

Abstract
It has been known since 1904 that, in humans, diverse cognitive traits are positively inter correlated. This forms the basis for the general factor of intelligence (g). Here, we directly test whether there is a partial genetic basis for individual differences in g using data from seven different cognitive tests (N = 11,263 to N = 331,679) and genome-wide autosomal single nucleotide polymorphisms. A genetic g factor accounts for an average of 58.4% (SE = 4.8%) of the genetic variance in the cognitive traits, with the proportion varying widely across traits (range: 9% to 95%). We distill genetic loci that are broadly relevant for many cognitive traits (g) from loci associated specifically with individual cognitive traits. These results contribute to elucidating the etiology of a long-known yet poorly-understood phenomenon, revealing a fundamental dimension of genetic sharing across diverse cognitive traits.
Scores on psychometric tests of cognitive abilities are prospectively associated with educational performance, socio-economic attainments, everyday functioning, health, and longevity1–3. In 1904, Charles Spearman identified a positive manifold of intercorrelations among school test results and estimates of intelligence, leading him to propose that they arise from a single general dimension of variation, which he termed general intelligence (and which he denoted as g)4. He theorized that most of the remaining variance in each cognitive test was accounted for by a factor specific to that test, which he called s. Thus, some variance in each cognitive test was thought to be shared with all other cognitive tests (g), and some was thought to be specific to that test (its s). Hundreds of studies have since replicated the finding that, when many diverse cognitive tests are administered to a sizeable sample of people, a g factor is found that accounts for between about 25% and 50% of the total test variance, depending on the specific composition of the participants and test battery5–7. Considerable efforts over the past century have been placed on identifying biological associations with g, spanning levels of analysis from molecular, to neuroanatomical, to cognitive8–12.
Psychometrically, a hierarchical structure of cognitive abilities is commonly agreed, with cognitive tests’ variance accounted for by three different strata of variation (Supplementary Figure 1), representing: (1) each test’s specific variance (s); (2) broad domains of cognitive function (e.g. reasoning, processing speed, memory); and (3) g5. All cognitive tests have some g loading, though this varies from test to test. Twin studies that have employed multivariate methods to examine genetic associations within the hierarchy of cognitive test score variance13 indicate a strong heritable basis for g, suggesting that cognitive traits are positively correlated substantially because of strongly-overlapping genetic architecture14–18. Multivariate approaches, however, have not yet been combined with modern molecular genetic methods needed to separate general from specific genetic associations with cognitive traits at the level of individual genetic loci.
Genome-Wide Association Studies (GWAS) have been applied to individual cognitive measures or composite scores formed from multiple such cognitive measures19–23. However, existing univariate approaches are limited in their capabilities to separate g from s variance. In the case of GWASs of individual cognitive tests, e.g. a measure of verbal declarative memory or processing speed, the identified loci could either be related to g and/or to the named cognitive property22,23. This is a common limitation in both phenotypic and genetic cognitive studies24. Here we sought to test for a genetic g factor directly, using Genomic Structural Equation Modelling (Genomic SEM25), a multivariate genome-wide molecular genetics approach. We model shared and unique genome-wide architecture in aggregate across the entirety of the genome and we distinguish individual variants that are broadly relevant for many cognitive traits (via genetic g) from those associated with only individual cognitive traits (via genetic s factors). Thus, this investigation attempts to provide insights into the shared genetic architecture across multiple cognitive traits and affords the explicit identification of genetic variants underlying g.
Results
Data for the present study came from the UK Biobank, a biomedical cohort study that collects a wide range of genetic and health-related measures from a population-based sample of community-dwelling participants from the UK. Participants were measured on up to seven cognitive traits using tests that often show substantial concurrent validity with established psychometric tests of cognitive abilities, and modest to good test-retest reliability26: Reaction Time (RT; n = 330,024; which assesses perceptual motor speed), Matrix Pattern Recognition (n = 11,356; nonverbal reasoning), Verbal Numerical Reasoning (VNR; n = 171,304; verbal and numeric problem solving; the test is called ‘Fluid intelligence’ in UK Biobank), Symbol Digit Substitution (n = 87,741; information processing speed), Pairs Matching Test (n = 331,679; episodic memory), Tower Rearranging (n = 11,263; executive functioning), and Trail Making Test–B (Trails-B; n = 78,547; executive functioning). Scores on all tests were coded such that higher scores represented more optimal (i.e., faster or more accurate) performance.
Phenotypic Covariance Structure
A positive manifold of phenotypic correlations was observed across the seven cognitive traits. All correlations were positive, ranging from 0.074 to 0.490, indicating that more optimal performance on a given test is associated with more optimal performance on the other tests (Supplementary Figure 1; Supplementary Table 1). The mean phenotypic correlation was 0.232. In principal components analysis (PCA), the first unrotated component accounted for a total of 35.8% of the phenotypic variance. A confirmatory factor model with a single common g factor (Figure 1, bottom panel) fit the phenotypic covariance matrix well (χ2(14) = 740.748, p < 0.001; SRMR=0.024; CFI=0.985; RMSEA = .013). Table 1 reports both the proportion of phenotypic g:phenotypic s variance for each cognitive trait, and the respective absolute contributions. The g factor accounted for 26.5% (SE = 0.2%) of the variance in the seven cognitive traits. That this proportion is appreciably lower than that obtained from that obtained from PCA highlights the distinction between factor analysis, which formally models the effects of factors on constellations of variables, from PCA, which simply seeks to maximize variance of a weighted linear composite of those variables. All of the standardized loadings were statistically significant, ranging from 0.231 to 0.766 (M = 0.48, SD = 0.19).
Figure 1.
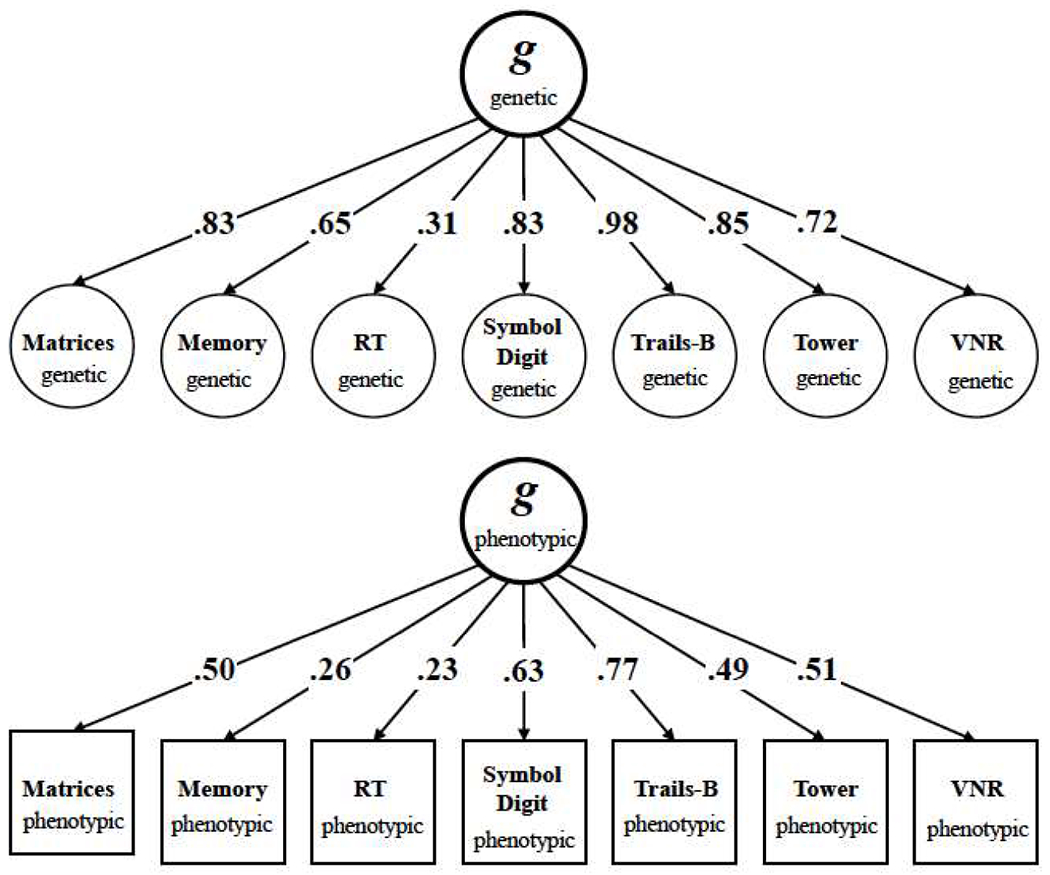
Standardized genetic (top) and phenotypic (bottom) factor solutions for the covariance structure of seven UK Biobank cognitive traits used in the present study. Squares represent observed variables, i.e. the phenotypes that are directly measured. Circles represent latent variables that are statistically inferred from the data, i.e. the genetic and phenotypic g factors that are inferred through factor analysis, and the genetic components of the observed phenotypes that are inferred through LD Score Regression. Arrows are standardized factor loadings, which can be interpreted as standardized regression relations with the arrow pointing from the predictor variable to the outcome variable. Genetic factor models were estimated using Genomic SEM (26), and phenotypic models were estimated using the lavaan package for R (41). Matrix = Matrix Pattern Completion task; Memory = Memory – Pairs Matching Test; RT = Reaction Time; Symbol Digit = Symbol Digit Substitution Task; Trails-B = Trail Making Test – B; Tower = Tower Rearranging Task; VNR = Verbal Numerical Reasoning Test. All variables are scaled such that higher scores indicate better cognitive performance. The genetic g factor accounts for an average of 58.37% of the genetic variance in the seven cognitive traits. The phenotypic g factor accounts for an average of 26.50% of the observed phenotypic variance in the seven cognitive traits.
Table 1.
Common factor solutions for the genetic (top section) and phenotypic (bottom section) covariance structure of seven UKB cognitive traits.
| Standardized Factor Loadings | Common (g) and specific (s) sources of genetic variation | ||||||
|
| |||||||
| Genetic g | Proportion of genetic variation explained by genetic g and genetic s | Proportion of phenotypic variation explained by genetic g and genetic s (HapMap3 Common Variants Only) | |||||
|
| |||||||
| Cognitive Trait | Estimate | SE | Common (g) | Specific (s) | Common (g) | Specific (s) | Total SNP h2 |
| Matrices | 0.826 | 0.070 | 68.23% | 31.77% | 10.60% | 4.90% | 15.50% |
| Memory | 0.651 | 0.031 | 42.38% | 57.62% | 1.70% | 2.30% | 4.00% |
| RT | 0.308 | 0.026 | 9.49% | 90.51% | 0.70% | 6.70% | 7.40% |
| Symbol | 0.831 | 0.034 | 7.60% | 3.40% | 11.00% | ||
| Digit | 69.06% | 30.94% | |||||
| Trails-B | 0.976 | 0.035 | 95.26% | 4.74% | 14.20% | 0.70% | 14.90% |
| Tower | 0.853 | 0.080 | 72.76% | 27.24% | 8.30% | 3.10% | 11.40% |
| VNR | 0.717 | 0.024 | 51.41% | 48.59% | 10.90% | 10.30% | 21.20% |
| Mean % | 58.36% | 41.64% | 7.71% | 4.49% | 12.20% | ||
|
| |||||||
| Phenotypic g | Proportion of phenotypic variation explained by phenotypic g and phenotypic s | ||||||
|
| |||||||
| Cognitive Trait | Estimate | SE | Common (g) | Specific (s) | |||
|
| |||||||
| Matrices | 0.501 | 0.009 | 25.10% | 74.90% | |||
| Memory | 0.257 | 0.003 | 6.60% | 93.40% | |||
| RT | 0.231 | 0.003 | 5.34% | 94.66% | |||
| Symbol Digit | 0.628 | 0.004 | 39.44% | 60.56% | |||
| Trails-B | 0.766 | 0.003 | 58.68% | 41.32% | |||
| Tower | 0.487 | 0.009 | 23.72% | 76.28% | |||
| VNR | 0.514 | 0.003 | 26.42% | 73.58% | |||
| Mean % | 26.50% | 73.50% | |||||
Note. Matrices = Matrix Pattern Completion task; Memory = Memory – Pairs Matching Test; RT = Reaction Time; Symbol Digit = Symbol Digit Substitution Task; Trails-B = Trail Making Test - B; Tower = Tower Rearranging Task; VNR = Verbal Numerical Reasoning Test. All traits are scaled such that higher scores indicate higher cognitive performance. Total SNP h2 = total proportion of phenotypic variance in the corresponding cognitive trait accounted for by all tagged common variants. By definition, the common (g) and specific (s) proportional contributions to total phenotypic variation sum to the total SNP h2, and the common (g) and specific (s) proportional contributions to genetic variation sum to 100%. Note: Standardized factor loadings indicate the standardized linear relationship between the factor and each of the cognitive outcomes. Models are fit to LDSC-derived genetic covariance matrices using Genomics SEM. As per best practices for LDSC, genetic covariance matrices were derived using HapMap3 SNPs with minor allele frequencies > 1%, excluding SNPs with INFO < 0.9 and those from the MHC region.
Multivariate Genome-Wide Architecture
We next aimed to estimate the extent of genetic sharing across the cognitive traits using molecular genetic data. We used a multivariable version of Linkage Disequilibrium Score Regression (LDSC)27 implemented in Genomic SEM25 to estimate genetic correlations among the cognitive traits. Prior to this formal modelling, we conducted descriptive analyses of the cognitive traits’ genetic correlations, similar to those often conducted on cognitive phenotypes. We report those descriptive analyses’ results first.
As was first reported at the phenotypic level by Spearman in 19044, we identified, using LDSC, a positive manifold of genetic correlations among the UK Biobank cognitive traits, ranging from 0.135 to 0.869 (M = 0.53, SD = 0.22; Supplementary Figure 3; Supplementary Tables 2–3). The mean genetic correlation was 0.530, and the first principal component accounted for a total of 62.17% of the genetic variance. Using genomic-relatedness based restricted maximum-likelihood (GCTA-GREML)28,29, a different estimator of the genetic correlations among the seven cognitive traits (Supplementary Figure 4), the mean genetic correlation was 0.502, and the first principal component accounted for 61.24% of the genetic variance. The correlation between LDSC- and GCTA-GREML-derived genetic correlations was r = 0.947, indicating very close correspondence between results of the two methods (Supplementary Figure 5).
We then proceeded with Genomic SEM to formally model the genetic covariance matrix. This allowed us to evaluate the fit of the genetic g factor model, estimate SEs for model parameters, estimate genetic correlations with collateral phenotypes, and incorporate genetic g explicitly into multivariate discovery. We applied Genomic SEM to fit a single common factor model to the LDSC-derived genetic covariance matrix among the seven cognitive traits. This model specified the genetic component of each cognitive trait to load on a single common factor, which we term genetic g. For each trait, we additionally estimated residual, trait-specific genetic variance components (genetic ss). Thus, we formally distill the molecular genetic contributions of g and s to heritable variation in each of the cognitive traits, and test the fit of this model. Fit indices (χ2(14) = 117.019, p < 0.001; CFI = 0.970; SRMR = 0.088) indicated that the factor model closely approximated the observed genetic covariance matrix (Supplementary Figures 7–8). Figure 1 displays the standardized estimates for this model (top panel) and the standardized estimates from a phenotypic factor model (bottom panel) fitted to the phenotypic covariance matrix (Supplementary Figure 5; Supplementary Table 1). A factor model that constrained the standardized genetic factor loadings to be equal to the standardized point estimates for loadings from the phenotypic model produced a substantial decrement in model fit (χ2(7) = 823.037, p < 0.001), indicating that the genetic and phenotypic factor structures were not strictly equivalent. Indeed, the standardized genetic factor loadings were consistently higher in magnitude than the standardized phenotypic factor loadings, but there was a strong linear association between them (Supplementary Figure 6). Table 1 reports both the proportions of genetic g and genetic s variance for each cognitive trait, and the respective absolute contributions. The genetic g factor accounted for 58.36% (SE = 4.84%) of the genetic variance in the seven cognitive traits. All of the standardized loadings on genetic g were statistically significant, ranging from 0.308 to 0.976 (M = 0.74, SD = 0.22).
The proportion of genetic variation in each trait accounted for by genetic g differed substantially across traits. Supporting this inference, a factor model that constrained the standardized genetic factor loadings to be equal across traits produced a substantial decrement in model fit (χ2(12) = 749.122, p < 0.001). Four of the cognitive traits have a genetic contribution to their variance that is principally from genetic g and much less from a genetic s; these are Trails-B (95.26% genetic g; 4.74% genetic s), Tower (72.76% genetic g; 27.20% genetic s), Symbol Digit (69.06% genetic g; 30.94% genetic s), and Matrices (68.23% genetic g; 31.77% genetic s). VNR (51.41% genetic g; 48.59% genetic s) and Memory (42.38% genetic g; 57.62% genetic s) are more evenly split. RT has the majority of its genetic influence from a genetic s (9.49% genetic g; 90.51% genetic s). We emphasize one important implication of these results, i.e. that univariate genetic analyses (e.g. GWAS) of some of these individual traits will largely reveal results relevant to g rather than to the specific abilities thought to be required to perform the test24. As the pre-specified model was parsimonious and the fit was close, we chose to forego implementing data-driven exploratory steps to further improve fit. Supplementary Tables 4–5 report full parameter estimates for genetic and phenotypic factor models.
Multivariate Genome-Wide Discovery
We next aimed to determine the contributions of individual genetic loci specifically to genetic g, and to distill those from loci associated with other levels of the cognitive hierarchy. We fit a multivariate GWAS of genetic g within Genomic SEM25 to distinguish loci relevant to genetic g from loci whose patterns of association across the individual traits is inconsistent with their operation on genetic g, as indexed by the heterogeneity statistic, Q. We provide detailed explication of the Q statistic and how it can be appropriately interpreted in the Interpreting the Heterogeneity Statistic section of the Supplementary Materials.
The GWAS results for genetic g and Q are displayed in Figure 2 as a Miami plot, with further information provided in Table 2. Our method distinguishes four types of genome-wide significant loci. First, highlighted in red are genome-wide significant loci for genetic g that are not genome-wide significant loci for the univariate GWAS analyses of the individual traits. These are loci influencing general intelligence identified by leveraging the joint genetic architecture of the traits. Second, highlighted in blue are genome-wide significant loci for g that are also genome-wide significant loci in the univariate GWAS analyses for at least one individual cognitive trait. These loci might otherwise have been interpreted as relevant specifically to the individual trait, when in fact the multivariate results indicate that they are relevant to genetic g24. Third, highlighted in green are genome-wide significant loci for the univariate phenotypes that are not genome-wide significant loci for g. These might be loci that are specific to the individual traits, but not genetic g. Fourth, highlighted in yellow are loci that evince genome-wide significant heterogeneity (Q), indicating that they show patterns of associations with the cognitive traits that depart from the pattern that would be expected if they were to act on the traits via genetic g. Q findings that exceed the genome-wide significance threshold for genetic g (yellow triangles) are implicated as false discoveries on genetic g that are likely driven by a strong signal in a subset of the cognitive traits or in a single cognitive trait. The Q statistic helps to safeguard against these false discoveries. The Q findings that do not surpass the genome-wide significance threshold for genetic g (yellow diamonds) are not significantly related to genetic g but are significantly heterogeneous in their patterns of associations with the cognitive traits. These loci may be relevant to specific cognitive traits, or to cognitive domains that are intermediate in specificity and generality between g and s, but not to general intelligence (see Supplementary Figure 1). Note that these four types of genome-wide significant loci are represented both in the top and bottom panels of the Miami plot, with their locations corresponding to the −log10(p) of their associations with genetic g in the top panel, and the −log10(p) of their Q statistic in the bottom panel.
Figure 2.
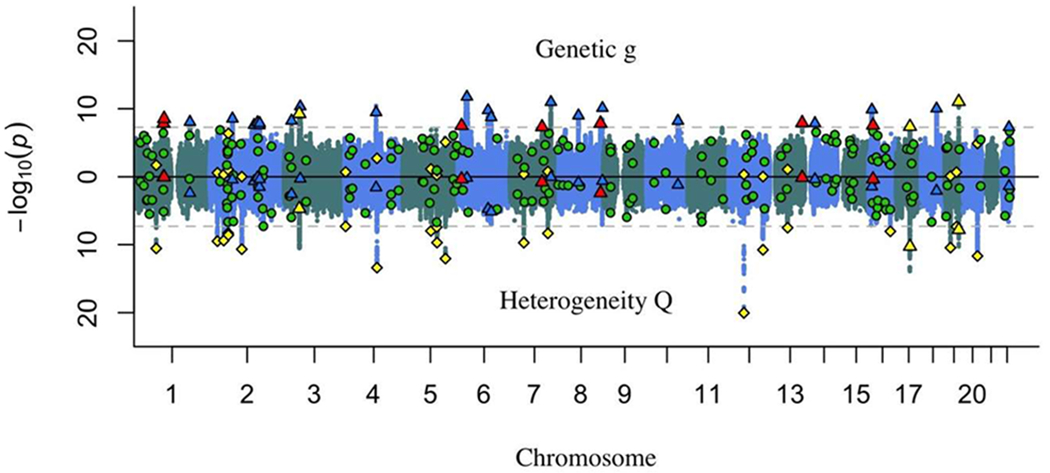
Miami plot of unique, independent hits for genetic g (top) and Q (bottom) (7,857,346 variants). The heterogeneity statistic (Q) indexes whether a SNP evinces patterns of associations with the cognitive traits that departs from the pattern that would be expected if it were to act on the traits via genetic g. Thus, genetic g loci in common with Q loci are false discoveries on genetic g. The dotted grey horizontal lines are the genome-wide significance threshold (p < 5×10−8). The genome-wide significant loci represented by the triangles, circles, and diamonds are represented both in the top and bottom panels of the Miami plot, with their locations corresponding to the −log10(p) of their associations with genetic g in the top panel, and the −log10(p) of their Q statistic in the bottom panel.
Red triangles  : genetic g loci unique of univariate loci.
: genetic g loci unique of univariate loci.
Blue triangles  : genetic g loci in common with univariate loci.
: genetic g loci in common with univariate loci.
Green circles  : univariate loci unique of genetic g loci.
: univariate loci unique of genetic g loci.
Yellow triangles  : genetic g loci in common with Q loci.
: genetic g loci in common with Q loci.
Yellow diamonds  : Q loci unique of genetic g loci.
: Q loci unique of genetic g loci.
Table 2.
Summary of Multivariate (Genetic g) and Univariate GWAS results.
| Multivariate GWAS | |||||||
|
| |||||||
| Outcome | Significant loci (p < 5x10-8) | Independent of univariate loci | Independent of Q loci | Common with loci previously reported for cognitive tests and cognitively-relevant traits1 in GWAS that did not include UKB | Mean χ2(1) | LDSC Intercept | λGC |
|
| |||||||
| Genetic g | 30 | 7 | 27 | 18 | 1.471 | 0.973 | 1.373 |
|
| |||||||
| Significant loci (p < 5x10-8) | Independent of univariate loci | Independent of genetic g loci | Common with loci previously reported for cognitive tests and cognitively-relevant traits1 in GWAS that did not include UKB | Mean χ2(1) | |||
|
| |||||||
| Heterogeneity (Q) | 24 | 9 | 21 | 5 | 1.337 | ||
|
| |||||||
| Univariate GWASs | |||||||
|
| |||||||
| Significant loci (p < 5x10-8) | Independent of genetic g loci | Independent of Q loci | Mean χ2 | ||||
|
| |||||||
| Matrices (n=11,356) | 0 | - | - | 1.040 | 1.013 | 1.033 | |
| Memory n = 331,679) | 10 | 9 | 7 | 1.223 | 1.002 | 1.187 | |
| Reaction Time (n = 330,024) | 39 | 34 | 23 | 1.420 | 1.021 | 1.318 | |
| Symbol Digit (n = 87,741) | 1 | 0 | 0 | 1.188 | 1.021 | 1.169 | |
| Trails-B (n = 78,547) | 7 | 1 | 5 | 1.203 | 1.005 | 1.177 | |
| Tower (n = 11,263) | 0 | - | - | 1.022 | 1.001 | 1.025 | |
| VNR (n = 171,304) | 89 | 71 | 83 | 1.640 | 1.025 | 1.471 | |
Note: The same 7,857,346 variants were used for univariate and multivariate GWAS. Genome-wide significant loci that were 250 kb or closer were merged into a single locus. Here we report loci that are independent from each other at r2 < 0.1.
For Q, mean χ2 was calculating by converting Q statistics (which are χ2 distributed with 6 degrees of freedom) to distributed test statistics with 1 degree of freedom, and then taking their mean.
Matrices = Matrix Pattern Completion task; Memory = Memory – Pairs Matching Test; Symbol Digit = Symbol Digit Substitution Task; Trails-B = Trail Making Test – B; Tower = Tower Rearranging Task; VNR = Verbal Numerical Reasoning Test.
Cognitive tests and cognitively relevant traits include performance-based cognitive test scores, educational attainment, self-reported math ability, and highest math course taken.
Supplementary Tables 8–28 report the individual SNPs, loci and, for those that have been reported in previous studies, the references for the studies, and the implicated phenotypes.
Overall, we identified 30 genome-wide significant (p < 5×10−8) loci associated with genetic g. Of these, 18 (60%) have been previously reported as hits for cognitive tests (5 loci) and/or cognitively-relevant phenotypes, such as educational attainment or highest math course taken (16 loci) in GWAS that did not include data from UKB. Of the 18 genetic g hits that replicated outside of UKB, 16 were also hits on at least 1 cognitive test included in the multivariate UKB analysis. Of the 30 total genetic g loci, 12 loci were discoveries specific to UKB, 5 of which were discoveries specific to the present study’s multivariate modeling, and 7 of which were also hits on at least 1 cognitive test included in the multivariate UKB analysis. Thus, of the 30 total loci that were found here to be associated with genetic g, 23 were common with the univariate GWAS of the individual cognitive traits that served as the basis for our multivariate analysis, indicating that 7 loci were discoveries specific to multivariate modelling (Supplementary Tables 15 and 22). An LDSC analysis of genetic g GWAS summary statistics yielded an intercept slightly below 1.0, indicating that inflation of the test statistics (Mean χ2(1) = 1.471; λGC = 1.373) was attributable to true polygenic signal, rather than under-controlled population stratification.
We identified a total of 24 genome-wide significant loci for Q, 3 of which were significantly associated with genetic g (and therefore likely to be relevant to more specific cognitive traits, and false discoveries on g) and 15 of which were significantly associated with at least one individual cognitive trait in the test-specific GWASs (and may therefore be interpreted as hits for more specific cognitive traits, rather than for a general dimension of cognitive function). Of the 24 loci for Q, 5 (21%) have been previously reported as hits for cognitive tests (1 locus) and/or cognitively-relevant phenotypes (4 loci) in GWAS that did not include data from UKB, and 19 loci were specific to UKB (Supplementary Tables 16 and 23). Two of the Q loci previously reported in non-UKB GWAS were also genetic g loci (but, because they were Q hits, are most likely to be false discoveries for genetic g and more relevant to specific cognitive traits).
Inspection of the univariate GWAS results for the individual traits may help to determine the sources of heterogeneity for the Q findings. For instance, a SNP (rs429358) within APOE, which is a known risk factor for Alzheimer’s Disease30, was a significant Q finding. With the exception of its association with VNR, this SNP displayed a pattern of associations with the traits that corresponded closely with the degree to which they represented genetic g. However, consistent with the inference that APOE is specifically relevant for cognitive aging, the SNP displayed a negligible null association with VNR (p = .142), which is a test that shows minimal age-related differences in the UK Biobank data31. Another example of a Q finding is located on Chromosome 17 (chr17: 44021960- 44852612), which was reported to be significantly associated with both general cognitive ability and Reaction Time19. From the univariate GWAS results, the largest association for this locus was with Reaction Time, a measure of psychomotor speed with a relatively low loading on genetic g. This locus may have a particularly pronounced association with speeded abilities, rather than a general association with genetic g. The third Q locus which is also significant for genetic g is located on chromosome 3 (chr3:49120040-50234126). This locus has previously-reported associations with general cognitive ability, educational attainment, intelligence, and math ability19–21,32. In the current study, this locus demonstrates significant heterogeneity and displays its largest associations with VNR, Tower, Matrices, and Trails-B, all measures of higher-order cognition. Its associations with measures of speed and episodic memory (arguably more basic cognitive processes) are negligible.
Genetic Correlations with External GWAS Traits
As expected, the genetic g factor identified here displayed strong but imperfect genetic correlations (as estimated with LDSC) with general cognitive function from Davies et al.,19 (rg = 0.90, SE = 0.02), and Savage et al.,21 (2018) (rg = 0.87, SE = 0.05), which were univariate GWASs of broad cognitive phenotypes, and that of Hill et al.,20 (rg = 0.80, SE = 0.02), which was a GWAS of intelligence that incorporated educational attainment GWAS summary statistics to boost power via multi-trait analysis of GWAS (MTAG)33. As reported in Supplementary Table 6, genetic g had a positive genetic correlation with Educational Attainment32 (rg = 0.48, SE = 0.02) that is lower than those found between Education and previous GWASs of cognitive ability (all estimated at r > 0.69)19–21. To determine whether this lower association was driven by the inclusion of RT, or more generally with speeded measures as indicators of genetic g, we re-estimated the genetic correlations using a genetic g factor formed from the cognitive traits that excluded either RT or all speeded measures (RT, Trails-B, and Symbol Digit). The version of the genetic g factor that excluded RT accounted for 66.76% (SE = 5.85%) of the genetic variance in the six remaining cognitive traits, and the version that excluded all speeded tests accounted for 69.66% (SE = 8.08%) of the genetic variance in the four remaining cognitive traits. These versions of genetic g produced somewhat higher genetic correlations between genetic g and Educational Attainment (rg = 0.50 when excluding RT, rg = 0.55 when excluding all three speeded measures) that continued to be lower than those found between Educational Attainment and previous GWASs of general cognitive ability.
Negative genetic correlations were found between genetic g and Alzheimer’s disease34 (rg = −0.34, SE = 0.06), Schizophrenia35 (rg = −0.38, SE = 0.03), and ADHD36 (rg = −0.23, SE = 0.04). Additionally, genetic g had significant positive genetic associations with total Brain Volume37 (rg = 0.20, SE = 0.04), and Longevity38 (rg = 0.26, SE = 0.03) (Supplementary Table 6). Notably, the negative genetic association between genetic g and Schizophrenia was substantially stronger than that obtained for associations between other GWASs of cognitive function and Schizophrenia, and contrasts substantially with the mild positive genetic correlation that has been reported between educational attainment and schizophrenia39. Genetic associations between genetic g and Alzheimer’s Disease, Autism Spectrum Disorder, ADHD, Total Brain Volume, and Longevity were similar to those obtained for other GWASs of cognitive function, particularly when the speeded tests were removed from the genetic g factor.
Replication of Positive Genetic Manifold and Polygenic Prediction in Generation Scotland
We sought to confirm key results in the independent Generation Scotland study (N=6,950 unrelated individuals). The cognitive measures in Generation Scotland were Wechsler Logical Memory (episodic memory), Mill Hill Vocabulary (crystallized knowledge), Wechsler Digit Symbol Substitution (processing speed), and Verbal Fluency (semantic fluency), as described previously40,41. Because the sample size of Generation Scotland is too small to produce stable estimates of heritability and genetic correlation within LDSC, is was not feasible to directly integrate these analyses into the above Genomic SEM models to estimate joint models with the UKB phenotypes and other external GWAS traits. Instead, we estimated a genetic correlation matrix for the four cognitive tests in Generation Scotland using GCTA-GREML28,29, which is more appropriate than LDSC for moderately-sized samples, such as this. The average genetic correlation in this matrix was 0.517 and an eigen decomposition indicated that the percentage of genetic variance explained by a single principal component was 64.90%. These two values are similar to those obtained for UKB, which we reiterate here for ease of comparison: mean rg LDSC = 0.530; mean rg GCTA-GREML = 0.502; percentage of genetic variance accounted for by PC1 LDSC = 62.17%; percentage of genetic variance accounted for by PC1 GCTA-GREML = 61.24%.
Using summary statistics from the above-described UK Biobank analyses, we next created polygenic scores (PGSs) for genetic g and the individual UK Biobank cognitive traits and used them to individually and simultaneously predict, in Generation Scotland, variance in performance on the individual cognitive tests, the first unrotated principal component of all tests (to index phenotypic g), the first unrotated principal component of all tests except Mill Hill Vocabulary (to index a more fluid g), and educational attainment (Supplementary Table 7). Consistent with the above findings that individual cognitive outcomes are associated with a combination of genetic g and specific genetic factors, we observed a pattern in which many of the regression models that included both the polygenic score (PGS) from genetic g and test-specific PGSs were considerably more predictive of the cognitive phenotypes in Generation Scotland than regression models that included only either a genetic g PGS or a PGS for a single test. A particularly relevant exception involved the Digit Symbol Substitution test in Generation Scotland, which is a similar test to the Symbol Digit Substitution test in UK Biobank, for which we derived a PGS. We found that the proportional increase in R2 in Digit Symbol by the Symbol Digit PGS beyond the genetic g PGS was <1%, whereas the genetic g PGS improved polygenic prediction beyond the Symbol Digit PGS by over 100%, reflecting the power advantage obtained from integrating GWAS data from multiple genetically correlated cognitive traits using a genetic g model. An interesting counterpoint is the PGS for the VNR test, which is unique in the UK Biobank cognitive test battery in partly indexing verbal knowledge26,31. Highlighting the role of domain-specific factors, a regression model that included this PGS and the genetic g PGS provided substantial incremental prediction relative to the genetic g PGS alone for those Generation Scotland phenotypes most directly related to verbal knowledge: Mill Hill Vocabulary (62.45% increase) and Educational Attainment (72.59%).
Discussion
Until now, research on the positive manifold of correlations among cognitive traits has been phenotypic in nature, or has made inferences regarding the roles of genes using twin approaches. Here we estimated and modeled the patterns of genetic sharing across diverse cognitive traits using genome-wide molecular data. Using data from seven different cognitive traits from UK Biobank, we identified a positive manifold of genetic correlations. We found that a genetic g factor accounts for an average of about 58% (SE = about 5%) of the genetic variance in the cognitive traits, with the proportion ranging widely (about 9% to about 95%) across the traits. We went on to distill specific genetic loci broadly relevant for many cognitive traits via genetic g from those displaying patterns of more associations with the individual cognitive traits.
The importance of our results may be seen by contrasting the results of Trails-B with Reaction Time. Analyses of multivariate genome-wide architecture indicated that, for Trails-B, 95% of the genetic variance is accounted for by genetic g, and only 5% of the genetic variance is specific to Trails-B. Moreover, all seven loci for Trails-B have been previously reported in GWAS of other cognitive phenotypes (Supplementary Table 19), and four of them were implicated as relevant for genetic g at nonsignificant levels of heterogeneity. In contrast, for Reaction Time, analyses of multivariate genome-wide architecture indicated that 9.5% of the genetic variance is accounted for by genetic g, and 90.5% of the genetic variance is specific to Reaction Time. Many of the 39 loci associated with RT have not been found in univariate GWASs of other cognitive traits (Supplementary Tables 17 and 24), and only four were implicated as relevant for genetic g at nonsignificant levels of heterogeneity. Therefore, when identifying loci associated with performance on an individual cognitive test, it is essential to know the extent to which its associations are broadly related to genetic g or specifically related to the phenotype under investigation.
Failure to take the multivariate structure of the cognitive traits into account may lead to incorrect inferences24—either that discoveries made in a univariate GWAS of a cognitive trait are generalizable to the broader universe of cognitive traits when they are in fact specific to that trait, or that discoveries made in a univariate GWAS of a cognitive trait are specific to that trait when they are in fact broadly associated with all traits that load on genetic g. For instance, our multivariate analysis indicates that a locus on chromosome 7 (chr7:104558814-104588161) is associated with genetic g. Similarly, we report an association of a locus on chromosome 8 (chr8:64496159-64842662) with Trails-B (an index of executive function, which is itself strongly genetically correlated with g14) in the univariate GWAS, and our multivariate analysis indicates that this locus is also related to genetic g. Lee et al.32 have previously reported both of these loci to be associated with math ability (Supplementary Table 22), but there are no previously-reported associations with general cognitive function or intelligence. The current results indicate that the loci are broadly relevant to many abilities via genetic g, not simply to math ability. Multivariate methods, such as that pioneered here, are necessary in order to distinguish whether a locus is narrowly relevant for an individual cognitive trait or broadly relevant to genetic g.
Genetic g was highly, but imperfectly, genetically correlated with previous univariate GWASs of general cognitive function and intelligence. Moreover, genetic g displayed lower (albeit still sizable) genetic correlations with educational attainment than have previous univariate GWASs of general cognitive function and intelligence. This pattern suggests that previous GWASs of general cognitive ability might have tapped more academic forms of cognitive function (i.e. crystallized abilities, such as verbal knowledge) than those tapped by the present group of cognitive tests. Consistent with this hypothesis, a version of genetic g that excluded speeded tests—which are known to be among the most culturally decontextualized of the cognitive traits42—produced somewhat higher genetic correlations with educational attainment, though they continued be lower than those found between Educational Attainment and the previous univariate GWASs of general cognitive function and intelligence. A priority for future research will be to distinguish between genetic correlates of cognitive abilities that are driven by forms of higher order thinking and academic knowledge from those that are driven more so by forms of arguably more basic neurocognitive processing43. Moreover, given that the phenotypic gs from different cognitive test batteries administered to the same sample correlate very highly6, it will be useful in future research to discover whether genetic gs obtained from different test batteries also have very high correlations. The advantage of modern genomic methods, such as those used here, is that it is not necessary for the same sample to be tested on both batteries, or even on the same tests within a given battery25.
Some researchers have adroitly argued that a positive manifold of test intercorrelations may, in principle, arise from a pattern in which individual genetic loci, biological mechanisms, or cognitive processes contribute to subsets of traits, with the subsets varying across loci, mechanisms, or processes44–46. Others have similarly argued that positive test intercorrelations may arise from reciprocal causation among abilities, or between abilities and external forces, and that genetic effects enter through specific points in the system and come to be correlated through dynamic propagation47–49. Here we have provided evidence not only of a positive manifold of genetic correlations at the aggregate, genome-wide, level of analysis, but also at the level of individual loci. Although these discoveries are themselves insufficient for determining the causes of the positive manifold, they do help to inform and constrain past and future accounts of the positive manifold and of the heritability of cognitive abilities.
It is important to consider this work in light of its key limitations. First, we had measures of different hierarchically-intermediate traits (e.g. processing speed, memory, reasoning), but we did not have multiple measures per intermediate trait. We were therefore unable formally to model genetic associations with intermediate traits as separate from those on s factors specific to the individual cognitive traits. In other words, based on the currently-available data, we have been well-positioned to discriminate between genetic loci that are broadly relevant for genetic g from those that display more heterogeneous patterns of relations with individual cognitive tests, but we are unable to distinguish loci relevant for very narrow traits captured by individual tests from those relevant to intermediately-broad traits. Future work that employs a denser battery of cognitive tests will be valuable for such discernment. Second, using data from Generation Scotland we closely replicated the positive manifold of genetic correlations observed in UKB, and we demonstrated the utility of polygenic scores for genetic g constructed on the basis of the UKB data, but Generational Scotland was not sufficiently powered to conduct individual SNP-level analyses. Several large-scale GWASs exist for intelligence, but UKB appears to be the only such large-scale dataset for which multiple tests spanning a broad range of cognitive traits is available. When large-scale datasets with multivariate cognitive data become available, it will be prudent to examine the replicability of the individual g and s loci identified here. Finally, our analyses were based exclusively on individuals of European ancestry residing in the United Kingdom. It may not be assumed that the results reported here will generalize beyond this population.
In summary, we have inferred a genetic g factor using molecular genetic data, and we have discerned genetic loci that are associated with genetic g from those that are associated with more specific cognitive traits. We emphasize the large extant explanatory gap between genetic variation and shared (i.e. general) variation in cognitive abilities.
Methods
Sample
Data from the UK Biobank study was used for the present study (https://www.ukbiobank.ac.uk/). The UK Biobank is a biomedical prospective cohort study, which collected a wide range of genetic and health related measures from a national sample of community dwelling participants from the UK. Ethical approval for the UK Biobank was granted from the Research Ethics Committee (11/NW/0382). This study uses European ancestry genome-wide genotyped data from seven cognitive tests with varying sample sizes across phenotypes. Individuals were removed sequentially based on non-British ancestry, high missingness, high relatedness (samples which have more than 10 putative third-degree relatives), and sex/gender mismatch between self-report and genetic data. Our analysis sample included 332,050 unrelated participants of European descent with high-quality genotyping. Participant ages ranged from approximately 40 to 70 years at the first assessment and approximately 45 to 75 years at later assessments in which further cognitive tests were administered. For each cognitive test, we included no more than one test administration per participant in analyses.
Cognitive Tests
Reaction Time (n = 330,024): This test was self-administered by participants at the baseline UK Biobank assessment. In this task, pairs of either identical or different cards were presented on a computer screen. If the two cards were identical, participants had to push a button as quickly as possible. Reaction time (RT) score corresponded with the time, in milliseconds, to identify the matching cards in four trials. Participants were presented with 12 trials in total. The first five trails were used as a practice. Of the remaining seven trials, four presented identical cards. The score is the mean time, in milliseconds, for these four trials. Whereas there are only a few trials, internal consistency is good (Cronbach α = 0.85). Scores were multiplied by −1 such that higher scored indicated more optimal performance.
Matrix Pattern Recognition (n = 11,356): The non-verbal fluid reasoning Matrix Pattern Recognition test is an adaptation of the Matrices test included in the COGNITO battery50, which is similar to the well-known raven’s Progressive Matrices test. This test was self-administered during the assessment centre imaging visit. This test involves inspection of an abstract grid pattern with a piece missing in the lower right-hand corner. The pattern has a logical order. The participant is asked to select the correct multiple-choice option at the bottom of the screen to complete the logical pattern both horizontally and vertically. This 15-item test aims at assessing the ability to solve non-verbal, non-numerical problems using novel and abstract materials. The score is the total number of correctly solved items in three minutes.
Verbal Numerical Reasoning (n = 171,304): At the baseline assessment center visit, a sub-sample of UK Biobank participants self-administered the verbal-numerical reasoning test. Participants were asked 13 multiple-choice questions that assessed verbal and numerical problem solving. The score was the number of questions answered correctly in two minutes. This test has been shown to have adequate test-retest reliability (r = 0.65)51 and internal consistency (Cronbach α = 0.62)31. The verbal-numerical reasoning test was also administered to three sub-samples of participants at the first repeat assessment visit, the assessment center imaging visit, and during the web-based cognitive assessment. In the web-based version of this test there was an additional question, thus the maximum score was 14. In the current analysis the verbal numerical reasoning score used is from the first testing occasion for each participant.
Symbol Digit Substitution (n = 87,741): The symbol digit substitution test was self-administered during both the assessment center imaging visit and the web-based cognitive assessment. Participants were shown a key, pairing shapes with numbers. Participants were asked to use the key to fill the maximum number of empty boxes with the corresponding number paired with shapes in a series of rows. The score is the number of correct symbol-digit matches made in 60 seconds. Those with a score coded as 0 and those with a score greater than 70 had their score set to missing. In this analysis, the scores used were from the first testing occasion for each participant.
Memory – Pairs Matching Test (n = 331,679): At the baseline UK Biobank assessment, memory was measured using a ‘pairs matching’ task. In this self-administered task, participants are shown a randomly arranged, four by three grid of 12 ‘cards’, with six pairs of matching symbols, for five seconds. The symbols were then hidden, and the participant was instructed to select, from memory, the locations of the pairs that matched, in the fewest possible number of attempts. There was no time limit for this task. The memory score was the total number of errors made during this task before all pairs were identified. Scores were multiplied by −1 such that higher scored indicated more optimal performance.
Tower rearranging (n = 11,263): This test was self-administered during the imaging assessment center visit. It is similar to the well-known ‘Tower of Hanoi’ task. Participants were presented with a display (display A) containing three different colored hoops arranged on three pegs (towers). Another display (display B) was shown underneath display A, with the three hoops arranged differently. The task involves deciding how many moves it would take to change display A into display B. The score was the number of correctly-completed trials achieved in three minutes.
Trail Making Test – B (n = 78,547): This test is a computerized version of the Halstead-Reitan Trail Making Test52. The trail making test was self-administered during both the assessment center imaging visit and the web-based cognitive assessment. In part B of the test, participants were presented with the numbers 1-13, and the letters A-L arranged quasi-randomly on a computer screen. The participants were instructed to switch between touching the numbers in ascending order, and the letters in alphabetical order (e.g., 1-A-2-B-3-C) as quickly as possible. The score was the time (in seconds) taken to successfully complete the test. Those with a score coded as 0 (denoting “Trail not completed”) had their score set to missing. Scores were multiplied by −1 such that higher scored indicated more optimal performance. In this analysis, the scores used were from the first testing occasion for each participant.
Genotyping
Prior to release of the UK Biobank genetic dataset, QC measures were applied; these are described in Bycroft et al53. Imputed dosage scores on up to 80,639,280 autosomal variants were analyzed (imputation reference panels included UK10K haplotype, 1000 Genomes Phase 3, and Haplotype Reference Consortium (HRC) panels); all variants had a minor allele frequency ≥ 0.000009 and an imputation quality (INFO) score of > 0.6.
Genome-wide association analyses
Univariate genome–wide association analyses were performed for the covariate-residualized scores on each UK Biobank cognitive phenotype using a linear association test in BGENIE53. As described above, the covariates were age, assessment center (where relevant), genotype batch, array, and 40 genetic principal components. For phenotypes which were collected across multiple testing occasions, a separate GWAS was performed for non-overlapping participants from each occasion and an inverse-variance weighted meta-analysis was implemented in METAL54. As the size of the subsets of individuals for each phenotype vary greatly, an additional QC filter to remove SNPs with a minor allele count < 25 was applied to all GWAS summary results prior to further analyses.
Factor Models
We performed genetic factor analysis on the UK Biobank cognitive phenotypes with Genomic SEM25 and phenotypic factor analysis with the lavaan package for R55. In both genetic and phenotypic factor analysis, a common factor model specifies that k phenotypes are described as linear functions of a smaller set of m (continuous) latent variables: y = Λη+ε. In this equation, y is a k×1 vector of indicators, ε is a k×1 vector of residuals, η is an m×1 vector of common factors, and Λ is a k×m matrix of factor loadings, i.e. regressions relating the common factors to the set of indicators. In the genetic factor model, y represents the genetic components of the GWAS phenotypes, whereas, in the phenotypic factor model, y represents the phenotypes themselves. The model-implied covariance matrix of a CFA is Σ(θ) = ΛΨΛ′+Θ, where Ψ is an m × m latent variable covariance matrix (in the case of a single common factor, Ψ is simply equal to the variance of the factor, which we fix to 1 for scaling identification purposes), and Θ is a k×k matrix of covariances among the residuals, ε (typically a diagonal matrix, to indicate that all indicator residuals are assumed to be independent of one another). A set of parameters (θ) is estimated such that the fit function indexing the discrepancy between the model-implied covariance matrix, Σ(θ), and the empirical covariance matrix, S, is minimized.
For genetic factor modelling in Genomic SEM25, S is a genetic covariance matrix estimated using a multivariable extension of Linkage Disequilibrium Score Regression (LDSC)27. For phenotypic factor modelling in lavaan, S is a phenotypic covariance matrix that is empirically estimated from the raw phenotypic data using full information maximum likelihood estimation. For genetic factor modelling in Genomic SEM the fit function used to estimate the model parameters takes into account the precision of the elements of the S matrix, along with their sampling dependencies (which are needed to appropriately account for sample overlap across the GWAS phenotypes) in the form of a sampling covariance matrix, V, that is estimated using a jackknife resampling procedure in the multivariable extension of LDSC available in Genomic SEM. Model fit is considered good when Σ(θ) closely approximates S. For Genomic SEM, the fit function used was Diagonally Weighted Least Squares (WLS), with a sandwich correction to adjust standard errors of model parameters based on the off-diagonal elements of V. For Phenotypic modeling, the fit function used was maximum likelihood.
In Genomic SEM, goodness-of-fit of the model is assessed by means of the standardized root mean square residual (SRMR), model χ2, Akaike Information Criterion (AIC), and Comparative Fit Index (CFI). For phenotypic factor modelling, we additionally consider Root Mean Square Error of Approximation (RMSEA). Hu and Bentler56 have proposed the following criteria for a good fit: Comparative Fit Index (CFI) >0.95; Root Mean Squared Error of Approximation (RMSEA) < 0.08.
Estimation of SNP-based heritability and genetic correlations using GCTA-GREML
Our primary means of estimating the UK Biobank’s cognitive phenotypes’ SNP-based heritability and genetic correlations was with the multivariable version of LDSC available in Genomic SEM, as described above. However, in order to verify that the estimated genetic correlation matrix was consistent across estimation methods relying on different assumptions, we additionally implemented GCTA-GREML to estimate SNP-based heritability29 and genetic correlations28. Due to computational requirements for the bivariate GCTA-GREML analyses a subset of individuals was created and used for all of the GCTA-GREML analyses. This subset was created by performing listwise deletion for reaction time, memory, VNR, symbol digit substitution, and TMT-B; n = 72,583. The same covariates were included in all GCTA-GREML analyses as for the SNP-based association analyses. One individual was excluded from any pair of individuals who had an estimated coefficient of relatedness of >0.05.
Genetic correlations with neural phenotypes and longevity
We extended the factor models in Genomic SEM to estimate the genetic correlations between the genetic g factor from the UK Biobank cognitive phenotypes and each of nine collateral phenotypes in turn: Educational Attainment32; general cognitive function from Davies et al.19, Savage et al.21, and Hill et al.20; total brain volume from UKB37; Alzheimer’s disease34, Schizophrenia35, Attention Deficit Hyperactive Disorder (ADHD)36, Autism Spectrum Disorder (ASD)57, and longevity38.
Multivariate GWAS in Genomic SEM
Using the univariate summary statistics for each of the seven UK Biobank cognitive phenotypes, Genomic SEM25 was used to conduct a multivariate GWAS, with the genetic g factor as the GWAS target (see left portion of Supplementary Figure 9). As the typical unit variance scaling cannot be directly specified in a model in which the latent factor is a dependent variable, we specified unit loading scaling (with Matrices as the reference indicator; Supplementary Table 29). Genomic SEM provides a SNP-specific heterogeneity statistic, Q, which indexes the extent to which the specified factor model is insufficient to account for the SNP effects on the individual traits analyzed. High Q values for a given SNP, indicate that a model which estimates SNP associations with each individual trait (see right portion of Supplementary Figure 9) fits better than a model which estimates a single SNP association with the factor. In other words, SNPs with genome-wide significant Q values may be interpreted as SNPs that do not affect the genetic g. A detailed explanation of the Q statistic is provided in the supplemental online text (“Interpreting the Heterogeneity Statistic”).
For multivariate GWAS in Genomic SEM, summary statistics for the individual tests were restricted to SNPs with a MAF > 1%, an INFO score > 0.6, and to SNPs that were present for all seven cognitive tests. The summary statistics were also filtered to SNPs present in the European only 1000 Genomes Phase 3 reference panel, as the SNP minor allele frequencies from the reference panel are necessary to obtain their variances for inclusion in the genetic covariance (S) matrix. Using these QC steps, 7,857,346 SNPs were present across all seven cognitive tests. Note that all cognitive phenotypes had already been residualized for covariates when conducting the univariate GWASs, summary statistics for which enter into multivariate analyses within Genomic SEM, such that further covariate-adjustment was not needed at this stage.
We adopt the field standard alpha threshold of 5×10−8 for GWAS. This threshold amounts to a Bonferroni correction for the theoretical number of independent tests in the genome given known LD structure58. As all seven phenotypes are highly genetically correlated, it is inappropriate to perform an additional Bonferroni correction for the 7 additional phenotypes, and we do not focus our interpretation on results of the seven univariate GWASs. Rather, we focus on results of the multivariate GWAS within Genomic SEM for which there are two families of theoretical tests, each constituting a single set of GWAS-type statistics: (1) genome-wide SNP associations with genetic g, and (2) genome-wide SNP-specific heterogeneity indices (Q).
Genome-wide significant loci using FUMA
Genome-wide significant loci were defined from the SNP-based association results, using Functional Mapping and Annotation of genetic associations (FUMA)59. The SNP2GENE function was used to identify independent significant SNPs defined as SNPs with a P-value of ≤5 × 10−8 and independent of other genome wide significant SNPs at r2 < 0.6. Tagged SNPs, for use in subsequent annotations, were then identified as all SNPs that had a MAF ≥ 0.0005 and were in LD of r2 ≥ 0.6 with at least one of the independent significant SNPs. These tagged SNPs included those from the 1000 genomes reference panel and need not have been included in the GWAS performed in the current study. Genome-wide significant loci that were 250 kb or closer were merged into a single locus. Lead SNPs were defined as independent significant SNPs that were independent from each other at r2 < 0.1. We performed lookups on all tagged SNPs (r2 > 0.6) within each locus, including all 1000 genomes SNPs; previously reported genome-wide significant findings are detailed in Supplementary Tables 22–28.
Polygenic prediction
Generation Scotland: the Scottish Family Health Study (GS) is a family-structured, population-based cohort study recruited between 2006 and 2011. Participant recruitment occurred in Glasgow, Tayside, Ayrshire, Arran, and North-East Scotland, yielding a total sample size of 24,084 with an age range between 18 and 100 years, and up to four generations per family, of which we selected one participant per family. A full cohort description is provided elsewhere40,41 and online at http://www.generationscotland.org/. Ethical approval for GS was obtained from the Tayside Committee on Medical Research Ethics (on behalf of the National Health Service). Genotyping, using the Illumina HumanOmniExpressExome-8 v1.0 chip, was performed at the Edinburgh Clinical Research Facility, University of Edinburgh60. Participants were removed from GS if they had contributed to both GS and UK Biobank (n = 622). For the PGS analyses 6,950 unrelated GS participants were retained.
The cognitive measures available in GS were Wechsler Logical Memory (episodic memory), Mill Hill Vocabulary (crystallized knowledge), Wechsler Digit Symbol Substitution (processing speed), and Verbal Fluency (semantic fluency), as described previously40,41. We created a phenotypic g using the first unrotated principal component of the cognitive measures, and also a fluid g using the first unrotated principal component of the cognitive measures excluding the Mill Hill Vocabulary scores.
Polygenic profile scores were created using PRSice version 2 (https://github.com/choishingwan/PRSice) using a pre-specified p-value threshold of 1.0, i.e. all SNPs61. Summary results from the genetic g and univariate cognitive test GWAS were used to create polygenic profile scores for the GS individuals. Prior to creating these scores SNPs with a MAF < 0.01 were removed and clumping was used to obtain SNPs in linkage disequilibrium with an r2 < 0.25 within a 250 kb window. Polygenic profile scores for the individual cognitive tests were created using all available SNPs from the GWAS summary results. For genetic g, all SNPs located within significant Q loci were removed from the GWA summary results prior to the profile scores being created.
Linear regression models were used to examine the associations between the polygenic profile scores and cognitive performance and educational attainment in GS. All models included age at measurement, sex, and 10 genetic principal components to adjust for population stratification. We created regression models fitting each polygenic score individually, a multivariate model including all eight polygenic scores (genetic g, reaction time, memory, matrix, symbol digit substitution, trail making test B, and tower rearranging) and, a series of models which fitted the genetic g polygenic score plus one individual cognitive test score. From these models we were able to determine contributions of genetic g and each individual UK Biobank cognitive test to prediction of variance in cognitive performance and educational attainment in an independent sample, GS.
Supplementary Material
Acknowledgements
This article benefited from valuable discussions with Michel Nivard.
Funding:
This work was supported by National Institutes of Health (NIH) grant R01AG054628. The Population Research Center at the University of Texas is supported by NIH grant P2CHD042849. I.J.D. and G.D. are within the Lothian Birth Cohorts group, which is funded by Age UK (Disconnected Mind grant), the Medical Research Council (grant MR/R024065/1), and the University of Edinburgh’s School of Philosophy, Psychology and Language Sciences. This research was conducted using the UK Biobank Resource (Application Nos. 10279 and 4844). We are grateful for the availability of data from Generation Scotland: Scottish Family Health Study. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates [CZD/16/6] and the Scottish Funding Council [HR03006]. Genotyping of the GS:SFHS samples was carried out by the Genetics Core Laboratory at the Edinburgh Clinical Research Facility, University of Edinburgh, Scotland and was funded by the Medical Research Council UK and the Wellcome Trust (Wellcome Trust Strategic Award “STratifying Resilience and Depression Longitudinally” (STRADL) Reference 104036/Z/14/Z).
Footnotes
Code Availability
Code to perform common factor modeling and multivariate GWAS within Genomic SEM can be found at https://github.com/MichelNivard/GenomicSEM/wiki.
Competing Interests
I.J.D is a participant in UK Biobank. All other authors declare no competing interests.
Data Availability
Complete summary GWAS results from this paper will be made available at the time of publication at https://www.lothianbirthcohort.ed.ac.uk/content/gwas-summary-data. Raw data for UK Biobank can be requested at https://www.ukbiobank.ac.uk/register-apply/. Raw data for Generation Scotland can be requested at https://www.ed.ac.uk/generation-scotland/using-resources/access-to-resources/access-process.
References
- 1.Deary IJ, Strand S, Smith P & Fernandes C Intelligence and educational achievement. Intelligence (2007) doi: 10.1016/j.intell.2006.02.001. [DOI] [Google Scholar]
- 2.Strenze T Intelligence and socioeconomic success: A meta-analytic review of longitudinal research. Intelligence (2007) doi: 10.1016/j.intell.2006.09.004. [DOI] [Google Scholar]
- 3.Calvin CM et al. Childhood intelligence in relation to major causes of death in 68 year follow-up: Prospective population study. BMJ (2017) doi: 10.1136/bmj.j2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spearman C ‘General Intelligence,’ Objectively Determined and Measured. Am. J. Psychol (1904) doi: 10.2307/1412107. [DOI] [PubMed] [Google Scholar]
- 5.Carroll JB The Identification and Description of Cognitive Abilities, in Human Cognitive Abilities: A Survey of Factor-Analytic Studies (Cambridge University Press, 1993). doi: 10.1017/CBO9780511486371. [DOI] [Google Scholar]
- 6.Johnson W, Nijenhuis J. te & Bouchard TJ Still just 1 g: Consistent results from five test batteries. Intelligence (2008) doi: 10.1016/j.intell.2007.06.001. [DOI] [Google Scholar]
- 7.Tucker-Drob EM Differentiation of Cognitive Abilities Across the Life Span. Dev. Psychol 45, 1097–1118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox SR, Ritchie SJ, Fawns-Ritchie C, Tucker-Drob EM & Deary IJ Structural brain imaging correlates of general intelligence in UK Biobank. Intelligence (2019) doi: 10.1016/j.intell.2019.101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deary IJ Looking Down on Human Intelligence: From Psychometrics to the Brain. (Cambridge University Press, 2000). [Google Scholar]
- 10.Deary IJ, Penke L & Johnson W The neuroscience of human intelligence differences. Nature Reviews Neuroscience (2010) doi: 10.1038/nrn2793. [DOI] [PubMed] [Google Scholar]
- 11.Haier RJ The Neuroscience of Intelligence. (Cambridge University Press, 2016). [Google Scholar]
- 12.Kovas Y & Plomin R Generalist genes: implications for the cognitive sciences. Trends Cogn. Sci (2006) doi: 10.1016/j.tics.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 13.Plomin R & Deary IJ Genetics and intelligence differences: Five special findings. Molecular Psychiatry (2015) doi: 10.1038/mp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelhardt LE et al. Strong genetic overlap between executive functions and intelligence. J. Exp. Psychol. Gen (2016) doi: 10.1037/xge0000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panizzon MS et al. Genetic and environmental influences on general cognitive ability: Is g a valid latent construct? Intelligence (2014) doi: 10.1016/j.intell.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrill SA Molarity versus modularity of cognitive functioning? A behavioral genetic perspective. Curr. Dir. Psychol. Sci (1997) doi: 10.1111/1467-8721.ep11512833. [DOI] [Google Scholar]
- 17.Petrill SA et al. The genetic and environmental relationship between general and specific cognitive abilities in twins age 80 and older. Psychol. Sci (1998) doi: 10.1111/1467-9280.00035. [DOI] [Google Scholar]
- 18.Rijsdijk FV, Vernon PA & Boomsma DI Application of hierarchical genetic models to raven and WAIS subtests: A Dutch twin study. Behav. Genet (2002) doi: 10.1023/A:1016021128949. [DOI] [PubMed] [Google Scholar]
- 19.Davies G et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun (2018) doi: 10.1038/s41467-018-04362-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hill WD et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol. Psychiatry (2019) doi: 10.1038/s41380-017-0001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savage JE et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet (2018) doi: 10.1038/s41588-018-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies G et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151). Mol. Psychiatry (2016) doi: 10.1038/mp.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Debette S et al. Genome-wide studies of verbal declarative memory in nondemented older people: The Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium. Biol. Psychiatry (2015) doi: 10.1016/j.biopsych.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmidt FL Beyond questionable research methods: The role of omitted relevant research in the credibility of research. Arch. Sci. Psychol (2017) doi: 10.1037/arc0000033. [DOI] [Google Scholar]
- 25.Grotzinger AD et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav (2019) doi: 10.1038/s41562-019-0566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fawns-Ritchie C & Deary IJ Reliability and validity of the UK Biobank cognitive tests. medRxiv (2019) doi: 10.30806/fs.24.3.201909.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet (2015) doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SH, Yang J, Goddard ME, Visscher PM & Wray NR Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics (2012) doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet (2011) doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marioni RE et al. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry (2018) doi: 10.1038/s41398-018-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hagenaars SP et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol. Psychiatry (2016) doi: 10.1038/mp.2015.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JJ et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet (2018) doi: 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turley P et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet (2018) doi: 10.1038/s41588-017-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert JC et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet (2013) doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee PH et al. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell (2019) doi: 10.1016/j.cell.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet (2019) doi: 10.1038/s41588-018-0269-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao B et al. Large-scale GWAS reveals genetic architecture of brain white matter microstructure and genetic overlap with cognitive and mental health traits (n = 17,706). Mol. Psychiatry (2019) doi: 10.1038/s41380-019-0569-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Timmers PR et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. Elife (2019) doi: 10.7554/eLife.39856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anttila V et al. Analysis of shared heritability in common disorders of the brain. Science (80-. ). (2018) doi: 10.1126/science.aap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith BH et al. Generation Scotland: The Scottish Pamily Health Study; a new resource for researching genes and heritability. BMC Med. Genet (2006) doi: 10.1186/1471-2350-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith BH et al. Cohort profile: Generation Scotland: Scottish family health study (GS: SLHS). The study, its participants and their potential for genetic research on health and illness. Int. J. Epidemiol (2013) doi: 10.1093/ije/dys084. [DOI] [PubMed] [Google Scholar]
- 42.Jensen AR Clocking the Mind: Mental Chronometry and Individual Differences. Pers. Psychol (2008) doi: 10.1111/j.1744-6570.2008.00111_7.x. [DOI] [Google Scholar]
- 43.Demange PA et al. Investigating the Genetic Architecture of Non-Cognitive Skills Using GWAS-by-Subtraction. bioRxiv (2020) doi: 10.1101/2020.01.14.905794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kovacs K & Conway ARA Process Overlap Theory: A Unified Account of the General Factor of Intelligence. Psychol. Inq (2016) doi: 10.1080/1047840X.2016.1153946. [DOI] [Google Scholar]
- 45.Bartholomew DJ, Deary IJ & Lawn M A New Lease of Life for Thomson’s Bonds Model of Intelligence. Psychol. Rev (2009) doi: 10.1037/a0016262. [DOI] [PubMed] [Google Scholar]
- 46.Ceci SJ On intelligence--more or less: A bio-ecological treatise on intellectual development. (Prentice Hall, 1990). [Google Scholar]
- 47.Van Der Maas HLJ et al. A dynamical model of general intelligence: The positive manifold of intelligence by mutualism. Psychol. Rev (2006) doi: 10.1037/0033-295X.113.4.842. [DOI] [PubMed] [Google Scholar]
- 48.Dickens WT What is g? (2007). [Google Scholar]
- 49.Tucker-Drob EM, Brandmaier AM & Lindenberger U Coupled cognitive changes in adulthood: A meta-analysis. Psychological Bulletin vol. 145 273–301 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karen R et al. COGNITO: Computerized Assessment of Information Processing. J. Psychol. Psychother (2014) doi: 10.4172/2161-0487.1000136. [DOI] [Google Scholar]
- 51.Lyall DM et al. Cognitive test scores in UK biobank: Data reduction in 480,416 participants and longitudinal stability in 20,346 participants. PLoS One (2016) doi: 10.1371/journal.pone.0154222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reitan RM & Wolfson D The Halstead-Reitan neuropsychological test battery: Theory and clinical interpretation (Vol. 4). (Neuropsychology Press, 1985). [Google Scholar]
- 53.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature (2018) doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Willer CJ, Li Y & Abecasis GR METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics (2010) doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosseel Y Lavaan: An R package for structural equation modeling. J. Stat. Softw (2012) doi: 10.18637/jss.v048.i02. [DOI] [Google Scholar]
- 56.Hu LT & Bentler PM Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct. Equ. Model (1999) doi: 10.1080/10705519909540118. [DOI] [Google Scholar]
- 57.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet (2019) doi: 10.1038/s41588-019-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pe’er I, Yelensky R, Altshuler D & Daly MJ Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol 32, 381–385 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Watanabe K, Taskesen E, Van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun (2017) doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kerr SM et al. Pedigree and genotyping quality analyses of over 10,000 DNA samples from the Generation Scotland: Scottish Family Health Study. BMC Med. Genet (2013) doi: 10.1186/1471-2350-14-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Euesden J, Lewis CM & O’Reilly PF PRSice: Polygenic Risk Score software. Bioinformatics (2015) doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete summary GWAS results from this paper will be made available at the time of publication at https://www.lothianbirthcohort.ed.ac.uk/content/gwas-summary-data. Raw data for UK Biobank can be requested at https://www.ukbiobank.ac.uk/register-apply/. Raw data for Generation Scotland can be requested at https://www.ed.ac.uk/generation-scotland/using-resources/access-to-resources/access-process.