

Abstract
Dementia in Alzheimer’s disease progresses alongside neurodegeneration1–4, but the specific events that cause neuronal dysfunction and death remain poorly understood. During normal ageing, neurons progressively accumulate somatic mutations5 at rates similar to those of dividing cells6,7 which suggests that genetic factors, environmental exposures or disease states might influence this accumulation5. Here we analysed single-cell whole-genome sequencing data from 319 neurons from the prefrontal cortex and hippocampus of individuals with Alzheimer’s disease and neurotypical control individuals. We found that somatic DNA alterations increase in individuals with Alzheimer’s disease, with distinct molecular patterns. Normal neurons accumulate mutations primarily in an age-related pattern (signature A), which closely resembles ‘clock-like’ mutational signatures that have been previously described in healthy and cancerous cells6–10. In neurons affected by Alzheimer’s disease, additional DNA alterations are driven by distinct processes (signature C) that highlight C>A and other specific nucleotide changes. These changes potentially implicate nucleotide oxidation4,11, which we show is increased in Alzheimer’s-disease-affected neurons in situ. Expressed genes exhibit signature-specific damage, and mutations show a transcriptional strand bias, which suggests that transcription-coupled nucleotide excision repair has a role in the generation of mutations. The alterations in Alzheimer’s disease affect coding exons and are predicted to create dysfunctional genetic knockout cells and proteostatic stress. Our results suggest that known pathogenic mechanisms in Alzheimer’s disease may lead to genomic damage to neurons that can progressively impair function. The aberrant accumulation of DNA alterations in neurodegeneration provides insight into the cascade of molecular and cellular events that occurs in the development of Alzheimer’s disease.
Alzheimer’s disease (AD) is a common, progressive and fatal age-associated neurodegenerative disorder that is characterized by neuron loss and stereotypic deposition of misfolded proteins2. The formation of oligomers of amyloid-β may initiate disease pathogenesis, triggering a cascade of events that include the development of tau neurofibrillary tangles and oxidative stress1. Tau deposition, which correlates most closely with clinical features, progresses topographically over the course of illness from medial temporal lobe structures to the neocortex, as delineated in the Braak staging system3. Despite substantial mechanistic knowledge of the formation of misfolded proteins, the core basis of cellular dysfunction in AD is not well understood.
Somatic mutations occur in healthy human tissues12–14, including post-mitotic neurons15,16, in which they accumulate during ageing in a process known as genosenium5,17. Analysis of somatic mutational signatures can identify the mutagenic forces responsible, including ultraviolet irradiation in sun-exposed cancers and tobacco-associated polycyclic aromatic hydrocarbons in lung cancers8,18. In human neurons, mutational signature analysis has revealed that somatic single-nucleotide variants (sSNVs) result from multiple mutagenic forces, potentially including the oxidation of DNA nucleotides5. AD shows increased oxidative stress and damaged nucleotides4, but the extent to which these damaged nucleotides are eliminated by manifold DNA repair processes, and whether they result in persistent DNA mutations, producing permanent effects on genome structure or transcription, are not known. Bulk methods, including targeted gene sequencing19 and single-molecule sequencing20, have profiled aspects of AD somatic genetics, but AD has not to our knowledge been examined at the level of individual cellular genomes. Here, to test the hypothesis that specific mechanisms of genomic damage affect AD neurons, we applied single-cell whole-genome sequencing (scWGS) to single neurons from the brains of individuals with AD and neurotypical control individuals to compare the number, genomic locations and classes of somatic mutations that are associated with AD.
Somatic mutations in neurons during ageing
We performed scWGS on pyramidal neurons isolated from the brains of individuals with AD and neurotypical control individuals (Fig. 1a, Supplementary Tables 1, 2). We stained for the pan-neuronal marker NeuN to mark neurons, and further gated only the largest NeuN-positive nuclei (Fig. 1b). This separates, to a purity greater than 99%, the nuclei of pyramidal, excitatory neurons—which are preferentially vulnerable to both neurofibrillary tangle formation21 and cell death in AD22—from those of glia and smaller, inhibitory neurons (Fig. 1c). Here, scWGS involves single-cell alkaline lysis on ice, whole-genome amplification using multiple displacement amplification (MDA) and then several screening and quality control steps, so that only genomes that are well amplified are finally sequenced. In total, using MDA, we analysed 91 neurons from 8 cases of AD and 159 neurons from 18 neurotypical control individuals (Table 1). We identified sSNVs using the LiRA pipeline23, which uses linkage to germline haplotypes to increase specificity and estimates the genome-wide somatic mutation rate by accounting for the cell-specific proportion of phaseable linked sites and false positive rate. For these MDA-amplified single-cell genomes, we performed additional filtration steps based on previously reported patterns of nucleotide substitution attributed to artefacts of genome amplification by MDA24 (see Methods, Extended Data Fig. 1). This set of filtered sSNV calls showed a variant allele fraction distribution that was very similar to that of germline heterozygous SNVs in single-cell data (Extended Data Fig. 2), which allowed us to confirm that, in neurotypical individuals, neuronal sSNVs increased with age at a rate of 16–21 sSNVs per year (Fig. 1d, Extended Data Fig. 3a–d)—consistent with previous work on neurons5,20,25. Studies using clonally expanded cells from other human tissues have shown comparable yearly increases in sSNVs, ranging from 13 to 55 sSNVs per year, with higher rates in more rapidly dividing cell types (Extended Data Table 1).
Fig. 1 |. Somatic mutations in single neurons in control individuals and individuals with AD.
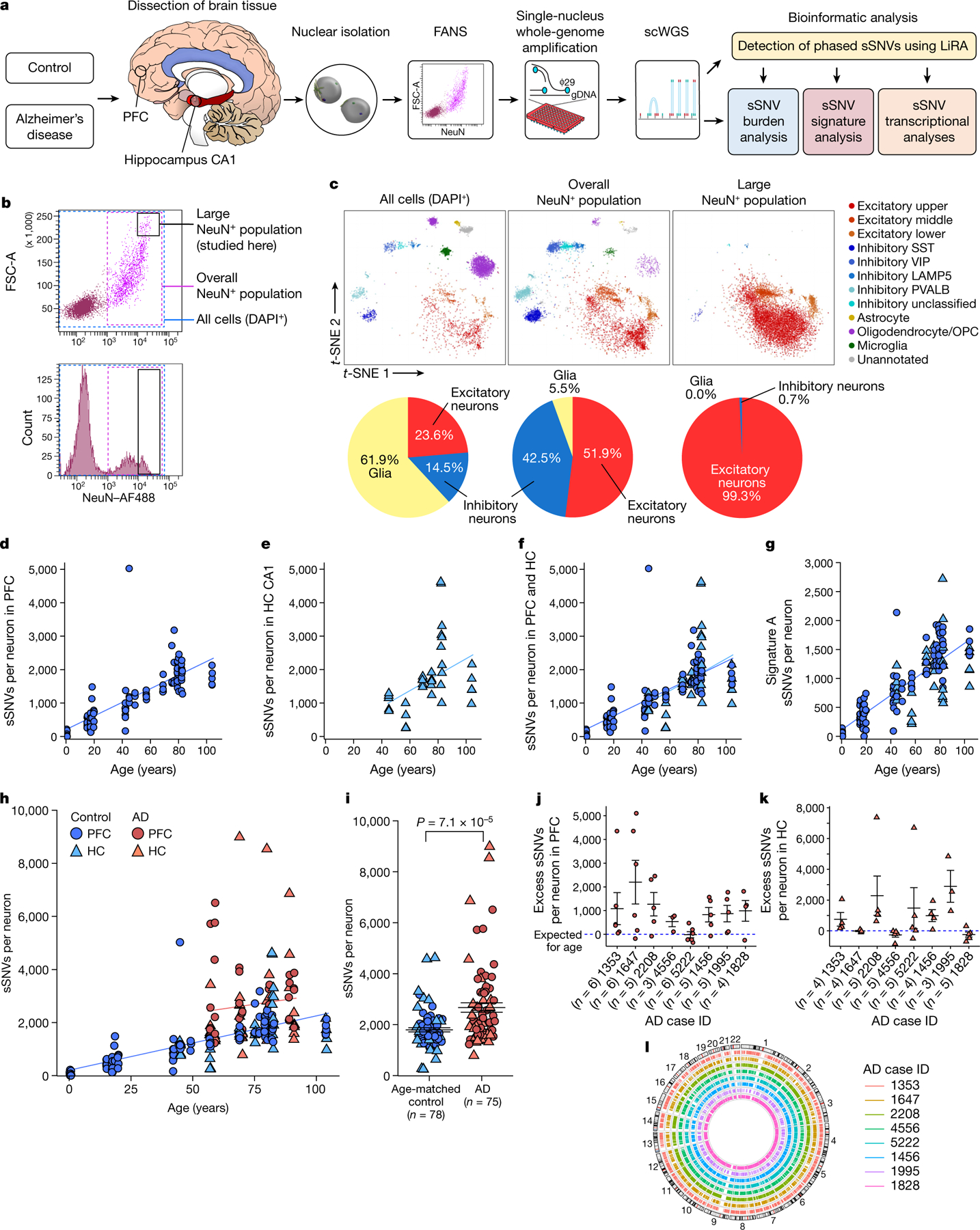
a, Experimental outline for scWGS. From human brain, large neurons were isolated and their genomes were amplified, sequenced, and analysed for sSNV. FANS, fluorescence-activated nuclear sorting. b, FANS using AF488-conjugated anti-NeuN antibodies to label candidate neurons for separation from glia and other cell types. Boxes show the full population of DAPI+ diploid cellular nuclei (blue dashed box); the overall population of NeuN+ nuclei (pink dashed box); and the large NeuN+ subset (black box; the subject of this study). c, Single-nucleus transcriptomic profiling of each population. Individual cells are plotted according to t-distributed stochastic neighbour embedding (t-SNE) coordinates, and clusters of 50 cells or more are annotated50 and labelled by colour, with a pie chart of the relative abundance of excitatory neurons, inhibitory neurons and glia in each population. OPC, oligodendrocyte precursor cell. d–l, sSNVs identified using MDA genome amplification. d–f, sSNVs in neurotypical control neurons. Data points represent single neurons; trend lines show linear mixed models (PFC: P = 3.3 × 10−7, R2 = 0.63; hippocampus (HC): P = 0.16, R2 = 0.18). g, Contribution of ageing signature A to sSNVs (P = 1.67 × 10−10, R2 = 0.68, linear mixed model). h, sSNVs as a function of age in neurotypical control individuals and individuals with AD (linear mixed model trend lines: blue, control: P = 6.8 × 10−7, R2 = 0.51; red, AD: P = 0.46, R2 = 0.01). AD contributes a significant excess of sSNVs in neurons relative to normal ageing (P = 6.5 × 10−5, linear mixed model). i, AD neurons show increased sSNVs compared with age-matched (over 50 years old) control neurons (874 sSNVs per neuron, P = 7.1 × 10−5, two-tailed Wilcoxon test). j, k, Excess sSNVs attributable to AD in the PFC (j) and the hippocampus (k). The dashed blue line shows sSNVs attributable to age (zero excess). For i–k, black bars show mean ± s.e.m. l, Circos plot showing the wide distribution of sSNVs across the genome in AD neurons.
Table 1 |.
Case information and number of neurons analysed in this study
| Case ID | Age (years) | Sex | Diagnosis | PFC neurons (MDA-amplified) | Hippocampus (HC) CA1 neurons (MDA-amplified) | PFC neurons (PTA-amplified) |
|---|---|---|---|---|---|---|
| Younger neurotypical controls | ||||||
| 1278 | 0.4 | M | Neurotypical | 9 | − | 3 |
| 5817 | 0.6 | M | Neurotypical | 4 | − | 3 |
| 4638 | 15.1 | F | Neurotypical | 10 | − | − |
| 1465 | 17.5 | M | Neurotypical | 24 | − | 4 |
| 5532 | 18.4 | M | Neurotypical | 4 | − | − |
| 5559 | 19.8 | F | Neurotypical | 5 | − | 3 |
| 4643 | 42.2 | F | Neurotypical | 10 | − | − |
| 5087 | 44 | M | Neurotypical | 4 | 5 | 3 |
| 936 | 49.2 | F | Neurotypical | 3 | − | 3 |
| 73 | 5 | 19 | ||||
| Aged neurotypical controls | ||||||
| 5451 | 57 | F | Neurotypical | 5 | 5 | 3 |
| 5666 | 65 | M | Neurotypical | − | − | 3 |
| 5943 | 69 | M | Neurotypical | 5 | 5 | 3 |
| 5572 | 70 | F | Neurotypical | − | − | 3 |
| 5840 | 75.3 | M | Neurotypical | 3 | 5 | − |
| 5219 | 77 | F | Neurotypical | 4 | − | − |
| 5171 | 79.2 | M | Neurotypical | 13 | − | − |
| 5511 | 80.2 | F | Neurotypical | 3 | − | − |
| 5657 | 82.2 | M | Neurotypical | 10 | 5 | 3 |
| 5823 | 82.7 | F | Neurotypical | 3 | 5 | 3 |
| 4976 | 104 | F | Neurotypical | 5 | 5 | 3 |
| 51 | 30 | 21 | ||||
| Alzheimer’s disease | ||||||
| 1353 | 57 | F | AD (Braak VI) | 7 | 5 | 4 |
| 1647 | 59 | F | AD (Braak VI) | 7 | 5 | 6 |
| 2208 | 69 | F | AD (Braak VI) | 5 | 5 | 4 |
| 4556 | 70 | F | AD (Braak VI) | 5 | 5 | − |
| 5222 | 80 | F | AD (Braak VI) | 6 | 5 | − |
| 1456 | 81 | M | AD (Braak VI) | 5 | 5 | 4 |
| 2207 | 83 | M | AD (Braak VI) | − | − | 3 |
| 1995 | 89 | F | AD (Braak V) | 8 | 4 | 4 |
| 1828 | 91 | F | AD (Braak VI) | 5 | 9 | 4 |
| 48 | 43 | 29 | ||||
| Total | 29 individuals | 172 PFC-MDA neurons | 78 HC-MDA neurons | 69 PFC-PTA neurons | ||
| Total: 319 neurons | ||||||
We next examined the accumulation of sSNVs in pyramidal neurons located in the CA1 subfield of Ammon’s horn of the normal hippocampus, as this is a critical region in AD and other diseases. Hippocampal CA1 neurons from individuals who died with no neurological diagnosis showed a trend towards the accumulation of sSNVs with age (Fig. 1e), which was not significantly different from the increase in sSNVs seen in prefrontal cortex (PFC) neurons from neurotypical control individuals (P = 0.72, linear mixed-effects regression model (linear mixed model); overlay in Fig. 1f). When considering the PFC and the hippocampus together (Extended Data Fig. 3a–d), this set of single cells highlights a common pattern of sSNV accumulation in the pyramidal neurons of neurotypical individuals.
Large-scale DNA sequencing studies in cancer have identified patterns and contexts of nucleotide substitution, termed ‘signatures’8, which often reveal mutagenic forces. In normal PFC neurons, the age-related increase in mutations is driven mainly by certain C>T and T>C changes, termed signature A5. This signature resembles the age-related ‘clock-like’ signature that is observed in other normal cells as well as in essentially all cancer cells9, designated as signature SBS5 in the COSMIC mutational signature database (https://cancer.sanger.ac.uk/cosmic/signatures). Signature decomposition analysis of sSNVs from the composite dataset of PFC and hippocampal pyramidal neurons showed that the contribution of signature A in each neuron increased with age, at a rate of 15.0 ± 1.2 sSNVs gained per year (Fig. 1g). This age-related increase in signature A mutations is similar for PFC and hippocampal pyramidal neurons (P = 0.18, linear mixed model), and is the major driver of age-related sSNV accumulation in normal neurons. Despite their universal presence in many cell types, and their accumulation in nondividing cells, the cellular mechanism of such clock-like mutations is not clear. Signature SBS5 exhibits a transcriptional strand bias9, which suggests that events leading to these mutations are associated with RNA transcription. During transcription, the double helix is unwound, exposing single DNA strands to cytosine and thymine deamination17, which are subject to transcription-coupled nucleotide excision repair (TC-NER). Transcription may therefore sensitize expressed loci to somatic mutagenesis through transcription-associated damage or ineffective repair.
Somatic mutations in AD
We next assessed the burden of sSNVs in neurons from the brains of eight individuals with AD and found that AD neurons showed significantly more called sSNVs than expected on the basis of age (P = 6.5 × 10−5, linear mixed model; Fig. 1h). This excess was variable between neurons, mirroring the variable presence of AD pathology within neurons of a given brain region. AD neurons also showed a significant increase in called sSNVs in MDA experiments when directly compared to age-matched neurotypical control neurons (P = 7.1 × 10−5, two-tailed Wilcoxon test; Fig. 1i). This increase remained after controlling for potential covariates including post-mortem interval, sample storage time, sample DNA quality, sequencing depth, sequencing quality score, library insert size and number of heterozygous germline SNVs, as well as technical metrics of scWGS evenness (see Methods, Extended Data Fig. 3e–h). In the PFC, we observed significant gains in sSNVs in AD relative to normal ageing in seven out of eight individual cases of AD (Fig. 1j). Several of the genomes with the highest sSNV counts in AD came from the hippocampus, in which five of eight cases also showed significant increases in sSNVs compared with normal ageing (Fig. 1k). However, in three cases, the assayed hippocampal neurons did not show a detectable increase in the handful of cells assayed. On the basis of tau (Braak) and amyloid-β (Consortium to Establish a Registry for Alzheimer’s Disease; CERAD) neuropathological staging, hippocampal pathology appears to precede PFC damage, and the hippocampus of these late-stage cases invariably showed widespread neuronal loss as well (not shown). Thus, it is possible that highly mutated neurons are lost before death and therefore not possible to assay here, so our results may reflect resilient neurons that have survived despite advanced AD22. These results show that neurons in AD contain hundreds of additional sSNVs beyond that expected for their age, indicating that the disease process produces a level of genomic damage that is on par with more than a decade of normal accumulation of sSNVs.
The somatic mutations identified in AD neurons are pervasively distributed across the genome (Fig. 1l), with a trend towards an excess in regions at least 1 kb upstream from the transcription start site—where DNA damage has been implicated during neuronal gene transcription26—that does not survive Bonferroni correction (P = 0.045, two-tailed t-test; Extended Data Fig. 4). The broad genomic distribution of variants suggests that, rather than constituting a specific initial event in disease pathogenesis, somatic mutations are more likely to be secondary, resulting from other events that initiate AD and instigate mutagenic processes. Specifically, we did not observe somatic instances of known pathogenic mutations in classic germline AD risk genes (APP, PSEN1, PSEN2 and APOE), concordant with a recent report27, nor did we observe somatic increases in copy number of the APP gene, contrary to a previous study28 and as we reported in detail separately29. We also observed no consistent effect of an individual’s ApoE status or sex on the accumulation of sSNVs.
Mutational signature analysis in AD neurons
We next performed mutational signature analysis to identify whether specific processes cause somatic alterations in AD neurons. De novo signature decomposition revealed mutational signatures concordant with those previously reported in human neurons5 (Extended Data Fig. 5). We focused our analysis on neuronal signatures A and C (Fig. 2a), as signature B contains clonal developmental mutations, but is also where artefactual C>T mutations created by MDA amplification aggregate24. Signature A mutations increase with age in all samples, which suggests that this clock-like signature (that is most similar to the clock-like signature SBS5 from cancer5) constitutes an inherent feature of genome ageing. Signature A also shows a marginal increase in AD relative to age-matched controls (Fig. 2b, c), which does not reach statistical significance in these MDA experiments, but suggests that these mutational mechanisms could be accentuated in the setting of disease. On the other hand, AD neurons show a pronounced increase in signature C compared to controls (Fig. 2d, e), which accounts for most of the observed excess in alterations. The signature C burden shows more variation between neurons than that for signature A (Extended Data Fig. 5d), which suggests that signature C could result from irregular ‘calamitous’ events, in contrast to the uniform ageing represented in signature A.
Fig. 2 |. Somatic mutational signatures and patterns in AD neurons by MDA.
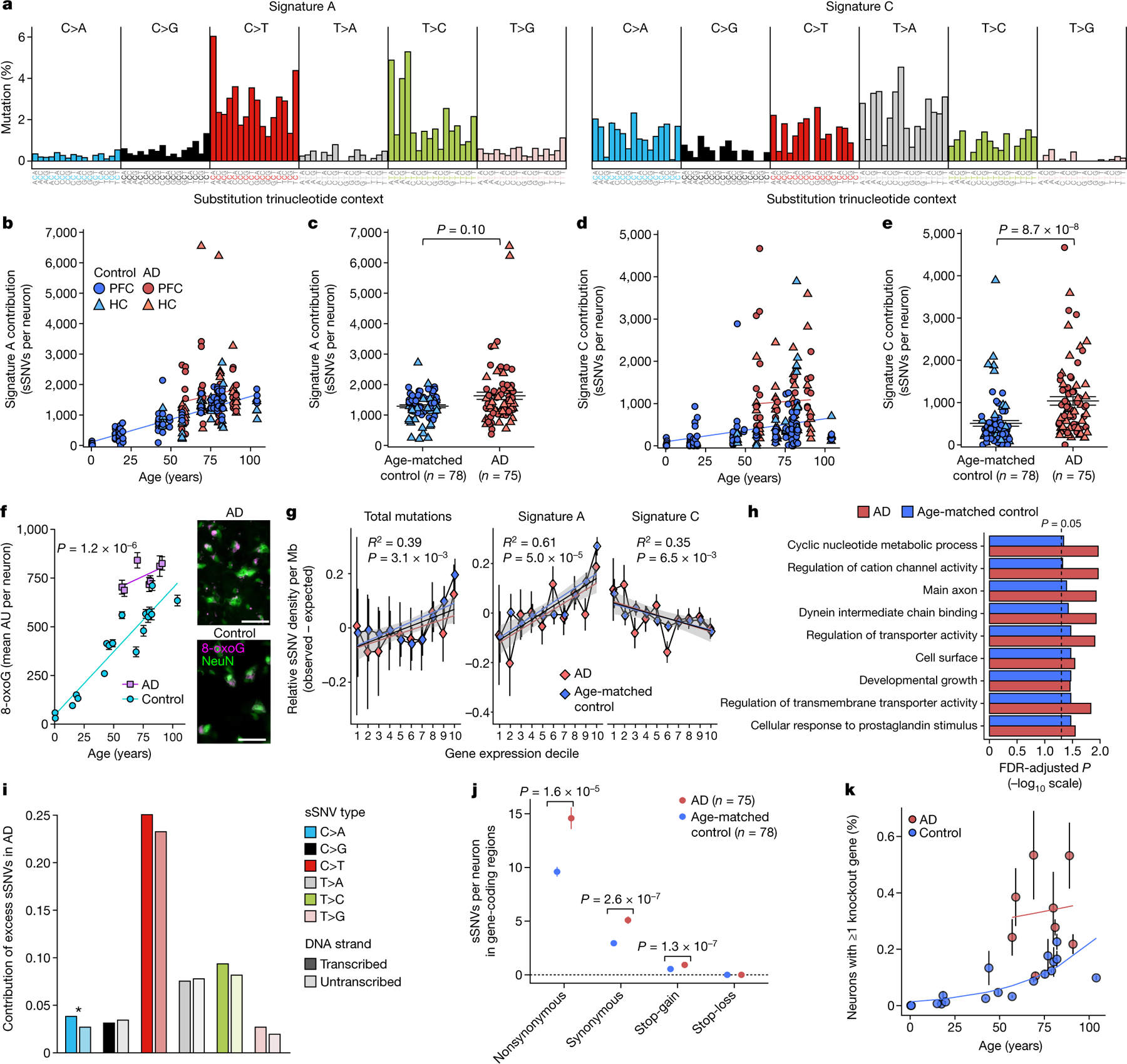
a, Somatic mutational signatures identified by NMF5. b, c, Signature A contribution by age (b; AD excess 418, P = 3.1 × 10−4, linear mixed model) and in individuals with AD versus age-matched control individuals (c; 27% increase in AD, P = 0.10, two-tailed Wilcoxon test). d, e, Signature C contribution by age (d; AD excess 549, P = 1.4 × 10−3, linear mixed model) and in individuals with AD versus control individuals (e; 104% increase in AD, P = 8.7 × 10−8, two-tailed Wilcoxon test). f, Oxidative damage in AD neurons, using 8-oxoG immunofluorescence. Data points represent mean absorbance units (AU) ± s.e.m. of n = 100 neurons per case in PFC (full data in Extended Data Fig. 7). Trend lines show linear mixed-effects regression (AD versus control: P = 1.2 × 10−6). Inset shows representative immunofluorescence images; neurons (NeuN; green) and oxidized guanine (8-oxoG; magenta). Scale bars, 60 μm. g, Genomic sSNV density as a function of gene expression in the brain. Diamonds represent mean relative sSNV density in single neurons (black vertical lines show s.d., n = 1,000 permutations). Overall trend line is shown in black (R2 and P value, Pearson correlation); 95% confidence interval (CI) in grey; and AD and control trend lines in colours. h, GO analysis of genes mutated in single neurons. i, sSNVs by DNA strand template status. sSNVs in transcribed regions exhibit a strand bias in the excess mutations in AD neurons, which is most pronounced in C>A variants (*P = 0.017, two-tailed Poisson test). j, Coding mutation subtypes, in which increased nonsynonymous mutations in AD (P = 1.6 × 10−5, two-tailed t-test) increase the propensity for presentation of neoantigen peptides. k, sSNVs that result in gene knockout cells. Model for the abundance of neurons with gene inactivation, affecting function. Circles represent mean for each individual, (n > 3 neurons each, see Source Data), with 95% CI. c, e, j, Data are mean ± s.e.m.
Signature C includes C>A substitutions, which have previously been associated with oxidative damage to guanine nucleotides18. Signature C also has a significant contribution from the cancer-associated signature SBS8 (ref. 5) (Extended Data Fig. 6a). This signature is increased in stem cells with disrupted TC-NER10,30, and we have observed an increase in signature C in single human neurons deficient in TC-NER owing to ERCC6 mutations, and in neurons deficient for global NER owing to XPA or XPD mutations5. Overlap between AD sSNVs and other cancer-derived signatures also suggests a potential role for NER in T>A, T>C and C>T mutations (Extended Data Fig. 6b). Signature C has been reported in normal neurons at low but highly variable levels5, with some accumulation with age in the normal PFC, and a similar signature has also been reported in ageing stem cells from the liver and intestine6. Given that increased reactive oxygen species (ROS) and oxidative nucleic acid lesions have been reported in AD4,31–33, a plausible mechanism for the accumulation of signature C in AD is that increased oxidative damage overwhelms NER, which could also be attenuated in AD.
The set of excess mutations in individuals with AD, represented as the trinucleotide spectrum of residual mutations when subtracting those present in control individuals, also includes contributions from the cancer signature SBS6 (Extended Data Fig. 6b), which is associated with defective DNA mismatch repair, raising the possibility that other repair mechanisms may further contribute to the generation of somatic mutations in AD neurons.
Oxidative damage in AD neurons
Because our mutational signature analysis suggested that DNA oxidation—previously observed in bulk analyses of brains from individuals with AD4,11—might contribute to the excess sSNVs in AD, we directly examined nucleotide oxidative damage in individual neurons. The most frequent oxidized nucleotide lesion due to oxidative stress is 8-oxoguanine (8-oxoG), and this is therefore used as a biomarker for cellular oxidative status and DNA damage. Immunofluorescence microscopy using an antibody targeting 8-oxoG showed that there were significantly higher levels of 8-oxoG in AD neurons than in neurotypical control neurons (P = 1.2 × 10−6, linear mixed model; Fig. 2f, Extended Data Fig. 7), indicating that increased levels of oxidative nucleotide damage contribute to C>A changes and to the increase in signature C in AD neurons.
Transcriptional influence on somatic SNVs
Mutations in genes that are critical for neuronal function and survival could directly affect cellular fitness. Despite the preferential repair of transcribed genes in human neurons34, the burden of sSNVs in transcribed regions of the genome correlated with gene expression levels in the brain (P = 3.1 × 10−3, Pearson correlation; Fig. 2g). When this observation was separated by signature, with increased expression we observed increased signature A mutations (P = 5.0 × 10−5, Pearson correlation), but decreased signature C mutations (P = 6.5 × 10−3, Pearson correlation). These findings provide further support for the hypothesis that ageing-associated signature A and AD-associated signature C arise from different mechanisms. For signature A, events during transcription appear to have a role in generating mutations, whereas signature C correlates inversely with expression and therefore may be more effectively repaired during transcription, including by TC-NER35.
Gene Ontology (GO) analysis of loci mutated in AD and control neurons revealed that genes involved in neuronal function were enriched for sSNVs (Fig. 2h). When considered together with the expression–sSNV findings, AD neurons show an influence of transcriptional processes on mutation generation. Such a transcriptional influence can produce an asymmetric pattern of mutations on the paired DNA strands. We therefore distinguished the sSNV sites by template status, between transcribed template strands and untranscribed strands (Fig. 2i). We found a significant strand bias for C>A mutations on the transcribed strand, along with a modest strand bias for C>T and T>C, providing further evidence that errors in transcription-related mechanisms have a role in the generation of sSNVs in AD neurons. As one example, an unrepaired oxidized guanine nucleotide, 8-oxoG, on an untranscribed strand could become a G>T mutation, which would be classified as a C>A mutation on the transcribed strand. In addition to the apparent protective role of NER processes against somatic mutation, the involvement of NER in signature C mutations also presents a potential mechanism for the accumulation of mutations in non-cycling cells, as NER involves the removal of an approximately 29-bp sequence by an exonuclease, followed by the replication of those 29 bp from the remaining DNA strand36; this allows for replication errors during repair if the template strand is also damaged.
Potential consequences of somatic mutations in AD
Somatic mutation or single-stranded damage that alters amino acids can contribute to neuronal dysfunction or loss by many mechanisms, including direct impairment of transcription, alterations in protein stability or creation of neoantigens. In protein-coding genes, AD neurons show more nonsynonymous mutations than age-matched control neurons (Fig. 2j), which has the potential to impair dosage-sensitive genes, or to create neoantigen peptides that could elicit T lymphocyte activation, immune attack and consequent cellular damage. Observations of clonal CD8+ T cells in cerebrospinal fluid and brain tissue in AD37 suggest that such autoactivation could be relevant in AD. Moreover, as somatic alterations accumulate in a genome, the likelihood of two deleterious exonic alterations in the same gene, producing a knockout cell, increases exponentially. We modelled the rate of sSNV-caused knockout neurons (Fig. 2k), and found a substantial projected increase in AD over controls (P = 0.022, generalized estimating equation model). This model suggests that dysfunctional neurons would be markedly more abundant in AD, which may be compounded by the length of certain AD-relevant genes38; compromising neuronal function may therefore be one way in which sSNVs affect cellular physiology39. The pronounced effect of genomic damage, even in non-dividing cells, is underscored by the observation that multiple defects in DNA repair result in neuronal dysfunction and degeneration5,40.
Interrogation of AD neuron genomes by PTA
The experiments discussed thus far, which used MDA to amplify the genomes of single neurons, used LiRA variant calling to counteract allele dropout23 and signature-based filtering of amplification artefacts (Extended Data Fig. 1), which are features of MDA-based methods. To corroborate our findings from MDA-amplified single neuron genomes, we applied a second single-cell amplification method that removes most or all amplification artefacts41,42 as an orthogonal approach. Primary template-directed amplification (PTA)41 achieves highly uniform genome amplification by using chain-terminating nucleotides to disfavour long amplification products that can be re-primed. PTA thus allows the identification of sSNVs in single human neurons while mitigating known single-cell artefacts that can be seen from MDA42, obviating the need for signature-based variant filtering. PTA-based scWGS of human neurons has confirmed that somatic mutations increase with age42. We performed PTA-based scWGS on a small sample of neurons from most brains profiled by MDA (29 neurons from 7 cases of AD and 40 neurons from 13 neurotypical control individuals; Table 1) and confirmed that AD neurons contain increased somatic alterations compared to controls (P = 3.9 × 10−4, linear mixed model; Fig. 3a). This effect remained after controlling for technical metrics (Methods, Extended Data Fig. 8c–f). The magnitude of the PTA-detected AD increase is somewhat lower than what was observed by MDA, which is likely to reflect in part residual amplification artefacts in MDA material. sSNVs detected by PTA show trinucleotide spectra (Extended Data Fig. 8a) and COSMIC signature contributions (Extended Data Fig. 8b) that are highly similar to those seen in multiplexed end-tagging amplification of complementary strands (META-CS), a recently reported duplex sequencing method that explicitly distinguishes double-stranded mutations and single-stranded DNA lesions25. PTA-identified mutational spectra closely cluster with META-CS-identified double-stranded mutations and are distinct from META-CS single-stranded lesions, which strongly suggests that PTA-detected sSNVs represent double-stranded somatic mutations.
Fig. 3 |. Profile of somatic mutations in single AD neurons by PTA.
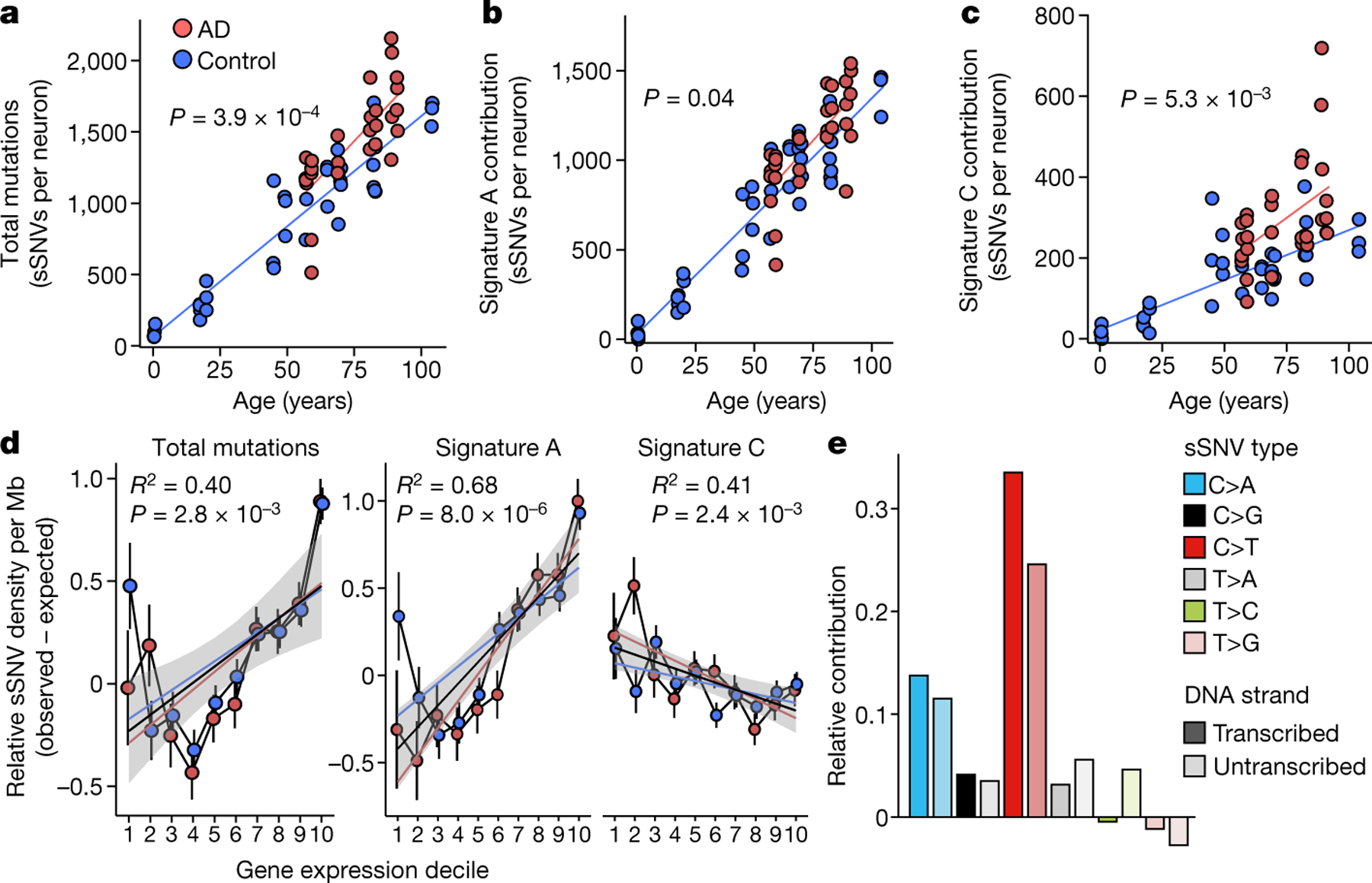
Single-neuronal nuclei were isolated from control and AD prefrontal cortex and subjected to PTA whole-genome amplification for scWGS. a, sSNVs as a function of age in neurotypical control individuals (blue) and individuals with AD (red). Blue and red lines show linear mixed model trend lines for each group (control: P = 2.0 × 10−16, R2 = 0.90; AD: P = 6.57 × 10−7, R2 = 0.59). By PTA, AD contributes a significant excess of sSNVs (196 per genome) in neurons compared to the normal ageing trend line (P = 3.9 × 10−4, linear mixed model). b, c, PTA-called sSNVs by mutational signature in each individual neuron. sSNV contributions are shown as a function of age for signature A (b; AD versus control P = 0.04, linear mixed model) and signature C (c; AD versus control P = 5.3 × 10−3, linear mixed model). d, Transcriptional influence on somatic mutation in neurons profiled by PTA. Genes with higher expression levels show increased overall and signature A density and decreased signature C density. Data points represent mean sSNV density relative to expected density based on the mutation trinucleotide context, with black vertical lines showing s.e.m. Controls represent age-matched (over 50 years old) neurotypical neurons. Overall trend line is shown in black; 95% CI in grey; and separate AD and control trend lines in colours. R2 and P values are shown for Pearson correlation. e, sSNVs by DNA strand template status. sSNVs in transcribed regions exhibit a strand bias in the excess mutations in AD neurons. For each nucleotide change, the proportional contributions of the transcribed and the untranscribed strand are shown. The strand bias ratio data in PTA-amplified neuron data showed a similar trend to that seen in MDA-amplified neurons.
We also examined PTA-detected mutations by signature decomposition, which again confirmed that signature A mutations increase with age in a clock-like manner (Fig. 3b), with a marginally significant increase in signature A in AD neurons (P = 0.04, linear mixed model). The AD-associated increase in mutations is most pronounced for signature C (P = 5.3 × 10−3, linear mixed model; Fig. 3c). As with the increase in total mutations in AD neurons, the PTA mutational signature findings mirrored the trends seen in MDA-amplified neuron genomes. The residual PTA-detected mutations in AD neurons show a distinct trinucleotide spectrum (Extended Data Fig. 8a), with an excess of C>A and C>T mutations that is also seen in MDA-amplified neurons. When analysed for contributions of COSMIC cancer mutation signatures, the residual mutations in AD neurons show a distinct pattern from that of control neurons (Extended Data Fig. 8b), including many signatures seen with MDA-detected AD residual mutations. Among these are SBS8 as well as SBS30, which is associated with the DNA repair enzyme NTHL1 that is involved in oxidative lesion repair. The PTA-detected burden of sSNVs in transcribed regions correlated with levels of gene expression in the brain (P = 2.8 × 10−3, Pearson correlation; Fig. 3d), whereas signature A and C mutations showed similar patterns to those seen with MDA-detected sSNVs, pointing to specific effects of transcriptional activity on mutation occurrence. We also noted a C>A strand bias in PTA-amplified AD neurons (Fig. 3e), further implicating transcription-related events in the generation of sSNVs in AD neurons. Thus, both scWGS approaches identified similar patterns, and suggest that the pathogenic mutational mechanisms in AD include DNA oxidation, NER DNA repair and transcriptional activity.
Although several studies have confirmed that neurons accumulate sSNVs with age5,20,25, one recent study using a single-molecule technique called NanoSeq did not find greater genome-wide mutation rates in AD-affected brains compared to aged brains of neurotypical control individuals, and actually reported a small but significant decrease in somatic mutations in AD20. There are a few potential reasons for this discrepancy as compared to our findings in single AD neurons. One possibility is that single-stranded lesions or variants contribute to our signal, although we have taken lengths to exclude this, including custom computational removal of known MDA artefacts and application of the PTA scWGS method. The NanoSeq study may also reflect an analysis of different cell populations from the individual cells that we studied here. The NanoSeq analysis studied bulk DNA from 15,000 pooled cells sorted using NeuN without size gating20, but we observed that sorting by NeuN alone includes excitatory and inhibitory neurons, as well as some glial cells (Fig. 1b, c). Therefore, the NanoSeq study does not enrich for the excitatory pyramidal neurons that are selectively vulnerable to AD21,22, which is likely to obscure the modest but consistent difference that we find when pyramidal neurons are enriched. The bulk NanoSeq method on all NeuN-expressing cells would also be susceptible to differences in cell-type abundance, which could account for the slightly decreased mutation count that was observed. Thus, increased somatic mutation burden in the AD brain may be limited to precisely the neuron subtypes that are most affected by the disease, potentially sparing some cell types.
Discussion
Our analysis reveals that excitatory neurons in the brains of individuals with AD accumulate genomic damage—and likely permanent mutations—beyond the levels that occur as a result of ageing alone. The pattern of genomic SNV accumulation in AD neurons appears to be distinct from an accentuation of normal ageing, as suggested by (1) the abundance of signature C, which is present but limited in the brain of neurotypical control individuals; and (2) signature-specific transcriptional influences. These genomic changes may include a spectrum of manifestations, including single-stranded DNA lesions and double-stranded mutations. Notably, putative mutations identified by PTA-based scWGS were molecularly similar to bone fide double-stranded mutations identified by duplex sequencing, but dissimilar to single-stranded lesions. These correlations, combined with the evenness of PTA genome coverage, suggest that the AD-specific somatic alterations are predominantly double-stranded mutations. Future studies that are specifically designed to compare DNA lesions with permanent mutations may shed further light on the differential effects these related phenomena have in AD. Other types of somatic alterations—such as short insertions and deletions, structural variants and retrotransposition events—can also be explored in greater depth as technologies improve.
Beyond abundance, the specific patterns of somatic alterations in AD neurons provide clues as to their causes and potential effects in AD pathogenesis (Fig. 4), and identify potential therapeutic targets. Signature C is notable for the presence of C>A variants, associated with oxidative damage, which has been observed previously in AD4 and which we found to be increased in AD neurons. This suggests that sSNVs occur downstream of ROS during disease pathogenesis. Signature C has a notable similarity to COSMIC signature SBS8, which is associated with the transcription-coupled repair of damaged guanine10, strongly suggesting that it accumulates either through disease-related defects in NER, or, more likely, from an accelerated accumulation of oxidized nucleotides that overwhelms the repair pathway. Oxidized nucleotides reflect the presence of increased ROS, which have previously been reported in the brain of individuals with AD, and which can be generated by a variety of processes—including inflammation and mitochondrial dysfunction, which have also been reported in AD43. Our data show how these oxidative lesions may impair genomic function by interacting with mutations that occur as a part of ageing.
Fig. 4 |. Model of the role of somatic mutations in AD pathogenesis.
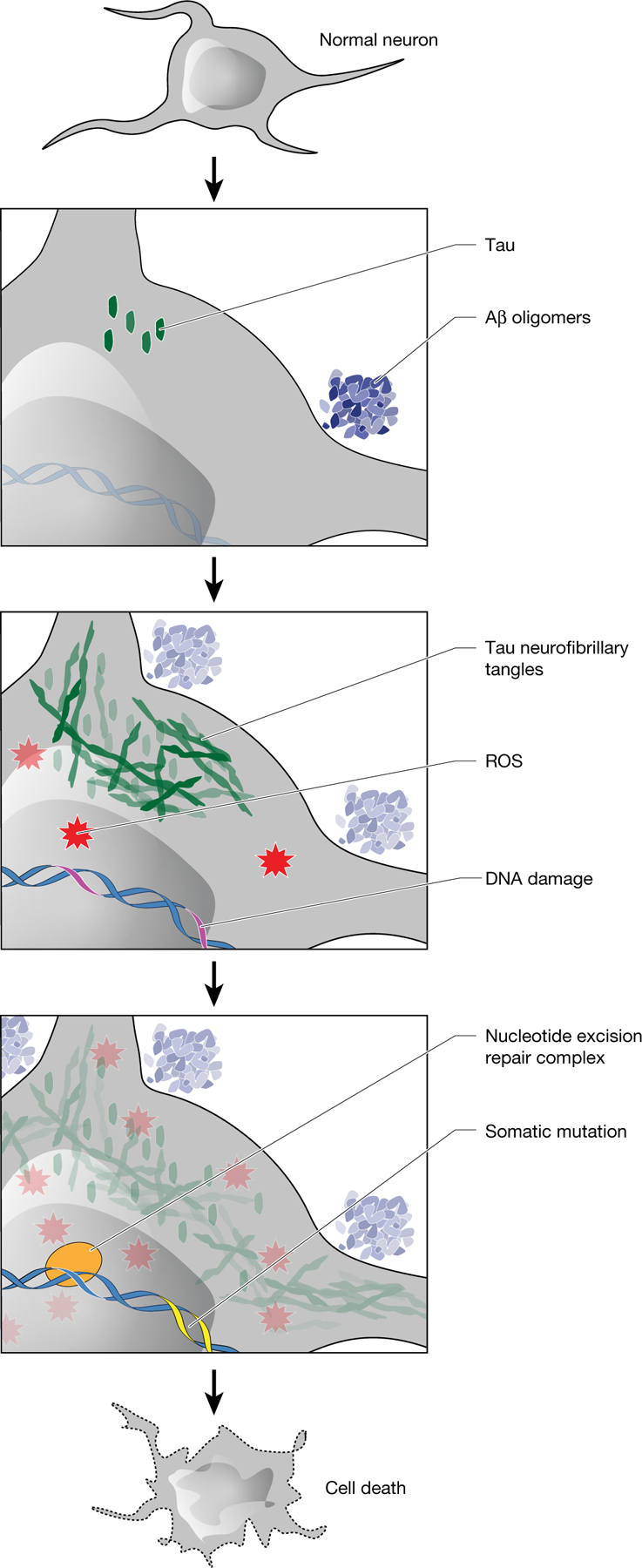
Amyloid-β(Aβ) oligomers initiate a cascade of events, including the conversion of tau to neurofibrillary tangles and the accumulation of ROS. After DNA damage by ROS or other mutagens, somatic mutations develop with characteristic features of signature C. NER affects the strand and gene distribution of somatic mutations, and rare base misincorporation during repair may also have a role in the progression from DNA damage to mutation. These somatic mutations stand to increase cellular vulnerabilitybymechanismsincludinggeneinactivationandneoantigenpresentation.
A major question that remains concerns how the buildup of AD-related genomic damage relates to the well-established accumulation of amyloid-β and tau proteins1,2. Indeed, both of these AD-associated misfolded proteins can induce ROS44,45, with the tau effect being mediated by mitochondrial dysfunction45. Furthermore, tau can trigger double-stranded DNA breaks46, thus further compounding the effects of sSNVs and potentially inducing more47. Many aspects of the oxidative stress induced by AD proteins are not clear, but this process may also include the amyloid-β-stimulated activation of microglia, which can produce ROS directly and can also indirectly initiate the generation of ROS through the release of pro-inflammatory cytokines48. Binding of amyloid-β to redox-active iron may also add oxidative stress49. It will be important to identify how protein misfolding and other known events in AD relate to the accumulation of somatic mutations in the pathogenesis of disease.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Human tissue samples and selection of cases of AD
Post-mortem frozen human tissues were obtained from the Massachusetts Alzheimer’s Disease Research Center (MADRC) at Massachusetts General Hospital and the NIH Neurobiobank at the University of Maryland Brain and Tissue Bank (UMBTB). Tissue collection and distribution for research and publication was conducted according to protocols approved by the Partners Human Research Committee (for MADRC: 1999P009556/MGH, expedited waiver category 5) and the University of Maryland Institutional Review Board (for UMBTB: 00042077), and after provision of written authorization and informed consent. Research on these de-identified specimens and data was performed at Boston Children’s Hospital with approval from the Committee on Clinical Investigation (S07-02-0087 with waiver of authorization, exempt category 4). Many neurotypical control tissues and datasets were obtained as part of a previous study5. Neurotypical control cases had no clinical history of dementia or other neurological disease. AD cases had a clinical history of dementia consistent with AD, pathologically confirmed AD pathological change (Braak stage V–VI) and no other notable neurodegenerative pathology. Age-matched cohorts included individuals who were over 50 years old (Table 1).
Isolation of individual pyramidal neurons for single-cell studies
The isolation of single neuronal nuclei using fluorescence-activated nuclear sorting (FANS) for the neuronal nuclear transcription factor NeuN and whole-genome amplification (WGA) using MDA51 have been described previously5,52. In brief, nuclei were prepared from unfixed frozen human brain tissue, previously stored at −80 °C, in a dounce homogenizer using a chilled tissue lysis buffer (10 mM Tris-HCl, 0.32 M sucrose, 3 mM Mg(OAc)2, 5 mM CaCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% Triton X-100, pH 8) on ice. Tissue lysates were layered on top of a sucrose cushion buffer (1.8 M sucrose 3 mM Mg(OAc)2, 10 mM Tris-HCl, 1 mM DTT, pH 8) and ultra-centrifuged for 1 h at 30,000g. Nuclear pellets were resuspended in ice-cold PBS supplemented with 3 mM MgCl2, filtered, then stained with anti-NeuN antibody directly conjugated to Alexa Fluor 488 (AF488) (Millipore MAB377X, clone A60, 1:1,250). NeuN staining produced a bimodal signal distribution (Fig. 1b, bottom), distinguishing NeuN+ and NeuN− nuclei. Large neuronal nuclei, representing excitatory pyramidal neurons, were then identified by flow cytometry (using software BD FACSDiva v.8.0.2) by targeting the nuclei with highest NeuN signal among the NeuN+ neuronal fraction, while also gating for the population with the highest forward scatter area (FSC-A) signal, designated by the black box in Fig. 1b. This high-FSC-A, high-NeuN population is intended to represent large neurons, comprising 2–5% of the total population of nuclei in each sample.
The composition of the targeted population of large neurons was assessed using single-nucleus RNA transcriptomic sequencing (snRNA-seq), along with two control populations: all cells and all NeuN+ cells (each shown with respective gating boxes in Fig. 1b). snRNA-seq of these three populations of cellular nuclei was performed on a representative tissue sample (control individual 1465, prefrontal cortex). Nuclei were isolated as described above, with the following modifications: 0.2 U μl−1 Protector RNAse inhibitor (Roche RNAINH-RO) and 0.2 U μl−1 SuPERase-IN RNAse inhibitor (Invitrogen) were both added to the tissue lysis buffer and to the immunostaining buffer, and MgCl2 was omitted from the immunostaining buffer. For each of the 3 populations, 16,000 nuclei were sorted into one well of a 96-well plate, then subjected to snRNA-seq using the 10X Genomics Next GEM Single Cell 3′ GEM Kit v3.1 and Chromium Controller. From these three populations, three libraries were prepared, each with dual indexes using the 10X Genomics Dual Index Plate. Each library was then sequenced on Illumina NovaSeq S4. The raw snRNA-seq data of three 10X libraries were analysed separately and then aggregated by Cell Ranger (v.6.0.0)53, followed by variance normalization, t-SNE clustering and visualization processed by Pagoda2 (v.0.1.0)54. Clusters with 50 or more cells were manually annotated as different neuronal and glial subtypes on the basis of the expression of marker genes using a similar protocol to that described in a previous study50 These snRNA-seq data (Fig. 1c) enabled the assessment of various sorting populations shown in Fig. 1b. The full population of cells (DAPI+) contained a mixture of excitatory neurons, inhibitory neurons and glia. The overall NeuN+ population was highly enriched for neurons, but contained many inhibitory neurons and some glia. The population of cells targeted in this study, large NeuN+ nuclei, was highly enriched in pyramidal neurons, consisting of 100% neurons, of which 99.3% were excitatory neurons (Fig. 1c), with minimal inhibitory neurons and glia.
scWGS of pyramidal neurons using MDA
Single nuclei, prepared as described above, were sorted one nucleus per well into 96-well plates, with each well containing 2.8 μl alkaline lysis buffer (200 mM KOH, 5 mM EDTA, 40 mM DTT) pre-chilled on ice. Nuclei were lysed on ice for 15–30 min, then neutralized on ice in 1.4 μl neutralization buffer (400 mM HCl, 600 mM Tris-HCl, pH 7.5). These cold temperatures appear to be important to limit artefacts55. MDA was then performed in a 20 μl total reaction volume by addition of an MDA master mix (12.18 μL QIAGEN REPLI-g reaction buffer, 2.675 μl H2O, 0.105 μl DTT, 0.84 μl REPLI-g Phi29 polymerase enzyme). MDA was performed at 30 °C for 2 h. This protocol was applied to all new MDA samples in this study, and was confirmed to yield equivalent results as a prior protocol using Phi29 polymerase from a different distributor (repliPHI, Epicentre).
Samples were subjected to quality control by DNA quantification (PicoGreen, 3 μg yield required) and multiplex PCR for four random genomic loci. For an additional quality control step, we performed low coverage (0.5×) WGS, and cells with sufficiently even genome coverage (median absolute pairwise difference, MAPD; and coefficient of variation, CoV) were processed for deep sequencing. For germline reference, bulk DNA was purified using phenol:chloroform:isoamyl alcohol extraction and isopropanol precipitation, without RNAse A treatment. Amplified single-neuron genomes were prepared for sequencing by DNA shearing and libraries generated by Psomagen (Macrogen) and Novogene using Illumina Tru-Seq kits and Illumina HiSeq X10 paired end sequencing (150 bp × 2) (Supplementary Table 1), as described previously5.
scWGS of pyramidal neurons using PTA
Single neurons, prepared as described above, were sorted one nucleus per well into 96-well plates and their genomes were amplified by PTA41,42, a method that pairs an isothermal DNA polymerase with a termination base to induce quasi-linear amplification. PTA reactions were performed using the ResolveDNA Whole Genome Amplification Kit (previously known as SkrybAmp EA WGA Kit) (BioSkryb Genomics). Nuclei were sorted into 3 μl Cell Buffer pre-chilled on ice. Nuclei were then lysed by addition of 3 μl MS Mix, with mixing at 1,400 rpm performed after each step. Lysed nuclei were then neutralized with 3 μl SN1 buffer. Three microlitres of SDX reagent was then added, followed by a 10-min incubation at room temperature. Eight microlitres of reaction mix (containing enzyme) was then added, for a total reaction volume of 20 μl. Amplification was carried out for 10 h at 30 °C, followed by enzyme inactivation at 65 °C for 3 min. Amplified DNA was then cleaned up using AMPure, and the yield was determined using PicoGreen binding (Quant-iT dsDNA Assay Kit, Thermo Fisher Scientific). Samples were then subjected to quality control by multiplex PCR for four random genomic loci as previously described5, and also by Bioanalyzer for DNA fragment size distribution. Amplified genomes showing positive amplification for all four multiplex PCR loci were prepared for Illumina sequencing. In contrast to MDA, a low-coverage WGS screening step was performed.
Libraries were prepared following a modified KAPA HyperPlus Library Preparation protocol described in the ResolveDNA EA Whole Genome Amplification protocol. In brief, end repair and A-tailing were performed for 500 ng amplified DNA input. Adapter ligation was then performed using the SeqCap Adapter Kit (Roche, 07141548001). Ligated DNA was cleaned up using AMPure and amplified through an on-bead PCR amplification. Amplified libraries were selected for a size of 300–600 bp using AMPure. Libraries were subjected to quality control using PicoGreen and TapeStation HS DS100 Screen Tape (Agilent PN 5067-5584) before sequencing. Single-cell genome libraries were sequenced on the Illumina NovaSeq platform (150 bp × 2) at 30× coverage (Supplementary Table 1). Data from PTA-amplified neuronal genomes in AD were analysed alongside data from control neurons that are reported elsewhere42.
Read-mapping and generation of BAM files
Reads generated from WGS were mapped onto the human reference genome (GRCh37 with decoy) by BWA (v.0.7.15)56 with default parameters. Duplicate reads were marked by MarkDuplicates of Picard tools (v.2.8) and post-processed with local realignment around indels and base quality score recalibration using Genome Analysis Toolkit (GATK) (v.3.5)57.
Calling of sSNVs from scWGS data
We used phasing-based linked read analysis (LiRA, v.2018Feb)23 to identify sSNVs against individual-specific bulk germline reference genomes, as described previously5. The initial somatic and germline variants were called using GATK’s HaplotypeCaller and germline variants were further phased by Shapeit 2 (v.904). sSNVs were called by LiRA and distinguished from technical artefacts when showing strong evidence for only two haplotypes with paired-end, read-backed linkage between the sSNV candidate and the adjacent germline heterozygous site. The autosomal genome-wide burden of sSNVs was then calculated by accounting for the proportion of phaseable sites and estimated false positive rate. We should emphasize that the raw LiRA calls are an intermediate step that requires scaling by a power ratio to calculate genome-wide somatic mutation rates that are comparable between cells (for example from MDA data, see Extended Data Fig. 1b). Of note, LiRA is only designed to call phased somatic variants in diploid genome regions, so we only considered sSNVs in autosomes for subsequent analyses to avoid potential detection bias in sex chromosomes between male and female individuals.
Because LiRA calling requires linked heterozygous germline sites for optimal specificity and false positive rate, it may limit its detection sensitivity in regions lacking phaseable germline variants. Therefore, to more comprehensively assess sSNVs in known AD risk genes (APP, PSEN1, PSEN2 or APOE) and the tau-encoding gene MAPT, we considered both the LiRA-called variants and the larger group of GATK calls that includes non-phaseable parts of these genes. In both LiRA-called variants and GATK calls, we identified no known pathogenic sSNVs in any of these AD-related genes. The question of clonal somatic mutations in these and other AD risk genes also has been examined in other studies by bulk gene sequencing19,58,59.
Given the more even genome coverage and potentially fewer artefacts that are produced by PTA42, we used Single Cell ANalysis of SNVs (SCAN-SNV, v.2019Oct)60, which does not require phasing information from adjacent germline variants and thus has more detection power in non-phaseable regions, to identify specific genomic sites of sSNVs for mutational signature and other downstream analyses.
Determining the evenness of single-cell genome amplification
The evenness of single-cell genome amplification was quantified using two different methods (Supplementary Table 4). First, the MAPD metric was calculated as reported previously61, which is the median value across all absolute differences between log2-transformed copy number ratio of neighbouring genome bins, and a higher MAPD score represents greater unevenness of amplification. Binning, GC normalization, segmentation and copy number estimation were performed to obtain copy number ratio per bin following a previous single-cell copy number analysis protocol62, and MAPD was then calculated by taking a median of absolute difference between neighbouring bins. Second, considering that MAPD cannot reflect the variance of the copy number ratio distribution within each neuron, the CoV was also calculated by normalizing the standard deviation of absolute difference between neighbouring bins by their mean. We also calculated a ‘power ratio’ metric, which is defined as the ratio between the LiRA-estimated genome-wide sSNV burden and the LiRA-called phaseable sSNV count, reflecting the proportion of the genome that has been adequately amplified for each single cell. Using mixed-effects modelling, we measured the effect of these three metrics of genome evenness on sSNV burden in well-characterized neurotypical PFC neurons. We then normalized the mutation burden in each cell and estimated the age and disease effects on sSNV burden, as described in the section ‘Mixed-effects modelling of somatic SNV burden’.
Mutational signature analysis
To discover mutational signatures of sSNVs, we calculated the frequency of mutations in the 96-trinucleotide contexts for all control and AD neurons from the identified single-neuron sSNVs (synthesized in Extended Data Fig. 5a for MDA, and in Extended Data Fig. 8a for PTA). Mutation signatures in MDA-amplified neurons were detected by fitting a non-negative matrix factorization (NMF)-based mutational signature framework63 using MutationalPatterns (v.1.8.0)64 (Extended Data Fig. 5b). As we increased the number of signatures, we estimated the signature stability and reconstruction error of each signature and identified four signatures (N1, N2, N3 and N4) (Extended Data Fig. 5c) that maximize the number of signatures while minimizing error (Extended Data Fig. 5b). We also used a second signature derivation method, SignatureAnalyzer (v.1.1)10,65, which can infer the optimal number of signatures from data by considering both model complexity and fitting accuracy. Under default parameters with half-normal distribution for priors and reducing effect of ultramutated samples, SignatureAnalyzer produced four signatures (W1–W4) with the greatest likelihood, which are nearly identical to signatures N1–N4 that were identified by MutationalPatterns (Extended Data Fig. 5c).
We observed a marked similarity between the de novo single-neuron signatures and previously published single-neuron signatures5 (Extended Data Fig. 5c), particularly when taking into account recently identified signatures of potential single-cell artefacts24. Each newly derived signature closely resembled a previously derived one: N4 with neuron signature A, N2 with neuron signature C, N1 with neuron signature B and potential artefact signature SBS scF, and N3 with SBS scE. To understand the underlying mechanisms for the identified mutational signatures, we further performed NMF analysis to decompose our signatures into the reported the COSMIC v3 signatures (https://cancer.sanger.ac.uk/cosmic/signatures/; Extended Data Fig. 6a). We also performed NMF analysis to fit the COSMIC signatures to our composite disease and control single-neuron mutational profiles, which is shown in Extended Data Fig. 6b.
Given the near identity between the de novo and prior neuron signatures, we used the prior signatures for our subsequent analyses. On the basis of the evidence that SBS scF (highly similar to signature B) represents potential single-cell artefacts24, we excluded the contributions from these signatures in our assessment of genome-wide sSNV burden for each single neuron.
Similarly, we used MutationalPatterns to determine mutational signature contributions in PTA-amplified neurons using the signatures we identified in MDA-amplified neurons. For PTA-amplified single-neuron genomes, we did not identify significant contributions from potential artefact signatures SBS scE and SBS scF, which prompted the filtering steps for data from MDA-amplified genomes. Therefore, for PTA-amplified genomes, we report unfiltered variant calling data.
Filtering of LiRA-called somatic SNVs from MDA-amplified genomes of single neurons
Previous studies and our observations have suggested additional measures beyond LiRA to further minimize experimental artefacts that may occur during MDA amplification of single-cell genomes24. Beginning with total LiRA-called sSNVs (Extended Data Fig. 1a), we undertook a series of analyses on our human neuron MDA scWGS data, examining the influence of uneven genome amplification and the value of identification of specific mutational signatures proposed as potential artefacts of single-cell genome amplification24. We found that cells with highly uneven genome amplification (MAPD > 2.0) show increased LiRA-called sSNV counts (Extended Data Fig. 1c), including sSNVs attributable to the potential artefact signature SBS scE, largely comprising GC>GT changes (Extended Data Fig. 1d). We also observed that a small subset of neurons, only seen in AD, show an ‘ultramutated’ profile (more than 20,000 LiRA-called sSNVs; Extended Data Fig. 1a), which is dominated by SBS scE (Extended Data Fig. 1d), suggesting that these amplified genomes may show LiRA sSNV calls that do not represent biological double-stranded fixed somatic mutations. The observed variants in these outlier cells may represent experimental artefacts, including false calls due to errors occurring early in genome amplification. Alternatively, the observed scE variants may also represent non-mutation biological events, such as unrepaired single-strand damaged nucleotides, which could be misread as sSNVs owing to strand dropout during genome amplification (Extended Data Fig. 1f). Although examination of the potential biological component of this phenomenon may provide important insights, we developed a computational filtering pipeline to generate a set of filtered sSNV calls, focusing our analysis on bona fide somatic mutations (Extended Data Fig. 1g).
Mixed-effects modelling of somatic SNV burden
To evaluate the relationships between somatic mutation and factors including age and disease status, we performed linear mixed-effects regression modelling using the lme4 (v.1.1-23) R package66, in a similar manner to our previous study5. Both genome-wide sSNV burden and signature-specific sSNV burden were considered as continuous outcomes in modelling. Disease status and other covariates of interest (for example, age and measurement of amplification evenness) were modelled as fixed effects, and donor–tissue groups were modelled as random effects, because neurons from a donor and each tissue type may be correlated owing to shared biological environment. Linear mixed-effects models were fitted using the maximum likelihood method, and P values from a t-test with the Satterthwaite approximation were calculated for each fixed effect as implemented in the lmerTest (v.3.1-2) R package67. Of note, we also used the marginal generalized least-squared method to fit the mixed-effects model, using the nlme (v.3.1-137) R package, which produced substantially similar results.
To test the age effect of sSNV burden in PFC and hippocampus from neurotypical individuals, we fitted the model yijk = (β + γj)× ρi + μ + θij + εijk, where yijk is the sSNV burden in neuron k from brain region j of donor i, β is the fixed-effect of age, γj is the fixed-effect of brain region j on age indicating interaction terms of age and brain region, ρi is the age of donor i, μ is the number of sSNVs at birth, θij is the random effect of the donor–tissue pair following a normal distribution with mean 0 and variance τ, and εijk is the measurement error of each neuron also following a normal distribution with mean 0 and variance σikj (Fig. 1d–f). To control for the potential confounding factor of genome amplification evenness, we further introduced another covariate, δijk, which represents the neuron-specific measurement of amplification evenness (for example, MAPD, CoV and power ratio) into the previous model, and re-estimated the age effect by subtracting the neuron-specific contribution of the amplification unevenness coefficient from yijk (Extended Data Fig. 3a–d). We found that PFC and hippocampus show no significant difference on the age effect before and after controlling for amplification evenness (all P > 0.25), therefore we did not consider the brain region covariate in downstream modelling. In addition to the genome-wide sSNV burden, we also analysed signature-specific sSNVs with similar models (Fig. 1g).
To test the difference of sSNV burden between AD and control neurons in an age-controlled manner, we fitted the model yijk = β × ρi + αi + μ + θij + εijk, where αi is the fixed-effect of disease status (AD versus control), whereas yijk, β, ρi, μ, θij and εijk are defined as previously (Fig. 1h). We further adjusted the sSNV burden by considering the contribution of amplification evenness δijk as we estimated above, and the difference of sSNV burden between AD and control neurons remained significant in both MDA- and PTA-amplified neurons (Extended Data Figs. 3e–h, 8c–f).
To exclude the possibility that the observed sSNV burden increase in AD can be driven by systemic differences in sample or sequencing quality metrics, we further introduced ωijk into the linear mixed-effects model: yijk = β × ρi + αi + μ + θij + εijk + ωijk, where ωijk denotes one of the potential confounding factors including sex, post-mortem interval, DNA quality (DIN), sample storage time, sequencing depth, library insert size, proportion of read bases with base quality at least 20, and number of heterozygous germline SNVs (an indicator of genomic size of phaseable region). We confirmed that, in both MDA- and PTA-amplified neurons, the increased sSNV burden in AD remained significant after controlling for each (all P < 0.01). For Fig. 1j, k, we also calculated AD-attributable excess somatic mutations as the residual value for each single neuron after subtracting the age effect (β × ρi + μ) estimated from neurotypical control neurons in prefrontal cortex.
To test whether sSNV burden is associated with ApoE genotype in patients with AD, we fit the model y′ijk = ωi + θij + εijk, where y′ijk is the age-corrected sSNV burden (yijk − β × ρi) for each neuron, and ωi is the ApoE genotype of risk allele ε4 under dominant, recessive and additive genetic models. No significant association was observed in any of the three genetic models in MDA- or PTA-amplified neurons (all P > 0.21).
Gene expression analysis
To test whether somatic mutation is associated with gene expression level, we extracted the brain PFC expression data from GTEx68. The per-gene expression value was normalized for each individual after controlling for age and gender using DESeq2 (v.1.24.0)69 and averaged across all the individuals. Genes were then assigned to 10 deciles on the basis of their PFC expression levels, and all sSNV density was calculated for each decile of genes after normalizing by per-neuron sSNV detection power ratio and total gene length. To control for potential bias due to trinucleotide context and the distribution of phaseable regions (areas with sufficient sequencing coverage and an adjacent heterozygous germline SNP), we permuted the per-neuron sSNV list for 1,000 rounds by randomly shuffling the sSNVs within the phaseable regions while keeping the trinucleotide context distribution the same. We calculated the mean and standard deviation of the per-decile density in the permuted dataset, and then measured the difference between observed and expected sSNV density for each decile of AD or age-matched control group. This analysis included all brain regions in each experiment (PFC and hippocampus for MDA-based scWGS; PFC for PTA-based scWGS).
We further performed an NMF-based mutational signature analysis for sSNVs located in each decile of genes, to estimate the relative contributions of signature A, signature C, SBS scE and SBS scF for each decile. The sSNV density for each signature was calculated by multiplexing the overall sSNV density by each signature contribution.
Functional enrichment analysis
Analysis for functional enrichment of GO terms was performed using GOseq (v.1.34.1)70. For each RefSeq gene, we assigned a binary value ‘0’ or ‘1’ according to whether any sSNVs are located in the corresponding gene. Of note, this analysis is based on the LiRA output of sSNVs (signature-based filtering cannot be applied to individual genes or variants), and therefore this list may contain a small proportion of artefactual sSNVs. A probability weighting function in GOseq was applied to control for potential gene length bias. The Wallenius approximation method was used to test the enrichment of sSNVs, and the false discovery rate (FDR) method was further applied for the correction of multiple hypothesis testing. Genes without any GO annotation were ignored when calculating the total gene count. GO terms with fewer than 10 hits were excluded to avoid ascertainment bias. Very large GO terms with more than 1,000 genes were also ignored. All the GO terms with P < 0.01 in either AD or control neurons are listed in Supplementary Table 6.
Strand bias analysis
Mutations in transcribed regions of the genome may show a different density between transcribed and untranscribed strands (so-called strand bias)71,72, resulting from asymmetric mutagenesis and/or repair activity between strands. The transcriptional strands of genic sSNVs were assigned on the basis of the UCSC TxDb annotations by MutationalPatterns64. Mutated bases (‘C’ or ‘T’) on the same strand as the gene direction were categorized as ‘untranscribed’ and on the opposite strand as ‘transcribed’. Strand bias analysis was performed on the set of mutations identified in PFC and hippocampal neurons together, on the net increase (residual) of mutations in AD neurons over control neurons. Statistical significance was determined by the Poisson test.
Location of sSNVs relative to genomic features
Annotations from ANNOVAR73 were used to identify sSNVs falling in the following positions: intergenic, upstream (within 1 kb region upstream of transcription start site), 5′ UTR, exonic (coding sequence, not including untranslated regions), 3′ UTR, downstream (within 1 kb region downstream of transcription start site), splicing (within intronic 2 bp of a splicing junction), intronic. The functional interpretation was classified using four categories of SNV annotation: synonymous (SNV that does not cause an amino acid change), nonsynonymous (SNV that causes an amino acid change, excluding stop-gain and stoploss SNVs), stop-loss (nonsynonymous SNV that eliminates a stop codon), and stop-gain (nonsynonymous SNV that creates a stop codon). For exonic and UTR sSNVs, we further grouped them into 10 deciles according to their position relative to the transcript length. Similar to gene expression analysis, we used the 1,000 rounds of permutation within phaseable regions by controlling for trinucleotide context distribution, and then calculated the normalized difference (D) between observed (Nobs) and expected (Nexp) sSNV counts as below:
Modelling the accumulation of gene knockouts in neurons
Many specific heterozygous mutations could damage neuronal function39. Biallelic, exonic, deleterious ‘gene knockout’ (KO) mutations in essential genes would be especially damaging, such that there may be a threshold for the accumulation of such KO mutations above which neuronal function would deteriorate. On the basis of the number of sSNVs we identified in this report, we estimated the accumulation of gene KOs in cortical neurons, using a method described previously5. In brief, we estimated the probability of a mutation causing a gene knockout in a cell. In a diploid genome this corresponds to calculating the probability that two or more damaging mutations fall on the same gene, given the number of damaging mutations observed in a sample. This probabilistic problem can be modelled by an approximation of the birthday problem:
where n is the expected number of deleterious mutations for a given neuron. The approximation used here is different from the one published previously5 to allow for more robust approximation when 0 < n < 1. This model was further expanded to include information about genes that are intolerant to heterozygous mutations, resulting in haploinsufficiency and functional knockout. This is captured by the probability of loss-of-function intolerance (pLI) metric, with genes with a high pLI score (pLI ≥ 0.90) being less tolerant74. ExAC reported that 17% of all genes have such high pLI scores. We then used this information for the final model, written a follows:
The average was taken across all cells per individual (n > 3 cells each, with specific n shown in the Source Data for Fig. 2k) and 95% CI on those point estimates were calculated for illustration purposes. A scale factor of 100 was used to convert probabilities into percentages. To test whether there was a higher probability of obtaining a KO in AD versus controls, we used generalized estimating equations with an exchangeable working correlation structure to model the probabilities using a probit link function using the geepack (v.1.3-1) R package. Namely, we fitted the model for each donor–tissue pairing k and neuron i as follows:
with the correlation between two neurons in a donor-tissue pair defined as Corr(κk,i, κk,i′) = ρ, where κijk is the probability of a neuron having a KO mutation with the function g() being the probit link function.
Immunofluorescence microscopy for 8-oxoG as a biomarker for neuron oxidative damage
To examine whole-cell oxidation status in individual neurons in post-mortem human brain, we performed immunofluorescence staining and quantification for cellular 8-oxoG, the most frequent oxidative nucleotide product caused by ROS, under conditions known as oxidative stress. Formation of 8-oxoG is an important biomarker for oxidative status and oxidative DNA damage lesions in the cell75.
Fresh-frozen human brain PFC tissue was embedded in OCT medium and then cryo-sectioned (20 μm), with sections applied to uncharged glass slides and fixed for 10 min using 4 °C Carnoy’s fixative (60% ethanol, 30% chloroform and 10% acetic acid). Slides were washed in cold 1× PBS 3 times for 10 min each. A circle was drawn around the tissue section using a grease pen and slides were placed into a humifying chamber. Primary antibody solution consisted of: 0.2% Tween-20, rabbit anti-NeuN (1:1,000, Abcam ab177487) and mouse anti-8-oxoG (1:500, Abcam ab206461, clone 2Q2311) in blocking solution (10 mg ml−1 bovine serum albumin, 0.02 % sterile normal donkey serum, 2 mg ml−1 glycine, 2 mg ml−1 lysine in 1× PBS). Primary antibody solution was applied, and slides were sealed in a humidifying chamber and incubated at 4 °C overnight. Slides were then washed with cold 1× PBS and secondary antibody solution was applied to each slide. Secondary antibody solution: 0.2 % Tween-20, donkey anti-rabbit Alexa Fluor 488 (1:250, Thermo Fisher Scientific A32790) and donkey anti-mouse Alexa Fluor 555 (1:250, Thermo Fisher Scientific A32773) in 1× PBS. Slides were sealed in a humidifying chamber and incubated at 4 °C overnight. Slides were washed in 1× PBS then put in a dehydration series consisting of 50% ethanol (5 min), 70% ethanol (3 min × 2), 95% ethanol (3 min × 2), 100% ethanol (3 min × 2), and xylenes (5 min × 2). After the xylene step, tissue was permanently mounted using DPX and a glass coverslip. Slides were allowed to dry overnight before microscopy.
Two staining batches were performed for all cases, using an antibody master mix to reduce staining differences between slides. A middle-aged individual (46-year-old woman; case 5773) was used to establish the fluorescence exposure setting for 8-oxoG and NeuN and used for the imaging of all cases. Tissue was visualized by using a Zeiss Axio Observer 7 fluorescent microscope equipped with an X-cite Exacte 120 LEDboost lamp, Zeiss Axiocam 506 mono camera, Zen Blue 2.5 pro software and a 20× objective lens. AF488 (499ex/520 em) was paired with a 530/30 nm bandpass filter and AF555 (553ex/568em) was paired with a 582/15 nm bandpass filter channel. The top and bottom of intracellular NeuN immunoreactivity were used to establish z-stack bounds using 0.24-μm steps at 2,752 × 2,208 resolution, pixel size 4.54 μm × 4.54 μm and 1 × 1 binning. Neuron cell body 8-oxoG immunofluorescence was quantified using Fiji (ImageJ) software. For each case, n = 100 total neurons were examined and quantified for 8-oxoG (50 neurons each from two independent staining experiment batches per case). For each cell, a single z-section was chosen representing the centre of the neuron in the Z-plane. A line was drawn around the perimeter of the neuron cell body, as visualized by NeuN 488 channel. The mean grey value (absorbance units, AU) was measured within the perimeter area in the 8-oxoG 555 channel and considered the ‘intracellular signal’. The neuron perimeter object was moved to an area adjacent to the neuron with no intracellular NeuN or 8-oxoG immunoreactivity and the mean grey value was measured. This value was considered ‘background signal’ and was subtracted from the intracellular signal value. The final value was used to represent mean 8-oxoG immunofluorescence signal for the cell.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Extended Data
Extended Data Fig. 1 |. Filtering of LiRA-called sSNVs to minimize single-cell artefacts from MDA amplification.
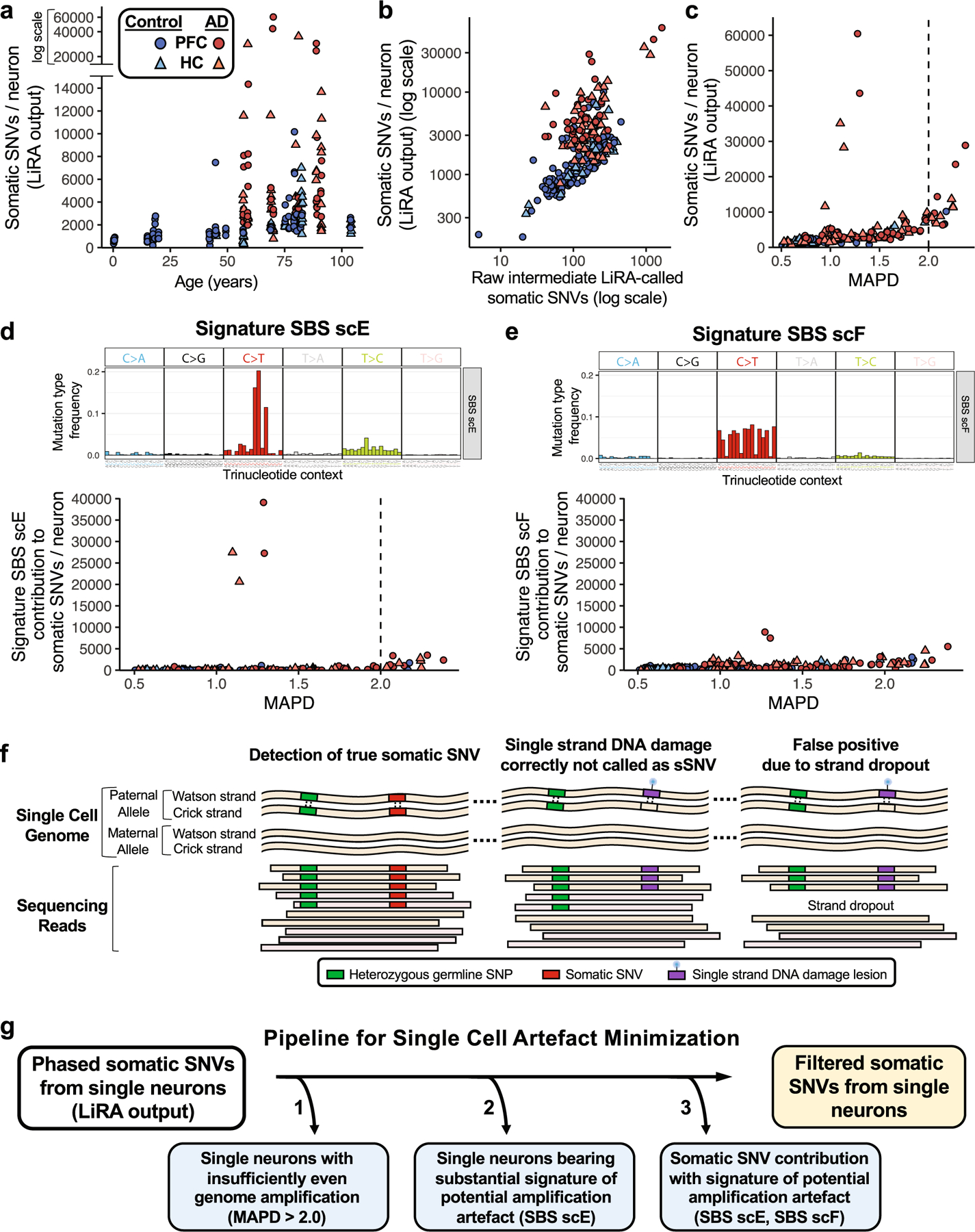
a, Total pre-filtering LiRA-called sSNV per genome for control and AD single neurons. Single neuronal nuclei from prefrontal cortex (PFC) and hippocampal CA1 (HC) underwent scWGS (45X targeted average coverage). Genome-wide counts of sSNV were determined using linked-read analysis (LiRA). Per genome sSNV counts for all control and AD neurons are shown here, prior to signature-based filtering. b, Total pre-filtering LiRA-called sSNV per genome plotted against raw LiRA-called sSNVs, an intermediate metric in the LiRA calling pipeline prior to power ratio adjustment for genome coverage and false positive rate. c, Single neuron sSNV counts in relation to coverage evenness of genome sequencing. Total pre-filtering LiRA-called sSNV counts from single neuronal nuclei are shown in relation to median absolute pairwise difference (MAPD) scores for the coverage evenness of each cell. At very high MAPD scores (>2.0), sSNV counts increase with MAPD, raising concern for artefactual sSNV calls in these cells owing to uneven genome coverage. d, e, Using NMF mutational signature analysis, the sSNV contribution was determined for two signatures potentially representing single-cell amplification artefacts: SBS scE and SBS scF24. For signature, the mutation type frequency for each trinucleotide context is shown above the sSNV plot. SBS scF is composed of C>T changes, while SBS scE is characterized by a particular subset of C>T, GC>GT. Signature SBS scE showed elevation in cells with MAPD >2.0. Signature SBS scF shows a relationship between uneven amplification (high MAPD) and SBS scF, perhaps owing to allele dropout causing single strand lesions to be read as somatic mutations. A subset of AD neurons showed LiRA-called pre-filtering sSNV counts >20,000/neuron and substantial component of potential artefact signature SBS scE. These neurons may represent an agonal ‘ultramutated’ state, but were not included in subsequent analyses owing to the abundance of potential artefact signature SBS scE (see g). f, Schematic for potential generation of artefactual sSNV in scWGS owing to uneven coverage. The scWGS LiRA platform calls sSNVs that are linked by sequencing reads to heterozygous germline single nucleotide polymorphisms (SNPs) (left). A single-stranded lesion of DNA damage, such as oxidation or alkylation, is paired with an unmodified base on the opposite genomic strand, such that LiRA would not call a sSNV under conditions of sufficiently even sequencing coverage (middle). However, if severe non-uniformity in strand-specific amplification (strand dropout) occurred, the single-stranded DNA lesion (or a polymerase error on one strand) could be erroneously called as an sSNV (right). For this reason, severely uneven single-cell genome amplification could produce artefactual LiRA sSNV calls. g, Analysis pipeline for minimization of potential artefacts of single-cell genome amplification and sequencing. Using our observations and advances reported in Petljak et al.24, we developed a computational pipeline to generate a set of higher-confidence filtered sSNV calls. This pipeline uses SNP-phased SNVs called by linked-read analysis (LiRA), and applies 3 additional specific steps to the initial variant call set: 1) Removal of single neurons which display widely uneven genome amplification, as indicated by MAPD score >2.0, above which the number of sSNVs increases (see c), raising concern for false positive variant calls due to uneven genome coverage; 2) Removal of single neurons whose mutational profile is dominated by the potential artefact mutational signature SBS scE (see d); and 3) Removal from each neuron the contribution of variants from the potentially artefactual signatures SBS scE and SBS scF. These steps produce counts of higher-confidence filtered sSNVs from single neurons. Although mutational signatures SBS scE and SBS scF have been previously reported as a potential artefact of single-cell genome amplification, the signal does potentially carry biological information. However, in this study we exclude these variants so as to minimize the influence of potential artefactual sSNV calls, to focus our analysis on the higher-confidence filtered sSNVs.
Extended Data Fig. 2 |. Single-cell variant calling identifies high-confidence sSNVs.
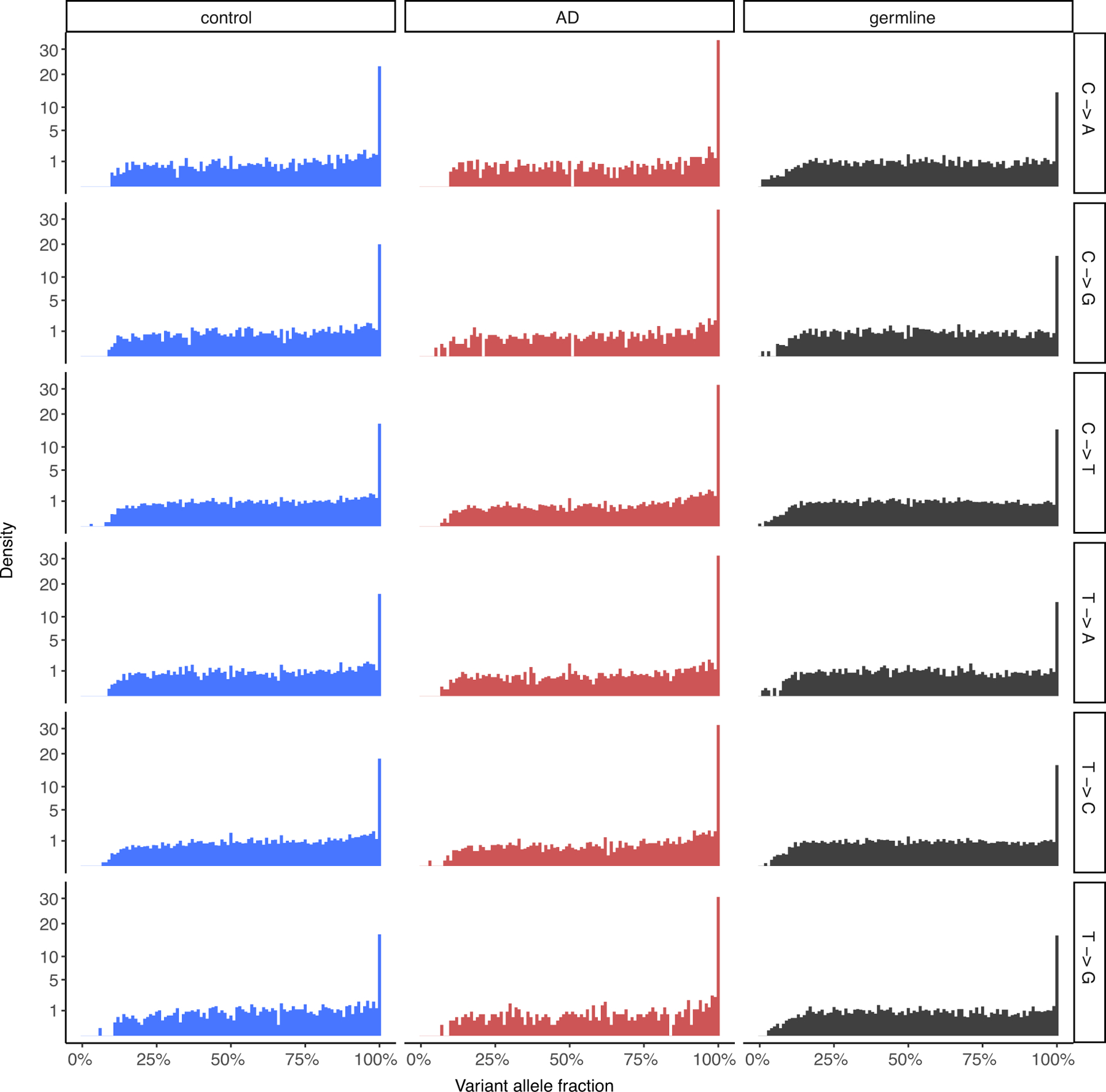
To assess the quality of the sSNVs identified from single-cell MDA-amplified WGS data, we compared their variant allele fractions in control and AD neurons to those of phaseable high-confidence heterozygous germline SNVs from the same neurons, shown for each base change type. The distributions between somatic and germline SNVs are comparable, indicating the validity of the somatic mutation calling method, as has been previously reported for the LiRA calling method5,23.
Extended Data Fig. 3 |. sSNVs in neurotypical control and AD neurons, normalized by evenness of genome amplification or LiRA caller power ratio.
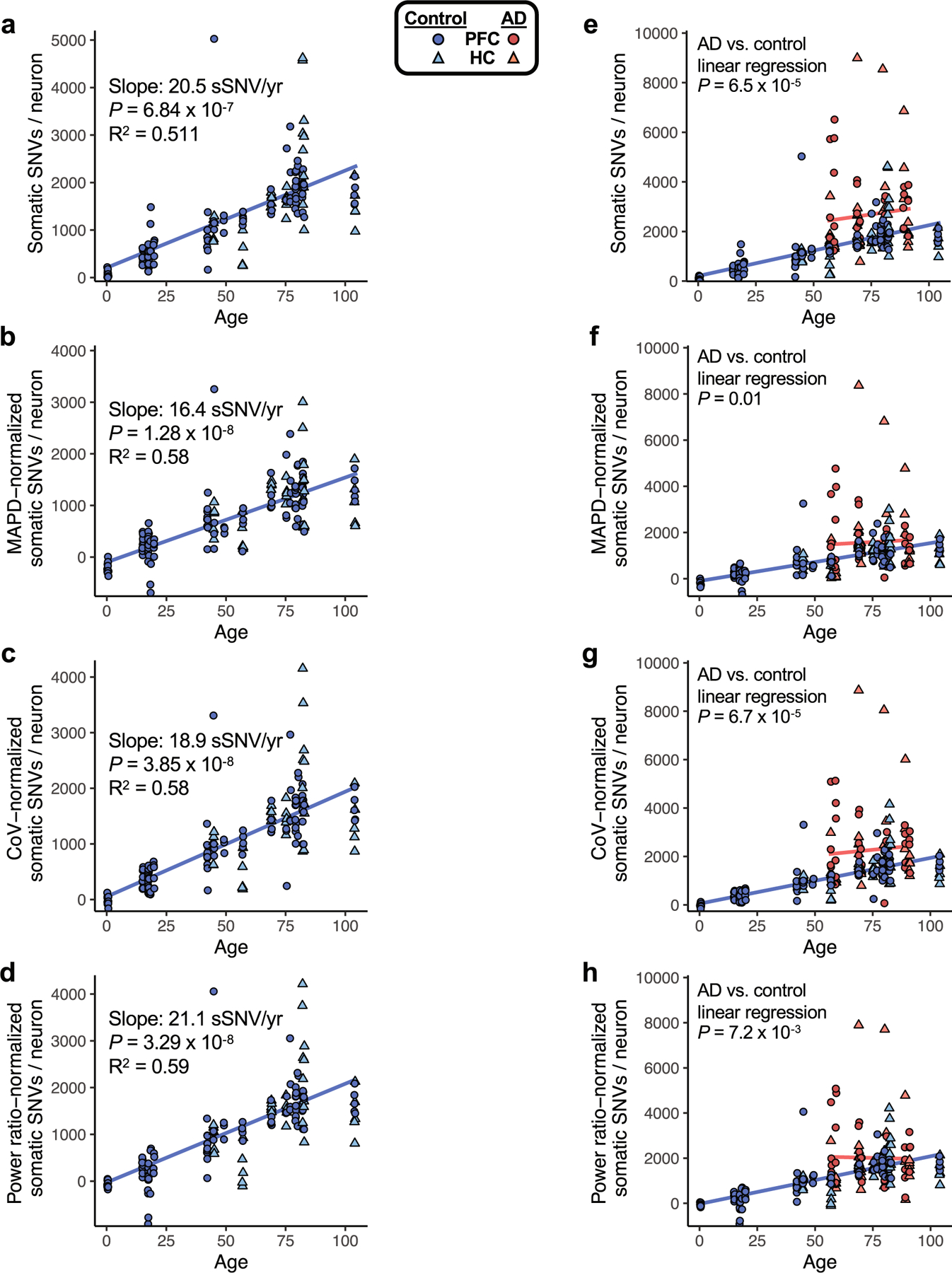
To assess the sSNV, as determined by the variant calling approach used in this study, we plotted sSNV counts from MDA-amplified single neurons against age, including using sSNV counts that were normalized for two distinct measures of evenness of genome coverage, median absolute pairwise difference (MAPD) and coefficient of variation (CoV). We also normalized by the power ratio used in LiRA phasing-based sSNV detection (see Methods). a–d, sSNVs per genome for neurotypical control neurons, with mixed-effects modelling trend lines for ageing. We observed a significant age-dependent increase of sSNV burden in each analysis, with the slope for human pyramidal neurons ranging from 16.4 sSNV/yr to 21.1 sSNV/yr, depending on the method of adjustment for genome coverage evenness. For analysis of PFC region cells alone, we observed a similar range of slopes by this analysis: 16.8 sSNV/yr to 21.3 sSNV/yr. e–h, sSNVs in AD compared to neurotypical control neurons. Unadjusted for evenness (e, reproduced from Fig. 1h, AD neurons show a mean of 2672 (range 783-8990) sSNVs, an excess of 971 over controls (P = 6.5 × 10−5, linear mixed model). f, Normalized for MAPD, AD neurons show a mean of 1582 (range 33-8366) sSNVs, an excess of 480 over controls (P = 0.01, linear mixed model). g, Normalized for CoV, AD neurons show a mean of 2264 (range 68-8861) sSNVs, an excess of 831 over controls (P = 6.7 × 10−5, linear mixed model). h, Normalized for power ratio, AD neurons show a mean of 2015 (range 162-7892) sSNVs, an excess of 511 over controls (P = 7.2 × 10−3, linear mixed model). In each analysis, AD neurons showed a significantly greater number of sSNV compared to control neurons. Although some normalizations may result in reduced detection of biological differences in AD specimens, we observed that sSNV differences are retained even after normalization, supporting a sSNV difference between AD and control neurons.
Extended Data Fig. 4 |. Distribution of sSNVs in relation to gene position comparing AD and age-matched control neurons.
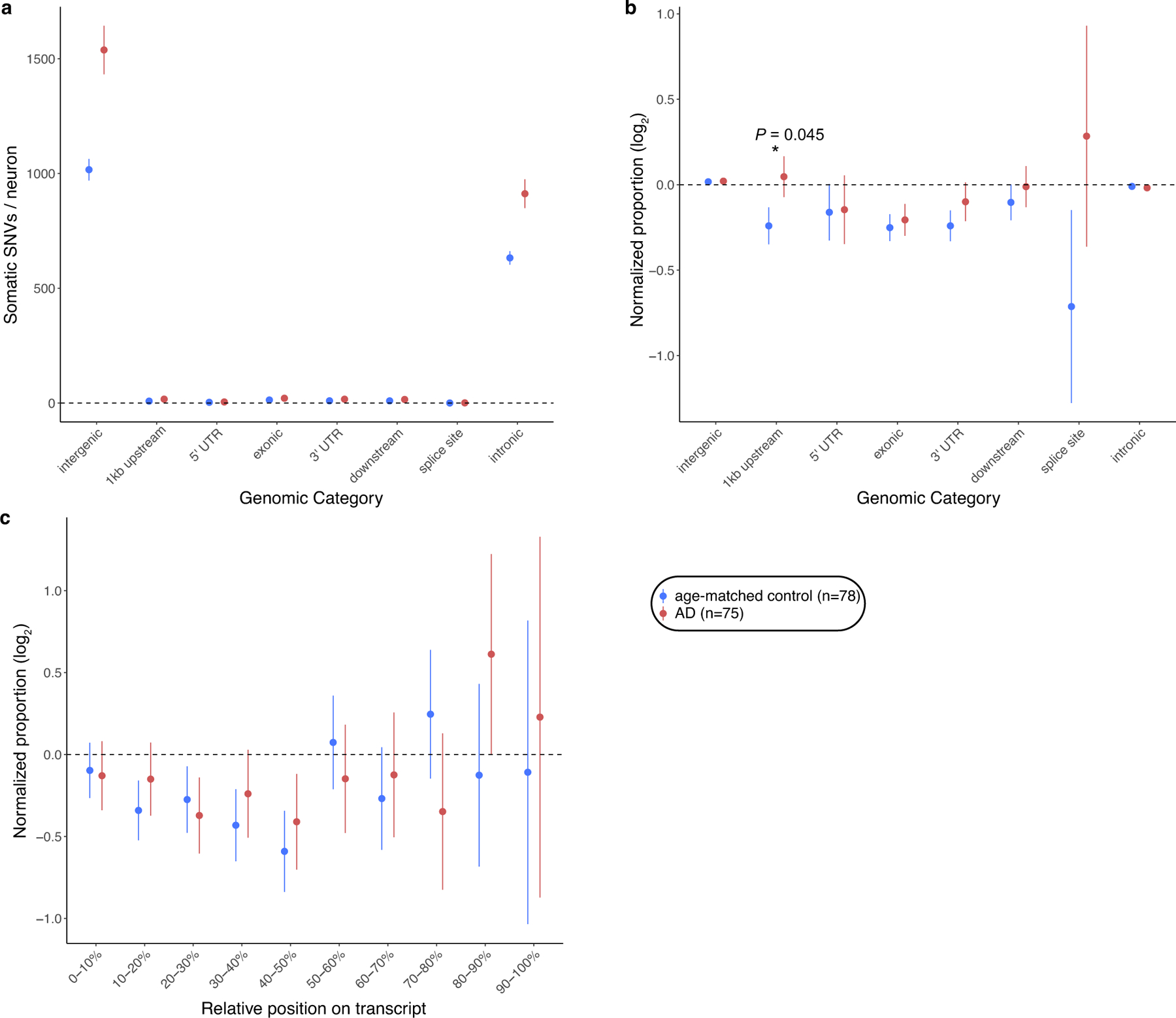
a, sSNVs per neuron across different categories of genomic regions, based on position relative to gene structure. b, Proportional distribution of sSNVs in AD and control cases across different categories of genomic regions. Upstream and downstream were defined as <1 kb genomic regions from the transcription start and end sites, respectively. Each proportion is normalized by the expected proportion after controlling for trinucleotide context of phaseable regions. c, Proportional distribution of sSNVs relative to gene transcript length. The proportions for control or AD sSNVs were normalized by the expected proportion after controlling for trinucleotide context of phaseable regions. For each set, mean ± SEM is shown. For b, c, P value is shown for the observation showing statistically significant difference between AD and control (two-tailed t-test). AD neurons show a trend of excess over controls in sSNVs in upstream positions (not surviving Bonferroni correction). Data in this figure were obtained by MDA amplification of single genomes of neurons.
Extended Data Fig. 5 |. Somatic mutation trinucleotide context profiles and signature derivation in MDA-amplified single-neuron genomes.
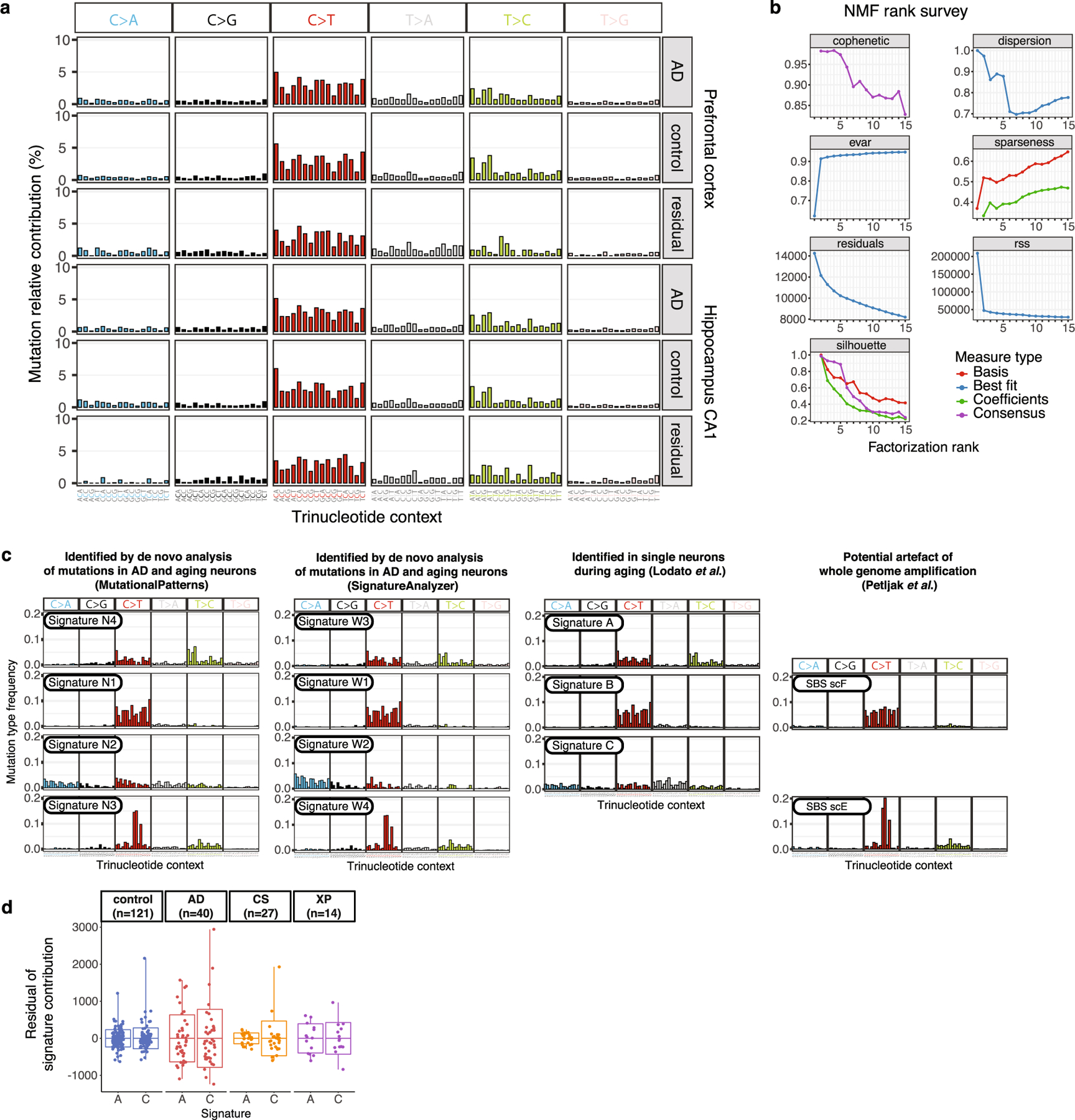
a, Trinucleotide context somatic mutation profiles in AD and control neurons. Mutations called by LiRA are shown by base substitution change (bar colour), separated for each of the 16 possible trinucleotide contexts for each substitution (96 total trinucleotide contexts). For each brain region profiled, the aggregate is shown for AD cases, neurotypical controls, and the difference (residual of cases mutations minus control mutations). b, Signature metrics for de novo mutational signature derivation from neurons in this study. Using the frequency of sSNV mutations in their trinucleotide context for all control and AD neurons, we fitted mutational signatures with a NMF-based framework. We identified four signatures, N1-N4, that maximize the cophenetic of the decomposition81. c, sSNV mutational signatures evaluated in this study. We performed de novo mutational signature generation using NMF (MutationalPatterns and SignatureAnalyzer) on the set of scWGS data from single neurons from AD and neurotypical controls, which each produced 4 highly similar signatures by best fit. Previously published analysis of single neurons (Lodato et al.)5 during ageing produced 3 signatures: A, B, and C. A recently published study of cultured cells (Petljak et al.)24 identified signatures thought to represent artefacts of scWGS, including SBS scE and SBS scF. d, Variation between neurons of mutational signature contributions. We performed linear regression for signature contribution with respect to age and disease status. The residual signature contribution of each neuron for signature A and signature C is shown here, for each disease group. Also shown are the mean (bar) ± standard deviation (boxes), with the range (whisker lines). In addition to the neurotypical control and AD neurons reported in this manuscript, we also performed this analysis on previously reported single human neuron data for two NER-deficiency diseases: Cockayne syndrome (CS) and xeroderma pigmentosum (XP)5. Because only PFC was studied for CS and XP, only the control and AD neurons from PFC were used for this analysis. For each disease group, signature C showed a greater standard deviation than signature A; standard deviation ratios between signatures C and A are as follows: 1.2 (control), 1.2 (AD), 3.2 (CS), and 1.1 (XP). Data were obtained from MDA amplification of single neuron genomes. Boxplots show mean ± SD, with whiskers denoting minima and maxima.
Extended Data Fig. 6 |. COSMIC mutational signature contributions to single-neuron signatures and disease-related mutational patterns.
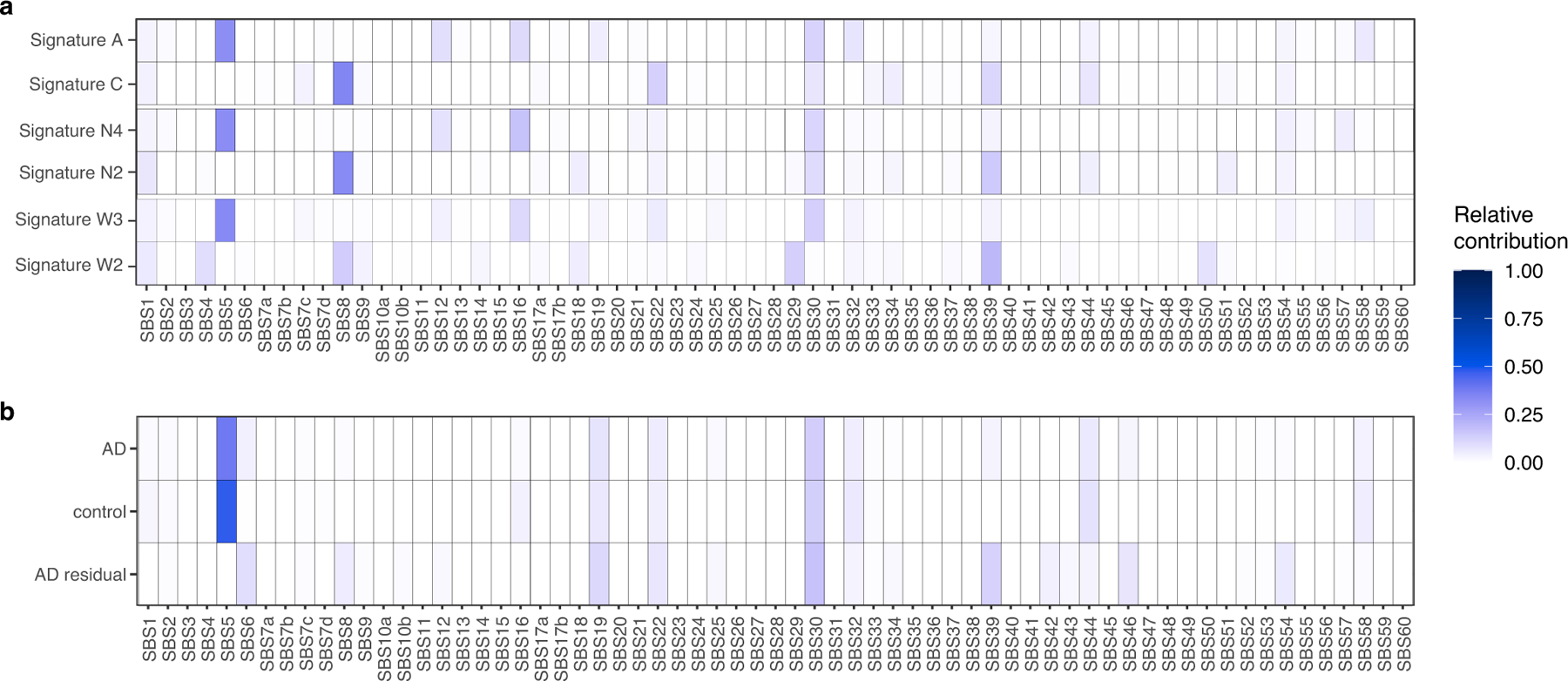
a, The set of trinucleotide contexts in single neuron signatures derived in the prior study (signatures A and C)5, along with single neuron signatures derived de novo from single AD and control neurons (signatures N4 and N2 derived using MutationalPatterns, and signatures W3 and W2 derived using SignatureAnalyzer) were analysed for contributions by COSMIC v3 single base substitution mutational signatures by NMF. The matching prior and de novo signatures show highly similar COSMIC signature contributions. b, The set of mutation trinucleotide contexts present in AD and control neuron genomes amplified by MDA, as well as the matrix of mutations obtained by subtracting control from AD (AD residual), were analysed for contributions by COSMIC signatures. Multiple COSMIC signatures identified here, many of which also contribute to signature C5, are associated with transcription-coupled nucleotide excision repair at particular damaged nucleotides with specific resultant base changes, including: SBS8 (guanine damage, C>A mutations), SBS22 (adenine damage, T>A mutations), SBS12 (adenine damage, T>C mutations), and SBS19 (guanine damage, C>T mutations). Other signatures have been associated with deficiencies of separate DNA repair processes: SBS6 (mismatch repair) and SBS30 (base excision repair). SBS5, associated with ageing, contributes significantly to the control and AD samples, but not to the AD residual mutations.
Extended Data Fig. 7 |. Immunofluorescent detection of nucleotide oxidation in neurons.
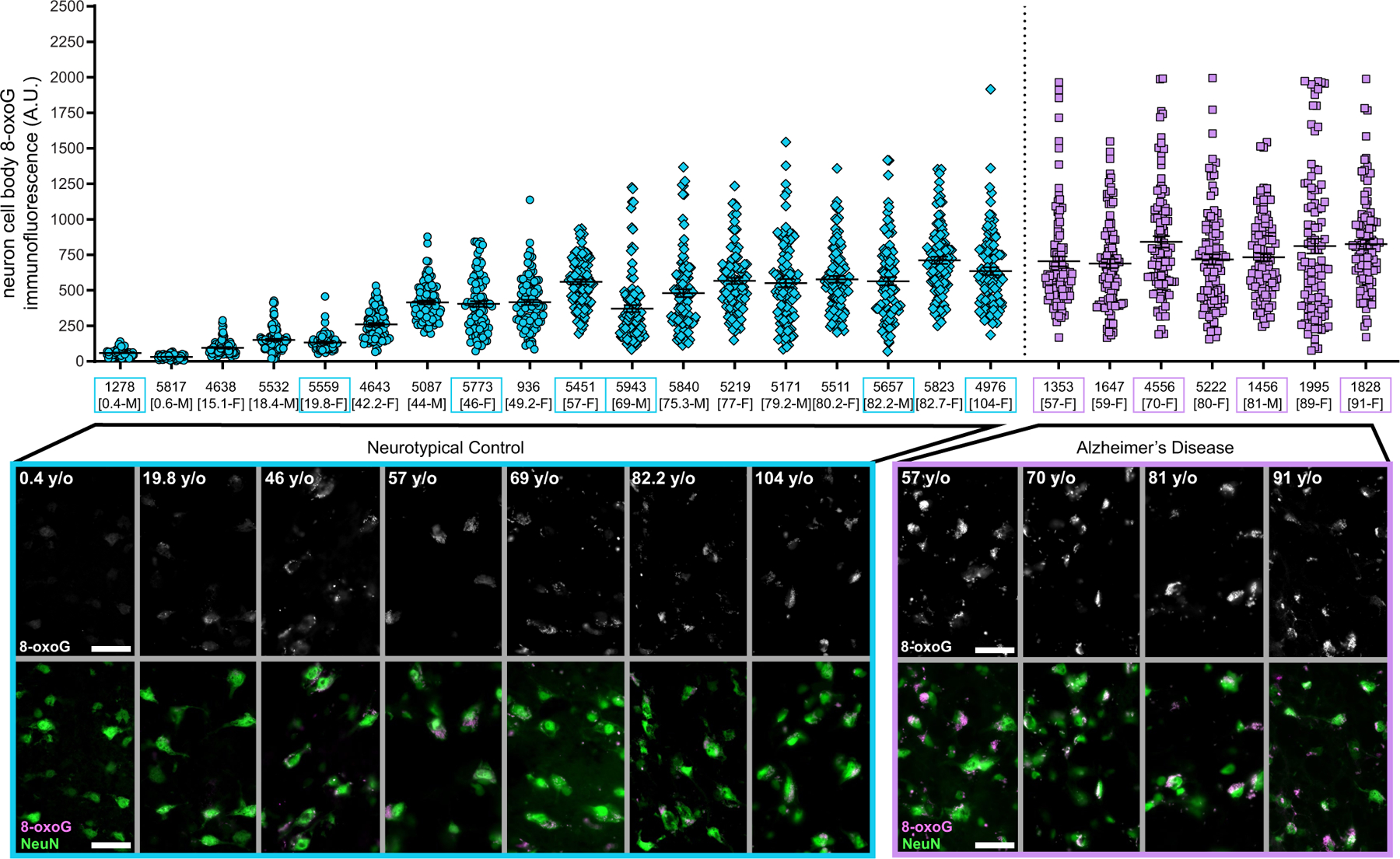
Immunofluorescence was performed on post-mortem human brain prefrontal cortex. NeuN (AF488) was used to label neurons and 8-oxoG (AF555) used to label oxidized guanine nucleotides. a, For each case sample, in a full microscopic field of up to 100 NeuN+ neurons, 8-oxoG signal was quantified per neuron. Here, each data point represents the 8-oxoG signal from one neuron, with mean and SEM shown in black for each case. Figure 2f shows mean 8-oxoG values of each case in relation to age and disease status. b, Representative microscopy images (turquoise or purple boxes) are shown for neurotypical control and AD samples from a. n = 100 total neurons examined (50 neurons each from two independent staining experiment batches per case). NeuN+ neurons are shown in green and 8-oxoG in greyscale or magenta. Scale bars represent 60 μm.
Extended Data Fig. 8 |. Features of somatic mutations in single neurons assessed by PTA.
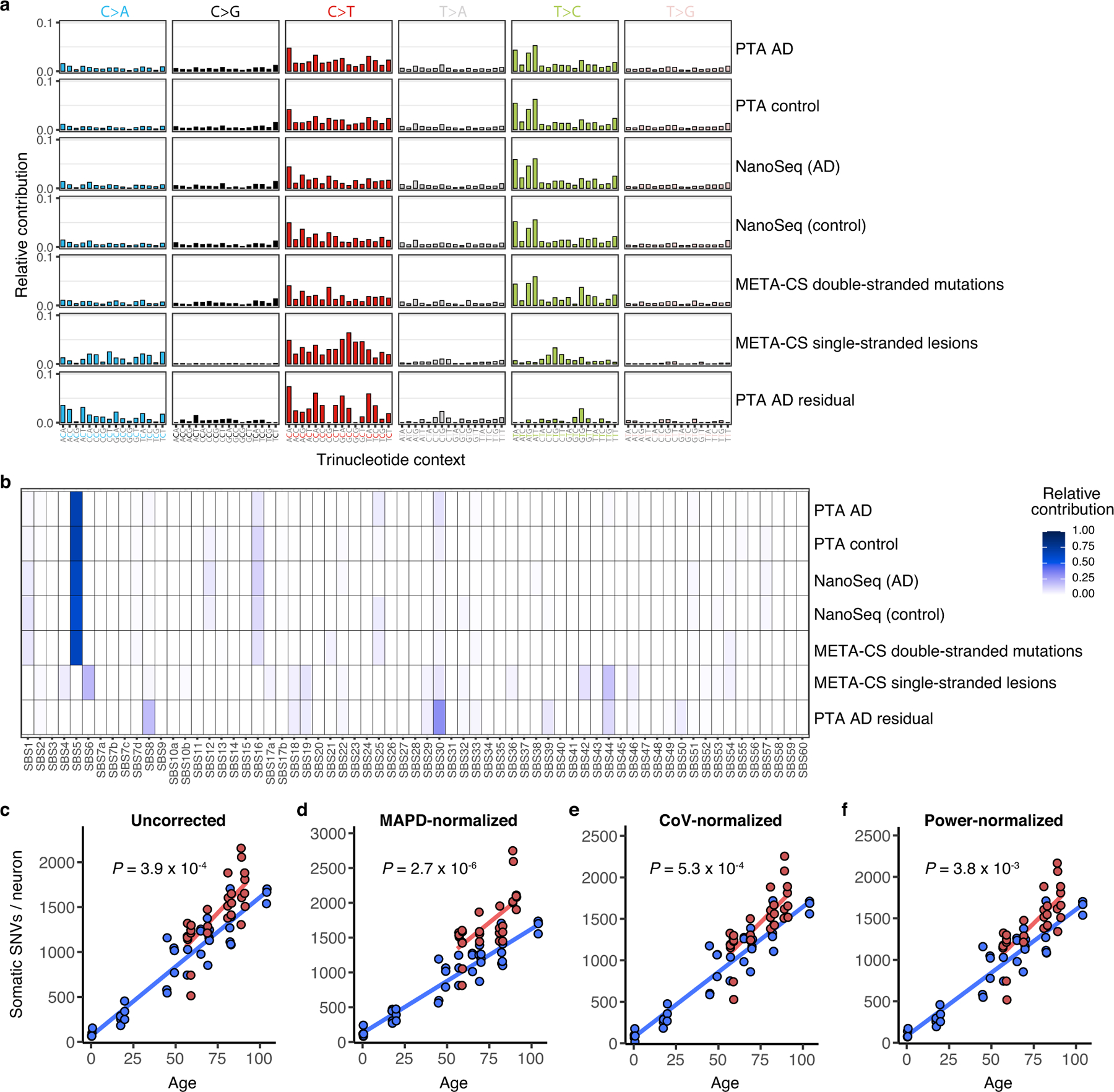
a, Trinucleotide somatic mutation spectra of cells or bulk samples studied by various methods were compared. For PTA-amplified single neurons, the aggregate of mutations is shown for AD cases, age-matched neurotypical controls, and the residual (net increase of case mutations over control mutations). Mutational spectra from other methods include NanoSeq-studied bulk samples from AD or controls and META-CS single neuron data for double-stranded mutations or single-stranded DNA lesions. Mutations are shown by base substitution change (bar colour). Of note, single-stranded DNA lesions show a distinct profile from mutations detected by PTA, NanoSeq, and META-CS. b, The spectra of mutations detected in PTA-amplified neurons (AD, control, and AD residual) and from other published methods were analysed for contributions by COSMIC cancer signatures. Elements of COSMIC signatures identified in the AD residual mutation set, including SBS8, also contribute to signature C5. Of note, single-stranded DNA lesions show a distinct profile from mutations detected by PTA, NanoSeq, and META-CS. c–f, sSNV detected using PTA in AD and neurotypical control neurons, normalized by evenness of genome amplification or LiRA caller power ratio. c, Total sSNVs per genome plotted against age (uncorrected, reproduced here from Fig. 3a for comparison). AD neurons show a mean of 1419 (range 514–2157) sSNVs, an excess of 196 over controls (P = 3.9 × 10−4, linear mixed model). d, MAPD-normalized sSNVs per genome, from which AD neurons show a mean of 1703 (range 814-2748) sSNVs, an excess of 453 over controls (P = 2.7 × 10−6, linear mixed model). e, CoV-normalized sSNVs per genome, from which AD neurons show a mean of 1440 (range 527-2255) sSNVs, an excess of 189 over controls (P = 5.3 × 10−4, linear mixed model). f, Power-normalized sSNVs per genome, from which AD neurons show a mean of 1423 (range 517–2166) sSNVs, an excess of 198 over controls (P = 3.8 × 10−3, linear mixed model). In each analysis, AD neurons showed a significantly greater number of sSNV compared to control neurons.
Extended Data Table 1 |.
| Study | Tissue/cell | Method | sSNV increase per year per cell | Mutational signatures of aging cells |
|---|---|---|---|---|
| Blokzijl et al. 20166 | adult stem cells of small intestine, colon, liver | WGS of clonal organoid cultures derived from primary multipotent cells | 35–40 | COSMIC Signature 5 |
| Hoang et al. 201676 | bulk brain (frontal cortex), colon, kidney | dilution followed by WGS (BotSeqS) | ~33 (in bulk brain) | |
| Lodato et al. 20185 | neurons (prefrontal cortex) | single-cell WGS | ~23 | COSMIC Signature 5 |
| Osorio et al. 20187 | hematopoietic stem cells | WGS on clonal cultures | 14 | COSMIC Signature 5 |
| Franco et al. 201877 | skeletal muscle resident progenitor/stem (satellite) cells | WGS on in vitro clonally expanded single cells | 13 | COSMIC Signatures 1,5,8 |
| Zhang et al. 201978 | B lymphocytes | single cell WGS | ~26 | COSMIC Signatures 1,5 |
| Lee-Six et al. 201979 | colon (crypts) | WGS of colorectal crypts, to represent clones from colorectal stem cells | >40 | COSMIC SBS5, SBS1 |
| Franco et al. 201980 | kidney tubules, epidermis, subcutaneous adipose, visceral adipose | WGS on in vitro clonally expanded single cells | ~55 (KT2) ~12 (KT1) ~20 (adipose) |
COSMIC SBS1, SBS3/8, SBS5, SBS40 |
| This study | neurons (prefrontal cortex and CA1 hippocampus) | single-cell WGS | 16–21 | COSMIC SBS5 |
Of note, COSMIC v3 single base substitution signatures SBS1 and SBS5 are similar and analogous to v2 signatures 1 and 5, respectively (https://cancer.sanger.ac.uk/cosmic/signatures). The table refers to several previous studies.
Supplementary Material
Acknowledgements
We thank R. Mathieu and L. Cheemalamarri at the Boston Children’s Hospital and Harvard Stem Cell Institute Flow Cytometry Research Facility, R. S. Hill, the Research Computing group at Harvard Medical School and the Boston Children’s Hospital Intellectual and Developmental Disabilities Research Center (IDDRC) Molecular Genetics Core for assistance. We thank C. L. Bohrson for mutational signature discussions. The brain and nuclei in Fig. 1 were illustrated by A. Lai with input from the authors, and Fig. 4 was illustrated by K. Probst (Xavier Studio) with input from the authors. Human tissue was obtained from the Massachusetts Alzheimer’s Disease Research Center (1P30AG062421-01) and the NIH Neurobiobank at the University of Maryland, and we thank the donors and families for their contributions, and J. Gonzalez and P. Dooley for assistance with tissue procurement. This work was supported by K08 AG065502 (M.B.M.); T32 HL007627 (M.B.M.); the Brigham and Women’s Hospital Program for Interdisciplinary Neuroscience through a gift from L. and T. Rand (M.B.M.); the donors of the Alzheimer’s Disease Research program of the BrightFocus Foundation A20201292F (M.B.M.); the Doris Duke Charitable Foundation Clinical Scientist Development Award 2021183 (M.B.M.); T32 GM007753 (E.A.M.); T15 LM007098 (E.A.M.); R00 AG054748 (M.A.L.); K01 AG051791 (E.A.L.); the Suh Kyungbae Foundation (E.A.L.), DP2 AG072437 (E.A.L.); R01 NS032457-20S1 (C.A.W.); R01 AG070921 (C.A.W. and E.A.L.); the F-Prime Foundation (C.A.W.); and the Allen Discovery Center program, a Paul G. Allen Frontiers Group advised program of the Paul G. Allen Family Foundation (C.A.W. and E.A.L.). C.A.W. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-022-04640-1.
Code availability
Custom Bash and R scripts used in this study are publicly available at https://gitlab.aleelab.net/august/ad-single-cell.git.
Competing interests C.A.W. is a paid consultant (cash, no equity) to Third Rock Ventures and Flagship Pioneering (cash, no equity) and is on the Clinical Advisory Board (cash and equity) of Maze Therapeutics. No research support is received. These companies did not fund and had no role in the conception or performance of this research project. The remaining authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-022-04640-1.
Peer review information Nature thanks Young Seok Ju and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
scWGS data have been deposited in the NIH Alzheimer’s disease genomic data repository, NIAGADS, under accession number NG00121. The data are available under controlled-use conditions established by the tissue banks and institutional review boards (see Methods), and can be obtained by qualified investigators at https://www.niagads.org/. Gene transcripts per million (TPM) data (V8) of GTEx samples were downloaded from https://www.gtexportal.org/home/datasets. Source data are provided with this paper.
References
- 1.Selkoe DJ & Hardy J The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 8, 595–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman BT et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braak H & Braak E Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 16, 271–278 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Gabbita SP, Lovell MA & Markesbery WR Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem 71, 2034–2040 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Lodato MA et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blokzijl F et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osorio FG et al. Somatic mutations reveal lineage relationships and age-related mutagenesis in human hematopoiesis. Cell Rep 25, 2308–2316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexandrov LB et al. Clock-like mutational processes in human somatic cells. Nat. Genet 47, 1402–1407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu T et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martincorena I et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martincorena I et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lodato MA et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hazen JL et al. The complete genome sequences, unique mutational spectra, and developmental potency of adult neurons revealed by cloning. Neuron 89, 1223–1236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhagwat AS et al. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl Acad. Sci. USA 113, 2176–2181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kucab JE et al. A compendium of mutational signatures of environmental agents. Cell 177, 821–836 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sala Frigerio C et al. On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimers Dement 11, 1265–1276 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Abascal F et al. Somatic mutation landscapes at single-molecule resolution. Nature 593, 405–410 (2021). [DOI] [PubMed] [Google Scholar]
- 21.Fu H et al. A tau homeostasis signature is linked with the cellular and regional vulnerability of excitatory neurons to tau pathology. Nat. Neurosci 22, 47–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng K et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci 24, 276–287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bohrson CL et al. Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nat. Genet 51, 749–754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petljak M et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell 176, 1282–1294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xing D, Tan L, Chang C-H, Li H & Xie XS Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl Acad. Sci. USA 118, e2013106118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madabhushi R et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min S et al. Absence of coding somatic single nucleotide variants within well-known candidate genes in late-onset sporadic Alzheimer’s disease based on the analysis of multi-omics data. Neurobiol. Aging 108, 207–209 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MH et al. Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563, 639–645 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J et al. APP gene copy number changes reflect exogenous contamination. Nature 584, E20–E28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jager M et al. Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Res 29, 1067–1077 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mecocci P, MacGarvey U & Beal MF Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol 36, 747–751 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Chun H et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H O − production. Nat. Neurosci 23, 1555–1566 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Pao PC et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun 11, 2484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nouspikel T & Hanawalt PC Terminally differentiated human neurons repair transcribed genes but display attenuated global DNA repair and modulation of repair gene expression. Mol. Cell. Biol 20, 1562–1570 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seplyarskiy VB et al. Error-prone bypass of DNA lesions during lagging-strand replication is a common source of germline and cancer mutations. Nat. Genet 51, 36–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang JC, Svoboda DL, Reardon JT & Sancar A Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5′ and the 6th phosphodiester bond 3′ to the photodimer . Proc. Natl Acad. Sci. USA 89, 3664–3668 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gate D et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soheili-Nezhad S, van der Linden RJ, Olde Rikkert M, Sprooten E & Poelmans G Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer’s disease: Implications for disease etiology. Alzheimers Dement 17, 489–499 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crabtree GR Our fragile intellect. Part I. Trends Genet 29, 1–3 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Fragola G et al. Deletion of topoisomerase 1 in excitatory neurons causes genomic instability and early onset neurodegeneration. Nat. Commun 11, 1962 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gonzalez-Pena V et al. Accurate genomic variant detection in single cells with primary template-directed amplification. Proc. Natl Acad. Sci. USA 118, e2024176118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luquette LJ et al. Ultraspecific somatic SNV and indel detection in single neurons using primary template-directed amplification Preprint at bioRxiv 10.1101/2021.04.30.442032 (2021). [DOI]
- 43.Kaur U et al. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: the NF-κB connection. Curr. Top. Med. Chem 15, 446–457 (2015). [DOI] [PubMed] [Google Scholar]
- 44.Butterfield DA, Castegna A, Lauderback CM & Drake J Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 23, 655–664 (2002). [DOI] [PubMed] [Google Scholar]
- 45.David DC et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem 280, 23802–23814 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Khurana V et al. A neuroprotective role for the DNA damage checkpoint in tauopathy. Aging Cell 11, 360–362 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Sakofsky CJ et al. Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PLoS Biol 17, e3000464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandrekar-Colucci S & Landreth GE Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 9, 156–167 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rottkamp CA et al. Redox-active iron mediates amyloid-beta toxicity. Free Radic. Biol. Med 30, 447–450 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Huang AY et al. Parallel RNA and DNA analysis after deep sequencing (PRDD-seq) reveals cell type-specific lineage patterns in human brain. Proc. Natl Acad. Sci. USA 117, 13886–13895 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dean FB, Nelson JR, Giesler TL & Lasken RS Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 11, 1095–1099 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Evrony GD et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng GXY et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fan J et al. Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong X et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods 14, 491–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keogh MJ et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun 9, 4257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park JS et al. Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat. Commun 10, 3090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luquette LJ, Bohrson CL, Sherman MA & Park PJ Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat. Commun 10, 3908 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai X et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep 8, 1280–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baslan T et al. Genome-wide copy number analysis of single cells. Nat. Protoc 7, 1024–1041 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ & Stratton MR Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3, 246–259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blokzijl F, Janssen R, van Boxtel R & Cuppen E MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 10, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim J et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet 48, 600–606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bates D, Mächler M, Bolker B & Walker S Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48 (2015). [Google Scholar]
- 67.Kuznetsova A, Brockhoff PB & Christensen RHB lmerTest Package: tests in linear mixed effects models. J. Stat. Softw 82, 1–26 (2017). [Google Scholar]
- 68.Consortium GT et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Young MD, Wakefield MJ, Smyth GK & Oshlack A Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11, R14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Green P et al. Transcription-associated mutational asymmetry in mammalian evolution. Nat. Genet 33, 514–517 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Polak P & Arndt PF Transcription induces strand-specific mutations at the 5′ end of human genes. Genome Res 18, 1216–1223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coppede F & Migliore L DNA damage and repair in Alzheimer’s disease. Curr. Alzheimer Res 6, 36–47 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Hoang ML et al. Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc. Natl Acad. Sci. USA 113, 9846–9851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Franco I et al. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nat. Commun 9, 800 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L et al. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl Acad. Sci. USA 116, 9014–9019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee-Six H et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Franco I et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol 20, 285 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brunet JP, Tamayo P, Golub TR & Mesirov JP Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl Acad. Sci. USA 101, 4164–4169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scWGS data have been deposited in the NIH Alzheimer’s disease genomic data repository, NIAGADS, under accession number NG00121. The data are available under controlled-use conditions established by the tissue banks and institutional review boards (see Methods), and can be obtained by qualified investigators at https://www.niagads.org/. Gene transcripts per million (TPM) data (V8) of GTEx samples were downloaded from https://www.gtexportal.org/home/datasets. Source data are provided with this paper.