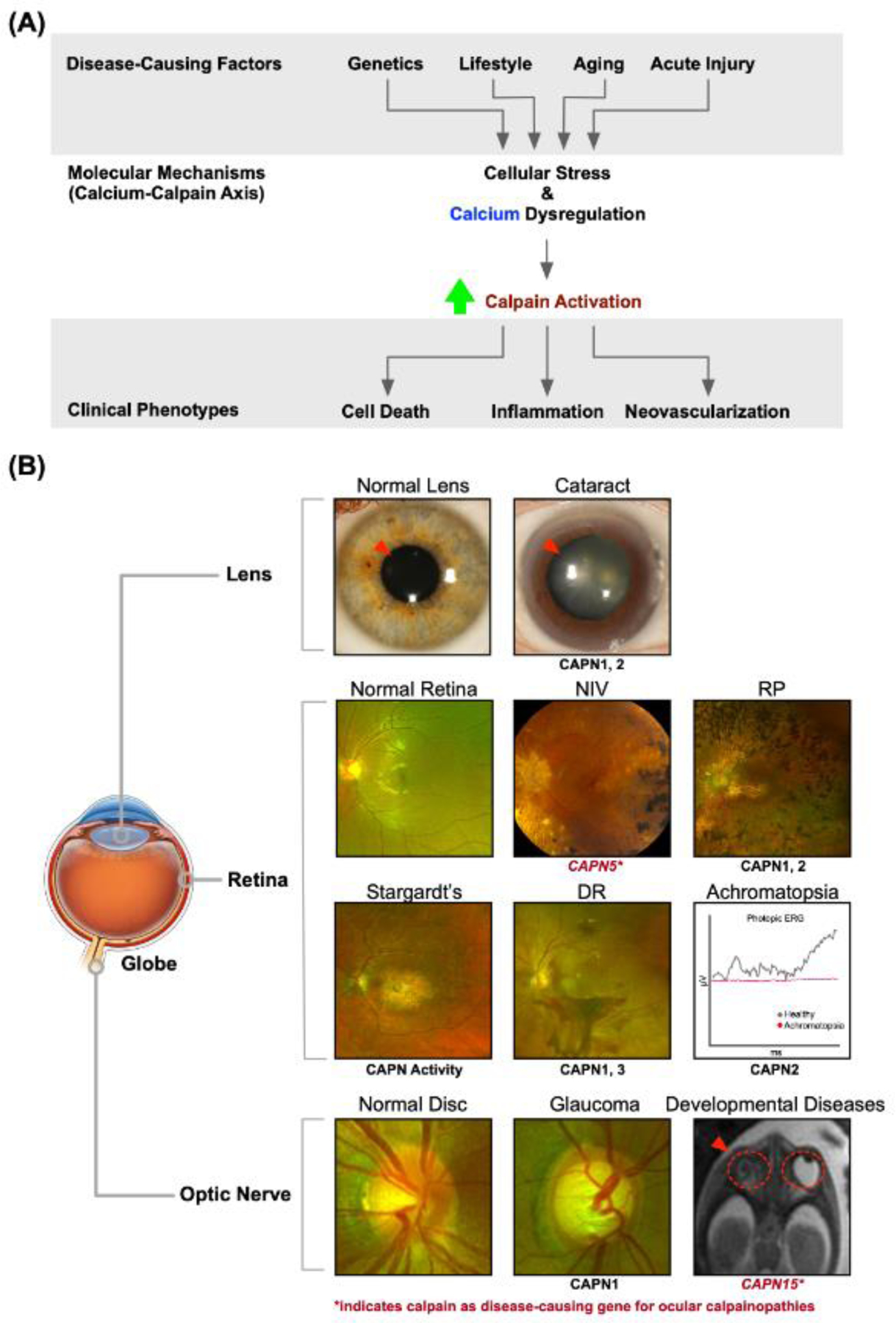
(A) Ocular diseases have a wide variety of causes (genetic, lifestyle-related, aging, and/or acute injury) that share an underlying molecular mechanism: calcium-calpain axis dysregulation. Increased cellular stress and intracellular calcium levels trigger calpain hyperactivation, which manifests in the eye as cell death, inflammation, and neovascularization. (B) Calpain involvement in pathologies affecting different anatomical regions of the eye (e.g., lens, retina, and optic nerve). Representative clinical images of these disorders are shown compared to a healthy eye. The top row shows two panels representing a healthy lens (left) and opacified lens (i.e., cataract; right). The next two rows demonstrate clinical exam findings (i.e., fundus photography and electroretinography) of calpain-associated retinal disorders: Top left, fundus photo for a healthy retina with normal vasculature; top middle, early neovascular inflammatory vitreoretinopathy (NIV) with vitreous haze and cystoid macular edema; top right, retinitis pigmentosa (RP) with characteristic bone-spicule pigment deposits; bottom left, Stargardt’s disease demonstrating atrophic macular changes and diffuse pisciform flecks; bottom middle, diabetic retinopathy (DR) demonstrating retinal neovascularization and pre-retinal hemorrhages; bottom right, electroretinogram of an eye effected by Achromatopsia compared to a healthy eye. Under photopic conditions, cones are observed to be unresponsive in achromatopsia compared to the normal activity of healthy eyes. The bottom row shows a healthy optic nerve and another with glaucoma, which has a larger cup-to-disc ratio indicating optic nerve damage. The bottom right image shows a fetal MRI demonstrating anophthalmia (i.e., absent eye). Written informed consent to publish clinical images was obtained from all participants under a Stanford University IRB approved protocol. The NIV image is republished with permission from Mahajan, et al. (2012).
Clinician’s Corner.
Calpains are a diverse family of calcium-dependent intracellular proteases that mediate a multitude of physiologic and pathologic processes through limited proteolysis, whereby they modulate, rather than destroy, their protein targets.
Calpain-related diseases have a considerable impact on public health. Calpains are involved in a multitude of neurodegenerative and systemic diseases where calcium homeostasis is perturbed (e.g., Alzheimer’s, cardiovascular and cerebral ischemia, diabetes, retinal degeneration, and malaria). Although some of these disorders are inherited, many are attributed to environmental and lifestyle factors. In particular, the neural retina is highly susceptible to aberrant calpain activation due to the high resting intracellular calcium concentrations in photoreceptors.
Several animal models of ophthalmic disorders related to elevated calpain activity have been generated (e.g., retinitis pigmentosa, neovascular inflammatory vitreoretinopathy, and diabetic retinopathy). These model organisms provide an ideal system to test calpain activity and therapeutic inhibition due to the ability to deliver drugs locally via intraocular injection.
Despite differences in domain architecture and function among calpains, the three-dimensional structure of the protease core domain is highly conserved, making the generation of specific therapeutic inhibitors targeting the active site challenging. Therefore, a more detailed understanding of the structural and functional diversity among the non-catalytic domains of calpains may provide better insight into how to target specific isoforms. Nevertheless, global calpain inhibition may also allow for broader treatment of calpain-related diseases in the eye.
Calpains and inherited eye disease
The increasing prevalence of genetic testing (i.e., via whole-exome sequencing [WES], whole-genome sequencing [WGS], and/or an ocular genetic-screening panel) has linked over 350 genes to hereditary eye diseases, and the number of pathogenic variants continues to increase [86]. To date, there are two ocular calpainopathies caused by genetic variants in either CAPN5 (NIV, OMIM #193235) or CAPN15 (developmental eye anomalies, OMIM #603267). Other genetic diseases in which dysregulation of the calpain-calcium axis was identified as a key molecular pathology are achromatopsia and retinitis pigmentosa (RP), both of which exhibit inherited retinal degeneration like CAPN5 vitreoretinopathy. Transgenic animal models of inherited retinal disease (IRD), including PDE6C, PDE6B, RHO, and FAM161A mutants, demonstrated increased expression of CAPN1 or 2 as well as calpain-induced apoptotic and necrotic pathways resulting in photoreceptor death. This suggests that even when calpain genes are not mutated in IRD, calpains may act as key mediators of disease (Box 1, Figure S2) [87–91].
Box 1. Inherited retinal degenerations and calpain-mediated cell death.
Inherited retinal degenerations (IRD) encompass a diverse group of ocular diseases, including achromatopsia (OMIM #216900) and retinitis pigmentosa (RP; OMIM #268000), characterized by progressive photoreceptor loss. Achromatopsia is clinically characterized by loss of color vision, which indicates defective cone photoreceptors [136]. The clinical hallmark of RP is a ‘bone spicule’ pigmentation in the peripheral retina attributed to failing rod photoreceptors.
Genetic animal models of achromatopsia (i.e., PDE6C; cone cGMP-specific 3’,5’-cyclic phosphodiesterase subunit alpha) and RP (i.e., PDE6A [rod cGMP-specific 3’,5’-cyclic phosphodiesterase subunit alpha], PDE6B [rod cGMP-specific 3’,5’-cyclic phosphodiesterase subunit beta], RHO [rhodopsin], and FAM161A) revealed that calpain-calcium axis dysregulation is a major contributor to the underlying pathogenesis of these diseases [87–89]. In mouse RP models, cone-photoreceptor-function loss has been phenocopied with mutations within either rod or cone-specific PDE6 genes (Figure S2A–B). In these models, mice exhibited photoreceptor death induced by apoptotic (mediated by CAPN1-induced activation of BCL2 Associated X; BAX, an apoptosis regulator [90,91]) and necrotic pathways (mediated by CAPN1-induced activation of apoptosis-inducing factor, AIF [90,91])), increased calpain activity, and CAPN1 or 2 expression [87,89,137–140]. In transgenic RHO-mutant RP models (RHO-p.P23H and RHO-p.S334ter; Figure S2C), increased calcium-mediated calpain activity led to CAPN1-induced activation of BAX- and AIF-dependent cell-death pathways [87,91,141]. Rhodopsin, encoded by RHO, is a G-protein coupled receptor (GPCR) expressed on rods that drives visual phototransduction. In Rho-mutant RP rats, intravitreal injection or topical application with Tat-μCL, a mitochondrial CAPN1 inhibitor, led to inhibited photoreceptor death and decreased nuclear AIF translocation [44,142]. In corroborating studies, calpain inhibition using calpastatin also reduced apoptotic cell death [91]. These results strongly suggest calpains’ association with RP and confirm that calpain inhibition is a viable therapeutic strategy for IRDs.
Unlike the above-described RP models which predominantly studied CAPN1 activity, other studies established an important role for CAPN2 over CAPN1 in photoreceptor degeneration [143]. Likewise, the FAM161Atm1b/tm1b knockout RP mouse model (Figure S4D) revealed that retinal CAPN2 expression peaks at 1 month of age, and the number of CAPN2-positive cells declines linearly over time [144]. In this model, stressed photoreceptors showed increased inflammatory cytokines (e.g., TNF and IL-6), reactive oxygen (ROS), and reactive nitrogen species (RNS) leading to elevated calcium levels and calpain activation [145]. Notably, in achromatopsia (Cpfl1) mice, CAPN2-driven retinal cell death peaks in early stages of the disease and delays postnatal development of cones [138], therefore, CAPN2 might be more critical than CAPN1 in driving early stages of retinal degeneration. This ambiguity in the role of CAPN1 and −2 in photoreceptor degeneration warrants further investigation.
CAPN5 vitreoretinopathy: dysregulation of cell death, inflammatory and neovascular pathways
NIV, an autosomal-dominant ocular disease exclusively caused by genetic defects in the CAPN5 gene [8], leads to CAPN5 hyperactivity in the retina resulting in retinal cell death, inflammation, neovascularization, intraocular fibrosis, and ultimately vision loss (Figure 3B; NIV panel) [92]. Despite its rarity (occurring in approximately one out of every million people), NIV shares clinical phenotypes with a plethora of more common ocular diseases (e.g. RP, AMD, and DR) [8] and may provide important mechanistic insights into pathogenesis of more common eye disease. Expressed in the signal-transducing neuronal cells of human, mouse, and zebrafish retina [93,94], CAPN5 is upregulated in response to photoreceptor degeneration induced by acute light exposure [93]. Importantly, because normally depolarized photoreceptor cells have high intracellular Ca2+ concentrations, hyperactivating CAPN5 mutations can lead to neuroretinal toxicity, retinal inflammation, and retinal neovascularization [95].
Currently, there are two well-studied in vivo NIV models (Figure 4A). One model used a lentivirus engineered to express human (h) CAPN5-p.R243L [28,96] to recapitulate NIV-like retinal phenotypes such as photoreceptor degeneration, retinal inflammation, and loss of b-wave amplitude (detected by electroretinography [ERG]) [96]. However, likely due to transient expression of the disease-causing CAPN5 mutant, the lentiviral model never developed the severe neovascularization or intraocular fibrosis characteristic of late-stage human NIV. In contrast, a transgenic mouse model expressing the hCAPN5-p.R243L mutant (hCAPN5R243L) recapitulated all of the main features of NIV [28] and serves as a compelling preclinical model to study NIV pathology (Figure 4A). CAPN5-specific inhibitors are currently unavailable [97,98], and may need to be developed for in vivo therapeutic investigation.
Figure 4. In vivo models and pathology of ocular calpainopathies.
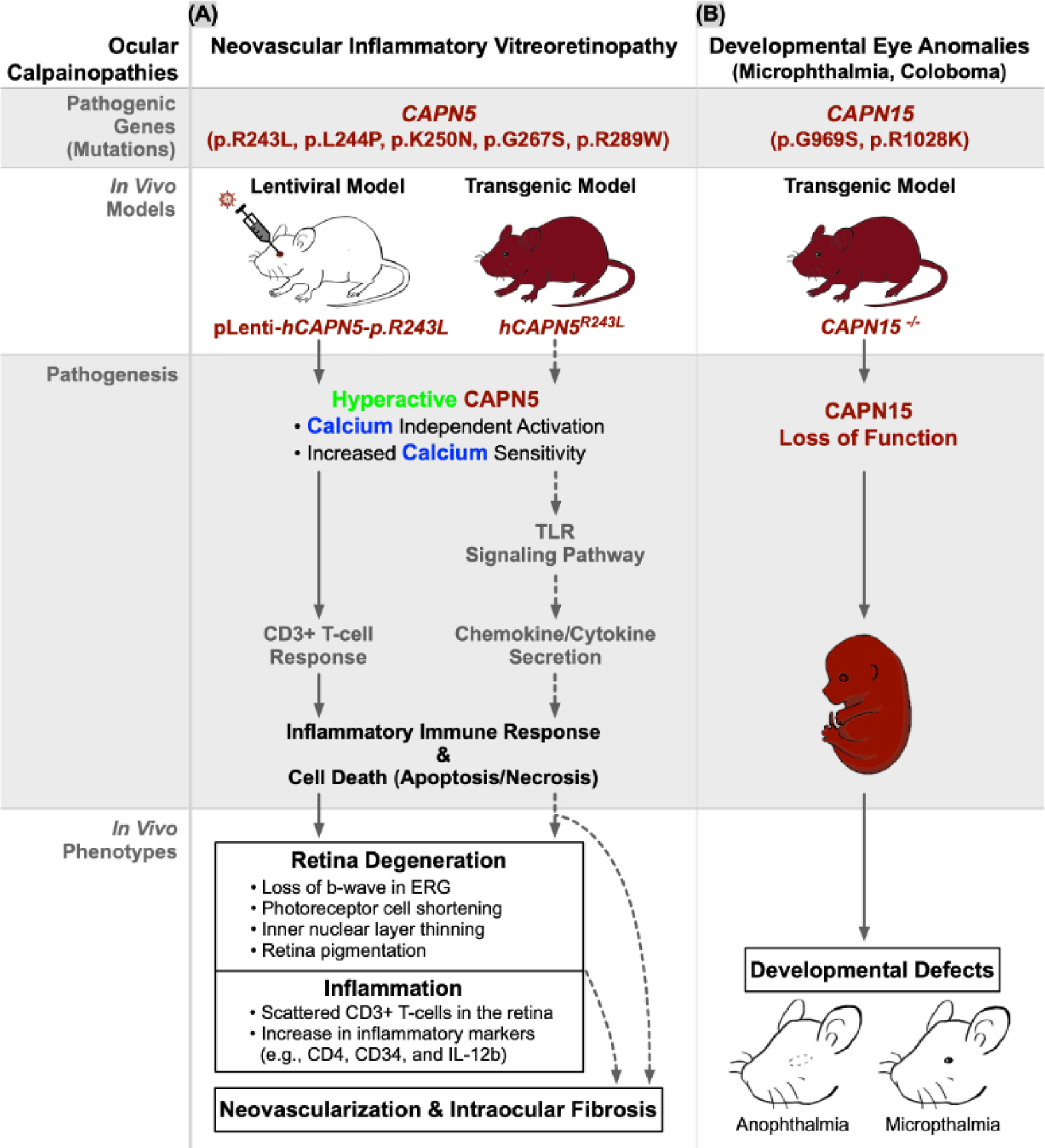
Calpains are involved in a large variety of ocular diseases called calpainopathies that lead to manifestations in (A) neovascular inflammatory vitreoretinopathy (NIV) and (B) developmental eye anomalies. CAPN5 mutations cause CAPN5 hyperactivity either by calcium independent activation or increasing its sensitivity to intracellular calcium concentrations. There are two reported mouse models of NIV to model CAPN5 mutations—a lentiviral model and a transgenic model. NIV models were created based on biochemical findings of five known NIV mutations in the catalytic PC domains that increase CAPN5 catalytic activity: three mutations (p.R243L, p.L244P, and p.K250N) located on the G1 gating loop of the PC2 subdomain (which may impede the G1-loop’s ability to gate substrate binding thus making it constantly available for target proteolysis [8,28,29]); a fourth, p.G267S, on the PC2L2 loop (distantly located from the active site suggesting the mutation works through allosteric effects [55]) and the fifth, p.R289W, located on the G2 gating loop (which disrupts a key conserved calpain structural regulatory motif—the tryptophan “wedge” leading to calcium-independent calpain hyperactivity[149]). A transgenic CAPN15 knockout model to model developmental eye anomalies resulted in developmental defects such as anophthalmia and microphthalmia.
CAPN15: developmental eye anomalies
Calpains control developmental pathways in a variety of in vivo models [93,99,100], yet their role in human eye development is unknown. Expressed throughout the retina, CAPN15 contains a catalytic PC domain and two non-catalytic domains: the zinc-finger and the SOLH domain [101] (Figure 2A). Pathogenic human mutations within the SOLH domain and their genetic relevance to other species collectively suggest the domain plays a key molecular role unique to the eye [71]. In 2020, human CAPN15 mutations were linked to microphthalmia (i.e., small eyes) and coloboma (i.e., incomplete optic-fissure closure) which both belong to a spectrum of developmental eye disorders [71]. Three homozygous-monoallelic variants of CAPN15 and a set of heterozygous-biallelic variants were identified in five individuals from four independent families, each with either eye anomaly. Two of the homozygous-monoallelic variants (i.e., p.G969S and p.R1028K) were predicted to decrease CAPN15 protein expression. This hypothesis was further supported by Capn15 knockout mice which showed developmental eye defects such as anophthalmia (i.e., no eye), microphthalmia or cataract (Figure 4B) [71]. Although there is strong evidence that genetic mutations in CAPN15 cause ocular and developmental defects, their molecular pathology and postnatal function have yet to be elucidated. Future research could utilize CAPN15-calpainopathic patient stem-cells alongside CAPN15 knockout mice. Selective deletion of various calpain isoforms, like Capn15, has given insight into their physiologic functions in different tissues. However, few studies have utilized ocular calpain knockout or knockdown techniques. Interrogation of the International Mouse Phenotyping Consortium (IMPC) and the Mouse Genome Informatics (MGI) databases revealed phenotyping data for one specific Capn12 KO mouse model: Capn12tm1a(EUCOMM)Hmgu [102,103]. Capn12 gene function is disrupted in these mice through tm1a allele insertion; mice homozygous for the tm1a allele have a loss-of-function of the Capn12 gene [102,103] (Figure S3).
Non-mendelian ophthalmic diseases of chronic cellular stress
Cataract: sustained calpain overactivation increases crystallin deposition in the lens
Cataracts are caused by progressive lens opacification (i.e., cataractogenesis) and are a leading cause of reversible blindness worldwide (Figure 3B; Cataract panel). Mechanistically, in a variety of human and animal cataract models, different calpains are involved in disease progression through cleavage of proteins like spectrin and vimentin. For instance, in human lenses, CAPN2 is the dominant isoform involved [104] while in a genetic ovine model (i.e., New Zealand Romney sheep with inherited cataract; Figure 5A), broad calpain activity contributes to cataractogenesis [47]. Interestingly, in rat (selenite-induced cataract model) and sheep lenses, overactivation of CAPN1, CAPN2, and the lens-specific splice variant of CAPN3 (Lp82) resulted in rapid truncation, insolubility, and precipitation of crystallin proteins leading to cataract formation (Figure 5A). These studies have also shown that calpain inhibitors delay cataract formation. For instance, in multiple in vivo bovine cataract models, topical application of the calpain family inhibitors CAT0059, CAT811, and SJA6017 impeded disease [47,105–107]. However, whether CAPN3 is involved in the pathogenesis of human cataractogenesis merits validation. Specifically, Lp82 is not expressed in humans due to the presence of deleted nucleotides in exon 1 of the human transcript that produce a stop codon. Therefore, although an Lp82-like cleavage site was identified in the human αA crystallin sequence, it is unclear if this site is amenable to cleavage by other proteases in the human lens [108].
Figure 5. In vivo models of chronic ocular diseases and their calpain-associated pathologies.
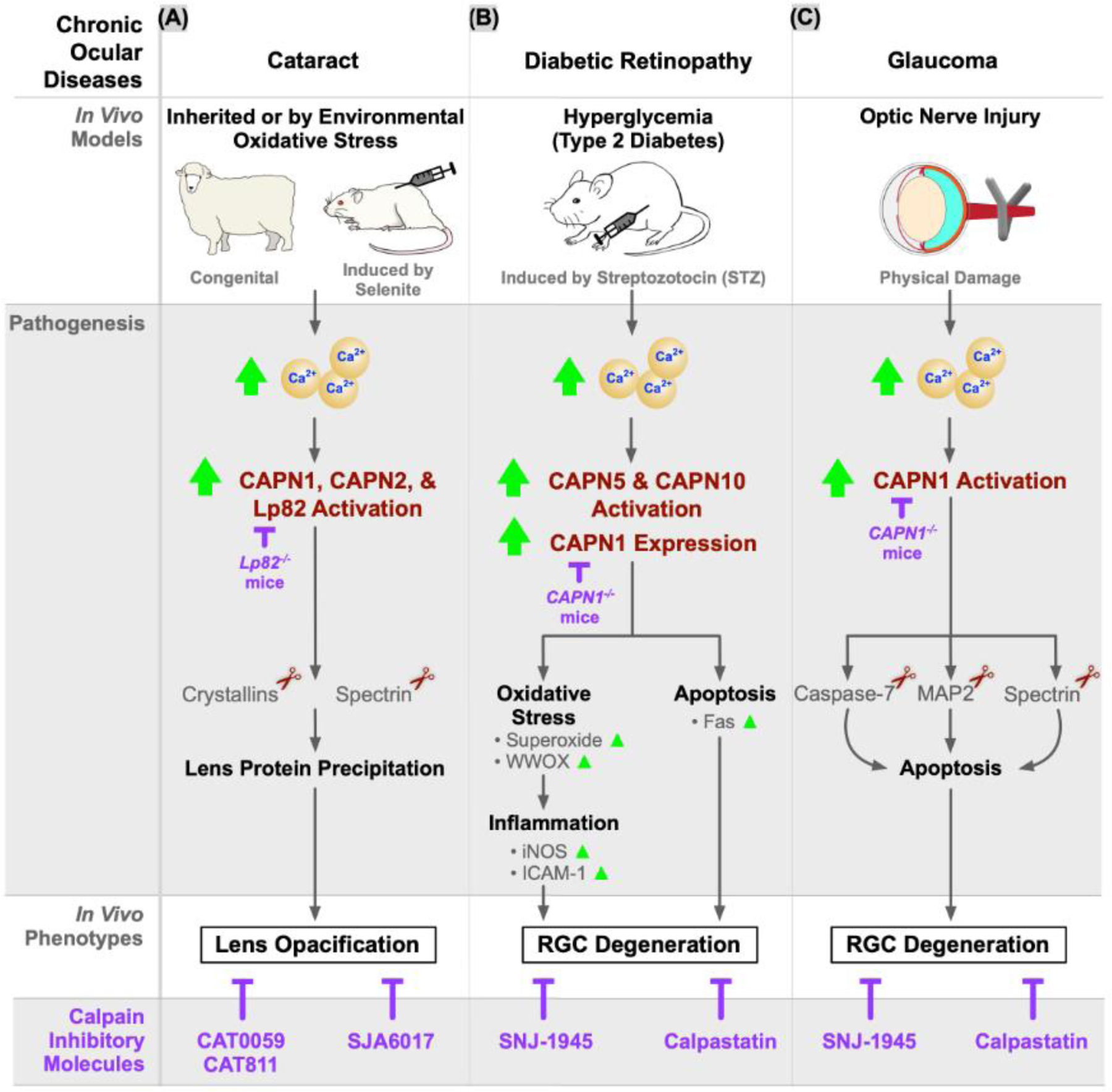
Calpains are involved in common chronic ocular diseases: (A) cataract, (B) diabetic retinopathy, and (C) glaucoma. A congenital cataract model using New Zealand Romney sheep displayed inherited cataract. Cataracts caused by environmental oxidative stress can be modeled by selenite injection into rats. In both cataract models, CAPN1, CAPN2, and a lens-specific splice variant of CAPN3 (Lp82) were increasingly activated, which causes lens opacification. The diabetic retinopathy mouse model is induced by streptozotocin (STZ) injection, which severely damages pancreatic beta cells to induce hyperglycemia. It demonstrated increased activation of CAPN5 and CAPN10 as well as increased expression of CAPN1. As a result, oxidative stress and apoptotic pathways were activated and led to retinal ganglion cell (RGC) degeneration. Glaucoma murine models employed optic nerve injury to induce RGC death by activating CAPN1 and its downstream apoptotic pathways. A multitude of calpain inhibitors were shown to ameliorate the disease phenotypes in these in vivo models. Additionally, knock-out Lp82 mice and knock-out CAPN1 mice exhibited decreased disease progression.
Diabetic retinopathy: calpains regulate hyperglycemia-induced inflammation and apoptosis
Diabetic retinopathy (DR; Figure 3B; DR panel) is a complex, blinding disease affecting more than 1/100 people worldwide with clinical manifestations including retinal inflammation, cell death, and neovascularization [109]. Mechanistically, hyperglycemia-induced oxidative stress leads to dysregulated calcium levels, increased calpain expression and in calpastatin knockout mice (CAST−/−), retinal ganglion cell (RGC) death [7,110]. In vivo DR models demonstrated increased retinal CAPN1, CAPN5, and CAPN10 expression in addition to increased basal intracellular calcium levels (Figure 5B) [7]. Notably, pharmacologic (calpastatin) or genetic loss of CAPN1 downregulated photoreceptor expression of oxidoreductase (WW domain-containing oxidoreductase [WWOX]) and pro-inflammatory markers of diabetes-induced stress such as iNOS and ICAM-1, highlighting CAPN1 as an upstream regulator of diabetic inflammatory pathways [7,111,112]. Finally, treating DR mice with SNJ-1945, a calpain inhibitor, rescued RGC density such that treated mice were indistinguishable from non-diabetic controls [110].
Additionally, Rt88 (a CAPN3 splice variant) and CAPN10 both correlate with degeneration of retinal and photoreceptor cells in a diabetic rodent model (WBN/Kob rat). However, this phenotype occurred before the onset of hyperglycemia, so Rt88 and CAPN10 may not be directly implicated in DR pathogenesis in this model. Instead, they might have roles in retinopathies independent from hyperglycemia-induced stress [113].
Glaucoma: calpains drive neurodegenerative pathways in retinal ganglion cells
Glaucoma is a heterogeneous group of neurodegenerative disorders that lead to death of RGCs and their axons which make up the optic nerve (ON; Figure 3B; Glaucoma panel). It is the second leading cause of blindness globally and clinically manifests as elevated intraocular pressure (IOP) and ON damage, leading to RGC loss and blindness [114]. Due to its complex pathogenesis, there are currently no animal models that can recapitulate the full spectrum of the human glaucoma phenotype, although experimentally induced animal models phenocopy many of the factors associated with glaucoma pathogenesis (e.g., increased intraocular pressure [IOP] and ON degeneration). Nevertheless, ON injury (e.g., simple acute ON crush injury) and inducible ocular hypertension models (e.g., intracameral silicon oil injections) have become useful methods to study glaucomatous optic neuropathy in rodents [115]. As such, these models have provided evidence that CAPN1 hyperactivation is associated with neurodegeneration through three major apoptotic pathways: caspase-7 (an apoptosis-related peptidase), acid-sensing ion channels (ASIC1) and endoplasmic reticulum (ER) stress (Figure 4C) [116–118]. In these studies, calpain-mediated cleavage of caspase-7 led to the progressive loss of RGCs, an effect that was rescued in caspase-7 KO mouse models [116]. In rat models for an investigation of the ASIC1 pathway, CAPN1 activation triggered the degradation of cytoskeletal proteins like spectrin, and eventually increased neuronal death [118]. Finally, in rat models of ER-stress-induced apoptosis, ON injury resulted in a rapid induction of CAPN1 activity, implicating calpains in glaucoma [117].
Calpain inhibition has been tested as a therapeutic intervention with promising results. For example, treating ON-crushed eyes with calpain inhibitors significantly protected against ganglion cell layer loss and axonal-damage-induced cell death, preserved the functional RGC ERG response, and improved visual function (Figure 4C) [33,35]. Interestingly, although CAPN1 was shown to promote neuroretinal cell death in glaucoma through multiple apoptosis pathways [116–118], another study reported the opposite. CAPN1-mediated cleavage of PH domain and leucine rich repeat protein phosphatase 1 (PHLPP1) was demonstrated to promote survival of RGCs in a model of glaucomatous neurodegeneration through activation of the pro-survival Akt (protein kinase B) pathway [33]. Conversely, CAPN2 activity promoted RGC death through cleavage of striatal-enriched protein tyrosine phosphatase (STEP) and activation of the pro-death STEP/p38 pathway following IOP elevation [33]. These conflicting findings suggest that multiple calpains might orchestrate the intricacies of apoptosis and suggest a role for selective calpain inhibition in glaucoma therapy.
Calpain activation has also been implicated in other models of ocular injury caused by retinal ischemia-reperfusion, retinal detachment, light exposure, and alkylating agents like N-methyl-N-nitrosourea (MNU) (Figure S4). However, since glaucoma in humans is typically associated with elevated IOP, the pathogenic mechanisms of ON-injury models based on physical injuries to the ON are limited. Thus, a recently developed glaucoma model which induces ocular hypertension with silicone oil [119] is more realistic to test calpain inhibition.
Macular degeneration: calpain inhibition rescues RPE degeneration
Age-related macular degeneration (AMD), caused by damage to the RPE and the photoreceptors within the macula, is a primary source of vision loss in elderly populations. One of the clinical hallmarks of AMD is drusen: protein and lipid aggregates that accumulate between the RPE and choroid. Notably, retinal explants from monkeys and humans under hypoxic stress (which models AMD in vitro) showed calpain activation due to accumulation of A2E (N-retinyledin-N-retinylethanolamine; a pyridinium bis-retinoid), a principal component of drusen) [120,121]. Importantly, the degenerating explants were rescued by application of a generic calpain inhibitor, SNJ-1945, suggesting that calpain activation is involved in AMD progression (Figure S5A) [122]. Because AMD models do not fully recapitulate various stages of the human disease, other models of early-onset macular degeneration like Stargardt’s disease (OMIM #248200) models are sometimes used by proxy (Figure S5B).
Therapeutic strategies to inhibit calpains in eye disease
Dysregulated calpain activity is implicated in various pathological conditions of the eye, and a myriad of in vivo studies has confirmed that calpain inhibitors hold immense therapeutic potential [7,33,35,123,124]. In most in vivo studies described in this review, calpain inhibitors were administered systemically (e.g., orally [35], subcutaneously [124], or intraperitoneally [7,33,123]). However, systemically administered drugs are prone to toxicity due to the lack of tissue- and/or calpain isoform-specificity, which may explain why ocular calpain inhibitors are currently neither in clinical trials nor FDA-approved.
The eye possesses unique advantages as a target organ to test therapeutic calpain inhibition. It is readily accessible to phenotypic evaluation of therapeutic effects in vivo. It is the only organ with neural tissue that can be visually inspected noninvasively, making it well suited for studying neurodegenerative diseases. In clinical practice, drugs are routinely delivered directly into the vitreous or subretinal space (Figure 1). The eye’s highly compartmentalized anatomy facilitates direct delivery to the diseased tissue while minimizing systemic dissemination and the potential for unwanted off-target effects.
While calpains have been largely described as aggravating factors in a number of pathophysiologic phenomena, the physiological relevance of calpains in the eye remains elusive. Calpains appear to recognize specific three-dimensional structure of their substrates (i.e., interdomain linkers and flexible loops) rather than a single specific amino acid sequence motif like trypsin, for example [125]. Additionally, each calpain species is fairly specific in terms of its cellular function, which can be attributed to the modular nature of their non-catalytic domains. Nevertheless, numerous studies have uncovered putative calpain substrates involved in a wide range of physiologic functions, including metabolism [126], visual transduction [127], cytoskeletal regulation [128], transcription [129], and apoptosis [130]. Hence, broad calpain inhibition may disrupt numerous important physiologic functions in eye. Creation of highly selective calpain inhibitors could be critical in developing clinically feasible therapies that avoid off-target effects.
Small molecule peptidomimetics, designed to mimic natural protein-peptide interactions, are attractive calpain inhibitors [131]. High-throughput screening of peptidomimetic candidates combined with their structural modifications to enhance specificity and stability (e.g., replacement of natural/unnatural amino acid side chains, cyclization, and/or non-peptide small molecules) can identify lead compounds for the drug development pipeline: hit-to-lead optimization, in vitro testing, and in vivo studies. SNJ-1945 and SJA6017 (Senju Pharmaceutical Co. Ltd) are orally available, water-soluble, and membrane-permeable peptidomimetic calpain inhibitors that block the active site of calpains [132]. Calpeptin and MG-101 are also cell-permeable peptidomimetic inhibitors that target cytosolic calpains. In SH-SY5Y human neuroblastoma cells, various peptidomimetics (e.g., calpeptin, MG-101, and leupeptin) can inhibit CAPN1 and CAPN5 activity by inhibiting their auto-proteolysis, which is an auto-activating mechanism [68]. Peptidomimetics in liquid form can be administered topically and intravitreally to target the anterior or posterior segment, respectively. To avoid frequent injections and drug overdose, drug release can be prolonged by the ever-advancing field of ocular drug formulations – e.g., micro-/nano-particles, intraocular implants, injectable hydrogels, prodrugs, penetration enhancers. For cell-specific delivery of calpain inhibitors, peptidomimetics could be conjugated to antibodies (Antibody-Drug Conjugates; ADCs) or other molecules that recognize specific surface proteins on target cells [133]. Cell specific therapy can also be achieved using cell-specific promoters in gene therapy vectors. While gene supplementation is used to treat recessive conditions, other forms of gene therapy, like RNA interference-mediated gene silencing, have the potential to treat dominant diseases by modulating calpain expression with superior specificity (Box 2). An alternate form of gene therapy is stimuli-triggering viral or non-viral vectors, which can facilitate the delivery of genomic materials to target cells under the disease conditions or the unique molecular environment of target tissues.
Box 2. Calpains and gene therapy.
In a proof-of-principle study, a CAPN5-specific, short hairpin RNA post-transcriptionally silenced target gene expression, effectively reduced CAPN5 expression in neuroblastoma SH-SY5Y cells [97], suggesting that a gene-silencing strategy could target CAPN5 specifically. Notably, the eyes of CAPN5 knock-out mice appear to function normally, as evaluated by retina-histology and ERG [56], supporting the concept that eliminating CAPN5 expression in the eye would pose little risk for off-target effects. To overcome gene-delivery hurdles (e.g., large molecular size, immense negative charge, and poor enzymatic stability), genetic material is often delivered by engineered non-viral or viral vectors. In most disease contexts, the major challenge of implementing gene therapy is its immunogenicity: viral proteins, transgene products, unmethylated CpG DNA in plasmid/bacterial DNA and non-viral vector components can stimulate innate and adaptive immunity which could lead to severe complications in patients [146]. Local toxicity and immune responses caused by non-viral or viral vectors can be prevented by using biocompatible materials or selecting less immunogenic viral vectors, respectively. Moreover, the biology of the eye renders it an ideal system for gene therapy, it is an immune-privileged organ with a naturally immunosuppressive intraocular microenvironment (such as blood-retina barriers) [147]. Indeed, recent clinical trials are proving the safety of gene therapy for retinal diseases, one example being Luxturna (Spark Therapeutics), an FDA-approved treatment of RPE65-associated inherited retinal dystrophy [148].
While peptidomimetics and gene therapy can target calpains directly, metabolite supplementation can provide an indirect, yet efficient, therapeutic strategy by preemptively reducing calpain activity through modulation of intracellular calcium levels and oxidative stress. For example, zinc and taurine were found to ameliorate calpain-related cellular damage in in vitro models using immortalized human RPE cells, primary monkey RPE cells, as well as primary RPE cells isolated from human AMD patients [122,134,135]. Antioxidants are involved in a wide variety of intricate physiological processes, ranging from enzyme regulation to nuclear regulatory elements, yet the precise underlying molecular pathways of metabolites supporting calpains are unknown. Thus, more research about the crosstalk of calpain in antioxidant pathways is needed before metabolite supplementation can be a viable therapeutic strategy.
Recent advancements in human gene therapy, genome surgery, and protein engineering in the eye have paved the way for future clinical trials for calpain-related disorders, both inherited and acquired. The advent of CRISPR-Cas systems for gene editing or modulation of gene expression could soon enable the correction of both recessive and dominant disorders. Nevertheless, dominantly inherited diseases, like NIV, may benefit from alternate strategies like antisense therapy (e.g., oligonucleotides and shRNA), CRISPRi, mAbs, or inhibitory receptor traps – all modalities that have been applied to eye disease. Additionally, although calpains are primarily considered intracellular proteins, recent studies have detected catalytically active calpains in the vitreous and aqueous (unpublished observations), suggesting that extracellular antibodies may have applications in calpain-related eye disease. The selectivity of small molecule inhibitors for calpains remains an issue due to the conserved PC domain structure. However, allosteric inhibition of calpain activity, through targeting of their unique non-catalytic domains, may be a viable strategy to achieve selectivity. Molecular therapies targeting the non-catalytic activities of calpains are also needed. CAPN15 calpainopathy, on the other hand, presents a distinct challenge since the pathology appears to present in early development. Like other congenital inherited diseases, family planning by pre-implantation genetic testing and in vitro fertilization may be a viable option for this disorder.
Concluding Remarks
Calcium dysregulation is the underlying molecular mechanism of calpain hyperactivity and ultimately manifests in a wide variety of ophthalmic pathologies. The unique and diverse calpain isoforms are well described through genomic, proteomic, and structure-function studies in human eyes and in vivo models. Although each isoform’s distribution may slightly differ among animal species, the general trends are comparable and support the reliability of in vivo models to simulate human ocular diseases [80]. Yet, more research is needed to determine the unique and/or complementary roles of calpain isoforms and whether their characteristics are variable depending on the specific eye tissue or cell, type of disease and disease stage (see Outstanding Questions).
Outstanding Questions.
How can apoptotic and necrotic cell death pathways in calpain-mediated diseases be further delineated in mammalian models to identify other biomarkers for targeted therapy?
Using in vitro methods including structure biology and biochemistry, which domains (PC, CBSW, PEF, and C2) are most sensitive to calcium binding and more likely to lead to a calpain-mediated disease?
Which of the specific calpain isoforms are disease-causing?
How can small molecule peptidomimetics be developed as calpain inhibitors for other systemic, calpain-mediated diseases while minimizing off-target effects in other organs that are not as compartmentalized as the eye?
Because rapid clearance is common challenge for drugs administered via intraocular injection, how can calpain inhibitors be developed with sustained delivery in mind?
How can calpain inhibitors be used synergistically with current ocular treatments such as anti-VEGF and steroid therapy?
Given the role of CAPN15 in causing developmental eye disorders, what implications should be considered and implemented during prenatal genetic screening for this gene?
Because calpains are involved in many other systemic and neurodegenerative diseases, how does knowledge gained from ocular calpain therapeutics improve the treatment for other organs and diseases?
New knowledge about calpain’s structure-function relationship can guide protease inhibitor research. In tandem, the development of peptidomimetics and calpain-targeting gene therapies holds broad implications to treat leading causes of blindness. In the future, specificity of small molecule peptidomimetics can be improved by modifying chemical structures; in addition, gene therapy efforts should focus on characterizing pathogenic mutations and minimizing immunogenicity. Minimally invasive surgical techniques will allow specialized drug administration to specific cells in the eye. Finally, given its immune-privileged attributes, the eye is an ideal test organ for therapeutics with promising applications in other organs (e.g., the brain, the thyroid, and the heart) that are similarly impacted by calpain-mediated diseases.
Supplementary Material
Highlights.
The eye is an attractive model organ to study molecular therapeutic interventions for neurodegenerative diseases caused by calpain hyperactivity.
Calcium-mediated calpain hyperactivity is a central molecular mediator of several ocular diseases, leading to cell death, inflammation, and neovascularization.
Calpainopathies caused by Mendelian defects in CAPN5 and CAPN15 result in eye-specific diseases, including neovascular inflammatory vitreoretinopathy and ocular developmental anomalies, respectively.
Novel structure-function studies and translational models of eye disease have shed light on mechanisms underlying calpain hyperactivity.
Small-molecule calpain inhibitors, protein, peptide and gene-based therapies are promising avenues of treatment for calpain-mediated ocular diseases, especially given the eye’s accessibility to targeted molecular therapy.
Acknowledgements
The authors would like to thank Teja Chemudupati and Aarushi Kumar for technical assistance. We thank Marcus Toral for providing the eye anatomy illustration used in Figure 1. We also thank Dr. Scott Lambert and Zachary Wennerg-Smith for clinical images of microphthalmia and a healthy lens, respectively. We thank Dr. Youn Soo Jung at the Stanford Epidemiology and Clinical Research Graduate Program for providing animal illustrations in our figures.
Funding/Support:
VBM is supported by NIH grants (R01EY031952, R01EY030151, R01EY024665, R01EY025225, and P30EY026877), Stanford ChEM-H IMA, the Stanford Center for Optic Disc Drusen, and Research to Prevent Blindness, New York, New York. YJS is supported by BrightFocus Foundation’s Macular Degeneration Research program. GV is supported by NIH grants [F30EYE027986 and T32GM007337].
Glossary
- Antisense Oligonucleotide
single-stranded deoxyribonucleotides that are complementary to their mRNA target and reduce target gene translation by induction of RNase H endonuclease activity
- Apoptosis
the physiological process of programmed cell death (PCD) that is tightly regulated by proteolytic enzymes including caspases and transcription factors
- Blood-Retina-Barrier (BRB)
a partially restrictive physiologic barrier composed of adherens and tight junctions that regulates the exchange of metabolites, proteins, and waste products between the neural retina and vascular lumen
- Calpainopathy
a diverse group of diseases known to be directly caused by genetic variants in one of the calpain genes. To date, the only ocular calpainopathies are neovascular inflammatory vitreoretinopathy (NIV) and developmental eye disorders (like microphthalmia, anophthalmia, coloboma) which induced by variants in CAPN5 and CAPN15, respectively
- Cysteine proteases
protein-degrading enzymes that share a catalytic mechanism involving a cysteine nucleophilic residue. All calpains are cysteine proteases
- Domain topology
a visual arrangement of a group of proteins’ structural and/or functional units drawn from the N terminus to C terminus. It is often used to align protein sequences to highlight conserved and unique regions
- Electroretinography
a diagnostic test involving electrodes used in human and mammalian models to measure the electrical activity of the retina, specifically the cones and rods, in response to a light stimulus
- Immune privilege
refers to tissues (e.g., eye, brain, testes, placenta, etc.) that exhibit a reduced inflammatory immune response compared to the rest of the body. In particular, the eye limits its inflammatory immune response to protect vision from edema and tissue changes associated with inflammation
- Necrosis
another mechanism of cell death once believed to be less regulated compared to apoptosis now known to be modulated by receptor-interacting protein kinases (RIPKs)
- Neovascularization
the growth of local blood vessels, often in response to ischemic conditions, that are prone to leaking which causes further inflammation
- Peptidomimetics
synthetic peptide analogs designed to mimic natural peptide structures/functions for drug discovery and therapeutic purposes
- Phototransduction
a process that converts light into electrical potential in the rod cells, cone cells and photosensitive ganglion cells of the retina
- Proteomics
the large-scale study of high-density protein datasets to track the expression, interaction, and modifications of proteins in a given organ or region of interest. Ocular proteomics often involve the investigation of proteomes within the vitreous or aqueous humor
- scRNA-seq
single-cell RNA sequencing is a high-resolution, genomic technique used to cluster distinct cell types in a tissue of interest based on their unique mRNA expression profiles
- Transgenic
a genetically modified or engineered organism with DNA from an external organism. Transgenic animal models are often used to simulate genetic diseases
- Vitreous
a clear, gel-like fluid that maintains the eye’s shape and integrity by occupying the space between the lens and the retina
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interest.
Resources
Data Availability Statement
Not applicable.
References
- 1.DiCarlo JE et al. (2018) Gene therapy and genome surgery in the retina. J Clin Invest 128, 2177–2188. 10.1172/JCI120429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodrigues EB et al. (2009) Therapeutic monoclonal antibodies in ophthalmology. Prog Retin Eye Res 28, 117–144. 10.1016/j.preteyeres.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 3.Sugar EA et al. (2017) Longitudinal Vision-Related Quality of Life for Patients with Noninfectious Uveitis Treated with Fluocinolone Acetonide Implant or Systemic Corticosteroid Therapy. Ophthalmology 124, 1662–1669. 10.1016/j.ophtha.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seal JR et al. (2019) Intracameral Sustained-Release Bimatoprost Implant Delivers Bimatoprost to Target Tissues with Reduced Drug Exposure to Off-Target Tissues. J Ocul Pharmacol Ther 35, 50–57. 10.1089/jop.2018.0067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ono Y et al. (2016) Calpain research for drug discovery: challenges and potential. Nat Rev Drug Discov 15, 854–876. 10.1038/nrd.2016.212 [DOI] [PubMed] [Google Scholar]
- 6.Goll DE et al. (2003) The calpain system. Physiol Rev 83, 731–801. 10.1152/physrev.00029.2002 [DOI] [PubMed] [Google Scholar]
- 7.Saadane A et al. (2021) Photoreceptor Cell Calcium Dysregulation and Calpain Activation Promote Pathogenic Photoreceptor Oxidative Stress and Inflammation in Prodromal Diabetic Retinopathy. Am J Pathol 10.1016/j.ajpath.2021.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahajan VB et al. (2012) Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet 8, e1003001. 10.1371/journal.pgen.1003001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nikkel AL et al. (2012) The novel calpain inhibitor A-705253 prevents stress-induced tau hyperphosphorylation in vitro and in vivo. Neuropharmacology 63, 606–612. 10.1016/j.neuropharm.2012.05.011 [DOI] [PubMed] [Google Scholar]
- 10.Letavernier E et al. (2008) Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res 102, 720–728. 10.1161/CIRCRESAHA.107.160077 [DOI] [PubMed] [Google Scholar]
- 11.Yokota M et al. (1999) Calpain inhibitor entrapped in liposome rescues ischemic neuronal damage. Brain Res 819, 8–14. 10.1016/s0006-8993(98)01334-1 [DOI] [PubMed] [Google Scholar]
- 12.Miyazaki T et al. (2011) m-Calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation 124, 2522–2532. 10.1161/CIRCULATIONAHA.111.021675 [DOI] [PubMed] [Google Scholar]
- 13.Spencer MJ et al. (1997) Absence of calpain 3 in a form of limb-girdle muscular dystrophy (LGMD2A). J Neurol Sci 146, 173–178. 10.1016/s0022-510x(96)00304-8 [DOI] [PubMed] [Google Scholar]
- 14.Gan-Or Z et al. (2016) Mutations in CAPN1 Cause Autosomal-Recessive Hereditary Spastic Paraplegia. Am J Hum Genet 98, 1038–1046. 10.1016/j.ajhg.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hata S et al. (2010) Calpain 8/nCL-2 and calpain 9/nCL-4 constitute an active protease complex, G-calpain, involved in gastric mucosal defense. PLoS Genet 6, e1001040. 10.1371/journal.pgen.1001040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kottyan LC et al. (2014) Genome-wide association analysis of eosinophilic esophagitis provides insight into the tissue specificity of this allergic disease. Nat Genet 46, 895–900. 10.1038/ng.3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sleiman PM et al. (2014) GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun 5, 5593. 10.1038/ncomms6593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li FZ et al. (2015) Crosstalk between calpain activation and TGF-beta1 augments collagen-I synthesis in pulmonary fibrosis. Biochim Biophys Acta 1852, 1796–1804. 10.1016/j.bbadis.2015.06.008 [DOI] [PubMed] [Google Scholar]
- 19.Randriamboavonjy V et al. (2017) Calpain 1 cleaves and inactivates prostacyclin synthase in mesenteric arteries from diabetic mice. Basic Res Cardiol 112, 10. 10.1007/s00395-016-0596-8 [DOI] [PubMed] [Google Scholar]
- 20.Horikawa Y et al. (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26, 163–175. 10.1038/79876 [DOI] [PubMed] [Google Scholar]
- 21.Li Y et al. (2011) Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60, 2985–2994. 10.2337/db10-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ni R et al. (2016) Mitochondrial Calpain-1 Disrupts ATP Synthase and Induces Superoxide Generation in Type 1 Diabetic Hearts: A Novel Mechanism Contributing to Diabetic Cardiomyopathy. Diabetes 65, 255–268. 10.2337/db15-0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grieve S et al. (2016) Calpain Genetic Disruption and HSP90 Inhibition Combine To Attenuate Mammary Tumorigenesis. Mol Cell Biol 36, 2078–2088. 10.1128/MCB.01062-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leloup L and Wells A (2011) Calpains as potential anti-cancer targets. Expert Opin Ther Targets 15, 309–323. 10.1517/14728222.2011.553611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCall K (2010) Genetic control of necrosis - another type of programmed cell death. Curr Opin Cell Biol 22, 882–888. 10.1016/j.ceb.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paquet-Durand F et al. (2007) Calpain activity in retinal degeneration. J Neurosci Res 85, 693–702. 10.1002/jnr.21151 [DOI] [PubMed] [Google Scholar]
- 27.Li AS et al. (2021) Whole-Exome Sequencing of Patients With Posterior Segment Uveitis. Am J Ophthalmol 221, 246–259. 10.1016/j.ajo.2020.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wert KJ et al. (2015) CAPN5 mutation in hereditary uveitis: the R243L mutation increases calpain catalytic activity and triggers intraocular inflammation in a mouse model. Hum Mol Genet 24, 4584–4598. 10.1093/hmg/ddv189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bassuk AG et al. (2015) Structural modeling of a novel CAPN5 mutation that causes uveitis and neovascular retinal detachment. PLoS One 10, e0122352. 10.1371/journal.pone.0122352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y et al. (2018) Calpain-2 as a therapeutic target for acute neuronal injury. Expert Opin Ther Targets 22, 19–29. 10.1080/14728222.2018.1409723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang W et al. (2010) Calpain activation in experimental glaucoma. Invest Ophthalmol Vis Sci 51, 3049–3054. 10.1167/iovs.09-4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cham A et al. (2016) Secondary glaucoma in CAPN5-associated neovascular inflammatory vitreoretinopathy. Clin Ophthalmol 10, 1187–1197. 10.2147/OPTH.S103324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y et al. (2016) Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol Dis 93, 121–128. 10.1016/j.nbd.2016.05.007 [DOI] [PubMed] [Google Scholar]
- 34.Qu J et al. (2010) Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp Eye Res 91, 48–53. 10.1016/j.exer.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryu M et al. (2012) Critical role of calpain in axonal damage-induced retinal ganglion cell death. J Neurosci Res 90, 802–815. 10.1002/jnr.22800 [DOI] [PubMed] [Google Scholar]
- 36.Ozaki T et al. (2016) The protection of rat retinal ganglion cells from ischemia/reperfusion injury by the inhibitory peptide of mitochondrial μ-calpain. Biochem Biophys Res Commun 478, 1700–1705. 10.1016/j.bbrc.2016.09.006 [DOI] [PubMed] [Google Scholar]
- 37.McKernan DP et al. (2007) A key role for calpains in retinal ganglion cell death. Invest Ophthalmol Vis Sci 48, 5420–5430. 10.1167/iovs.07-0287 [DOI] [PubMed] [Google Scholar]
- 38.Biswas S et al. (2004) Calpains: targets of cataract prevention? Trends Mol Med 10, 78–84. 10.1016/j.molmed.2003.12.007 [DOI] [PubMed] [Google Scholar]
- 39.Biswas S et al. (2004) The in vitro retardation of porcine cataractogenesis by the calpain inhibitor, SJA6017. Mol Cell Biochem 261, 169–173. 10.1023/b:mcbi.0000028752.89886.43 [DOI] [PubMed] [Google Scholar]
- 40.GUROFF G (1964) A NEUTRAL, CALCIUM-ACTIVATED PROTEINASE FROM THE SOLUBLE FRACTION OF RAT BRAIN. J Biol Chem 239, 149–155 [PubMed] [Google Scholar]
- 41.Lon HK et al. (2019) Pharmacokinetics, Safety, Tolerability, and Pharmacodynamics of Alicapistat, a Selective Inhibitor of Human Calpains 1 and 2 for the Treatment of Alzheimer Disease: An Overview of Phase 1 Studies. Clin Pharmacol Drug Dev 8, 290–303. 10.1002/cpdd.598 [DOI] [PubMed] [Google Scholar]
- 42.Hannan K and McKinney W (2020) An Overview of Current Clinical Trials of Agents for the Treatment and Prevention of COVID-19 in the United States. The University of Louisville Journal of Respiratory Infections 4. 10.18297/jri/vol4/iss1/49 [DOI] [Google Scholar]
- 43.Suzuki R et al. (2014) Degeneration and dysfunction of retinal neurons in acute ocular hypertensive rats: involvement of calpains. J Ocul Pharmacol Ther 30, 419–428. 10.1089/jop.2013.0100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozaki T et al. (2013) Inhibitory peptide of mitochondrial μ-calpain protects against photoreceptor degeneration in rhodopsin transgenic S334ter and P23H rats. PLoS One 8, e71650. 10.1371/journal.pone.0071650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukiage C et al. (1997) SJA6017, a newly synthesized peptide aldehyde inhibitor of calpain: amelioration of cataract in cultured rat lenses. Biochim Biophys Acta 1361, 304–312. 10.1016/s0925-4439(97)00043-4 [DOI] [PubMed] [Google Scholar]
- 46.Tamada Y et al. (2001) Calpain inhibitor, SJA6017, reduces the rate of formation of selenite cataract in rats. Curr Eye Res 22, 280–285. 10.1076/ceyr.22.4.280.5505 [DOI] [PubMed] [Google Scholar]
- 47.Robertson LJ et al. (2005) Calpain may contribute to hereditary cataract formation in sheep. Invest Ophthalmol Vis Sci 46, 4634–4640. 10.1167/iovs.04-1291 [DOI] [PubMed] [Google Scholar]
- 48.Tamada Y et al. (2005) Proteolysis of neuronal cytoskeletal proteins by calpain contributes to rat retinal cell death induced by hypoxia. Brain Res 1050, 148–155. 10.1016/j.brainres.2005.05.048 [DOI] [PubMed] [Google Scholar]
- 49.Nakajima E et al. (2006) Calpain-specific proteolysis in primate retina: Contribution of calpains in cell death. Invest Ophthalmol Vis Sci 47, 5469–5475. 10.1167/iovs.06-0567 [DOI] [PubMed] [Google Scholar]
- 50.Shirasaki Y et al. (2006) Retinal penetration of calpain inhibitors in rats after oral administration. J Ocul Pharmacol Ther 22, 417–424. 10.1089/jop.2006.22.417 [DOI] [PubMed] [Google Scholar]
- 51.Trifunović D et al. (2012) Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr Mol Med 12, 598–612. 10.2174/156652412800620048 [DOI] [PubMed] [Google Scholar]
- 52.Pietsch M et al. (2010) Calpains: attractive targets for the development of synthetic inhibitors. Curr Top Med Chem 10, 270–293. 10.2174/156802610790725489 [DOI] [PubMed] [Google Scholar]
- 53.Carragher NO (2006) Calpain inhibition: a therapeutic strategy targeting multiple disease states. Curr Pharm Des 12, 615–638. 10.2174/138161206775474314 [DOI] [PubMed] [Google Scholar]
- 54.Donkor IO (2011) Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin Ther Pat 21, 601–636. 10.1517/13543776.2011.568480 [DOI] [PubMed] [Google Scholar]
- 55.Velez G et al. (2020) Structural Insights into the Unique Activation Mechanisms of a Non-classical Calpain and Its Disease-Causing Variants. Cell Rep 30, 881–892.e885. 10.1016/j.celrep.2019.12.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wert KJ et al. (2019) CAPN5 genetic inactivation phenotype supports therapeutic inhibition trials. Hum Mutat 40, 2377–2392. 10.1002/humu.23894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ono Y and Sorimachi H (2012) Calpains: an elaborate proteolytic system. Biochim Biophys Acta 1824, 224–236. 10.1016/j.bbapap.2011.08.005 [DOI] [PubMed] [Google Scholar]
- 58.Sorimachi H et al. (2011) Impact of genetic insights into calpain biology. J Biochem 150, 23–37. 10.1093/jb/mvr070 [DOI] [PubMed] [Google Scholar]
- 59.Croall DE and Ersfeld K (2007) The calpains: modular designs and functional diversity. Genome Biol 8, 218. 10.1186/gb-2007-8-6-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell RL and Davies PL (2012) Structure-function relationships in calpains. Biochem J 447, 335–351. 10.1042/BJ20120921 [DOI] [PubMed] [Google Scholar]
- 61.Moldoveanu T et al. (2002) A Ca(2+) switch aligns the active site of calpain. Cell 108, 649–660. 10.1016/s0092-8674(02)00659-1 [DOI] [PubMed] [Google Scholar]
- 62.Corbalán-García S and Gómez-Fernández JC (2010) The C2 domains of classical and novel PKCs as versatile decoders of membrane signals. Biofactors 36, 1–7. 10.1002/biof.68 [DOI] [PubMed] [Google Scholar]
- 63.Rizo J and Südhof TC (1998) C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem 273, 15879–15882. 10.1074/jbc.273.26.15879 [DOI] [PubMed] [Google Scholar]
- 64.Ohno S et al. (1986) Nucleotide sequence of a cDNA coding for the small subunit of human calcium-dependent protease. Nucleic Acids Res 14, 5559. [PMC free article] [PubMed] [Google Scholar]
- 65.Hastings MH et al. (2018) The zinc fingers of the small optic lobes calpain bind polyubiquitin. J Neurochem 146, 429–445. 10.1111/jnc.14473 [DOI] [PubMed] [Google Scholar]
- 66.Low KE et al. (2016) Rational Design of Calpain Inhibitors Based on Calpastatin Peptidomimetics. J Med Chem 59, 5403–5415. 10.1021/acs.jmedchem.6b00267 [DOI] [PubMed] [Google Scholar]
- 67.Hanna RA et al. (2008) Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 456, 409–412. 10.1038/nature07451 [DOI] [PubMed] [Google Scholar]
- 68.Bondada V et al. (2021) The C2 domain of calpain 5 contributes to enzyme activation and membrane localization. Biochim Biophys Acta Mol Cell Res 1868, 119019. 10.1016/j.bbamcr.2021.119019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bruening J et al. (2018) Hepatitis C virus enters liver cells using the CD81 receptor complex proteins calpain-5 and CBLB. PLoS Pathog 14, e1007111. 10.1371/journal.ppat.1007111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tonami K et al. (2007) Calpain 6 is involved in microtubule stabilization and cytoskeletal organization. Mol Cell Biol 27, 2548–2561. 10.1128/MCB.00992-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zha C et al. (2020) Biallelic variants in the small optic lobe calpain CAPN15 are associated with congenital eye anomalies, deafness and other neurodevelopmental deficits. Hum Mol Genet 29, 3054–3063. 10.1093/hmg/ddaa198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zha C et al. (2021) MRI of Capn15 Knockout Mice and Analysis of Capn 15 Distribution Reveal Possible Roles in Brain Development and Plasticity. Neuroscience 465, 128–141. 10.1016/j.neuroscience.2021.04.023 [DOI] [PubMed] [Google Scholar]
- 73.Yang Y et al. (2018) Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Brief Bioinform 19, 482–494. 10.1093/bib/bbw129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dill KA et al. (2008) The protein folding problem. Annu Rev Biophys 37, 289–316. 10.1146/annurev.biophys.37.092707.153558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carpentier M and Chomilier J (2019) Protein Multiple Alignments: Sequence-based vs Structure-based Programs. Bioinformatics. 10.1093/bioinformatics/btz236 [DOI] [PubMed] [Google Scholar]
- 76.Sun YJ et al. (2021) Structure-based phylogeny identifies avoralstat as a TMPRSS2 inhibitor that prevents SARS-CoV-2 infection in mice. J Clin Invest 131. 10.1172/JCI147973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sneath PHA and Sokal RR (1973) Unweighted Pair Group Method with Arithmetic Mean. In Numerical taxonomy. The principles and practice of numerical classification., pp. 230–234, San Francisco, W.H. Freeman and Company. [Google Scholar]
- 78.Gakhar L et al. (2016) Small-angle X-ray scattering of calpain-5 reveals a highly open conformation among calpains. J Struct Biol 196, 309–318. 10.1016/j.jsb.2016.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schaefer KA et al. (2016) Calpain-5 Expression in the Retina Localizes to Photoreceptor Synapses. Invest Ophthalmol Vis Sci 57, 2509–2521. 10.1167/iovs.15-18680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Menon M et al. (2019) Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat Commun 10, 4902. 10.1038/s41467-019-12780-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan W et al. (2020) Cell Atlas of The Human Fovea and Peripheral Retina. Sci Rep 10, 9802. 10.1038/s41598-020-66092-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakajima T et al. (2001) Different expression patterns for ubiquitous calpains and Capn3 splice variants in monkey ocular tissues. Biochim Biophys Acta 1519, 55–64. 10.1016/s0167-4781(01)00212-3 [DOI] [PubMed] [Google Scholar]
- 84.Skeie JM et al. (2015) Proteomic insight into the molecular function of the vitreous. PLoS One 10, e0127567. 10.1371/journal.pone.0127567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Velez G et al. (2018) Proteomic analysis of the human retina reveals region-specific susceptibilities to metabolic- and oxidative stress-related diseases. PLoS One 13, e0193250. 10.1371/journal.pone.0193250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mustafi D et al. (2019) Retinal Gene Distribution and Functionality Implicated in Inherited Retinal Degenerations Can Reveal Disease-Relevant Pathways for Pharmacologic Intervention. Pharmaceuticals (Basel) 12. 10.3390/ph12020074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Power MJ et al. (2020) Systematic spatiotemporal mapping reveals divergent cell death pathways in three mouse models of hereditary retinal degeneration. J Comp Neurol 528, 1113–1139. 10.1002/cne.24807 [DOI] [PubMed] [Google Scholar]
- 88.Sothilingam V et al. (2015) Retinitis pigmentosa: impact of different Pde6a point mutations on the disease phenotype. Hum Mol Genet 24, 5486–5499. 10.1093/hmg/ddv275 [DOI] [PubMed] [Google Scholar]
- 89.Power M et al. (2020) Cellular mechanisms of hereditary photoreceptor degeneration - Focus on cGMP. Prog Retin Eye Res 74, 100772. 10.1016/j.preteyeres.2019.07.005 [DOI] [PubMed] [Google Scholar]
- 90.Kutluer M et al. (2020) Targeting molecular pathways for the treatment of inherited retinal degeneration. Neural Regen Res 15, 1784–1791. 10.4103/1673-5374.280303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Comitato A et al. (2014) Activation of Bax in three models of retinitis pigmentosa. Invest Ophthalmol Vis Sci 55, 3555–3562. 10.1167/iovs.14-13917 [DOI] [PubMed] [Google Scholar]
- 92.Mahajan VB and Lin JH (2013) Lymphocyte infiltration in CAPN5 autosomal dominant neovascular inflammatory vitreoretinopathy. Clin Ophthalmol 7, 1339–1345. 10.2147/OPTH.S46450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Coomer CE and Morris AC (2018) Capn5 Expression in the Healthy and Regenerating Zebrafish Retina. Invest Ophthalmol Vis Sci 59, 3643–3654. 10.1167/iovs.18-24278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schaefer K et al. (2017) Calpain-5 gene expression in the mouse eye and brain. BMC Res Notes 10, 602. 10.1186/s13104-017-2927-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krizaj D and Copenhagen DR (2002) Calcium regulation in photoreceptors. Front Biosci 7, d2023–2044. 10.2741/A896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wert KJ et al. (2014) Functional validation of a human CAPN5 exome variant by lentiviral transduction into mouse retina. Hum Mol Genet 23, 2665–2677. 10.1093/hmg/ddt661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nelson NG et al. (2014) CAPN5 gene silencing by short hairpin RNA interference. BMC Res Notes 7,642. 10.1186/1756-0500-7-642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Y et al. (2017) An anti-CAPN5 intracellular antibody acts as an inhibitor of CAPN5-mediated neuronal degeneration. Oncotarget 8, 100312–100325. 10.18632/oncotarget.22221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dutt P et al. (2006) m-Calpain is required for preimplantation embryonic development in mice. BMC Dev Biol 6, 3. 10.1186/1471-213X-6-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Delaney SJ et al. (1991) Molecular cloning and analysis of small optic lobes, a structural brain gene of Drosophila melanogaster. Proc Natl Acad Sci U S A 88, 7214–7218. 10.1073/pnas.88.16.7214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kamei M et al. (1998) SOLH, a human homologue of the Drosophila melanogaster small optic lobes gene is a member of the calpain and zinc-finger gene families and maps to human chromosome 16p13.3 near CATM (cataract with microphthalmia). Genomics 51, 197–206. 10.1006/geno.1998.5395 [DOI] [PubMed] [Google Scholar]
- 102.Koscielny G et al. (2014) The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res 42, D802–809. 10.1093/nar/gkt977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Eppig JT (2017) Mouse Genome Informatics (MGI) Resource: Genetic, Genomic, and Biological Knowledgebase for the Laboratory Mouse. ILAR J 58, 17–41. 10.1093/ilar/ilx013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biswas S et al. (2005) Calpains: enzymes of vision? Med Sci Monit 11, RA301–310 [PubMed] [Google Scholar]
- 105.Morton JD et al. (2013) A macrocyclic calpain inhibitor slows the development of inherited cortical cataracts in a sheep model. Invest Ophthalmol Vis Sci 54, 389–395. 10.1167/iovs.12-11088 [DOI] [PubMed] [Google Scholar]
- 106.Lee HY et al. (2008) Evaluation of a novel calpain inhibitor as a treatment for cataract. Clin Exp Ophthalmol 36, 852–860. 10.1111/j.1442-9071.2009.01925.x [DOI] [PubMed] [Google Scholar]
- 107.Lee HY et al. (2008) The involvement of calpains in opacification induced by Ca2+-overload in ovine lens culture. Vet Ophthalmol 11, 347–355. 10.1111/j.1463-5224.2008.00655.x [DOI] [PubMed] [Google Scholar]
- 108.Fougerousse F et al. (2000) Human-mouse differences in the embryonic expression patterns of developmental control genes and disease genes. Hum Mol Genet 9, 165–173. 10.1093/hmg/9.2.165 [DOI] [PubMed] [Google Scholar]
- 109.Yau JW et al. (2012) Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35, 556–564. 10.2337/dc11-1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shanab AY et al. (2012) Metabolic stress response implicated in diabetic retinopathy: the role of calpain, and the therapeutic impact of calpain inhibitor. Neurobiol Dis 48, 556–567. 10.1016/j.nbd.2012.07.025 [DOI] [PubMed] [Google Scholar]
- 111.Du Y et al. (2013) Photoreceptor cells are major contributors to diabetes-induced oxidative stress and local inflammation in the retina. Proc Natl Acad Sci U S A 110, 16586–16591. 10.1073/pnas.1314575110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gao BB et al. (2008) Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res 7, 2516–2525. 10.1021/pr800112g [DOI] [PubMed] [Google Scholar]
- 113.Azuma M et al. (2004) Involvement of calpain isoforms in retinal degeneration in WBN/Kob rats. Comp Med 54, 533–542 [PubMed] [Google Scholar]
- 114.Quigley HA and Broman AT (2006) The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 90, 262–267. 10.1136/bjo.2005.081224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ishikawa M et al. (2015) Experimentally Induced Mammalian Models of Glaucoma. Biomed Res Int 2015, 281214. 10.1155/2015/281214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Choudhury S et al. (2015) Caspase-7: a critical mediator of optic nerve injury-induced retinal ganglion cell death. Mol Neurodegener 10, 40. 10.1186/s13024-015-0039-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Agudo M et al. (2009) Immediate upregulation of proteins belonging to different branches of the apoptotic cascade in the retina after optic nerve transection and optic nerve crush. Invest Ophthalmol Vis Sci 50, 424–431. 10.1167/iovs.08-2404 [DOI] [PubMed] [Google Scholar]
- 118.Stankowska DL et al. (2018) Neuroprotective effects of inhibitors of Acid-Sensing ion channels (ASICs) in optic nerve crush model in rodents. Curr Eye Res 43, 84–95. 10.1080/02713683.2017.1383442 [DOI] [PubMed] [Google Scholar]
- 119.Zhang J et al. (2019) Silicone oil-induced ocular hypertension and glaucomatous neurodegeneration in mouse. Elife 8. 10.7554/eLife.45881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nakajima E et al. (2017) Contribution of Calpain and Caspases to Cell Death in Cultured Monkey RPE Cells. Invest Ophthalmol Vis Sci 58, 5412–5420. 10.1167/iovs.17-22325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kobayashi-Otsugu M et al. (2020) Activation of Cytosolic Calpain, Not Caspase, Is Underlying Mechanism for Hypoxic RGC Damage in Human Retinal Explants. Invest Ophthalmol Vis Sci 61, 13. 10.1167/iovs.61.13.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tamada Y et al. (2007) Contribution of calpain to cellular damage in human retinal pigment epithelial cells cultured with zinc chelator. Curr Eye Res 32, 565–573. 10.1080/02713680701359633 [DOI] [PubMed] [Google Scholar]
- 123.Koriyama Y et al. (2014) Heat shock protein 70 induction by valproic acid delays photoreceptor cell death by N-methyl-N-nitrosourea in mice. J Neurochem 130, 707–719. 10.1111/jnc.12750 [DOI] [PubMed] [Google Scholar]
- 124.Viringipurampeer IA et al. (2019) Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Mol Neurobiol 56, 1637–1652. 10.1007/s12035-018-1192-8 [DOI] [PubMed] [Google Scholar]
- 125.Tompa P et al. (2004) On the sequential determinants of calpain cleavage. J Biol Chem 279, 20775–20785. 10.1074/jbc.M313873200 [DOI] [PubMed] [Google Scholar]
- 126.Dahlqvist-Edberg U and Ekman P (1981) Purification of a Ca2+-activated protease from rat erythrocytes and its possible effect on pyruvate kinase in vivo. Biochim Biophys Acta 660, 96–101. 10.1016/0005-2744(81)90113-3 [DOI] [PubMed] [Google Scholar]
- 127.Azarian SM et al. (1995) Selective proteolysis of arrestin by calpain. Molecular characteristics and its effect on rhodopsin dephosphorylation. J Biol Chem 270, 24375–24384. 10.1074/jbc.270.41.24375 [DOI] [PubMed] [Google Scholar]
- 128.Roberts-Lewis JM et al. (1994) Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci 14, 3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Watt F and Molloy PL (1993) Specific cleavage of transcription factors by the thiol protease, m-calpain. Nucleic Acids Res 21, 5092–5100. 10.1093/nar/21.22.5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gil-Parrado S et al. (2002) Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem 277, 27217–27226. 10.1074/jbc.M202945200 [DOI] [PubMed] [Google Scholar]
- 131.Parsons DE et al. (2021) Peptidomimetics Therapeutics for Retinal Disease. Biomolecules 11. 10.3390/biom11030339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Azuma M and Shearer TR (2008) The role of calcium-activated protease calpain in experimental retinal pathology. Surv Ophthalmol 53, 150–163. 10.1016/j.survophthal.2007.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen J and Attar M (2015) Antibody-Drug Conjugate (ADC) Research in Ophthalmology--a Review. Pharm Res 32, 3572–3576. 10.1007/s11095-015-1728-9 [DOI] [PubMed] [Google Scholar]
- 134.Nakajima E et al. (2014) Activation of the mitochondrial caspase pathway and subsequent calpain activation in monkey RPE cells cultured under zinc depletion. Eye (Lond) 28, 85–92. 10.1038/eye.2013.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang Y et al. (2017) Inhibition of Starvation-Triggered Endoplasmic Reticulum Stress, Autophagy, and Apoptosis in ARPE-19 Cells by Taurine through Modulating the Expression of Calpain-1 and Calpain-2. Int J Mol Sci 18. 10.3390/ijms18102146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hirji N et al. (2018) Achromatopsia: clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet 39, 149–157. 10.1080/13816810.2017.1418389 [DOI] [PubMed] [Google Scholar]
- 137.Chang B et al. (2009) A homologous genetic basis of the murine cpfl1 mutant and human achromatopsia linked to mutations in the PDE6C gene. Proc Natl Acad Sci U S A 106, 19581–19586. 10.1073/pnas.0907720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Trifunović D et al. (2010) cGMP-dependent cone photoreceptor degeneration in the cpfl1 mouse retina. J Comp Neurol 518, 3604–3617. 10.1002/cne.22416 [DOI] [PubMed] [Google Scholar]
- 139.Sanges D et al. (2006) Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc Natl Acad Sci U S A 103, 17366–17371. 10.1073/pnas.0606276103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paquet-Durand F et al. (2006) Calpain is activated in degenerating photoreceptors in the rd1 mouse. J Neurochem 96, 802–814. 10.1111/j.1471-4159.2005.03628.x [DOI] [PubMed] [Google Scholar]
- 141.Newton F and Megaw R (2020) Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes (Basel) 11. 10.3390/genes11101120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ozaki T et al. (2012) Intravitreal injection or topical eye-drop application of a μ-calpain C2L domain peptide protects against photoreceptor cell death in Royal College of Surgeons’ rats, a model of retinitis pigmentosa. Biochim Biophys Acta 1822, 1783–1795. 10.1016/j.bbadis.2012.07.018 [DOI] [PubMed] [Google Scholar]
- 143.Das S et al. (2022) Redefining the role of Ca. Cell Death Dis 13, 47. 10.1038/s41419-021-04482-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beryozkin A et al. (2021) A new mouse model for retinal degeneration due to Fam161a deficiency. Sci Rep 11, 2030. 10.1038/s41598-021-81414-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dannhausen K et al. (2018) Microglia Analysis in Retinal Degeneration Mouse Models. Methods Mol Biol 1753, 159–166. 10.1007/978-1-4939-7720-8_10 [DOI] [PubMed] [Google Scholar]
- 146.Zhou HS et al. (2004) Challenges and strategies: the immune responses in gene therapy. Med Res Rev 24, 748–761. 10.1002/med.20009 [DOI] [PubMed] [Google Scholar]
- 147.Keino H et al. (2018) Immune Privilege and Eye-Derived T-Regulatory Cells. J Immunol Res 2018, 1679197. 10.1155/2018/1679197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Prado DA et al. (2020) Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease. Curr Opin Ophthalmol 31, 147–154. 10.1097/ICU.0000000000000660 [DOI] [PubMed] [Google Scholar]
- 149.Velez G et al. (2018) A novel de novo. Cold Spring Harb Mol Case Stud 4. 10.1101/mcs.a002519 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.