

Abstract
Culture expanded bone marrow stromal cells (BMSCs) are easily isolated, can be grown rapidly en masse, and contain both skeletal stem cells (SSCs) and multipotent mesenchymal progenitors (MMPs). Despite this functional heterogeneity, BMSCs continue to be utilized for many applications due to the lack of definitive and universally accepted markers to prospectively identify and purify SSCs. Isolation is widely based on adherence to tissue culture plastic; however, high hematopoietic contamination is a significant impediment in murine models. Remarkably, when cultured at a physiological oxygen tension of 1% O2, a 10-fold reduction in CD45+ hematopoietic cells associated with a concomitant increase in PDGFRα+ stromal cells occur. This is due, in part, to a differential response of the two populations to hypoxia. In standard tissue culture conditions of 21% O2, CD45+ cells showed increased proliferation coupled with no changes in cell death compared to their counterparts grown at 1% O2. In contrast, PDGFRα+ stromal cells responded to hypoxia by increasing proliferation and exhibiting a 10-fold decrease in cell death. In summary, we describe a simple and reliable method exploiting the divergent biological response of hematopoietic and stromal cells to hypoxia to significantly increase the PDGFRα+ stromal cell population in murine BMSC cultures.
INTRODUCTION
The skeleton houses SSCs, bona fide stem cells, as defined by their in vivo ability to both self-renew and to differentiate into hematopoietic supporting stroma, osteogenic, chondrogenic and adipogenic lineages, thus regenerate bone tissue [1–3]. While SSCs have been associated with the expression of PDGRα, leptin receptor (LepR), and CD146; currently, there remains a lack of definitive cell surface markers to identify and/or prospectively isolate a homogenous population of SSCs [4–6]. In lieu of this, BMSCs which contain both SSCs and MMPs have been used as surrogates for a homogenous population of SSCs. Importantly, while MMP which lack the definitive properties of bone fide stems, presumably as they contain more committed downstream progenitors, they have been shown to support hematopoiesis and contribute to functions of the skeleton. BMSCs were first isolated based on their rapid adherence to tissue culture (TC) plastic [7]. At non-clonal density, they can be quickly expanded and are capable of differentiating into distinct mesodermal lineages in vitro demonstrating the presence of MMPs [8]. When plated at clonogenic density, some BMSCs exhibit density independent growth, forming discrete colonies derived from single cells, referred to as colony forming units-fibroblastic (CFU-F) [9–11]. Upon transplantation, some CFU-Fs have the capacity for in vivo multipotent differentiation and self-renewal, the defining features of SSCs [12, 13]. For these reasons, BMSCs serve as a powerful tool for cell-based therapies to treat musculoskeletal disorders. Moreover, with the breadth of transgenic and genetically engineered mouse models, BMSCs also serve as an invaluable means to harness information regarding the cellular and molecular mechanisms regulating MMP and SSC function. Importantly, the successful use of BMSCs for cell-based therapies and meaningful interpretation of genetic studies is dependent upon the ability to isolate pure populations of mesenchymal cells.
Preferential attachment to TC plastic can be used with success for the isolation of mesenchymal populations from human sources; however, for murine models, this technique presents several challenges [14, 15]. Most notably, this includes high hematopoietic contamination because murine hematopoietic cells can directly adhere to TC plastic and both hematopoietic stem cells (HSCs) and their differentiated progeny can attach directly to stromal cells [16, 17]. Moreover, stromal cells have the capacity to support in vitro granulopoiesis in the absence of exogenous cytokines [18]. Collectively, these factors contribute to the persistent contamination of hematopoietic cells in murine BMSC cultures even after prolonged culturing [16, 19]. Several methodologies such as subculturing, immunodepletion, and low-density seeding have been tested to eliminate hematopoietic cells from BMSC cultures. While frequent and prolonged subculturing improves purity, it also results in a replicative senescent phenotype with the possibility of altered differentiation potential [20, 21] and can select for clones carrying inactivating mutations in p53 increasing the likelihood of cellular transformation [22, 23]. Immunodepletion can result in a limited proliferative capacity of BMSCs [24] while initiating cultures at low densities is prohibitively time consuming [25]. These limitations demonstrate that hematopoietic contamination continues to serve as a significant impediment to the isolation of BMSCs based on plastic adherence [26].
The bone microenvironment is characterized by vascular heterogeneity associated with regional areas of hypoxia [27–29]. Under hypoxic conditions, cells upregulate the hypoxia inducible factor (HIF) signaling pathway to drive expression of genes which facilitate cellular adaptation to low oxygen [30]. Importantly, activation of HIF target genes is highly context dependent with cellular responses exquisitely sensitive to oxygen concentration [31]. Imaging analysis of HSC and SSC cell surface markers demonstrate that these cells are localized to regions where oxygen tensions range from 4–1% [1, 32]. While both human and murine BMSCs demonstrate improved proliferation capacity at 5% O2, the differential response of mesenchymal stromal cells and hematopoietic progenitors to physiologic oxygen tensions in the bone marrow is less well defined [22]. We discovered that 1% O2 strikingly reduced the percentage of CD45 expressing hematopoietic cells and this reduction remained consistent over time. Here, we report an easy and reliable method by which hematopoietic cells can be selectively eliminated while simultaneously enriching for stromal cells during BMSC preparations.
RESULTS
CD45+ Cells Are Prevalent When Isolation is Based on Adherence to TC Plastic
We isolated BMSCs based on the rapid adherence of cells to TC plastic and noted that high density BMSC (1.3 × 105 cm2) cultures were phenotypically heterogeneous when incubated under 21% O2 (FIG 1A, C). Given this morphological heterogeneity, we evaluated the expression of a known bone marrow stromal cell marker which enriches for SSCs, the leptin receptor (LepR). LepR marks a population of bone marrow stromal cells which arise perinatally and contribute to osteoblast, chondrocyte and adipocyte lineages in vivo, suggesting that it marks a population of SSCs [33]. To visualize and analyze LepR Cre expressing cells, LepRCre mice were crossed to Rosa26-CAG-loxp-stop-loxp-tdTomtao (Rosa26tdTm/+) mice to generate LeprCre;Rosa26tdTm/+ conditional reporter animals [33]. Here, CRE recombinase is driven by the activity of the LepR promoter thereby mediating the expression of tdTomato in LepRCre expressing cells. Flow cytometry and imaging analysis of BMSCs isolated from LeprCre;Rosa26tdTm/+ conditional reporter mice revealed only a small proportion of cells expressed tdTomato (Td/Tm) (FIG 1A). To confirm our analysis we evaluated the expression of PDGFRα, an early mesenchymal marker which is broadly expressed in the bone marrow stroma and highly expressed in CFU-Fs [34]. Consistent with other reports, LepR+ bone marrow stromal cells uniformly express PDGFRα, supporting the concept that bone marrow stromal cells express both PDGFRα and LepR (FIG 1A). Notably, the proportion of tdTomato+, PDGFRα+or tdTomato+;PDGFRα+, double positive cells constituted only a minority of the total cell population when using standard BMSC isolation methods (FIG 1A). Remarkably, flow cytometry analysis revealed that the majority of cells in BMSCS cultures express the hematopoietic cell surface marker CD45, (FIG 1A).
FIG 1. CD45+ cells constitute the majority of cells in BMSC cultures.
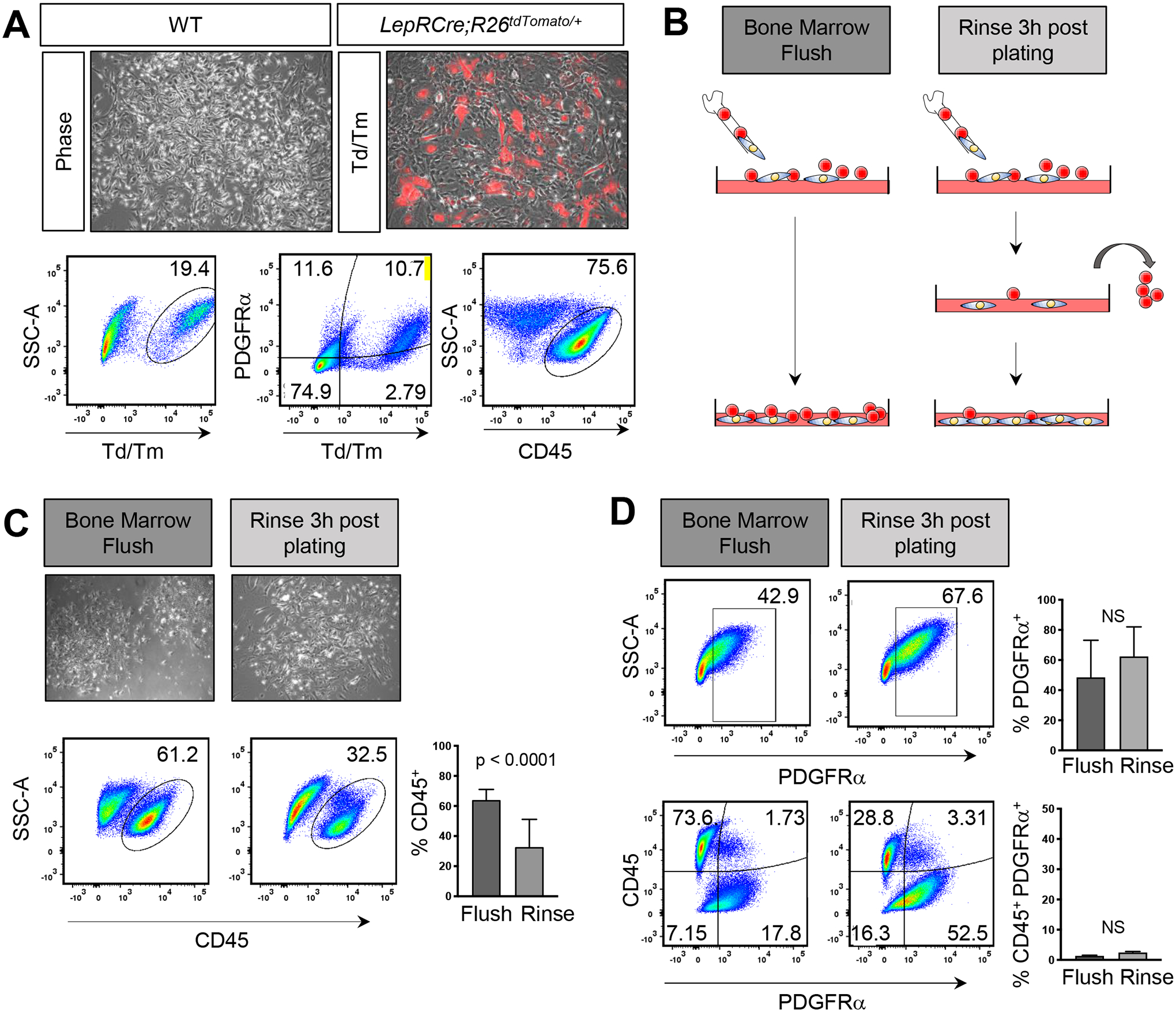
A) Phase contrast and overlay of immunofluorescent images of BMSCs isolated from LepRCre;Rosa26tdTm/+ mice. Red depicts cells in which CRE recombinase is active. Representative flow analysis plots and quantification of the percentage of CD45+, tdTomato+ (Td/Tm), and PDGFRα+;tdTomato+ double positive cells isolated from LepRCre;Rosa26tdTm/+ mice. B) Schematic representation of isolation methods. C) Phase contrast images of passage 0 BMSCs after 7 days in culture. Representative flow analysis plots and quantification of the percentage C) CD45+, D) PDGFRα+, and PDGFRα+;CD45+ double positive cells in WT BMSCs. Each graph represents an individual trial using BMSCs isolated from 2–3 month male and female mice. n≥3, unpaired two tailed t-tests, significance p < 0.05.
We sought to determine if vigorously rinsing dishes 3 hours after plating would reduce the number of CD45+ cells (FIG 1B) [10]. While the majority removed cells were CD45+ (FIG S1B); a high percentage of contaminating CD45+ cells remained in our BMSC cultures (FIG 1C). Strikingly, in unrinsed samples, only 48.43 ± 24.71% SD of cells expressed PDGFRα (FIG 1D). Vigorous rinsing after plating led to an increasing trend of PDGFRα+ cells (62.35 ± 19.65% SD); however, these findings were not statistically significant suggesting that rinsing may remove only CD45 expressing cells that take longer to adhere to TC plastic or to stromal cells and not enrich for PDGFRα+ cells (FIG 1D, S1B). Notably, PDGFRα expression was primarily restricted to CD45− cells (FIG 1D). Hence, isolation of BMSCs with conventional techniques leads to a high contamination of CD45+ hematopoietic cells coupled with moderately low numbers of mesenchymal stromal cells, highlighting the need for methods to increase the purity of stromal cells during BMSC isolation.
Hypoxia Reduces CD45+ Cells While Increasing PDGFRα+ Cells
Given the physiological relevance of hypoxia in regulating cell function within the bone microenvironment, we sought to determine if low oxygen could influence the cellular composition of BMSC preparations. BMSCs were isolated, rinsed and grown either at atmospheric (21%; normoxia) or physiologic (1%; hypoxia) oxygen tensions. In contrast to normoxic cultures, phase microscopy revealed that hypoxic cultures consisted of a homogeneous population of large bipolar spindle shaped cells (FIG 2A). Consistent with the observed changes in cellular morphology, cultures grown in hypoxic conditions showed a significant reduction in CD45+ cells coupled with a substantial increase in PDGFRα+ cells when compared to cells grown in normoxic conditions (FIG 2B). Supporting these findings, BMSCs isolated from LepRCre;Rosa26tdTm/+ mice and grown at 1% O2 also showed a significant decrease in CD45+ cells which was associated with an increase in both tdTomato+, PDGFRα+ and tdTomato+;PDGFRα+ double positive cells (FIG 2C, D). Hence, modifying culture conditions by decreasing oxygen levels to 1% profoundly diminishes the percentage of hematopoietic contaminants while enriching for PDGFRα+/LepRCre+ derived stromal cells in BMSC cultures.
FIG 2. Hypoxia depletes CD45+ cells in BMSC cultures.
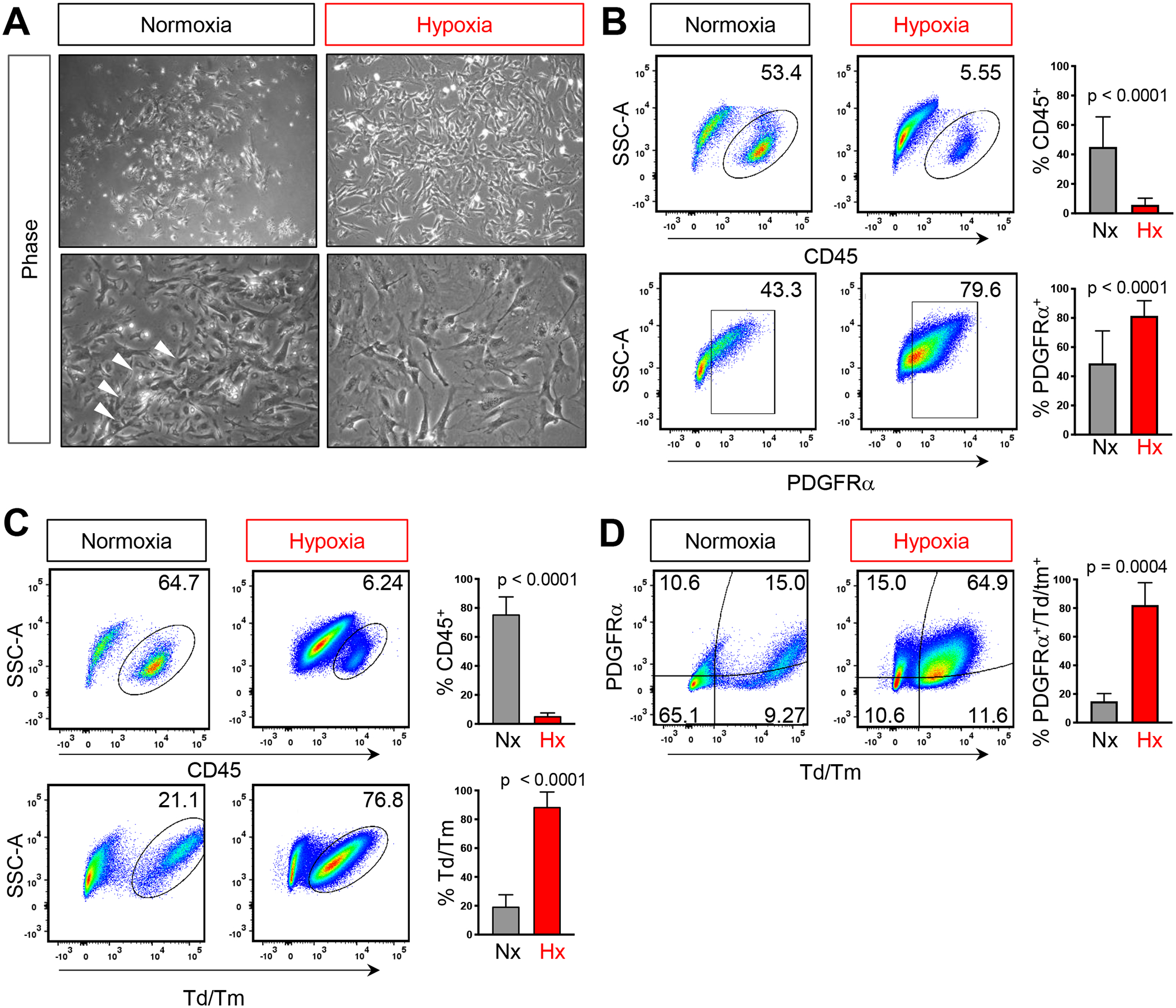
A) Phase contrast images of passage 0 BMSCs after 7 days in either 21% O2 (normoxia, Nx) or 1% O2 (hypoxia, Hx). Representative flow analysis plots and quantification of the percentage of CD45+, PDGFRα+ and PDGFRα+;tdTomato+ double positive cells in B) WT or C) LepRCre;Rosa26tdTm/+. Each graph represents an individual trial using BMSCs isolated from 2–3M male mice were used for analysis, n≥3, unpaired two tailed t-tests, significance p < 0.05
Hypoxic Reduction of CD45+ Cells Remains Constant Over Time, Passage Number, and Cell Density
We noted that over a period of seven days, CD45+ cells remained consistently high in normoxic cultures but remained below 6% in hypoxic cultures at all time points (FIG 3A). Moreover, after two rounds of trypsinization, CD45+ cells continued to persist in normoxic cultures but were reduced to 1.36 ± 0.19% of cells in hypoxic cultures (FIG 3B). While there was an increase in PDGFRα cells cultured under normoxic conditions in passage 2 (P2) when compared to passage 1 (P1), this was likely due to an increase in CD45+;PDGFRα+ double positive cells, suggesting that over time in normoxia, CD45+ cells increase their expression of the PDGFRα receptor (FIG 3B). Interestingly, PDGFRα can be expressed on macrophages which adhere to stromal cells and to TC plastic. Moreover, normoxic oxygen tensions enhance macrophage cell adhesion via activation of RhoB. Hence, PDGFRα+CD45+ double positive cells may represent macrophages that have increased adhesive properties in normoxic cultures (Huang et al., 2015) which may ultimately contribute to an increasing population CD45+ overtime in 21% oxygen tension.
FIG 3. Hypoxic depletion of CD45+ cells is maintained over time and passage number.
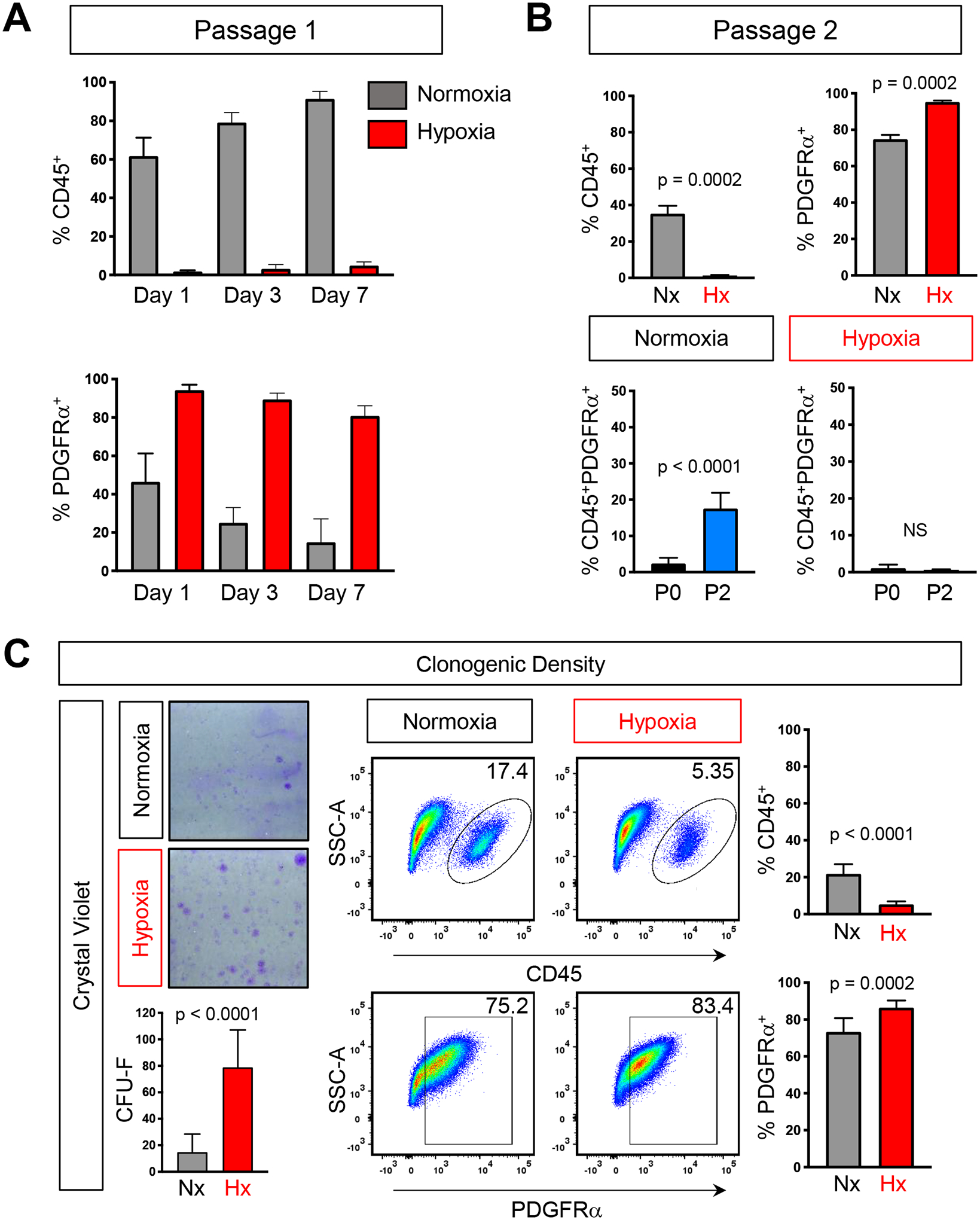
A) Quantification of flow analysis showing the percentage of CD45+ and PDGFRα cells over 7 days in either normoxia (grey) or hypoxia (red). B) Quantification of the percentage of CD45+, PDGFRα+, and CD45+;PDGFRα+ double positive cells after two passages. C) Crystal violet staining and quantification of BMSCs plated at clonogenic density (left) and representative flow analysis plots and quantification of the percentage of CD45+ and PDGFRα+ (right). Each graph represents an individual trial using BMSCs isolated from 2–3M male mice were used for analysis, n≥3, unpaired two tailed t-tests, significance p < 0.05
Culturing cells at low density has been shown to reduce hematopoietic contaminates [25]. As such, we next sought to determine if hypoxia would also enrich the cellular composition of stromal cells in BMSC cultures grown at clonogenic density. Consistent with previous reports, when cells were plated at 6 × 105 cells/cm2 with rinsing, crystal violet staining highlighted the presence of discrete colonies, derived from single cells (FIG 3C). Flow cytometry analysis revealed a significant reduction in the proportion of hematopoietic cells when compared to high density BMSC cultures, however, 21.82% ± 5.19% of cells were still of hematopoietic origin. In contrast, when grown in hypoxia, only 5.04 % ± 1.59% of cells expressed CD45 (FIG 3C). Additionally, the percentage of PDGFRα+ cells in hypoxia was also increased relative to normoxia (FIG 3C). While at clonogenic density, the number of CD45+ cells are fewer than when plated at high density cultures; importantly, hypoxia is still able to reduce hematopoietic cells in these conditions. Taken together, our data demonstrates that BMSCs grown under physiological hypoxia results in a dramatic decrease of contaminating CD45+ hematopoietic cells while increasing PDGFRα+ cells which is consistent over time, passage number and cell density.
Hypoxia Reduces Proliferation of CD45+ Cells While Inhibiting Apoptosis of PDGFRα+ Cells.
While the effect and hypoxia on hematopoietic and stromal cells have been previously examined as independent populations, the differential effect between the two populations is not clearly understood. Given the observation that hypoxia increased both the total number of CFU-F and their overall size (FIG 3C), we first assessed proliferation by incubating cells with EdU. Flow cytometry analysis revealed CD45+ cells exhibit increased EdU incorporation in normoxia when compared to hypoxia (FIG 4 A,C). Conversely, in normoxic conditions we noted a decrease in PDGFRα+;EdU+ double positive cells when compared to hypoxic conditions (FIG 4B,C). Importantly, in normoxia, CD45+ cells had higher levels of EdU incorporation when compared to PDGFRα+ cells; while in hypoxia, CD45+ cells were less proliferative than PDGFRα+ cells (FIG 4C). We next evaluated apoptosis by performing TUNEL staining assays to assess DNA fragmentation. We did not detect statistically significant differences between CD45+ cells subjected to varying oxygen tensions (FIG 4D, F). However, there was a 10-fold decrease of PDGFRα+ cells undergoing programmed cell death in hypoxia when compared to normoxia (FIG 4E, F). Moreover, in normoxia, we detected a 6-fold increase in PDGFRα+ cells undergoing apoptosis when compared to CD45+ cells (FIG 4F) but no differences between the two cell populations were detected under hypoxia. Taken together, these data indicate that CD45+ hematopoietic cells are more proliferative while PDGFRα+ cells undergo much greater cell death at 21% oxygen. However, in hypoxia, PDGFRα+ cells are more proliferative while both populations undergo cell death at the same low rate. Hence, this differential response in proliferation and apoptosis likely contributes to diminished CD45+ cells and the associated expansion of PDGFRα+ cells at low oxygen tensions.
Figure 4. Differential effect of hypoxia on proliferation and apoptosis within CD45 and PDGFRα subsets.
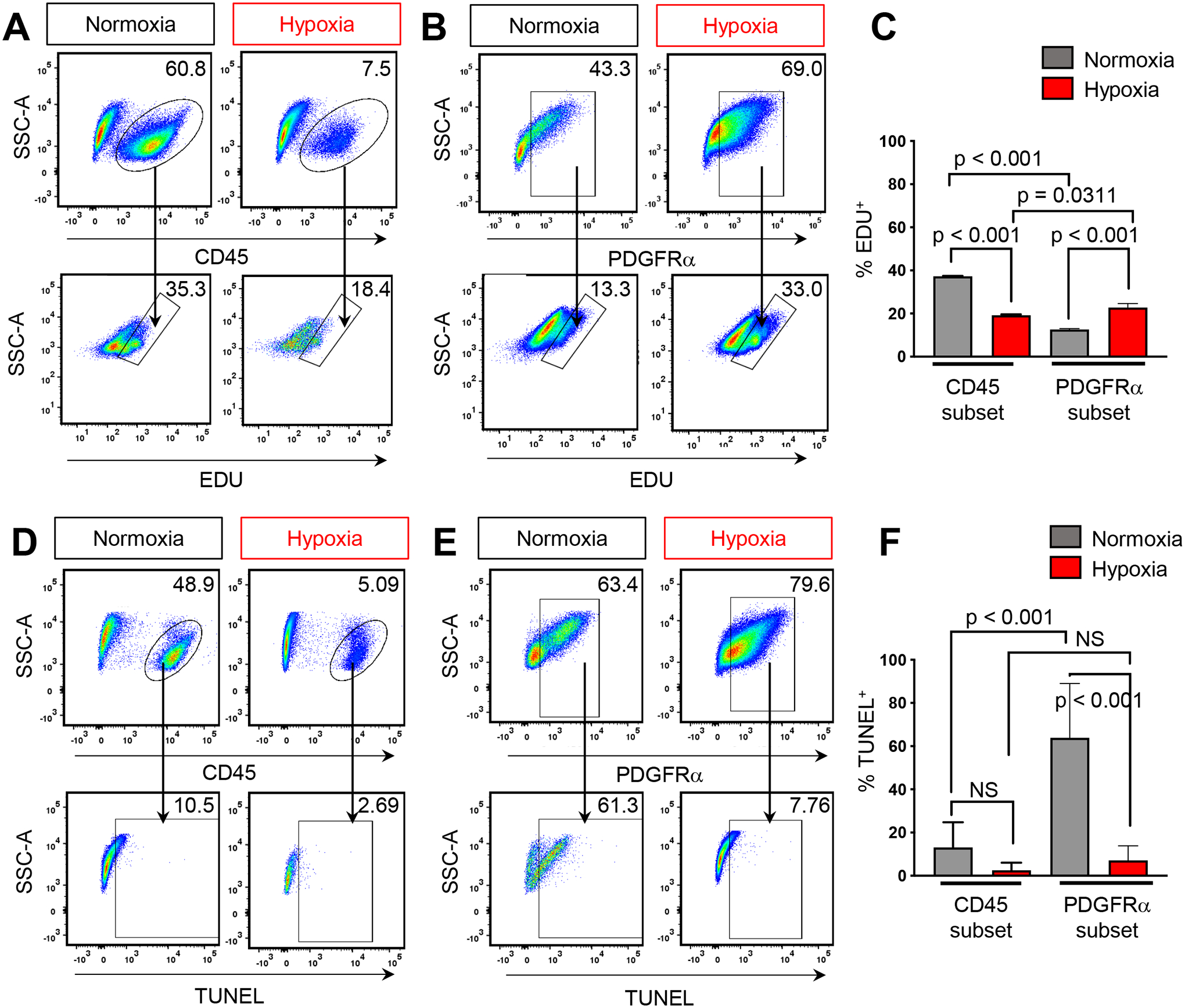
A) Representative flow analysis plots of CD45+ cells or B) PDGFRα+ and percentage of cells which have incorporated EdU C) Quantification of flow analysis for CD45+;EdU+ and PDGFRα+;EdU+ cells. D) Representative flow analysis plots of CD45+ cells or E) PDGFRα+ and percentage of TUNEL+ cells F) Quantification of flow analysis for CD45+;TUNEL+ and PDGFRα+;TUNEL+ cells. BMSCs isolated from 2–3M old male and female mice, n≥3 for each condition. One-way ANOVA, post-hoc Tukey’s multiple comparisons test, significance p < 0.05.
DISCUSSION
Although physiologic oxygen tensions in the bone microenvironment range from 4%–1%, incubators are standardly set to mimic atmospheric oxygen conditions of 21% [28]. Consistent with other reports, we found at this oxygen tension, the majority of cells in murine BMSC cultures expressed the hematopoietic marker, CD45 [22, 24]. Standard protocols that incorporate a vigorous rinsing step three hours after plating reduced CD45+ hematopoietic contaminating cells by ~50%. Here, we demonstrate incubation at a physiological oxygen tension of 1% reduced hematopoietic contaminants by over 90% during murine bone marrow preparations. Importantly, culturing at 1% oxygen also had the added benefit of selecting for PDGFRα+ stromal cells. A previous study culturing BMSCs at 5% O2 demonstrated improved murine BMSCs lifespan and differentiation potential; however, CD45 expressing cells remained prevalent, constituting 49% of the total cell population and serial immunodepletion was performed [22, 35]. In contrast, our data indicates culturing BMSCs directly in 1% O2 is sufficient to dramatically reduce contaminating CD45+ cells to less than 6% of the total cellular population, thus potentially eliminating the need for immunodepletion, reducing the need for additional passaging and decreasing the possibility of cellular transformation or immortalization associated with increased passage number [36, 37].
The bone microenvironment provides the supportive niche for HSCs and SSCs while also housing their differentiated progeny. During HSC differentiation and maturation, hematopoietic cells switch from glycolysis to oxidative phosphorylation to allow for cells to respond to increasing energy demands associated with differentiation [38–40]. Accordingly, this differentiation process is associated with an increase in mitochondrial reactive oxygen species (ROS) created during oxidative phosphorylation [41, 42]. We noted in normoxic conditions, CD45 expressing cells exhibited an increase in EdU incorporation when compared to cells grown in hypoxic conditions with no observed differences in TUNEL+ staining. It is possible that differentiated hematopoietic cells present in BMSC cultures are inherently resistant to ROS generated at 21% O2 due to the metabolic reprogramming that occurs during differentiation. However, murine HSCs, are localized to areas of low oxygen tension, are defined by strong retention of the hypoxia marker, pimonidazole, and require HIF-1α for survival and quiescence [43, 44]. As such, we cannot rule out the possibility that these biological characteristics leads to the retainment of HSC in hypoxia, contributing to a persistent low level of CD45+ cells at 1% O2.
Supraphysiologic levels of oxygen can result in a several fold increase in ROS causing extensive damage to DNA leading to genomic instability, cell cycle arrest and cell death [45]. Indeed, high oxygen tension has been shown to inhibit proliferation and survival of both human and murine BMSCs, attributed in part, to p53 dependent mitochondrial ROS production [22]. Consistent with these studies, our data indicates PDGFRα+ BMSC are exquisitely sensitive to high oxygen tension, undergoing rapid cell death as indicated by the high percentage of TUNEL positive cells when compared to CD45+ hematopoietic cells. BMSC cultured in 1% oxygen were characterized by a 10-fold reduction in programmed cell death when compared to their normoxic counterparts. These data are consistent with studies demonstrating reduced BMSC apoptosis when cultured at 3–5% oxygen [22, 46]. Collectively, the differential response seen between CD45+ and PDGFRα+ cells suggests that inherent differences in biological response to oxygen can be exploited to enhance purification of BMSC cultures.
Previous reports in murine models demonstrated that hematopoietic cells are required to provide paracrine factors necessary for stromal cell growth. Specifically, after 8 days of culture, immunodepleted BMSCs isolated from FVB/N and Balb/C mice exhibited limited growth potential relative to non-immunodepleted cells [11, 13, 24]. However, this growth inhibition was not observed in immunodepleted cells in hypoxic conditions [22]. In our study, hypoxic cultures passaged after 7 days showed incorporation of EdU suggesting the retainment of proliferative capacity. Moreover, our studies were performed using BMSCs isolated from C57BL/6 mice which have growth rates much lower than both FVB and Balb/C mice suggesting that this technique could be used to purify and overcome the inherent growth limitations in BMSCs isolated form C57BL/6 stain of mice [47]. However, we cannot rule out the possibility that the low level of CD45+ cells present in hypoxic cultures was sufficient to provide paracrine factors for stromal cells.
In summary, several methodologies have been developed for the isolation of more homogeneous populations of BMSCs including prospective isolation using cell surface markers [32], plating at low densities [13], and immunodepletion; however, these protocols have not been widely adopted. Difficulties in purifying BMSCs arise from limited lifespan, the capacity to undergo transformation and high hematopoietic content. In this report, we identified that isolating and culturing BMSCs at 1% O2 affords an easy and reproducible method to improve the purity of BMSC cultures that can be universally adopted.
EXPERIMENTAL PROCEDURES
Tissue Culture:
Bone marrow stromal cells were isolated from 8–16-week-old male and female C57BL/6J mice (Jackson Laboratory, 000664). All experimental procedures were approved by the Institutional Animal Care and Use Committee at Duke University. BMSCs were isolated by flushing the bone marrow from long bones and incubating with red blood cell lysis buffer (Sigma-Aldrich, R7757). Cells were cultured in αMEM (Gibco, 12571–063) supplemented with 20% fetal bovine serum (FBS) (GE Healthcare Life Sciences, SH30071.03), and 1% Penicillin-Streptomycin (P/S) (Gibco, 15140–122) at 37°C in 5% CO2 atmosphere. For rinsed cells three hours post-plating, the BMSCs cultures were subjected to a 3x PBS rinse to remove non-adherent cells. Media was changed 2 days post-plating and cells were grown for 7 days in their respective conditions. To assess clonogenic densities, cells were cultured and grown for 7 days as described above and seeded at 6 × 104 per cm2. For the passage 0 time point, cells were seeded at a density of 40 million cells per 150 mm dish for normoxic (21% O2) cultures and 13 million cells per 100 mm dish for hypoxic cultures prior to rinsing. Hypoxic cell cultures were grown in an InvivO2 400 chamber (Baker Ruskinn) at 1% oxygen tension. For passage 1 time points, after 7 days, cells were fully trypsinized using 0.25% Trypsin-EDTA (Gibco, 25200-056) gently scraped to collect all adherent cells, cells were plated in 100mm dishes at a density of 3.0 × 105, 2.0 × 105, and 1.5 × 105 for days 3, 5, and 7 respectively under normoxic conditions. For hypoxic conditions, cells were plated in 100mm dishes at a density of 1.1 × 105, 0.7 × 105, and 0.35 × 105 for days 3, 5, and 7 respectively. Stated densities were chosen so that cells were 80% confluent at the time of harvest.
Animal Models:
Both LepRCre (Jackson Laboratory, 032457) and CAG-loxp-stop-loxp-tdTomtao (Rosa26tdTm/+) (Jackson Laboratory, 007909) strains were purchased from The Jackson Laboratory and were maintained on a C57BL/6 background. To generate LepRCre;Rosa26tdTm/+ mice, male LepRCre mice were crossed to female Rosa26tdTm/+ mice. All experimental procedures were approved by the Institutional Animal Care and Use Committee at Duke University.
Flow cytometry:
At the indicated end points, BMSCs were harvested and stained with the LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Invitrogen, L10119) for 30 minutes at room temperature. For antigen staining, antibodies were used at the following dilutions: CD45-PE (1:250, Invitrogen, 12–0451-81), CD45-PerCP-Cyanine5.5 (1:250, Invitrogen, 45–0451-82), PDGFRα-APC (1:250, Invitrogen, 17–1401-81). A complete list of antibodies can be found in Table S1. Cells were stained with antibodies at room temperature for 30 minutes. Flow cytometry was performed on a BD FACSCanto II (BD Biosciences, 338962) and analyzed using FlowJo software (version 10.6.1). Compensation was conducted using OneComp eBeads Compensation Beads (Invitrogen, 01-1111-41) and ArC Amine Reactive Compensation Beads (Invitrogen, A10346) according to manufacturer’s instructions.
EdU staining:
Proliferation was assessed through incorporation of 5-ethynyl-2’-deoxyuridine (EdU) using the Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Invitrogen, C10425). Cells were incubated with 50mM EdU for 24 hours afterwhich cells were trypsinized and dishes were gently scraped to collect all adherent cells. Cells were fixed with 4% paraformaldehyde, permeabilized, and incorporated EdU was detected by a click reaction using a fluorescent Alexa Fluor 488 dye according to the manufacturer’s protocol. Following EdU detection, cells were stained with additional antibodies and subsequently analyzed with flow cytometry using the parameters described above.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL):
Apoptosis was assessed through the incorporation of BrdUTP at the sites of DNA damage and detected with an Alexa Fluor 488 dye–labeled anti-BrdU antibody using the APO-BrdU TUNEL Assay Kit (Invitrogen, A23210). At the desired endpoints, cells were trypsinized and dishes were gently scraped to collect all adherent cells. Following harvest, cells were treated according to the manufacturer’s instructions for the detection of TUNEL-positive cells. Briefly, cells were fixed with 1% paraformaldehyde, permeabilized in ice cold 70% ethanol at −20°C for 60 minutes, and incubated in a mixture of TdT and BrdUTP for DNA labeling for 60 mins in a 37°C water bath. Afterward, cells were incubated for 30 minutes at room temperature in an antibody staining solution containing Alexa 488 labeled anti-BrdU. During this incubation step, antibodies against markers of interest were also added at the concentrations indicated previously.
Statistical Analysis:
Statistical differences were determined by one-way ANOVA followed by Tukey’s post-hoc testing or unpaired Student’s t-test, as appropriate (GraphPad, Prism 7, version 7.04). Results are expressed as mean ± SD p<0.05 was considered significant.
Supplementary Material
REFERENCES
- 1.Sacchetti B, et al. , Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell, 2007. 131(2): p. 324–36. [DOI] [PubMed] [Google Scholar]
- 2.Chan CK, et al. , Identification and specification of the mouse skeletal stem cell. Cell, 2015. 160(1–2): p. 285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan CKF, et al. , Identification of the Human Skeletal Stem Cell. Cell, 2018. 175(1): p. 43–56 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen KG, Johnson KR, and Robey PG, Mouse Genetic Analysis of Bone Marrow Stem Cell Niches: Technological Pitfalls, Challenges, and Translational Considerations. Stem Cell Reports, 2017. 9(5): p. 1343–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baryawno N, et al. , A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell, 2019. 177(7): p. 1915–1932 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tikhonova AN, et al. , The bone marrow microenvironment at single-cell resolution. Nature, 2019. 569(7755): p. 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedenstein AJ, Chailakhjan RK, and Lalykina KS, The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet, 1970. 3(4): p. 393–403. [DOI] [PubMed] [Google Scholar]
- 8.Bianco P, et al. , Multipotential cells in the bone marrow stroma: regulation in the context of organ physiology. Crit Rev Eukaryot Gene Expr, 1999. 9(2): p. 159–73. [DOI] [PubMed] [Google Scholar]
- 9.Kuznetsov SA, et al. , Enumeration of the colony-forming units-fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res, 2009. 2(1): p. 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robey PG, et al. , Bone marrow stromal cell assays: in vitro and in vivo. Methods Mol Biol, 2014. 1130: p. 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedenstein AJ, Gorskaja JF, and Kulagina NN, Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol, 1976. 4(5): p. 267–74. [PubMed] [Google Scholar]
- 12.Bianco P, et al. , Postnatal skeletal stem cells. Methods Enzymol, 2006. 419: p. 117–48. [DOI] [PubMed] [Google Scholar]
- 13.Kuznetsov SA, et al. , Single-colony derived strains of human marrow stromal fibroblasts form bone after transplantation in vivo. J Bone Miner Res, 1997. 12(9): p. 1335–47. [DOI] [PubMed] [Google Scholar]
- 14.Sotiropoulou PA, et al. , Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells, 2006. 24(2): p. 462–71. [DOI] [PubMed] [Google Scholar]
- 15.Schallmoser K, et al. , Rapid large-scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng Part C Methods, 2008. 14(3): p. 185–96. [DOI] [PubMed] [Google Scholar]
- 16.Bearpark AD and Gordon MY, Adhesive properties distinguish sub-populations of haemopoietic stem cells with different spleen colony-forming and marrow repopulating capacities. Bone Marrow Transplant, 1989. 4(6): p. 625–8. [PubMed] [Google Scholar]
- 17.Kerk DK, et al. , Two classes of primitive pluripotent hemopoietic progenitor cells: separation by adherence. J Cell Physiol, 1985. 125(1): p. 127–34. [DOI] [PubMed] [Google Scholar]
- 18.Iguchi A, et al. , Selective stimulation of granulopoiesis in vitro by established bone marrow stromal cells. Cell Struct Funct, 1997. 22(3): p. 357–64. [DOI] [PubMed] [Google Scholar]
- 19.Gordon MY, et al. , Haemopoietic stem cell subpopulations in mouse and man: discrimination by differential adherence and marrow repopulating ability. Bone Marrow Transplant, 1990. 5 Suppl 1: p. 6–8. [PubMed] [Google Scholar]
- 20.Krebsbach PH, et al. , Bone formation in vivo: comparison of osteogenesis by transplanted mouse and human marrow stromal fibroblasts. Transplantation, 1997. 63(8): p. 1059–69. [DOI] [PubMed] [Google Scholar]
- 21.Krebsbach PH, et al. , Bone marrow stromal cells: characterization and clinical application. Crit Rev Oral Biol Med, 1999. 10(2): p. 165–81. [DOI] [PubMed] [Google Scholar]
- 22.Boregowda SV, et al. , Atmospheric oxygen inhibits growth and differentiation of marrow-derived mouse mesenchymal stem cells via a p53-dependent mechanism: implications for long-term culture expansion. Stem Cells, 2012. 30(5): p. 975–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey DM and Levine AJ, p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev, 1991. 5(12B): p. 2375–85. [DOI] [PubMed] [Google Scholar]
- 24.Baddoo M, et al. , Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J Cell Biochem, 2003. 89(6): p. 1235–49. [DOI] [PubMed] [Google Scholar]
- 25.Wang QR and Wolf NS, Dissecting the hematopoietic microenvironment. VIII. Clonal isolation and identification of cell types in murine CFU-F colonies by limiting dilution. Exp Hematol, 1990. 18(4): p. 355–9. [PubMed] [Google Scholar]
- 26.Suire C, et al. , Isolation of the stromal-vascular fraction of mouse bone marrow markedly enhances the yield of clonogenic stromal progenitors. Blood, 2012. 119(11): p. e86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kusumbe AP, Ramasamy SK, and Adams RH, Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature, 2014. 507(7492): p. 323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer JA, et al. , Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature, 2014. 508(7495): p. 269–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parmar K, et al. , Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A, 2007. 104(13): p. 5431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayer C and Vaupel P, Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther Onkol, 2012. 188(7): p. 616–27. [DOI] [PubMed] [Google Scholar]
- 31.Hu CJ, et al. , Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol, 2003. 23(24): p. 9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morikawa S, et al. , Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med, 2009. 206(11): p. 2483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou BO, et al. , Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell, 2014. 15(2): p. 154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takakura N, et al. , PDGFR alpha expression during mouse embryogenesis: immunolocalization analyzed by whole-mount immunohistostaining using the monoclonal anti-mouse PDGFR alpha antibody APA5. J Histochem Cytochem, 1997. 45(6): p. 883–93. [DOI] [PubMed] [Google Scholar]
- 35.Boregowda SV, Krishnappa V, and Phinney DG, Isolation of Mouse Bone Marrow Mesenchymal Stem Cells. Methods Mol Biol, 2016. 1416: p. 205–23. [DOI] [PubMed] [Google Scholar]
- 36.Prowse KR and Greider CW, Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci U S A, 1995. 92(11): p. 4818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wadhwa R, et al. , The ARF-p53 senescence pathway in mouse and human cells. Histol Histopathol, 2004. 19(1): p. 311–6. [DOI] [PubMed] [Google Scholar]
- 38.Takubo K, et al. , Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell, 2013. 12(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu WM, et al. , Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell, 2013. 12(1): p. 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kohli L and Passegue E, Surviving change: the metabolic journey of hematopoietic stem cells. Trends Cell Biol, 2014. 24(8): p. 479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klimmeck D, et al. , Proteomic cornerstones of hematopoietic stem cell differentiation: distinct signatures of multipotent progenitors and myeloid committed cells. Mol Cell Proteomics, 2012. 11(8): p. 286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simsek T, et al. , The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell, 2010. 7(3): p. 380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takubo K, et al. , Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell, 2010. 7(3): p. 391–402. [DOI] [PubMed] [Google Scholar]
- 44.Nombela-Arrieta C, et al. , Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol, 2013. 15(5): p. 533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parrinello S, et al. , Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol, 2003. 5(8): p. 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fehrer C, et al. , Reduced oxygen tension attenuates differentiation capacity of human mesenchymal stem cells and prolongs their lifespan. Aging Cell, 2007. 6(6): p. 745–57. [DOI] [PubMed] [Google Scholar]
- 47.Phinney DG, et al. , Plastic adherent stromal cells from the bone marrow of commonly used strains of inbred mice: variations in yield, growth, and differentiation. J Cell Biochem, 1999. 72(4): p. 570–85. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.