

Abstract
Interleukin-17 receptor (IL-17R) signaling is required for control of extracellular pathogens and is also implicated in development of chronic inflammatory processes. The response to the human pathogen Helicobacter pylori results in Th1 and Th17 cell activation and a chronic inflammatory process which can lead to adverse outcomes such as gastric cancer. Previously, we identified IL-17RA as a requirement for the recruitment of neutrophils and control of H. pylori colonization in the gastric mucosa. Unexpectedly, H. pylori infected Il17ra−/− mice had significantly more chronic inflammation than H. pylori infected WT mice. In this study, human epithelial cell lines and murine models were used to investigate differential roles for IL-17A, IL-17F and IL-17A/F during H. pylori infection. Moreover, the hypothesis that IL-17RA signaling, specifically in lymphocytes, provides an autocrine feedback loop that downregulates Th17 cytokine production was investigated. The data indicate that epithelial cells exhibit a stronger response to IL-17A and IL-17A/F than IL-17F and that IL-17A and IL-17A/F can synergize with TNF and IL-22 to induce antimicrobial genes of gastric epithelial cells. In vivo deficiencies of IL-17A or IL-17F alone did not significantly change the immunopathological response to H. pylori, but if both cytokines were absent, a hyper inflammatory lymphocytic response developed. Using a cre/flox targeting approach for IL-17RA combined with infection, our findings demonstrate that increased chronic inflammation in Il17ra−/− mice was not attributed to a T cell intrinsic defect. These data imply that IL-17A and IL-17F may have overlapping roles in maintenance of the gastric mucosal response to infection.
Keywords: IL-17 cytokines, T cell cytokines, Helicobacter pylori, gastritis, inflammation
Introduction
Chronic inflammation contributes to carcinogenesis in many tissues (1). The development of chronic inflammation can be activated by factors, such as infection, smoking, obesity, high-calorie diet, genetic polymorphisms or even dysbiosis; these triggers are also recognized risk factors for the development of cancer (2). Although numerous viruses are known to be associated with cancer pathogenesis, bacteria are also emerging as significant drivers of neoplasia. Global estimates suggest that infectious agents cause an estimated 2.2 million new cancer cases annually (13% of all malignancies); of these infectious agents, H. pylori was the most prominent, accounting for about 800,000 cases of gastric cancer annually (3–6). H. pylori is the single most common risk factor for gastric cancer. Infection with H. pylori is responsible for nearly 90% of stomach cancers worldwide and approximately 37% of all infection-related cancers (6). In 1994, the International Agency for Research on Cancer (a World Health Organization agency) classified H. pylori as a Class I carcinogen. Incredibly, about 90% of non-cardia gastric cancers are also attributable to H. pylori infection. Gastric cancer is the third most common cause of cancer-related deaths in the world, killing approximately 783,000 people in 2018 (7). Other risk factors which are believed to augment the development of gastric cancer can be divided into two categories- those the host can control and those the host cannot control. Host controlled risk factors include consumption of high salt or low antioxidant diet, and smoking. Risk factors that may be imposed on the host include colonization with a strain of H. pylori which expresses the cag pathogenicity island or host genetic variations which could drive a strong immune response (i.e. polymorphism in Il1b) (8–10).
H. pylori colonization is believed to result in mild inflammation in all people. However, in some individuals this inflammation contributes to the development of peptic ulcers and gastric cancer (11). An effective adaptive immune response is essential to minimize colonization of H. pylori, but the immune response can also contribute to gastric inflammation (termed gastritis) (12). Wild-type (WT) mice infected with H. pylori also develop inflammation in response to infection. Previously, it has been shown that lymphocyte deficient Rag1−/− mice and Prkdcscid mice colonized with H. pylori develop very little inflammation (13–15), despite carrying a high level of colonization with H. pylori compared to WT mice. These data suggested that the adaptive immune response is required for the development of gastritis and the control of bacterial proliferation. In these animals, a T cell response was sufficient to drive chronic inflammation (13, 15). In humans and in animal models the proinflammatory CD4+ Th cell response includes a mixed Th1 and Th17 response (reviewed in (16, 17)). Further, regulatory T cells (Treg cells) and their production of IL-10 play an anti-inflammatory role which may limit gastritis (13, 18, 19). In fact, it has been demonstrated that when children are infected with H. pylori, they have a stronger Treg response with a concomitant decrease in Th17 responses (19, 20). Nevertheless, it should be noted that downregulating inflammation may contribute to disease persistence through inhibition of the antimicrobial activities of Th1 and Th17 responses.
Increased levels of Th17 cytokines, including production of IL-17A, IL-17F, IL-21 and IL-22 (and IL-26 in humans), are associated with more detrimental outcomes of H. pylori infection (reviewed in (16)). The Th17 response contributes to recruitment of neutrophils by activating nonhematopoietic cells to produce neutrophil recruiting chemokines, especially IL-8 (CXCL8, and its homologs) (21). IL-17A expression is commonly associated with the inflammatory response to extracellular pathogens and fungi (reviewed in(22)). Further, it is well-established that IL-17A and IL-22 act synergistically to activate several antimicrobials including S100 proteins, lipocalin 2 and some β-defensins (23, 24). Collectively, the data on Th17 cells suggests that Th17 responses are important for regulating neutrophil migration (acute inflammation) and antimicrobial responses in the gastric mucosa (16, 25, 26).
Previously, we reported on the impact of H. pylori infection in IL-17 receptor subunit A deficient mice (Il17ra−/−). In those studies, unlike H. pylori infected WT mice, H. pylori infected Il17ra−/− mice did not develop a neutrophilic infiltrate. Additionally, the Il17ra−/− mice carry a higher H. pylori burden of colonization than WT mice (27, 28), suggesting that the IL-17 signaling pathway was required for control of infection. Surprisingly, the Il17ra−/− mice had high levels of chronic inflammation. This inflammation was the result of the Il17ra−/− mice having a significant increase in infiltrating CD4+ T cells and a remarkably higher number of B cells that organized into lymphoid follicles with germinal centers. This was not observed in WT mice. Conversely, when Il17a−/− mice were infected with H. pylori by Shiomi, et al., they did not develop extensive chronic inflammation, increased H. pylori colonization, nor did they exhibit increased mononuclear cell infiltration compared to WT mice (28). Comparably, they found that H. pylori infected Il17a−/− mice had reduced neutrophils in their tissue and reduced myeloperoxidase activity (a marker of PMNs).
To create a functioning IL-17 receptor, IL-17RA complexes with IL-17RC or IL-17RB to mediate signaling (29, 30). IL-17A, IL-17F, and the IL-17A/F heterodimer mediate signaling through IL-17RA/RC complexes. Although IL-17F’s binding affinity to IL-17RA is reported to be much lower than IL-17A, the homodimers have similar affinity for the IL-17RC component of the signaling complex (31). In addition to this, IL-17A/F and IL-17F are reported to have similar immune activation profiles as IL-17A in fibroblasts, but IL-17F is reported to be less potent at similar concentrations (32). While IL-17E (also known as IL-25) signals through IL-17RA/RB complexes (33), our previously published data on H. pylori infected IL-17RB−/− mice suggest that the phenotype observed in H. pylori infected I17ra−/− is not due to a lack of IL-17E signaling (34).
This study was designed to investigate the differential roles of IL-17A, IL-17F, and IL-17A/F in controlling H. pylori colonization and H. pylori-induced gastritis. This is an area of interest, since IL-17RA is utilized by IL-17A, IL-17F and the heterodimer IL-17A/F. In addition to this, IL17a−/− and IL17ra−/− exhibit very different phenotypes with chronic H. pylori infection (27). Therefore, this study aimed also to determine if IL-17RA may contribute to the control of Th17 cytokine production during H. pylori infection. The data herein suggests a role for IL-17R signaling in downregulating the inflammatory response which can be mediated by either IL-17A or IL-17F, and further that this anti-inflammatory role is not a direct effect of IL-17RA signaling in T cells.
Methods and Materials
Ethics statement
This study was accomplished under protocol numbers V/15/130 and V1800070-00 and was approved by Institutional Animal Care and Use Committee (IACUC) of Vanderbilt University Medical Center and the Research and Development Committee of the Veterans Affairs Tennessee Valley Healthcare System. Experiments were executed in accordance with AAALAC guidelines, the AVMA Guidelines on Euthanasia, NIH regulations (Guide for the Care and Use of Laboratory Animals), and the United States Animal Welfare Act. All animals were housed in an accredited research animal facility that is fully staffed with trained personnel.
Bacterial strain and growth conditions
The Pre-mouse Sydney Strain 1 (PMSS1) of Helicobacter pylori was used in this study. PMSS1 contains a functional type 4 secretion system, which is often referred to as the major oncogenic factor of H. pylori. Bacteria were grown on Trypticase soy agar plates containing 5% sheep blood incubated at 37°C in 5% CO2 and subcultured every two days. In order to infect mice, PMSS1 was grown in liquid culture made of Brucella broth with 10% heat-inactivated FBS and 10μg/ml vancomycin. Liquid cultures were incubated in microaerophilic conditions generated by a GasPak™ EZ Campy Container System (BD), shaking at 150 rpm for 18 hrs. The H. pylori burden in the mice was measured by spreading stomach homogenate on TSA plates containing sheep blood (5%), nalidixic acid (10 μg/ml), vancomycin (50 μg/ml), amphotericin (2 μg/ml) and bacitracin (100 μg/ml) in microaerophilic conditions at 37°C for 3–5 days.
Cytokine Treatments and other Stimulants
Recombinant human IL-17A (Invitrogen), IL-17F (Tonbo), IL-22 (Peprotech) and TNFα (Tonbo) were reconstituted according to the manufacturers’ instructions in 0.01% bovine serum albumin (BSA). These were used at a range of concentrations as described in the results section and figure legends.
Epithelial Cell Culture
Gastric adenocarcinoma epithelial cells MKN-28 cells or AGS cells were grown and maintained in Gibco RPMI medium with L-Glutamine, 10% fetal bovine serum, 25 mM HEPES. The cells were maintained in tissue culture treated flasks and passaged every 3-4 days. During this seeding process, TrypLE Express [−] Phenol Red is used from Gibco were used according to the protocol on the manufacturer’s website. Cells were grown at 37°C in a 5% CO2 humidified incubator.
For the gene expression experiments, cell lines were plated in a 24-well tissue-culture treated plate. The cells were seeded in cell media at a concentration of 2.5x104 cells/well. Once confluent (24-48 hrs later) the cells were then stimulated in triplicates or quadruplicates. The PMSS1 H. pylori liquid culture, and recombinant cytokines IL-17A, IL-17F, IL-17A/F, TNFα, and/or IL-22 were diluted and resuspended in serum-free media. Upon administration of stimulant(s), the 24-well plates were placed back in the 5% CO2 humidified incubator and incubated for 8 hours before performing RNA extraction.
Animals and experimental challenge
Breeders for each species of mice were procured from multiple sources in order to generate the experimental groups for this project. Mice with genetic deficiency in interleukin-17A (Il17a−/−), interleukin-17F (Il17f−/−) and interleukin-17A/F double knockout (Il17a/f−/−) were generated by Yoichiro Iwakura and obtained through a material transfer agreement (MTA) with Research Institute for Biomedical Sciences Tokyo University of Science. Interleukin-17 receptor A deficient (Il17ra−/−) mice were generated by Jay Kolls and obtained from Amgen (MTA). CD4-Cre transgenic mice (Cd4cre) and wild-type breeders were originally from The Jackson Laboratory and have been breeding in colonies at Vanderbilt for several years. Il17rafl/fl mice were procured through an MTA with Michael Karin, University of California, San Diego. In order to generate the experimental group and littermate controls, Cd4creIl17rafl/fl were mated with Il17rafl/fl. To sort the mice, genomic DNA from the mouse tail was analyzed using standard PCR followed by gel electrophoresis. The Cre transgene was established by a strong band present at 330bp using GCCTGCATTACCGGTCGATGCAACGA and GTGGCAGATGGCGCGGCAACACCATT forward and reverse primers respectively. Il17rafl/fl mice were identified by a band located at 572bp using primer set GGGGTTTTTGTTGTTGTTGG (P1), GCAGCTGTTCTCAACCTTCC (P2) and GGCCAGGATCTACCACAAAG (P3) (35). These mice were all on the C57BL/6J background, and Helicobacter-free prior to infection. Feces from sentinel mice housed in the same room consistently tested negative for pinworms, mouse parvovirus, and several other murine pathogens. At age 8-10 weeks, mice were orogastrically inoculated with a suspension of 5×108 CFU or 1x109 CFU of PMSS1 strain of H. pylori in 0.5 ml of Brucella broth. Each dose was given twice, two days apart. The mice were then euthanized after 1 month or 3 months post infection and tissue collected for analyses.
Harvest and stomach processing
With an excision at the duodenum and esophagus, the stomach was removed from each mouse. A cross-sectional incision was made at the gastroesophageal border separating the forestomach (nonglandular portion) from the glandular stomach (antrum and corpus). The forestomach was then discarded. Another incision was made along the lesser curvature of the glandular stomach, and its contents were removed and rinsed gently in cold PBS. The organ was then divided into three longitudinal strips that included the duodenum, pylorus, antrum, corpus and squamocolumnar junction. The first strip was used to ascertain bacterial load and was placed into Brucella broth-10% FBS for immediate processing. The middle strip was used to investigate changes in histology and was placed in 10% normal buffered formalin for 24 hours, before being embedded in paraffin and processed routinely for hematoxylin and eosin (H&E) staining. The third strip was used for genetic analysis and was stored in RNALater solution at −80°C for subsequent RNA isolation. Indices of inflammation were scored using the updated Sydney System by a single pathologist (MBP) who was blind to the identity of the mice. Acute inflammation was graded based on density of neutrophils, while chronic inflammation was graded based on the density of lamina propria mononuclear cell infiltration. Both acute and chronic inflammation were graded on a 0-3 scale as follows: absent inflammation (Grade 0), mild inflammation (Grade 1), moderate inflammation (Grade 2), and severe inflammation (Grade 3) with both the antrum and the corpus scored separately. The total inflammation was calculated as a sum of acute and chronic inflammation in the corpus and the antrum, which allowed for quantification of total inflammation on a scale from 0–12.
Quantitative Real-time PCR
Tissue samples and cell were processed and RNA isolated using TRIzol™ reagent as detailed in past work (23). cDNA was synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), and diluted 10-fold for subsequent rtPCR. Using a StepOne Plus PCR machine (Applied Biosystems), TaqMan®™ assays (ThermoFisher Scientific) were used to ascertain cumulative gene expression, relative to uninfected or unstimulated control samples and normalized by Gapdh expression. Mouse primer sets were as follows: Gapdh (Mm99999915_g1), Defb14 (Mm00806979_m1), Ifng (Mm99999071_m1), Il17a (Mm00439619_m1), Il17f (Mm00521423_m1), Il21 (Mm00517640_m1) and Pigr (Mm00465049_m1). Primer sets used to analyze human cell lines are as follows: GAPDH (Hs99999905_m1), CXCL8 (Hs00174103_m1), PIGR (Hs00922561_m1), NOX1 ( Hs00246589_m1), S100A8 (Hs00374264_g1) and S100A9 (Hs00610058_m1).
Nanostring Analysis
For assessing gene expression using Nanostring, RNA was isolated from gastric tissue using the Trizol method. RNA quality and quantity were assessed using a spectrophotometer (NanoDrop™), and total RNA concentrations and size distributions were confirmed with the Bioanalyzer (Agilent Technologies) in the VANTAGE Core Facility at Vanderbilt University Medical Center. For NanoString hybridization, the input RNA was hybridized overnight (65°C) to nCounter NanoString mouse Immunology panel capture and barcoded reporter probes (CodeSet XT-CXO-MIM1). This nCounter NanoString Mouse Immunology panel analyzed the expression of 561 genes (including 14 internal reference genes). The samples were then loaded onto an nCounter SPRINT Cartridge, and the expression of the genes was determined by counting the barcodes using the nCounter SPRINT Profiler (NanoString Technologies) in the VANTAGE Core. The NanoString nSolver software (version 4.0, 2016) was used to normalize each sample based on the geometric mean of positive controls and the geometric mean of selected housekeeping genes. All NanoString counts were log2 transformed before further analyses.
Flow cytometry
To confirm that crossing the Cd4cre x Il17rafl/fl mice resulted in selective deletion of Il17ra on T cells, splenocytes from IL-17RAfl/fl mice, Cd4creIl17rafl/fl mice and Il17ra−/− mice were stained with fluorochrome-conjugated monoclonal antibodies specific for cell surface antigens. Anti-CD8a (clone 53-6.7), anti-CD4 (clone H129.19), anti-CD19 (clone 1D3), anti-IL-17RA (clone PAJ-17R), and anti-rat IgG2a (eBR2a) monoclonal antibodies were purchased from BD Biosciences. The gating strategy used for these experiments was to gate on lymphocytes by FSC/SSC, and then gate on either CD19+ cells (reported in Fig. 5) or B220−. From the CD19− gated cells, CD4+CD8a− or CD8a+CD4− cells were gated on to identify CD4+ and CD8+ cells respectively (reported in Fig. 5)
Figure 5.
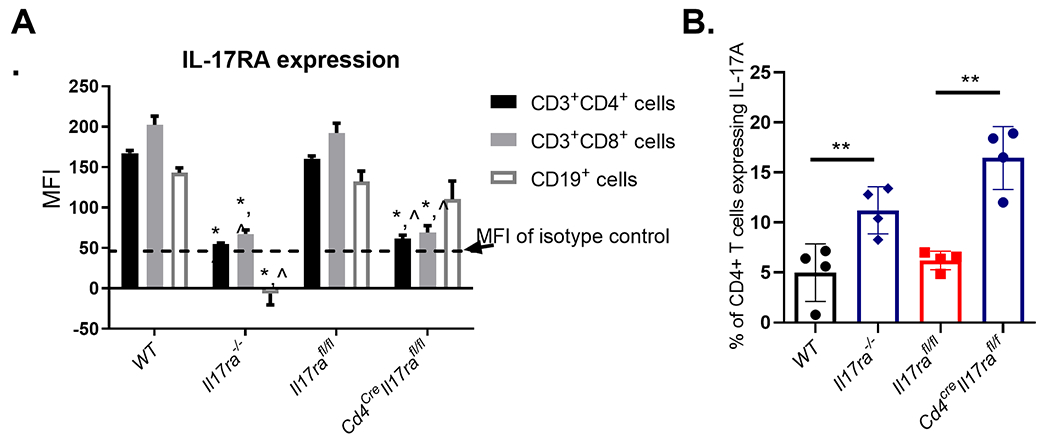
T cell specific deletion of IL-17RA results in increased IL-17A production and reduces expression of IL-17RA on CD4+ and CD8+ T cells but not B cells. A) Flow cytometry was used to measure IL-17RA expression on different white blood cell populations in the spleens of Il17rafl/fl mice compared to IL17ra−/− and Cd4creIl17rafl/fl mice. (n=3 per genotype, representative of 2 experiments). * is statistically (p<0.05) different than WT and ^ is statistically different than Il17rafl/fl using a 2way ANOVA followed by Tukey’s Multiple comparisons test. Data are representative of 3 independent experiments. B) Naïve T cells were differentiated into Th17 cells in vitro and the production of IL-17A was measured on day 3 of differentiation by intracellular cytokine staining. (n= 4 per genotype, Error bars represent the SEM.) Statistical significance compared to Il17rafl/fl samples was determined with Dunnett’s multiple comparison. Data representative of 3 experiments. *P ≤ 0.05
For intracellular cytokine staining, the single-cell suspensions were prepared from spleens of Il17rafl/fl mice, Cd4cre Il17rafl/fl mice and Il17ra−/− mice. Suspensions were treated with GolgiStop protein transport inhibitor (BD Bioscience) and restimulated with PMA-ionomycin for 6 hrs. Subsequently, surface staining with fluorochrome-conjugated monoclonal antibodies specific for cell surface antigens was performed, followed by fixation with Cytofix/Cytoperm solution (BD Biosciences, San Jose, CA, USA). The cells were then stained with anti-IL-17 in Cell Perm/Wash buffer for 30 min and washed prior to collection. Anti-CD3 (clone 145-2C11), anti-CD4 (clone GK1.1), anti-IL-17A (clone TC11-18H10), and anti-rat Ig (clone eBRG1) monoclonal antibodies were purchased from BD Biosciences. Cells were acquired in a BD LSR II instrument (Vanderbilt Flow Cytometry Core), and data were analyzed using FlowJo software (FlowJo, LCC, a BD Company). The gating strategy here was to gate on the lymphocytes by FSC/SSC followed by exclusion of doublets. CD3+CD4+ cells were gated on and the percent of those cells expressing IL-17A were then identified.
Th17 differentiation of naïve murine T cells
With modifications, Th17 differentiation was carried out as described by Bedoya et al (36) Naïve CD4 (CD4+CD25−) T cells were isolated from the lymph nodes and spleens of WT and Il17ra−/− mice using MACS Naïve CD4+ T cell isolation kit (Miltenyi Biotec) following the given protocol. The naïve CD4 T cells were cultured in a 96-well round bottom tissue culture plate bound with anti-CD3 (2 μg/ml) and anti-CD28 (0.5 μg/ml) and polarized in the presence of IL-6 (20 ng/ml), TGFβ1 (1 ng/ml), anti-IL-12 (1 μg/ml), anti-IFNγ (1 μg/ml); anti-IL-4 (1 μg/ml), and at day 2 IL-23 (10 ng/ml). Cells were incubated at 37°C and 5% CO2 for 96 hrs, then stimulated with 50 μg/ml PMA (Sigma Aldrich) and 1 mg/ml ionomycin (Fisher Scientific). GolgiStop was added 2 hours post-stimulation and the cells were incubated for another 2 hours before harvested for flow cytometric analysis or (without golgi stop) cells were processed for RNA isolation.
Results
The roles of IL-17F and the IL-17A/F heterodimer have not been investigated in the context of H. pylori infection. Although IL-17F and the IL-17A/F heterodimer have been shown to signal the IL-17 receptor (comprised of IL-17RA and IL-17RC), their contributions to the antimicrobial responses in the gastric mucosa have not be identified. In this study, MKN-28 and AGS cells, human gastric epithelial cell lines, were treated with IL-17A, IL-17F, or IL-17A/F to compare the activation of known IL-17A induced genes, including CXCL8, S100A8, and antimicrobial genes induced in the gastric mucosa, including PIGR and NOX1. Additionally, the effects of H. pylori co-culture (MOI 50) and co-stimulation with TNF (2ng/mL) or IL-22 (100ng/mL) on cytokine stimulation was investigated, due to converging of TRAF signaling and the NFκB pathway. In the MKN cells, both IL17RA and IL17RC were expressed, and IL-17A stimulation significantly down regulated IL17RC and IL17RA expression after PMSS1 treatment, but there was no significant difference with other stimuli (Table I). When measuring CXCL8 and NOX1 expression in the cell lines, responsiveness to recombinant IL-17 cytokines was low, but there was some upregulation in expression of CXCL8 with IL-17A and IL-17A/F stimulation (at 100ug/mL) (Fig. 1A, Supplemental Fig. 1). On the other hand, the polymeric immunoglobulin receptor gene, PIGR, was upregulated to significant level with IL-17A or IL-17A/F stimulation in AGS cells but not MKN cells. Despite this, expression of S100A8 was not induced in AGS cultures with IL-17A, IL-17F or IL-17A/F alone and only minimally induced in MKN cells (Supplemental Fig. 1); this is consistent with previous studies which reported that a co-stimulus may be necessary for activation of the IL-17R pathway and subsequent antimicrobial expression (23).
Table I.
Expression of IL17RA and IL17RC on MKN cells with and without stimulation.1
| PMSS1 (moi 50) | PMSS1 + IL-17A | TNF (2ng/mL) | TNF + IL-17A | IL-17A (50ng/ml) | |
|---|---|---|---|---|---|
| Gene | RU (± SD) | ||||
| IL17RA | 1.04 (± 0.18) | 0.60 (±0.15) | 1.32 (±0.18) | 0.80 (±0.20) | 0.74 (±0.28) |
| IL17RC | 1.26 (± 0.53) | 0.64 (±0.13) | 1.32 (±0.14) | 0.91 (±0.17) | 0.55 (±0.21) |
Relative Units of gene expression of IL17RA and IL17RC in human gastric epithelial cell line, MKN cells with different stimuli (or co-stimulation). RU (± Standard Deviation) are presented. RU are relative to untreated controls.
Figure 1.
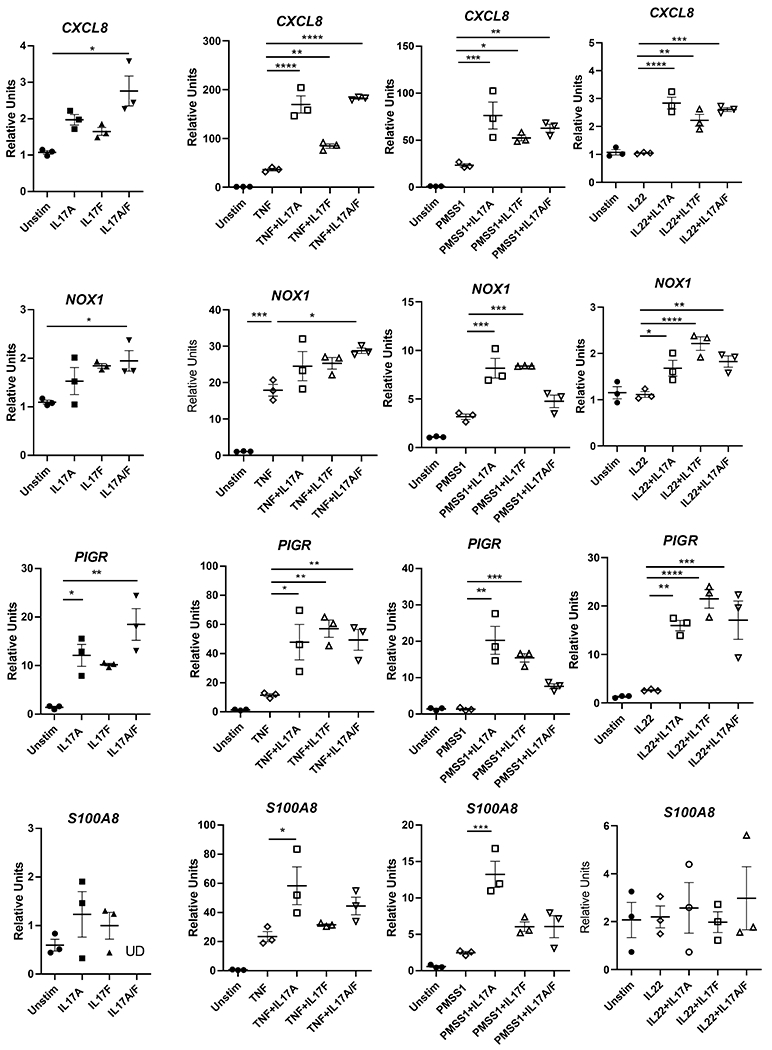
Gastric epithelial cells, AGS cells, response to IL-17A, IL-17F or IL-17A/F. A. AGS cells were simulated with IL-17A, IL-17F or IL-17A/F (100ng/mL). B. Various stimuli (IL-17A at 100ug/mL, IL-17F at 100ug/ml, TNFa at 2ng/mL, IL-22 at 200ug/ml, or PMSS1 at an MOI of 50) were added to serum-starved AGS cells for 8 hours. Quantitative real-time PCR was performed to measure expression of CXCL8, PIGR, NOX1 and S100A8 in response to these stimuli or co-stimuli. Expression is shown as relative units and is relative to RNA expression from cells treated with equal concentration of the carrier (BSA). Data shown as ± SEM and are representative of 3 independent experiments. Statistical analysis was performed with one-way ANOVA and Tukey’s test for multiple comparisons.
To explore how the IL-17 cytokines impact epithelial cell activation in response to a co-stimulus, gastric epithelial cells were stimulated with TNF, PMSS1 or IL-22 in concert with the IL-17 cytokines. These data indicate that TNF is a potent, synergizing stimulus for CXCL8 and PIGR expression. While TNF activated low expression of NOX1 and S100A8, the synergistic response was not as pronounced compared to CXCL8 or PIGR. Using H. pylori (strain PMSS1) as a co-stimulus resulted in greater synergy with IL-17A than the other IL-17 cytokines (i.e. CXCL8, PIGR, NOX1, S100A8). In general, it appears that IL-17A/F provides the strongest stimulus alone, but when a co-stimulus is used IL-17A has similar or more potency. For example, in the case of PIGR expression, while PMSS1 and IL-17A still induced PIGR expression, the combination of PMSS1 and IL-17A/F reduced PIGR expression compared to IL-17A or IL-17F. Further, CXCL8 gene expression is upregulated more with IL-17A or IL-17A/F than IL-17F when TNF or IL-22 provides a co-stimulus. Further, expression of S100A8 and CXCL8 in the gastric epithelial cells were activated upon stimulation by PMSS1 alone, IL-17A alone, and upon co-stimulation by TNF and IL-17A. IL-17F did not induce these genes in MKN cells. (Fig 1B).
In vivo, the potential for differential roles of these cytokines was assessed by infecting IL-17A, IL-17F, IL-17A/F and IL-17RA deficient mice with H. pylori strain PMSS1. Since H. pylori infection chronically persists and chronic gastritis is commonly assessed at 3 months post infection, we have focused these studies on the 3-month post infection time point. The ability of these genotypes of mice to control H. pylori colonization was measured by serial plating of stomach homogenates. At 3 months post infection, there was no significant difference in the ability of the knockout mice to control H. pylori infection compared to WT mice.
In our previous findings, we reported that Il17ra−/− mice develop significantly lower levels of PMNs and their acute inflammation scores are significantly reduced compared to WT mice (27). To determine if deficiency of IL-17A or IL-17F alone lead to a change in acute inflammation (or the presence of extravascular polymorphonuclear neutrophils on a scale of 0-3), the levels were scored by a blinded pathologist at 3 months post infection in the antrum and the corpus as previously described (37). Deficiency in IL-17A, IL-17F or both IL-17A/F lead to a significant decrease in acute inflammation scores compared to WT mice (Fig. 2B).
Figure 2.
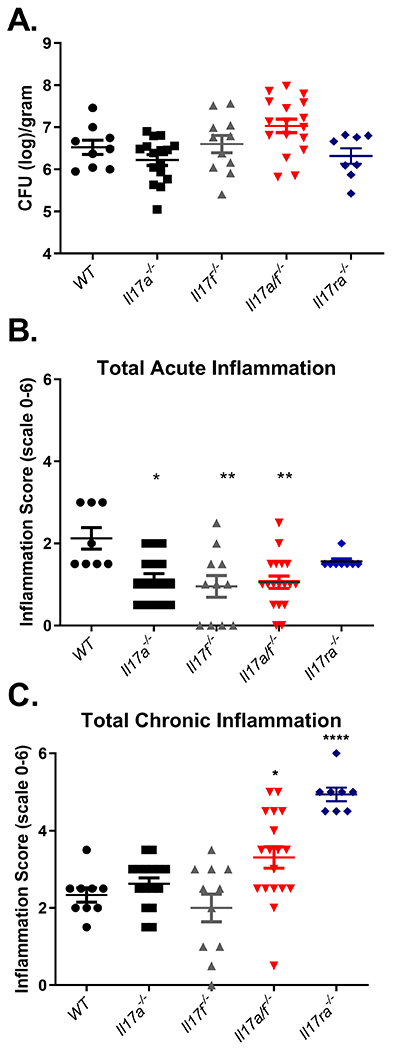
During Helicobacter pylori infection, both IL-17A and IL-17F can independently mediate acute inflammation, but the presence of either regulates chronic inflammation. A) Bacterial burden was measured in WT, Il17a−/−, Il17f−/−, Il17a/f−/− and Il17ra−/− mice that were infected with PMSS1 for 3 months (8-18 mice per genotype). Colony forming units (CFU) per gram of stomach tissues was calculated and is presented in the graph. Statistical analysis was performed using Dunnett’s Multiple comparisons test. B and C) Levels of acute and chronic inflammation were scored on stomach tissue (in the corpus and antrum) at 3 months post infection with strain PMSS1. Statistical analysis was performed using Kruskal-Wallis test, and error bars represented mean ± SEM. See methods for scoring system (scale is 0–12). *P ≤ 0.05, **P≤0.01, ****P≤0.0001. Data combines 2 independent experiments.
Chronic inflammation associated with H. pylori was scored using the Sydney system and to assess the ability of these different strains of mice to develop chronic gastritis as a marker of T and B cell infiltration. Deficiencies in IL-17A or IL-17F alone (Il17a−/− or Il17f−/−) did not significantly impact H. pylori induced chronic inflammation during infection, but a deficiency in both IL-17A and IL-17F (Il17a/f−/−) or a deficiency in IL-17 receptor signaling (Il17ra−/−), led to significantly increased chronic inflammation during H. pylori infection compared to wild-type infected mice (Fig 2C). Lymphoid follicles and plasma cells are significantly increased in abundance in the Il17ra−/− mice compared to other genotypes (Table II), and while there was a trend towards increased lymphoid follicles in Il17a−/− and Il17a/f−/− mice, the differences were not significant.
Table II.
Lymphoid Follicles and Plasma Cells during H. pylori infection.1
| WT | IL-17A−/− | IL-17F−/− | IL-17A/F−/− | IL-17RA−/− | |
|---|---|---|---|---|---|
| Pathological finding | |||||
| Lymphoid Follicles(% of animals in the group) | 10% | 50% | 0% | 20% | *75% |
| Ave. Score Plasma Cells (Scale is 0-3 ±SEM) | 0.9 (±0.87) | 1.0 (+0.26) | 0.417 (±0.154) | 0.955 (±0.157) | *2.5 (±0.283) |
A fisher’s exact test was used to determine if lymphoid follicles formed in the different mouse genotypes more frequently than in WT mice at 3 mo post infection. There was a significant increase in lymphoid follicle formation in IL-17RA−/− mice compared to WT mice. The pathology score for plasma cells is significantly different when data analyzed by 1way ANOVA followed by a Dunn’s multiple comparison test, WT vs. IL-17RA−/−
While control of H. pylori colonization has often been inversely correlated to acute inflammation, that is not consistent in these experiments. This led us to investigate whether host factors involved in defense of the mucosal barrier were impacted in Il17a−/−, Il17f−/− and Il17a/f−/−. Quantitative real-time rtPCR (qPCR) was perform on RNA isolated from the stomachs of mice at 3 months post infection. Expression of Pigr, which encodes the polymeric immunoglobulin receptor, was reduced in Il17ra−/− and Il17a/f−/− compared to WT mice, but Il17f−/− and Il17a−/− mice levels were not significantly reduced (Fig. 3A). There was a trend toward reduced expression of Defb14 in Il17ra−/− and Il17a/f−/− but there was no significant change. Interestingly, Il17a−/− produced significantly higher levels of Defb14 (Fig. 3B). These data suggest that Defb14 may be activated by either IL-17A or IL-17F, but also may suggest that IL-17F is a strong activator of Defb14 since the Il17a−/− mice had high levels of this mRNA. Further, these data suggest that Pigr may be regulated either by IL-17 signaling or through the pathological response.
Figure 3.
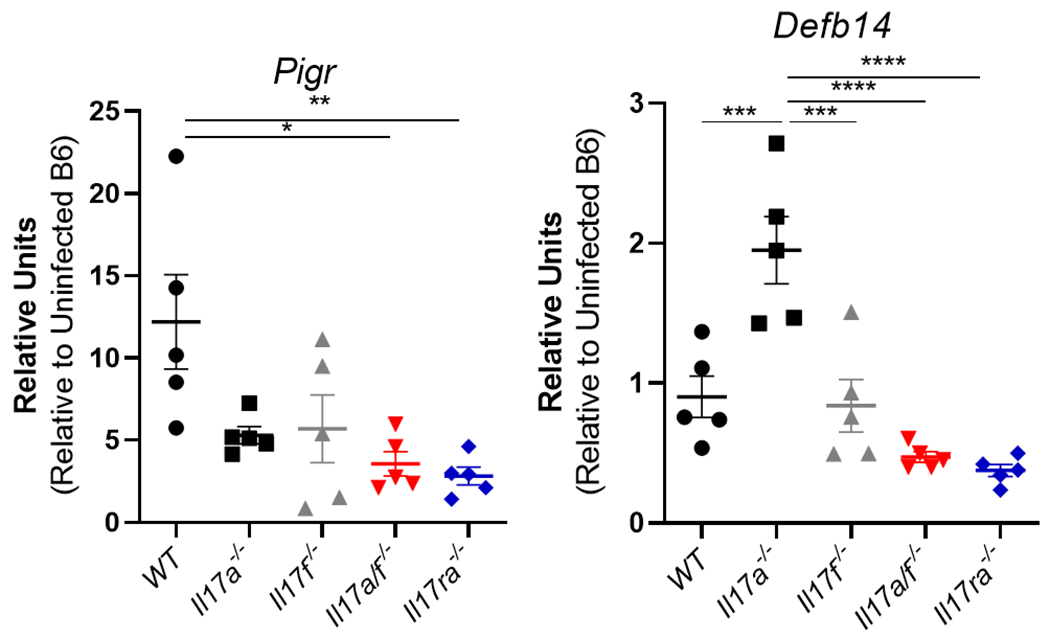
Antimicrobial response including Pigr expression are lower in Il17ra−/− and Il17a/f−/− mice. A) At 3 months post infection, Pigr was measured in gastric tissue of H. pylori–infected WT, Il17a−/−, Il17f−/−, Il17a/f−/− and Il17ra−/− mice (n=5 per genotype). Relative units are calculated as described in the methods, relative to Gapdh expression and calibrated to uninfected WT mice. These data were not performed on additional mice (See Table III for reproducibility) B) At 3 months post infection, Defb14 was measured in H. pylori–infected WT and these other genotypes (n=5 per genotype). Relative units are calculated as described in the methods, relative to Gapdh and calibrated to uninfected WT mice. Statistical significance among groups was determined with Dunnett’s multiple comparison. Error bars represent ± SEM; *P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001 compared to WT.
H. pylori infected Il17ra−/− mice were reported to have very high levels of T helper cytokines including Il21, Il17a and Il17f in the gastric mucosa by 3 months post infection (27). To investigate if T cell cytokine responses in the H. pylori infected Il17a−/−, ll17f−/−, Il17a/f−/− and Il17ra−/− mice were elevated compared to H. pylori infected WT mice, qPCR was performed on gastric tissue at 3 months post infection. There are no significant differences in Ifng expression. Gene expression of Il21 is only significantly higher in the Il17ra−/− mice compare to WT mice (Fig. 4A, 4C). Due to the variability in Il21 levels in these mice at this chronic time point, the correlation between Il21 expression and chronic inflammation was tested. There is a significant correlation between chronic inflammation and relative units of Il21 expression in the gastric tissue (Fig. 4B). Further, H. pylori infected Il17ra−/− mice had significantly increased expression of Il17a and Il17f compared to H. pylori infected WT mice which is similar to what had been observed previously when Il17ra−/− were infected with SS1 (a type 4 secretion system deficient H. pylori strain) ((27), Fig. 4D).
Figure 4.
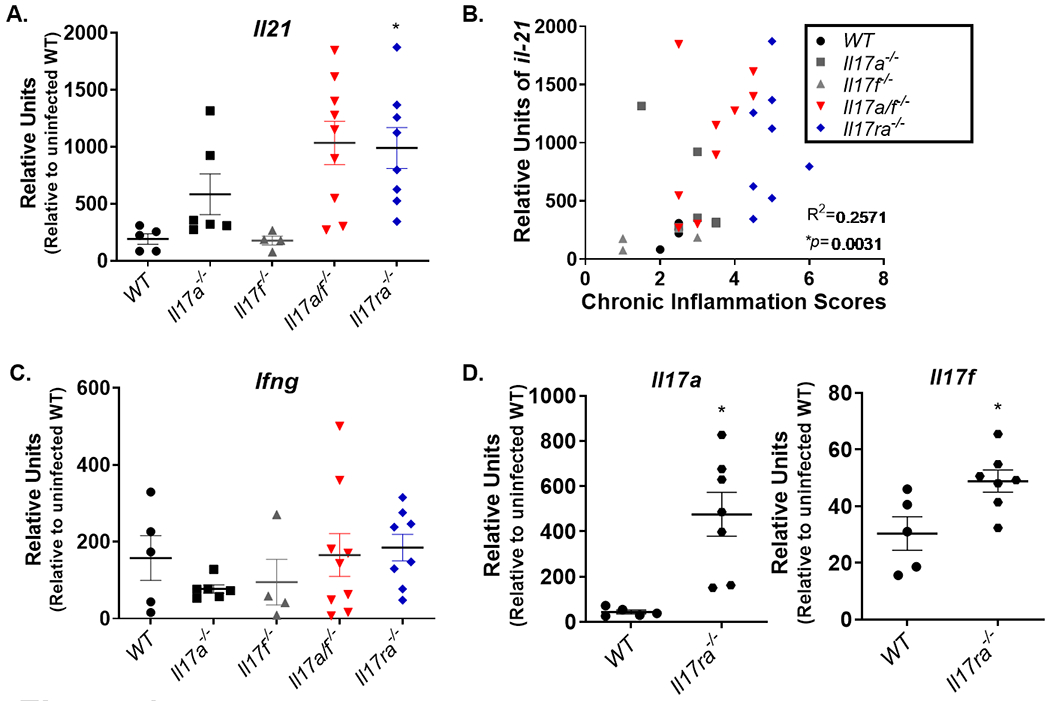
The absence of both IL-17A and IL-17F signaling results in a hyper inflammatory response to H. pylori infection. A) At 3 months post infection, Il21 expression was measured in gastric tissue of H. pylori–infected WT, Il17a−/−, Il17f−/−, Il17a/f−/− and Il17ra−/− mice (n=5-8 mice per genotype). Relative units are calculated as described in the methods, relative to Gapdh and calibrated to uninfected WT mice. B) The correlation was measured between Il21 expression and inflammation scores, using the nonparametric Spearman correlation test. C) At 3 months post infection, Ifng expression was measured in gastric tissue of H. pylori–infected WT, Il17a−/−, Il17f−/−, Il17a/f−/− and Il17ra−/− mice (n=5-8 mice per genotype). D) Gene expression of Il17a and Il17f was measured by qPCR in WT and Il17ra−/− mice. Relative units are calculated as described in the methods, relative to Gapdh and calibrated to uninfected WT mice. Statistical analysis for realtime rtPCR was performed using one-way ANOVA with Tukey’s multiple comparisons test Error bars represent ± SEM; Data are representative of two independent experiments.
To investigate the immune activation in the gastric mucosa and to consider other pathways contributing to the exacerbated immunopathological outcome in the Il17ra−/− mice, we performed a broader gene expression analysis on gastric tissue from Il17ra−/− mice and WT mice using NanoString (Ms_Immunology Gene Panel of >500 genes, Table III and Supplemental Table I). Many of these findings are consistent with previous observations (27); for example, there were significant decreases in expression of neutrophil markers/antimicrobials S100a8 and S100a9 in Il17ra−/− mice compared to WT mice. Further, these data support the hypothesis that several subsets of T cells are more activated and/or increased in abundance in the gastric tissue of Il17ra−/− compared to WT mice, including Tfh cells (Cxcr5, Maf, Pdcd1), Th2 cells (Gata3, Irf4), and Th17 (Rorc). There is no evidence of increased innate lymphocytes in the gastric tissues of Il17ra−/− mice (Supplemental Table I). These data suggest that in the absence of IL-17RA, H. pylori infected mice develop lymphoid follicles with germinal centers activating Tfh cells, IL-21 expression, B cells and antibody production (27).
Table III.
Nanostring (Ms_Immunology Panel) was used to determine the Ratio and Log2Fold change of abundance of transcripts of genes associated with T and B cell responses in Il17ra−/− vs WT (C57BL/6J) gastric tissue at 3 months post infection with PMSS1 strain of H. pylori. Values highlighted in red were significantly different between genotypes.
| T CELL SUBSET | GENE | % SAMPLES ABOVE THRESHOLD | RATIO | LOG2 FOLD CHANGE | P-VALUE |
|---|---|---|---|---|---|
| T-CELLS | Cd3ε | 91.67 % | 3.16 | 1.66 | 0.00615821 |
| T-CYTOTOXIC CELLS | Cd8a | 66.6.7 % | 2.4 | 1.26 | 0.08565527 |
| T-HELPER CELLS | Cd4 | 100 % | 2.53 | 1.34 | 0.00077244 |
| TFH | Bcl6 | 100 % | 1.22 | 0.29 | 0.13560879 |
| TH1 | Tbx21 | 8.33 % | 1.09 | 0.12 | 0.75548154 |
| TH2 | Gata3 | 100 % | 1.5 | 0.58 | 0.02776563 |
| TH17 | Rorc | 100 % | 1.23 | 0.3 | 0.04490001 |
| TREG | Foxp3 | 41.67 % | 1.25 | 0.32 | 0.3662084 |
| Gene | T cell subset | % samples above threshold | Ratio | Log2Ratio fold change | p-value |
|---|---|---|---|---|---|
| Tbx21 | Induces Ifng (Th1) | 8.33 % | 1.09 | 0.12 | 0.75548154 |
| Stat1 | Induces Ifng (Th1) and Tbx21 | 100 % | 2.9 | 1.54 | 0.00953628 |
| Irf1 | Induces Ifng | 100 % | 1.76 | 0.82 | 0.0477938 |
| Eomes | Induces Ifng | 66.67 % | 1.18 | 0.24 | 0.45388737 |
| Gata3 | Regulates Th2 | 100 % | 1.5 | 0.58 | 0.02776563 |
| Stat5b | Th2 differentiation | 100 % | 1.11 | 0.16 | 0.12631282 |
| Stat6 | Induces Gata3 | 100 % | 1.14 | 0.19 | 0.18275337 |
| Irf4 | Upregulates Gata3 | 100 % | 2.6 | 1.38 | 0.00053553 |
| Rorc | Promotes Th17 | 100 % | 1.23 | 0.3 | 0.04490001 |
| Stat3 | Induces Th17 | 100 % | 1.3 | 0.38 | 0.09895591 |
| Il17a | Th17 hallmark cytokine | 100% | 4.53 | 2.18 | 0.00007938 |
| Foxp3 | Induces Treg differentiation | 41.67 % | 1.25 | 0.32 | 0.3662084 |
| Bcl6 | Tfh mature | 100 % | 1.22 | 0.29 | 0.13560879 |
| Cxcr5 | Tfh mature | 50 % | 7.61 | 2.93 | 0.00025311 |
| Maf | Tfh | 100 % | 1.69 | 0.75 | 0.00020007 |
| Pdcd1 | Tfh or exhausted T cell | 83.33 % | 2.73 | 1.45 | 0.00062396 |
| Il21 | Th17/Tfh cell cytokine | 100% | 2.25 | 1.17 | 0.01160472 |
| B CELL RESPONSES |
GENE | % SAMPLES ABOVE THRESHOLD | RATIO | LOG2 FOLD CHANGE | P-VALUE |
|---|---|---|---|---|---|
| B CELLS | Cd19 | 100 % | 17.1 | 4.1 | 0.00003543 |
| Cd22 | 100 % | 6.02 | 2.59 | 0.00051727 | |
| Cd79b | 100 % | 8.22 | 3.04 | 0.00001381 | |
| Pax5 | 100 % | 9.45 | 3.24 | 0.00000503 | |
| RECRUITING | Ccl19 | 100 % | 4.66 | 2.22 | 0.00015125 |
| CHEMOKINES | Ccl20 | 100 % | 3.82 | 1.93 | 0.00175557 |
| POLYMERIC IG RECEPTOR | Pigr | 100 % | −5.22 | −2.38 | 0.00002739 |
It was reported previously that IL-17A controls the expansion of IL17A-producing naïve T cell populations through IL-17 receptor (38) and in the gastric mucosa of uninfected Il17ra−/− there was a 2-3 fold increase in IL-17A levels compared to uninfected WT mice (27). T cells were differentiated in vitro to Th17 cells and then restimulated with or without recombinant IL-17A to investigate whether this impacted the expression of Il17a and Il21. The exacerbated cytokine expression was not seen in the Il17ra−/− T cells in this in vitro model, nor did recombinant IL-17A impact the gene expression in WT T cells (Supplemental Figure 2). Because the H. pylori infected Il17ra−/− mice express remarkable levels of the Il17a gene (Fig. 4D and (27)), we hypothesized that IL-17RA on T cells is facilitating an autocrine inhibitory signal to down regulate cytokine production. This was tested in vivo utilizing the cre-lox model. Cd4creIl17rafl/fl mice were generated, thereby retaining the anti-microbial and chemotactic impact of IL-17R signaling on non-hematopoietic cells. To confirm knockout of IL-17RA on T cells, IL-17RA expression was measured by flow cytometry and expression was down on all T cells (CD4+ and CD8+) but not down on the CD19+ B cell subset (Fig. 5A). Further, IL-17A expression was measured in splenocytes from the Cd4creIl17rafl/fl and Il17rafl/fl mice using intracellular cytokine staining. There was a 2-fold increase in IL-17A expression in the CD4+ cells deficient for IL-17RA signaling (Fig. 5B).
Next, Cd4creIl17rafl/fl and Il17rafl/fl mice were infected with H. pylori, and their ability to control H. pylori infection and inflammation was assessed. Interestingly, many of the Cd4creIl17rafl/fl mice had evidence of yeast outgrowth in their stomachs independent of H. pylori infection (Supplemental Fig. 3A). This was evident in the histological examination of the tissue (Supplemental Fig. 3A). In this cohort of mice when many had yeast colonization in their stomachs, the data indicate that CFU levels were higher in Cd4creIl17rafl/fl compared to Il17rafl/fl mice suggesting that IL-17R signaling on T cells was required for control of H. pylori colonization with this complication (Supplemental Fig 3B). This finding is unexpected because the epithelial cell/stromal cell response to IL-17 is attributable to the antimicrobial response. While infection of these mice with H. pylori did induce inflammation and T cell cytokine gene expression, the T cell specific deficiency in IL-17RA did not lead to increased chronic inflammation nor did it lead to any increased Il17a, Il21, or Ifng expression compared to H. pylori infected control mice (Il17rafl/fl) (Supplemental Fig. 3B and C). It cannot be ruled out that the yeast did not impact the host’s immune response, but it is unlikely that the yeast would have decreased the inflammation or inhibited the T cell response.
After treating breeders with fluconazole and successfully removing this yeast from the colony, more cohorts of Cd4creIl17rafl/fl and Il17rafl/fl mice were infected with H. pylori. There was no significant difference in the ability of mice with T cells deficient for IL-17RA to control H. pylori colonization (Fig. 6A), no change in total inflammation scores (Fig. 6B), and no significant differences in Ifng, Il17a, or Il21 expression in the gastric mucosa (Fig. 6C). These data suggest that IL-17RA deficiency in T cells is not sufficient to induce exacerbated chronic inflammation and hyper Th17 responses during H. pylori infection.
Figure 6.
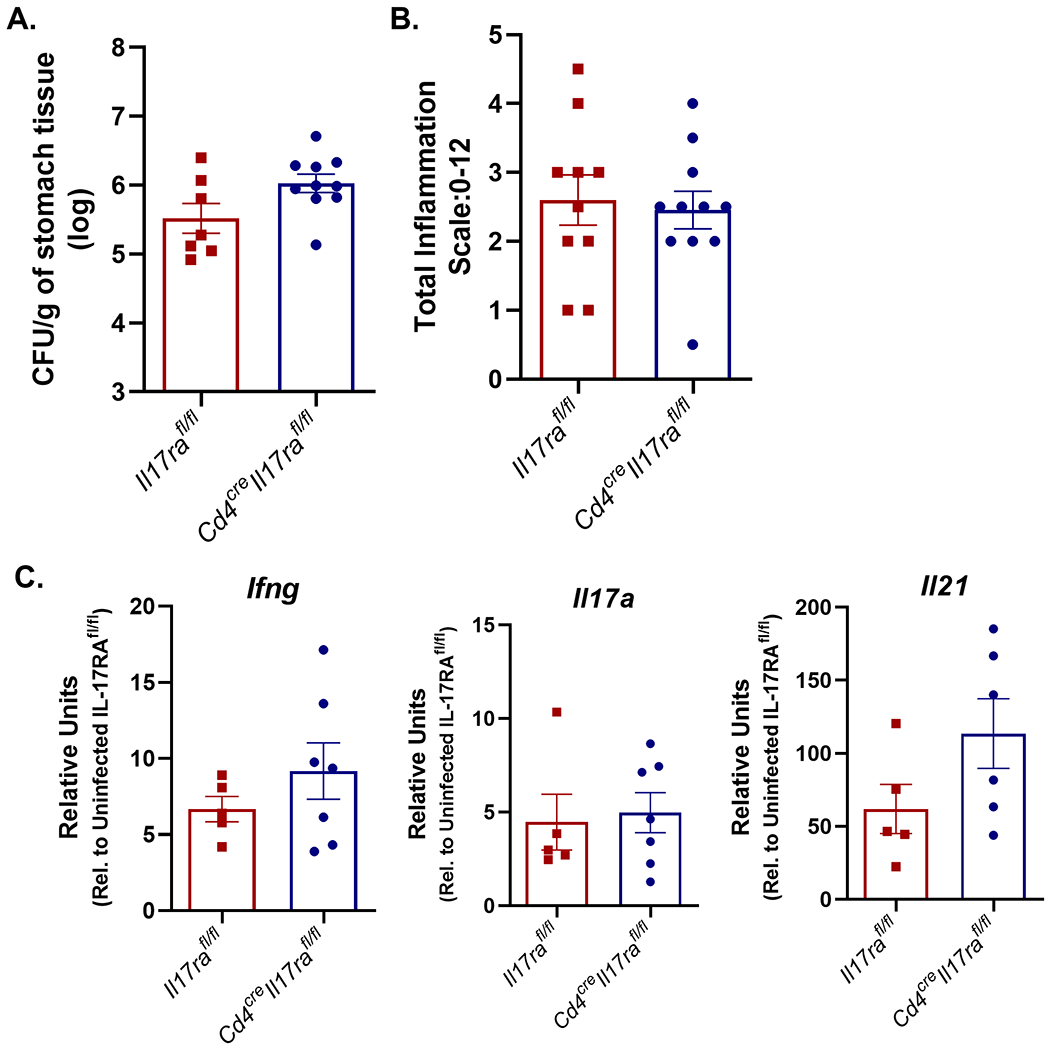
IL-17RA expression on T cells is required to control bacterial burden but not required to control gastric inflammation in the mouse model. A) Bacterial burden was measured in Il17rafl/fl (WT) and Cd4creIl17rafl/fl mice that were infected with PMSS1 for 3 months (8-11 per genotype). Colony forming units (CFU) per gram of stomach tissues was calculated and is presented in the graph ±SEM. Statistical analysis was performed using Mann Whitney U unpaired t test on log transformed CFU/gram data. These data are representative of 2 independent experiments. B) Levels of acute and chronic inflammation were scored on stomach tissue (in the corpus and antrum) at 3 months post infection with strain PMSS1. Total inflammation as presented is the sum of acute and chronic inflammation (8-10 per genotype). Statistical analysis was performed using Kruskal-Wallis test’s and the Dunn’s multiple comparisons test which resulted in no significant difference between genotypes. See methods for scoring system (scale is 0–12). Error bars represented mean ± SEM and are representative of 2 independent experiments. C) qPCR was used to measure Il21, Il17a and Ifng transcripts in the gastric tissue of H. pylori–infected Il17rafl/fl and infected Cd4creIl17rafl/fl mice (5-7 per genotype). Relative units are calculated as described in the methods, relative to Gapdh and calibrated to uninfected WT mice. pPCR data is representative of 2 independent experiments. Statistical analysis was performed using an ANOVA analysis and Tukey’s multiple comparisons test. Error bars represent ± SEM; *P ≤ 0.05, **, P ≤ 0.01 compared to the uninfected group.
Discussion
A critical role for IL-17A in host immunity has been well defined. IL-17A signals through IL-17RA/RC multimeric receptors in epithelial cells to activate several transcription factors including AP-1, C/EBP and NFκB. These transcription factors then upregulate mRNA expression of cxcl8 and several antimicrobial products especially in the presence of a co-stimulator like IL-22. The role for IL-17F or the heterodimer IL-17A/F in this process has been less clear despite their use of the same multimeric receptor. Moreover, in several models, including H. pylori infection models, Il17a−/− mice do not respond similarly to Il17ra−/− mice suggesting that there is either a differential role for IL-17F or compensatory impact of having one of the cytokines.
Working with gastric epithelial cell cultures, it was determined that MKN cells and AGS cells can be used to measure IL-17 responsiveness, but they do not respond similarly to the different co-stimuli tested. For example, AGS cells respond by producing NOX1 and PIGR when co-stimuli are present, especially in response to IL-17A. The binding affinity of IL-17F to IL-17RA is reported to be much lower than IL-17A, yet the homodimers have similar affinity for the IL-17RC component of the signaling complex (39). Recent studies have suggested that the IL-17RA signaling pathway may also play a role in downregulating the inflammatory response; it is possible that this process is mediated by IL-17A and/or IL-17F. Interestingly, our data suggest that it is not as strong of a synergistic activator of either MKN or AGS cells.
The S100A9 protein forms a heterodimer with S100A8, known as calprotectin (40). Calprotectin can affect the growth and virulence of bacteria, such as H. pylori, by sequestering nutrient metals, such as zinc, making it harder for the bacteria to survive in the gastric mucosa (23, 40). Although S100A8 and S100A9 proteins have been characterized as antimicrobial, their role during bacterial infection may be multi-dimensional (40). This is because S100A9 also plays a prominent role in the regulation of inflammatory processes and immune response; S100A9 can induce neutrophil chemotaxis and adhesion, acting as a potent amplifier of inflammation. In addition to this, ligation of calprotectin to TLR4 can activate MAP-kinase and NFkB signaling pathways, which can also drive transcription of proinflammatory genes (41, 42). This activity leads to calprotectin’s classification as a Damage Associated Molecular Pattern (DAMP). Therefore, understanding the effects of calprotectin, an antimicrobial protein and a pro-inflammatory DAMP, in the context of H. pylori can be challenging (43–45). Consistent with the role for IL-17A or IL-17F in mediating acute inflammation and neutrophilic responses, acute inflammation was significantly reduced in Il17a−/−, Il17f−/− and Il17a/f−/− mice in comparison to WT mice. It is evident that the IL-17A/F double knockout and the Il17ra−/− exhibit high levels of chronic inflammation, suggesting that IL-17A or IL-17F plays a role in modulating a protective immune response during the control of chronic inflammation. Levels of chronic inflammation correlate to increases in Il21 and Ifng, suggesting that exacerbated T cell responses are down regulated by IL-17A or IL-17F signaling through IL-17RA.
The hypothesis that IL-17A or IL-17F modulate T cell cytokine production directly leading to downregulation of proinflammatory cytokines such as IL-17A and IL-21 was addressed. Naïve T cells from both WT and Il17ra−/− mice were differentiated into Th17 cells and gene expression was assessed by qPCR. We were unable to see an impact of IL-17A on naïve T cells differentiated into Th17 cells and there was no inherit difference in Il17a or Il21 expression in the Il17ra−/− T cells in these in vitro models (Supplemental Figure 2). The rationale for moving in vivo with these models was that the microenvironment of in vitro T cell cultures is very isolated and cannot recapitulate the chronic time course in which this pathological response is observed in the H. pylori infected l17ra−/− and Il17a/f−/− mice. Utilizing the Cre/lox recombination system to develop a T cell specific deficiency in the IL-17 receptor, it is evident that the absence of IL-17RA on T cells does not lead to an exacerbated T cell response compared to WT mice or control models. These data suggest that the impact of congenic deficiency of IL-17RA may have an indirect impact the T cell response or requires two hits. In the case of a two-hit hypothesis, the first hit or primary driver of the hyper Th17 response in the Il17ra−/− mice may be driven by a shift in the gastric microbiome and/or an impact on gastric permeability or wound healing while the second hit may be activation of an aberrant T cell response against the shifted microbiome or microbes gaining access to the submucosa. IL-17 impacting expression of CXCL8, PIGR and NOX1 support the potential for IL-17R signaling being a requirement for preventing dysbiosis. These hypotheses are the focus of future investigations.
The role of the polymeric immunoglobulin receptor has only minimally been investigated in the gastric mucosa in response to H. pylori infection. Expression of PIGR is low in healthy adults and increases with gastric inflammation and development of intestinal metaplasia(46–48). This is the first report that we are aware of that links IL-17 cytokines to induction of PIGR expression in human gastric epithelial cells. Gorrell et al., describe the course of H. pylori infection in pIgR KO mice infected with the SS1 strain (49). In the study there was no differences in the gastric bacterial loads of WT and pIgR KO mice up to 3 mpi. Their experiments were carried out to 12 mpi, and by 6 mpi, they report a reduction in bacterial load in the WT mice compared to that in the pIgR KO mice. Since gene expression of Pigr is reduced in the Il17ra−/− mice and not completely absent, a different outcome is possible.
Noteworthy, the T cell specific deficiency in IL-17RA did result in at least one known shift in the gastric microbiome- colonization with a yeast (Supplemental Figure 3A). Evidence of a yeast, likely Kazachstania pintolopesii, was observed in many of the Cd4creIl17rafl/fl mice. Our pathologist noted that the colonization was more abundant in the corpus. This yeast has been reported in the literature occasionally in immunocompromised animals (50, 51). Increased susceptibility to commensal fungi, mainly manifesting as mucocutaneous candidiasis, has been reported in individuals with mutations in IL-17F and IL-17RA (52), and anti-IL-17 treatment in Crohn’s patients lead to an increased number of fungal infections compared to patients that did not receive anti-IL-17 (53). While these connections between IL-17 signaling and fungal infections has been noted, there has not been any indication that this would be due to a T cell specific response.
Supplementary Material
Acknowledgements
We thank Danyvid Olivares-Villagomez and other members of our Mucosal Immunology Group for helpful discussions through the Center for Mucosal Inflammation and Cancer.
This work was supported by Office of Medical Research, Veterans Affairs Merit Review grant IBX000915A (to H.M.S.A.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veteran’s Affairs. Core services are supported by NIH grants P30DK058404 (through Vanderbilt University Medical Center’s Digestive Disease Research Center) and P30CA068485 (through a Vanderbilt-Ingram Cancer Center support grant). We acknowledge the Translational Pathology Shared Resource, supported by NCI/NIH Cancer Center support grant 5P30 CA68485-19 and Vanderbilt Mouse Phenotyping Center grant 2 U24 DK059637-16.
Abbreviations:
- IL-17R
Interleukin-17 receptor
- WT
Wild-type
- PMSS1
pre-mouse Sydney strain 1
- pigr
polymeric immunoglobulin receptor
- qPCR
quantitative real-time PCR
References
- 1.Karin M 2006. Nuclear factor-kappaB in cancer development and progression. Nature 441: 431–436. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal BB, Vijayalekshmi RV, and Sung B. 2009. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 15: 425–430. [DOI] [PubMed] [Google Scholar]
- 3.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, and Jemal A. 2015. Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108. [DOI] [PubMed] [Google Scholar]
- 4.Plummer M, Franceschi S, Vignat J, Forman D, and de Martel C. 2015. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer 136: 487–490. [DOI] [PubMed] [Google Scholar]
- 5.Plummer M, de Martel C, Vignat J, Ferlay J, Bray F, and Franceschi S. 2016. Global burden of cancers attributable to infections in 2012: a synthetic analysis. Lancet Glob Health 4: e609–616. [DOI] [PubMed] [Google Scholar]
- 6.de Martel C, Georges D, Bray F, Ferlay J, and Clifford GM. 2020. Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob Health 8: e180–e190. [DOI] [PubMed] [Google Scholar]
- 7.Organization, W. H. 2018. Key Facts on Cancer World Health Organization.
- 8.Poorolajal J, Moradi L, Mohammadi Y, Cheraghi Z, and Gohari-Ensaf F. 2020. Risk factors for stomach cancer: a systematic review and meta-analysis. Epidemiol Health 42: e2020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Negovan A, Iancu M, Fulop E, and Banescu C. 2019. Helicobacter pylori and cytokine gene variants as predictors of premalignant gastric lesions. World J Gastroenterol 25: 4105–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caston RR, Loh JT, Voss BJ, McDonald WH, Scholz MB, McClain MS, and Cover TL. 2019. Effect of environmental salt concentration on the Helicobacter pylori exoproteome. J Proteomics 202: 103374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ansari S, and Yamaoka Y. 2019. Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity. Toxins (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Arnold IC, and Muller A. 2020. Mechanisms of persistence, innate immune activation and immunomodulation by the gastric pathogen Helicobacter pylori. Curr Opin Microbiol 54: 1–10. [DOI] [PubMed] [Google Scholar]
- 13.Eaton KA, Mefford M, and Thevenot T. 2001. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol 166: 7456–7461. [DOI] [PubMed] [Google Scholar]
- 14.Eaton KA, Ringler SR, and Danon SJ. 1999. Murine splenocytes induce severe gastritis and delayed-type hypersensitivity and suppress bacterial colonization in Helicobacter pylori-infected SCID mice. Infect Immun 67: 4594–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray BM, Fontaine CA, Poe SA, and Eaton KA. 2013. Complex T cell interactions contribute to Helicobacter pylori gastritis in mice. Infect Immun 81: 740–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon B, Hossain R, Patel RV, and Algood HMS. 2019. Th17 Cells in Helicobacter pylori Infection: a Dichotomy of Help and Harm. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ikuse T, Blanchard TG, and Czinn SJ. 2019. Inflammation, Immunity, and Vaccine Development for the Gastric Pathogen Helicobacter pylori. Curr Top Microbiol Immunol 421: 1–19. [DOI] [PubMed] [Google Scholar]
- 18.Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, Reindl W, Dossumbekova A, Friedrich M, Saur D, Wagner H, Schmid RM, and Prinz C. 2006. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131: 525–537. [DOI] [PubMed] [Google Scholar]
- 19.Gil JH, Seo JW, Cho MS, Ahn JH, and Sung HY. 2014. Role of Treg and TH17 cells of the gastric mucosa in children with Helicobacter pylori gastritis. J Pediatr Gastroenterol Nutr 58: 245–251. [DOI] [PubMed] [Google Scholar]
- 20.Serrano C, Wright SW, Bimczok D, Shaffer CL, Cover TL, Venegas A, Salazar MG, Smythies LE, Harris PR, and Smith PD. 2013. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Mucosal Immunol 6: 950–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, and Kolls JK. 2001. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 194: 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen K, and Kolls JK. 2017. Interluekin-17A (IL17A). Gene 614: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon BR, Radin JN, Piazuelo MB, Contreras DC, and Algood HM. 2016. IL-17a and IL-22 Induce Expression of Antimicrobials in Gastrointestinal Epithelial Cells and May Contribute to Epithelial Cell Defense against Helicobacter pylori. PLoS One 11: e0148514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, and Fouser LA. 2006. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203: 2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lina TT, Alzahrani S, Gonzalez J, Pinchuk IV, Beswick EJ, and Reyes VE. 2014. Immune evasion strategies used by Helicobacter pylori. World J Gastroenterol 20: 12753–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cremniter J, Bodet C, Tougeron D, Dray X, Guilhot J, Jegou JF, Morel F, Lecron JC, Silvain C, and Burucoa C. 2018. Th-17 response and antimicrobial peptide expression are uniformly expressed in gastric mucosa of Helicobacter pylori-infected patients independently of their clinical outcomes. Helicobacter 23: e12479. [DOI] [PubMed] [Google Scholar]
- 27.Algood HM, Allen SS, Washington MK, Peek RM Jr., Miller GG, and Cover TL. 2009. Regulation of gastric B cell recruitment is dependent on IL-17 receptor A signaling in a model of chronic bacterial infection. J Immunol 183: 5837–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiomi S, Toriie A, Imamura S, Konishi H, Mitsufuji S, Iwakura Y, Yamaoka Y, Ota H, Yamamoto T, Imanishi J, and Kita M. 2008. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter 13: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, and Peschon J. 2006. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 177: 36–39. [DOI] [PubMed] [Google Scholar]
- 30.Gu C, Wu L, and Li X. 2013. IL-17 family: cytokines, receptors and signaling. Cytokine 64: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ely LK, Fischer S, and Garcia KC. 2009. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol 10: 1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, and Dong C. 2008. Regulation of inflammatory responses by IL-17F. J Exp Med 205: 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, and Budelsky AL. 2008. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol 181: 4299–4310. [DOI] [PubMed] [Google Scholar]
- 34.Horvath DJ Jr., Radin JN, Cho SH, Washington MK, and Algood HM. 2013. The interleukin-17 receptor B subunit is essential for the Th2 response to Helicobacter pylori, but not for control of bacterial burden. PLoS One 8: e60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, Wan J, Zhang W, Zhong Z, Sanchez-Lopez E, Wu LW, Taniguchi K, Feng Y, Fearon E, Grivennikov SI, and Karin M. 2014. Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 41: 1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedoya SK, Wilson TD, Collins EL, Lau K, and Larkin J 3rd. 2013. Isolation and th17 differentiation of naive CD4 T lymphocytes. J Vis Exp: e50765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dixon MF, Genta RM, Yardley JH, and Correa P. 1996. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 20: 1161–1181. [DOI] [PubMed] [Google Scholar]
- 38.Smith E, Stark MA, Zarbock A, Burcin TL, Bruce AC, Vaswani D, Foley P, and Ley K. 2008. IL-17A inhibits the expansion of IL-17A-producing T cells in mice through “short-loop” inhibition via IL-17 receptor. J Immunol 181: 1357–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Onishi RM, and Gaffen SL. 2010. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaddy JA, Radin JN, Loh JT, Piazuelo MB, Kehl-Fie TE, Delgado AG, Ilca FT, Peek RM, Cover TL, Chazin WJ, Skaar EP, and Scott Algood HM. 2014. The host protein calprotectin modulates the Helicobacter pylori cag type IV secretion system via zinc sequestration. PLoS Pathog 10: e1004450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai SY, Segovia JA, Chang TH, Morris IR, Berton MT, Tessier PA, Tardif MR, Cesaro A, and Bose S. 2014. DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathog 10: e1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahman MT, Myles A, Gaur P, Misra R, and Aggarwal A. 2014. TLR4 endogenous ligand MRP8/14 level in enthesitis-related arthritis and its association with disease activity and TLR4 expression. Rheumatology (Oxford) 53: 270–274. [DOI] [PubMed] [Google Scholar]
- 43.Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, Restrepo NA, Hosking MP, Edwards RA, Battistoni A, Pasquali P, Lane TE, Chazin WJ, Vogl T, Roth J, Skaar EP, and Raffatellu M. 2012. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11: 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro KA, Chazin WJ, and Skaar EP. 2011. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10: 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, and Skaar EP. 2008. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319: 962–965. [DOI] [PubMed] [Google Scholar]
- 46.Takemura K, Hirokawa K, Esaki Y, and Mishima Y. 1990. Distribution of immunoglobulins and secretory component in gastric cancer of the aged. Cancer 66: 2168–2173. [DOI] [PubMed] [Google Scholar]
- 47.Kaneko T, Ota H, Hayama M, Akamatsu T, and Katsuyama T. 2000. Helicobacter pylori infection produces expression of a secretory component in gastric mucous cells. Virchows Arch 437: 514–520. [DOI] [PubMed] [Google Scholar]
- 48.Gologan A, Acquafondata M, Dhir R, and Sepulveda AR. 2008. Polymeric immunoglobulin receptor-negative tumors represent a more aggressive type of adenocarcinomas of distal esophagus and gastroesophageal junction. Arch Pathol Lab Med 132: 1295–1301. [DOI] [PubMed] [Google Scholar]
- 49.Gorrell RJ, Wijburg OL, Pedersen JS, Walduck AK, Kwok T, Strugnell RA, and Robins-Browne RM. 2013. Contribution of secretory antibodies to intestinal mucosal immunity against Helicobacter pylori. Infect Immun 81: 3880–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurtzman CP, Robnett CJ, Ward JM, Brayton C, Gorelick P, and Walsh TJ. 2005. Multigene phylogenetic analysis of pathogenic candida species in the Kazachstania (Arxiozyma) telluris complex and description of their ascosporic states as Kazachstania bovina sp. nov., K. heterogenica sp. nov., K. pintolopesii sp. nov., and K. slooffiae sp. nov. J Clin Microbiol 43: 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treuting PM, Clifford CB, Sellers RS, and Brayton CF. 2012. Of mice and microflora: considerations for genetically engineered mice. Vet Pathol 49: 44–63. [DOI] [PubMed] [Google Scholar]
- 52.Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, Migaud M, Hauck F, Al Ali A, Cyrus C, Vatte C, Patiroglu T, Unal E, Ferneiny M, Hyakuna N, Nepesov S, Oleastro M, Ikinciogullari A, Dogu F, Asano T, Ohara O, Yun L, Della Mina E, Bronnimann D, Itan Y, Gothe F, Bustamante J, Boisson-Dupuis S, Tahuil N, Aytekin C, Salhi A, Al Muhsen S, Kobayashi M, Toubiana J, Abel L, Li X, Camcioglu Y, Celmeli F, Klein C, AlKhater SA, Casanova JL, and Puel A. 2016. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A 113: E8277–E8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J, Pezous N, Bek S, Bruin G, Mellgard B, Berger C, Londei M, Bertolino AP, Tougas G, Travis SP, and Secukinumab G in Crohn’s Disease Study. 2012. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.