

Abstract
Type 2 diabetic hearts are more vulnerable to myocardial ischemia reperfusion (MIR) injury, which involves decreased mitophagy status with unknown mechanisms. MitoQ, a mitochondria-targeted antioxidant, has been shown to have protection against ischemia reperfusion injury through upregulating mitophagy. The aim of this study was to investigate the effects of MitoQ on myocardium during MIR injury in type 2 diabetes (T2D). Herein, this study discovered that type 2 diabetic hearts with PINK1/Parkin downregulation suffered more MIR injury accompanied by reduced mitophagy. Treatment with MitoQ significantly decreased the levels of CK-MB, LDH, myocardial infarction, myocardial pathological damage, and cardiomyocytes apoptosis, while it improved cardiac function, mitophagy status, and PINK1/Parkin pathway in vivo study. Furthermore, MitoQ significantly reduced high glucose/high fat and hypoxia/reoxygenation induced injury in H9C2 cells as evidenced by reduced cardiomyocytes apoptosis and ROS production, and increased cell viability, the level of mitochondrial membrane potential, PINK1/Parkin expression. However, mitochondrial division inhibitor (mdivi-1), an inhibitor of mitophagy, reversed the improvement and protein expression levels of PINK1/Parkin pathway in vitro models. In conclusion, MIR induced more severe damage in T2D by reduction of mitophagy. MitoQ can confer cardioprotection following MIR in T2D by mitophagy up-regulation via PINK1/Parkin pathway.
Keywords: Type 2 diabetes, Myocardium, Ischemia reperfusion, Mitophagy, MitoQ
Introduction
Diabetes is one of the most influential and representative chronic metabolic diseases; its global incidence has been estimated to be 9.3% (463 million people), and the prevalence will rise to 10.2% (578 million) by 2030 and 10.9% (700 million) by 2045 (Saeedi et al. 2019). Type 2 diabetes (T2D) accounts for more than 90% of patients with diabetes, and patients with T2D are more vulnerable to cardiovascular complications, causing considerable psychological and physical distress to patients and caregivers (Packer 2018). Among the complications, ischemic heart disease, especially acute myocardial infarction (AMI), remains a leading cause of disability and mortality in diabetic patients (Bragg et al. 2017). After coronary revascularization, the diabetic heart with AMI is more likely to get damage than in the nondiabetic population, which may be caused by impaired mitophagy (Guan et al. 2019).
Mitophagy is a specific autophagy that clears damaged mitochondria, playing an important role in maintaining mitochondrial homeostasis and cell survival (Shefa et al. 2019). Numerous studies have demonstrated that mitochondrial dysfunction is the basis of many epidemic diseases, including heart disease caused by myocardial ischemia/reperfusion (MIR) injury (Morales et al. 2019; W. Zhang et al. 2016). Various intracellular signaling pathways were proven to participate in regulating mitophagy, including PINK1/Parkin, FUNDC1, BNIP3, and BNIP3L/NIX signaling pathway (M. Yang et al. 2019). Furthermore, increasing evidence suggest that PINK1/Parkin pathway is well known to be involved in the process of MIR, but they were impaired in diabetes (Xiao et al. 2017; Y. Zhang et al. 2019). Thus, it is a promising way to attenuate MIR injury in diabetes by restoring PINK1/Parkin-mediated mitophagy.
The mitochondria-targeted antioxidant MitoQ, a compound obtained by the covalent combination of the endogenous antioxidant coenzyme Q10 and triphenylphosphine, has been widely used in the prevention and treatment of many diseases (Rodriguez-Cuenca et al. 2010). Previous studies have shown that MitoQ attenuates the damage of liver during the period of IR (Mukhopadhyay et al. 2012). Moreover, it should be noted that the protective effects of MitoQ were found in diabetic hearts of rats (Escribano-Lopez et al. 2016; Mackenzie et al. 2013). Furthermore, Xiao et al. (Xiao et al. 2017) reported that MitoQ ameliorated tubular injury mediated by mitophagy, as was evident from increased levels of PINK1 and Parkin. Therefore, further studies are necessary to better define whether MitoQ ameliorates MIR injury by restoring mitophagy in STZ-induced type 2 diabetic rats via PINK1/Parkin.
In this study, STZ and high-fat diet (HFD)-induced T2D and MIR models were established, and we aimed to assess if MitoQ could ameliorate MIR injury in type 2 diabetic rats by up-regulation of mitophagy via PINK1/Parkin pathway.
Methods
Experimental animals
The protocol of this experiment was in compliance with the Principles of Laboratory Animal Care of Wuhan University and was approved by the Committee for the Use of Live Animals in Teaching and Research. Seventy male Sprague–Dawley (SD) rats, which were in SPF level (aged 4–5 weeks and obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd.), were housed in an environment with a maintained temperature of 22 ℃, relative humidity of 50 ± 10%, and a fixed light/dark schedule (12 h light/12 h dark). All rats were given free access to standard chow and water.
Diabetes and MIR model
After 7 days of acclimatization, the rats were randomly divided into T2D rats and normal rats. From weeks 2 to 14, normal rats were fed a standard normal chow diet and the T2D rats were fed an HFD consisting of 66.5% normal diet, 20% sucrose, 10% lard, 2.5% cholesterol, and 1% sodium cholate (Zeng et al. 2019). On the first day of week 6, all rats were fasted for 12 h for diabetes induction as normal (Qiu et al. 2021), which could enhance the injury of pancreatic β-cells with STZ. T2D rats were induced by a single intraperitoneal (i.p.) injection of 35 mg/kg STZ (Sigma, S0130-1G) dissolved in freshly prepared citrate buffer (0.1 mol/L, pH 4.5) at a dose of 40 mg/kg, while rats in the nondiabetic group were injected with an equal volume of sodium citrate buffer (X. X. Li et al. 2019). Rats on HFD with low STZ were recognized as T2D rats, while the fasting blood glucose (FBG) level reached 16.7 mM after 1 week of STZ injection, at the same time normal rats received an equivalent volume of citrate buffer. 8 weeks after diabetes induction, the MIR injury model was established as previously described (Leng et al. 2018; Wu et al. 2017). Briefly, the rats were anesthetized by i.p. injection of pentobarbital sodium (50 mg/kg), while subjected to tracheotomy and artificial ventilation with an animal ventilator after endotracheal intubation. Electrocardiogram and heart rate (HR) were continuously monitored by an electrophysiolograph (BioPAC, MH150, USA). A thoracotomy was performed at the fourth intercostal space of the left subclavian midline to expose the heart, and then MIR was achieved by occluding the left anterior descending coronary artery (LAD) for 45 min followed by reperfusion for 2 h with a 6–0 silk suture. The sham-operated groups underwent the same surgical procedures without occlusion in the corresponding location. To keep the operation stable and safe, we used heat lamps and a heating pad. A gauze with warm saline was taken to cover rats to avoid loss of temperature and moisture. Successful MIR was confirmed by observing the pale myocardium in the left ventricle beneath the suture, and the electrocardiogram showed an elevated ST segment. After the operation, animals were sacrificed to obtain blood and tissue samples for the subsequent experiments.
Experimental protocols
Eight weeks after STZ injection, both T2D and normal rats were randomly divided into four groups (eight in each group) as follows: (1) normal shame (NS), (2) normal IR (NIR), (3) diabetic shame (DS), and (4) diabetic IR(DIR). Besides, to determine the cardioprotection of MitoQ, another protocol was under performance as the following groups: (1) DM + sham group (DS), (2) DM + I/R group (DIR), and (3) diabetic IR + MitoQ (MitoQ). In the MitoQ group, MitoQ (2.8 mg/kg; added as MitoQ adsorbed to β-cyclodextran in 100 μl 0.9% saline) was injected into the tail vein 15 min before the onset of ischemia (Liu et al. 2018). For the in vitro study, H9C2 cardiomyocytes were randomly divided into the three groups as follows: (1) high-glucose/high-fat control (HG/HF) (30 mM glucose, palmitate 300 μM); (2) high-glucose/high-fat culture followed by hypoxia-reoxygenation (HR) (HG/HF-HR); (3) high-glucose/high-fat culture followed by HR pretreated with MitoQ (HG/HF-HR + MitoQ) (MitoQ, 1 µM, Focus Biomolecules, 10–3914) (Kelso et al. 2001); and (4) high-glucose/high-fat culture followed by HR pretreated with MitoQ and mitochondrial division inhibitor (mdivi-1) (mdivi-1, 50 µM, Abcam, ab144589) (Wendt et al. 2020). Rat cardiomyocyte-derived H9C2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco Laboratories) containing 10% fetal bovine serum (FBS) (Gibco Laboratories) and 100 μg/ml penicillin/streptomycin (Gibco Laboratories) at 37 °C in a humidified atmosphere of 10% CO2. To simulate the condition of T2D patients suffering MIR injury, rat cardiomyocyte-derived H9C2 cells were firstly cultured with high glucose/high fat(He et al. 2018). When the density of cells reached 60–70% confluence, they were exposed to HG conditions for 24 h and HR were established by 4 h of hypoxia (94% N2 and 5% CO2) and 2 h reoxygenation to simulate the process of MIR as described previously (Leng et al. 2018). MitoQ was administered 2 h before HR, and mdivi-1 was managed 24 h before HR.
Cardiac function evaluation
For the in vivo study, 8 weeks after diabetic condition, rats were anesthetized with pentobarbital sodium and fastened in the supine position. Then, data were collected at 120 min reperfusion. At the beginning of the operation, a heparin-saline-filled catheter was inserted into the left ventricle through an incision in the right common carotid artery, and the other end of the catheter was connected to a pressure sensor (Yixinda, Shenzhen, China). Left ventricular systolic blood pressure (LVSP) and LVSP maximum increase and decrease rates (±dp/dtmax) were monitored by an electrophysiological instrument (BioPAC). Finally, AcqKnowledge 4.0 software was used for data processing.
Measurement of CK-MB and LDH
At the end of the operation, arterial blood samples were collected and then centrifuged at 2000 rpm for 10 min to measure the level of serum creatine kinase-MB (CK-MB) and lactate dehydrogenase (LDH) with a CK-MB assay kit (Jiancheng, Nanjing, China) and LDH assay kit (Jiancheng, Nanjing, China).
Determination of myocardial infarct size
After reperfusion, six rats were selected randomly from the respective group to measure the myocardial infarct size with 2 ml of 2% Evans Blue dye (Sigma, E2129-10G) and 2,3,5-triphenyltetrazolium chloride staining (Sigma, T8877-5G) as described previously (Leng et al. 2018; Wu et al. 2017). The infarct size was determined by using an image analysis system (Image-Pro plus; Media Cybernetics). The blue area was normal myocardium, red indicated ischemic myocardium, pale denoted myocardial infarction, and the infarct size and the percentage of myocardial area at risk were calculated.
Transmission electron microscopy
After reperfusion, approximately 1 mm3 tissue of the left ventricle from each heart was collected and fixed in 2.5% glutaraldehyde for 6 h as described by Qiu et al. (2017). The samples were prepared by professional teachers of the Electron Microscopy Centre of Renmin Hospital of Wuhan University and then detected by TEM (TEM, HT7700, Japan).
Histopathological analysis
After reperfusion, a part of the left ventricular tissue was taken to be washed with 4 ℃ sterile normal saline, fixed and dehydrated in 4% paraformaldehyde for 24 h. Then, it was embedded in paraffin and sliced into 4-µm-thick sections which were stained with hematoxylin and eosin. Finally, histopathological analysis was performed under light microscopy.
Determination of apoptosis
In vivo study, the rate of myocardial apoptosis was evaluated by a Colorimetric TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, China). After operation, a small part of the ventricle was taken and incubated with 4% paraformaldehyde overnight at room temperature, and TUNEL staining was performed according to the manufacturer instruction of this kit. In in vitro study, the apoptosis rate of H9C2 cells was assessed with a PE Annexin V Apoptosis Detection Kit (BD Biosciences, USA). After HR, cells were collected and resuspended in binding buffer. Then, PE Annexin V and 7-amino-actinomycin(7-AAD) were used to incubate the cells in the dark for 15 min. Cellular fluorescence was measured with a CytoFLEX instrument (BD Biosciences, USA), and the data were analyzed with the Cell Quest Pro software (BD Biosciences, USA).
ROS measurement
Dichloro-dihydro-fluorescein diacetate (DCFH-DA) assay (Sigma, USA) was used to measure the intracellular level of ROS. In short, cells were incubated with 50 μM DCFH-DA probes for 30 min at 37 °C in the dark. Then, fluorescence images were recorded by a fluorescence microscope (Olympus IX51) after washing the cells twice with cold PBS.
Mitochondrial membrane potential measurement
Mitochondrial membrane potential of H9C2 was measured with a JC-1 Kit (Beyotime, China). After HR, cells were incubated with JC-1 staining working solution for 20 min at 37 °C. Then JC-1 staining buffer was used to wash the cells for twice, and the red/green fluorescence was detected with a fluorescence microscope (Olympus Bx 50-FLA).
Cell viability assay
Cell viability was evaluated with a cell counting kit-8 (CCK-8) (Dojindo, Kumamoto, Japan). After treatment, H9C2 cells were incubated with CCK-8 solution for 3 h. The microplate reader was used to assess the percentage of viable cells.
Western blot analysis
The expressions of PINK, Parkin, P62, and LC3II/I in left ventricular tissue or cells were determined by Western blot. After 2 h reperfusion or reoxygenation, the tissue or cell was homogenized with a mixture of RIPA lysis buffer, PMSF (Beyotime, China), and a protease inhibitor cocktail (Roche, USA), and then transferred to 1.5 ml tubes and centrifuged at 1.2 × 103 g for 15 min at 4 ℃. The supernatant was collected, and protein concentration was determined with BCA kit (Beyotime, China). Proteins were separated on a 10% SDS polyacrylamide gel and electrotransferred onto a PVDF membrane. The PVDF membrane was blocked with 5% milk for 1 h and incubated overnight at 4 ℃ with the appropriate primary antibodies, respectively (anti-PINK1, 1:1000, CST, 6946S; anti-Parkin, 1:1000, CST,4211S; anti-P62, 1:1000, CST, 88588S; anti-LC3II/I, 1:1000, CST, 2775S; anti-GAPDH, 1:1000, CST,5174S) diluted in 5% bovine serum albumin(BSA). Then, the membranes were washed by TBST for 3 times (5 min each) and incubated with HRP-labelled antibody (1:5000 goat anti-rabbit/mouse IgG(H + L), AntGene, China,ANT019, ANT020) for 1 h at room temperature. The protein bands were detected by Bio-Rad imaging system.
Statistical analysis
All results are expressed as the mean ± SD. Differences among experimental groups were evaluated by one-way ANOVA followed by the Tukey test. Statistical analysis was performed using R software (version 3.60) and GraphPad Prism 8.0 (GraphPad Software, USA). P < 0.05 was considered statistically significant.
Results
Characteristics of the experimental rats
After 8 weeks of induction of type 2 diabetes, the rats showed increased blood glucose, water intake, and food consumption, and decreased body weight as compared with the control rats (Table 1). Apart from these, diabetic symptoms, including hyperglycemia, polydipsia, polyphagia, and weight loss are fully presented in T2D rats (Table 1).
Table 1.
General characteristics of each group after 8 weeks
| Parameters/group | Non-T2D | T2D |
|---|---|---|
| Blood glucose (mM) | 5.98 ± 0.71 | 18.4 ± 1.23* |
| Body weight (g) | 438.3 ± 29.72 | 340.0 ± 25.68* |
| Water intake (ml/kg/day) | 121.4 ± 11.32 | 324.2 ± 30.10* |
| Food consumption (g/kg/day) | 61.75 ± 5.85 | 140.4 ± 13.56* |
The results are expressed as the mean ± SD
Non-T2D nondiabetic rats of type 2, T2D type 2 diabetic rats
*P < 0.05 versus non-T2D group; n = 12
T2D aggravated post-ischemic injury of myocardial tissues in rats suffered MIR
To investigate the effects of diabetes on MIR injury, we evaluated biochemical markers and cardiac function. Compared with the NS group, the levels of LDH (Fig. 1a) and CK-MB (Fig. 1b) in serum from the DS group significantly increased. At the time point of reperfusion for 120 min (R120), LVSP (Fig. 1c), + dP/dt (Fig. 1d), and − dP/dt (Fig. 1e) notably decreased in DS group. Higher serum levels of LDH (Fig. 1a), CK-MB (Fig. 1b), and larger infarct size (Fig. 1f) were exhibited in T2D rats suffered MIR than non-diabetic rats, accompanied with decreased levels of LVSP (Fig. 1c), + dP/dt (Fig. 1d), and − dP/dt (Fig. 1e) at the R120 time point. The data above suggest that T2D rats are more vulnerable to MIR injury.
Fig. 1.

T2D aggravated the degree of MIR in rats. NS, nondiabetic rats; DS, T2D rats; NIR and DIR, nondiabetic and T2D rats were subjected to 30 min of ischemia followed by 2 h of reperfusion. Serum levels of myocardial injury marker LDH (a) and CK-MB (b) were assessed with assay kits. Left ventricle hemodynamic parameters including LVSP (c), + dp/dt max (d), and − dp/dt max (e) were measured in each group. The infarct size was detected by TTC staining (f). All values are presented as the mean ± SD, n = 5/group. ∗∗P < 0.01; ∗P < 0.05; #P < 0.05 versus NIR group
T2D reduced the increased activity of PINK1/Parkin pathway caused by MIR
Meanwhile, we assessed the expression of PINK1, Parkin, P62, and the ratio of LC3II/I, which are associated with mitophagy. As shown in Fig. 2a–d, T2D decreased the expression of PINK1, Parkin, the ratio of LCC3II/I, and increased expression of P62 compared with nondiabetic controls at baseline. MIR increased the expression of PINK1 (Fig. 2a), Parkin (Fig. 2b), and the ratio of LC3II/I (Fig. 2c) in nondiabetic rats, accompanied with the decreased expression of P62 (Fig. 2d). The level of P62 and ratio of LC3II/I are typical markers to evaluate autophagy or mitophagy (Xu et al. 2020). However, the alteration caused by MIR was removed in T2D condition compared with nondiabetic condition in Fig. 2a–d.
Fig. 2.

Effects of MIR injury on expressions of PINK1, Parkin, ratio of LC3II/I, and P62 in nondiabetic and T2D rats. Representative myocardial PINK1 (a), Parkin (b), ratio of LC3II/I (c), and P62 (d) Western blots images and protein levels analysis. GAPDH served as the loading control. All values are expressed as the mean ± SD, n = 5/group. ∗P < 0.05 versus NS group; #P < 0.05 versus NIR group
Treatment with MitoQ improved cardiac function and reduced myocardial injury in T2D rats suffered MIR
We next investigated whether MitoQ could attenuate MIR injury in T2D rats. As shown in Fig. 3a–c, MitoQ treatment significantly attenuated the serum levels of LDH (Fig. 3a), CK-MB (Fig. 3b), and infarct size (Fig. 3c) as compared to the diabetic rats without treatment. Meanwhile, LVSP (Fig. 3d), + dP/dt (Fig. 3e), and − dP/dt (Fig. 3f) at the R120 time point were notably reversed in the MitoQ group. Besides, histopathological changes are well-established evidence for evaluating injury of heart. There were some necrotic cells, and the cell arrangement was relatively uniform in the DS group (Fig. 3g). After ischemia reperfusion, horizontal stripes and myocardial microstructure were more severe in the DIR group (Fig. 3g). Treatment with MitoQ largely attenuated the structural disorder of the myocardium (Fig. 3g). Moreover, cardiomyocytes apoptosis was another critical marker to determine the degree of myocardial tissue damage. Compared with the DS group, we found that MIR significantly increased cell apoptosis (Fig. 3h). MitoQ exhibited significantly decreased cell apoptosis compared with the DIR group (Fig. 3h). Treated with MitoQ, notable improvements were observed in these indicators, which suggested that MitoQ could attenuate the MIR injury in T2D rats.
Fig. 3.

Effects of MitoQ on MIR injury in T2D rats. NS, nondiabetic rats; DS, T2D rats; NIR and DIR, nondiabetic and T2D rats were subjected to 30 min of ischemia followed by 2 h of reperfusion; MitoQ, T2D rats were subjected to 30 min of ischemia followed by 2 h of reperfusion and were given MitoQ 15 min before ischemia. Serum levels of myocardial injury marker LDH (a) and CK-MB (b) were assessed with assay kits. The infarct size was detected by TTC staining (c). Left ventricle hemodynamic parameters including LVSP (d), + dp/dt max (e), and − dp/dt max (f) were measured in each group. The myocardial tissue structure and cell necrosis were observed by HE staining (g), scale bar 100 μm. Representative TUNEL staining images were observed (h), scale bar 100 μm. All values are presented as the mean ± SD, n = 5/group. ∗∗P < 0.01; ∗P < 0.05; #P < 0.05 versus DIR group
MitoQ conferred cardioprotection by up-regulation mitophagy via PINK1/Parkin pathway in T2D rats suffered MIR
To evaluate the mitophagy in cardiomyocytes, we used TEM to observe autophagosome-engulfing mitochondria, the indicator of mitophagy in myocardium. In the DS group, the structure and form of mitochondria were relatively normal and elongated cylindrical-shaped, whereas they were fragmented and swollen in the DIR group, which was partially reversed by the treatment of MitoQ (Fig. 4a). In the MitoQ group, we found some autophagosome-engulfing mitochondria in the cells, but there were rarely observed in the DIR and DS groups. Then, we measured the expression of PINK1, Parkin, P62, and the ratio of LC3II/I, which is represented by mitophagy. The expressions of PINK1, Parkin, and the ratio of LC3II/I and P62 were not significantly changing between the DS and DIR groups. There was a great increase in the levels of PINK1, Parkin, and the ratio of LC3II/I in the MitoQ group compared with the DIR group (Fig. 4b–e). Meanwhile, the level of P62 was decreased in the MitoQ group. These results suggest that MitoQ enhanced mitophagy by high expression of PINK1, Parkin, and the ratio of LC3II/I and low expression of P62.
Fig. 4.

Effects of MitoQ on mitophagy in T2D rats suffered MIR. Representative electron micrographs of each group (a): autophagosome-engulfing mitochondria ( →), mitochondrial fragmentation and swelling (►), Scale bar: 1 μm. Representative immune blot images and assessment of PINK1 (b), Parkin (c), ratio of LC3II/I (d), and P62 (e). All results are expressed as the mean ± SD, n = 5/group. ∗P < 0.05 versus DS group; #P < 0.05 versus DIR group
Mdivi-1 eliminated the cardioprotection against HG/HF-HR injury induced by MitoQ
Whether MitoQ could provide effective treatment was also tested in H9C2 cells during HG/HF-HR conditions. As shown in Fig. 5, HG/HF-HR conditions notably increased the apoptosis rate (Fig. 5a, b) but decreased the cell viability (Fig. 5c), the level of mitochondrial membrane potential (Fig. 5d), and ROS (Fig. 5e) in H9C2 cells. On the contrary, treatment with MitoQ remarkably recovered from HG/HF-HR injury, which indicated the protective effect of MitoQ. We further investigated whether the improvement of MitoQ was via up-regulation of mitophagy. Compared with the HG/HF-HR + MitoQ group, cell viability (Fig. 5c), the level of mitochondrial membrane potential (Fig. 5d), and ROS (Fig. 5e) were significantly decreased in the Mdivi-1 group, accompanying with increased apoptosis rate (Fig. 5a, b). These results collectively suggest that MitoQ confers cardioprotection against HG/HF-HR injury, and the improvement is removed by the mitophagy inhibitor mdivi-1, demonstrating the critical role of mitophagy in the process.
Fig. 5.
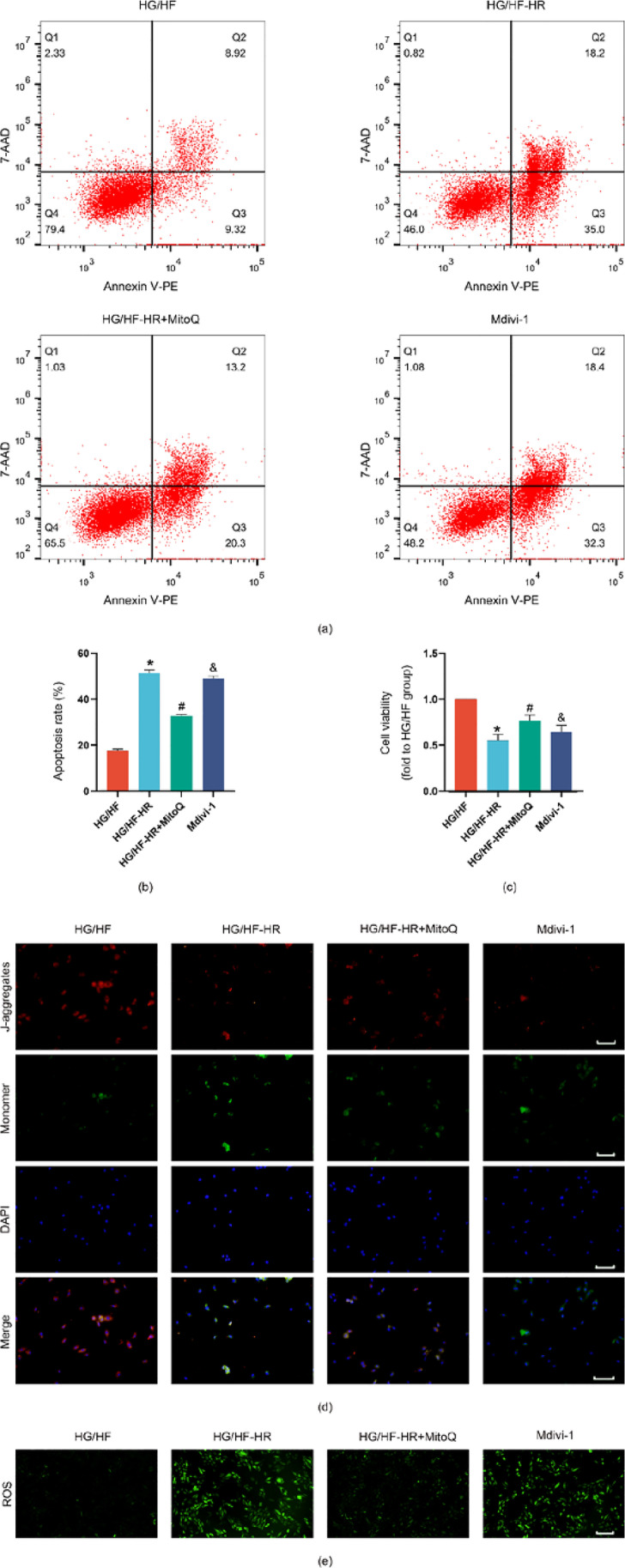
Effects of MitoQ and mdivi-1 on cellular injury, mitochondrial membrane potential, ROS apoptosis rats in H9C2 cells exposed to HG/HF-HR. HG/HF, high glucose and high fat (30 mM glucose, palmitate 300 μM); HG/HF-HR: H9C2 cardiomyocytes were cultured with high glucose and high fat and were subjected to 4 h of hypoxia followed by 2 h of reoxygenation; HG/HF-HR + MitoQ, cells were administrated with MitoQ 2 h before hypoxia; Mdivi-1, cells were administrated with MitoQ 2 h before hypoxia and mdivi-1 24 h before hypoxia. Apoptosis rate (a, b), cell viability (c), mitochondrial membrane potential (d), and ROS (e) were detected by representative kits, scale bar 100 μm. All values are presented as the mean ± SD, n = 5/group. ∗P < 0.05 versus HG/HF group; #P < 0.05 versus HG/HF-HR group; &P < 0.05 versus HG/HF-HR + MitoQ group
Effects of MitoQ and Mdivi-1 on PINK1/Parkin expression and mitophagy in H9C2 cells exposed to HG/HF-HR
Combined with the results of TEM, we next determined whether PINK1/Parkin pathway, which is involved in mitophagy, was critical in the process. There was no significant difference between HG and HG/HF-HR condition in PINK1 (Fig. 6a), Parkin, P62 (Fig. 6d) expression (Fig. 6b), and the ratio of LC3II/LC3I (Fig. 6c). Treatment with MitoQ remarkably activated the PINK1/Parkin signalling pathway in H9C2 cells with HG/HF-HR exposure, as illustrated by increased expression of PINK1, Parkin expression and the ratio of LC3II/LC3I and decreased P62 expression. The administration of mdivi-1 suppressed the relative protein expression of PINK1/Parkin pathway. By doing these above, we proved MitoQ could provide myocardial protection against MIR injury by upregulation mitophagy in T2D rats through PINK1/Parkin pathway.
Fig. 6.

Effects of MitoQ and mdivi-1 on expressions of PINK1, Parkin, ratio of LC3II/I and P62. Representative myocardial PINK1 (a), Parkin (b), ratio of LC3II/I (c) and P62 (d) Western blot images and protein level analysis. GAPDH served as the loading control. All values are expressed as the mean ± SD, n = 5/group. ∗P < 0.05 versus HG/HF group; #P < 0.05 versus HG/HF-HR group; &P < 0.05 versus HG/HF-HR + MitoQ group
Discussion
In this study, consistent with the pathological results, myocardial injury suffering MIR rats was found to be more severe in the NIR and DIR groups. Meanwhile, we demonstrated that decreased mitophagy in the diabetic heart is accompanied by decreased levels of PINK1 and Parkin. MitoQ attenuated MIR-induced cellular injury, which is implicated in the reduction of mitophagy in T2D and HG/HF-HR conditions. To the best of our knowledge, this was the first study to explore the mechanism of cardioprotection by MitoQ and examine the role of PINK1/Parkin pathway in mitophagy in this process.
As a kind of antioxidant targeted at mitochondria, MitoQ consists of ubiquinol, which is a natural antioxidant attached to lipophilic cations and its characteristics of lipophilicity and positive charge enable it to cross cell membranes and accumulate on the matrix facing the surface of the mitochondrial inner membrane to reduce mtROS properly (Rossman et al. 2018). Initially, MitoQ, as an antioxidant was widely used to reduce the level of ROS to play a protective role in organs (Skulachev 2005). With the researches of MitoQ deepen, Escribano et al. (Escribano-Lopez et al. 2016) had found that MitoQ could take action with NFκB to reduce the production of ROS, leukocyte-endothelium interactions, and TNFα, turning to anti-inflammatory and antioxidant action in the leukocytes of T2D patients to prevent cardiovascular events. Compared with other conventional antioxidants which may not confer effective clinically cardioprotection, MitoQ could exhibit beneficial effects in clinic by targeting at mitochondria (Rossman et al. 2018). Recently, the cardioprotection of MitoQ was found to attenuate the IR jury in nondiabetic heart by regulation of mitophagy (Hansson et al. 2015; Xiao et al. 2017). Meanwhile, the function of pancreatic β-cells and vascular endothelial in diabetes were found to recover with treatment of MitoQ (J. Yang et al. 2021a, b). Therefore, there are questions that whether MitoQ could also confer cardioprotection in T2D subjected to MIR and whether protection is associated with mitophagy. To confirm the hypothesis that MitoQ could provide protection against MIR injury in T2D, experiments in vivo and vitro were performed. Our in vivo study exhibited that both T2D and MIR could aggravate the injury in the myocardial tissue of rats by analyzing histopathological changes, cardiac function, serum levels of LDH and CK-MB, and apoptosis rate. MitoQ attenuated the MIR-induced myocardial injury in T2D rats. Similar results were found in our vitro studies with observations in the changes of cell viability, mitochondrial membrane potential, level of ROS, and apoptosis rate in H9C2 cells.
Mitophagy, a kind of selective autophagy, is a process of clearing damage or excessive mitochondria, activating the autophagosome-lysosome pathway under the stimulation of ischemia, hypoxia, oxidative stress, and aging to complete the degradation of damaged mitochondria and maintain the homeostasis of the cellular environment (Bueno et al. 2015). Mitochondrial dysfunction plays crucial role in reducing insulin sensitivity, disrupting its production and secretion to cause the development of T2D (X. Yang et al. 2020). Numerous studies revealed that impaired mitophagy occurs in T2D condition and mitophagy with upregulation confers protection on T2D by limiting oxidative stress and inflammasome activation (Durga Devi et al. 2017; Mu et al. 2020; Tong et al. 2019; Yu et al. 2021). During MIR injury, activated mitophagy occurs when ROS increase, electron transport chain activity is impaired, mitochondrial dynamics is aberrant, Ca2+ overload, mitochondrial permeability transition pore opens, and mitochondria dysfunctions (Paradies et al. 2018). Mitophagy is activated largely by the condition of ischemia, and the phase of reperfusion furtherly upregulates mitophagy (M. Yang et al. 2019). It is well known that insufficient or excessive mitophagy is harmful to cells and organs suffering IR. Therefore, it is of great necessity for cells and organs to protect against IR injury by a proper level of mitophagy. However, it remains somewhat controversial with regard to whether mitophagy level of up-regulation or down-regulation provides protection against IR injury. Li et al. (Q. Li et al. 2018) demonstrated that up-regulation of mitophagy could play an important role in protecting against IR injury. A similar result was made out by Zhang et al. (Y. Zhang et al. 2019). In contrast, Feng et al. (Feng et al. 2017) and Jin et al. (Jin et al. 2018) demonstrated that IR injury could be alleviated by inhibiting mitophagy. The differences in experimental models, animal species, and methods among studies may be the reason for these incongruous findings. In this study, we found that PINK1/Parkin pathway was remarkably activated by MIR in nondiabetic rats, which indicated the mitophagy was up-regulated and was consistent with previous studies. Whereas, activity of PINK1/Parkin pathway and TEM showed that the effective increase of mitophagy by MIR was removed in T2D, demonstrating that the vanishment of mitophagy may be responsible for the severe damage caused by MIR in diabetic condition.
To confirm the hypothesis that MitoQ could provide protection associated with the alteration of mitophagy levels, we firstly used the TEM to investigate the changes of mitophagy in T2D subjected to MIR. In our study, it was obviously found that autophagosome-engulfing mitochondria was rarely found in the DIR group, while it could be easier to be observed in the MitoQ group. What is more, the changes of its structure and form showed that it was more fragmented and swollen in the DIR group compared to the MitoQ group. Although MitoQ is the antioxidant, it can exert uncoupling of oxidative phosphorylation and result in lowering the mitochondrial membrane potential that drives mitophagy (Lyamzaev et al. 2018). These findings indicated that the level of mitophagy was removed in T2D rats after MIR which could be reversed by MitoQ. We demonstrated for the first time that mitophagy indeed participated in the process of MIR in STZ-induced T2D rats and MitoQ conferred myocardial protection by the up-regulation of mitophagy. In our in vitro study, the improvement of MitoQ was removed with administration of the mitophagy inhibitor mdivi-1. These results are consistent with previous reports involving cardioprotection by up-regulation of mitophagy and are also consistent with the hypothesis based on previous findings regarding protection of MitoQ (Escribano-Lopez et al. 2016; Hansson et al. 2015; Q. Li et al. 2018; Xiao et al. 2017). The incongruous finding of mitophagy by Feng (Feng et al. 2017) and Jin (Jin et al. 2018) may be the proper level of mitophagy hard to determine caused by the different time of ischemia and reperfusion and animal model.
Three different types of mechanism exist in mitophagy as follows. First type is the mitochondrial outer membrane receptor-mediated mechanism: proteins including Bnip3, Nix, FUNDC1, and Bcl-Rambo were localized, and part of the outer membrane of the mitochondria can combine with lipidated LC3 (LC3-II) to form autophagosomes and regulate the phosphorylation of the LIR domain directly. Second type is the mechanism of PINK1/Parkin pathway: it begins with the activation of PINK1 by the changes in mitochondrial membrane potential (DΨm), and then E3 ligase Parkin is recruited by the target proteins of PINK1 (ubiquitin/Ub and Mfn2) to the mitochondrial outer membrane and ubiquitinate many proteins in the mitochondrial outer membrane which can form autophagosomes by the recruitment of specific autophagy-related receptors (P62/SQSTM1, NBR1, and optineurin/optin) and binding LC3-II. The last one is the lipid receptor-mediated mechanism: a specific transporter (MtCK, NDPK-D) can transfer cardiolipin to the mitochondria outer membrane and then cardiolipin plays the role of eliminating damaged mitochondria by binding to LC3-II (Qiu et al. 2019). In response to MIR, mitochondrial dysfunction, low membrane potential, and more mitochondrial permeability transition pore (mPTP) opening lead to increased PINK1/Parkin activity and upregulation of mitophagy (Zhou et al. 2017). Among these three types of mechanism, PINK1/Parkin is the best-characterized pathway to maintain a healthy mitochondrial network (Turkieh, El Masri, Pinet, & Dubois-Deruy, 2022). Furthermore, PINK1/Parkin pathway is impaired in many diabetic diseases and cardiomyopathy (M. Yang et al. 2021a, b; Y. Zhang et al. 2022). Yang et al. (M. Yang et al. 2021a, b) found that knockout of Parkin exacerbated the mitochondrial dysfunction and cardiac contractile defects with chronic alcohol exposure. As the previous study said, the protection of MitoQ is upregulation of mitophagy accompanied with increased levels of PINK1 and Parkin (Xiao et al. 2017). Therefore, there is a reasonable hypothesis based on the above result, remaining to be explored that PINK1/Parkin pathway relates to changes of mitophagy in T2D rats subjected to MIR.
Therefore, we next assessed the protein expression levels to determine the hypothesis. In our in vivo study, the results showed that there was no significant difference of PINK1 and Parkin expressions between the DS and DIR groups. Notably, the levels of PINK1 and Parkin were increased with the treatment of MitoQ. Similar findings were shown in in vitro study. P62 is an adaptor of autophagy and mitophagy to target selected cargoes to autophagosomes and lead to autophagic degradation by recognizing polyubiquitin chains and interacts with LC3. The accumulation of P62 may depend on lack of autophagosome degradation (Emanuele et al. 2020). Combining with our findings above, the data strongly demonstrated that T2D removed the upregulation of mitophagy and the protective effects provided by MitoQ involved in PINK1/Parkin pathway through upregulating mitophagy. It is noteworthy that in our study, the protein expression of PINK1/Parkin pathway relapsed into the levels of HG/HF-HR conditions when we used mdivi-1. The alteration of protein expression may be that the mechanism of mdivi-1 is inhibition of GTPase dynamin-related protein 1 (Drp1)-dependent fission, and Parkin-independent mitophagy requires Drp1 to play synergistic roles in mitochondrial homeostasis and survival(Bordt et al. 2017; Kageyama et al. 2014).Although correlation between MitoQ and PINK1/Parkin pathway made us explore this signal pathway in this process, further studies are necessary to better define whether other two mechanisms of mitophagy participate in the MIR with T2D or not.
To summarize, the results of our present study found compelling evidence that MitoQ confers cardioprotection against MIR in STZ-induced type 2 diabetic rats. And the protective effects of treatment with MitoQ are implicated in up-regulation of mitophagy during MIR via PINK1/Parkin pathway. This finding provides new evidence for the cardioprotection of MitoQ and provides new potential therapy for patients with T2D suffered ischemic heart disease.
Acknowledgements
The authors would like to thank the central laboratory at Renmin Hospital of Wuhan University (Wuhan, Hubei, China) for their support of our study.
Abbreviations
- CCK-8
Cell counting kit-8
- CK-MB
Creatine kinase-MB
- DCFH-DA
Dichloro-dihydro-fluorescein diacetate
- DMEM
Dulbecco’s modified Eagle’s medium
- FBG
Fasting blood glucose
- FBS
Fetal bovine serum
- HG
High glucose
- HR
Hypoxia/reoxygenation
- HFD
High-fat diet
- HR
Heart rate
- LAD
Left anterior descending coronary artery
- LDH
Lactate dehydrogenase
- LVSP
Left ventricular systolic blood pressure
- + dp/dtmax
LVSP maximum increase rates
- − dp/dtmax
LVSP maximum decrease rates
- MIR
Myocardial ischemia reperfusion
- TEM
Transmission electron microscopy
- T2D
Type 2 diabetes
- 7-AAD
7-Amino-actinomycin
Author contribution
Zhongyuan Xia designed the overall project and supervised the study; Xiazhong Yuan, Yelong Ji, Yang Wu, and Yan Leng designed the experiments; Yelong Ji and Zhen Qiu performed in vivo experiments; Yelong Ji and Yi Zhang performed in vitro experiment; Yelong Ji, Hao Ming, and Aining Zhang performed formal analysis; Yelong Ji, Yang Wu, and Yan Leng wrote the paper with input from other authors. Zhongyuan Xia and Shaoqing Lei reviewed and supervised the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (NSFC 81970722, 81671891, and 81901947) and the Basic Scientific Research of Central University Fund (2042019kf0056).
Data availability
All the data can be obtained from the corresponding authors.
Declarations
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yelong Ji and Yan Leng equally contributed to this article.
Contributor Information
Yang Wu, Email: wywytwin@163.com.
Zhongyaun Xia, Email: xiazhongyuan2005@aliyun.com.
References
- Bordt EA, Clerc P, Roelofs BA et al (2017) The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell 40(6):583–594.e58610.1016/j.devcel.2017.02.020 [DOI] [PMC free article] [PubMed]
- Bragg F, Holmes MV, Iona A et al (2017) Association between diabetes and cause-specific mortality in rural and urban areas of China. Jama 317(3):280–289. 10.1001/jama.2016.19720 [DOI] [PMC free article] [PubMed]
- Bueno M, Lai YC, Romero Y et al (2015) PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest 125(2):521–538. 10.1172/jci74942 [DOI] [PMC free article] [PubMed]
- Durga Devi T, Babu M, Mäkinen P et al (2017) Aggravated postinfarct heart failure in type 2 diabetes is associated with impaired mitophagy and exaggerated inflammasome activation. Am J Pathol 187(12):2659–2673. 10.1016/j.ajpath.2017.08.023 [DOI] [PubMed]
- Emanuele S, Lauricella M, D'Anneo A et al (2020) p62: friend or foe? Evidences for oncojanus and neurojanus roles. Int J Mol Sci 21(14). 10.3390/ijms21145029 [DOI] [PMC free article] [PubMed]
- Escribano-Lopez I, Diaz-Morales N, Rovira-Llopis S et al (2016) The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol 10:200–205. 10.1016/j.redox.2016.10.017 [DOI] [PMC free article] [PubMed]
- Feng Y, Madungwe NB, da Cruz Junho CV et al (2017) Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br J Pharmacol 174(23):4329–4344. 10.1111/bph.14033 [DOI] [PMC free article] [PubMed]
- Guan L, Che Z, Meng X et al (2019) MCU Up-regulation contributes to myocardial ischemia-reperfusion Injury through calpain/OPA-1-mediated mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med 23(11):7830–7843. 10.1111/jcmm.14662 [DOI] [PMC free article] [PubMed]
- Hansson MJ, Llwyd O, Morin D et al (2015) Differences in the profile of protection afforded by TRO40303 and mild hypothermia in models of cardiac ischemia/reperfusion injury. Eur J Pharmacol 760:7–19. 10.1016/j.ejphar.2015.04.009 [DOI] [PubMed]
- He Y, Zhou L, Fan Z et al (2018) Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by N-acetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell Death Dis 9(5):568. 10.1038/s41419-018-0593-y [DOI] [PMC free article] [PubMed]
- Jin Q, Li R, Hu N et al (2018) DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol 14:576–587. 10.1016/j.redox.2017.11.004 [DOI] [PMC free article] [PubMed]
- Kageyama Y, Hoshijima M, Seo K et al(2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J 33(23):2798–2813. 10.15252/embj.201488658 [DOI] [PMC free article] [PubMed]
- Kelso GF, Porteous CM, Coulter CV et al(2001) Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem 276(7):4588–4596. 10.1074/jbc.M009093200 [DOI] [PubMed]
- Leng Y, Wu Y, Lei S et al (2018) Inhibition of HDAC6 Activity Alleviates myocardial ischemia/reperfusion injury in diabetic rats: potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid Med Cell Longev 2018:9494052. 10.1155/2018/9494052 [DOI] [PMC free article] [PubMed]
- Li Q, Gao S, Kang Z et al (2018) Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front Neurosci 12:865. 10.3389/fnins.2018.00865 [DOI] [PMC free article] [PubMed]
- Li XX, Ling SK, Hu MY et al (2019) Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic Biol Med 145:175–186. 10.1016/j.freeradbiomed.2019.09.015 [DOI] [PubMed]
- Liu X, Murphy MP, Xing W et al (2018) Mitochondria-targeted antioxidant MitoQ reduced renal damage caused by ischemia-reperfusion injury in rodent kidneys: Longitudinal observations of T2 -weighted imaging and dynamic contrast-enhanced MRI. Magn Reson Med 79(3):1559–1567. 10.1002/mrm.26772 [DOI] [PMC free article] [PubMed]
- Lyamzaev KG, Tokarchuk AV, Panteleeva AA et al (2018) Induction of autophagy by depolarization of mitochondria. Autophagy 14(5):921–924. 10.1080/15548627.2018.1436937 [DOI] [PMC free article] [PubMed]
- Mackenzie RM, Salt IP, Miller WH et al (2013) Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin Sci (Lond) 124(6):403–411. 10.1042/cs20120239 [DOI] [PMC free article] [PubMed]
- Morales PE, Arias-Duran C, Avalos-Guajardo Y et al (2019) Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med 100822. 10.1016/j.mam.2019.09.006 [DOI] [PubMed]
- Mu J, Zhang D, Tian Y et al (2020) BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J Mol Cell Cardiol 149:1–14. 10.1016/j.yjmcc.2020.09.003 [DOI] [PMC free article] [PubMed]
- Mukhopadhyay P, Horvath B, Zsengeller Z et al (2012) Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic Biol Med 53(5):1123–1138. 10.1016/j.freeradbiomed.2012.05.036 [DOI] [PMC free article] [PubMed]
- Packer M. Heart failure: the most important, preventable, and treatable cardiovascular complication of type 2 diabetes. Diabetes Care. 2018;41(1):11–13. doi: 10.2337/dci17-0052. [DOI] [PubMed] [Google Scholar]
- Paradies G, Paradies V, Ruggiero FM et al (2018) Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. Am J Physiol Heart Circ Physiol 315(5):H1341–h1352. 10.1152/ajpheart.00028.2018 [DOI] [PubMed]
- Qiu Z, Lei S, Zhao B et al (2017) NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid Med Cell Longev 2017:9743280. 10.1155/2017/9743280 [DOI] [PMC free article] [PubMed]
- Qiu Z, Ming H, Lei S et al (2021) Roles of HDAC3-orchestrated circadian clock gene oscillations in diabetic rats following myocardial ischaemia/reperfusion injury. Cell Death Dis 12(1):43. 10.1038/s41419-020-03295-y [DOI] [PMC free article] [PubMed]
- Qiu Z, Wei Y, Song Q et al (2019) The role of myocardial mitochondrial quality control in heart failure. Front Pharmacol 10:1404. 10.3389/fphar.2019.01404 [DOI] [PMC free article] [PubMed]
- Rodriguez-Cuenca S, Cocheme HM, Logan A et al (2010) Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med 48(1):161–172. 10.1016/j.freeradbiomed.2009.10.039 [DOI] [PubMed]
- Rossman MJ, Santos-Parker JR, Steward CAC et al (2018) Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71(6):1056–1063. 10.1161/hypertensionaha.117.10787 [DOI] [PMC free article] [PubMed]
- Saeedi P, Petersohn I, Salpea P et al (2019) Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract 157:107843. 10.1016/j.diabres.2019.107843 [DOI] [PubMed]
- Shefa U, Jeong NY, Song IO et al (2019) Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen Res 14(5):749–756. 10.4103/1673-5374.249218 [DOI] [PMC free article] [PubMed]
- Skulachev VP. How to clean the dirtiest place in the cell: cationic antioxidants as intramitochondrial ROS scavengers. IUBMB Life. 2005;57(4–5):305–310. doi: 10.1080/15216540500092161. [DOI] [PubMed] [Google Scholar]
- Tong M, Saito T, Zhai P et al (2019) Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 124(9):1360–1371. 10.1161/circresaha.118.314607 [DOI] [PMC free article] [PubMed]
- Turkieh A, El Masri Y, Pinet et al (2022) Mitophagy regulation following myocardial infarction. Cells 11(2). 10.3390/cells11020199 [DOI] [PMC free article] [PubMed]
- Wendt L, Vider J, Hoe LES et al (2020) Complex effects of putative DRP-1 inhibitors on stress responses in mouse heart and rat cardiomyoblasts. J Pharmacol Exp Ther 372(1):95–106. 10.1124/jpet.119.258897 [DOI] [PubMed]
- Wu Y, Leng Y, Meng Q et al (2017) Suppression of excessive histone deacetylases activity in diabetic hearts attenuates myocardial ischemia/reperfusion injury via mitochondria apoptosis pathway. J Diabetes Res 2017:8208065. 10.1155/2017/8208065 [DOI] [PMC free article] [PubMed]
- Xiao L, Xu X, Zhang F et al (2017) The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol 11:297–311. 10.1016/j.redox.2016.12.022 [DOI] [PMC free article] [PubMed]
- Xu Y, Shen J, Ran Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy. 2020;16(1):3–17. doi: 10.1080/15548627.2019.1603547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Suo H, Song J. Protective role of mitoquinone against impaired mitochondrial homeostasis in metabolic syndrome. Crit Rev Food Sci Nutr. 2021;61(22):3857–3875. doi: 10.1080/10408398.2020.1809344. [DOI] [PubMed] [Google Scholar]
- Yang M, Linn BS, Zhang Y et al (2019) Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim Biophys Acta Mol Basis Dis 1865(9):2293–2302. 10.1016/j.bbadis.2019.05.007 [DOI] [PubMed]
- Yang M, Wang S, Fu S et al (2021b) Deletion of the E3 ubiquitin ligase, Parkin, exacerbates chronic alcohol intake-induced cardiomyopathy through an Ambra1-dependent mechanism. Br J Pharmacol 178(4):964–982. 10.1111/bph.15340 [DOI] [PubMed]
- Yang X, Pan W, Xu G et al (2020) Mitophagy: A crucial modulator in the pathogenesis of chronic diseases. Clin Chim Acta 502:245–254. 10.1016/j.cca.2019.11.008 [DOI] [PubMed]
- Yu LM, Dong X, Xue XD et al (2021) Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: Role of SIRT6. J Pineal Res 70(1):e12698. 10.1111/jpi.12698 [DOI] [PubMed]
- Zeng Z, Yuan Q, Yu R et al (2019) Ameliorative effects of probiotic Lactobacillus paracasei NL41 on insulin sensitivity, oxidative stress, and beta-cell function in a type 2 diabetes mellitus rat model. Mol Nutr Food Res 63(22):e1900457. 10.1002/mnfr.201900457 [DOI] [PubMed]
- Zhang W, Ren H, Xu C et al (2016) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5. 10.7554/eLife.21407 [DOI] [PMC free article] [PubMed]
- Zhang Y, Wang S, Chen X et al (2022) Liraglutide prevents high glucose induced HUVECs dysfunction via inhibition of PINK1/Parkin-dependent mitophagy. Mol Cell Endocrinol 545:111560. 10.1016/j.mce.2022.111560 [DOI] [PubMed]
- Zhang Y, Wang Y, Xu J et al (2019) Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J Pineal Res 66(2):e12542. 10.1111/jpi.12542 [DOI] [PubMed]
- Zhou H, Zhang Y, Hu S et al (2017) Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res 63(1). 10.1111/jpi.12413 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data can be obtained from the corresponding authors.