

ABSTRACT
Abnormalities in the regulation of gene expression are associated with various pathological conditions. Among the distal regulatory elements in the genome, the activation of target genes by enhancers plays a central role in the formation of cell type–specific gene expression patterns. Super-enhancers are a subclass of enhancers that frequently contain multiple enhancer-like elements and are characterized by dense binding of master transcription factors and Mediator complexes and high signals of active histone marks. Super-enhancers have been studied in detail as important regulatory regions that control cell identity and contribute to the pathogenesis of diverse diseases. In cancer, super-enhancers have multifaceted roles by activating various oncogenes and other cancer-related genes and shaping characteristic gene expression patterns in cancer cells. Alterations in super-enhancer activities in cancer involve multiple mechanisms, including the dysregulation of transcription factors and the super-enhancer–associated genomic abnormalities. The study of super-enhancers could contribute to the identification of effective biomarkers and the development of cancer therapeutics targeting transcriptional addiction. In this review, we summarize the roles of super-enhancers in cancer biology, with a particular focus on hematopoietic malignancies, in which multiple super-enhancer alteration mechanisms have been reported.
Key Words: super-enhancer, oncogene, transcription factor, genome, cancer therapy
INTRODUCTION
Recent advanced sequencing technologies in cancer research enable the establishment of a complete registry of recurrent mutational targets. These technologies also enable analysis of the molecular basis of epigenetic regulation in cancer cells, which includes DNA methylation, histone modification, and higher-order chromatin structure regulation. A better understanding of this molecular basis has greatly expanded knowledge of cancer-associated alterations in gene expression programs. Among genomic distal regulatory elements, enhancer-mediated activation of target genes is key to the establishment of cell type–specific gene expression patterns. Enhancers are genomic regions that are bound by transcription factors (TFs) and transcriptional coactivators to promote gene transcription. Enhancers regulate spatiotemporal gene expression patterns through chromatin looping-based interactions with target promoters. In general, ~10,000 putative enhancers can be identified in a single cell type. Among them, a subset of enhancer regions near cell type–specific genes contains multiple putative enhancers densely bound by master TFs and transcriptional coactivators, including the Mediator complex and bromodomain-containing protein 4 (BRD4).1 This enhancer subclass, called super-enhancers, was distinguished from typical enhancers in a study reanalyzing standard chromatin immunoprecipitation sequencing (ChIP-seq) datasets.1
Super-enhancers have been studied in detail as important regulatory regions that control cell identity and contribute to the pathogenesis of diverse diseases.2,3 In the field of cancer biology, super-enhancers activate various oncogenes and other cancer-related genes and shape characteristic gene expression patterns in cancer cells. Alterations in super-enhancer activity in cancer involve multiple mechanisms, including TF dysregulation and super-enhancer–associated genomic abnormalities. TF dysregulation includes the generation of aberrant chimeric TFs and the altered expression, function, stability, and crosstalk of transcriptional regulators. Some genomic abnormalities in cancer, such as translocations, amplifications, and insertions, can be considered examples of super-enhancer dysregulation. A better understanding of super-enhancers could enable the identification of effective biomarkers and the development of therapeutics targeting transcriptional addiction.4-6 Here, we review the roles of super-enhancers in cancer, particularly in hematopoietic malignancies, in which various super-enhancer alteration mechanisms have been documented.
DEFINITION AND COMPILATION OF SUPER-ENHANCERS
The concept of super-enhancers was proposed by Richard A. Young and colleagues in 2013 after they reinterpreted the ChIP-seq results of master TFs, Mediator complexes, and histone modifications (H3K27ac) in mouse embryonic stem cells (mESCs).1 In the original study, super-enhancers were identified in mESCs via the following steps: (1) Putative enhancer sites bound by all three master TFs, Oct4, Sox2, and Nanog, were identified. (2) Enhancer peaks within 12.5 kb were stitched. (3) Stitched enhancer regions were then ranked according to the ChIP-seq signal of the Mediator complex component MED1. (4) The MED1 signal was plotted against the enhancer rank, and super-enhancers were distinguished from typical enhancers by identifying the inflection point of the curve. The computational pipeline is called Rank Ordering of SE (ROSE). In mESCs, approximately 40% of the MED1 signal associated with enhancers was found in 231 super-enhancer domains among ~10,000 enhancer regions.
Based on the substantial overlap between Oct4, Sox2, Nanog, and MED1-defined super-enhancers and H3K27ac-defined super-enhancers, H3K27ac ChIP-seq datasets have been widely used to identify super-enhancers in various cell types.2 Other cell type–specific TFs or transcriptional coactivators, such as MED1 and BRD4, have also been used as surrogate markers.7,8 While ~10,000 enhancers can be identified per cell type, approximately 200 to 800 super-enhancers are typically identified. Super-enhancers are frequently found in the vicinity of cell type–specific genes, including those encoding cell type–specific master TFs (eg, Oct4, Sox2, and Nanog in mESCs), and are associated with high levels of target gene expression. In addition, a small subset of cell type-specific microRNAs (miRNAs), whose expression and function are highly dominant in each cell type, is frequently associated with super-enhancers.9
Information on super-enhancers in various ChIP-seq datasets is currently available in several databases, such as dbSUPER (https://asntech.org/dbsuper/), SEdb (http://www.licpathway.net/sedb/), SEanalysis (licpathway.net/SEanalysis/), and SEA (http://sea.edbc.org/).10-14 These databases provide resources for investigating the relationships among super-enhancers, target genes, TF association, and disease-associated single nucleotide polymorphisms (SNPs).
MOLECULAR FEATURES OF SUPER-ENHANCERS
Super-enhancers are co-occupied by various TFs crucial for the relevant cell type and occupied by high levels of transcriptional regulators, including Mediator, p300, CBP, BRD4, RNA polymerase II (RNA Pol II), cohesin, and chromatin remodelers (Figure 1).2 Recently developed platforms for super-enhancer prediction based on machine learning and convolutional neural networks have demonstrated that several components, including H3K27ac, MED1, MED12, BRD4, p300, CDK7, CDK8, and CDK9, are important factors that characterize super-enhancers.15,16
Fig. 1.

Comparison of typical enhancers and super-enhancers
Super-enhancers are enriched with more transcription factors, Mediator complexes, and RNA Pol II molecules than typical enhancers. Hence, super-enhancers have higher transcription activity levels than typical enhancers. Super-enhancers activate cell identity–related gene expression programs.
TF: transcription factor
Pol II: RNA polymerase II
Among these coactivators, the Mediator complex plays a central role in super-enhancer function and cell identity control by mediating enhancer-promoter communication and RNA Pol II transcription.17,18 BRD4 is another key player in super-enhancer function and a bromodomain and extraterminal (BET) domain protein family member that binds to histone acetylated lysines.19 BRD4 contributes to cell type–specific gene expression by preferentially binding to activated enhancers and activating the target promoters; the primary functions of BRD4 are promoting RNA Pol II phosphorylation and mediating RNA Pol II pause release and elongation.20,21 Recent studies have demonstrated that BRD4 interacts with multiple factors associated with 5'-elongation control and 3'-RNA processing.22,23 Additionally, BRD4 supports recruitment of 3'-RNA processing factors during a 5'-elongation checkpoint.22,23 Overall, these observations are consistent with reports that super-enhancer–associated genes have low levels of transcriptional pausing, suggesting that high transcription levels are achieved by rapid pause release, despite stronger recruitment of RNA Pol II.24,25 This would also explain the exquisite sensitivity of super-enhancer–associated genes to inhibition of CDK7, which regulates multiple transcription steps by promoting RNA Pol II phosphorylation.26-28
A series of recent studies have suggested the involvement of liquid-liquid phase separation (LLPS) processes in the formation and activity of super-enhancers.29,30 In recent years, LLPS has attracted attention as the main principle underlying the organization of diverse membraneless organelles, collectively called “biomolecular condensates,” within the cell.31 Intracellular condensate formation is mediated by cooperative interactions among multivalent molecules, such as RNA, DNA, and intrinsically disordered regions (IDRs) in proteins.31 Such cooperatively has been documented in the enhancer biology as various TFs cooperatively bind to and activate enhancers.7,8 In addition, functional studies of super-enhancers have highlighted that multiple enhancer-like elements within super-enhancers act cooperatively.9,32,33 Together with imaging studies showing clustering of multiple RNA Pol II molecules, these findings led to the proposal of a phase separation (transcriptional condensate) model. In this model, multiple enhancer-like elements and associated multivalent proteins, such as TFs, Mediator, BRD4, and RNA Pol II, facilitate phase-separated multimolecular assembly and compartmentalized transcription reactions.29,30,34 This concept has been supported by subsequent studies showing the widespread LLPS capacity of transcriptional regulators, including RNA Pol II, Mediator, BRD4, and various TFs.35-38 In addition, alterations in biomolecular condensates have multiple roles in cancer pathogenesis.39
ROLES OF SUPER-ENHANCERS IN CANCER
Super-enhancers play a central role in cell type–specific gene expression programs and are involved in the pathogenesis of a wide variety of tumors. Cancers of different tissue origins and the different subtypes exhibit specific gene expression patterns, which are associated with prognosis and distinct biological behaviors, including drug resistance. As changes in enhancer elements reportedly drive a specific transcriptional program in cancer,40,41 super-enhancers contribute to characteristic gene expression patterns in cancer cells by activating various oncogenes and modulating other cancer-related genes. Compared to normal cells, cancer cells have altered super-enhancer usage patterns, which are partly responsible for the activation of various oncogenes and other genes associated with “hallmarks of cancer”.2 Super-enhancers drive the expression of not only protein-coding genes but also non-coding RNAs in cancer cells. A comprehensive comparison of super-enhancers between normal cells and cancer cells showed that cancer cells exhibit the formation of super-enhancers associated with cancer-promoting miRNAs and the loss of super-enhancers associated with cancer-suppressing miRNAs.9 An analysis using The Cancer Genome Atlas (TCGA) database showed that high expression of cancer-promoting miRNAs along with super-enhancer formation correlates with poor prognosis in certain cancer types.9 In addition, several super-enhancer–driven long non-coding RNAs (lncRNAs) contribute to cancer pathogenesis.42-45
Alterations in super-enhancer activities in cancer involve multiple mechanisms, including dysregulation of transcriptional regulators and super-enhancer–associated genomic abnormalities. These events have been referred to as “enhancer hijacking”. Transcriptional regulator dysregulation includes the generation of aberrant chimeric TFs; altered expression, function, and stability of transcriptional regulators; and altered crosstalk among various transcriptional regulators. Some super-enhancer–associated genomic abnormalities, such as translocations, amplifications, and insertions, can be recognized as super-enhancer dysregulation mechanisms. In the following sections, we summarize findings regarding the mechanisms of super-enhancer dysregulation in hematological malignancies. These mechanisms include transcriptional regulator abnormalities that accumulate in enhancers and genomic abnormalities of enhancers themselves.
DYSREGULATION OF SUPER-ENHANCERS BY ABNORMALITIES IN TRANSCRIPTIONAL REGULATORS
The hundreds of genes regulated by super-enhancers often include those encoding master TFs and transcriptional regulators that define the characteristics of each cell type. This is also the case in hematological malignancies. Examples of super-enhancer–associated TFs and transcriptional regulators in various hematological malignancies include OCA-B (POU2AF1), BCL6, PAX5, and IRF8 in diffuse large B-cell lymphoma (DLBCL); PAX5 in chronic lymphocytic leukemia (CLL); HOXA cluster, HOXB cluster, and MEIS1 in NPM1-mutated acute myeloid leukemia (AML); BATF3 and IRF4 in adult T-cell leukemia-lymphoma (ATLL); and TCF4 in blast plasmacytoid dendritic cell neoplasm (BPDCN).46-50 These TFs serve as the core of the transcriptional network in each tumor type. In addition, super-enhancer–activated TOX, an HMG box–containing protein, regulates the proliferation and non-homologous end joining (NHEJ) repair in T-cell acute lymphoblastic leukemia (T-ALL).51
Abnormalities in transcriptional regulators can be recognized as a combination of the following mechanisms: (1) production of chimeric TFs; (2) altered expression, function, and stability of transcriptional regulators; and (3) altered crosstalk among transcriptional regulators. These mechanisms have been reported in hematological malignancies (Figure 2).
Fig. 2.

Dysregulation of super-enhancers by abnormalities in transcription factors and transcriptional regulators
Abnormalities in transcriptional regulators reported in hematological malignancies are categorized as a combination of mechanisms, including (1) production of chimeric TFs, (2) altered expression, function, and stability of transcriptional regulators, and (3) altered crosstalk among various transcriptional regulators. Abnormalities in transcriptional regulators result in dysregulation of super-enhancers observed in normal cells (black) and/or formation of novel super-enhancer near oncogenes (red).
The first scenario, production of chimeric TFs, is frequently observed in hematological malignancies. The TCF3-HLF chimeric TF in pediatric acute lymphoblastic leukemia (ALL) binds to the HLF binding sites in hematopoietic stem cell/myeloid lineage-associated super-enhancers and activates a conserved MYC-driven transformation program.52 Inhibition of p300 decommissions TCF3-HLF enhancer programs and exerts a profound anti-leukemic effect. In pediatric acute megakaryoblastic leukemia (AMKL), the ETO2-GLIS2 chimeric TF also accumulates in the super-enhancers of leukemic cells.53 The binding sites of ETO2-GLIS2 are enriched with the DNA-binding motifs of GLIS2 and several known ETO2 partners, including ERG (ETS), GATA, and RUNX. Importantly, more than 50% of the binding sites are not bound by ETO2 partners in normal cells, suggesting that the chimeric TFs bind to specific, novel sites in cancer cells.53 The importance of such altered TF properties has been reinforced by recent reports that NUP98-fusion chimeric TFs in pediatric AML have altered condensate formation properties to drive leukemogenic gene expression.54,55 In addition, CBFβ-SMMHC, found in AML with chromosome 16 inversion, inhibits the function of RUNX1, a master TF in the hematopoietic system, and neutralizes RUNX1-mediated repression of MYC expression.56
As the second and third scenarios, multiple cancer types have been frequently associated with alterations in the expression of oncogenic TFs, mutations in transcriptional regulators, and altered functional crosstalk. AML is often associated with elevated HOXA9 expression, which contributes to leukemogenesis by driving the activity of de novo enhancers characteristic of leukemic cells, including super-enhancers.57 The pseudokinase TRIB1, a myeloid oncoprotein, modulates HOXA9-bound super-enhancers by suppressing C/EBPα p42, and accelerates HOXA9-induced leukemia onset.58 The NUP214-ABL1 fusion kinase found in ALL promotes the cooperative binding of TLX1 and STAT5 to enhancers and activates key proto-oncogenes, such as MYC and BCL2.59 Loss-of-function mutations in CREBBP acetyltransferase and its paralogue p300, which accumulate in super-enhancers, are highly frequent in follicular lymphoma (FL) and DLBCL.60 Mutations in CREBBP have been suggested to affect the function of the transcriptional repressor BCL6 and gene regulation in the germinal center, which is controlled by super-enhancers.60 In addition, FL and DLBCL are associated with frequent mutations in MEF2B, a TF that similarly accumulates in germinal center-specific super-enhancers.61 Mutations in MEF2B have been suggested to perturb gene expression by decreasing the DNA-binding capacity and protein stability of MEF2B, as well as by evading negative regulation via the HUCA complex.61 Viral-associated tumorigenesis could also be associated with alterations in super-enhancers; in primary effusion lymphoma (PEL), Kaposi’s sarcoma-associated herpesvirus (KSHV)-driven TF, viral interferon regulatory factor 3 (vIRF3), reportedly drives super-enhancer–mediated survival gene expression programs in cooperation with host TFs, such as IRF4 and BATF.62,63
Super-enhancers are also enriched with cohesin complexes, which mediate enhancer-promoter communication.2 STAG2, a gene encoding a cohesin subunit, is frequently mutated in myeloid neoplasms along with other genes, such as RUNX1, SRSF2, and ASXL1.25 In mice, the combined loss of Stag2 and Runx1 synergistically attenuates enhancer-promoter loops, particularly at sites with enriched levels of RNA Pol II and Mediator, and causes myelodysplastic syndromes (MDS).25 Super-enhancer–associated genes in hematopoietic stem/progenitor cells (HSPCs) are downregulated upon single knockout of Stag2 and double knockout of Stag2 and Runx1. In addition to super-enhancer perturbation, downregulation of genes with high basal transcriptional pausing, which are important for HSPC regulation, has also been observed in Stag2 single and double knockout mice. Downregulation of high-pausing genes has been confirmed in samples of primary leukemia with STAG2/cohesin mutations.25
DYSREGULATION OF SUPER-ENHANCERS BY GENOMIC ABNORMALITIES
In addition to aberrant transcriptional networks, a variety of genomic abnormalities, including translocations, inversions, amplifications, and insertions/deletions, can cause aberrant super-enhancer formation near oncogenes in cancer cells (Figure 3).
Fig. 3.
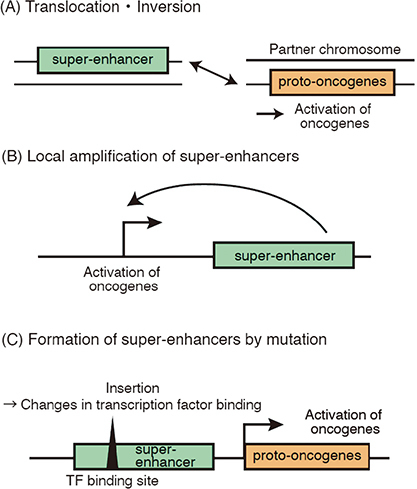
Dysregulation of super-enhancers by genomic aberrations
Fig. 3A: Translocations, inversions, and interchromosomal deletions rearrange the positional relationships between super-enhancers and proto-oncogenes, leading to abnormal transcription and cancer development.
Fig. 3B–3C: Other mechanisms include focal amplifications of super-enhancer regions (B), point mutation, or small insertions or deletions (indels) that create new super-enhancers (C). These mechanisms can also increase transcriptional output and perturb the regulation of oncogenes.
In well-known oncogenes such as MYC and N-MYC, different abnormalities cause aberrant super-enhancer formation and oncogene dysregulation in different cancer types.2 In malignant lymphomas, some chromosomal translocations reposition super-enhancers at immunoglobulin loci in proximity to MYC, leading to high MYC expression levels (Figure 3A). In AML, translocations or inversions of chromosome 3 reposition a distal GATA2 enhancer so that it ectopically activates EVI1 oncogene through translocation-derived super-enhancers and simultaneously confers GATA2 haploinsufficiency.64,65 This type of “enhancer hijacking” event has been reported in solid tumors, such as medulloblastoma, adenoid cystic carcinoma, and thyroid cancer, in recent years.66-68 A high-throughput approach for the simultaneous detection of enhancer activity and chromosome rearrangements, called pinpointing enhancer-associated rearrangements by chromatin immunoprecipitation (PEAR-ChIP), has been used to identify various rearrangements, including translocations and interchromosomal deletions, that involve known cancer-associated genes, including CCND1, BCL2, MYC, PDCD1LG2, NOTCH1, CIITA, and SGK1.69 PEAR-ChIP has also been used to identify novel enhancer duplication events and lymphoma subtype–specific enhancers at the MYC locus.69
In some cases of ALL and AML, MYC is activated by focally amplified super-enhancers in the 3’ region (Figure 3B).70,71 Focally amplified lineage-specific super-enhancers of MYC and other genes have also been identified in human epithelial cancers.72 Super-enhancers typically contain multiple discrete enhancer RNA (eRNA) loci. The eRNA loci are regulated by evolutionarily conserved, well-positioned nucleosomes and are frequently dysregulated in cancer.73 Pan-cancer analysis of the eRNA loci in super-enhancers has revealed that super-enhancers are globally activated in many cancers and that a substantial portion of eRNA loci is affected by somatic copy number alteration and CpG methylation in human cancers.73
In addition to chromosomal aberrations, mutations in super-enhancer regions are an important genomic aberration scenario (Figure 3C). In a subset of T-ALL, a short base insertion occurs upstream of the TAL1 oncogene.74 This mutation creates a binding sequence for the MYB TF, which induces the binding of various other TFs and the formation of super-enhancers, leading to TAL1 activation.74 Other cancer-associated mutations in enhancers and CTCF binding sites have gradually been revealed by recent studies.75,76 A recent integrative analysis, which combined targeted resequencing of hematopoietic lineage–associated cis-regulatory elements (CRE) with CRISPR/dCas9-based perturbation screening, comprehensively identified recurrently mutated oncogenic and tumor suppressive CREs in AML, lymphoma, and ALL.77 This study demonstrated that CRE variants at KRAS and PER2 enhancers reside in proximity to the binding sites of nuclear receptors (NRs), affecting responsiveness to NR signaling and expression levels of target genes. Furthermore, the colocalization of CRE and NR binding sites was shown to be widespread.77
POTENTIAL OF CANCER THERAPY VIA SUPER-ENHANCER INHIBITION
The super-enhancer concept is partially based on the observation that cell identity maintenance in mESCs is highly sensitive to inhibition of the transcriptional coactivator Mediator and cohesin complex, as well as to perturbation of master TFs.78 A similar high dependency on transcriptional regulators for survival is frequently observed in cancer cells (“transcriptional addiction”), and this is generally consistent with the super-enhancer concept.79-81 Several drugs targeting super-enhancer components have been shown to affect the transcriptional machinery of cells and produce anti-tumor effects (Figure 4).
Fig. 4.

Therapeutic targeting of super-enhancers in cancers
The discovery of JQ1 and other bromodomain inhibitors, as well as the preferential targeting of super-enhancers, has led to the development of several first- and second-generation bromodomain inhibitors. CDK7 (CDK12/CDK13) is also attracting attention as a target for cancer therapy; CDK7 inhibitors have been reported to be effective against various types of cancer.
BETi: bromodomain and extraterminal domain inhibitor
JQ1, I-BET151 (GSK1210151A), and other bromodomain inhibitors selectively bind to bromodomains and exert anti-cancer effects in a wide range of hematological malignancies, including AML, ALL, multiple myeloma, Burkitt lymphoma, and other tumor types.82-89 Early studies have shown that bromodomain inhibitors elicit anti-tumor effects by selectively inhibiting the MYC oncogene. Later, these gene-specific selective effects were partly explained by the preferential targeting of super-enhancers associated with MYC, other key oncogenes, and lineage-specific TFs in cancer cells.3,90 As described earlier, BRD4 plays important roles in transcriptional pause release, 3'-end processing, and transcriptional termination. Mechanisms of resistance to bromodomain inhibitors have been also reported.91-93 Another super-enhancer-targeting drug, THZ1, is a covalent inhibitor of CDK7, CDK12, and CDK13 that selectively kills cancer cells by inhibiting super-enhancer–induced oncogenic transcription.27 Anti-tumor activities of THZ1 have been reported in multiple tumor types, including hematological malignancies, MYCN-driven neuroblastomas, small cell lung cancer (SCLC), triple-negative breast cancer, and esophageal squamous cell carcinoma.27,94-97 THZ1 inhibits phosphorylation of the carboxyl-terminal domain (CTD) of RNA Pol II and attenuates multiple transcription processes, including capping, pausing, and productive elongation.28 A recent combined analysis of drug sensitivity and gene dependency screens unexpectedly revealed that small-cell neuroendocrine cancers (SCNC) and hematological malignancies have common gene expression profiles, protein expression profiles, and drug sensitivity profiles. This study also showed that CDK7 inhibition is more effective in SCLC and blood cancers than in other tumor types.98 This is consistent with other reports showing that THZ1 is highly effective at perturbing super-enhancers in these tumor types.27,95 These transcription-perturbing drugs are currently being evaluated in several phase I/II clinical trials.81
While the inhibition of super-enhancers is a promising approach in multiple cancer types, the potential side effects and off-target effects have not been fully explored. Because the inhibition of super-enhancers in cancer cells also affects super-enhancers in normal cells, it is necessary to further investigate transcriptional addiction mechanisms in cancer cells and identify selective targets. TAF12, a subunit of TFIID and SAGA coactivator complexes, has been identified as a target that selectively regulates AML progression.99 Peptide-based squelching of TAF12 and MYB interactions has been shown to exhibit potent anti-leukemia effects without harming normal tissues.99 Similarly, given that different TFs downstream of distinct signaling pathways interact with different Mediator subunits,17 targeting context-dependent interactions of super-enhancer components may also be promising. In addition, it has been reported that super-enhancers are prone to double-strand breaks and susceptible to defects in cellular DNA repair mechanisms.100 Therefore, the combined use of super-enhancer inhibitors and drugs targeting DNA damage repair mechanisms may improve anti-tumor effects.
CONCLUSIONS
In this review, we summarized the roles of super-enhancers in cancer, especially in hematopoietic malignancies. Super-enhancers are useful tools for understanding cancer-specific gene and miRNA expression mechanisms and have potential as therapeutic targets. In addition, alterations in super-enhancer activities in cancer involve multiple mechanisms, including dysregulation of transcriptional regulators and super-enhancer–associated genomic abnormalities. Targeting super-enhancers is a promising therapeutic strategy. However, the development of methodologies for targeting super-enhancers more selectively will require a deeper understanding of the basic molecular mechanisms.
ACKNOWLEDGMENTS
We thank Dr Koichi Ogami, Dr Koh Onimaru, Dr Shintaro Komatsu, and members of the Suzuki laboratories for their discussions. We apologize to the many researchers whose work has not been cited owing to space limitations. This work was supported in part by the JSPS KAKENHI (20K16318 (SY), 19K24694 (HIS)), Mochida Memorial Foundation for Medical and Pharmaceutical Research (HIS), Grant for Basic Science Research Projects from the Sumitomo Foundation (HIS), Mitsubishi Foundation (HIS), Daiichi Sankyo Foundation of Life Science (HIS), Uehara Memorial Foundation (SY, HIS), and Takeda Science Foundation (HIS).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Abbreviations
- TF
transcription factor
- BRD4
bromodomain-containing protein 4
- ChIP-seq
chromatin immunoprecipitation sequencing
- mESC
mouse embryonic stem cell
- miRNA
microRNA
- RNA Pol II
RNA polymerase II
- AML
acute myeloid leukemia
- ALL
acute lymphoblastic leukemia
REFERENCES
- 1.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed]
- 2.Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed]
- 3.Lovén J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed]
- 4.He Y, Long W, Liu Q. Targeting super-enhancers as a therapeutic strategy for cancer treatment. Front Pharmacol. 2019;10(361):1–10. doi: 10.3389/fphar.2019.00361. [DOI] [PMC free article] [PubMed]
- 5.Zheng C, Liu M, Fan H. Targeting complexes of super-enhancers is a promising strategy for cancer therapy (Review). Oncol Lett. 2020;20(3):2557–2566. doi: 10.3892/ol.2020.11855. [DOI] [PMC free article] [PubMed]
- 6.Jia Y, Chng WJ, Zhou J. Super-enhancers: Critical roles and therapeutic targets in hematologic malignancies. J Hematol Oncol. 2019;12(1):1–17. doi: 10.1186/s13045-019-0757-y. [DOI] [PMC free article] [PubMed]
- 7.Pott S, Lieb JD. What are super-enhancers? Nat Genet. 2015;47(1):8–12. doi: 10.1038/ng.3167. [DOI] [PubMed]
- 8.Blobel GA, Higgs DR, Mitchell JA, Notani D, Young RA. Testing the super-enhancer concept. Nat Rev Genet. 2021;22(12):749–755. doi: 10.1038/s41576-021-00398-w. [DOI] [PubMed]
- 9.Suzuki HI, Young RA, Sharp PA. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell. 2017;168(6):1000–1014.e15. doi: 10.1016/j.cell.2017.02.015. [DOI] [PMC free article] [PubMed]
- 10.Khan A, Zhang X. DbSUPER: A database of Super-enhancers in mouse and human genome. Nucleic Acids Res. 2016;44(D1):D164–D171. doi: 10.1093/nar/gkv1002. [DOI] [PMC free article] [PubMed]
- 11.Jiang Y, Qian F, Bai X, et al. SEdb: A comprehensive human super-enhancer database. Nucleic Acids Res. 2019;47(D1):D235–D243. doi: 10.1093/nar/gky1025. [DOI] [PMC free article] [PubMed]
- 12.Qian FC, Li XC, Guo JC, et al. SEanalysis: A web tool for super-enhancer associated regulatory analysis. Nucleic Acids Res. 2019;47(W1):W248–W255. doi: 10.1093/nar/gkz302. [DOI] [PMC free article] [PubMed]
- 13.Wei Y, Zhang S, Shang S, et al. SEA: A Super-enhancer Archive. Nucleic Acids Res. 2016;44(D1):D172–D179. doi: 10.1093/nar/gkv1243. [DOI] [PMC free article] [PubMed]
- 14.Chen C, Zhou D, Gu Y, et al. SEA version 3.0: A comprehensive extension and update of the Super-Enhancer archive. Nucleic Acids Res. 2020;48(D1):D198–D203. doi: 10.1093/nar/gkz1028. [DOI] [PMC free article] [PubMed]
- 15.Khan A, Zhang X. Integrative modeling reveals key chromatin and sequence signatures predicting super-enhancers. Sci Rep. 2019;9(1):1–15. doi: 10.1038/s41598-019-38979-9. [DOI] [PMC free article] [PubMed]
- 16.Bu H, Hao J, Gan Y, Zhou S, Guan J. DEEPSEN: A convolutional neural network based method for super-enhancer prediction. BMC Bioinformatics. 2019;20(Suppl 15):598. doi: 10.1186/s12859-019-3180-z. [DOI] [PMC free article] [PubMed]
- 17.Allen BL, Taatjes DJ. The Mediator complex: A central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16(3):155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed]
- 18.Soutourina J. Transcription regulation by the Mediator complex. Nat Rev Mol Cell Biol. 2018;19(4):262–274. doi: 10.1038/nrm.2017.115. [DOI] [PubMed]
- 19.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol Cancer. 2018;17(1):1–13. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed]
- 20.Zhang W, Prakash C, Sum C, et al. Bromodomain-containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem. 2012;287(51):43137–43155. doi: 10.1074/jbc.M112.413047. [DOI] [PMC free article] [PubMed]
- 21.Itzen F, Greifenberg AK, Bösken CA, Geyer M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014;42(12):7577–7590. doi: 10.1093/nar/gku449. [DOI] [PMC free article] [PubMed]
- 22.Winter GE, Mayer A, Buckley DL, et al. BET Bromodomain Proteins Function as Master Transcription Elongation Factors Independent of CDK9 Recruitment. Mol Cell. 2017;67(1):5–18. doi: 10.1016/j.molcel.2017.06.004. [DOI] [PMC free article] [PubMed]
- 23.Arnold M, Bressin A, Jasnovidova O, Meierhofer D, Mayer A. A BRD4-mediated elongation control point primes transcribing RNA polymerase II for 3’-processing and termination. Mol Cell. 2021;81(17):3589–3603. doi: 10.1016/j.molcel.2021.06.026. [DOI] [PubMed]
- 24.Henriques T, Scruggs BS, Inouye MO, et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;32(1):26–41. doi: 10.1101/gad.309351.117. [DOI] [PMC free article] [PubMed]
- 25.Ochi Y, Kon A, Sakata T, et al. Combined Cohesin–RUNX1 deficiency synergistically perturbs chromatin looping and causes myelodysplastic syndromes. Cancer Discov. 2020;10(6):836–853. doi: 10.1158/2159-8290.CD-19-0982. [DOI] [PMC free article] [PubMed]
- 26.Eick D, Geyer M. The RNA Polymerase II Carboxy-Terminal Domain (CTD) Code. Chem Rev. 2013;113(11):8456–8490. doi: 10.1021/cr400071f. [DOI] [PubMed]
- 27.Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed]
- 28.Nilson KA, Guo J, Turek ME, et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol Cell. 2015;59(4):576–587. doi: 10.1016/j.molcel.2015.06.032. [DOI] [PMC free article] [PubMed]
- 29.Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A Phase Separation Model for Transcriptional Control. Cell. 2017;169(1):13–23. doi: 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed]
- 30.Sharp PA, Chakraborty AK, Henninger JE, Young RA. RNA in formation and regulation of transcriptional condensates. RNA. 2021;rna.078997. doi: 10.1261/rna.078997.121. [DOI] [PMC free article] [PubMed]
- 31.Bergeron-Sandoval LP, Safaee N, Michnick SW. Mechanisms and Consequences of Macromolecular Phase Separation. Cell. 2016;165(5):1067–1079. doi: 10.1016/j.cell.2016.05.026. [DOI] [PubMed]
- 32.Hnisz D, Schuijers J, Lin CY, et al. Convergence of Developmental and Oncogenic Signaling Pathways at Transcriptional Super-Enhancers. Mol Cell. 2015;58(2):362–370. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed]
- 33.Shin HY, Willi M, Yoo KH, et al. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat Genet. 2016;48(8):904–911. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed]
- 34.Cisse II, Izeddin I, Causse SZ, et al. Real-time dynamics of RNA polymerase II clustering in live human cells. Science. 2013;341(6146):664–667. doi: 10.1126/science.1239053. [DOI] [PubMed]
- 35.Sabari BR, Dall’Agnese A, Boija A, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361(6400):eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed]
- 36.Cho WK, Spille JH, Hecht M, et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018;361(6400):412–415. doi: 10.1126/science.aar4199. [DOI] [PMC free article] [PubMed]
- 37.Boija A, Klein IA, Sabari BR, et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 2018;175(7):1842–1855.e16. doi: 10.1016/j.cell.2018.10.042. [DOI] [PMC free article] [PubMed]
- 38.Chong S, Dugast-Darzacq C, Liu Z, et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 2018;361(6400):eaar2555. doi: 10.1126/science.aar2555. [DOI] [PMC free article] [PubMed]
- 39.Suzuki HI, Koh O. Biomolecular Condensates in Cancer Biology. Cancer Sci. Published online 2021. doi: 10.1111/cas.15232. [DOI] [PMC free article] [PubMed]
- 40.Akhtar-Zaidi B, Cowper-Sallari R, Corradin O, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336(6082):736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed]
- 41.Jin Y, Chen K, De Paepe A, et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood. 2018;131(19):2138–2150. doi: 10.1182/blood-2017-09-808063. [DOI] [PMC free article] [PubMed]
- 42.Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24(5):513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed]
- 43.Xie JJ, Jiang YY, Jiang Y, et al. Super-Enhancer-Driven Long Non-Coding RNA LINC01503, Regulated by TP63, Is Over-Expressed and Oncogenic in Squamous Cell Carcinoma. Gastroenterology. 2018;154(8):2137–2151. doi: 10.1053/j.gastro.2018.02.018. [DOI] [PubMed]
- 44.Jiang Y, Jiang YY, Xie JJ, et al. Co-activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat Commun. 2018;9(1):3619. doi: 10.1038/s41467-018-06081-9. [DOI] [PMC free article] [PubMed]
- 45.Peng L, Jiang B, Yuan X, et al. Super-enhancer-associated long noncoding RNA HCCL5 is activated by ZEB1 and promotes the malignancy of hepatocellular carcinoma. Cancer Res. 2019;79(3):572–584. doi: 10.1158/0008-5472.CAN-18-0367. [DOI] [PubMed]
- 46.Chapuy B, McKeown MR, Lin CY, et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell. 2013;24(6):777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed]
- 47.Ott CJ, Federation AJ, Schwartz LS, et al. Enhancer Architecture and Essential Core Regulatory Circuitry of Chronic Lymphocytic Leukemia. Cancer Cell. 2018;34(6):982–995. doi: 10.1016/j.ccell.2018.11.001. [DOI] [PMC free article] [PubMed]
- 48.Brunetti L, Gundry MC, Sorcini D, et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell. 2018;34(3):499–512.e9. doi: 10.1016/j.ccell.2018.08.005. [DOI] [PMC free article] [PubMed]
- 49.Nakagawa M, Shaffer AL, Ceribelli M, et al. Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell. 2018;34(2):286–297.e10. doi: 10.1016/j.ccell.2018.06.014. [DOI] [PMC free article] [PubMed]
- 50.Ceribelli M, Hou ZE, Kelly PN, et al. A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell. 2016;30(5):764–778. doi: 10.1016/j.ccell.2016.10.002. [DOI] [PMC free article] [PubMed]
- 51.Lobbardi R, Pinder J, Martinez-Pastor B, et al. TOX regulates growth, DNA repair, and genomic instability in T-cell acute lymphoblastic leukemia. Cancer Discov. 2017;7(11):1336–1353. doi: 10.1158/2159-8290.CD-17-0267. [DOI] [PMC free article] [PubMed]
- 52.Huang Y, Mouttet B, Warnatz HJ, et al. The Leukemogenic TCF3-HLF Complex Rewires Enhancers Driving Cellular Identity and Self-Renewal Conferring EP300 Vulnerability. Cancer Cell. 2019;36(6):630–644. doi: 10.1016/j.ccell.2019.10.004. [DOI] [PubMed]
- 53.Thirant C, Ignacimouttou C, Lopez CK, et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell. 2017;31(3):452–465. doi: 10.1016/j.ccell.2017.02.006. [DOI] [PubMed]
- 54.Terlecki-Zaniewicz S, Humer T, Eder T, et al. Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nat Struct Mol Biol. 2021;28(2):190–201. doi: 10.1038/s41594-020-00550-w. [DOI] [PMC free article] [PubMed]
- 55.Ahn JH, Davis ES, Daugird TA, et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021;595(7868):591–595. doi: 10.1038/s41586-021-03662-5. [DOI] [PMC free article] [PubMed]
- 56.Pulikkan JA, Hegde M, Ahmad HM, et al. CBFβ-SMMHC Inhibition Triggers Apoptosis by Disrupting MYC Chromatin Dynamics in Acute Myeloid Leukemia. Cell. 2018;174(1):172–186.e21. doi: 10.1016/j.cell.2018.05.048. [DOI] [PMC free article] [PubMed]
- 57.Sun Y, Zhou B, Mao F, et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell. 2018;34(4):643–658. doi: 10.1016/j.ccell.2018.08.018. [DOI] [PMC free article] [PubMed]
- 58.Yoshino S, Yokoyama T, Sunami Y, et al. Trib1 promotes acute myeloid leukemia progression by modulating the transcriptional programs of Hoxa9. Blood. 2021;137(1):75–88. doi: 10.1182/blood.2019004586. [DOI] [PMC free article] [PubMed]
- 59.Vanden Bempt M, Demeyer S, Broux M, et al. Cooperative Enhancer Activation by TLX1 and STAT5 Drives Development of NUP214-ABL1/TLX1-Positive T Cell Acute Lymphoblastic Leukemia. Cancer Cell. 2018;34(2):271–285.e7. doi: 10.1016/j.ccell.2018.07.007. [DOI] [PMC free article] [PubMed]
- 60.Zhang J, Vlasevska S, Wells VA, et al. The CREBBP acetyltransferase is a haploinsufficient tumor suppressor in B-cell lymphoma. Cancer Discov. 2017;7(3):323–337. doi: 10.1158/2159-8290.CD-16-1417. [DOI] [PMC free article] [PubMed]
- 61.Brescia P, Schneider C, Holmes AB, et al. MEF2B Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell. 2018;34(3):453–465.e9. doi: 10.1016/j.ccell.2018.08.006. [DOI] [PMC free article] [PubMed]
- 62.Wang C, Jiang S, Zhang L, et al. TAF Family Proteins and MEF2C Are Essential for Epstein-Barr Virus Super-Enhancer Activity. J Virol. 2019;93(16):e00513–19. doi: 10.1128/jvi.00513-19. [DOI] [PMC free article] [PubMed]
- 63.Manzano M, Günther T, Ju H, et al. Kaposi’s sarcoma-associated herpesvirus drives a super-enhancer-mediated survival gene expression program in primary effusion lymphoma. MBio. 2020;11(4):e01457–20. doi: 10.1128/mBio.01457-20. [DOI] [PMC free article] [PubMed]
- 64.Gröschel S, Sanders MA, Hoogenboezem R, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in Leukemia. Cell. 2014;157(2):369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed]
- 65.Yamazaki H, Suzuki M, Otsuki A, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25(4):415–427. doi: 10.1016/j.ccr.2014.02.008. [DOI] [PMC free article] [PubMed]
- 66.Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511(7510):428–434. doi: 10.1038/nature13379. [DOI] [PMC free article] [PubMed]
- 67.Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48(3):265–272. doi: 10.1038/ng.3502. [DOI] [PMC free article] [PubMed]
- 68.Yun JW, Yang L, Park HY, et al. Dysregulation of cancer genes by recurrent intergenic fusions. Genome Biol. 2020;21(1):166. doi: 10.1186/s13059-020-02076-2. [DOI] [PMC free article] [PubMed]
- 69.Ryan RJH, Drier Y, Whitton H, et al. Detection of enhancer-associated rearrangements reveals mechanisms of oncogene dysregulation in B-cell lymphoma. Cancer Discov. 2015;5(10):1058–1071. doi: 10.1158/2159-8290.CD-15-0370. [DOI] [PMC free article] [PubMed]
- 70.Herranz D, Ambesi-Impiombato A, Palomero T, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat Med. 2014;20(10):1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed]
- 71.Shi J, Whyte WA, Zepeda-Mendoza CJ, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013;27(24):2648–2662. doi: 10.1101/gad.232710.113. [DOI] [PMC free article] [PubMed]
- 72.Zhang X, Choi PS, Francis JM, et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat Genet. 2016;48(2):176–182. doi: 10.1038/ng.3470. [DOI] [PMC free article] [PubMed]
- 73.Chen H, Liang H. A High-Resolution Map of Human Enhancer RNA Loci Characterizes Super-enhancer Activities in Cancer. Cancer Cell. 2020;38(5):701–715. doi: 10.1016/j.ccell.2020.08.020. [DOI] [PMC free article] [PubMed]
- 74.Mansour MR, Abraham BJ, Anders L, et al. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346(6215):1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed]
- 75.Elliott K, Larsson E. Non-coding driver mutations in human cancer. Nat Rev Cancer. 2021;21(8):505–509. doi: 10.1038/s41568-021-00371-z. [DOI] [PubMed]
- 76.Hnisz D, Weintrau AS, Day DS, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351(6280):1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed]
- 77.Li K, Zhang Y, Liu X, et al. Noncoding variants connect enhancer dysregulation with nuclear receptor signaling in hematopoietic malignancies. Cancer Discov. 2020;10(5):724–745. doi: 10.1158/2159-8290.CD-19-1128. [DOI] [PMC free article] [PubMed]
- 78.Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed]
- 79.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell. 2017;168(4):629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed]
- 80.Wang X, Cairns MJ, Yan J. Super-enhancers in transcriptional regulation and genome organization. Nucleic Acids Res. 2019;47(22):11481–11496. doi: 10.1093/nar/gkz1038. [DOI] [PMC free article] [PubMed]
- 81.Vervoort SJ, Devlin JR, Kwiatkowski N, Teng M, Gray NS, Johnstone RW. Targeting transcription cycles in cancer. Nat Rev Cancer. Published online 2021. doi: 10.1038/s41568-021-00411-8. [DOI] [PubMed]
- 82.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010; 468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed]
- 83.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed]
- 84.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed]
- 85.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed]
- 86.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed]
- 87.Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed]
- 88.Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed]
- 89.Wu Q, Heidenreich D, Zhou S, et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat Commun. 2019;10(1):1915. doi: 10.1038/s41467-019-09672-2. [DOI] [PMC free article] [PubMed]
- 90.Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015;58(6):1028–1039. doi: 10.1016/j.molcel.2015.04.011. [DOI] [PMC free article] [PubMed]
- 91.Fong CY, Gilan O, Lam EYN, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–542. doi: 10.1038/nature14888. [DOI] [PMC free article] [PubMed]
- 92.Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed]
- 93.Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed]
- 94.Chipumuro E, Marco E, Christensen CL, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed]
- 95.Christensen CL, Kwiatkowski N, Abraham BJ, et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell. 2014;26(6):909–922. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed]
- 96.Wang Y, Zhang T, Kwiatkowski N, et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163(1):174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed]
- 97.Jiang YY, Lin DC, Mayakonda A, et al. Targeting super-enhancer-Associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66(8):1358–1368. doi: 10.1136/gutjnl-2016-311818. [DOI] [PMC free article] [PubMed]
- 98.Balanis NG, Sheu KM, Esedebe FN, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell. 2019;36(1):17–34. doi: 10.1016/j.ccell.2019.06.005. [DOI] [PMC free article] [PubMed]
- 99.Xu Y, Milazzo JP, Somerville TDD, et al. A TFIID-SAGA Perturbation that Targets MYB and Suppresses Acute Myeloid Leukemia. Cancer Cell. 2018;33(1):13–28.e8. doi: 10.1016/j.ccell.2017.12.002. [DOI] [PMC free article] [PubMed]
- 100.Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of Oncogenic Super-Enhancers Is Coupled with DNA Repair by RAD51. Cell Rep. 2019;29(3):560–572.e4. doi: 10.1016/j.celrep.2019.09.001. [DOI] [PMC free article] [PubMed]