

Abstract
BACKGROUND:
Cellular redox control is maintained by generation of reactive oxygen/nitrogen species balanced by activation of antioxidative pathways. Disruption of redox balance leads to oxidative stress, a central causative event in numerous diseases including heart failure. Redox control in the heart exposed to hemodynamic stress, however, remains to be fully elucidated.
METHODS:
Pressure overload was triggered by transverse aortic constriction (TAC) in mice. Transcriptomic and metabolomic regulations were evaluated by RNA-sequencing and metabolomics, respectively. Stable isotope tracer labeling experiments were conducted to determine metabolic flux in vitro. Neonatal rat ventricular myocytes (NRVMs) and H9c2 cells were used to examine molecular mechanisms.
RESULTS:
We show that production of cardiomyocyte NADPH, a key factor in redox regulation, is decreased in pressure overload-induced heart failure. As a consequence, the level of reduced glutathione is downregulated, a change associated with fibrosis and cardiomyopathy. We report that the pentose phosphate pathway (PPP) and mitochondrial serine/glycine/folate metabolic signaling, two NADPH-generating pathways in the cytosol and mitochondria, respectively, are induced by TAC. We identify activating transcription factor 4 (ATF4) as an upstream transcription factor controlling the expression of multiple enzymes in these two pathways. Consistently, joint pathway analysis of transcriptomic and metabolomic data reveals that ATF4 preferably controls oxidative stress and redox-related pathways. Overexpression of ATF4 in NRVMs increases NADPH-producing enzymes whereas silencing of ATF4 decreases their expression. Further, stable isotope tracer experiments reveal that ATF4 overexpression augments metabolic flux within these two pathways. In vivo, cardiomyocyte specific deletion of ATF4 exacerbates cardiomyopathy in the setting of TAC and accelerates heart failure development, attributable, at least in part, to an inability to increase the expression of NADPH-generating enzymes.
CONCLUSIONS:
Our findings reveal that ATF4 plays a critical role in the heart under conditions of hemodynamic stress by governing both cytosolic and mitochondrial production of NADPH.
Keywords: Basic Science Research, Cell Signaling/ Signal Transduction, Myocardial Biology, Oxidative Stress
Graphical Abstract
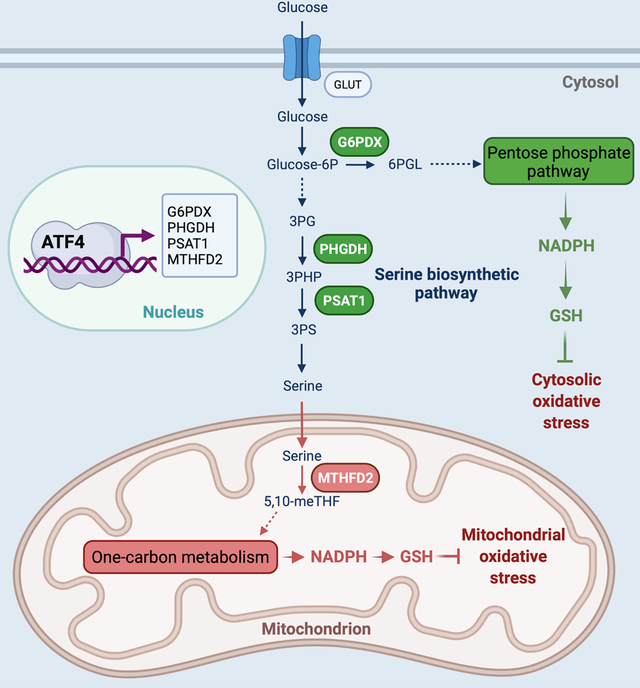
INTRODUCTION
Cellular redox control, governed by the balanced production of reactive oxygen/nitrogen species and endogenous antioxidant defense mechanisms, is essential for virtually all biological activities in a cell. Oxidative stress ensues when this balance is broken. Whereas an appropriate level of ROS (reactive oxygen species) plays a critical role in cell proliferation and survival,1 excessive ROS cause protein and lipid peroxidation, DNA damage, and cell death.2–4
Cardiovascular disease is a leading cause of death worldwide, and heart failure is a common route of convergence for various forms of cardiovascular disease.5 Oxidative stress participates in the development and progression of heart failure under a wide range of pathological conditions.6, 7 In cardiomyocytes, as in most other cell types, major endogenous components of antioxidant defense mechanisms consist of antioxidant enzymes such as superoxide dismutase, catalase, glutathione reductase, and antioxidant co-factors. The steady state levels of antioxidant molecules are largely dependent on the generation of NADPH (reduced nicotinamide adenine dinucleotide phosphate), which is used to maintain glutathione (GSH) and thioredoxin in the reduced state.8, 9 NADPH biosynthesis is controlled by a number of metabolic pathways and enzymes, including the pentose phosphate pathway (PPP), cytosolic and mitochondrial folate-mediated one-carbon metabolism, glutamine metabolism, NAD kinase, malic enzyme, nicotinamide nucleotide transhydrogenase, cytosolic or mitochondrial NADP-dependent isocitrate dehydrogenase, and fatty acid oxidation. Whereas the role of NADPH governed by these pathways and enzymes is well established in cancer,10, 11 contributions of these pathways and enzymes to NADPH biosynthesis in heart failure remain elusive.
ATF4 (activating transcription factor 4), a key element of both the unfolded protein response and the integrated stress response, belongs to the basic leucine zipper transcription factor family and governs transcription of genes involved in antioxidative response, autophagy, and nutrient sensing.12, 13 Its function in the heart, however, is largely uncharacterized.
The causal role of oxidative stress in heart failure, coupled with the intimate connections among ATF4, redox balance, and cell survival, prompted us to hypothesize that ATF4 is essential to maintain cardiac function under pathological conditions through oxidative stress control. To test this hypothesis, both in vivo and in vitro approaches were undertaken to overexpress or inhibit ATF4 expression. We show that ATF4 stimulates transcription of genes coding for multiple key enzymes involved in cardiac NADPH production both in the cytosol and in mitochondria. This, in turn, improves cardiomyocyte adaptation and survival during the initiation and progression of heart failure triggered by hemodynamic stress.
METHODS
Data Availability
Detailed experimental procedures are described in the Supplemental Materials. Data, methods, and study materials are available from the corresponding author upon reasonable request. Key research materials are listed in the Major Resources Table in the Supplemental Materials.
Animals
Mouse protocols were approved by the Institutional Animal Care and Use Committee of University of Texas Southwestern Medical Center (UTSW). Genotyping primers are listed in Supplemental Table S1.
Statistical Analyses
All data are represented as mean±SEM. Normality of distribution was assessed by using Shapiro-Wilk test. Normally distributed data were then analyzed with unpaired, 2-tailed Student’s t test to compare differences between two groups. For multiple group comparisons with 1 variable, one-way ANOVA was performed, followed by Dunnett or Tukey’s multiple comparisons test. For multiple group comparisons with more than 2 variables, two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test. For data not following a normal distribution, unpaired 2-tailed Mann-Whitney test (2 groups) or Kruskal-Wallis test (3 or more groups) was used, followed by Dunn post hoc test. A P value of <0.05 was considered statistically significant. Statistical analyses were conducted with Graphpad Prism 8.4.3.
RESULTS
Oxidative stress is increased in pressure overload-induced heart failure.
Although it is well established that oxidative stress contributes to the pathogenesis of heart failure,7 stress-induced dynamic changes in cardiac redox balance remain to be characterized. To address this question, we subjected wild-type C57BL/6 mice to transverse aortic constriction (TAC) to trigger dilated cardiomyopathy and heart failure. TAC is a widely used surgical procedure to induce pathological cardiac remodeling by imposing sustained pressure overload.14 We collected the heart 3 days, 1 week, and 3 weeks after the operation for morphological and molecular analyses (Figure 1A). TAC induced profound pathological hypertrophic growth (Figure S1A and S1B) and contractile dysfunction (Figure 1B and Figure S1C through S1E). Moreover, cell death in the heart was elevated by TAC as revealed by an increase in cleaved caspase 3 (Figure 1C) and TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling)-positive cells (Figure 1D).
Figure 1. Oxidative stress is elevated by pressure overload-induced heart failure.
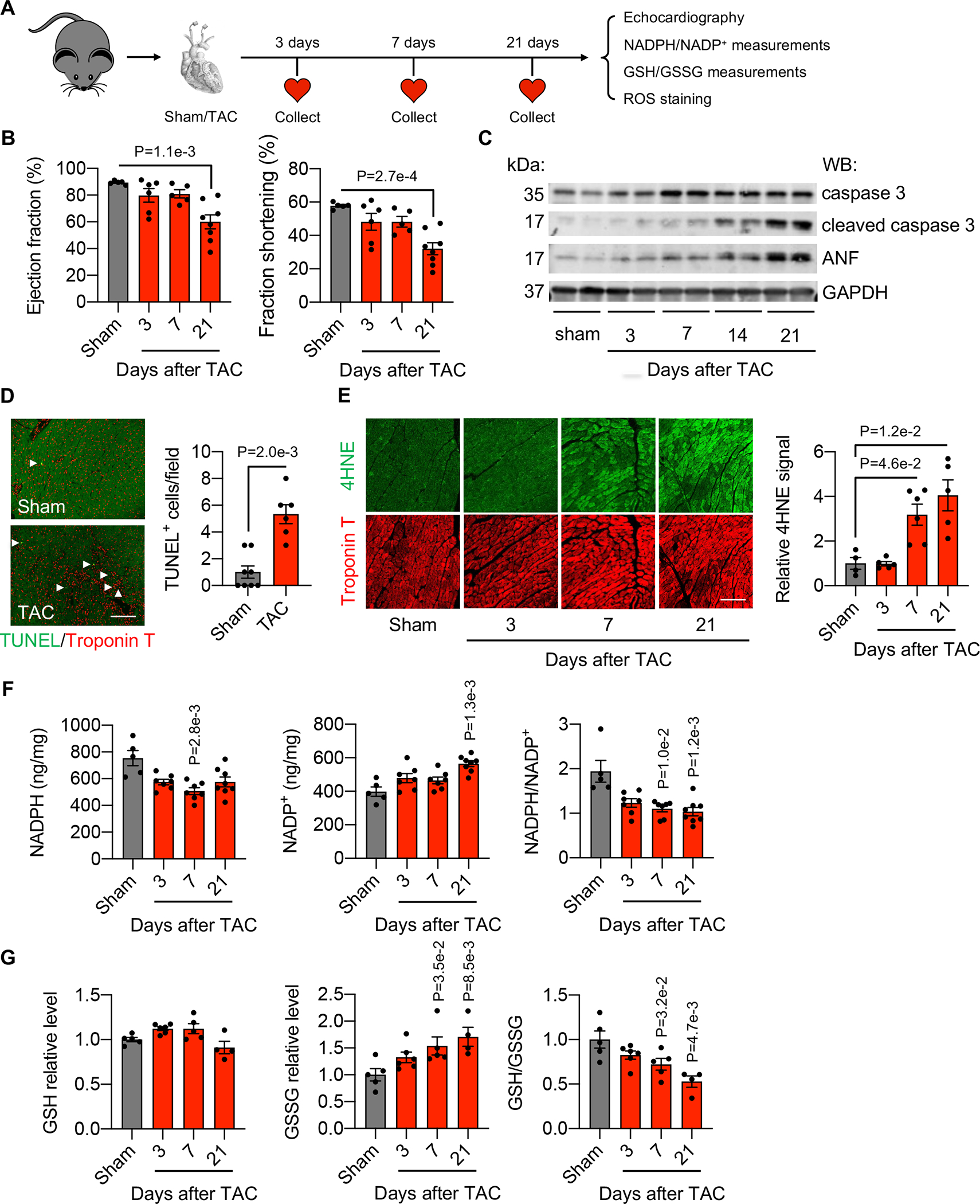
A. Schematic of experimental design. Wild-type C57BL/6 mice were subjected to sham or transverse aortic constriction (TAC). A series of measurements were made 3, 7, and 21 days after TAC. NADPH, reduced nicotinamide adenine dinucleotide phosphate; NADP+, nicotinamide adenine dinucleotide phosphate; GSSG, glutathione disulfide; GSH, glutathione; ROS, reactive oxygen species.
B. TAC caused cardiac dysfunction in mice. Ejection fraction and fractional shortening (%) were determined by echocardiography. Sham, n=5; 3 days after TAC, n=6; 7 days after TAC, n=5; 21 days after TAC, n=8.
C. Western blotting showed that cleaved caspase 3 and ANP were upregulated in the heart after TAC.
D. TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) staining identified apoptotic cardiac cells 3 weeks after TAC. Scale: 100 μm. Sham, n=8; TAC, n=6.
E. The hypertrophied heart at 7 and 21 days after TAC showed an elevated level of 4-HNE (4-hydroxynonenal)-positive signal. Scale: 100 μm. N=4–6. Sham, n=4; 3 days after TAC, n=5; 7 days after TAC, n=6; 21 days after TAC, n=5.
F. Both NADPH level and the NADPH/NADP+ ratio were significantly decreased in mice 3, 7, and 21 days after TAC. NADP+ level was significantly increased in mice 3, 7, and 21 days after TAC. Sham, n=5; 3 days after TAC, n=7; 7 days after TAC, n=7; 21 days after TAC, n=8.
G. GSSG was increased in the heart 7 and 21 days after TAC. The GSH/GSSG ratio was decreased 7 and 21 days after TAC. Sham, n=5; 3 days after TAC, n=6; 7 days after TAC, n=5; 21 days after TAC, n=4.
Kruskal-Wallis test was conducted, followed by Dunnett’s test to determine statistical significance for comparisons of the means of each surgical group (3, 7, and 21 days after TAC) with the mean of the sham group (B, E-G). Unpaired Student’s t test was conducted for D. GSH (F) and GSSG (G) levels from each surgical group (3, 7, and 21 days after TAC) were normalized to the mean of the sham group, which was set to 1. Data are represented as mean±SEM.
Next, we used this heart failure model to investigate whether cell death and cardiomyopathy are associated with increased oxidative stress. We performed 4-HNE (4-hydroxynonenal) staining to detect lipid peroxidation as a surrogate of ROS levels. The hypertrophied heart 7 days and 21 days after TAC manifested an elevated 4-HNE signal (Figure 1E). These findings suggest that pressure overload-induced cardiac dysfunction is accompanied by an increase in oxidative stress.
We then hypothesized that endogenous antioxidant mechanisms may be induced as a defense response during the development of heart failure. Glutathione (GST) is a prominent reducing factor and the major antioxidant in cells.15 NADPH is required to convert oxidized glutathione disulfide (GSSG) to reduced GSH, and increased production of NADPH promotes GSH regeneration. We found that the levels of NADPH in the heart were significantly reduced 3 days, 7 days, and 21 days after TAC (Figure 1F). As a consequence, the NADPH/NADP+ ratio was decreased (Figure 1F). Consistent with the essential role of NADPH to regenerate GSH, cardiac GSSG was significantly higher and the ratio between GSH and GSSG was reduced 7 days and 21 days after TAC (Figure 1G). Taken together, these findings suggest that pressure overload-induced cardiomyopathy is associated with an increase in oxidative stress and reduced production of NADPH and GSH in the heart.
NADPH production pathways are increased in the heart by pressure overload.
Given the significant decrease in NADPH in heart failure that we observed, we asked whether NADPH production pathways may be altered. NADPH is generated by various pathways in the cytosol and mitochondria (Figure 2A). Among them, we found that MTHFD2 and G6PDX were significantly upregulated in the heart 3 days, 7 days, and 21 days after TAC as compared to sham controls at both protein (Figure 2B and Figure S2A) and mRNA levels (Figure 2C). MTHFD2 in mitochondria generates NADPH by catalyzing the conversion of formyl-THF to methylene-THF in the one-carbon metabolic pathway.16 G6PDX is the rate-limiting enzyme in the pentose phosphate pathway (PPP), an ancillary glucose metabolic pathway for cytosolic NADPH production and nucleotide synthesis.17 Serine is a key substrate for NADPH production mediated by MTHFD2 in mitochondria. Consistent with elevations in MTHFD2, we found that PHGDH and PSAT1, two enzymes in the de novo serine biosynthetic pathway, were also increased by TAC (Figure 2B and 2C, and Figure S2A). Together, these data suggest that pressure overload stimulates both mitochondrial and cytosolic NADPH generation in the heart.
Figure 2. NADPH production pathways are stimulated in the heart by pressure overload.
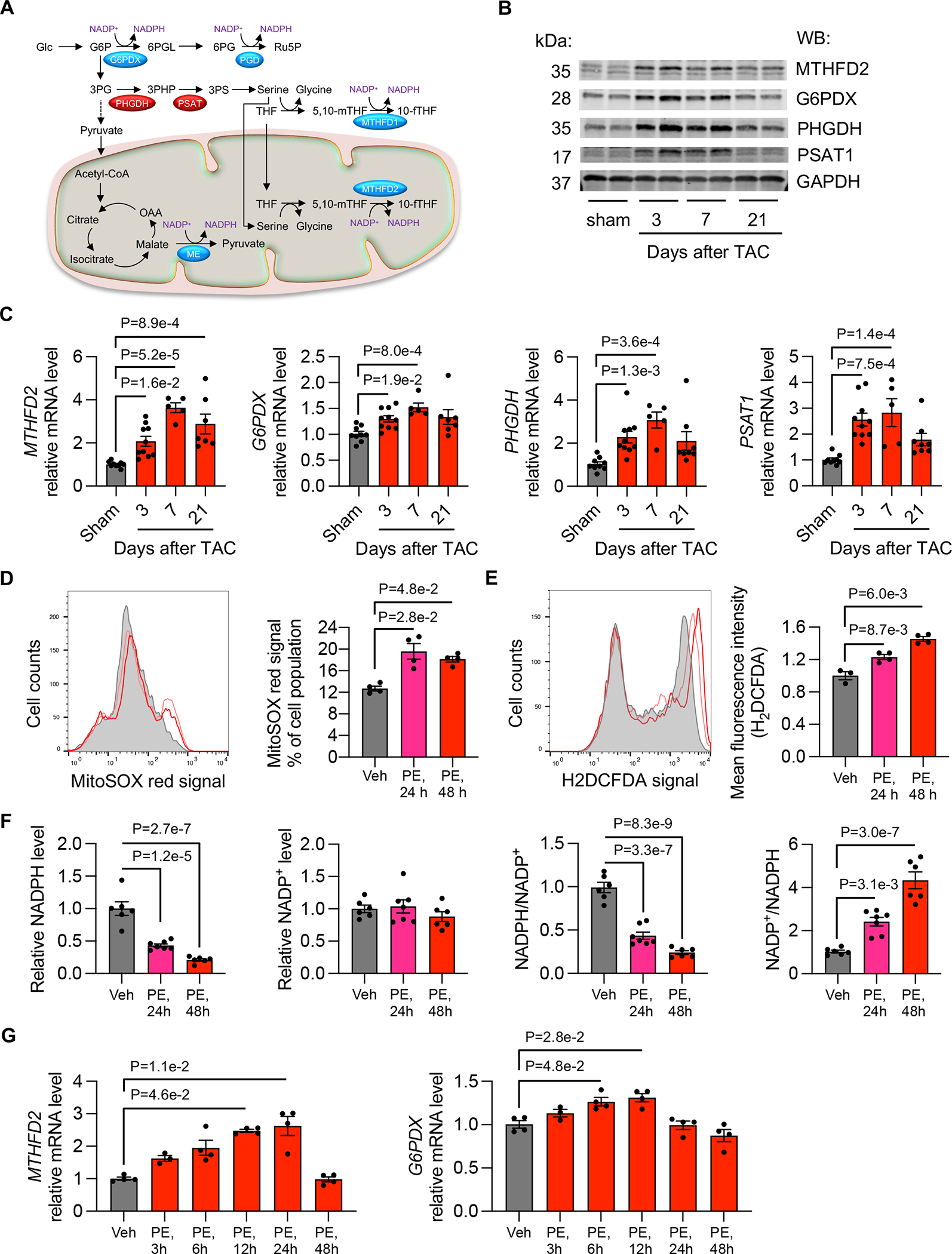
A. Schematic of NADPH-generation pathways.
B. TAC increased the protein levels of MTFFD2, G6PDX, PHGDH, and PSAT1 in the heart. GAPDH was used as a loading control.
C. TAC elevated the mRNA levels of MTHFD2, G6PDX, PHGDH, and PSAT1 in the heart. Sham, n=9; 3 days after TAC, n=10; 7 days after TAC, n=5; 21 days after TAC, n=8.
D. Mitochondrial ROS production was measured by oxidation of the fluorescent probe MitoSOX red in NRVMs (neonatal rat ventricular myocytes) after PE treatment for 24 and 48 hours, respectively. N=4 for each group.
E. Cytosolic ROS level was determined by H2DCFDA staining and flow cytometry. Vehicle (Veh), n=3; PE, 24 h, n=4; PE, 48 h, n=4.
F. NADPH was reduced in NRVMs after PE treatment, whereas the ratio of NADP+/NADPH was increased. Vehicle (Veh), n=6; PE, 24 h, n=7; PE, 48 h, n=6.
G. PE treatment in NRVMs stimulated the mRNA expression of MTHFD2 and G6PDX, respectively. Vehicle (Veh), n=3; PE, 24 h, n=4; PE, 48 h, n=4.
Kruskal-Wallis test was conducted, followed by Dunn’s test to determine statistical significance for comparisons of the means of each surgical group (3, 7, and 21 days after TAC) with the mean of the sham group (C). Gene expression at the mRNA level from each surgical group (3, 7, and 21 days after TAC) was normalized to the mean of the sham group, which was set to 1 (C). One-way ANOVA was conducted, followed by Dunnett’s test to determine statistical significance for comparisons of the means of each treatment group (PE treatment for various times) with the mean of the vehicle (Veh) group (D-E, G). Mean fluorescent intensity of each PE treatment group was normalized to the mean of the vehicle (Veh) group, which was set to 1 (E). Gene expression at the mRNA level from each PE treatment group was normalized to the mean of the vehicle (Veh) group, which was set to 1 (G). One-way ANOVA was conducted, followed by Tukey’s test to determine statistical significance for comparisons of the means of each treatment group (PE treatment for various times) with the mean of the vehicle (Veh) group (F). NADPH and NADP+ levels from each PE treatment were normalized to the mean of the vehicle (Veh) group, which was set to 1 (F). Data are represented as mean±SEM.
To determine whether the correlation between ROS and NADPH generation in the heart is cardiomyocyte-autonomous, we turned to cultured neonatal rat ventricular myocytes (NRVMs) in vitro. We isolated NRVMs from 1–2-day old Sprague-Dawley rats. Phenylephrine (PE) is an α1-adrenergic receptor agonist commonly used to stimulate cardiomyocyte hypertrophic growth in vitro.18, 19 We found that PE (50 μM) led to a significant increase in mitochondrial ROS levels in NRVMs 24 hours and 48 hours after treatment (Figure 2D). In addition, cytosolic ROS levels were elevated by PE (Figure 2E). Moreover, the level of NADPH and the ratio of NADPH to NADP+ were decreased, whereas the ratio of NADP+ to NADPH was increased in NRVMs by PE treatment (Figure 2F). Along these lines, transcripts for MTHFD2 and G6PDX, two enzymes that generate NADPH, were significantly upregulated by PE (Figure 2G). However, we observed no consistent change in transcript levels of other genes in antioxidant pathways in vivo or in vitro (Figure S2B and S2C). Collectively, these findings suggest that NADPH generation in both mitochondria and the cytosol, via the one-carbon metabolic pathway and PPP, respectively, is activated in response to oxidative stress. This, however, is apparently insufficient to curb the progression of cardiomyopathy in the setting of persistent hemodynamic stress.
Transcription of key enzymes in the one-carbon metabolic pathway and PPP is governed by ATF4.
We observed that the expression of MTHFD2, G6PDX, PHGDH, and PSAT1 are acutely increased at an early stage in the heart after TAC or in NRVMs exposed to PE. We next hypothesized that these genes may be controlled by a common signaling pathway to coordinately generate NADPH in both mitochondria and the cytosol. To identify shared transcription factor(s), we screened the promoters of these 4 genes, finding that the promoters of MTHFD2 and PHGDH genes harbored conserved binding sites for ATF4 (Figure S3A). Consistent with this, previous studies by Manning and colleagues identified a highly conserved ATF4-binding site in the MTHFD2 promoter.20 Additional analysis revealed similar sites in the promoters of G6PDX and PSAT1 (Figure S3A). Further, a chromatin immunoprecipitation (ChIP) assay showed that ATF4 bound the promoters of MTHFD2 and G6PDX (Figure S3B). These data suggest that ATF4 may be an upstream transcription factor controlling both the PPP and one-carbon metabolic pathway in the heart.
ATF4 is a stress-induced transcription factor that is frequently upregulated in cancer cells.13 ATF4 controls the expression of a wide range of adaptive genes, positioning cells to endure periods of stress, such as hypoxia or limited access to amino acids. However, the role of ATF4 in the heart remains undefined. We first sought to determine whether ATF4 expression is altered during the initiation and progression of heart failure. Compared to sham animals, TAC led to a significant increase in ATF4 mRNA levels in the heart 3 days and 7 days after the onset of pressure overload (Figure 3A). Moreover, we conducted a reverse experiment by removing aortic constriction after 3 days of TAC. This deTAC procedure led to a decrease of hypertrophic marker gene expression (Figure S3C). Importantly, the induction of ATF4 was also diminished by deTAC (Figure S3D), along with the decrease of the expression of MTHFD2, G6PDX, PHDGH, and PSAT1 (Figure S3E). In contrast, volume overload did not affect ATF4 expression in the heart (Figure S3F). Consistent with the induction of ATF4 by TAC in vivo, ATF4 expression was also increased in NRVMs 6, 12, and 24 hours after PE treatment in vitro (Figure 3B).
Figure 3. ATF4 expression is elevated under cardiomyocyte hypertrophic growth.
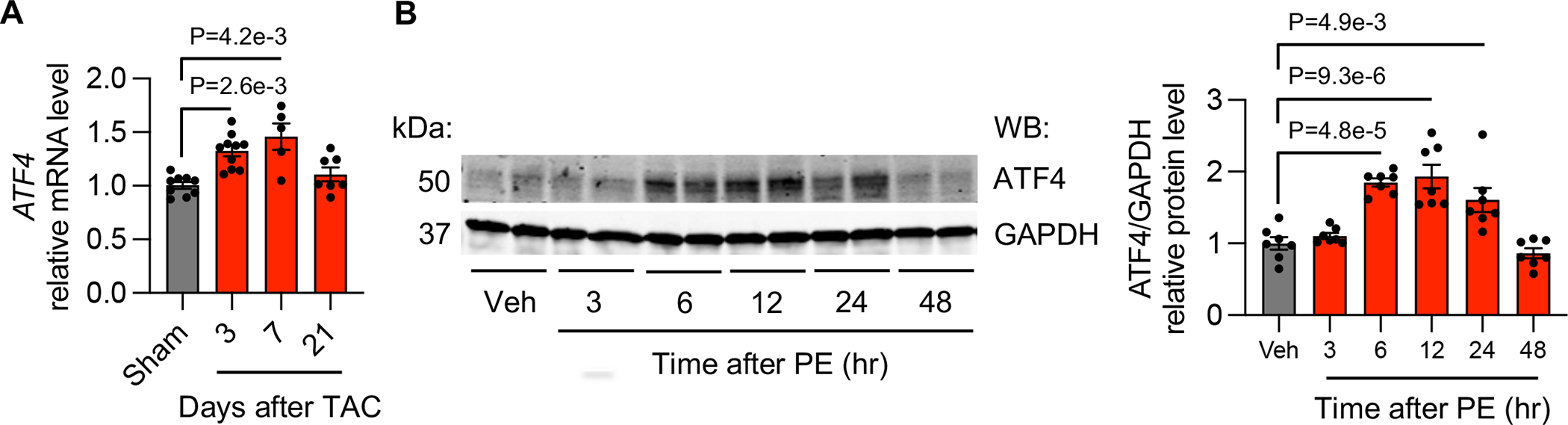
A. ATF4 expression was upregulated in the heart 3 and 7 days after TAC. Sham, n=9; 3 days after TAC, n=10; 7 days after TAC, n=5; 21 days after TAC, n=8.
B. ATF4 was elevated at the protein level in NRVMs 6, 12, and 24 hours after PE treatment. N=7 for each group.
Kruskal-Wallis test was conducted, followed by Dunn’s test to determine statistical significance for comparisons of the means of each surgical group (3, 7, and 21 days after TAC) with the mean of the sham group (A). Gene expression at the mRNA level from each surgical group (3, 7, and 21 days after TAC) was normalized to the mean of the sham group, which was set to 1 (A). One-way ANOVA was conducted, followed by Tukey’s test to determine statistical significance for comparisons of the means of each treatment group (PE treatment for various times) with the mean of the vehicle (Veh) group (B). Gene expression at the protein level from each PE treatment group was normalized to the mean of the vehicle (Veh) group, which was set to 1 (B). Data are represented as mean±SEM.
To further assess the role of ATF4 in promoting expression of genes in the one-carbon metabolic pathway and PPP, we overexpressed ATF4 in NRVMs by adenoviral infection (Figure S4A). Overexpression of ATF4 was sufficient to stimulate transcription of MTHFD2, G6PDX, PHGDH, and PSAT1 in a dose-dependent manner (Figure S4B). Conversely, silencing of ATF4 in NRVMs reduced the mRNA levels of these enzymes (Figure S4C). Transcripts for MTHFD1L, SHMT2, and ALDH1L2 also showed consistent changes under ATF4 overexpression and silencing (Figure S4D and S4E), suggesting that ATF4 directly promotes expression of multiple enzymes of the one-carbon metabolic pathway.
After establishing transcriptional regulation by ATF4 of enzymes in the one-carbon metabolic pathway and PPP, we next asked whether ATF4 directly controls flux through these pathways. We first overexpressed ATF4 by adenoviral infection of NRVMs, using GFP as a control. We then conducted two tracing assays using 1,2-13C-glucose and U-13C-glucose to determine the flux through the PPP and serine biosynthesis pathway, respectively. Consistent with the increase in gene expression mediated by ATF4, fluxes through the PPP (Figure S5A) and serine biosynthetic pathway (Figure S5B) showed a trend of increase in ATF4-overexpressing NRVMs. Collectively, these data support a model in which ATF4 directly governs the transcription of multiple enzymes involved in metabolic flux through the PPP and serine biosynthesis in cardiomyocytes.
ATF4 controls redox balance in cardiomyocytes.
To further evaluate the role of ATF4 in cardiac remodeling, we overexpressed ATF4 in NRVMs and carried out RNA-sequencing and metabolomics analyses. Our goal was to evaluate pathways co-regulated by ATF4 at both transcriptional and metabolomic levels in an unbiased manner. Principal component analysis (PCA) of transcriptomic data revealed distinct profiles between the control group and the ATF4 overexpression group (Figure 4A). Metabolite profiles were analyzed by PCA using MetaboAnalyst 5.0,21 which uncovered a clear separation between the two treatment groups (Figure 4B). Next, we employed a combined bioinformatic analytical approach to dissect pathways co-regulated by ATF4 at both transcriptomic and metabolomic levels. A total of 1,550 transcripts were significantly downregulated whereas 1,317 transcripts were upregulated between the GFP and ATF4 overexpression samples (Figure 4C and Figure S6A). In addition, metabolomic analysis identified 44 metabolites that were significantly changed (≥1.2 or ≤0.8 with adjusted P value <0.1), 40 of which were elevated (Figure 4C and Figure S6B).
Figure 4. ATF4 controls the one-carbon metabolic pathway and PPP.
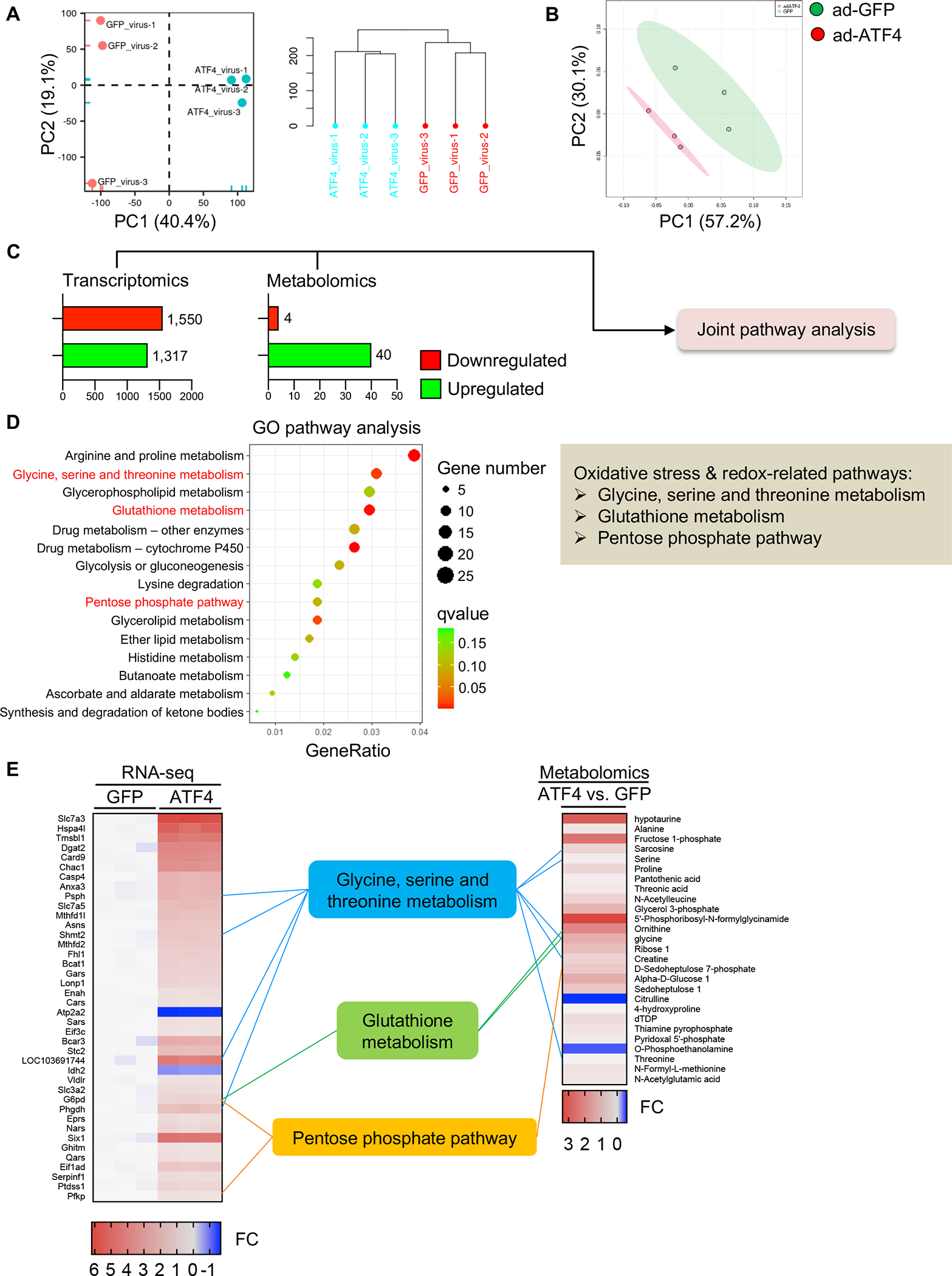
A. NRVMs were infected by adenoviruses expressing ATF4 or control GFP. RNA-sequencing was conducted and PCA analysis was performed. N=3 for each group.
B. Metabolomics were performed with NRVMs expressing control GFP or ATF4. N=3 for each group.
C. Significantly changed genes (RNA-sequencing) and metabolites (metabolomics) were subjected to the joint pathway analysis (JPA).
D. Multi-omics analysis using transcriptomic and metabolomic data was performed according to the analytical scheme shown in C. JPA was used to calculate the bubble chart, which is a combination of enrichment P values and gene numbers of individual pathways.
E. The top 40 transcripts (left, RNA-sequencing) and 27 metabolites (right, metabolomics) with cutoff threshold of fold change>1.2 or <0.8 and P value adjusted <0.05 are shown. FC, fold change.
We next used these differentially regulated transcripts and metabolites as input for multi-omics analysis with the joint pathway analysis (JPA) module from MetaboAnalyst 5.0.21 JPA is commonly used for pathway topology analyses and functional enrichment from a combination of different omics data sources, such as transcriptomics and metabolomics. The 15 top ranked pathways revealed by JPA are shown (Figure 4D and Figure S6C). Importantly, among these were factors involved in oxidative stress control and redox regulation, such as glycine, serine, and threonine metabolism, glutathione metabolism, and the PPP (Figure 4D and 4E). Taken together, these results are consistent with our promoter analysis findings, which further supports a role for ATF4 as a common upstream transcription factor promoting expression of both the one-carbon metabolic pathway and PPP to govern redox control.
ATF4 deficiency exacerbates pressure overload-induced heart failure.
We went on to investigate the role of ATF4 in the heart in vivo under pathological conditions. To selectively eliminate ATF4 from cardiomyocytes, we crossed conditional ATF4F/F animals22 with cardiomyocyte specific Cre transgenic mice (ɑMHC-Cre). Conditional knockout (cKO) mice did not manifest appreciable differences at the time of birth, weaning, or as an adult (data not shown).
We subjected cKO mice and littermate controls to surgical pressure overload. There was no decline in cardiac function 7 days after TAC in ATF4F/F control mice (Figure 5A and 5B). However, loss of ATF4 from cardiomyocytes exacerbated the progression to heart failure, as evidenced by decreases in ejection fraction (EF) (Figure 5B) and fractional shortening (FS) (Figure 5B) and increases in ventricular dimensions (Figure S7A) in cKO mice. Moreover, Masson’s trichrome staining revealed excessive collagen deposition in cKO hearts after TAC (Figure 5C), indicating elevated cardiac fibrosis. TUNEL staining identified more apoptotic cells in ATF4 cKO hearts than ATF4F/F controls 7 days after TAC (Figure 5D). Furthermore, Western blots to detect cleaved caspase 3 displayed a significant increase in ATF4 cKO hearts 7 days after TAC (Figure 5E). Gene expression of markers for heart failure (ANP and BNP) and fibrosis (TGFβ2, CTGF, and COLa1) were upregulated in ATF4-deficient hearts 7 days after TAC (Figure 5F and 5G). Collectively, these data suggest that cardiomyocyte specific deletion of ATF4 increases the severity of cell death and cardiomyopathy in the setting of pressure overload.
Figure 5. ATF4 deficiency exacerbates heart failure under pressure overload.
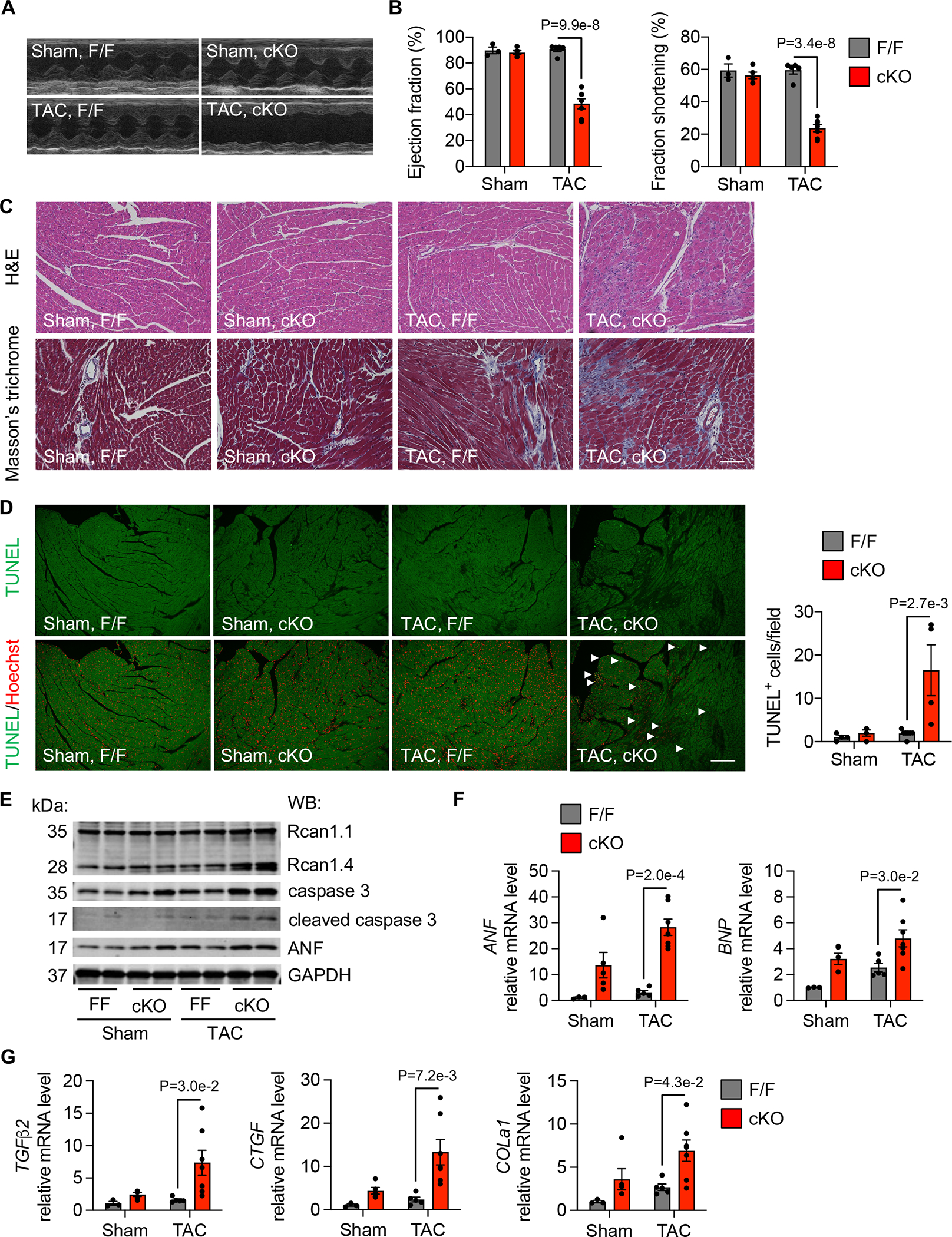
A. After 1 week of sham or TAC, control ATF4F/F (F/F) and ATF4 cardiomyocyte specific conditional knockout (cKO) mice were used for echocardiography measurements. Representative M-mode images are shown.
B. ATF4 cKO led to deterioration of cardiac function 1 week after TAC, as revealed by decreases in ejection fraction and fraction shortening. Sham, F/F, n=3; Sham, cKO, n=5; TAC, F/F, n=5; TAC, cKO, n=7.
C. ATF4-deficient hearts displayed profound immune cell infiltration after TAC, which was clustered as patches and distributed in myocardium. Fibrosis was increased in cKO hearts compared to controls. Scale: 100 μm.
D. TUNEL staining revealed more apoptotic cell death in the ATF4 cKO heart after TAC. Scale: 100 μm. Sham, F/F, n=4; Sham, cKO, n=4; TAC, F/F, n=7; TAC, cKO, n=4.
E. ATF4 knockout in the heart caused pathological cardiac remodeling after TAC as revealed by increases of expression of Rcan1.4, caspase 3, cleaved caspase 3, and ANP.
F. The mRNA levels of heart failure markers, ANF and BNP, were augmented in cKO hearts. Sham, F/F, n=3; Sham, cKO, n=5; TAC, F/F, n=6; TAC, cKO, n=7.
G. Cardiac fibrosis marker genes including TGFβ2, CTGF, and COLa1 were elevated by ATF4 deletion in the heart. Sham, F/F, n=3; Sham, cKO, n=5; TAC, F/F, n=6; TAC, cKO, n=7.
Two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (B, D, F-G). Gene expression at the mRNA level from each group was normalized to the mean of the sham/ATF4F/F group, which was set to 1 (F-G). Data are represented as mean±SEM.
ATF4 mediates protection from oxidative stress.
We have shown that ATF4 governs multiple pathways involved in NADPH production and antioxidative control. In addition, we found that loss of ATF4 from cardiomyocytes exacerbates the development of heart failure under pressure overload. We next asked whether reduction in oxidative stress is a mechanism underlying ATF4-mediated protection of cardiac function.
Cardiac GSSG levels were significantly increased in ATF4 cKO hearts 3 days after TAC compared to controls (Figure 6A), a time before functional evidence of cardiomyopathy emerges. The ratio of GSH to GSSG was reduced, whereas the ratio of GSSG to GSH was increased (Figure 6A). Along this line, the ratio of NADPH to NADP+ was reduced in cKO hearts (Figure S7B). Furthermore, 4HNE staining revealed a higher level of signal in cKO hearts compared to control hearts 3 days after TAC (Figure 6B). Consistent with the role of ATF4 in controlling expression of enzymes in the one-carbon metabolic pathway and PPP, ATF4 cKO resulted in a significant decrease in the mRNA levels of MTHFD2, G6PDX, PHGDH, and PSAT1 (Figure 6C). Beside oxidative stress control, ATF4 as a versatile transcription factor may regulate other cellular processes that participate in cardiac functional maintenance. To address this question, we conducted RNA-sequencing analysis using cardiac samples 7 days after sham or TAC. KEGG analysis showed that in addition to carbon metabolism that includes the PPP and one-carbon metabolic pathway, fatty acid metabolism was highly enriched that may contribute to ATF4 actions in the heart (Figure S7C).
Figure 6. ATF4 maintains oxidative stress control.
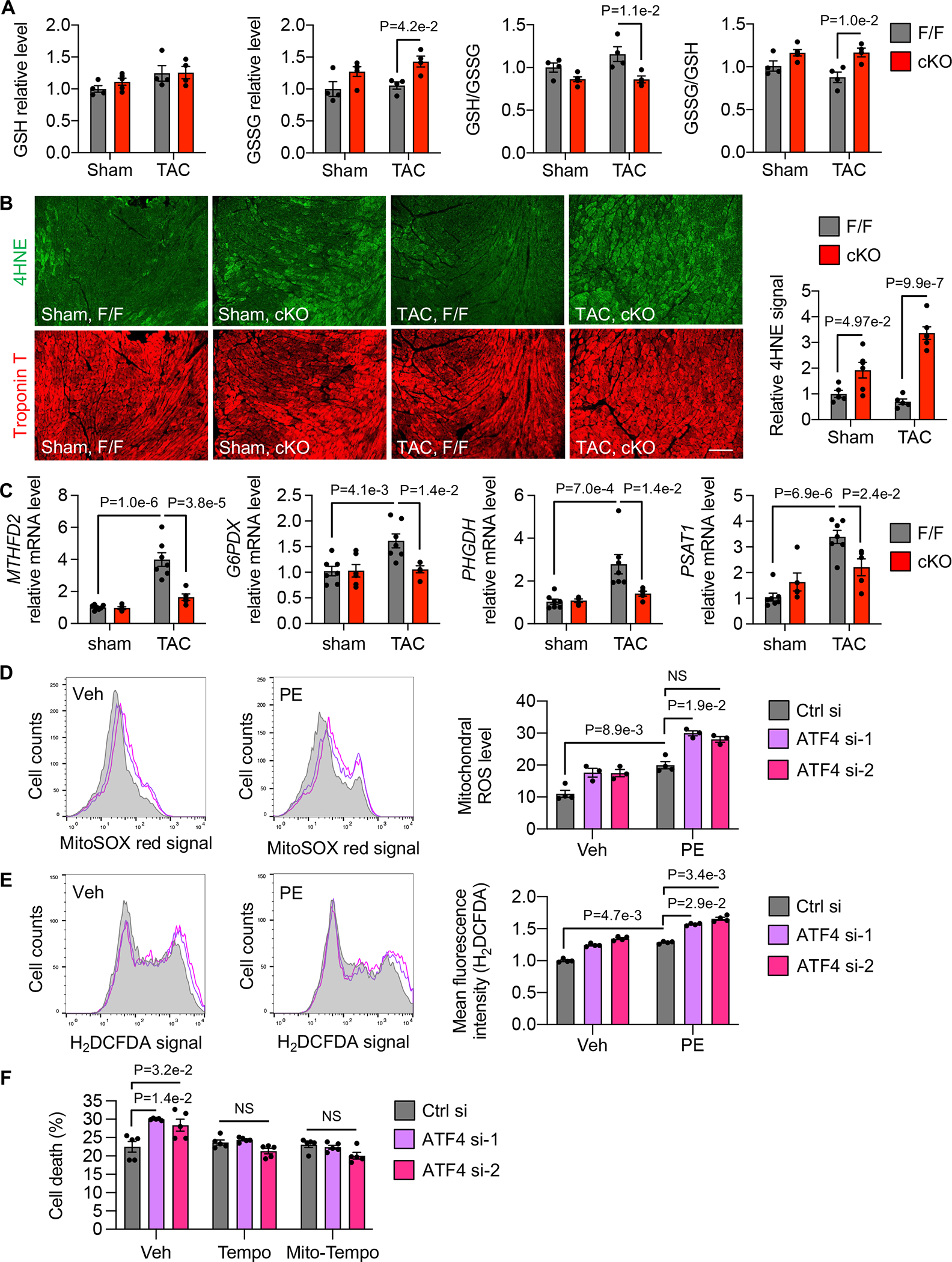
A. Cardiac specific ATF4 deficiency led to a decrease in the ratio of GSH to GSSG and an increase in GSSG level after TAC. ATF4 cKO mice along with F/F controls were subjected to sham or TAC. After 3 days, cardiac tissues were harvested for various measurements. Sham, F/F, n=4; Sham, cKO, n=5; TAC, F/F, n=4; TAC, cKO, n=4.
B. ATF4 cKO hearts showed increased oxidative stress in the heart. Sham, F/F, n=5; Sham, cKO, n=6; TAC, F/F, n=5; TAC, cKO, n=6.
C. ATF4 deletion in the heart caused a failure of induction of MTHFD2, G6PDX, PHGDH, and PSAT1 by TAC. Sham, F/F, n=5; Sham, cKO, n=6; TAC, F/F, n=5; TAC, cKO, n=6.
D. ATF4 silencing in NRVMs increased mitochondrial ROS as measured by flow cytometry. Two independent siRNA oligos against ATF4 were used. NS, not significant. Vehicle (Veh), Ctrl si, n=4; PE, Ctrl si, n=4; all others, n=3.
E. ATF4 knockdown in NRVMs caused an increase in cytosolic ROS as examined by H2DCFDA staining. N=4 for each group.
F. ATF4 silencing exacerbated NRVM death after PE treatment, which was significantly rescued by either Tempo or Mito-Tempo treatment. Note that all groups were treated by PE. N=5 for each group.
Two-way ANOVA was conducted, followed by Tukey’s multiple comparisons test (A-C). GSH and GSSG levels from each group were normalized to the mean of the sham/ ATF4F/F group, which was set to 1 (A). The 4HNE level from each group was normalized to the mean of the sham/ ATF4F/F group, which was set to 1 (B). Gene expression at the mRNA level from each group was normalized to the mean of the sham/ATF4F/F group, which was set to 1 (C). Kruskal-Wallis test was conducted, followed by Dunn’s test to determine statistical significance for comparisons of the means of each group with the mean of the vehicle (Veh)/Control si (Ctrl si) group (D-F). Mean fluorescent intensity (H2DCFDA) of each treatment group was normalized to the mean of the vehicle (Veh)/Control si (Ctrl si) group, which was set to 1 (E). Data are represented as mean±SEM.
At the in vitro level, overexpression of ATF4 in NRVMs increased NADPH production and the ratio of NADPH to NADP+ (Figure S7D). We further showed that knockdown of ATF4 using 2 sequence-independent siRNAs increased both mitochondrial and cytosolic ROS (Figure 6D and 6E). Consistent with the essential role of redox control in cell survival, ATF4 silencing led to an increase in cell death in NRVMs under PE stimulation (Figure 6F), and either Tempo or Mito-Tempo treatment significantly rescued cell death (Figure 6F). Taken together, these findings suggest that ATF4 is essential for maintaining NADPH and GSSH levels to control oxidative stress by transcriptionally regulating multiple enzymes in the one-carbon metabolic pathway and PPP, thereby maintaining redox balance in the heart.
DISCUSSION
Heart failure is a major public health crisis that affects more than 26 million people worldwide.23 Cumulative evidence has firmly established that oxidative stress participates in the initiation and progression of heart failure.7, 24 However, the dynamic regulation of redox control and intrinsic antioxidative mechanisms in the heart under pathophysiological conditions remain poorly defined. Here, we report that cardiac ROS are significantly elevated in pressure overload-induced heart failure in vivo and in PE-induced cardiomyocyte hypertrophic growth in vitro. Importantly, multiple enzymes of the one-carbon metabolic pathway and PPP that generate NADPH, the main antioxidant co-factor, are upregulated in response to pressure overload. Mechanistically, we identify ATF4 as a key upstream transcription factor that governs the expression of these NADPH-generating enzymes in both mitochondria and the cytosol for coordinated redox maintenance. Manipulation of ATF4 in the heart therefore represents a potential route for therapeutic exploration to tackle heart failure triggered by hemodynamic stress.
ROS production and antioxidative mechanisms in heart failure
Cardiac ROS are produced primarily by mitochondria, NADPH oxidase, xanthine oxidase, and uncoupled nitric oxide synthase (NOS).7 Previous studies have shown that exposure to several stimuli, including mechanical stretch, angiotensin II, endothelin-1, and tumor necrosis factor (TNF)-α, leads to an increase in ROS in cardiomyocytes due to elevated expression and activation of NADPH oxidase.25–27 Heart failure is associated with an elevation in serum uric acid levels likely deriving from an increase in xanthine oxidase.28 In addition, abnormal activation of NOS has been implicated in the development of heart failure, and modulation of NOS represents a promising treatment for heart disease.29
Consistent with the increase in oxidative stress, heart failure is also characterized by exhaustion of the inherent antioxidant defense mechanisms, including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), NADPH, and GSH. Here, we show that GSSG is increased and the ratio of GSH to GSSG is decreased in mouse hearts 21 days after TAC. NADPH is required to convert GSSG to GSH. Importantly, NADPH is decreased and the ratio of NADPH to NADP+ is reduced more rapidly after TAC, consistent with an essential role for NADPH in controlling GSH levels and oxidative stress. Therefore, these findings reveal detrimental consequences of an imbalance between ROS production and GSH generation in the initiation and development of heart failure.
Molecular mechanisms controlling NADPH levels in heart failure
Recent studies have shown that cellular NADPH can be generated by the PPP, folate-mediated one-carbon metabolism, and malic enzyme (ME) in tumors and proliferating cells.30, 31 In addition, fatty acid oxidation increases TCA cycle activity to produce citrate, which is exported to the cytosol to engage in NADPH production by ME1 and isocitrate dehydrogenase 1 (IDH1).32 However, current understanding of the mechanisms governing NADPH biosynthesis in heart failure is incomplete. We show here that NADPH levels are controlled by the one-carbon metabolism pathway and PPP in the setting of load-induced heart failure. In addition, expression of PHGDH and PSAT1, the rate-limiting enzymes for coupling the serine/glycine metabolic pathway with one-carbon metabolism, is elevated in response to pressure overload. These two pathways are not only major sources of NADPH in the cytosol and mitochondria, but also supply metabolites to other major metabolic pathways in cells, including nucleotide biosynthesis, amino acid production, methylation reaction, and antioxidative defense. Therefore, NADPH production is coupled with both redox control and macromolecule biosynthesis in cardiomyocytes under stress, highlighting the importance of homeostatic regulation of metabolic pathways in maintaining cellular function and survival.
Metabolic roles of ATF4 in heart failure
Glucose metabolism, including glycolysis and ancillary pathways, plays critical roles in the etiology of heart failure.33 Recent studies show that both pyruvate production34 and import35–38 are essential in maintaining homeostatic heart function under pressure overload. On the other hand, the hexosamine biosynthetic pathway is acutely activated by afterload stress, and chronic stimulation exacerbates pathological cardiac remodeling.39–41 Moreover, serine biosynthesis, a downstream pathway of glucose metabolism, has been implicated in multiple cancers;42 however, its relevance and function in cardiovascular disease remain to be fully illustrated.
ATF4 has been implicated in the survival of cancer cells experiencing a deficit of oxygen or nutrients and in alleviating oncogene-induced stress.43, 44 Previous studies from Manning and colleagues have shown that ATF4 acts downstream of mTORC1 and stimulates MTHFD2 for purine biosynthesis in both wild-type mouse embryonic fibroblasts and cancer cells, using a mechanism that is independent of the canonical signaling from either the unfolded protein response or the integrated stress response.20 More recently, Torrence et al. report that ATF4 promotes glutathione synthesis through SLC7A11-dependent uptake of cystine.45 In addition, Tran et al. show that proline deficiency due to the lack of mitochondrial NADP+ strongly augments ATF4 level in Hela cells,46 indicating a connection between NADP+/NADPH biosynthesis and ATF4 expression. Here, we report that ATF4 activates the one-carbon metabolic pathway and PPP in NRVMs in vitro. At in vivo, ATF4 is cardioprotective, necessary for the improved antioxidative capacity and the maintenance of cardiac function in the setting of pressure overload. Consistently, ATF4 deficiency in the heart leads to reduced expression of multiple enzymes within the one-carbon metabolic pathway, the PPP, and serine synthesis under conditions of hemodynamic stress, suggesting that activation of these pathways governed by ATF4 is necessary for the maintenance of NADPH homeostasis and cardiac function. Molkentin and colleagues recently show that AAV9-mediated overexpression of ATF4 in the heart per se causes cardiomyopathy due to excessive activation of autophagy.47 The discrepancy may stem from different animal models used (cardiac specific knockout vs. virus-mediated overexpression) and dose of manipulation (deletion vs. overexpression). More work is warranted to further dissect the role of ATF4 in cardiovascular disease.
Conclusions and future perspectives
Heart failure is a common outcome of multiple forms of cardiovascular disease. Despite extensive interest and overwhelming clinical relevance, our understanding of mechanisms underlying heart failure remains incomplete. Oxidative stress plays a central role in the etiology of heart failure. Here, we report that heart failure elicited by hemodynamic stress is associated with an increase in oxidative stress in the myocardium. At the cellular level, GSH production and NADPH levels are diminished at the early stage of disease progression. We further demonstrate that ATF4 is a transcription factor acting upstream to directly govern expression of genes in the one-carbon metabolic pathway and PPP. Deficiency of ATF4 exacerbates cardiomyopathy in response to pressure overload. In summary, ATF4 represents a potential target to curb pathological accumulation of ROS and control oxidative stress in the setting of heart disease.
Supplementary Material
Novelty and Significance.
What Is Known?
Redox control is essential for cardiac cell function and survival, disruption of which causes cardiomyopathy and heart failure.
Redox balance is maintained by production of reactive oxygen/nitrogen species and activation of endogenous antioxidant mechanisms.
NADPH (nicotinamide adenine dinucleotide phosphate) and GSH (glutathione) are key molecules in cellular redox control.
What New Information Does This Article Contribute?
Heart failure in the setting of hemodynamic stress is accompanied by a decrease in NADPH and GSH in the heart and an increase in cardiac cell death.
ATF4 (activating transcription factor 4) is a common upstream transcription factor for multiple key enzymes of NADPH biosynthesis in both the mitochondria and the cytosol.
ATF4 governs the metabolic flux of the pentose phosphate pathway and the one-carbon metabolic pathway in cardiomyocytes.
Cardiac specific deficiency of ATF4 precipitates the development of heart failure in response to pressure overload.
Heart failure is a leading cause of death worldwide. Homeostatic redox control is essential in maintaining cardiac function and ensuring cardiomyocyte survival under various disease conditions. Cellular redox balance is governed by production of reactive oxygen/nitrogen species and activation of antioxidant mechanisms. The dynamic regulation of redox control in the heart under pressure overload remains to be fully defined. We show that reduced NADPH and GSH, key metabolites in oxidative stress control, are decreased in hypertrophic hearts. Importantly, multiple essential enzymes of the pentose phosphate pathway (PPP) and one-carbon metabolic pathway, two signaling axes of NADPH biosynthesis, are downregulated in the heart under hemodynamic stress. Further, we demonstrate that activating transcription factor 4 (ATF4) is an upstream transcription factor of these enzymes and overexpression of ATF4 stimulates metabolic flux of the PPP and the one-carbon metabolic pathway. Additionally, joint pathway analysis using transcriptomic and metabolic results confirms a critical role of ATF4 in redox control in cardiomyocytes. Consistently, cardiomyocyte specific deletion of ATF4 exacerbates cardiomyopathy and heart failure in response to pressure overload. In summary, these findings reveal an essential role of ATF4 in maintaining redox balance and ensure cardiomyocyte survival in the heart under hemodynamic stress.
Acknowledgements
We thank the Molecular Pathology Core (John Shelton) of University of Texas Southwestern Medical Center (UTSW) for help with histology. We are grateful to the Animal Resources Center of UTSW for mouse generation, breeding, and care. Graphic abstract and schematic diagram were created with Biorender.com.
Sources of Funding
This work was supported by funds from the American Heart Association (17IRG33460191 to Z.V.W., 20POST35210756 to X.W., and 19TPA34920001 to B.A.R.).
Nonstandard Abbreviations and Acronyms
- αMHC
α-myosin heavy chain
- ATF4
activating transcription factor 4
- ChIP
chromatin immunoprecipitation
- cKO
cardiac specific conditional knockout
- EF
ejection fraction
- FS
fractional shortening
- G6PDX
glucose-6-phosphate dehydrogenase
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- 4-HNE
4-hydroxynonenal
- JPA
joint pathway analysis
- KEGG
Kyoto encyclopedia of genes and genomes
- MTHFD2
methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2
- NADP+
oxidized nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NRVMs
neonatal rat ventricular myocytes
- PCA
principal component analysis
- PE
phenylephrine
- PHGDH
phosphoglycerate dehydrogenase
- PPP
pentose phosphate pathway
- PSAT1
phosphoserine aminotransferase 1
- TAC
transverse aortic constriction
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
Footnotes
Disclosures
None.
Supplemental Materials
REFERENCES
- 1.Ray PD, Huang BW and Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nogueira V and Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res. 2013;19:4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rolo AP, Teodoro JS and Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med. 2012;52:59–69. [DOI] [PubMed] [Google Scholar]
- 4.Hayes JD, Dinkova-Kostova AT and Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38:167–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP, Commodore-Mensah Y, Elkind MSV, Evenson KR, Eze-Nliam C, Ferguson JF, Generoso G, Ho JE, Kalani R, Khan SS, Kissela BM, Knutson KL, Levine DA, Lewis TT, Liu J, Loop MS, Ma J, Mussolino ME, Navaneethan SD, Perak AM, Poudel R, Rezk-Hanna M, Roth GA, Schroeder EB, Shah SH, Thacker EL, VanWagner LB, Virani SS, Voecks JH, Wang NY, Yaffe K and Martin SS. Heart disease and stroke statistics-2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639. [DOI] [PubMed] [Google Scholar]
- 6.Karimi Galougahi K, Antoniades C, Nicholls SJ, Channon KM and Figtree GA. Redox biomarkers in cardiovascular medicine. Eur Heart J. 2015;36:1576–1582, 1582a-b. [DOI] [PubMed] [Google Scholar]
- 7.Tsutsui H, Kinugawa S and Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–h2190. [DOI] [PubMed] [Google Scholar]
- 8.Ying W NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. [DOI] [PubMed] [Google Scholar]
- 9.Xiao W, Wang RS, Handy DE and Loscalzo J. NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid Redox Signal. 2018;28:251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ju HQ, Lin JF, Tian T, Xie D and Xu RH. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patra KC and Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. 2014;39:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitale PM, Gorbatyuk O and Gorbatyuk M. Neurodegeneration: Keeping ATF4 on a tight leash. Front Cell Neurosci. 2017;11:410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wortel IMN, van der Meer LT, Kilberg MS and van Leeuwen FN. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J Jr. and Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–8281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forman HJ, Zhang H and Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ducker GS and Rabinowitz JD. One-carbon metabolism in health and disease. Cell Metab. 2017;25:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puleston DJ, Villa M and Pearce EL. Ancillary activity: beyond core metabolism in immune cells. Cell Metab. 2017;26:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang G, Wang X, Bi X, Li C, Deng Y, Al-Hashimi AA, Luo X, Gillette TG, Austin RC, Wang Y and Wang ZV. GRP78 (glucose-regulated protein of 78 kDa) promotes cardiomyocyte growth through activation of GATA4 (GATA-binding protein 4). Hypertension. 2019;73:390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Deng Y, Zhang G, Li C, Ding G, May HI, Tran DH, Luo X, Jiang DS, Li DL, Wei X, Xu L, Ferdous A, Gillette TG, Scherer PE, Jiang X and Wang ZV. Spliced X-box binding protein 1 stimulates adaptive growth through activation of mTOR. Circulation. 2019;140:566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM and Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pang Z, Chong J, Zhou G, de Lima Morais DA, Chang L, Barrette M, Gauthier C, Jacques P, Li S and Xia J. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021;49:W388–w396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED and Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem. 2012;287:27290–27301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savarese G and Lund LH. Global public health burden of heart failure. Card Fail Rev. 2017;3:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grieve DJ and Shah AM. Oxidative stress in heart failure. More than just damage. Eur Heart J. 2003;24:2161–2163. [DOI] [PubMed] [Google Scholar]
- 25.Doughan AK, Harrison DG and Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. [DOI] [PubMed] [Google Scholar]
- 26.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G and Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol. 2003;41:2164–2171. [DOI] [PubMed] [Google Scholar]
- 27.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marbán E and Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–2411. [DOI] [PubMed] [Google Scholar]
- 28.Packer M Uric acid Is a biomarker of oxidative stress in the failing heart: lessons learned from trials with allopurinol and SGLT2 inhibitors. J Card Fail. 2020;26:977–984. [DOI] [PubMed] [Google Scholar]
- 29.Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B and Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal. 2013;18:1078–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB and Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Shah S, Fan J, Park JO, Wellen KE and Rabinowitz JD. Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat Chem Biol. 2016;12:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Q, Zeng F, Liu X, Wang QJ and Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibb AA and Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. 2018;123:107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Q, Li C, Elnwasany A, Sharma G, An YA, Zhang G, Elhelaly WM, Lin J, Gong Y, Chen G, Wang MH, Zhao S, Dai C, Smart CD, Liu J, Luo X, Deng Y, Tan L, Lv SJ, Davidson SM, Locasale JW, Lorenzi PL, Malloy CR, Gillette TG, Vander Heiden MG, Scherer PE, Szweda LI, Fu G and Wang ZV. PKM1 exerts critical roles in cardiac remodeling under pressure overload in the heart. Circulation. 2021;144:712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCommis KS, Kovacs A, Weinheimer CJ, Shew TM, Koves TR, Ilkayeva OR, Kamm DR, Pyles KD, King MT, Veech RL, DeBosch BJ, Muoio DM, Gross RW and Finck BN. Nutritional modulation of heart failure in mitochondrial pyruvate carrier-deficient mice. Nat Metab. 2020;2:1232–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Taufalele PV, Cochran JD, Robillard-Frayne I, Marx JM, Soto J, Rauckhorst AJ, Tayyari F, Pewa AD, Gray LR, Teesch LM, Puchalska P, Funari TR, McGlauflin R, Zimmerman K, Kutschke WJ, Cassier T, Hitchcock S, Lin K, Kato KM, Stueve JL, Haff L, Weiss RM, Cox JE, Rutter J, Taylor EB, Crawford PA, Lewandowski ED, Des Rosiers C and Abel ED. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat Metab. 2020;2:1248–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cluntun AA, Badolia R, Lettlova S, Parnell KM, Shankar TS, Diakos NA, Olson KA, Taleb I, Tatum SM, Berg JA, Cunningham CN, Van Ry T, Bott AJ, Krokidi AT, Fogarty S, Skedros S, Swiatek WI, Yu X, Luo B, Merx S, Navankasattusas S, Cox JE, Ducker GS, Holland WL, McKellar SH, Rutter J and Drakos SG. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. 2021;33:629–648.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandez-Caggiano M, Kamynina A, Francois AA, Prysyazhna O, Eykyn TR, Krasemann S, Crespo-Leiro MG, Vieites MG, Bianchi K, Morales V, Domenech N and Eaton P. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat Metab. 2020;2:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tran DH, May HI, Li Q, Luo X, Huang J, Zhang G, Niewold E, Wang X, Gillette TG, Deng Y and Wang ZV. Chronic activation of hexosamine biosynthesis in the heart triggers pathological cardiac remodeling. Nat Commun. 2020;11:1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, Gauthier C, Olson AK, Bouchard B, Des Rosiers C, Viollet B, Sakamoto K, Balligand JL, Vanoverschelde JL, Beauloye C, Horman S and Bertrand L. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun. 2018;9:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umapathi P, Mesubi OO, Banerjee PS, Abrol N, Wang Q, Luczak ED, Wu Y, Granger JM, Wei AC, Reyes Gaido OE, Florea L, Talbot CC Jr., Hart GW, Zachara NE and Anderson ME. Excessive O-GlcNAcylation causes heart failure and sudden death. Circulation. 2021;143:1687–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geeraerts SL, Heylen E, De Keersmaecker K and Kampen KR. The ins and outs of serine and glycine metabolism in cancer. Nat Metab. 2021;3:131–141. [DOI] [PubMed] [Google Scholar]
- 43.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, Li Y, Gao Y, Liu H, Li C, Maity A, Thomas-Tikhonenko A, Perl AE, Koong A, Fuchs SY, Diehl JA, Mills IG, Ruggero D and Koumenis C. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, Salas Salinas C, Chinga F, Monroy AM, Fu W, Wang P, Kossenkov A, Ye J, Amaravadi RK, Ignatova Z, Fuchs SY, Diehl JA, Ruggero D and Koumenis C. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol. 2019;21:889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torrence ME, MacArthur MR, Hosios AM, Valvezan AJ, Asara JM, Mitchell JR and Manning BD. The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. Elife. 2021;10:e63326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tran DH, Kesavan R, Rion H, Soflaee MH, Solmonson A, Bezwada D, Vu HS, Cai F, Phillips JA 3rd, DeBerardinis RJ and Hoxhaj G. Mitochondrial NADP(+) is essential for proline biosynthesis during cell growth. Nat Metab. 2021;3:571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ, Accornero F, Sargent MA and Molkentin JD. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nature Commun. 2021;12:3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei CC, Lucchesi PA, Tallaj J, Bradley WE, Powell PC and Dell’Italia LJ. Cardiac interstitial bradykinin and mast cells modulate pattern of LV remodeling in volume overload in rats. Am J Physiol Heart Circ Physiol. 2003;285:H784–h792. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Zhang Y, Ding G, May HI, Xu J, Gillette TG, Wang H and Wang ZV. Temporal dynamics of cardiac hypertrophic growth in response to pressure overload. Am J Physiol Heart Circ Physiol. 2017;313:H1119–h1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du D, Tan L, Wang Y, Peng B, Weinstein JN, Wondisford FE, Su X and Lorenzi PL. ElemCor: accurate data analysis and enrichment calculation for high-resolution LC-MS stable isotope labeling experiments. BMC Bioinformatics. 2019;20:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed experimental procedures are described in the Supplemental Materials. Data, methods, and study materials are available from the corresponding author upon reasonable request. Key research materials are listed in the Major Resources Table in the Supplemental Materials.