

Three article in this issue present preclinical data aimed at improving immunotherapeutic approaches for relapsed B- and T-cell acute lymphoblastic leukemia (T-ALL). Chimeric antigen receptor T (CAR-T) cell therapy directed against relapsed T-ALL is hampered by difficulty in identifying a target antigen that is not also expressed on healthy T cells, risking “fratricide” with T-cell aplasia and loss of the CAR-T cells. Maciocia and colleagues identify CCR9 as an antigen expressed on >85% of relapsed T-ALL and on <5% of normal T cells, demonstrating in cell lines and patient-derived xenografts that CAR-T cells targeting CCR9 have potent anti-leukemic activity without any evidence of fratricide. In the second article, Müller et al explore the potential of dual antibody targeting of CD47 and CD38 in a preclinical models of T-ALL; although CD47 does not avoid the risk of aplasia, it shows efficacy in combination with daratumumab in treating relapsed T-ALL and eradicating MRD. Finally, the third article addresses relapse following CD19-directed immunotherapies for B-ALL, which may be mediated by recurrence of CD19-negative leukemia. Bueno et al identify CD34+CD19−CD22+ cells in patients at diagnosis and relapse; they further show that CD22 expression precedes CD19 expression and that this population harbors the same genetic abnormalities of the B-ALL clone. They propose that this may represent a progenitor population that can give rise to B-ALL recurrence, supporting the use of combined CAR-T therapies, perhaps directed at both CD19 and CD22.
Key Points
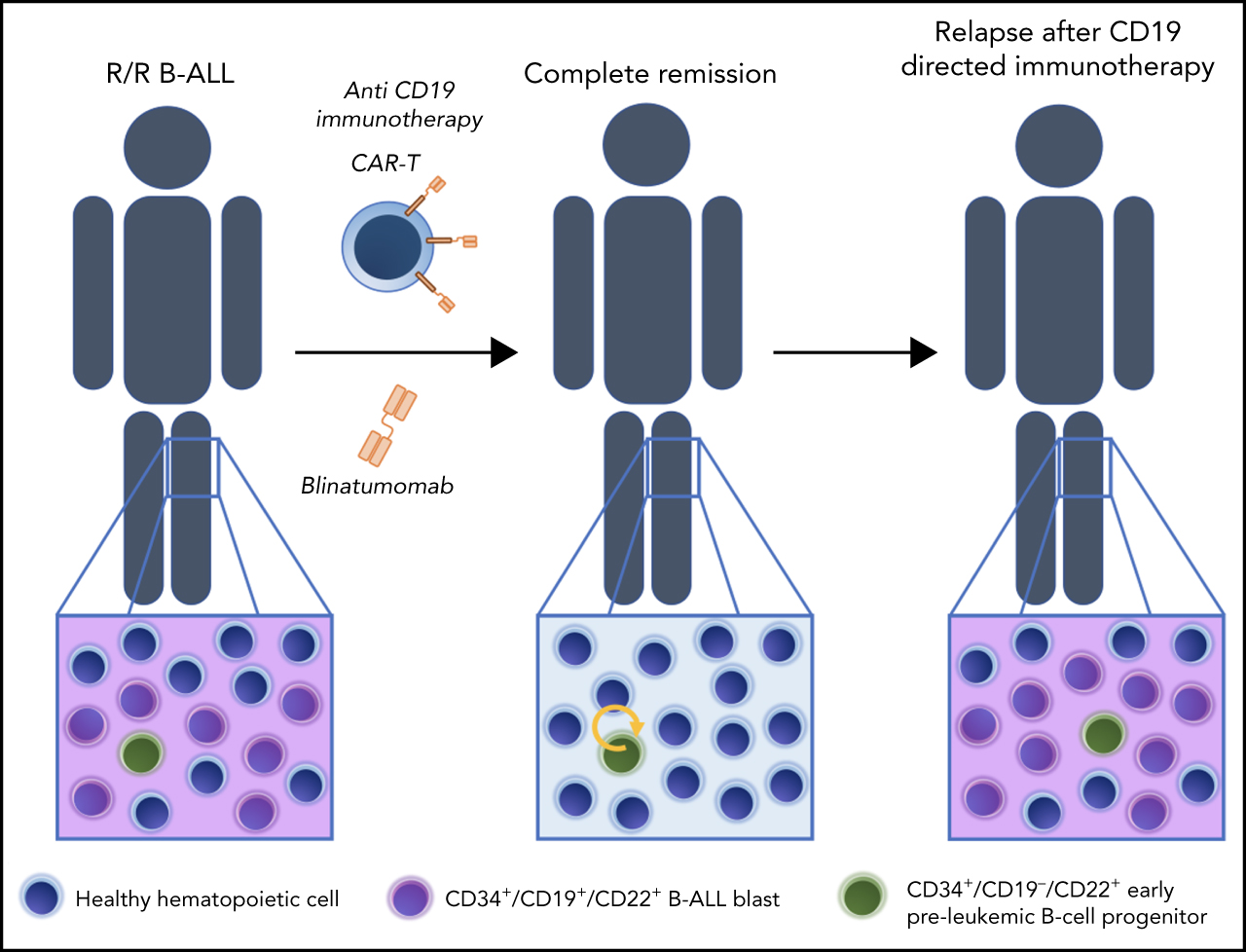
Preleukemic CD34+CD19−CD22+ immature progenitors may underlie phenotypic escape in patients treated with CD19-directed immunotherapies.
The study contributes to identifying patients with B-ALL at risk of failure of CD19-targeted therapy.
Visual Abstract

Abstract
CD19-directed immunotherapies have revolutionized the treatment of advanced B-cell acute lymphoblastic leukemia (B-ALL). Despite initial impressive rates of complete remission (CR) many patients ultimately relapse. Patients with B-ALL successfully treated with CD19-directed T cells eventually relapse, which, coupled with the early onset of CD22 expression during B-cell development, suggests that preexisting CD34+CD22+CD19− (pre)-leukemic cells represent an “early progenitor origin-related” mechanism underlying phenotypic escape to CD19-directed immunotherapies. We demonstrate that CD22 expression precedes CD19 expression during B-cell development. CD34+CD19−CD22+ cells are found in diagnostic and relapsed bone marrow samples of ∼70% of patients with B-ALL, and their frequency increases twofold in patients with B-ALL in CR after CD19 CAR T-cell therapy. The median of CD34+CD19−CD22+ cells before treatment was threefold higher in patients in whom B-ALL relapsed after CD19-directed immunotherapy (median follow-up, 24 months). Fluorescence in situ hybridization analysis in flow-sorted cell populations and xenograft modeling revealed that CD34+CD19−CD22+ cells harbor the genetic abnormalities present at diagnosis and initiate leukemogenesis in vivo. Our data suggest that preleukemic CD34+CD19−CD22+ progenitors underlie phenotypic escape after CD19-directed immunotherapies and reinforce ongoing clinical studies aimed at CD19/CD22 dual targeting as a strategy for reducing CD19− relapses. The implementation of CD34/CD19/CD22 immunophenotyping in clinical laboratories for initial diagnosis and subsequent monitoring of patients with B-ALL during CD19-targeted therapy is encouraged.
Introduction
Both bispecific antibody-based (blinatumomab) and cell-based (chimeric antigen receptor [CAR]), CD19-directed immunotherapies have been transformational in the treatment of relapsed or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL).1 Despite high rates of complete response (CR), many patients eventually relapse over time.2 On the one hand, relapses that retain CD19 expression (CD19+ relapses) may be observed and are commonly associated with poor T-cell function or loss of CAR T cells, most likely because of T-cell intrinsic immune-regulatory exhaustion signals.3-5 On the other hand, relapses where the disease recurs with loss of CD19 expression (CD19− relapses) have also emerged as a major clinical challenge. Mechanisms of lineage switch or restrained membrane expression of CD19 (ie, splice variants or truncating mutations in the CD19 gene) underlie the loss of CD19, which represents a widely accepted immune escape mechanism to CD19-directed immunotherapies.6-12 The intrinsic properties of leukemic cells and the tumor microenvironment surely contribute to the mechanisms underlying resistance to CD19-directed immunotherapy pressure.13
Normal human B-cell development occurs through multistep commitment to B cells from hematopoietic stem/progenitor cells (HSPCs),14,15 which is characterized by serial acquisition of B-cell–associated cell surface antigens, with CD19 being the definitive marker of B-cell commitment, almost always strongly expressed on B-ALL cells.16 Human fetal B-lymphoid development has been recently described with CD34+CD19+CD10− PreProB progenitors identified as the earliest B-committed progenitors (Figure 1A). However, the panspecific B-cell surface protein CD22 is considered to be a very early marker for B-cell commitment and its expression precedes that of CD19.17 In fact, dual targeting of CD19 and CD22 is being clinically investigated as a strategy to overcome single target-associated immune escape.18,19
Figure 1.

CD22 expression precedes CD19 expression during normal human B-cell differentiation. (A) Cartoon depicting the immunophenotypes used to flow sort the indicated HSPC populations across human fetal B-cell differentiation. Lineage markers: Lin: CD2/3/14/16/19/56/235a; Lin2: CD2/3/14/16/56/235a. (B) Bulk RNA-seq–normalized gene expression (transcripts per million [TPM]) of CD19 (left) and CD22 (right) in flow-sorted hematopoietic stem cells (HSCs), multipotent progenitors (MPPs), lymphoid-primed multipotent progenitors (LMPPs), early lymphoid progenitors (ELPs), Pre-ProB progenitors, ProB progenitors, and PreB cells from human FBM (n = 3). (C) Percentage of CD22+ cells within these FBM-derived HSPC populations analyzed by flow cytometry. (D) scRNA-seq UMAP plots showing CD22 expression within flow-sorted Lin−CD19−CD34+ cells from matched human FL (left) and human FBM (right) (n = 2; total, 30 000 cells). (E) Violin plots showing the log normalized median expression levels of CD22 in CD19−Lin−CD34+ cells from human FL and FBM. (F) Frequency of CD34+CD19−CD22+ cells in healthy FL (n = 8), FBM (n = 9), neonatal CB (n = 10), and adult BM (n = 13). Red horizontal lines represent the median. Inset: a representative FACS plot of CD22 and CD19 within the gated CD34+ cells. Blue cells are non-B-progenitor cells (mainly myeloid). Red cells are CD19+CD22+ bona fide B-cell progenitors. Green cells are early CD22+CD19− B-cell progenitors. CB, cord blood; FACS, fluorescence activated cell sorting; UMAP, Uniform Manifold Approximation and Projection.
Relapse of patients with B-ALL who had achieved CR after successful anti-CD19–directed CAR T-cell therapy coupled to the very early onset of CD22 expression during B-cell development prompted us to hypothesize that preexisting CD22+CD19− (pre)-leukemic cells may represent an early/immature progenitor origin-related mechanism that underlies phenotypic escape to CD19-directed immunotherapies. In fact, an elegant single-cell RNA-sequencing (scRNA-seq) study profiled leukemic cells from 1 patient with B-ALL who experienced CD19− B-ALL relapse after CD19-directed CAR T-cell therapy and showed that CD19− leukemic cells were already present before CAR T-cell therapy, suggesting that relapse emerges from the selection of rare CD19− B-ALL clones.20
In this study, using bulk and scRNA-seq on multiple cell populations across fetal human B-cell development and immunophenotyping characterization of healthy fetal, neonatal, and postnatal CD34+progenitors, we demonstrate that CD22 precedes CD19 in normal B-cell development and that CD34+CD19−CD22+ early/immature progenitors are found in most patients with relapsed B-ALL. A trend toward an increased frequency of these cells before treatment was observed in patients with R/R B-ALL who had a relapse after CD19-directed immunotherapy. Fluorescence in situ hybridization (FISH) studies in flow-sorted cell populations, together with in vivo xenograft modeling, revealed that CD34+CD19−CD22+ cells harbor the genetic abnormalities present at diagnosis and initiate leukemogenesis in vivo, phenocopying the primary diagnostic B-ALL. We suggest that preleukemic CD34+CD19−CD22+ immature progenitors underlie phenotypic escape in patients treated with CD19-directed immunotherapies. Our study helps to identify patients at risk of CD19-targeted therapy failure.
Study design
Detailed information on B-ALL and healthy primary samples, clinical correlations, bulk RNA-seq, scRNA-seq, immunophenotyping, FISH on flow-sorted CD34+ subpopulations, in vivo xenografts, targeted next-generation sequencing, and optical genome mapping (OGM) can be found in the supplemental Methods (available on the Blood Web site).
The study was approved by the institutional review board of the Ethics Committee on Clinical Research of the Clinic Hospital of Barcelona (HCB/2013/868) and the Barcelona Biomedical Research Park Ethics Committee on Animal Experimentation (HRH-17-0029-P1). Ethics regarding use of human fetal samples were also covered (18/NE/0290 and 18/LO/0822).
Results and discussion
CD22 expression precedes CD19 in normal B-cell development
To define the earliest HSPC fractions that express CD19 and CD22, both at gene and protein level, we examined specific HSPC populations from second-trimester fetal bone marrow (FBM) (Figure 1A) by bulk and scRNA-seq and by multiparametric flow cytometry. There was weak expression of CD19 on cells of the lymphoid-primed multipotent progenitor (%s) and the early lymphoid progenitor (ELP) stages, but its expression progressively increased with B-cell commitment in PreProB and ProB progenitors. However, CD22 was readily expressed at the LMPP stage and was robustly upregulated in ELPs, which showed CD22 expression levels similar to those of PreProB and ProB progenitors (Figure 1A-B). This finding was confirmed by flow cytometry where CD22 expression was positive in 1.5% ± 1.2% of HSCs/MPPs, 9% ± 5.4% of LMPPs, and 77% ± 8.9% of ELPs (Figure 1C). Using our own scRNA-seq transcriptomic dataset,21 CD22 expression was confirmed in Lin−CD19−CD34+ fetal liver (FL)- and FBM-HSPCs at a single-cell level, with 2% of FL and 8% of FBM Lin−CD19−CD34+ cells expressing CD22, respectively (Figure 1D-E). In addition, ∼13% of FL- and FBM-CD34+ HSPCs and 3% to 5% of postnatal (cord blood and adult BM)–derived CD34+ cells are CD19−CD22+, as confirmed by flow cytometry (Figure 1F). These data confirm that CD22 precedes CD19 in normal human B-cell development in very early lymphoid/B progenitors.
CD34+CD19−CD22+ (pre)-leukemic progenitors are present in patients with B-ALL, harbor the diagnostic genetic abnormalities, and initiate leukemogenesis in vivo phenocopying the disease
The relapse of patients with B-ALL who had achieved CR after successful anti-CD19–targeted immunotherapy and our data showing the very early onset of CD22 expression during B-cell development suggest that preexisting CD34+CD19−CD22+ (pre)-leukemic cells represent early lymphoid/B progenitors underlying the phenotypic escape of B-ALL in response to CD19-directed immunotherapies. Accordingly, Rabilloud et al have very recently shown in a patient with B-ALL that CD19− leukemic cells are already present before CAR T-cell therapy, suggesting that relapse emerges from the selection of rare CD19− B-ALL clones.20 We next investigated the presence of CD34+CD19−CD22+ cells in nonmatched BM samples from 237 patients with B-ALL. CD34+CD19−CD22+ early lymphoid/B progenitors were found in 72% (mean, 2.1% ± 7.6%), 92% (mean, 6.1% ± 17%), and 66% (mean, 1.6% ± 3.4%) of diagnostic, CR, and relapsed B-ALL BM samples, respectively (Table 1; Figure 2A). This CD34+CD19−CD22+ population was similarly represented among the major cytogenetic/molecular groups (data not shown). In addition, the frequency of CD34+CD19−CD22+ cells was 1.7-fold higher (3.8 ± 2.9 vs 2.1 ± 1.8; P = .016) in BM on day +30 than before CAR T-cell infusion, in 15 of 20 (75%) patients with B-ALL who achieved CR after CD19-CAR T-cell therapy (Figure 2B). This results is indicative of a relative increase in the proportion of CD34+CD19−CD22+ cells as a result of the targeted elimination of CD19+ blasts. We next analyzed 53 patients with R/R B-ALL treated with CD19-directed immunotherapy (n = 37 CD19− CAR T-cells; n = 16 blinatumomab) to assess the impact of CD34+CD19−CD22+ cells on disease relapse risk within a median follow-up of 24 months (Table 2). The median of CD34+CD19−CD22+ cells before treatment was threefold higher in patients with B-ALL who had a relapse after CD19-directed immunotherapy (1% vs 0.3%; P > .05; Figure 2C). Furthermore, the cumulative incidence of relapse was analyzed by splitting our cohort into 2 groups, according to the frequency of CD34+CD19−CD22+ cells. Although not statistically significant, a trend toward a higher cumulative incidence of relapse was found in patients with a frequency of CD34+CD19−CD22+ cells above the mean (supplemental Figure 1), suggesting that the presence of CD34+CD19−CD22+ cells negatively impacts relapse-free survival. This premise must be validated on a larger, more homogeneous series of patients.
Table 1.
Clinical characteristics of patients with B-ALL according to flow cytometry immunophenotyping
| Age, y (range) | 40 (17-60) |
|---|---|
| Sex, % | |
| Male | 50.7 |
| Female | 49.3 |
| Genetics, n | |
| t(9;22)+ | 26 |
| Aneuploid | 24 |
| TCF3* | 12 |
| Normal karyotype | 18 |
| AML1 Gain | 10 |
| t(14q32)(IGH) | 8 |
| MLL† | 6 |
| TELdel | 5 |
| del(9) | 4 |
| t(12;21)+ | 5 |
| t(9;12)+ | 3 |
| CRLF2mut | 2 |
| t(4;17)+ | 1 |
| t(12;13)+ | 1 |
| t(2;4)+ | 1 |
| t(X;10)+ | 1 |
| RB/p53mut | 1 |
| Failed karyotype | 20 |
| NA | 89 |
N = 237. NA, not available.
Deletion/translocation.
Translocation/gain.
Figure 2.

CD34+CD19−CD22+ (pre)-leukemic progenitors are present in patients with B-ALL, harbor the genetic abnormalities present at disease presentation, and initiate leukemogenesis in NSG mice, phenocopying the primary B-ALL sample. (A) Frequency of CD34+CD19−CD22+ cells in nonmatched samples from patients with B-ALL at diagnosis (n = 159), CR (n = 63), and relapse (n = 15). The black horizontal line represents the mean. FACS analysis (top) of 3 representative patients with a high proportion (left), low proportion (middle), and absence (right) of CD34+CD19−CD22+ cells. (B) Frequency of CD34+CD19−CD22+ cells in 20 patients with B-ALL before (pre) and after (post, BM day +30) treatment with CD19-directed CAR T-cells. Top panel shows representative FACS analysis of CD22 and CD19 within the gated CD34+ cells from patients with B-ALL before and after CD19-directed CAR T-cell infusion. Orange dots identify CD34+CD19−CD22+ cells. (C) Frequency of CD34+CD19−CD22+ cells in 53 patients with R/R B-ALL treated with CD19-directed immunotherapy (n = 37 CD19−CAR T-cells; n = 16 blinatumomab) who did or did not have a relapse after a median follow-up of 24 months. (D) Representative FISH analysis performed in flow-sorted CD34+CD19-CD22+ and CD34+CD19+CD22+ cells from 3 patients with B-ALL revealing the presence of the diagnostic genetic abnormality in preleukemic CD34+CD19−CD22+ progenitors. (E) Summary of the FISH analysis in diagnosed whole-BM and flow-sorted CD34+CD19−CD22+ and CD34+CD19+CD22+ cells. (F) Scheme of the experimental plan designed to study the ability of the preleukemic CD34+CD19−CD22+ progenitors and the bona fide CD34+CD19+CD22+ blasts to initiate B-ALL in vivo in NSG mice. Leukemic onset was evaluated every other week by PB bleeding. When leukemia was evident, mice were euthanized and the leukemic cells were characterized by FACS immunophenotyping and by NGSeq/Bionano technology to confirm the presence of the patient-specific genetic/molecular diagnostic alterations. (G) Representative B-ALL engraftment in mouse transplant recipients of CD34+CD19−CD22+ or CD34+CD19+CD22+ B-ALL cells. Blue cells represent the CD45+HLA−ABC+ human graft. (H) Targeted NGSseq (left) and circus plot from Bionano (right) confirming the identity of the B-ALL graft from CD34+CD19−CD22+ cells. NGSeq, next generation sequencing.
Table 2.
Clinical features and type of CD19-directed immunotherapy received by the 53 patients with R/R B-ALL and used for clinicobiological correlations
| Group | n |
|---|---|
| Adult vs Pediatric | |
| Pediatric* | 15 |
| Adult | 38 |
| CD19-directed immunotherapy | |
| Blinatumomab | 16 |
| Kymriah | 14 |
| ARI001† | 23 |
| Clinical outcome | |
| Relapse | 27 |
| No relapse | 26 |
Adapted from Ortiz-Maldonado et al.22,23
Age < 18 years.
Academic local CD19 CAR phase 1/2 trial (completed).
We next assessed whether CD34+CD19−CD22+ cells were normal hematopoietic progenitors or harbored the molecular oncogenic lesions observed at disease presentation. CD34+CD19−CD22+ and CD34+CD19+CD22+ cells from 3 patients with B-ALL harboring different genetic lesions (11q23, t(8;14), MLL+) were purified by fluorescence-activated cell sorting at disease presentation, immediately fixed, and subjected to interphase FISH analysis with commercial chromosomal probes routinely used in diagnostic laboratories. CD34+CD19−CD22+ cells from all 3 patients harbored the molecular lesion (15%, 32%, and 93%, respectively; Figure 2D-E). These data strongly support previous studies demonstrating that the initiating molecular lesions in B-ALL arise very early during B-cell development, in pre-VDJ progenitors that are likely to be CD19−.24-26
The cell of origin and the leukemia-initiating cell may represent different entities. We finally wanted to assess whether these CD34+CD19−CD22+ (pre)-leukemic progenitors can initiate leukemia in vivo. CD34+CD19−CD22+ or CD34+CD19+CD22+ cells (1.3 × 105) from pooled samples (because of the rarity of these CD34+CD19−2CD22+ cells) from CD19 CAR T-cell–treated patients with B-ALL were flow sorted and transplanted into sublethally irradiated NSG mice by intraosseous transplantation (Figure 2F). Strikingly, both CD34+CD19−CD22+ and CD34+CD19+CD22+ populations initiated leukemia in vivo that phenocopied the primary diagnostic B-ALL (CD45+CD19+CD22+; Figure 2G). The leukemic identity of the human graft from CD34+CD19−CD22+ cells was confirmed by detecting the pathogenic alterations found at diagnostic by targeted next-generation sequencing and optical genome mapping (Figure 2H). This, together with the B-lymphoid–skewed graft in the absence of a myeloid graft (Figure 2G), confirms that the leukemic graft stems from a (pre)-leukemic B-cell clone and not from residual healthy HSPCs.
We report a possible “phenotypic escape” mechanism of resistance to CD19-directed immunotherapies. We describe the existence of (pre)-leukemic CD34+CD22+CD19− immature B-cell progenitors that are not recognized by CD19-directed CAR T cells. This phenotypic escape mechanism differs from, but does not necessarily lead to, “immune escape” mechanisms driven by alternative splicing or CD19 mutations, resulting in CD19 downmodulation as a response to the antigenic pressure by CD19-directed T-cell therapies. Our data reinforce the ongoing therapeutic clinical studies aimed at simultaneously targeting CD19 and CD22 as a strategy for reducing the likelihood of CD19− relapses and encourage the implementation of CD34/CD19/CD22 panels in flow cytometry diagnostic laboratories for initial diagnosis and follow-up monitoring of patients with B-ALL during CD19-targeted therapy.
Supplementary Material
The online version of this article contains a data supplement.
{kind=link}
Acknowledgments
The authors thank CERCA/Generalitat de Catalunya and Fundació Josep Carreras-Obra Social la Caixa for core support. The work in P.M. and C.B.’s Laboratory was supported by the European Research Council (CoG-2014-646903, PoC-2018-811220) and the Spanish Ministry of Economy and Competitiveness (SAF2016-80481R, PID2019-108160RB-I00) (P.M.); the ISCIII (ISCIII/FEDER, PI17/01028 and PI20/00822), the Spanish Association against Cancer (AECC), and the FERO Foundation (C.B.). P.M. and J.M.R. acknowledge the support of ISCIII-RICORS within the Next Generation EU program (Plan de Recuperación, Transformación y Resiliencia). N.E. is supported by Cancer Research UK and a Children and Young People’s Cancer Innovation Award (DRCPGM\100058). A.R. is supported by a Wellcome Trust Clinical Research Career Development Fellowship (216632/Z/19/Z). A.O. was supported by ISCIII (PI19/011183). F.S. was supported by AGAUR/Generalitat de Catalunya (SGR288). The single-cell transcriptomic analysis was supported by MRC Discovery award MRCDA 0816-11 and the MRC WIMM Single Cell Facility and MRC-funded Oxford Consortium for Single-Cell Biology (MR/M00919X/1) provided assistance. The human fetal tissue was provided by the Joint MRC/Wellcome Trust (grant MR/R006237/1) Human Developmental Biology Resource (http://hdbr.org).
Footnotes
All the transcriptomic data have already been published, and the data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the following accession numbers: 10× single-cell data: GSE1552592521; FBMA-seq data: GSE122982.27
Original data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.B. conceived the study, designed and performed the experiments, analyzed and interpreted the data, prepared the figures, wrote the manuscript, and financially supported the study; S.B., F.G.-A., J.L.T., M.G., P.B., S.R.Z., N.V., M. Mallo, F.S., N.E., and S.O. performed the experiments and analyzed the data; G.W., B.P., and S.T. performed the computational analysis and designed the pipelines for the transcriptomic analysis; A.B. provided the clinical data and analyzed the data; V.O.-M., M.R., M. Morgades, M.S., M.T., S.R., M.J., J.D., A.U.-I., J.M.R., and A.O. provided the human primary samples and the clinical data; A.J.M. and I.R provided the financial supported for the study (MR/M00919X/1); A.R. performed the experiments, prepared the figures, and wrote the manuscript; P.M., conceived the study, designed the experiments, interpreted data, wrote the manuscript, and financially supported the study; and all authors agreed to publish the manuscript.
Conflict-of-interest disclosure: P.M. is a cofounder of the spinoff One Chain Immunotherapeutics. S.O. is now an employee of Becton Dickinson (BD). Her contributions to the work were made before her employment at BD commenced. The remaining authors declare no competing financial interests.
Correspondence: Pablo Menendez, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Carrer Casanova 143, 4th Floor, 08036 Barcelona, Spain; e-mail: pmenendez@carrerasresearch.org; and Clara Bueno, Josep Carreras Leukemia Research Institute, School of Medicine, University of Barcelona, Carrer Casanova 143, 4th Floor, 08036 Barcelona, Spain; e-mail: cbueno@carrerasresearch.org.
REFERENCES
- 1.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capitini CM. CAR-T immunotherapy: how will it change treatment for acute lymphoblastic leukemia and beyond? Expert Opin Orphan Drugs. 2018;6(10):563-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruella M, Maus MV. Catch me if you can: leukemia escape after CD19-directed T cell immunotherapies. Comput Struct Biotechnol J. 2016;14:357-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16(6):372-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song MK, Park BB, Uhm JE. Resistance mechanisms to CAR T-cell therapy and overcoming strategy in B-cell hematologic malignancies. Int J Mol Sci. 2019;20(20):E5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagashev A, Sotillo E, Tang CH, et al. CD19 alterations emerging after CD19-directed immunotherapy cause retention of the misfolded protein in the endoplasmic reticulum. Mol Cell Biol. 2018;38(21):e00383-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braig F, Brandt A, Goebeler M, et al. Resistance to anti-CD19/CD3 BiTE in acute lymphoblastic leukemia may be mediated by disrupted CD19 membrane trafficking. Blood. 2017;129(1):100-104. [DOI] [PubMed] [Google Scholar]
- 8.Fischer J, Paret C, El Malki K, et al. CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother. 2017;40(5):187-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacoby E, Nguyen SM, Fountaine TJ, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7:12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504-1506. [DOI] [PubMed] [Google Scholar]
- 12.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zanetti SR, Velasco-Hernandez T, Gutierrez-Aguera F, et al. A novel and efficient tandem CD19- and CD22-directed CAR for B-cell ALL. Mol Ther. 2022;30(2):550-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardy RR. B-cell commitment: deciding on the players. Curr Opin Immunol. 2003;15(2):158-165. [DOI] [PubMed] [Google Scholar]
- 15.van Zelm MC, van der Burg M, de Ridder D, et al. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol. 2005;175(9):5912-5922. [DOI] [PubMed] [Google Scholar]
- 16.McGregor S, McNeer J, Gurbuxani S. Beyond the 2008 World Health Organization classification: the role of the hematopathology laboratory in the diagnosis and management of acute lymphoblastic leukemia. Semin Diagn Pathol. 2012;29(1): 2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menéndez P, Prósper F, Bueno C, et al. Sequential analysis of CD34+ and CD34− cell subsets in peripheral blood and leukapheresis products from breast cancer patients mobilized with SCF plus G-CSF and cyclophosphamide. Leukemia. 2001;15(3):430-439. [DOI] [PubMed] [Google Scholar]
- 18.Cordoba S, Onuoha S, Thomas S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiegel JY, Patel S, Muffly L, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27(8):1419-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabilloud T, Potier D, Pankaew S, Nozais M, Loosveld M, Payet-Bornet D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat Commun. 2021;12(1):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roy A, Wang G, Iskander D, et al. Transitions in lineage specification and gene regulatory networks in hematopoietic stem/progenitor cells over human development. Cell Rep. 2021;36(11)109698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortiz-Maldonado V, Rives S, Castella M, et al. CART19-BE-01: a multicenter trial of ARI-0001 cell therapy in patients with CD19 relapsed/refractory malignancies. Mol Ther. 2021;29(2):636-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ortiz-Maldonado V, Rives S, Español-Rego M, et al. Factors associated with the clinical outcome of patients with relapsed/refractory CD19+ acute lymphoblastic leukemia treated with ARI-0001 CAR T19-cell therapy. J Immunother of Cancer 2021;9:e003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agraz-Doblas A, Bueno C, Bashford-Rogers R, et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica. 2019;104(6):1176-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bueno C, Tejedor JR, Bashford-Rogers R, et al. Natural history and cell of origin of TC F3-ZN F384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood. 2019;134(11):900-905. [DOI] [PubMed] [Google Scholar]
- 26.Menendez P, Catalina P, Rodríguez R, et al. Bone marrow mesenchymal stem cells from infants with MLL-AF4+ acute leukemia harbor and express the MLL-AF4 fusion gene. J Exp Med. 2009;206(13):3131-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Byrne S, Elliott N, Sobhan R, et al. Discovery of a CD10-negative B-progenitor in human fetal life identifies unique ontogeny-related developmental programs. Blood. 2018;134(13)1069-1071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.