

Abstract
Activation of the stimulator of interferon genes (STING) pathway promotes antitumor immunity but STING agonists have yet to achieve clinical success. Increased understanding of the mechanism of action of STING agonists in human tumors is key to developing therapeutic combinations that activate effective innate antitumor immunity. Here, we report that malignant pleural mesothelioma cells robustly express STING and are responsive to STING agonist treatment ex vivo. Using dynamic single-cell RNA sequencing of explants treated with a STING agonist we observed CXCR3 chemokine activation primarily in tumor cells and cancer-associated fibroblasts, as well as T-cell cytotoxicity. In contrast, primary NK cells resisted STING agonist–induced cytotoxicity. STING agonists enhanced migration and killing of NK cells and mesothelin-targeted chimeric antigen receptor (CAR)-NK cells, improving therapeutic activity in patient-derived organotypic tumor spheroids. These studies reveal the fundamental importance of using human tumor samples to assess innate and cellular immune therapies. By functionally profiling mesothelioma tumor explants with elevated STING expression in tumor cells, we uncovered distinct consequences of STING agonist treatment in humans that support testing combining STING agonists with NK and CAR-NK cell therapies.
Keywords: STING, innate immunity, NK cells, combination immunotherapy, organotypic tumor spheroids
Introduction
Activation of innate antitumor immunity, including the stimulator of interferon genes (STING) pathway, can overcome barriers to immunotherapy response such as immune exclusion and exhaustion. STING agonist clinical development has focused primarily on myeloid-cell priming of CD8+ T cells that reject transplanted mouse syngeneic tumors (1–3). However, STING activation induces stress, cell-cycle arrest, and death in T cells (4–6), which may limit its clinical activity. Human tumors also undergo months to years of immune editing (7), rendering cross-species extrapolation of STING-induced, T cell–killing mechanisms tenuous. Despite these limitations, mouse studies have explored the complex interplay of STING signaling in the tumor immune microenvironment (TIME), identifying novel effector mechanisms including NK cells and juxtaposing the importance of immune-cell versus tumor-cell STING activity (2,8–11).
Advances in studying the human TIME using patient samples allow for development of immune therapies by treating patient-derived organotypic tumor spheroids (PDOTS) and tissue fragment explants in short-term culture (12,13). To date, these platforms have been used mainly to study anti–PD(L)-1 immune checkpoint inhibitors and can parallel patient response, but they also offer promise in designing novel cancer immunotherapies. In contrast to patient-derived xenografts grown in humanized mice, these systems provide the ability to interrogate the immune response within the native human tumor immune contexture and have the potential to allow the study of cell therapies without interference from the murine microenvironment. Furthermore, insights gained from modeling the direct human TIME may also help to close the translational gap for immune therapies that are effective in syngeneic mouse models but fail in clinical trials.
Multiple types of human cancer have recently been shown to silence STING and the downstream interferon response to avoid immune detection (14–16), demonstrating an important role for tumor-cell STING signaling in human cancer. In addition, higher STING expression correlates with better response to treatment across cancer types (17–20). Yet how STING agonists impact tumor cells and different cell types in the human TIME has not been carefully examined and such information could inform development of novel therapeutic combinations, including cell therapy. Here, we address this question by pursuing a large-scale study using PDOTS and developing methodology to conduct dynamic single-cell RNA sequencing in tumor explants, as well as dissecting STING agonist response in an inflamed histotype.
Materials and Methods
No sample size calculations were performed to predetermine group sizes, and investigators were not blinded during randomization and outcome assessments.
Patient samples
Formalin-fixed, paraffin-embedded (FFPE) tissue-microarray slides from patients with small-cell lung carcinoma (SCLC), non–small cell lung carcinoma (NSCLC), and thymoma (with normal thymus) were purchased from Biomax (LC245, LC817, LC2081, THY761) and used exclusively for STING immunohistochemistry (IHC) in Fig. 1A. In addition, FFPE slides were collected from DFCI/BWH patients with SCLC (n=58), malignant pleural mesothelioma (MPM) (n=68), and benign pleura (n=9) under Dana-Farber/Harvard Cancer Center protocols 02–180 and 98–063. Sections of normal brain, liver, and lung were analyzed from metastases to these organs. These IRB-approved protocols invite all patients seen in clinic to participate in tumor tissue collection/banking. Tumors from patients with MPM treated at DFCI/BWH between December 2014 and October 2021 were collected after surgery under protocol 98–063. FFPE tissue was used for IHC as above. Fresh tumor tissue (refrigerated but not frozen and studied within 24-hours of collection) was used for the flow cytometry experiments in Fig. 1B–D. A total of 66 samples were analyzed by flow cytometry between 2014 and 2021; as new immune checkpoints were identified the flow panel was expanded leading to different total numbers of patients for each checkpoint analyzed in Fig. 1B–D. A total of 37 MPM tumors were treated with STING agonists ex vivo in Figs. 2–5 (Supplmentary Table S1). Limited demographic information was available for these patients and is included in Supplementary Tables S1 and S2. Neoadjuvant therapy consisted of platinum/pemetrexed. One patient (#6) received neoadjuvant checkpoint immunotherapy. For a comparison with NK cells from MPM, NK cells were tested by flow cytometry of blood collected from patients with head & neck squamous cell carcinoma or oral proliferative verrucous leukoplakia (n=20) under protocols 17–255 and 18–387 (Fig. 1D). There was unspecified overlap between the patient samples analyzed by IHC, flow cytometry, and ex vivo treatment, as investigators were blinded to patient information organized by study staff. All patient studies were conducted according to the Declaration of Helsinki and approved by DFCI and BWH institutional review boards. Written informed consent was obtained from all patients whose tumors were studied.
Figure 1: STING is highly expressed in immune-exhausted MPM.
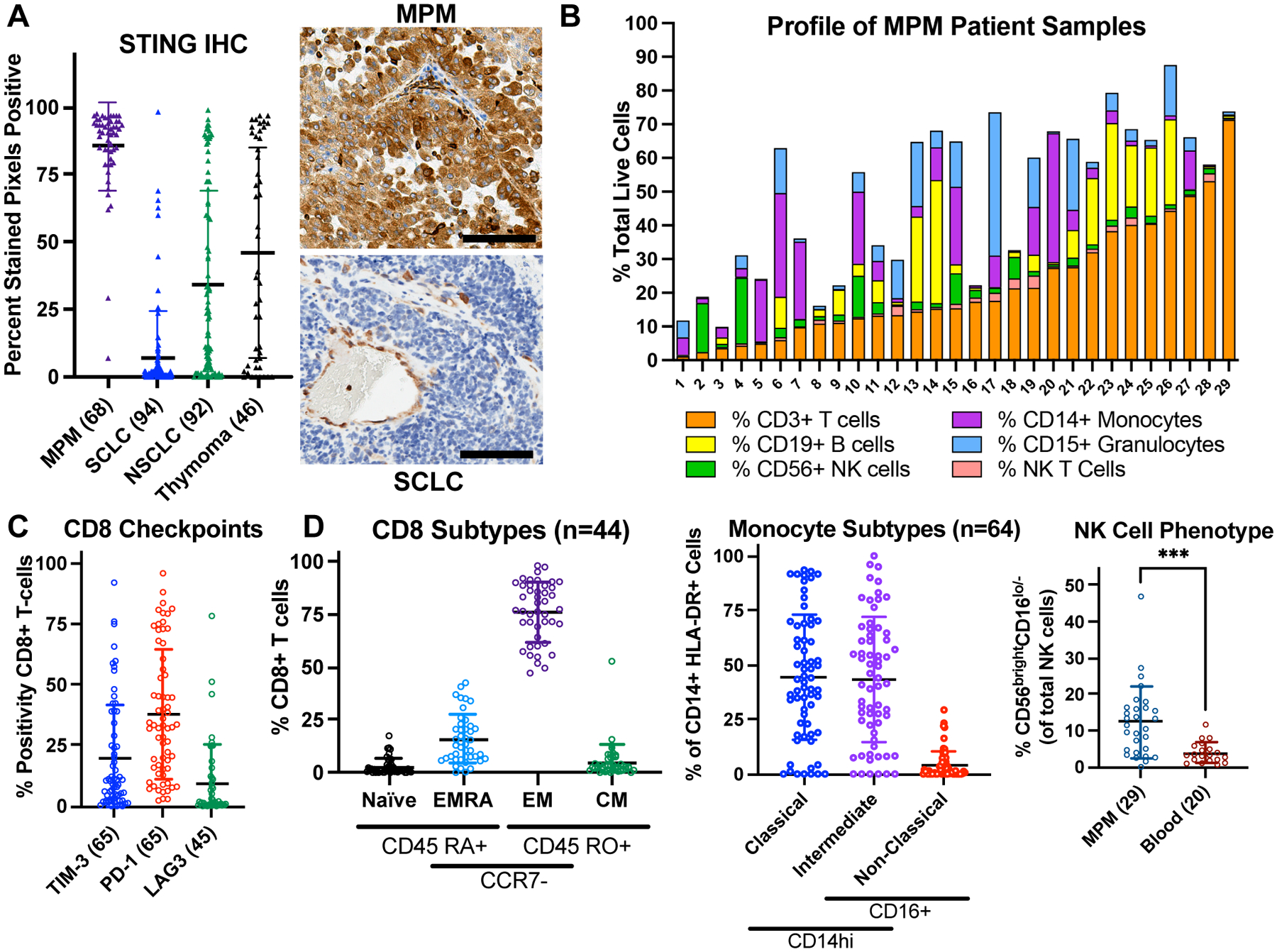
A, STING immunohistochemistry (IHC) quantified using QuPath software. MPM = malignant pleural mesothelioma; SCLC = small-cell lung carcinoma; NSCLC = non-small cell lung carcinoma. Scale bar = 100 μm. B, Immune cell flow cytometry analysis of MPM specimens (n = 29). C and D, Flow cytometry from freshly resected MPM specimens with number of samples specified. Blood for NK-cell analysis was collected from patients with head & neck squamous cell carcinoma or oral proliferative verrucous leukoplakia. Mann-Whitney test: ***p< 0.001. Flow cytometry antibody details provided in Supplementary Table 3 TIM-3 = T-cell immunoglobulin and mucin domain-containing protein 3; PD-1 = programmed cell death protein 1; LAG3 = lymphocyte activation gene 3; EMRA = effector memory re-expressing CD45 RA; EM = effector memory; CM = central memory.
Figure 2: STING agonists promote antitumor immunity in MPM.
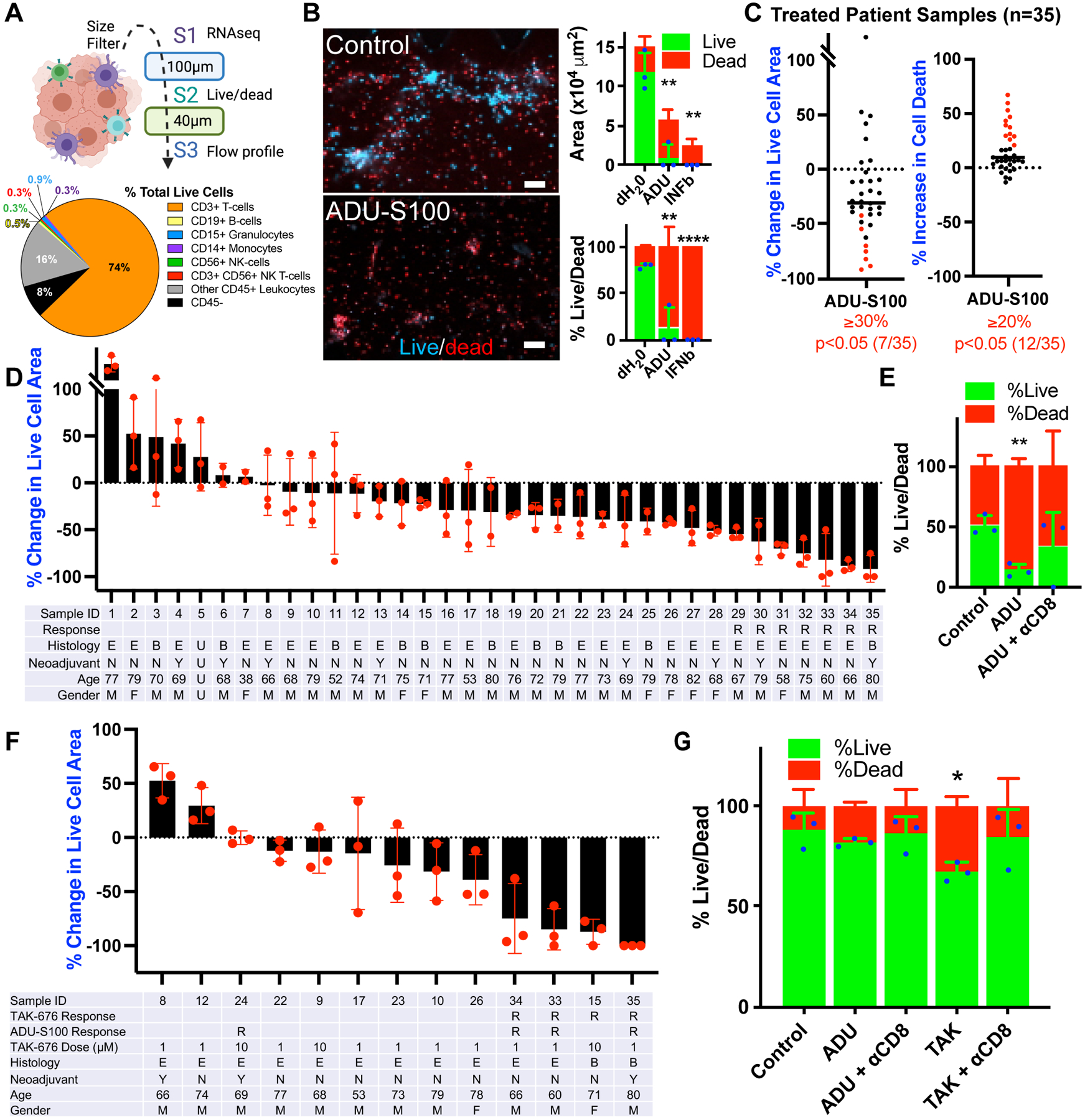
A, Schematic for generation of patient-derived organotypic spheroids (PDOTS) by size filtration and representative flow cytometry profiling (n=16) for MPM case #35. B, Hoechst/propidium iodide live/dead IF from MPM case #35 after 6-day treatment with 50 μM ADU-S100 (ADU) or dH20 and 100ng/mL IFNβ controls. Scale bars = 100 μm. Cell area and percent live/dead quantification of each stain. T-test vs. dH20 control: **p<0.01, ****p<0.0001. C, Summary of percent change in live cell area (Hoechst/acridine orange) and cell death (propidium iodide) in 35 MPM patient specimens treated for 6 days with 50 μM ADU-S100 or dH20 control. Response shown by criteria for live cell area (≥30% decrease) and ≥20% increase in cell death, with p<0.05 by t-test between treated triplicate wells, ADU-S100 vs. dH20 control. D, Waterfall plot from the 35 patient specimens treated for 6 days with 50 μM ADU-S100 or dH20 control and analyzed in C. R = response by reduced live cell area, E = epithelioid MPM, B = biphasic MPM, Y/N = yes/no neoadjuvant treatment, M/F = male/female. E, Percent live/dead for MPM case #34 after 6-day treatment with 50 μM ADU-S100 or dH20 control and CD8 blocking antibody (αCD8). T-test vs. dH20 control: **p<0.01. F, Waterfall plot from 13 MPM patient specimens treated for 6 days with 50 μM ADU-S100, TAK-676, or dH20 control. G, Percent live/dead for MPM case #26 after 6-day treatment with 50 μM ADU-S100, 1 μM TAK-676, or dH20 control and CD8 blocking antibody (αCD8). T-test vs. dH20 control: *p<0.05.
Figure 5: STING agonists enhance NK-cell therapies.
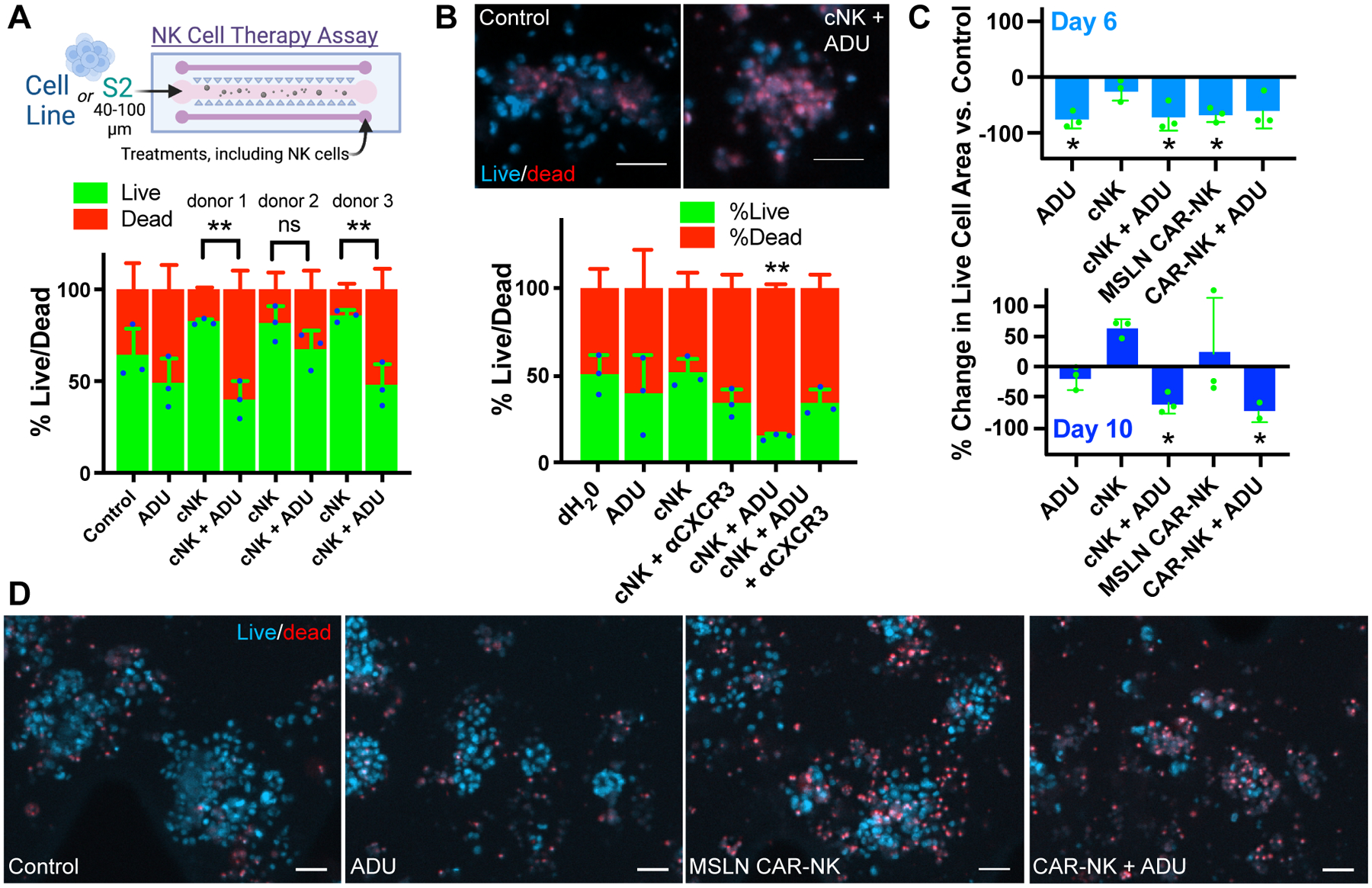
A, NK-cell therapy schematic. Quantification of Hoechst/propidium iodide live/dead IF from PDOTS sample #36 treated for 6 days with 50 μM ADU-S100 or dH20 control −/+ primary NK cells (cNK). One-way ANOVA p<0.001 with corrected pairwise comparisons: **p<0.01. B, Representative live/dead IF and quantification from sample #37 treated for 6 days with 50 μM ADU-S100 or dH20 control −/+ primary NK cells and/or 5 μg/mL CXCR3 blocking antibody (αCXCR3). T-test: **p<0.01. Scale bar = 50 μm. C, Quantification of percent change in live cell area (Hoechst IF) in PDOTS from MPM case #32 after 6- or 10-day treatment with 50 μM ADU-S100, cNK, or MSLN CAR-NK cells. One-sample t-test with expected difference of zero: *p<0.05. D, Representative live/dead IF from MPM case #32 after 10-day treatment. Scale bar = 50 μm.
Immunohistochemistry
IHC staining for STING and phospho-IRF3 was performed on the Leica Bond III automated staining platform. The antibody specific for STING (Cell Signaling Technology #13647, clone D2P2F) was run at 1:50 dilution using the Leica Biosystems Refine Detection Kit (#DS9800) with citrate antigen retrieval. The antibody specific for phospho-IRF3 (Cell Signaling Technology #29047, clone D6O1M) was run at 1:100 dilution using the Leica Biosystems Refine Detection Kit (# DS9800) with EDTA antigen retrieval (n=31). This was optimized from a range of dilutions from 1:50 to 1:200 and comparison of citrate vs. EDTA antigen retrieval on MPM cell lines treated in vitro with 50 μM ADU-S100 (Chemietek #CT-ADUS100) for 24-hours prior to paraformaldehyde fixation and paraffin embedding. STING IHC staining was quantified using the QuPath software (version 0.2.3) (21). Positive Pixel Detection analysis was used with default settings for DAB staining to detect and quantify positive pixels in each of three individual, randomly selected fields per tumor, which were then averaged. Phospho-IRF3 levels were too low to quantify systematically with this software.
Flow-cytometric immune profiling
Fresh tumors were mechanically and enzymatically disaggregated in dissociation buffer consisting of RPMI (Life Technologies 72400120) +10% fetal bovine serum (FBS; HyClone SH30088.03), 100 U/ml collagenase type IV (Life Technologies #17104019), and 50 μg/ml DNase I (Roche #10104159001). The suspension was incubated at 37°C for 45 minutes and then further mechanically dissociated. Red blood cells were removed from samples using red blood cell lysis buffer (Biolegend 420301). Samples were pelleted and then resuspended in fresh RPMI +10% FBS and strained through a 40μm filter. Cells were incubated with the Live/Dead Zombie NIR (Biolegend 423105) for 5 minutes in the dark at room temperature. Fc receptors were blocked prior to surface antibody staining using Human TruStain FcX Blocking Reagent (Biolegend 422301). Cells were stained with antibodies (see Supplementary Table 3) for 15 minutes on ice in the dark and washed 2x with PBS + 2% FBS. Cells were analyzed on a BD LSRFortessa with FACSDiva software (BD Biosciences). Data were analyzed using FlowJo software version 10.5.3. Representative dot plots and gating strategies are shown in Supplementary Fig. S1.
Patient-derived organotypic tumor spheroids (PDOTS)
PDOTS were generated as previously described (12,22). Briefly, fresh MPM tumor specimens (n=37; Supplementary Tables S1 and S2) were minced in a 15 mL falcon tube in prewarmed to 37°C full media (DMEM from Thermo Fisher Scientific #10013CV + 10% FBS) + 100 U/mL collagenase type IV (Thermo Fisher Scientific #17104019) and 50 μg/mL DNase I (Roche #10104159001) for approximately 20 minutes using sterile scissors and pipetting. Dissociated material was strained over 100-μm filter and 40-μm filters to generate S1 (>100 μm), S2 (40–100 μm), and S3 (<40 μm) spheroid fractions, which were subsequently maintained in ultralow-attachment (ULA) tissue culture plates (Corning). S1 fractions were treated with 50 μM ADU-S100 for cytokine analysis and single-cell RNA sequencing (scRNA-seq) (see below). S2 fractions were used for ex vivo culture by resuspending them in type I rat tail collagen (Corning #354236) at a concentration of 2.8 mg/mL prior to loading into the center gel region of the 3-D microfluidic culture device (AIM Biotech #DAX-1) and incubation for 40 minutes at 37°C in humidity chambers to allow for polymerization. Collagen hydrogels containing PDOTS were hydrated with media with or without indicated treatments. TAK-676 was provided by Takeda (23) and diluted in dH20. Recombinant human IFNβ (100 ng/mL; R&D Systems #8499-IF) was used as a positive control downstream of STING for STAT1 pathway activation. CD8a was blocked with 50 μg/mL InVivoMAb antibody (Bio X Cell #BE0004–2) vs. IgG control (Bio X Cell #BE0092). CXCR3 was blocked with 5 μg/mL human CXCR3 antibody (R&D MAB160).
PDOTS immunofluorescence and live/dead quantification
Dual labeling was performed by loading microfluidic devices with Nexcelom ViaStain acridine orange/propidium iodide (AO/PI) Staining Solution (Nexcelom #CS2–0106) or 10 μg/mL solution of Hoechst 33342 (Thermo Fisher Scientific #H3570) and 1 μg/mL solution of PI (Thermo Fisher Scientific #P3566). Following incubation with the dyes (20 minutes at room temperature in the dark for AO/PI or 45 minutes for Hoechst 33342/PI), images were captured using 4x objective of a Nikon Eclipse 80i fluorescence microscope equipped with automated motorized stage (Proscan), Z-stack (Prior), and Zyla 5.5 sCMOS camera (Andor). Image capture and analysis were performed using NIS-Elements AR software package version 5.00.00 64-bit. Live and dead cell quantitation was performed by measuring total cell area of each dye. For additional immunofluorescence studies, PDOTS were washed with PBS and blocked with FcR blocking reagent (Miltenyi #130–059-901) for 30 minutes at room temperature. Directly conjugated antibodies CD326 EpCAM-AlexaFluor647 (clone 9C4, BioLegend), CD45-AlexaFluor647 (HI30, BioLegend), and mesothelin-PE (clone REA1057, Miltenyi) were diluted 1:50 in 10 μg/mL solution of Hoechst 33342 in PBS and loaded into microfluidic devices for 1-hour incubation at room temperature in the dark. Spheroids were washed twice with PBS with 0.1% Tween20 followed by PBS. For viability assessment, microfluidic devices were loaded with 1:1,000 solution of calcein AM (Thermo Fisher Scientific #C34858) in PBS. For IRF3 immunofluoresecence (IF) analysis, PDOTS were treated for 3 hours with dH20 control or 50 μM ADU-S100, washed with PBS, fixed with 4% paraformaldehyde for 15 minutes, and permeabilized with 0.1% Triton-X for 10 minutes. Cell Signaling Antibody #11904 (clone D6I4C) was diluted 1:50 in PBS and incubated for 45 minutes, washed, and subsequently incubated in FITC-conjugated anti-rabbit secondary antibody (Thermo Fisher Scientific #A27034) diluted 1:100 for 30 minutes. PDOTS were washed twice with PBS containing 0.1% Tween20 and counterstained with 1μg/mL solution of Hoechst 33342. Images were captured as mentioned above for live/dead dual staining, using 20x objective.
Cytokine analysis
CXCL10 ELISA (R&D Systems DIP100) and granzyme B ELISA (R&D systems DY008) were performed according to the manufacturer’s instructions on conditioned media collected from cell culture. Cytokine analysis of conditioned media after 3 days of explant (S1) culture utilized the MSD U-PLEX Viral Combo 1 assay (Hu: K15343K-2), which was performed according to manufacturer’s instructions.
2’3’-cGAMP ELISA
The Cayman Chemical 2’3’-cGAMP ELISA kit (#501700) was used according to manufacturer’s instructions to detect levels of 2’3’-cGAMP in the supernatant of MPM cell lines. For these experiments, 3–5 ×105 cells were first plated in a 6-well plate and transfected with 1μg poly (dG:dC) (Invivogen #tlrl-pgcn) using X-tremeGENE HP DNA Transfection Reagent (Sigma Aldrich #6365787001) combined with Opti-MEM Reduced-Serum media (Thermo Fisher Scientific #31985070) for a 30 minute incubation. 2’3’-cGAMP (Invivogen #tlrl-nacga23–1) was used as a positive control.
Cell Culture
MPM cell lines were obtained in 2020 and cultured in RPMI-1640 (Thermo Fisher Scientific #11875–119) supplemented with 10% FBS. MPM cell lines were not authenticated recently. H226, H28, MSTO-211H, H2452 and H2052 were purchased from ATCC. MS428 was provided by the laboratory of Dr. William Richards at Brigham and Women’s Hospital. H2461 and H2591 were provided to Dr. Pasi Jänne by the NIH (24). JMN1B (25) and MS589 (26) were derived at BWH/DFCI and shared internally by Dr. Richards with permission. For NK-cell experiments described below, HEK-293 and Jurkat cells were purchased from ATCC. HEK293 were cultured in DMEM supplemented with 10% FBS, 1% Penicillin-Streptomycin (Thermo Fisher Scientific #15140–163), 1% L-Glutamine (Thermo Fisher #25030149) and HEPES (Thermo Fisher #1344041). Jurkat cells were cultured in RPMI-1640 supplemented with 10% FBS, 1% Penicillin-Streptavidin, 1% Glutamine and HEPES. For 3D vessel experiments detailed below using human umbilical vein endothelial cells (HUVECs; C2519AS, Lonza), cells were cultured in Vasculife (Lifeline #LL-0003). All experiments were performed before reaching 10 passages. Mycoplasma infection was regularly checked by PCR using the conditioned media derived from each cell line with primers as previously described (15).
Immunoblotting
Cells were lysed in RIPA buffer containing 1x protease inhibitors (Roche, 11–836-145–001) and phosphatase inhibitors (50 mmol/L NaF and 100 mmol/L Na3VO4; Sigma Aldrich #201154 and #450243, respectively). Immunoblotting was performed as previously described (15) using the antibodies listed in Supplementary Table 3. Secondary antibodies were from LI-COR Biosciences: IRDye 680LT Goat anti-Mouse IgG (#926–68020) and IRDye 800CW Goat anti-Rabbit IgG (#926–32211). Imaging of blots and was performed using the LI-COR Odyssey system.
Dynamic single-cell RNA sequencing and data analysis
Our previously published protocol (27) was adapted to test S1 explants from MPM PDOTS. The sample tested (#26) demonstrated baseline viability of 63% and 18-hour cytokine release in response to treatment. After 24 hours of treatment with ADU-S100 in a ULA dish, tumor spheroids were digested with trypsin in a 37°C incubator for 5 minutes to obtain single-cell suspensions. Cells were loaded onto a 10x chromium instrument (10x Genomics) per the manufacturer’s instructions. ScRNA libraries were generated using the single cell 3′ reagent kit (10x Genomics #PN-1000268) per the manufacturer’s instrctions. Quality control of the completed libraries was performed using a bioanalyzer high sensitivity DNA kit (Agilent #5067–4626) and then sequenced using the Illumina NextSeq 500 platform.
Raw sequencing reads were processed using the 10x Genomics CellRanger bioinformatics pipeline v6.0.1. The assembled matrix was then fed into the standard workflow of the R package, Seurat v4.0.4. Genes that were expressed in at least 3 cells, and only cells that expressed at least 2 genes, were kept for downstream processing. Additionally, cells expressing more than 7,000 genes and cells with more than 10% of unique molecular identifiers (UMIs) mapping to mitochondrial genes were removed from the analysis. All the samples were prepared and sequenced together on the same platform.
The filtered matrix was log-normalized using global scaling in Seurat. UMI and mitochondrial transcript content were used as regression parameters. The normalized matrix was scaled and centered gene-wise, and then underwent dimensionality reduction using principal component analysis (PCA) on the highly varying genes. After visual inspection of the PCA elbow plot, the top 10 principle components (PCs) were chosen for further analysis. Clustering was performed on the chosen PCs using the shared nearest neighbor algorithm in Seurat with default parameters.
A Uniform Manifold Approximation and Projection (UMAP) map was computed and plotted with the DimPlot module of Seurat. Cluster differential expression analysis was performed in Seurat using the FindMarkers command using the Wilcoxon rank sum test without thresholds. Contour plots overlayed onto UMAPs were generated with R package ggplot2 (28).
Cell types were identified based on comparative analysis with signatures published previously (29–32), as well as marker genes identified in this study, which were used to remove genes ubiquitously expressed across cell subpopulations. Collagen-encoding genes were added to the fibroblast signature. The list of gene signatures used for enrichment analysis is provided in Supplementary Table 4.
Isolation of tumor-infiltrating lymphocytes (TILs)
TILs were isolated from lung cancer patient specimens (n=3; Supplementary Table S1) under IRB protocol 02–180 and filtered as described above for PDOTS. The S3 fraction was expanded using T-cell growth medium (TCGM): RPMI-1640 with L-glutamine, 1% Penicillin-Streptomycin solution, 1mM Na Pyruvate (Thermo Fisher Scientific #J61840.22), 0.0375% Na Bicarbonate (Thermo Fisher Scientific #25080094), 50nM mercaptoethanol (Thermo Fisher Scientific #21985023), 10% Human AB Serum (Sigma Aldrich #H4522–100ML) and 6000U/mL IL2 (Miltenyi #130–097-746) in a 24-well plate and split 1:2 every other day over a period of 8–10 days. Upon expansion they were frozen/stored in liquid nitrogen.
Expansion and transduction of primary T cells
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors after informed consent and isolated by using Ficoll density centrifugation. Isolated PBMCs were activated with TransAct (1:100, Miltenyi #130–111-160) in complete medium (RPMI-1640 supplemented with 10% FBS, in the presence of IL2 at 10ng/ml). Two days after activation, the T cells were lentivirally transduced by spinoculation with the BCMA CAR virus (1% virus volume) in the presence of Lentiboost (1:100, Sirion Biotech). The BCMA CAR sequence has been previously described (33) and was cloned into the pHAGE lentiviral vector (a gift from the laboratory of Dr. Darrell Kotton at Boston University; Addgene plasmid #24526) and the generated plasmid was subjected to sequencing verification. For packaging and production of lentivirus particles, 293 Lenti-X packaging cells (Takara) were seeded into a 15 cm plate (8 × 106 cells/plate) for 24h, then transfected with plasmids encoding CAR, pMD.2 G encoding VSV-G envelope, and a packaging vector psPAX2 using PEI transfection reagent (Polysciences #23966). Virus supernatants were harvested at 24 hours and 48 hours after transfection, filtered through a 0.45 μm membrane, and concentrated by ultracentrifugation and stored at −80°C prior to transduction. After transduction, T cells were expanded with cytokines, IL2 (10ng/ml), IL7 (3ng/ml; #130–095-361), and IL15 (10ng/ml; Miltenyi #130–095-762), in RPMI-1640 supplemented 10% FBS, and their transduction efficiency was determined by FACS three days after transduction using a BCMA protein conjugated to PE (Acro Biosystems #BCA-HP2H2).
Expansion and transduction of primary NK cells
For experiments using unmanipulated primary NK cells, CD56+CD3− NK cells were expanded from human PBMCs (Lonza #4W-270) using the CellXVivo Human NK Cell Expansion Kit (R&D Systems #CDK015). Following 14 days of expansion, cells were transitioned to culture in CTS OpTmizer T-cell expansion media (Thermo Fisher #A1048501) supplemented with 5% human AB serum, 1% GlutaMAX (Thermo Fisher #35050061), 1% HEPES, and 1% Penicillin-Streptomycin in the presence of IL2 (200 U/mL for flow cytometry experiments, 500U/mL for killing experiments including PDOTS).
For experiments using transduced and control-processed primary NK cells, the NK cells were extracted from whole blood leukapheresis using RosetteSep (StemCell technologies) and Ficoll-Paque density gradient centrifugation under the approved Crimson Study protocol T0197. The isolated NK cells were inspected for purity and cultured for 2 days in RPMI supplemented with 10% heat-inactivated (HI)-FBS (Thermo Fisher #10082147), 1% Penicillin-Streptomycin, 2 mM L-Glutamine and HEPES in the presence of IL15 (1 ng/mL). Isolated NK cells were subsequently transduced as below or cultured in NK MACs media (Miltenyi #130–114-429) supplemented with 5% human serum and 1% v/v Penicillin-Streptomycin in the presence of IL2 (500 U/mL).
An anti-mesothelin CAR was custom synthesized by Integrated DNA Technologies in a pHIV backbone with the mesothelin-specific ScFv derived from YP218 antibody, followed by transmembrane domain and co-stimulatory domains (4–1BB and CD3ζ) as previously described (34,35) (Supplementary Fig. S2). The construct also contains EGFP fragment separated from the CAR fragment by self-cleaving P2A. The CAR gene construct was packaged into BaEV-pseudotyped lentiviral system by transfecting HEK-293 cells with pCMV-BaEV, pCMV-Δ8.9 and pAdv plasmids as previously described (36). The viral particles were titrated using Jurkat cells. Assuming a multiplicity of infection (MOI) of 1 for Jurkat cells, the viral titers were calculated to transduce NK cells with MOI of 10. NK cells were transduced using Retronectin (Takara Bio #T100B) and vectofusin-1 (Miltenyi #130–111-163) followed by spinfection +/− active lentivirus (cNK control without virus) two days after extraction and subsequently cultured in NK MACs media supplemented with 5% human serum and 1% Penicillin-Streptomycin in the presence of IL2 (500 U/mL; Miltenyi). The percentage of NK cells expressing CAR was determined via flow cytometric analysis of GFP and surface expression of ScFv using APC-conjugated Human agglutinin (HA).
Immune cell toxicity assays
For flow cytometry immune cell toxicity assays, primary NK cells and TILs were seeded at 200,000 cells per well (NK or TILs alone or 1:1 with 100,000 cells of each type) in a 96-well plate alone or in co-culture and treated with 10 μM or 50 μM ADU-S100 or dH20 control with or without IL2 (Miltenyi or PeproTech) at the indicated concentrations for 72 hours. Following treatment, samples were stained with antibodies specific for CD45, CD3, CD4, CD8, and CD56 (Supplementary Table 3), as well as Zombie Green live/dead (Biolegend #423111) and analyzed by flow cytometry as described above. Data were analyzed using FlowJo software version 10.5.3. As an orthogonal measure of viability, CellTiter-Glo was performed on primary T-cells and NK cells as below.
CellTiter-Glo luminescent cell viability assay
Cell viability was assessed using the CellTiter-Glo Luminescent Cell Viability assay (Promega, G7571) according to the manufacturer’s instructions. For untransduced primary T cells and BCMA CAR T cells, 25,000 cells per well were seeded in a 96-well plate and treated with ADU-S100 or dH2O as control for 24 hours at the indicated concentrations. For NK cells, 25,000 cells per well were seeded and treated with ADU-S100 or dH2O as control for 24 hours at the indicated concentrations. For MPM cell lines, 10,000 cells per well (MS428) or 12,500 cells per well (H2461, H2591) were seeded and treated with 50μM ADU-S100 or media as control for indicated times. All conditions were tested in triplicate and plates were read on a Tecan Infinite Mplex Microplate Reader.
Autophagy Staining
Autophagy was assessed by vacuole staining to identify autophagolysosomes using the CYTO-ID Autophagy detection kit 2.0 (Enzo ENZ-51031–0050) according to the manufacturer’s instructions. Briefly, 5 × 105 isolated primary NK cells or TILs were incubated in T-cell growth media (TCGM) with 500U/mL IL2, which was refreshed every time the media was changed to ensure proper growth and selection. Chloroquine (CLQ) from the kit (Enzo 51005-CLQ) was used starting at the recommended initial dose of 10μM compared with DMSO control. After 24 hours the media was changed, and the cells were treated for another 24 hours with CLQ + 10μM ADU-S100. The media was collected, and flow cytometry was performed after staining following manufacturer’s instructions with CYTO-ID Green Detection Reagent 2.
NK cell–killing assay
Target cells (MPM cell lines) were detached via trypsinization, labelled with CellTrace Violet (CTV; Thermo Fisher Scientific #C34571) and then seeded in a 96-well plate at a cell density of 25,000 cells per well. Target cells were allowed to adhere for 12–16 hours and NK or mesothelin-targeted CAR-NK cells were then added at different effector to target (E:T) ratios (1:1, 2:1, 5:1 and 10:1) with or without ADU-S100 (50 μM). After 6 hours of co-culture, the cells were harvested and incubated with an antibody for the apoptosis marker Annexin V (PE; Biolegend #640947) and the live/dead stain 7-AAD (Biolegend #420404). Cells were analyzed on a BD LSRFortessa with FACSDiva software (BD Biosciences). Data were analyzed using FlowJo software version 10.5.3. The apoptotic cells were evaluated by gating on the CTV+ population and represented as percentage live or dead (late apoptotic) cells. Apoptotic cell analysis was conducted using NK cells extracted from as many as 4 different healthy donors per target MPM cell line to incorporate baseline donor variability.
NK cell–infiltration assay
Immune cell infiltration was assessed as previously described (15,37). Briefly, mesothelioma cancer cell spheroids (H2591, H2461, H226) were generated by seeding 5 × 105 cells in suspension in a ULA dish for 24 hours. H226 cells were treated with 50 μM ADU-S100 during the final 6 hours of spheroid formation to establish a cytokine gradient. Samples were then pelleted and resuspended in type I rat tail collagen at a concentration of 2.5 mg/mL following the addition of 10x PBS with phenol red and pH adjustment using NaOH. pH 7.0–7.5 was confirmed using PANPEHA Whatman paper (Sigma-Aldrich). Cells and collagen were kept on ice to prevent polymerization. The spheroid–collagen suspension was then injected into the central gel region of the 3D DAX-1 microfluidic cell culture chip (AIM Biotech). Microfluidic devices were utilized as previously described (22), with a central region containing the cell–collagen mixture in a 3D microenviroment (3 × 104 cells H2591 and H2461, 2 × 104 cells H226 in 10 μL), flanked by 2 media channels. After injection, collagen hydrogels containing cells were incubated for 40 minutes at 37°C in humidity chambers, then hydrated with culture media, with labeled primary NK cells (E:T ratio 2:1) added to one of the side channels. Primary NK cells were labeled with Cell Tracker Red (Thermo Fisher Scientific #C34552) following the manufacturer’s instructions. After 96 hours of incubation, viability staining of cancer cell spheroids and infiltrated immune cells was performed (20-minute incubation with 1μg/mL solution of Propidium Iodide). For the experiment with the CXCR3 blocking antibody (R&D MAB160), NK cells were pre-treated for 30 minutes prior to loading.
For quantification, images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and CoolSNAP CCD camera (Roper Scientific). Image capture and analysis was performed using NIS-Elements AR software package. Whole device images were achieved by stitching in multiple captures. Quantification of immune cell infiltration into the 3D tumor microenvironment was performed by measuring the total cell area of cell tracker dye in the entire gel region. For the experiment with the CXCR3 blocking antibody, staining was quantified in a square region in the center of the channel to focus on effects of CXCR3 ligands released by tumor cells. Percent dead cell quantification was performed as described above (see PDOTS immunofluorescence and live/dead quantification).
3D vascular model
To generate the tumor-vascular model, H226 spheroids were mixed with collagen rat tail hydrogel (2.5 mg/ml) and injected into the center gel region of the 3D microfluidic chamber (10–15 μL per each microfluidic chamber). After incubation for 30 minutes at 37°C in sterile humidity chambers, the side wall of one flanked channel (media channel) was coated with a 150 μg/ml collagen solution in PBS to allow for better adhesion of ECs to the channel. After 15 minutes, the channel was washed once with media. To create the 3D vessel, 25 μL cell suspension of 3×106 cells/ml HUVECs were injected in the media channel coated with collagen. The channel was rotated twice to create a confluent hollow-lumen 3D vessel. To allow the cells to attach to the media–gel interface and form a monolayer, the chip was incubated with cells face down for 15 minutes. Next, 50 μL cell suspension was reinjected, and the chip was flipped to cover the upper part of the 3D vascular channel. After 90 minutes of incubation in the humidity chamber at 37°C, cell culture media was gently added to both channels and further incubated to form a confluent monolayer. After vessel formation, NK cells (labelled with Cell Tracker Red) were added to the 3D vessel at 2:1 E:T ratio. STING agonists (ADU-S100 and TAK-676) were added to the fluidic channel opposite the vascular barrier. NK-cell migration +/− vessel was quantified at 24 hours. The 3D vascular channels were rinsed in PBS and fixed with 4% PFA for 15 minutes at room temperature. Cell membranes were permeabilized with 0.1% Triton X-100 for 5 min at room temperature and washed twice with PBS. HUVEC cells were stained for F-actin with green phalloidin (Thermo Fisher Scientific A12379) and Hoechst 33342. Images were captured on a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior) and CoolSNAP CCD camera (Roper Scientific).
Statistical Analysis
Statistical significance was assessed using unpaired two-tailed Student t-test for pairwise comparisons, one-sample t-test against an expected value of 0% change or 100% control, or one-way ANOVA followed by Tukey post hoc test. Kruskal Wallis global test followed by Dunn’s multiple comparisons post hoc test was used for non-parametric analysis of IHC scores and normalized mRNA expression in cell line data obtained from the Cancer Cell Line Encyclopedia at the Broad Institute: https://sites.broadinstitute.org/ccle/ (17 MPM cell lines, 51 SCLC, 137 NSCLC). P values less than 0.05 were considered significant. Asterisks used to indicate significance correspond with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Columns and horizontal lines added to data points represent mean ± SD error bars. In one-way ANOVA followed by post hoc tests, we showed asterisks only in pairs of our interest. GraphPad Prism (version 9.2.0) was used for all statistical analysis.
Data Availability
The datasets generated and/or analyzed during the current study are available within the article and its supplementary data files or are available from Dr. Barbie on reasonable request. ScRNA-seq data are available in Gene Expression Omnibus (accession # GSE201925).
Results
STING is primed for activation in MPM
To identify human tumor histotypes with intact STING, we performed IHC profiling of 300 archival samples from diverse thoracic malignancies (Fig. 1A). Among the malignancies evaluated, MPM expressed the highest levels of STING. MPM arises from the serosal lining of the lungs and carries a poor prognosis despite incorporation of combined checkpoint immunotherapy into standard care (38). MPM demonstrated near-universal high expression of STING protein in tumor and stroma cells, in contrast to NSCLC, thymoma, and SCLC (37,39) (Fig. 1A, Supplementary Fig. S3A). STING was also highly expressed in benign pleura, consistent with its baseline upregulation in mesothelial cell types (Supplementary Fig. S3B). Flow cytometry–based immune profiling of a large panel of resected MPM specimens demonstrated robust immune infiltration in most tumors, but with features of exhaustion across multiple immune-cell subsets including heterogeneous expression of the checkpoint proteins PD-1, TIM-3, and LAG-3 (Fig. 1B–D; Supplementary Fig. S3D and S3E) (40). T-cell characterization revealed terminal differentiation consistent with exhaustion; monocyte/macrophage subtyping showed an abundance of intermediate cells; NK-cell characterization showed diminished cytotoxic capacity (increased CD56 bright/CD16 low compared with circulating NK cells; Fig. 1D). Thus, MPM express high levels of STING and demonstrate an inflamed but exhausted TIME.
We confirmed that MPM-derived cell lines expressed high levels of STING protein, but that both cell lines and tumors failed to exhibit baseline cGAS-STING pathway activation, as measured by phospho-IRF3, CXCL10 and IFIT1 expression, and secretion of 2’3’-cGAMP (Supplementary Figs. S3 and S4). Despite lacking baseline STING pathway activation, multiple MPM cell lines treated with the clinical STING agonist ADU-S100 (1,3) exhibited potent pathway activation, secreting high levels of CXCL10 (Supplementary Fig. S4). MPM cell lines with minimal response to clinical STING agonists in vitro exhibited reduced IRF3 transcription factor expression (Supplementary Fig. S4). Overall, however, the high levels of inactive basal STING and pronounced induction of CXCL10 release by STING agonism suggested that STING signaling is primed to respond in MPM. In addition, STING activation in MPM cell lines cultured in vitro did not cause cytotoxicity, suggesting a contribution from the TIME (Supplementary Fig. S4F). We therefore analyzed STING agonism in human tumor specimens using freshly resected MPM tumor explant models that retain the associated TIME (Fig. 2A) (12). After processing, 40–100 μm (S2) PDOTS were suspended in collagen and treated for 6 days to assess response by live/dead immunofluorescence and cytokine production (12) (Fig. 2A). We confirmed ADU-S100 induced CXCL10 release and robust killing of PDOTS after 6 days of treatment in specific samples (Fig. 2B; Supplementary Fig. S5). Overall, treatment of 35 patient specimens with ADU-S100 showed statistically significant cell death >20% above control (p<0.05) in 12 of the 35 (34%) patient specimens, with 7 specimens approximating clinical criteria for response with >30% decrease in cell area, p<0.05 (Fig. 2C and D; Supplementary Fig. S5). There was a non-significant trend toward higher response to STING agonism in PDOTs in patients who received neoadjuvant chemotherapy (HR 1.4, 95% CI 0.23–7.27, Chi Square p=0.72) and this response did not correlate with histology (epithelioid and biphasic specimens were tested), age, or gender (Supplementary Tables 1 and 2). We observed a potential correlation between tumor CD8 abundance and treatment response to ADU-S100 ex vivo (R2=0.35, p<0.05; Supplementary Fig. S6A), prompting us to test the impact of CD8 blockade. We found that anti-CD8 treatment partially rescued ADU-S100 cytotoxicity in three patient specimens (Fig. 2E; Supplementary Fig. S6A and S6B). Furthermore, treatment with a second clinical-stage next generation systemic STING agonist, TAK-676 (23), also showed activity ex vivo, with responses seen in 4 of 13 (31%) patient specimens (Fig. 2F), blunted by anti-CD8 treatment (Fig. 2G). These results motivated development of a higher resolution approach to further understand the impact of STING agonism on different cells within the TIME.
Dynamic scRNAseq of MPM explants
We adapted methodology to conduct dynamic scRNA-seq (27) following 24-hours of STING agonist treatment, focusing on a specimen (#26) that exhibited modest, dose-dependent killing in response to ADU-S100, which was rescued by CD8 blockade (Fig. 3; Supplementary Figs. S5–S7). Flow cytometry profiling prior to treatment demonstrated an average percentage of T cells and an above average monocyte/macrophage population (Supplementary Fig. S6C and Fig. 1B). We used >100 μm tumor fragments suspended in media for this short-term scRNA-seq analysis, confirming that size filtration did not change the leukocyte composition of each fraction (12) (Supplementary Fig. S6D). IRF3 immunofluorescence also showed nuclear translocation following ADU-S100 treatment, confirming effective STING activation in PDOTS (Supplementary Fig. S6E). UMAP clustering showed broad representation of tumor cell, fibroblast, and immune cell populations (Fig. 3A, Supplementary Fig. S7). ADU-S100 strongly induced expression of CXCR3 ligands (CXCL9, CXCL10, CXCL11) primarily in a subset of mesothelin (MSLN)-positive tumor cells (cluster 1) and cancer-associated fibroblasts (CAFs; clusters 4, 5), as compared with myeloid cell populations (clusters 7, 8; Fig. 3B and C). This analysis also revealed potent and unique STING agonist induction of IL33 expression in CAFs, whereas other IFN-stimulated genes (ISGs) such as IFIT1 exhibited more widespread expression across cell populations, confirming broad target engagement (Fig. 3C). Differential expression analysis showed that the subset of MPM cells most highly expressing CXCR3 ligands in response to ADU-S100 (cluster 1) also displayed increased expression of numerous ISGs relative to other MPM cells (clusters 0, 3) and downregulated TGFB1 expression (Supplementary Fig. S5C, Supplementary Table 5. We identified multiple granzyme- and perforin-positive T cells and NK cells within cluster 2 (Supplementary Fig. S5D), consistent with the impact of CD8 blockade and potential contribution of T and/or NK cell–mediated killing. However, analysis of the abundance of cells originating from each individual sample (contour and fraction plots in Fig. 3D, Supplementary Fig. S8) demonstrated depletion of CD8+ cell populations, including Tregs, following high-dose STING agonist treatment, contrasting with increased STING agonist response in tumor-cell cluster 1. Analysis of NK-cell ligands in tumor cells showed ADU-S100 dose-dependent increases in expression of HLA-A/B/C (inhibitory) coupled with increased NECTIN2 (CD112; activating) alongside decreased expression of the predominantly inhibitory ligand PVR (CD155) (41) (Supplementary Fig. S8B). These data reveal a prominent role for tumor cells and CAFs as targets of STING agonism, promoting release of T- and NK-cell chemotactic factors and alteration of tumor/CAF cell state. Yet they are also consistent with reports that excess STING activity can be toxic to T cells (4–6), suggesting that STING-mediated enhancement of tumor-cell release of CXCR3-binding chemokines may be countered by cytotoxicity in immune effector cells.
Figure 3: STING agonists activate tumor cells and fibroblasts.
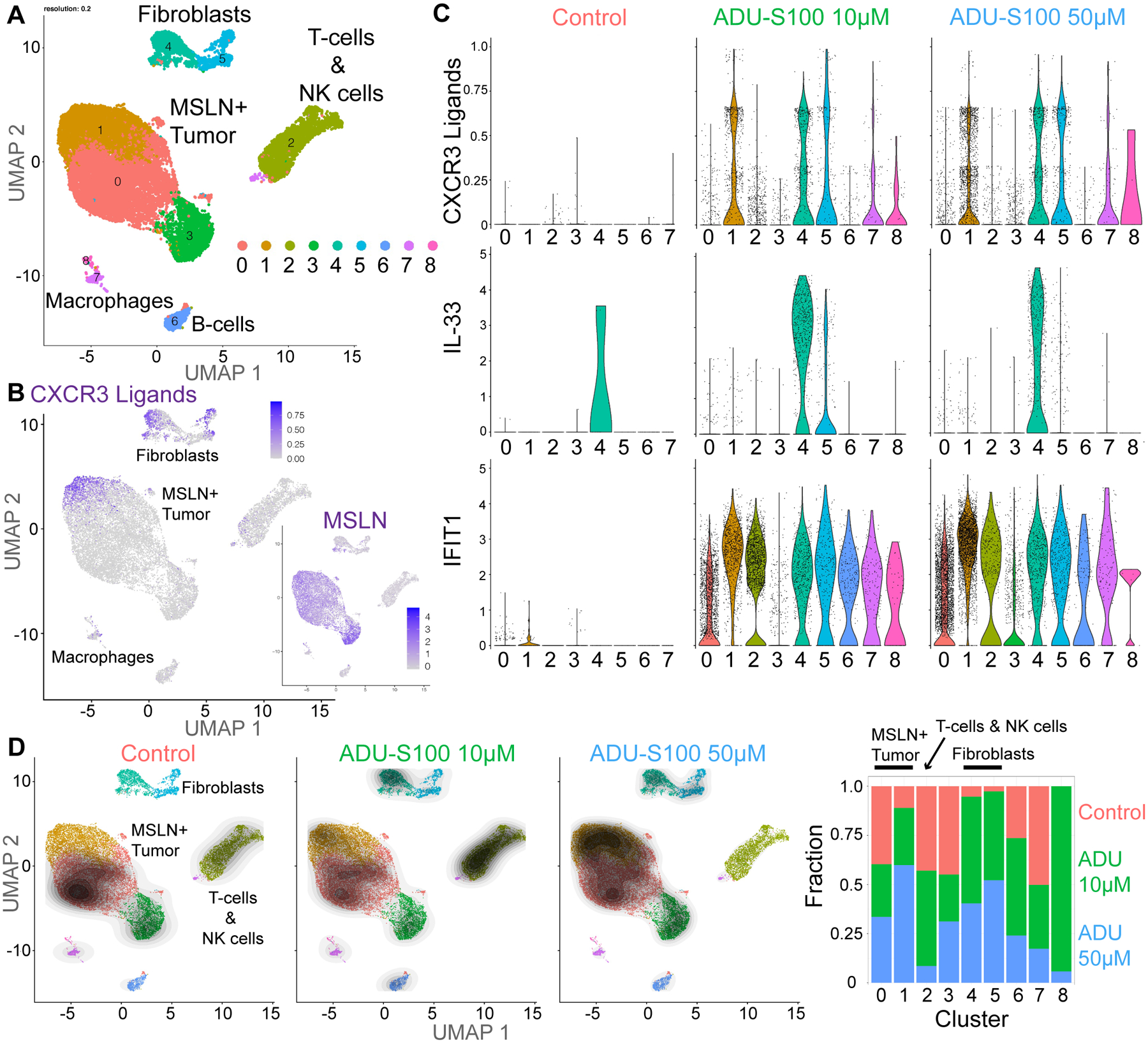
A, Combined UMAP plots from broad clustering of scRNA-seq of MPM case #26 after 24 hours of treatment with dH20 control, 10 μM ADU-S100 or 50 μM ADU-S100. B, Combined UMAP plot for CXCR3 ligands (CXCL9/CXCL10/CXCL11) and mesothelin (MSLN). C, Violin plots for select ISG transcripts from combined broad clustering split by treatment condition/dose of ADU-S100. D, UMAP plots from combined samples overlayed with contour plots showing the density of cells in each individual sample, normalized to number of cells per sample. Fraction bar graph for each cluster by treatment, normalized to number of cells per sample.
STING agonists are toxic to human T cells
To explore the idea that STING agonists are cytotoxic to T cells, we evaluated STING-induced cytotoxicity in T cells (4–6) using our models, as well as cytotoxicity in other immune cell types. ADU-S100 treatment, in contrast to downstream IFNβ exposure, was cytotoxic to T cells as measured by flow cytometry in MPM tumor explants (Fig. 4A, Supplementary Fig. S9A). T cells purified from peripheral blood with or without expression of a BCMA CAR also demonstrated dose-dependent cytotoxicity after STING agonist treatment (Fig. 4B, Supplementary Fig. S9B), likely limiting the efficiacy of combining STING agonists with CAR T cells, as recently proposed (42,43).
Figure 4: STING agonists are toxic to T cells but not NK cells.
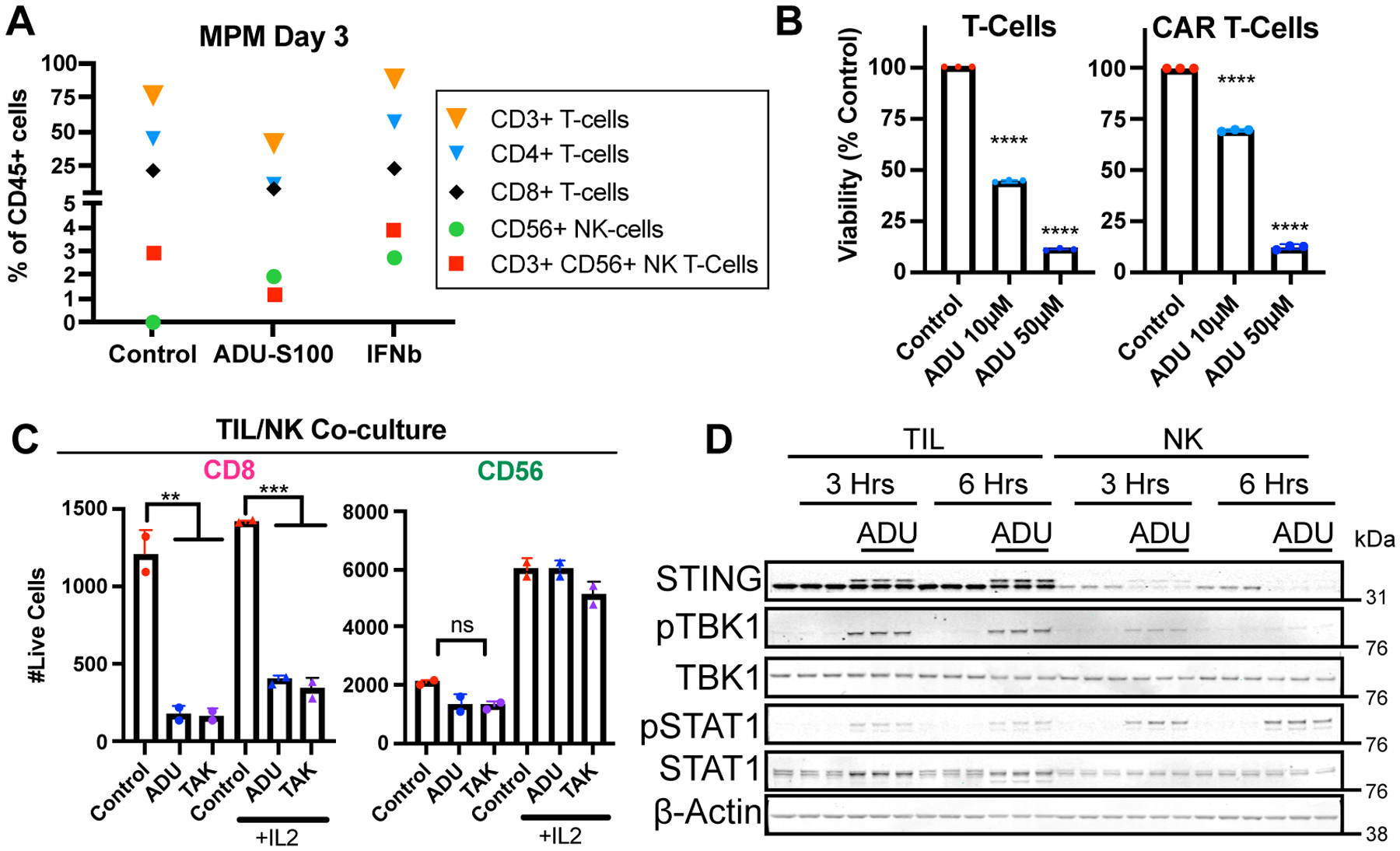
A, Immune flow cytometry in MPM case #12 after 3-day treatment with dH20 control, 50 μM ADU-S100, or 100ng/mL IFNβ. B, Cell-titer glow proliferation after 24-hours of treatment of MPM case #12 with dH20 control, 50 μM ADU-S100, or 100ng/mL IFNβ in the presence of primary T cells or BCMA CAR T cells. T-test vs. dH20 control: ****p<0.0001. C, Flow cytometry after 72-hour treatment of MPM case #12 with 50 μM ADU-S100, 10 μM TAK-676, or dH20 control +/− 200 U/mL IL2 in the presence of TILs or NK cells, gating for live cells out of 10,000 total events expressing CD8 or CD56. Batch 3 primary NK cells expanded from PBMCs and batch 3 TILs. One-way ANOVA p<0.01 with corrected pairwise comparisons: **p<0.01, ***p<0.001. D, Western blot for STING, phospho-TBK1 (pTBK1), TBK1, phospho-STAT1 (pSTAT1), STAT1, and beta-actin loading control in TILs and NK cells from C treated for 3 or 6 hours with 50 μM ADU-S100 or dH20 control in triplicate.
Although NKT cells were sensitive to STING agonism (Fig. 4A), human NK cells showed no significant cytotoxicity from STING agonist treatment, regardless of culture in IL2 or co-culture with TILs (Figs. 4A and C; Supplementary Fig. S9A–D). These findings held across human tumors and expanded primary NK cells, even after 72 hours of high dose ADU-S100 or TAK-676 exposure. We noted that NK cells principally rely on metabolism via oxidative phosphorylation (44) requiring ongoing autophagic flux (45), whereas T cells depend on glycolysis and tolerate defective autophagy (46). Consistent with their elevated autophagic flux following ADU-S100 treatment (Supplementary Fig. S9E), levels of STING protein were lower in NK cells and STING was rapidly degraded within 3–6 hours of STING agonism (Fig. 4D, Supplementary Figs. S9F and S9G). In contrast, T-cell STING was phosphorylated and activated by ADU-S100, but minimally degraded (Fig. 4D; Supplementary Fig. S7F). We confirmed autophagy-dependent STING recycling in NK cells, since treatment with CLQ, which blocks autophagic flux by inhibiting autophagosome fusion with the lysosome (47), prevented ADU-S100–induced STING degradation (Supplementary Fig. S9G). Thus, whereas STING agonist treatment causes effector T-cell cytotoxicity, primary NK cells remain largely unscathed.
STING agonists enhance NK-cell therapies
NK cells are generally low in number in MPM specimens (Fig. 1B), and also potentially restrained by inhibitory signals on tumor cells such as MHC class I, which may increase following STING agonist treatment (Supplementary Fig. S8B). We therefore examined whether STING agonism combined with adoptive transfer of primary or engineered NK cells might represent a promising therapeutic strategy by coupling tumor-cell release of CXCR3-binding chemokines with an effector-cell type resistant to STING agonist cytotoxicity. Whereas addition of primary NK cells alone to the treatment channel of microfluidic devices failed to kill MPM PDOTS, combined treatment with ADU-S100 significantly enhanced primary NK-cell responses using cells from 2 out of 3 donors (Fig. 5A, Supplementary Fig. S10A). Moreover, ADU-S100–mediated enhancement of NK cell–mediated cytotoxicity in MPM PDOTS was impaired by CXCR3 blockade (Fig. 5B). To further overcome a potentially inhibitory role of MHC class I, we next focused on MSLN-targeted CAR strategies being developed clinically for MPM (38), utilizing NK cells instead of T cells as an alternative vector for the CAR. Using an MPM specimen brightly positive for MSLN, we found that anti-MSLN CAR NK cells significantly augmented ADU-S100 activity at 6 days in a PDOTS sample that was minimally responsive to ADU-S100 treatment alone (Supplementary Fig. S10B). Furthermore, combined addition of NK and especially CAR NK cell therapy with ADU-S100 promoted deep growth suppression of MLSN+ PDOTS over time in culture, contrasting with day 10 rebound that occurred following single agent ADU-S100 therapy (Fig. 5C; Supplementary Fig. S10C). These data confirm that continuous STING agonist exposure is not toxic to NK-cell therapies and suggest that it may enhance activity, especially in combination with anti-MSLN CAR NK cells.
To isolate the role of tumor cells and further validate these findings, we used MPM cell lines that highly express STING and secrete CXCL10 over time during STING agonist treatment (H2591, H226, MS428) or uniquely lack STING expression and do not respond to STING agonism (H2461; Supplementary Figs. S4A and S11A) and compared NK-cell migration and killing −/+ ADU-S100 treatment in vitro (Fig. 6; Supplementary Figs. S11 and S12). STING agonism enhanced granzyme release by NK cells (Supplementary Fig. S11B) and killing of tumor cells (Fig. 6A) only in co-culture with MPM cells expressing STING (Supplementary Figs. S11C and S12C). These findings were consistent across E:T ratios (Supplementary Fig. S11C and S12B), in 2D and 3D culture (Fig. 6), and varied somewhat with NK-cell donors, like our experiments using patient specimens (Figs. 5 and 6; Supplementary Figs. S10, S11C, and S12C). STING agonist–induced increases in NK-cell migration toward tumor cells were rescued by treatment with a CXCR3 blocking antibody (Fig. 6B, Supplementary Fig. S11E), similar to our results for NK cell–mediated cytotoxicity in MPM patient specimens (Fig. 5B). To model NK-cell migration across a vascular barrier, we cultured HUVEC in 3D to form a vessel before assessing physiologic NK-cell migration out of the vessel and through collagen to reach MPM tumor cell lines (Supplementary Fig. S11F). Both ADU-S100 and especially TAK-676 enhanced NK-cell migration in the presence and absence of the vascular barrier, with expected decreases in total migration through the vessel (Fig. 6C).
Figure 6: STING agonists enhance adoptive NK-cell migration and killing.

A, Representative overlayed IF and brightfield images of labeled primary NK cells (blue) migrating toward H226 MPM cells with propidium iodide dead stain (red) after 4-day treatment with 50 μM ADU-S100 or dH20 control. Scale bar = 100 μm. Quantification of triplicate NK-cell migration (T-test: *p<0.05) and cell death (one-way ANOVA p<0.01 with corrected pairwise comparisons: *p<0.05). B Quantification of triplicate NK-cell migration towards H226, focused on the center of the chamber after 4-day treatment with 50 μM ADU-S100 +/− 5 μg/mL CXCR3 blocking antibody (αCXCR3). T-test: *p<0.05. C, Representative immunofluorescence modeling NK-cell migration through vasculature over 24-hours of STING agonist treatment (ADU-S100 or TAK-676) to reach H226 tumor cells. Scale bar = 200 μm. Quantification of triplicate migration +/− vasculature during 24-hour treatment with 50 μM ADU-S100, 1 μM TAK-676, or dH20 control. T-test: **p<0.01, ***p<0.001. D, Flow cytometry for annexin V and live/dead with H2591 MPM cells in co-culture with control NK (cNK) or MSLN CAR-NK cells (E:T 2:1) following 6-hour treatment with 50 μM ADU-S100 or dH20 control. Quantification of four NK-cell donors, graphed as percent change with ADU-S100 vs. dH20 control treatment. One-sample t-test with expected difference of zero: *p<0.05, **p<0.01.
Finally, since our PDOTS data suggested that MSLN CAR construct expression could enhance adoptive NK-cell therapy in MPM when combined with STING agonists (Fig. 5C), we evaluated this treatment combination in vitro to assess cytotoxicity. Anti-MSLN CAR expression further enhanced in vitro NK cell–mediated killing and combined with ADU-S100 treatment to cause the most tumor cell death (Fig. 6D and Supplementary Fig. S12). Taken together, these data reveal that STING agonism in STING+ human tumor models promotes release of CXCR3 ligands from tumor cells, enhances NK-cell recruitment and cytotoxicity, and may have potent combinatorial activity with NK-cell therapies.
Discussion
Evaluating human tumors in short-term cultures that preserve the TIME can overcome some of the limitations of mouse models, patient-derived xenografts, and passaged organoids to potentially inform clinical trials of next-generation immunotherapy combinations including cell therapies. Here we performed dynamic scRNA-seq of ADU-S100–treated human tumor explants to dissect the mechanism of action of a clinical stage STING agonist. We showed that STING agonism engages its target in most cells of the TIME, but principally drives release of CXCR3-binding chemokines by tumor cells and cancer-associated fibroblasts, while causing T-cell cytotoxicity. Blunting of effector T-cell activity is an unexpected consequence that could contribute to the disappointing clinical activity of STING agonists to date in humans. However, our studies reveal that this drawback can be overcome with the addition of NK-cell therapies (48), which benefit from STING agonist enhancement of NK-cell migration and killing.
More generally, available data from mouse models and clinical trials of injectable STING agonists support a complex interplay of STING activation in the TIME. Indeed, cell types other than CD8+ T cells such as monocytes and NK cells could be involved in the infrequent clinical responses to STING agonists reported in patients (49,50). Moreover, work in syngeneic mouse models has uncovered an important role for NK cells in tumor control mediated by the endogenous STING agonist ligand 2’3’-cGAMP (8,9). These data implicate NK cells in murine STING agonist response in vivo, which is otherwise difficult to model using artificial humanized mouse xenografts. In a related manuscript describing the preclinical activity of TAK-676, we observe enhanced trafficking and activation of NK cells following systemic TAK-676 administration in murine models (23). TAK-676 treatment was also especially potent at overcoming a human vascular barrier in our ex vivo model. These data are consistent with work from our group and others demonstrating that cyclic dinucleotides (CDNs) alter vascular permeability and upregulate adhesion molecules that can facilitate NK-cell extravasation (51), and further highlight the translational potential of TAK-676 and other clinical STING agonists to facilitate intra-tumoral trafficking of NK-cell therapies across vascular barriers.
Adding to the complexity of injectable STING agonist clinical trials is a potential threshold effect for cytokine release whereby tumor-cell STING activation crosses from metastasis promoting (10,52) to immune rejection. In MPM, we observe minimal baseline phosphorylation of downstream IRF3 in patient specimens and negligible extracellular 2’3’-cGAMP released from cell lines, suggesting potentially low contribution from basal cGAS-STING signaling to the observed immune exhaustion. But our data suggest instead that elevated tumor-cell STING expression in MPM creates a particular vulnerability to therapeutic STING agonism, especially when coupled with NK-cell therapy. This vulnerability could also extend to other tumor types with high basal STING expression, or to STING-silenced tumors treated with epigenetic inhibitors (53).
Clinical development of STING agonists is further limited by the narrow therapeutic window for injectable agents, which are rapidly cleared (49,50). Although novel slow-release and systemic formulations of STING agonists could solve some of these issues (3,23), our data indicate that constant exposure is likely to kill endogenous effector T cells, and also limit combinations with adoptively transferred transgenic TCR T or CAR T–cell therapies (42,43). Instead, our findings that NK cells are resistant to constant high-dose STING agonist exposure, and in fact are activated and recruited to kill MPM cells, support this novel immunobiology and provide a combinatorial approach with NK-cell therapies to develop clinically. We also observed potential STING agonist toxicity in Tregs, which should be validated in future studies, and this may contribute to the effects of NK cell–based immunotherapies in combination with STING agonists. Furthermore, we show that the benefits of adding a STING agonist to NK-cell therapies may not necessarily depend on the CAR construct, allowing for combinations with a variety of emerging NK effector cells (48) to be tested in future clinical trials. Treatments to enhance native NK-cell activation could also be effective in combination with STING agonists. Interestingly, although our scRNA-seq data do show upregulation of MHC class I, which is inhibitory to NK cells, they also reveal specific modulation of CD112 and CD155 that could converge to activate NK cells, especially when coupled with adoptive NK-cell therapies.
Timing and sequencing of combination immune therapies remain critical, as burst-dose STING agonism (perhaps alongside NK-cell infusion) could prevent T-cell cytotoxicity and allow for later cross-priming of T cells via NK cell to dendritic cell to T-cell crosstalk that enhances antitumor immunity. Alternatively, adoptive T-cell therapies could be engineered to resist STING agonist cytotoxicity, either by restoring effective autophagy or inhibiting STING pathway components. Indeed, we have previously reported that a potent/specific TBK1 inhibitor activates T cells on its own (12), although whether this would translate to improved therapeutic efficacy in combination with STING agonists is unclear.
Combination immunotherapy remains challenging to translate to the clinic. Our study suggests that utilizing patient-derived tumor samples to study innate/adaptive immune crosstalk and the effects of activating one pathway on the broader TIME may inform the best approaches to enhance emerging cell therapies and overcome immune exhaustion.
Supplementary Material
Synopsis.
Generating antitumor immunity via innate immune signaling represents an emerging therapeutic strategy. Functional profiling of human mesothelioma tumor explants expressing elevated STING uncovers distinct consequences of STING agonism that support combining such treatment with NK cell–based therapies.
Acknowledgements
We are grateful to Grace Birch, Roman Shapiro, Yurie Yamamoto, David Severson, and Matthew Couger for insightful discussions on this work. We thank the Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry service. DFHCC is supported in part by an NCI Cancer Center Support Grant P30CA06516. This work was also supported by NIH/NCI R01 CA190294, U01 CA214381, P01 CA154303, The Parker Institute for Cancer Immunotherapy, Schaubert Family Funds, the Depoian Family Fund for Mesothelioma Research, and the Polly and Ming Tsai Lung Cancer Research Fund (D.A.B.). Further support was provided by the Gross-Loh Research Fellowship and a Lung Cancer Developmental Research Project Award (E.H.K.). TAK-676 was provided under a Sponsored Research Agreement. We are grateful to KhanhLinh Dinh, Benjamin Ferland, Thomas Thayer, and Teri Bowman for technical assistance and Erin Boudreau for exceptional administrative assistance.
Conflicts of Interest:
E.H.K., C.P.P, and D.A.B. report a shared grant from Takeda during the conduct of the study. A.O.A., V.A.A., R.C.G, A.P., and N.L. are employees of Takeda and own stock. R. R. reports research grants from Crispr Therapeutics and Skyline Therapeutics and serves on the scientific advisory board of Glycostem Therapeutics and xNK Therapeutics. C.P.P. reports personal fees from Dropworks and personal fees from BioRad outside the submitted work; Scientific Advisory Board Member for Dropworks and Scientific Advisory Board Member/Co-Founder of XSphera Biosciences; research funding from Daiichi Sankyo, Bicycle Therapeutics, Transcenta, Bicara Therapeutics, AstraZeneca, Intellia Therapeutics, Janssen Pharmaceuticals, Array Biopharma, Takeda, KSQ therapeutics, Ideya Biosciences, BOLT therapeutics, Lilly Pharmaceuticals, Thermo Fisher Scientific and Bristol Myers Squibb. R. Bueno reports research grants and clinical trials support from MedGenome, Roche, Verastem, Genentech, Merck, Gritstone, Epizyme, Siemens, Celsius as well as equity in Navigation Sciences. D.A.B. reports personal fees from Qiagen, Exo Therapeutics and Tango Biosciences outside the submitted work; he is also a Scientific Advisory Board Member/Co-Founder of XSphera Biosciences, and has received grants from Gilead, Novartis, BMS, and Lilly/Loxo Oncology outside the submitted work.
References
- 1.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 2015;11(7):1018–30 doi 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep 2018;25(11):3074–85 e5 doi 10.1016/j.celrep.2018.11.047. [DOI] [PubMed] [Google Scholar]
- 3.Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING Agonists as Cancer Therapeutics. Cancers (Basel) 2021;13(11) doi 10.3390/cancers13112695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J Exp Med 2017;214(6):1769–85 doi 10.1084/jem.20161674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J Immunol 2017;199(2):397–402 doi 10.4049/jimmunol.1601999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun 2017;8(1):427 doi 10.1038/s41467-017-00573-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 2019;16(3):151–67 doi 10.1038/s41571-018-0142-8. [DOI] [PubMed] [Google Scholar]
- 8.Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, Raulet DH. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018;49(4):754–63.e4 doi 10.1016/j.immuni.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolai CJ, Wolf N, Chang IC, Kirn G, Marcus A, Ndubaku CO, et al. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci Immunol 2020;5(45) doi 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016;533(7604):493–8 doi 10.1038/nature18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 2019;9(5):646–61 doi 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 2018;8(2):196–215 doi 10.1158/2159-8290.Cd-17-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voabil P, de Bruijn M, Roelofsen LM, Hendriks SH, Brokamp S, van den Braber M, et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat Med 2021;27(7):1250–61 doi 10.1038/s41591-021-01398-3. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 2021;39(4):494–508.e5 doi 10.1016/j.ccell.2021.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov 2019;9(1):34–45 doi 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau L, Gray EE, Brunette RL, Stetson DB. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015;350(6260):568–71 doi 10.1126/science.aab3291. [DOI] [PubMed] [Google Scholar]
- 17.Hayman TJ, Baro M, MacNeil T, Phoomak C, Aung TN, Cui W, et al. STING enhances cell death through regulation of reactive oxygen species and DNA damage. Nature Communications 2021;12(1):2327 doi 10.1038/s41467-021-22572-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zugazagoitia J, Gupta S, Liu Y, Fuhrman K, Gettinger S, Herbst RS, et al. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin Cancer Res 2020;26(16):4360–8 doi 10.1158/1078-0432.Ccr-20-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi Z, Yan F, Chen D, Xing W, Li Q, Zeng W, et al. Identification of prognostic biomarkers and correlations with immune infiltrates among cGAS-STING in hepatocellular carcinoma. Biosci Rep 2020;40(10) doi 10.1042/bsr20202603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chon HJ, Kim H, Noh JH, Yang H, Lee WS, Kong SJ, et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J Cancer 2019;10(20):4932–8 doi 10.7150/jca.32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, et al. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 2018;18(20):3129–43 doi 10.1039/c8lc00322j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cunniff EC, Sato Y, Mai D, Appleman VA, Iwasaki S, Kolev V, et al. TAK-676: A Novel Stimulator of Interferon Genes (STING) Agonist Promoting Durable Interferon-Dependent Anti-Tumor Immunity in Preclinical Studies. Cancer Research Communications (2022) 2 (7): XXX–XXX. 10.1158/2767-9764.CRC-21-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pass HI, Stevens EJ, Oie H, Tsokos MG, Abati AD, Fetsch PA, et al. Characteristics of nine newly derived mesothelioma cell lines. Ann Thorac Surg 1995;59(4):835–44 doi 10.1016/0003-4975(95)00045-m. [DOI] [PubMed] [Google Scholar]
- 25.Demetri GD, Zenzie BW, Rheinwald JG, Griffin JD. Expression of colony-stimulating factor genes by normal human mesothelial cells and human malignant mesothelioma cells lines in vitro. Blood 1989;74(3):940–6. [PubMed] [Google Scholar]
- 26.Gordon GJ, Rockwell GN, Jensen RV, Rheinwald JG, Glickman JN, Aronson JP, et al. Identification of novel candidate oncogenes and tumor suppressors in malignant pleural mesothelioma using large-scale transcriptional profiling. Am J Pathol 2005;166(6):1827–40 doi 10.1016/s0002-9440(10)62492-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sehgal K, Portell A, Ivanova EV, Lizotte PH, Mahadevan NR, Greene JR, et al. Dynamic single-cell RNA sequencing identifies immunotherapy persister cells following PD-1 blockade. J Clin Invest 2021;131(2) doi 10.1172/jci135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wickham H ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York; 2016. [Google Scholar]
- 29.Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, et al. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 2018;172(5):1091–107.e17 doi 10.1016/j.cell.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Muhl L, Genové G, Leptidis S, Liu J, He L, Mocci G, et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun 2020;11(1):3953 doi 10.1038/s41467-020-17740-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Correia MP, Stojanovic A, Bauer K, Juraeva D, Tykocinski LO, Lorenz HM, et al. Distinct human circulating NKp30(+)FcεRIγ(+)CD8(+) T cell population exhibiting high natural killer-like antitumor potential. Proc Natl Acad Sci U S A 2018;115(26):E5980–e9 doi 10.1073/pnas.1720564115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gueugnon F, Leclercq S, Blanquart C, Sagan C, Cellerin L, Padieu M, et al. Identification of novel markers for the diagnosis of malignant pleural mesothelioma. Am J Pathol 2011;178(3):1033–42 doi 10.1016/j.ajpath.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Works M, Soni N, Hauskins C, Sierra C, Baturevych A, Jones JC, et al. Anti-B-cell Maturation Antigen Chimeric Antigen Receptor T cell Function against Multiple Myeloma Is Enhanced in the Presence of Lenalidomide. Mol Cancer Ther 2019;18(12):2246–57 doi 10.1158/1535-7163.Mct-18-1146. [DOI] [PubMed] [Google Scholar]
- 34.Xie G, Ivica NA, Jia B, Li Y, Dong H, Liang Y, et al. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat Biomed Eng 2021;5(5):399–413 doi 10.1038/s41551-020-00625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, et al. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis 2019;10(7):476 doi 10.1038/s41419-019-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 1997;15(9):871–5 doi 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- 37.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigí M, Campisi M, et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov 2021;11(8):1952–69 doi 10.1158/2159-8290.Cd-20-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janes SM, Alrifai D, Fennell DA. Perspectives on the Treatment of Malignant Pleural Mesothelioma. New England Journal of Medicine 2021;385(13):1207–18 doi 10.1056/NEJMra1912719. [DOI] [PubMed] [Google Scholar]
- 39.Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24(8):1143–50 doi 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Awad MM, Jones RE, Liu H, Lizotte PH, Ivanova EV, Kulkarni M, et al. Cytotoxic T Cells in PD-L1–Positive Malignant Pleural Mesotheliomas Are Counterbalanced by Distinct Immunosuppressive Factors. Cancer Immunology Research 2016;4(12):1038–48 doi 10.1158/2326-6066.Cir-16-0171. [DOI] [PubMed] [Google Scholar]
- 41.Lupo KB, Matosevic S. CD155 immunoregulation as a target for natural killer cell immunotherapy in glioblastoma. J Hematol Oncol 2020;13(1):76 doi 10.1186/s13045-020-00913-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 2021;218(2) doi 10.1084/jem.20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest 2017;127(6):2176–91 doi 10.1172/jci87624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol 2015;194(4):1954–62 doi 10.4049/jimmunol.1402099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Xia P, Huang G, Zhu P, Liu J, Ye B, et al. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat Commun 2016;7:11023 doi 10.1038/ncomms11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 2019;19(3):170–83 doi 10.1038/s41577-018-0095-2. [DOI] [PubMed] [Google Scholar]
- 47.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018;14(8):1435–55 doi 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol 2021;18(2):85–100 doi 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harrington KJ, Brody J, Ingham M, Strauss J, Cemerski S, Wang M, et al. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Annals of oncology : official journal of the European Society for Medical Oncology 2018;29 Suppl 8:viii712. [Google Scholar]
- 50.Meric-Bernstam F, Sandhu SK, Hamid O, Spreafico A, Kasper S, Dummer R, et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. Journal of Clinical Oncology 2019;37(15_suppl):2507- doi 10.1200/JCO.2019.37.15_suppl.2507. [DOI] [Google Scholar]
- 51.Campisi M, Sundararaman SK, Shelton SE, Knelson EH, Mahadevan NR, Yoshida R, et al. Tumor-Derived cGAMP Regulates Activation of the Vasculature. Frontiers in Immunology 2020;11(2090) doi 10.3389/fimmu.2020.02090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018;553(7689):467–72 doi 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Falahat R, Berglund A, Putney RM, Perez-Villarroel P, Aoyama S, Pilon-Thomas S, et al. Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc Natl Acad Sci U S A 2021;118(15) doi 10.1073/pnas.2013598118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available within the article and its supplementary data files or are available from Dr. Barbie on reasonable request. ScRNA-seq data are available in Gene Expression Omnibus (accession # GSE201925).