

Abstract
Purpose:
To identify molecular predictors of grade 3/4 neutropenic or leukopenic events (NLEs) after chemotherapy using a genome-wide association study (GWAS).
Patients and Methods:
A GWAS was performed on patients in the phase III chemotherapy study SUCCESS-A (n = 3322). Genotyping was done using the Illumina HumanOmniExpress-12v1 array. Findings were functionally validated with cell culture models and the genotypes and gene expression of possible causative genes were correlated with clinical treatment response and prognostic outcomes.
Results:
One locus on chromosome 16 (rs4784750; NLRC5; P = 1.56E-8) and another locus on chromosome 13 (rs16972207; TNFSF13B; P = 3.42E-8) were identified at a genome-wide significance level. Functional validation revealed that expression of these two genes is altered by genotype-dependent and chemotherapy-dependent activity of two transcription factors. Genotypes also showed an association with disease-free survival in patients with an NLE.
Conclusions:
Two loci in NLRC5 and TNFSF13B are associated with NLEs. The involvement of the major histocompatibility complex I regulator NLRC5 implies the possible involvement of immuno-oncological pathways.
Keywords: chemotherapy, leukopenia, GWAS, NLRC5, TNFSF13B
INTRODUCTION
Chemotherapy remains one of the main options in the treatment of many cancers. Due to its adverse effects and limited efficacy in some cancers, however, its use should be limited to patients who have an excellent risk–benefit ratio. Myelotoxicity is the most relevant side effect, resulting in anemia, thrombopenia, and leukopenia. Severe neutropenic or leukopenic events (NLEs) may be complicated by life-threatening infections (febrile neutropenia, FN), requiring hospitalization and antibiotic therapy (1).
Dose reductions and treatment delays in patients with NLEs were considered as a possible reason for a worse prognosis (2). More recently, effects of chemotherapy on the immune system that consequently affect cancer therapy have been explored (3,4). In breast cancer (BC), for example, the importance of immunoregulatory genes for prognosis and treatment efficacy has been shown in several studies, and a PD-L1 antibody has been approved for the treatment of advanced BC (5).
Apart from clinical predictors for FN (6), very few molecular markers have been reported to be associated with either FN or NLEs. Our group previously published a report based on a genome-wide association study in lymphoblastoid cell lines that identified genetic variants in PIGB (phosphatidylinositol glycan anchor biosynthesis, class B) as a predictor for NLEs (7). Other, mostly retrospective, studies have described genetic risk factors for chemotherapy-induced leukopenia, neutropenia, or FN in BC patients (8–10). The largest fluorouracil, epirubicin, and cyclophosphamide (FEC) chemotherapy study, including around 1000 BC patients, concluded that adding single nucleotide polymorphisms (SNPs) to clinical predictors of FN might improve prediction of the events (8,9). A smaller report from a genome-wide association study (GWAS) in 270 Asian patients with various solid tumor histologies (11) found that SNPs in MCPH1 were predictive for chemotherapy-induced neutropenia or leukopenia.
In the present study, we conducted a GWAS embedded in a large prospective and randomized chemotherapy study in patients with early BC, investigating associations with the occurrence of grade 3/4 NLEs. Two genome-wide significant (P < 5E-8) SNP signals were identified. One, rs4784750, mapped to the NLR family CARD domain containing 5 gene (NLRC5) and the other, rs16972207, to tumor necrosis factor superfamily member 13 beta (TNFSF13B, also known as BAFF, B-cell activating factor). The role of these genetic variants was also investigated in relation to prognosis and drug efficacy.
METHODS
Patients and Treatment
The multicenter SUCCESS-A study (12,13) included a prospective subprotocol concerned with the influence of germline genetic variants on side effects and efficacy of the chemotherapy. Patients were eligible if they had a histologically confirmed invasive BC with an increased risk for recurrence. Inclusion and exclusion criteria and patient characteristics are provided in Supplementary Tables S4A and S4B. The SUCCESS-A study was conducted in 251 study centers in all regions of Germany. The main study and all prespecified translational research projects, including the one reported here, were approved by all the ethics committees responsible and conducted in accordance with the Declaration of Helsinki. All patients gave written informed consent.
Patients in the SUCCESS-A study were treated with three cycles of fluorouracil–epirubicin–cyclophosphamide (500/100/500 mg/m2, FEC) followed by three cycles of docetaxel (100 mg/mg2) every 3 weeks (q3w) versus three cycles of FEC followed by three cycles of gemcitabine (1000 mg/m2 d1,8)–docetaxel (75 mg/m2) q3w. HER2 positive patients were additionally treated with a 12-month treatment of adjuvant trastuzumab. After completing chemotherapy, the patients were further randomized to receive either 2 or 5 years of zoledronic acid. Premenopausal hormone receptor–positive women received tamoxifen alone or in combination with goserelin for 2 years if they were under 40 years of age. Postmenopausal patients were treated with tamoxifen for 2 years, followed by anastrozole for 3 years.
Primary surgery consisted of either breast conservation or mastectomy, leading to R0 resection in all cases. Sentinel-node dissection (SND) was performed in all cN0 patients (with SND as the only axillary intervention), followed by complete axillary node dissection in patients with positive sentinel nodes. The cN1 patients primarily received axillary node dissection. Radiotherapy was performed in accordance with national guidelines.
Clinicopathologic Information and Follow-Up
During the treatment phase blood cell counts were required at least twice per week. Hematological toxicity was documented according to NCI-CTCAE Version 3.0 at the end of every three weekly therapy cycle. The patients were followed at the study sites at 3-month intervals for the first 3 years and every 6 months thereafter. Follow-up included clinical examinations (each visit), mammography (every 6 months), and symptom-driven examinations if necessary. Disease-free survival was defined as the time from randomization to censoring without event or to a local recurrence, a distant recurrence or death of any cause, whichever occurred first. All data were obtained from the SUCCESS-A study electronic case record forms. The quality of the data was ensured through electronic data management, including automated plausibility checks and regular monitoring visits to the study site by an independent clinical research organization (Alcedis GmbH, Giessen, Germany) and a data monitoring committee (DMC).
Biomaterial Sampling and Patient Selection
A total of 3754 patients were randomized between September 2005 and March 2007. Whole blood samples were retrieved from 3584 patients (initial biomarker cohort) at the time of randomization. An initial quality check with 2% agarose gel electrophoresis of all samples showed that 1751 of the samples were not good enough for genotyping. The patients were therefore recalled, and blood was again drawn from 1102 patients. A total of 493 samples with DNA quality assessed as good enough were restored using the Illumina FFPE restoration kit, resulting in 3428 patients for genotyping, which was successful in 3328 individual patients, of which 2 had withdrawn consent and 4 were unexpected duplicates (all removed). Five patients were removed because they were related, nine patients of non-European heritage were also removed. DNA samples from 3308 patients (final biomarker cohort) were therefore used for GWAS analyses. Patient data for the final biomarker cohort and all randomized patients did not differ from each other (Supplementary Tables S4B and S5).
SNP Genotyping, Quality Control, and Imputation
The Illumina HumanOmniExpress-12v1 G FFPE array and the Infinium HD assay, in accordance with the manufacturer’s recommendations, were considered as the best option for the restored DNA samples. Therefore, all samples were genotyped for 693,543 SNPs using the HumanOmniExpress-12v1 G FFPE array (genome build 37) regardless of whether they were restored or not.
For calling, the algorithm GenomeStudio, RRID:SCR_010973, version 2011.1, Genotyping Module 1.9.4, and GenTrain version 1.0 were used. Hardy Weinberg equilibrium was tested using two sets of unrelated subjects. Autosomal SNPs deviated from expectation at about 0.01 and the X-chromosome SNPs showed deviations between 0.01 and 0.001. SNPs were excluded using a filter threshold of 0.0001. Quality assurance and quality control were performed in accordance with Laurie et al. (14). As a consequence of this QC process the following number of SNPs were excluded in hierarchical order: 9,400 SNP assays failed; 17,134 SNPs had a MAF of zero; 26,652 SNPs had a missing call rate of >2%; for 160 SNPs, mendelian errors were observed in more than one HapMap trio/duo; 1,330 SNPs were excluded with HWE p-values < 1E-4, and 46 SNPs had more than one discordant call in 46 pairs of duplicated stidy samples, resulting in 638,837 SNPs remaining after the QC SNP filters. Finally, SNPs with a MAF < 0.01 were excluded resulting in genotyped 604,785 SNPs.
Median missing call rates per sample were 0.12% for genotypes from the original samples, 0.09% for new blood draws and 0.42% for the restored samples. No sample had a missing call rate >5%, and no sample was excluded because of post genotyping release QC failure. There were no statistically significant different call rates comparing patients with and without a NLE.
Variants were imputed from the 1000 Genomes Project, RRID:SCR_008801, data using the v3 April 2012 release35 as the reference panel. Imputation was based on the 1000 Genomes Project data with singletons removed. Genotype data (≈12.66 million SNPs) were imputed in a two-step procedure, with prephasing using SHAPEIT software and imputation of the phased data in the second step with IMPUTEv2. SNPs with MAF<0.01 and SNPs with the IMPUTEv2 “info” metric <0.3 were excluded, resulting in ≈8.86 million SNPs for further analysis. The “info” metric is highly correlated with the squared correlation r2 from BEAGLE, RRID:SCR_001789, and MARCH, and for convenience will be denoted r2 here too.
Statistical Analysis
The primary end point was grade 3/4 NLEs in the first three cycles of chemotherapy (yes vs. no), during which all patients were treated uniformly with 5-FU, epirubicin, and cyclophosphamide. Multiple logistic regression models were fitted for each SNP (ordinal; count of minor alleles) with age (continuous), body surface area (BSA, continuous), DNA type (restored vs. not restored), estrogen-receptor status, and HER2 status as additional predictors. Covariables were selected to account for general population differences (age, BSA), treatment differences (trastuzumab for HER2 positive patients), and possible differences concerning the influence of inherited genotypes on the molecular biology of breast cancer (e.g, estrogen receptor status). Adjusted odds ratios (ORs) per minor alleles and P values from likelihood ratio tests for each SNP were obtained from these logistic regression models.
A principal component analysis (PCA) was done using the R package SNPRelate. In order to avoid a strong influence of SNP clusters on the PCA, we used a LD-based pruned SNP set (ld.threshold = 0.2). The variance proportion was below 0.01% for each of the first ten principal components (PCs) indicating that the PCs have hardly any influence on the data variation. Therefore, PCs were not used as predictors for the logistic regression analyses. As a sensitivity analysis, however, the first and second PC were added to the regression models for the top SNPs and the ORs were recalculated.
The GWAS SNPs with a P value below the commonly accepted threshold of 5E-8 were regarded as having genome-wide significance (15). Only individuals with complete observations were considered (3276 of 3308 patients). Statistical analyses were conducted using the R statistical computing package. The Q–Q plot is shown in Supplementary Fig. S3.
As an exploratory study aim, the influence of the top SNP and NLE on disease-free survival was analyzed using the Kaplan-Meier method.
Cell Culture
The HL-60 and Jurkat cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The “Human Variation Panel” of lymphoblastoid cell lines (LCLs) was obtained from the Coriell Institute (Camden, NJ). DNA from these 287 LCLs had been genotyped in the Coriell Institute using the Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, CA), and in our laboratory using Illumina HumanHap 550K and HumanExon 510S-Duo BeadChips (Illumina, San Diego, CA). Imputation was then performed using 1000 Genomes data (http://www.1000genomes.org/data). We also generated gene expression data for these LCLs with Affymetrix U133/2.0/Plus GeneChip expression arrays, as described previously (16). Jurkat cells were cultured in Roswell Park Memorial Institute (RPMI) media with 10% fetal bovine serum (FBS), and the HL-60 and LCL cells were cultured in the same media with 15% FBS.
NLRC5 and TNFSF13B Knockdown and qRT-PCR
Green fluorescent protein (GFP)-labeled vectors that contained short hairpin RNA (shRNA) and small interfering RNA (siRNA) for NLRC5 as well as scrambled controls were obtained from OriGene Technologies, Inc. (Rockville, MD). For transfection of HL-60, Jurkat, and LCL cell lines, the Lonza (Basel, Switzerland) Anexa Nucleofector II Electroporation System was utilized. The knockdown efficiency was determined by quantitative reverse transcriptase polymerase chain reaction (qRT-PCR). mRNA was isolated with the DNA-free RNA kit (ZYMO Research Inc., Irvine, CA) and 100 ng/well total RNA was added for qRT-PCR assay using the Power SYBR® Green RNA-to-CT™ 1-Step Kit (Life Technologies, Carlsbad, CA) and predesigned PrimeTime primers obtained from Integrated DNA Technologies (Coralville, IA).
Cytotoxicity, Proliferation and Apoptosis assays
5-Fluorouracil and epirubicin were purchased from Sigma-Aldrich (St. Louis, MO). Mafosfamide (MFF) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). MFF can spontaneously decompose to 4-hydroxycyclophosphamide, the active metabolite of cyclophosphamide, when added in culture media. To assay drug cytotoxicity, varying concentrations were added at 2–10-fold dilutions based on the half-maximal effective concentration (EC50) values for each cell line. Cell viability was determined by MTS proliferation assay performed after drug treatment with the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) Reagent (Promega, Madison, WI), followed by analysis with an Infinite® M1000 PRO microplate reader (Tecan Systems Inc., San Jose, CA). Detailed methods for the cytotoxicity assay can be found in our previously publications (16–18). Briefly, cells were seeded in 96 well plates (Corning, Corning, NY) at a density of 5 × 105 cells/mL (100 μL/well). 10 μL of 5-FU (500–0.01 μM), epirubicin (10–0.0005 μM), or mafosfamide (100–0.005 μM) were added into the wells and incubated at 37°C for 72 hrs. 20 μL of MTS buffer was then added and plates were read in an Infinite M1000 PRO plate reader (Tecan AG, Switzerland) after incubation for 3 hrs. Relative cell viability was then plotted against drug concentration to derive cytotoxicity curves and EC50 values using GraphPad Prism (RRID:SCR_001789. GraphPad Software, La Jolla, CA). The drug concentrations (10 μM of 5-FU, 0.5 μM of EPI, and 5 μM of MFF) that were used to treat LCLs for TNFSF13B mRNA quantification were chosed based on their EC50 values determined by the cytotoxicity assay. For apoptosis assays, APC annexin V was purchased from BD Bioscience and propidium iodide (PI) from ThermoFisher Scientific. Samples were run on a BD FACSCanto™ flow cytometry system (San Jose, CA).
Western Blot Analyses
For Western blot experiments, cells were centrifuged at 200×g, washed with phosphate-buffered saline (PBS), and lysed with a hypotonic buffer that consisted of 10 mM Tris-HCl, pH 7.5, 10 mM NaCl, 2 mM ethylenediamine tetraacetic acid (EDTA), and 0.5% Triton X-100, to which “complete Mini EDTA free” tablets (Roche Applied Science, Indianapolis, IN) had been added. The buffer was maintained at 4°C and was added to the cells to initiate lysis. The mixture was incubated on ice for 15 minutes, and the lysed cell suspension was then centrifuged at 4°C at 12,000×g for 5 minutes. Protein concentrations in the supernatant were measured using the “Protein Assay Dye” Reagent (Bio-Rad, Hercules, CA) with bovine serum albumin (Sigma-Aldrich, St. Louis, MO) as a standard. Samples of the supernatant were then denatured with 4× Lamelli buffer (Bio-Rad), heated for 3 minutes, and cooled to 4°C before loading onto Mini-PROTEAN gels (Bio-Rad). Gels were transferred using the Turboblot system (Biorad), and blots were incubated with appropriate antibodies. NLRC5 (Anti-NOD4, produced in a rabbit) was purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal vinculin antibody was purchased from Sigma-Aldrich. Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA). Chemiluminescence was determined using Pierce SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher Scientific, Rockford, IL) and was assayed using a Geldoc XR+ system (Biorad, Hercules, CA).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed using the “Epitect ChIP OneDay Kit” (Qiagen) procedure, with the following modifications: as LCLs are nonadherent, they were centrifuged at 200×g and washed with PBS. Fresh 1% formaldehyde was added to cross-link proteins to DNA, and cell lysis was performed. Chromatin shearing by sonication was performed using a Misonix XL sonication system (Qsonica L.L.C., Newtown, CT). Protein/DNA immunoprecipitation, DNA isolation and purification, ChIP DNA detection, and data analysis were then performed. All antibodies used were ChIP-grade and were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Primers used for ChIP were purchased from IDT and had the following sequence: 5′-CAGGGCCTCATCTCCCA-3′ was the forward primer and 5′-TCCGAGCTCCTTCAGAAA-3′ was the reverse primer for the NLRC5 rs4784751 SNP site to which SRF was bound; 5′-GGGTGAGGAAGGG-AAAGAAAT-3′ was the forward primer and 5′-CCTACCCATGTCTGCAATGT-3′ was the reverse primer for the TNFSF13B rs16972207 SNP site to which PXR was bound.
Patients for Testing the Effect of NLRC5 Leukocyte Expression on the Efficacy of Neoadjuvant Chemotherapy
In order to explore possible roles of NLRC5 in the context of breast cancer treatment, we included an additional neoadjuvant study in which the therapeutic response of the tumors to chemotherapy could be assessed in relation to leukocyte NLRC5 expression. To test the effect of NLRC5 leukocyte expression on the rate of pCR after neoadjuvant chemotherapy, a patient cohort was selected from the iMODE-B/TilGen Study (19). The first consecutive triple-negative BC (TNBC) patients treated with carboplatin and paclitaxel were selected, as well as healthy control individuals. Patient and tumor characteristics, including therapy and surgery results, were documented prospectively (Supplementary Table S6). A pCR was defined as complete disappearance of all tumor cells (pT0/pN0). The ethics committee of the medical faculty of Friedrich-Alexander-University, Erlangen, approved the study and all patients provided written informed consent.
RT-PCR of NLRC5 from Leukocyte RNA for Predicting Pathological Complete Response
Full blood samples were collected in PaxGene® tubes from control individuals (n=21) and TNBC patients (n=21). All patient samples were collected before primary diagnosis. RNA was isolated according to the Maxwell® RSC miRNA tissue kit (Promega, Mannheim, Germany) with minor modifications. After centrifugation, the cell pellet was homogenized with 1-thioglycerol. Samples were denatured for 5 minutes at 80°C and treated with proteinase K for 10 minutes at 56°C. Lysates were centrifuged with QIAshredder® tubes (QIAGEN, Hilden, Germany) at full speed for 3 minutes. The flow-through of the sample lysates was taken for the Maxwell® extraction. After the automated run, samples were centrifuged at full speed for 5 minutes and supernatants were taken and incubated for 5 minutes at 65°C. RNA concentrations and purity were determined with the QuantiFluor® RNA Sample Kit (Promega, Mannheim, Germany) and PicoDrop® (Biozym, Hessisch Oldendorf, Germany).
cDNA synthesis (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems, Darmstadt, Germany) was performed in a thermal cycler (ABI2720, Applied Biosystems, Darmstadt, Germany) for 2 h at 37 °C. Gene expression of NLRC5 (TF 5′-AGCAGTGCAAGAAGCAGCAGC-3′; BR 5′-GCTGATGCCGCGGGCAGTG-3′) was measured with SYBR Green®-based technology (Applied Biosystems, Darmstadt, Germany). The internal standards OAZI, Calm2, and RPL37 had also been determined in order to achieve semiquantitative results for gene expression. For data evaluation, the CT values were transformed into ratios using the 2-ΔΔ-CT method.
The nonparametric Mann–Whitney U test for independent samples was performed. A P value below 0.05 was considered as statistically significant.
Data Availability
Data of this GWAS is available under the dbGaP (Study Accession: phs000547.v1.p1).
RESULTS
GWAS for NLEs in Breast Cancer
A total of 1679 patients (51.3%) had a grade 3 or 4 NLE at any time during FEC chemotherapy. Two loci were associated with grade 3/4 NLEs at a genome-wide significance level (P < 5E-8) in women with BC after chemotherapy. One (rs4784750) mapped to the NLRC5 gene on chromosome 16q12.2, and the other (rs16972207) to the TNFSF13B gene on chromosome 13q33.3 (Fig. 1A). The 10 SNPs with the lowest P values are shown in Table 1.
Figure 1.
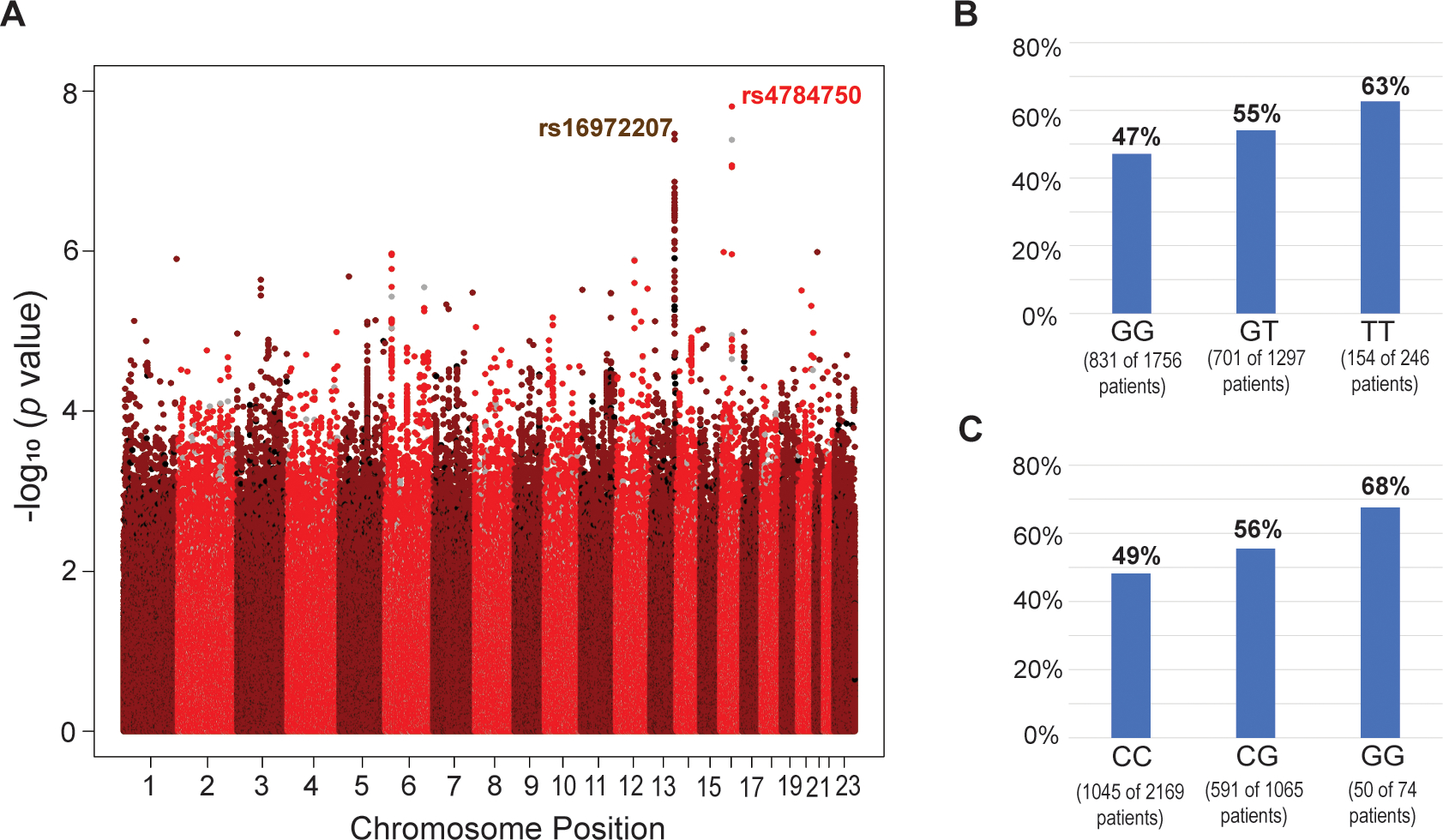
(A) Manhattan plot for the association between neutropenic or leukopenic events (NLEs) and the genotypes from genome-wide genotyping for SNPs with a minor allele frequency > 0.01 (imputed: red/dark red; genotyped: black/grey). (B and C) Distribution of genotypes and neutropenic and leukopenic events (NLEs) among all genotyped patients for the top SNPs in the 16q13 (rs4784751; P = 1.56E-8) locus (B) and 13q33.3 (rs16972207; P = 3.42E-8) locus (C).
Table 1.
Genetic variants with the lowest P values
| SNP | Chr | Position | MAF | Gene | Common allele | Minor allele | Type | Odds ratio (95% CI) | P value |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| rs4784750 | 16 | 57056064 | 0.273 | NLRC5 | G | T | I | 1.38 (1.23–1.54) | 1.56E-8 |
| rs16972207 | 13 | 108929066 | 0.185 | TNFSF13B | C | G | I | 1.54 (1.32–1.79) | 3.42E-8 |
| rs17564816 | 13 | 108927503 | 0.185 | TNFSF13B | G | A | I | 1.52 (1.31–1.77) | 4.01E-8 |
| rs4784751 | 16 | 57056574 | 0.270 | NLRC5 | C | T | O | 1.36 (1.22–1.52) | 4.07E-8 |
| rs12444396 | 16 | 57057194 | 0.264 | NLRC5 | G | A | I | 1.36 (1.21–1.52) | 8.43E-8 |
| rs12445252 | 16 | 57057679 | 0.264 | NLRC5 | C | T | I | 1.36 (1.21–1.52) | 8.90E-8 |
| rs61972007 | 13 | 108918701 | 0.191 | TNFSF13B | A | G | I | 1.42 (1.25–1.62) | 1.37E-7 |
| rs61971976 | 13 | 108889127 | 0.216 | TNFSF13B | G | A | I | 1.39 (1.23–1.58) | 1.60E-7 |
| rs61971980 | 13 | 108899416 | 0.186 | TNFSF13B | G | A | I | 1.40 (1.23–1.60) | 1.87E-7 |
| rs3900097 | 13 | 108905819 | 0.186 | TNFSF13B | C | T | I | 1.40 (1.23–1.60) | 1.88E-7 |
Abbreviations: Chr, chromosome; I, imputed single nucleotide polymorphism; MAF, minor allele frequency; O, originally genotyped single nucleotide polymorphism.
13q33.3 Locus
On chromosome 13, two imputed SNPs (rs16972207 and rs17564816) showed a genome-wide significant association with grade 3/4 NLEs (Figs. 2A and 2B). Both SNPs are located in intron 2 of TNFSF13B (Fig. 2B, red arrows). A SNP cluster in high linkage disequilibrium (LD) with the TNFSF13B rs16972207 SNP also showed low P values (P<1E-6). These SNPs mapped across this locus, which includes two other genes, LIG4 and ABHD13 (Fig. 2B). None of these SNPs is a nonsynonymous or nonsense SNP for either of these two genes. Frequencies of grade 3/4 NLEs according to rs16972207 are shown in Fig 1C.
Figure 2.
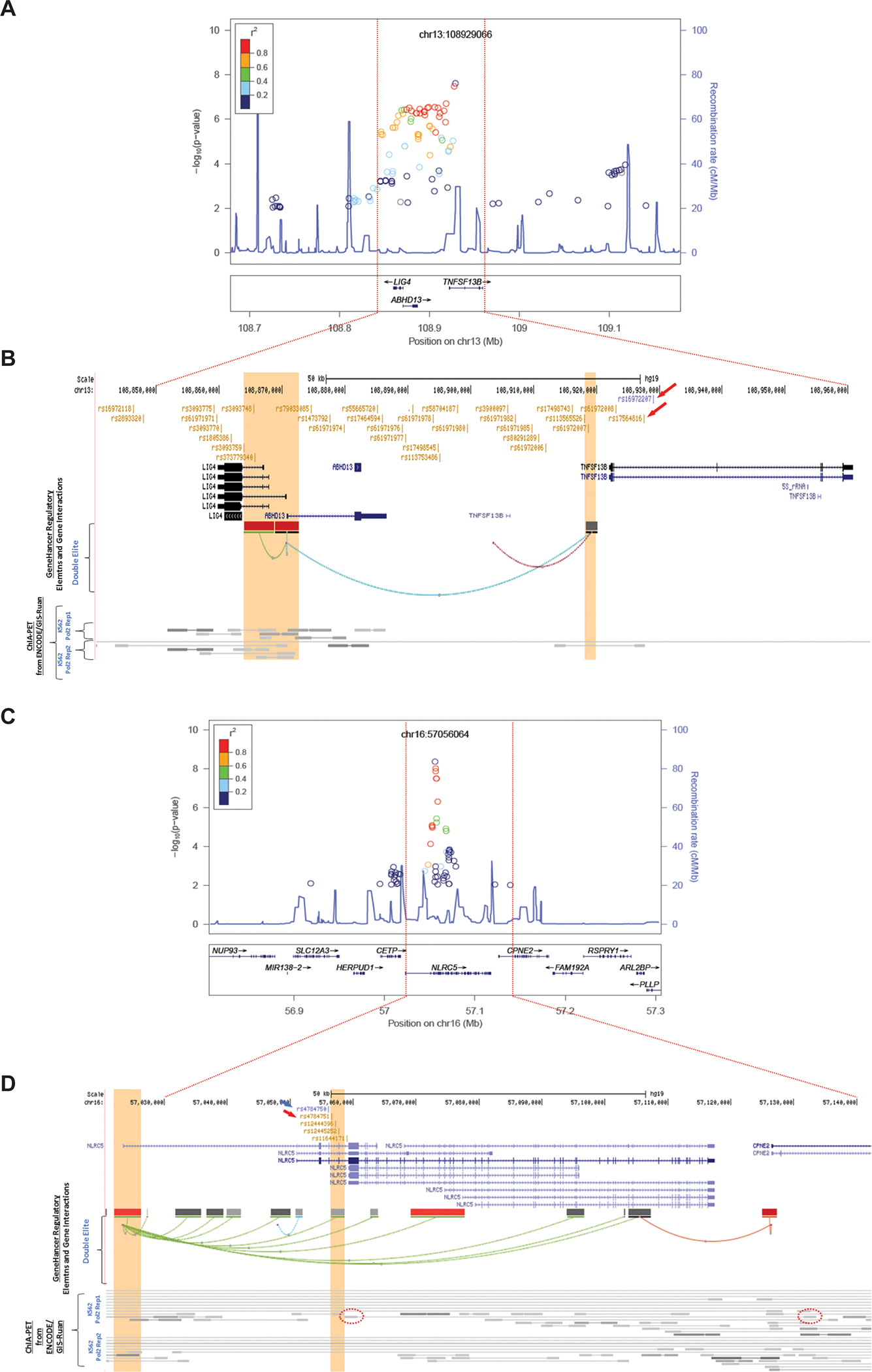
(A and B) Architecture of the 13q33.3 NLE susceptibility locus; (C and D) Architecture of the 16q13 NLE susceptibility locus.
Functional Analysis of the 13q33.3 Locus
To identify a set of credibly causal SNPs operating at the locus, we retrieved all SNPs in high (r2≥0.8) linkage disequilibrium (LD) with rs16972207 using LDlink (https://ldlink.nci.nih.gov/) in CEU (Utah residents from north and west Europe), generating a list of 32 additional SNPs located in genic (TNFSF13B, ABHD13, and LIG4) and intergenic regions (Supplementary Table 1). Interestingly, 17 SNPs in the set showed significant expression-quantitative trait locus (eQTL) associations with LIG4, including the two SNPs with the lowest (3a) RegulomeDB score (rs61972007 and rs61971985) (Supplementary Table 1). In addition, several SNPs showed interactions between enhancers and promoters, as identified by GeneHancer (which links enhancers to genes using tissue co-expression correlation between genes and enhancer RNAs, as well as enhancer-targeted transcription factor genes; expression quantitative trait loci for variants within enhancers; and capture Hi-C) and ChIA-PET (Chromatin Interaction Analysis by Paired-End Tag Sequencing) — suggesting that TNFSF13B, LIG4, and ABHD13 constitute plausible target genes at the locus (Supplementary Table 1; Fig. 2B).
It has been reported that patients with late-onset neutropenia after rituximab therapy have a very high level of BAFF in serum (20,21). We therefore set out to determine whether expression of the TNFSF13B gene changes after therapy with the drugs used to treat patients enrolled in the SUCCESS trial (13). To determine whether the TNFSF13B SNPs affect gene expression after drug treatment, LCLs that were homozygous reference (n=4) and homozygous variant (n=4) for the TNFSF13B “top” SNP, rs16972207 (C>G), were used. These LCLs are B-lymphocytes in origin and they highly express TNFSF13B. After 48 hours of epirubicin alone and epirubicin plus 5-fluorouracil (5-FU) treatment, the TNFSF13B mRNA level was increased in LCLs that were homozygous variant (G/G), in comparison with the reference genotype (C/C) for rs16972207 (Fig. 3A). TNFSF13B mRNA levels were more highly induced after treatment with the 5-FU/EPI combination than after EPI alone, indicating a synergistic effect on TNFSF13B induction by 5-FU/EPI combined treatment (Fig. 3A). No differences in TNFSF13B mRNA levels were observed after treatments with 5-FU or mafosfamide (MFF) alone or in combination (data not shown). Since the TNFSF13B protein is cleaved and is present in the extracellular milieu as a cytokine, we sought to determine whether levels of BAFF in cell media differ in an SNP-dependent manner by performing an enzyme-linked immunosorbent assay (ELISA). The BAFF levels did not differ in an allele-specific fashion at baseline after 48 hours when charcoal-stripped conditioned media incubated with LCLs was analyzed. However, after drug exposure for 24 and 48 hours, there was an allele-specific difference in the level of BAFF released by LCLs with the homozygous variant genotype (G/G) in comparison with LCLs with the reference genotype (C/C) for rs16972207 (Fig. 3B). To determine the possible mechanism for this difference, we analyzed the DNA sequences of TNFSF13B SNPs in the TRANSFAC database. These in silico analyses indicated that the rs16972207 SNP was located in a binding site for the pregnane X receptor (PXR), a ligand-activated transcription factor that can be activated by xenobiotics, including chemotherapy drugs (22) (Fig. 3C). To test the possibility that the SNP altered PXR binding to the nearby DNA sequence, a ChIP assay using anti-PXR antibody was performed in LCLs with homozygous variant or WT SNP genotypes after combined epirubicin and 5-fluorouracil treatment. The variant SNP sequence showed a 4.5-fold increase in the PXR bound (Fig. 3D), which might explain the higher TNFSF13B transcription in variant LCLs after combined epirubicin and 5-fluorouracil treatment.
Figure 3. Functional studies for the TNFSF13B signal.
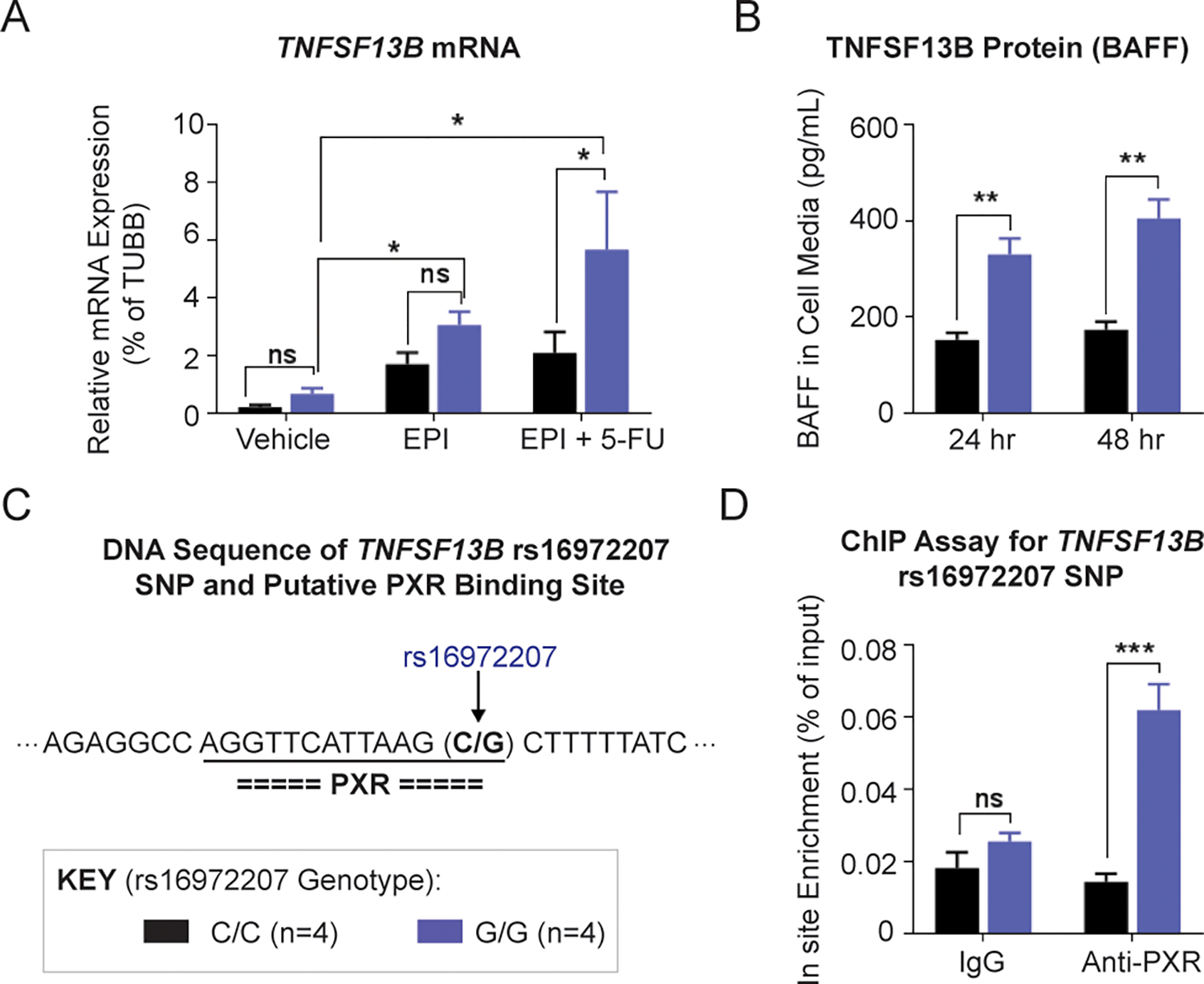
(A) TNFSF13B mRNA levels in lymphoblastoid cell lines (LCLs) with WT (C/C) and homozygous variant (G/G) rs16972207 SNP genotype after 48 h of EPI and EPI plus 5-FU treatment. Concentrations of 5-FU and EPI that were used were 10 μM and 0.5 μM, respectively, concentrations which are approximately equal to their EC50 values. (B) Secreted TNFSF13B protein (BAFF) in LCL cell media after EPI plus 5-FU treatment. (C) DNA sequence near the TNFSF13B rs16972207 SNP and putative transcriptional factor binding sites predicted by the TRANSFAC. The rs16972207 SNP was predicted, which maps to a site that binds pregnane X receptor (PXR). (D) ChIP assay with anti-PXR antibody for the rs16972207 SNP site in LCLs with WT (C/C) and homozygous variant (G/G) genotypes. * P < 0.05, ** P < 0.01, *** P < 0.001, ns = not significant.
16q13 Locus
At chromosome 16, the most strongly associated SNP (rs4784750, P=1.56E-8) was imputed (imputation r2=0.99), while the second most strongly associated one was an originally genotyped SNP (rs4784751, P=4.07E-8) (Fig. 2C). These SNPs, including the two additional SNPs at 16q12.2 (rs12444396 and rs12445252), are in tight linkage disequilibrium (LD). All SNPs with the lowest P values mapped to introns in NLRC5 (Fig. 2D).
With regard to rs4784751, Grade 3/4 NLEs occurred in 47.2% of patients with the common genotype during the FEC-containing chemotherapy cycles, increasing to 54.6% in heterozygous patients and 62.9% in patients who had two minor alleles. Corresponding odds ratios were 1.34 (95%CI: 1.16–1.55) and 1.89 (95%CI: 1.43–2.49) when heterozygous or homozygous variant patients were compared with patients carrying two reference alleles (Fig. 1B). Adding the first and second principal component to the regression model as a sensitivity analysis did not change those results.
As per protocol, prophylactic granulocyte colony-stimulating factor (G-CSF) was not required in the first chemotherapy cycle. Prophylactic G-CSF is usually given in the first week after chemotherapy regardless of white blood cell counts. After the occurrence of a grade 3 or 4 NLE, prophylactic G-CSF use is recommened during all subsequent chemotherapy cycles. While not being required in the first chemotherapy cycle, 10.9% of the patients received prophylactic G-CSF during the first chemohtherapy cycle and 16.7% and 19.8% inc cycles 2 and 3 respectively. To examine the effects of G-CSF on our results, we performed a sensitivity analysis for rs4784751, and restricted the outcome measure of grade 3/4 NLEs to the first chemotherapy cycle, and introduced G-CSF as a covariate. A total of 39.2% had a grade 3/4 NLE in the first chemotherapy cycle. rs4784751 maintained its predictive value both in patients who received G-CSF (10.9% of the patient population) and patients who did not. Nor were there any differences in effect size between the two groups (data not shown).
Functional Analysis of the 16q13 Locus
To identify a set of credible causal SNPs operating at the locus, we retrieved all SNPs in high (r2≥0.8) linkage disequilibrium with rs4784750 using LDlink (https://ldlink.nci.nih.gov/) in CEU (Utah residents from north and west Europe), generating a list of four additional SNPs, all located in an intronic region of NLRC5. This 3-kb region shows evidence of regulatory activity in blood (Haploreg v4.1) (Supplementary Table 1). Interestingly, chromatin interaction analysis paired-end tags (ChIA-PET) data from ENCODE/GIS-Ruan for RNA polymerase II in K562 cells show that three SNPs (rs4784751, rs12444396, and rs12445252) interact with the promoter region of NLRC5, while rs11644171 interacts with an intronic region of CPNE2 (Fig. 2D). Notably, all five SNPs displayed eQTL associations to NLRC5. Taken together, functional assessment of the credible causal SNPs in the locus strongly suggests that NLRC5 is the likely target gene in this locus.
As NLEs are mainly caused by the effect of chemotherapy on hematopoietic cells, functional studies were conducted on cell lines from the hematopoietic system to understand the role of NLRC5 in NLEs. We used three leukemia cell lines expressing NLRC5, representing different origins such as promyeloblasts, B lymphocytes and T lymphocytes.
We first performed cytotoxicity assays to determine whether silencing of NLRC5 might influence the cell response to the chemotherapy drugs used in the SUCCESS-A trial, including epirubicin (EPI), cyclophosphamide, and 5-fluorouracil (5-FU). Since cyclophosphamide is a prodrug that requires metabolic activation, mafosfamide (MFF) — which spontaneously decomposes to form the cyclophosphamide active metabolite (4-hydroxycyclophosphamide) when added in culture media (23) — was used in the cytotoxicity assay. After 72 hours of drug treatment, no significant changes were observed in the cytotoxicity of these drugs in HL-60, LCL, or Jurkat cells after NLRC5 knockdown (Supplementary Fig. 1). These results suggested that NLRC5 expression in these cell lines does not directly dramatically affect the cytotoxicity of the chemotherapy drugs used in the SUCCESS-A trial.
Next, we investigated whether NLCR5 silencing might affect cell viability in HL-60 cells — a cell line of promyelocytic origin that most closely represents hematopoietic stem cells. An immediate effect on cell viability after NLRC5 silencing was observed. Apoptosis increased after NLRC5 silencing, as analyzed by flow cytometry with staining of apoptotic markers by APC annexin-V (APC-A) and propidium iodide (PI) or PI alone (Fig. 4A). Cells that were in the process of undergoing apoptosis displayed annexin-V and PI, and cells that had already undergone apoptosis only displayed PI alone (Fig. 4A).
Figure 4. Functional studies for the NLRC5 signal.
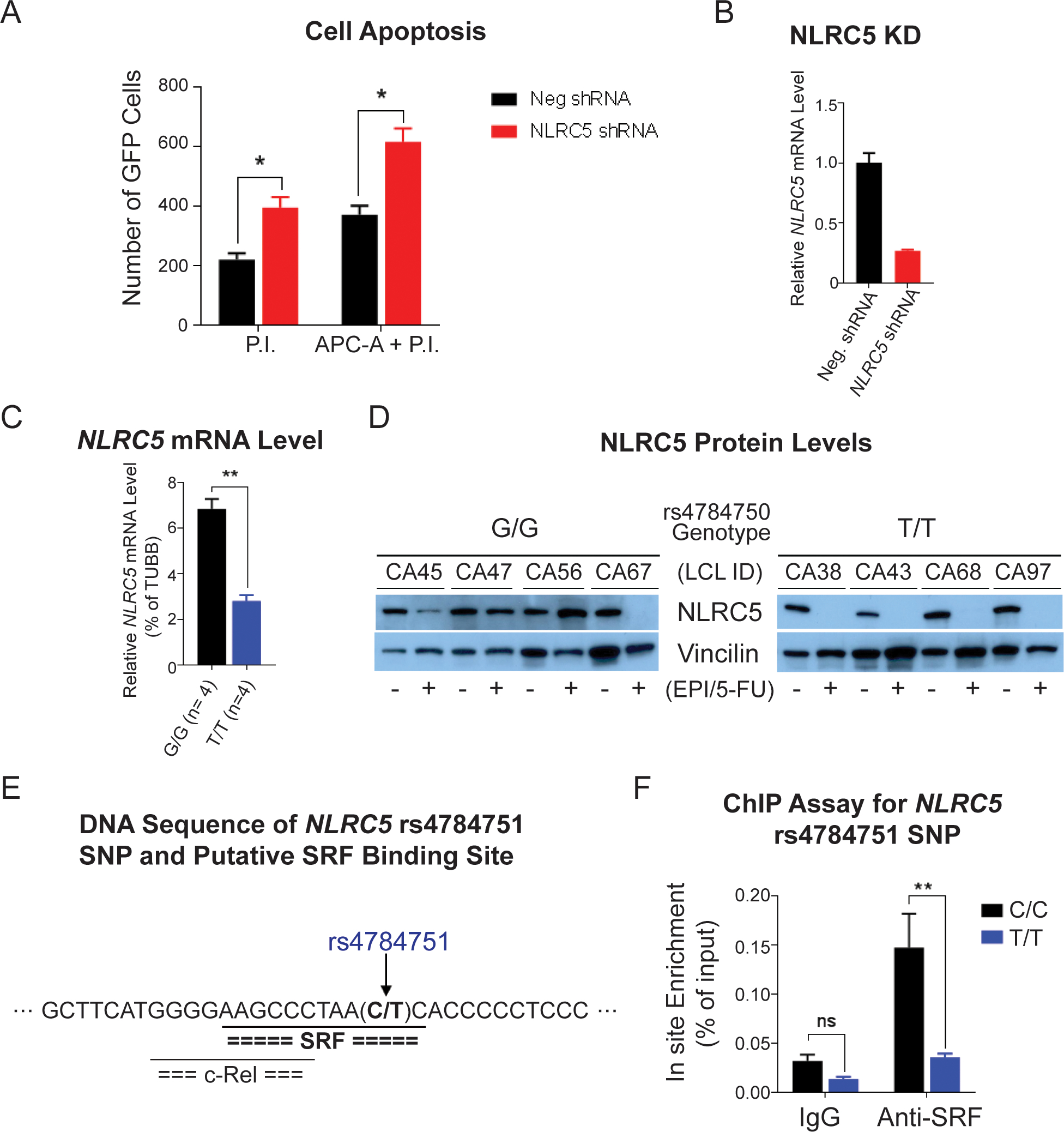
(A) HL-60 cell apoptosis measured by flow cytometry. The bar graph shows the numbers of cells that were stained with apoptosis markers by propidium iodide (P.I.) and P.I. plus APC annexin V (APC-A) 12 h after transfection of shRNAs. (B) Relative NLRC5 mRNA level in HL-60 cells after 12 h of shRNA transfection. (C) The NLRC5 mRNA level in lymphoblastoid cell lines (LCLs) with WT (G/G) and homozygous variant (T/T) rs4784750 SNP genotypes after 48 h of EPI and 5-FU combined treatment. (D) Western blot for NLRC5 protein in LCLs with WT and homozygous variant rs4784750 SNP genotypes after 72 h of EPI and 5-FU combined treatment. (E) DNA sequence near the NLRC5 rs4784751 SNP and putative transcriptional factor binding sites predicted by the TRANSFAC. The rs4784751 SNP, which is in LD (r2 = 0.99) with the rs4784750, was predicted in a serum response element that binds serum response factor (SRF). (F) ChIP assay with anti-SRF antibody for the rs4784751 SNP site in LCLs with WT (C/C) and homozygous variant (T/T) genotypes. * P < 0.05, ** P < 0.01, ns = not significant.
Since we observed that silencing of NRLC5 promotes cell apoptosis, we further determined the drug effects on NLRC5 expression and how the SNPs might contribute to the changes. To do this, the LCLs from which the genome-wide SNP genotype data were generated were used. Based on the genotype of the NLRC5 rs4784750 SNP, four each reference and variant LCLs were treated with the chemotherapy drugs at concentrations equivalent to their EC50 values. After 72 hours of drug exposure, we observed a genotype-dependent difference in NLRC5 mRNA levels that corresponded to a difference in NLRC5 protein levels in the same LCLs (Figs. 4C and 4D). Specifically, LCLs homozygous for the variant alleles (T/T) showed a greater decrease in NLRC5 mRNA and protein after drug exposure than did LCLs homozygous for the reference alleles (G/G) alleles, and this was statistically significant after 72 hours (Fig. 4D).
To determine the possible cause of this genotype-dependent and drug exposure–dependent difference in expression of NLRC5 mRNA and protein, we analyzed the DNA sequences of the top 5 NLRC5 SNPs with low P values in the GWAS using the TRANSFAC database. TRANSFAC suggested that the rs4784751 (C>T) SNP variant allele disrupted a serum response element (SRE) motif to which the serum response factor (SRF) transcription factor was predicted to bind (Fig. 4E). The rs4784751 SNP is in linkage disequilibrium (r2 = 0.99 in Caucasians, based on the 1K Genomes data) with the rs4784750 SNP that shows the lowest P value in the GWAS. To test the TRANSFAC prediction, a ChIP assay was performed with anti-SRF antibody, and the wild-type sequence showed 3.8-fold greater SRF binding than did the variant sequence (Fig. 4F).
These functional studies suggested a possible mechanism in which the chemotherapy dramatically decreased NLRC5 expression in patients with a variant genotype for the rs4784751 SNP, which might result from decreased SRF binding and NLRC5 transcription; thus, the decrease in NLRC5 expression was associated with increased cell apoptosis. Further mechanistic studies need to be pursued in order to understand how decreased levels of NLRC5 may lead to apoptosis.
eQTL Analysis in Whole Blood Samples for both loci
To determine whether the two loci on chromosome 16 and chromosome 13 are expression-quantitative trait loci (eQTLs), we associated the genotypes of the “top” SNPs in both loci (rs4784750 in NLRC5 and rs16972207 in TNFSF13B) with gene expression in a publicly available GTEx (v7) database (http://www.gtexportal.org/home/). As our phenotype, NLE, occurs in whole blood, we examined gene expression in the tissue “whole blood.” All sufficiently expressed genes (n = 23,076) were tested for eQTL analysis. The “top” ten genes with expression mostly associated with rs4784750 and rs16972207 SNP genotypes are shown in Supplementary Tables 2 and 3, respectively. The rs4784750 SNP genotype is most significantly associated with NLRC5 expression, with a beta value of –0.26 and a P value of 2.2E-7 in whole blood samples (Supplementary Table 2). The variant genotype was associated with decreased NLRC5 expression (Supplementary Fig. 2A). The rs4784750 SNP is a trans-eQTL for other genes in whole blood samples, such as PANX1, expression of which correlated positively with the variant genotype of rs4784750 (β=0.28, P=7.9E-6) (Supplementary Fig. 2B).
The rs16972207 SNP was an eQTL for TNFSF13B mRNA expression (Z-score=4.60, P=4.22E-6) in whole blood samples from a study with large cohort (n=5,311) while it was not associated withTNFSF13B mRNA expression in GTEx in which a relatively small sample size was tested (n=407) (24,25). Our functional study demonstrated that rs16972207 was significantly associated with TNFSF13B mRNA expression after drug exposure, a situation which has been referred to as a “pharmacogenomic-eQTL” (26–28). The rs16972207 genotype was associated with LIG4 gene expression (β=0.13, P=4E-4), which mapped centromerically to TNFSF13B (Fig. 2A and Supplementary Table FIg3).
Combined effects of NLRC5 and TNFSF13B SNPs
Since SNPs in both the NLRC5 and TNFSF13B genes were strongly associated with NLEs, we investigated possible correlations of mRNA expression for these two genes in our “Human Variation Panel” and other datasets (Fig. 5A–5C). It was found that NLRC5 and TNFSF13B mRNA expression was highly correlated in the LCLs (r2=0.51, P<1E-21) (Fig. 5B) and in BC datasets listed in Oncomine (r2=0.645, n=160 and r2=0.578, n=55) (29,30). We also assessed the “combined effect” of SNPs in these two genes relative to the risk for NLEs. This analysis showed that the difference between odds ratio (OR) values for patients who were homozygous for both risk alleles was 4.8, compared to patients homozygous for both protective alleles (Fig. 5B). It is interesting to note that the trend observed at baseline in healthy untreated LCLs as well as untreated BCs from Oncomine suggest that these genes may be under a similar type of regulation at baseline. However, upon treatment with chemotherapy and induction of these transcription factors, these genes become regulated in an inverse way (Fig. 5C).
Figure 5.
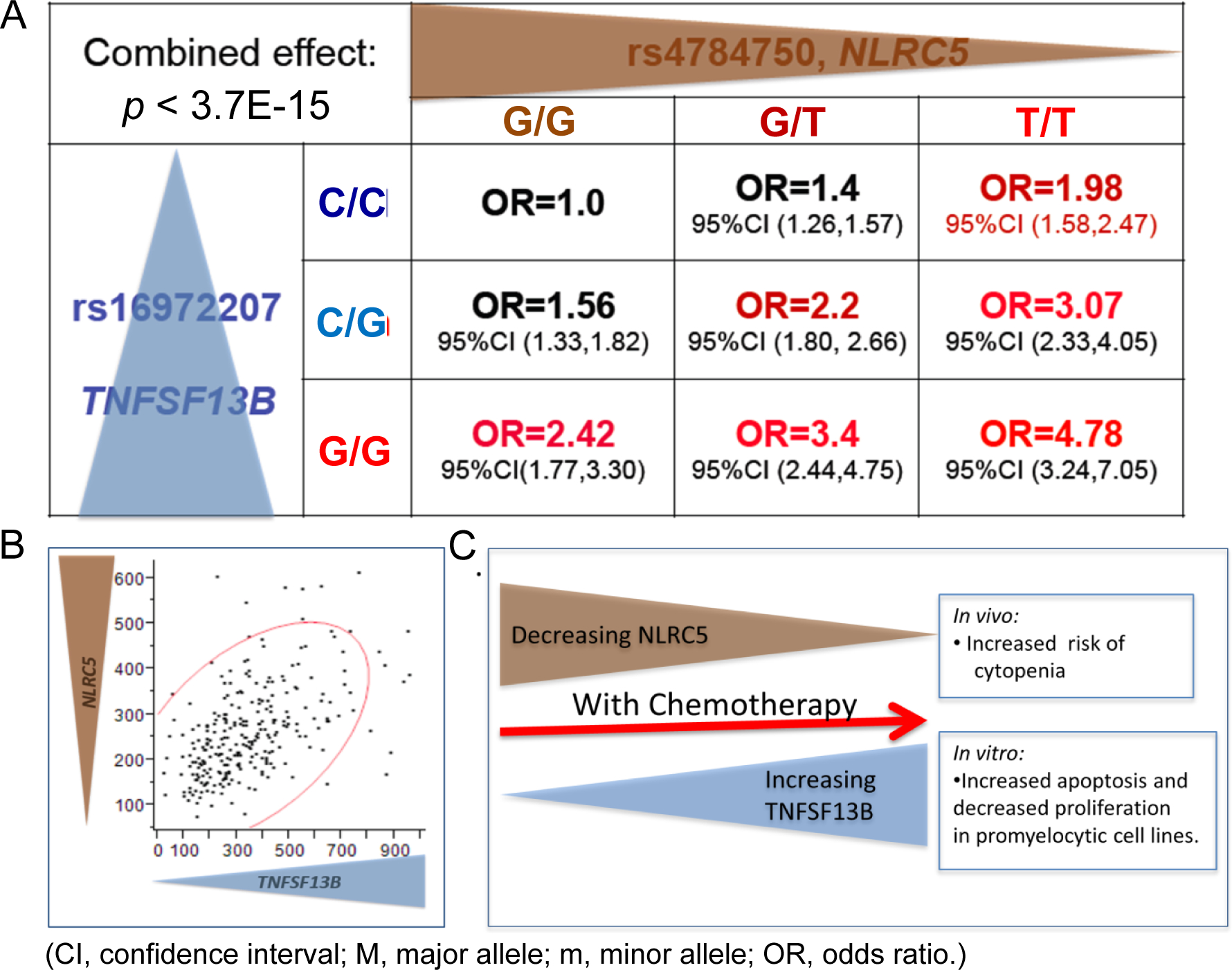
(A) Combined effect of NLRC5 and TNFSF13B top SNPs calculated as the odds ratio for developing cytopenia/toxicity with chemotherapy. (B) Correlation of the gene expression between NLRC5 and TNFSF13B in the Mayo Human Variation Panel of lymphoblastoid cell lines (LCLs) derived from 287 individuals. (C) Putative model of the interaction of NLRC5 and TNFSF13B and their influence on the risk concerning cytopenia.
Results for Clinical Outcome Parameters
Association with prognosis in the SUCCESS study.
In an exploratory analysis, we associated the genotypes and NLE with disease-free survival in the SUCCESS-A study (Fig. 6A). Median follow up time was 5.2 years and the number of events was 414. There appeared to be an effect on the prognosis in the group of women who had a homozygous variant effect. The group of women with NLEs and a genotype associated with down-regulation of NLRC5 after chemotherapy appeared to perform worst, while women without NLE after chemotherapy performed better.
Figure 6.
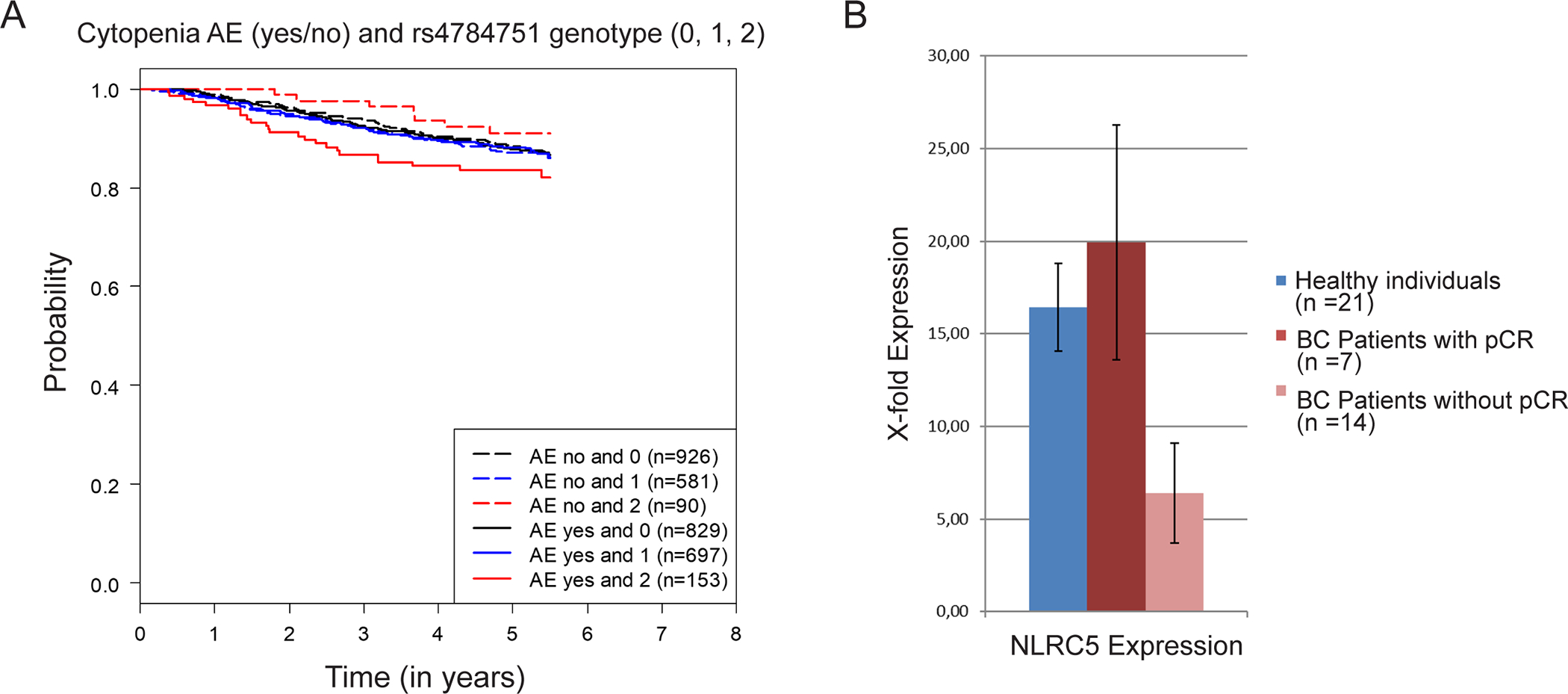
(A) Effect of NLRC5 genotype on the prognosis in the SUCCESS-A study: Kaplan–Meier curves for disease-free survival relative to neutropenic or leukopenic events (AE) and the NLRC5 rs4784751 genotype (0, zero minor alleles; 1 one minor allele; 2, two minor alleles). (B) Effect of NLRC5 leukocyte expression on the pathological complete response (pCR) in a group of triple-negative breast cancer (TNBC) patients: comparison of white blood cell NLRC5 expression between healthy individuals, patients who achieved a pCR after neoadjuvant chemotherapy, and patients who did not achieve a pCR.
Influence of white blood cell NLRC5 expression on neoadjuvant chemotherapy responsiveness.
As the eQTL analysis showed that NLRC5 genotype could have an influence on NLRC5 expression in whole blood (Supplementary Figure 2), we tested the possible influence of leukocyte NLRC5 expression on chemotherapy responsiveness. To do that, we conducted a small neoadjuvant study in which white blood cell RNA was collected prospectively before the start of chemotherapy. Blood was also collected from healthy control individuals. Although no differences were detectable between healthy individuals and triple-negative BC patients before chemotherapy (Fig. 6B), patients who did not achieve a pathological complete response (pCR) after chemotherapy had significantly lower (P=0.02) NLRC5 expression than patients who did achieve a pCR after chemotherapy (Fig. 6B).
DISCUSSION
We identified two chromosomal loci associated with grade 3/4 neutropenic or leukopenic events after chemotherapy with epirubicin, cyclophosphamide, and 5-fluorouracil. At the 13q33.3 locus, ABHD13, LIG4, and TNFSF13B (also known as BAFF, B cell activating factor) emerged as plausible target genes after functional annotation using publicly available data. ABHD13 has been associated through GWAS with the monocyte count (31), and LIG4 is essential for V(D)J recombination and DNA double-strand break (DSB) repair through nonhomologous end joining (NHEJ) — processes known to affect the response to therapeutic drugs. TNFSF13B is expressed by many cells such as antigen-presenting cells (B cells, macrophages, dendritic cells), neutrophils, epithelial cells, T lymphocytes, and stromal cells (32,33). Most functional knowledge of this gene relates to its role as a survival factor for peripheral B cells.
Impaired B cell maturation, decreased immunoglobulin levels, decreased T cell dependent and T cell independent immune responses have been observed in Tnfsf13b knockout mice (34). On the other hand, transgenic Tnfsf13b mice develop B cell hyperplasia, glomerulonephritis, and destruction of the salivary glands, as well as expansion of the effector and regulatory T cell compartments (35,36). Our top SNP in TNFSF13B (rs16972207) was observed to be an eQTL for TNFSF13B mRNA expression in a large cohort of subjects. We also demonstrated that this SNP is significantly associated with TNFSF13B expression after drug exposure, which appeared to be related to an influence on the binding of transcription factor PXR with the variant genotype, which showed greater affinity for the transcription factor than did the wild type.
There have been reports of ‘late-onset’ neutropenia after treatment with rituximab, with high levels of BAFF being found in patients with neutropenia. The neutropenia might be the consequence of hematopoietic lineage competition due to excessive B cell recovery in the bone marrow (20,21). Taken together, the data suggest that TNFSF13B is a likely target gene at this locus, but a contribution of ABHD13 or LIG4 cannot be ruled out.
At the 16q13 locus, NLRC5 emerged as the most likely target gene. The top SNPs at 16q13 showed an eQTL association with NLRC5 expression in whole blood samples, supporting the hypothesis that NLRC5 is the target gene. NLRC5 has been described as a key regulator of major histocompatibility complex (MHC) class I gene expression (37–40) as well as other genes in the antigen-presenting system (37,38). In contrast to MHC class II molecules, which are mainly expressed on hematopoietic cells, MHC class I molecules are expressed in all cells that contain a nucleus (41). This is observed in all immune tissues and organs such as the spleen, lymph nodes, bone marrow, and thymus. Although the transcription of NLRC5 has been described as being increased by interferon gamma (IFN-γ) and activation of STAT1 (42,43), our in silico analysis of the locus in NLRC5 implied a change in a region that might serve as a binding site for the transcription factor SRF. SRF has indeed been shown to indirectly regulate type I interferon signaling in macrophages (44) without interfering with the classic JAK/STAT pathway (44). It has also been demonstrated that in macrophages, lipopolysaccharide administration induces high levels of NLRC5 through the type I interferon pathway (45,46). The interaction between the genotype discovered and the effect of the chemotherapy might be mediated by an SRF-dependent effect after the interferon type I pathway. Recently, SRF has been described as an essential transcription factor in hematopoiesis (47). The present functional in vitro assessment showed that chemotherapy modulates the expression of NLRC5 and TNFSF13B in an allele-specific manner, down-regulating NLRC5 after chemotherapy and up-regulating TNFSF13B. Both NLRC5 and TNFSF13B are known genes with functions in innate and adaptive immune responses.
A more toxic effect of chemotherapy on the white blood cell count in patients with the NLRC5 variant genotype may be mediated through PANX1, which was the top trans-eQTL finding for the SNP rs4784750. Although formally not having a false discovery rate of ≤0.05 as required by the GTEx project (48,49), PANX1 has been reported to drive inflammation (50) and facilitate apoptosis, pyroptosis, and autophagy (50,51).
An interesting aspect of the association between these two genes and chemotherapy-related neutropenia or leukopenia is their relation to recent immuno-oncological findings. Particularly because NLRC5 regulates MHC class I gene expression, its role in immune evasion by cancer cells has been analyzed (52,53). For several histologies, high NLRC5 expression is associated with a favorable prognosis (52). It has also been shown that NLRC5 expression is associated with increased activation of CD8+ cytotoxic T cells (52). This makes NLRC5 an interesting target for possible cancer therapies, as well as an interesting prognostic marker. The present analysis in relation to the clinical outcome in the SUCCESS-A study did not show that rs4784750 was associated with prognosis. Nor was the occurrence of grade 3/4 NLEs associated with the prognosis. However, when the analysis for neutropenia and rs4784750 genotype was stratified, there was some indication that patients who suffer neutropenia after chemotherapy in the variant genotype group have an unfavorable prognosis. This effect may correlate with NLRC5 expression in white blood cells, but could also be a consequence of differential NLRC5 expression in the tumor. Our small neoadjuvant chemotherapy study also showed that chemotherapy responsiveness correlates with NLRC5 expression in white blood cells — implying a possible interaction of NLRC5 with immuno-oncological mechanisms and chemotherapy response. Of note, the effects of genotypes on prognosis and the effect of NLRC5 gene expression on chemotherapy responsiveness do not validate the GWAS findings. However, they shed light on the possible role of this gene in relation to breast cancer treatment.
With regard to treatment implications, one possible clinical application might be up-regulation of NLRC5 during chemotherapy — e.g., with interferon. For tumor cells in vivo, it has already been demonstrated that increasing NLRC5 activity restores tumor immunogenicity and stimulates antitumor immunity (54).
This study has both strengths and limitations. It is the first study to examine neutropenia and leukopenia as part of a prospective phase III chemotherapy study. This ensures high data quality, with on-site monitoring and auditing as well as pre-specified data management and statistical analysis procedures. Cumulative NLE events were available as a variable. While documentation according to NCI-CTCAE criteria as in our study is a standard for capturing this phenotype, more detailed data such as time to NLE were not available. With NLE grade 3/4 occurring in more than 40% of patients, the phenotype is also frequent enough to provide adequate statistical power. Having more than 3300 patients, the sample size should, therefore, have been sufficient to discover relevant genetic variants. Despite extensive in vitro functional validation of the findings, we acknowledge the need for in depth mechanistic studies. Although the functional experiments provide a degree of confidence for accurate findings, empirical validation would be desirable. At the time when the study was conducted (2005–2007), combined treatment with epirubicin, cyclophosphamide, 5-fluorouracil, docetaxel, and gemcitabine was reasonable, but gemcitabine never became a standard treatment in the adjuvant setting for BC. 5-FU, which appeared to play a role at least in molecular effects in relation to TNFSF13B, is no longer administered to BC patients, as its effectiveness was not confirmed (55). With regard to generalization of the data, it is noteworthy that minor allele frequencies differ widely among Caucasian, Han Chinese, and African-American individuals. The minor allele frequencies (MAFs) for the SNPs in our two genes were compared with the HapMap data. The variant alleles for rs4784751 in NLRC5 and rs16972207 in TNFSF13B were most prevalent in the Caucasian population, with MAFs of 0.32 and 0.18, respectively. In our study these MAFs were 0.27 and 0.19 respectively. In comparison with other ethnicities, the Han Chinese population had MAFs of 0.04 and 0.00, and the African-American population had MAFs of 0.02 and 0.192, respectively. While there have been reports on ethnicity-specific differences in the occurrence of neutropenia after chemotherapy (56), it is unclear whether genotypes might play a major role in these differences, in part because other factors might play a role like pre-chemotherapy baseline white blood cell count (57,58). Also, studies of this question are scarce. With regard to the clinical meaning of our results, the top SNPs could differentiate patient groups with 47–49% to 63–68% of patients experiencing grade 3 or 4 neutropenie/leukopenia. While these differences might seem clinically relevant, it has to be noted, that even with the protective alleles a large proportion of patients still experiences NLE, warranting further studies designed to examine the reasons for NLE after chemotherapy.
In summary, this study provides evidence that genetic variants of the key regulator of MHC class I expression may be involved in chemotherapy-induced neutropenia through genotype-dependent down-regulation of the gene. In addition, NLRC5 genotypes may be involved in differences in the efficacy of chemotherapy and in the prognosis in BC patients.
Supplementary Material
Statement of translational relevance.
Grade 3 or 4 neutropenic or leukopenic events are the most relevant side effects after chemotherapy but molecular factors associated with the occurrence are unclear. This study identifies loci in NLRC5 and TNFSF13B that are associated with post-chemotherapy neutropenia and leukopenia. Genotypes also showed an association with the prognosis in patients with a neutropenic or leukopenic event. Thus, NLRC5 is suggested as a prognostic and predictive marker for breast cancer patients receiving chemotherapy. With regard to treatment implications, one possible clinical application might be up-regulation of NLRC5 during chemotherapy — e.g., with interferon. For tumor cells in vivo, it has already been demonstrated that increasing NLRC5 activity restores tumor immunogenicity and stimulates antitumor immunity.
Acknowledgments
We are grateful to Sonja Oeser and Silke Landrith for handling of the samples and to Luanne Wussow for infrastructure support. Furthermore we would like to thank Teri Manolio, Corinne Boehm, Cathy C. Laurie for her support during the conduct of this consotrial effort. This study was conducted as part of the Genomics and Randomized Trials Network (GARNET), U01 HG005137/HG/NHGRI NIH HHS (RMW, PAF) and U01 HG004438 (KFD, JMR, CDB). The research is also supported, in part, by the following United States National Institutes of Health grants: U19 GM61388 (The Pharmacogenomics Research Network), P50 CA116201 (Mayo Clinic Breast Cancer Specialized Program of Research Excellence), R01 CA196648, U10 CA180868, and UO1 CA18967. It is also supported by the Breast Cancer Research Foundation, the Nan Sawyer Breast Cancer Fund, the Eisenberg Foundation, and the Moffitt Foundation. The NIH Clinical Pharmacology Training Grant T32 GM08685 supported the work of Dr. Scully. In addition, the research was supported by institutional funding from the Department of Gynecology and Obstetrics, Erlangen University Hospital, Comprehensive Cancer Center Erlangen–EMN (PAF, MR). Mayo Clinic grant CA15083 supported sample handling and processing at the Mayo Clinic Genotyping Shared Resource (JMC). Conduct of the main clinical phase III study was supported by grants from Novartis, AstraZeneca, Chugai, Sanofi-Aventis, and Lilly (WJ).
Financial support: U01 HG005137/HG/NHGRI NIH HHS; further funding information is provided at the end of the article.
Footnotes
Conflict of interests
Dr Fasching reports personal fees from Novartis, grants from Biontech, personal fees from Pfizer, personal fees from Daiichi-Sankyo, personal fees from AstraZeneca, personal fees from Eisai, personal fees from Merck Sharp & Dohme, grants from Cepheid, personal fees from Lilly, personal fees from Pierre Fabre, personal fees from seagen, personal fees from Roche, personal fees from Agendia, personal fees from Sanofi Aventis, and personal fees from Gilead outside the submitted work. Drs. Wang and Weinshilboum are co-founders of and stockholders in OneOme, LLC, a pharmacogenomic decision support company. Dr. Tesch, Prof. Dr. reports personal fees and non-financial support from Novartis, personal fees and non-financial support from Roche, personal fees and non-financial support from Astra Zeneca, personal fees and non-financial support from Pfizer, personal fees and non-financial support from Amgen, outside the submitted work; Dr. Schneeweiss reports grants from Celgene, grants from Roche, grants from AbbVie, grants from Molecular Partner, personal fees from Roche, personal fees from AstraZeneca, personal fees from Celgene, personal fees from Roche, personal fees from Roche, personal fees from Celgene, personal fees from Pfizer, personal fees from AstraZeneca, personal fees from Novartis, personal fees from MSD, personal fees from Tesaro, personal fees from Lilly, personal fees from Pfizer, other from Roche, outside the submitted work; all other authors have declared that they have no conflicts of interest.
REFERENCES
- 1.Lyman GH, Abella E, Pettengell R. Risk factors for febrile neutropenia among patients with cancer receiving chemotherapy: A systematic review. Critical reviews in oncology/hematology 2014;90(3):190–9 doi 10.1016/j.critrevonc.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Schwenkglenks M, Jackisch C, Constenla M, Kerger JN, Paridaens R, Auerbach L, et al. Neutropenic event risk and impaired chemotherapy delivery in six European audits of breast cancer treatment. Support Care Cancer 2006;14(9):901–9 doi 10.1007/s00520-006-0034-9. [DOI] [PubMed] [Google Scholar]
- 3.Shurin MR, Naiditch H, Gutkin DW, Umansky V, Shurin GV. ChemoImmunoModulation: immune regulation by the antineoplastic chemotherapeutic agents. Curr Med Chem 2012;19(12):1792–803. [DOI] [PubMed] [Google Scholar]
- 4.Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 2014;21(1):15–25 doi 10.1038/cdd.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, et al. Current Landscape of Immunotherapy in Breast Cancer: A Review. JAMA Oncol 2019. doi 10.1001/jamaoncol.2018.7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aapro MS, Bohlius J, Cameron DA, Dal Lago L, Donnelly JP, Kearney N, et al. 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 2011;47(1):8–32 doi 10.1016/j.ejca.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Fasching PA, Haberle L, Rack B, Li L, Hein A, Ekici AB, et al. Clinical validation of genetic variants associated with in vitro chemotherapy-related lymphoblastoid cell toxicity. Oncotarget 2017;8:78133–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeil AM, Vulsteke C, Paridaens R, Dieudonne AS, Pettengell R, Hatse S, et al. Multivariable regression analysis of febrile neutropenia occurrence in early breast cancer patients receiving chemotherapy assessing patient-related, chemotherapy-related and genetic risk factors. BMC Cancer 2014;14:201 doi 10.1186/1471-2407-14-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vulsteke C, Lambrechts D, Dieudonne A, Hatse S, Brouwers B, van Brussel T, et al. Genetic variability in the multidrug resistance associated protein-1 (ABCC1/MRP1) predicts hematological toxicity in breast cancer patients receiving (neo-)adjuvant chemotherapy with 5-fluorouracil, epirubicin and cyclophosphamide (FEC). Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 2013;24(6):1513–25 doi 10.1093/annonc/mdt008. [DOI] [PubMed] [Google Scholar]
- 10.Okishiro M, Kim SJ, Tsunashima R, Nakayama T, Shimazu K, Shimomura A, et al. MDM2 SNP309 and TP53 R72P associated with severe and febrile neutropenia in breast cancer patients treated with 5-FU/epirubicin/cyclophosphamide. Breast Cancer Res Treat 2012;132(3):947–53 doi 10.1007/s10549-011-1637-5. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasan Y, Sasa M, Honda J, Takahashi A, Uno S, Kamatani N, et al. Genome-wide association study of epirubicin-induced leukopenia in Japanese patients. Pharmacogenet Genomics 2011;21(9):552–8 doi 10.1097/FPC.0b013e328348e48f. [DOI] [PubMed] [Google Scholar]
- 12.Rack B, Schindlbeck C, Juckstock J, Andergassen U, Hepp P, Zwingers T, et al. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J Natl Cancer Inst 2014;106(5) doi 10.1093/jnci/dju066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Gregorio A, Haberle L, Fasching PA, Muller V, Schrader I, Lorenz R, et al. Gemcitabine as adjuvant chemotherapy in patients with high-risk early breast cancer-results from the randomized phase III SUCCESS-A trial. Breast Cancer Res 2020;22(1):111 doi 10.1186/s13058-020-01348-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laurie CC, Doheny KF, Mirel DB, Pugh EW, Bierut LJ, Bhangale T, et al. Quality control and quality assurance in genotypic data for genome-wide association studies. Genet Epidemiol 2010;34(6):591–602 doi 10.1002/gepi.20516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panagiotou OA, Ioannidis JP, Genome-Wide Significance P. What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. Int J Epidemiol 2012;41(1):273–86 doi 10.1093/ije/dyr178. [DOI] [PubMed] [Google Scholar]
- 16.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009;16(3):259–66 doi 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, et al. Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res 2008;68(17):7050–8 doi 10.1158/0008-5472.CAN-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bidadi B, Liu D, Kalari KR, Rubner M, Hein A, Beckmann MW, et al. Pathway-Based Analysis of Genome-Wide Association Data Identified SNPs in HMMR as Biomarker for Chemotherapy- Induced Neutropenia in Breast Cancer Patients. Front Pharmacol 2018;9:158 doi 10.3389/fphar.2018.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wurfel F, Erber R, Huebner H, Hein A, Lux MP, Jud S, et al. TILGen: A Program to Investigate Immune Targets in Breast Cancer Patients - First Results on the Influence of Tumor-Infiltrating Lymphocytes. Breast Care (Basel) 2018;13(1):8–14 doi 10.1159/000486949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terrier B, Ittah M, Tourneur L, Louache F, Soumelis V, Lavie F, et al. Late-onset neutropenia following rituximab results from a hematopoietic lineage competition due to an excessive BAFF-induced B-cell recovery. Haematologica 2007;92(2):e20–3. [DOI] [PubMed] [Google Scholar]
- 21.Ishida H, Inui M, Furusawa M, Tanabe K. Late-onset neutropenia (LON) after low-dose rituximab treatment in living related kidney transplantation--single-center study. Transpl Immunol 2013;28(2–3):93–9 doi 10.1016/j.trim.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Zhang B, Xie W, Krasowski MD. PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 2008;9(11):1695–709 doi 10.2217/14622416.9.11.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwon CH, Borch RF, Engel J, Niemeyer U. Activation mechanisms of mafosfamide and the role of thiols in cyclophosphamide metabolism. J Med Chem 1987;30(2):395–9 doi 10.1021/jm00385a023. [DOI] [PubMed] [Google Scholar]
- 24.Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45(10):1238–43 doi 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franke L. Blood eQTL browser. https://wwwgenenetworknl/bloodeqtlbrowser/ 2019:accessed September 30, 2019.
- 26.Ingle JN, Liu M, Wickerham DL, Schaid DJ, Wang L, Mushiroda T, et al. Selective estrogen receptor modulators and pharmacogenomic variation in ZNF423 regulation of BRCA1 expression: individualized breast cancer prevention. Cancer Discov 2013;3(7):812–25 doi 10.1158/2159-8290.CD-13-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neavin DR, Lee JH, Liu D, Ye Z, Li H, Wang L, et al. Single Nucleotide Polymorphisms at a Distance from Aryl Hydrocarbon Receptor (AHR) Binding Sites Influence AHR Ligand-Dependent Gene Expression. Drug Metab Dispos 2019;47(9):983–94 doi 10.1124/dmd.119.087312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu D, Nguyen TTL, Gao H, Huang H, Kim DC, Sharp B, et al. TCF7L2 lncRNA: a link between bipolar disorder and body mass index through glucocorticoid signaling. Mol Psychiatry 2021. doi 10.1038/s41380-021-01274-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginestier C, Cervera N, Finetti P, Esteyries S, Esterni B, Adelaide J, et al. Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res 2006;12(15):4533–44 doi 10.1158/1078-0432.CCR-05-2339. [DOI] [PubMed] [Google Scholar]
- 30.Bonnefoi H, Potti A, Delorenzi M, Mauriac L, Campone M, Tubiana-Hulin M, et al. Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00–01 clinical trial. Lancet Oncol 2007;8(12):1071–8 doi 10.1016/S1470-2045(07)70345-5. [DOI] [PubMed] [Google Scholar]
- 31.Crosslin DR, McDavid A, Weston N, Zheng X, Hart E, de Andrade M, et al. Genetic variation associated with circulating monocyte count in the eMERGE Network. Hum Mol Genet 2013;22(10):2119–27 doi 10.1093/hmg/ddt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Youinou P, Pers JO. Disturbance of cytokine networks in Sjogren’s syndrome. Arthritis Res Ther 2011;13(4):227 doi 10.1186/ar3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol 2009;9(7):491–502 doi 10.1038/nri2572. [DOI] [PubMed] [Google Scholar]
- 34.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol 2003;21:231–64 doi 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 35.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med 2007;204(8):1959–71 doi 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackay F, Leung H. The role of the BAFF/APRIL system on T cell function. Semin Immunol 2006;18(5):284–9 doi 10.1016/j.smim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 37.Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, et al. NLRC5 exclusively transactivates MHC class I and related genes through a distinctive SXY module. PLoS Genet 2015;11(3):e1005088 doi 10.1371/journal.pgen.1005088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proceedings of the National Academy of Sciences of the United States of America 2010;107(31):13794–9 doi 10.1073/pnas.1008684107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robbins GR, Truax AD, Davis BK, Zhang L, Brickey WJ, Ting JP. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. The Journal of biological chemistry 2012;287(29):24294–303 doi 10.1074/jbc.M112.364604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Y, Wang Y, Chen F, Huang Y, Zhu S, Leng Q, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell research 2012;22(5):836–47 doi 10.1038/cr.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Elsen PJ, Holling TM, Kuipers HF, van der Stoep N. Transcriptional regulation of antigen presentation. Curr Opin Immunol 2004;16(1):67–75. [DOI] [PubMed] [Google Scholar]
- 42.Downs I, Vijayan S, Sidiq T, Kobayashi KS. CITA/NLRC5: A critical transcriptional regulator of MHC class I gene expression. Biofactors 2016;42(4):349–57 doi 10.1002/biof.1285. [DOI] [PubMed] [Google Scholar]
- 43.Yao Y, Qian Y. Expression regulation and function of NLRC5. Protein & cell 2013;4(3):168–75 doi 10.1007/s13238-012-2109-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie L, Sullivan AL, Collier JG, Glass CK. Serum response factor indirectly regulates type I interferon-signaling in macrophages. J Interferon Cytokine Res 2013;33(10):588–96 doi 10.1089/jir.2012.0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010;141(3):483–96 doi 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staehli F, Ludigs K, Heinz LX, Seguin-Estevez Q, Ferrero I, Braun M, et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. Journal of immunology 2012;188(8):3820–8 doi 10.4049/jimmunol.1102671. [DOI] [PubMed] [Google Scholar]
- 47.Taylor A, Halene S. The regulatory role of serum response factor pathway in neutrophil inflammatory response. Curr Opin Hematol 2015;22(1):67–73 doi 10.1097/MOH.0000000000000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.GTex Project. Documentation Page. http://wwwgtexportalorg/home/documentationPage 2016:accessed Jan 10, 2017.
- 49.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America 2003;100(16):9440–5 doi 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crespo Yanguas S, Willebrords J, Johnstone SR, Maes M, Decrock E, De Bock M, et al. Pannexin1 as mediator of inflammation and cell death. Biochim Biophys Acta 2017;1864(1):51–61 doi 10.1016/j.bbamcr.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015;43(5):923–32 doi 10.1016/j.immuni.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proceedings of the National Academy of Sciences of the United States of America 2016;113(21):5999–6004 doi 10.1073/pnas.1602069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chelbi ST, Guarda G. NLRC5, a promising new entry in tumor immunology. J Immunother Cancer 2016;4:39 doi 10.1186/s40425-016-0143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez GM, Bobbala D, Serrano D, Mayhue M, Champagne A, Saucier C, et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8(+) T lymphocytes. Oncoimmunology 2016;5(6):e1151593 doi 10.1080/2162402X.2016.1151593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Del Mastro L, De Placido S, Bruzzi P, De Laurentiis M, Boni C, Cavazzini G, et al. Fluorouracil and dose-dense chemotherapy in adjuvant treatment of patients with early-stage breast cancer: an open-label, 2 × 2 factorial, randomised phase 3 trial. Lancet 2015;385(9980):1863–72 doi 10.1016/S0140-6736(14)62048-1. [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa Y, Kawaguchi T, Kubo A, Ando M, Shiraishi J, Isa S, et al. Ethnic difference in hematological toxicity in patients with non-small cell lung cancer treated with chemotherapy: a pooled analysis on Asian versus non-Asian in phase II and III clinical trials. J Thorac Oncol 2011;6(11):1881–8 doi 10.1097/JTO.0b013e31822722b6. [DOI] [PubMed] [Google Scholar]
- 57.Hershman D, Weinberg M, Rosner Z, Alexis K, Tiersten A, Grann VR, et al. Ethnic neutropenia and treatment delay in African American women undergoing chemotherapy for early-stage breast cancer. J Natl Cancer Inst 2003;95(20):1545–8 doi 10.1093/jnci/djg073. [DOI] [PubMed] [Google Scholar]
- 58.Smith K, Wray L, Klein-Cabral M, Schuchter L, Fox K, Glick J, et al. Ethnic disparities in adjuvant chemotherapy for breast cancer are not caused by excess toxicity in black patients. Clin Breast Cancer 2005;6(3):260–6; discussion 7–9 doi 10.3816/CBC.2005.n.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data of this GWAS is available under the dbGaP (Study Accession: phs000547.v1.p1).