

Abstract
Purpose:
Prostate-specific membrane antigen (PSMA) targeting therapies such as Lutetium-177 (177Lu)–PSMA-617 are affecting outcomes from metastatic castration-resistant prostate cancer (mCRPC). However, a significant subset of patients have prostate cancer cells lacking PSMA expression, raising concerns about treatment resistance attributable at least in part to heterogeneous PSMA expression. We have previously demonstrated an association between high PSMA expression and DNA damage repair defects in mCRPC biopsies and therefore hypothesized that DNA damage upregulates PSMA expression.
Experimental Design:
To test this relationship between PSMA and DNA damage we conducted a screen of 147 anticancer agents (NCI/NIH FDA-approved anticancer “Oncology Set”) and treated tumor cells with repeated ionizing irradiation.
Results:
The topoisomerase-2 inhibitors, daunorubicin and mitoxantrone, were identified from the screen to upregulate PSMA protein expression in castration-resistant LNCaP95 cells; this result was validated in vitro in LNCaP, LNCaP95, and 22Rv1 cell lines and in vivo using an mCRPC patient-derived xenograft model CP286 identified to have heterogeneous PSMA expression. As double-strand DNA break induction by topoisomerase-2 inhibitors upregulated PSMA, we next studied the impact of ionizing radiation on PSMA expression; this also upregulated PSMA protein expression in a dose-dependent fashion.
Conclusions:
The results presented herein are the first, to our knowledge, to demonstrate that PSMA is upregulated in response to double-strand DNA damage by anticancer treatment. These data support the study of rational combinations that maximize the antitumor activity of PSMA-targeted therapeutic strategies by upregulating PSMA.
Translational Relevance.
The VISION and TheraP trials compared Lutetium-177 (177Lu)–PSMA-617 with best supportive care and cabazitaxel, respectively, and demonstrated improved outcomes from mCRPC with this PSMA-targeting radionuclide. PSMA protein expression on tumor cells determines the anticancer activity of this and other PSMA-targeted therapies in development, with studies demonstrating both intra- and interpatient heterogeneity in PSMA expression. Strategies that increase PSMA expression prior to such targeted therapy may increase their anticancer activity. Herein, we demonstrate a novel relationship between DNA damage induction and PSMA upregulation that merits further exploration to increase the therapeutic benefits imparted by PSMA-targeted therapy against mCRPC, supporting the pursuit of clinical trials of combined topoisomerase-2 inhibition with PSMA-targeting agents including radionuclides.
Introduction
Therapeutic strategies targeting prostate-specific membrane antigen (PSMA) are showing significant promise in the treatment of advanced prostate cancer (1–3), with lutetium-177 (177Lu)–PSMA-617 improving overall survival in phase II and III clinical trials. Several radioligand therapies, radio-immunoconjugates, immunoconjugates, and antibody constructs targeting this protein are currently under clinical investigation with preliminary evidence of antitumor activity. These commonly carry cytotoxic or radioactive payloads or comprise bi- or trispecific antibodies that instigate antitumor immune responses by recruiting T cells and/or natural killer (NK) cells (4, 5). Such PSMA-directed agents have demonstrated varying degrees of antitumor activity, with this associating with PSMA expression; tumors low in PSMA are much less likely to respond to this therapeutic strategy (6–8). This has led to efforts to elucidate why prostate cancer cells express PSMA, to identify the prostate cancer subtype with high PSMA expression, as well as the pursuit of strategies that increase PSMA expression to improve the efficacy of PSMA-directed therapies.
PSMA is generally highly expressed in prostate cancer cells, and its expression can increase with the development of metastatic castration-resistant prostate cancer (mCRPC), but can ultimately be lost with the emergence of treatment-induced AR-negative or neuroendocrine cancer (9–12). Nonprostatic and benign tissues express very low or no PSMA. Taken together, this makes PSMA an excellent theranostic target; however, some patients are excluded from PSMA-targeting trials due to the lack of PSMA expression detection on imaging. Methods to upregulate PSMA expression are under investigation to improve PSMA-targeted therapy dose (13). Revealing the regulatory mechanisms and the underlying function of PSMA will accelerate this as it is currently unclear. Such methods could be adopted to drive PSMA expression in patients who would be otherwise excluded from PSMA-targeted therapies. This would provide clear treatment benefits for mCRPC patients if the upregulation is specific to prostate cancer cells. Furthermore, all patients eventually develop secondary resistance to these treatments, with some deriving limited clinical benefits. This resistance may be due to heterogeneous PSMA expression and the emergence or expansion of resistant cancer cell clones expressing little PSMA, as well as radioresistance mechanisms in PSMA-positive cells (8, 14). We have previously shown that PSMA expression is heterogeneous in most mCRPC biopsies (15), with substantial intrapatient and interpatient heterogeneity. Therefore, elucidating the functions of PSMA, and alongside this the mechanisms that regulate its expression, remains critical to developing strategies to improve therapeutic efficacy.
PSMA is a predominantly extracellular protein with glutamate-carboxypeptidase and folate-hydrolase enzymatic activity (16, 17). The enzymatic function of PSMA links it with various oncogenic signaling pathways including the phosphatidylinositol-3-kinase and the mammalian target of the rapamycin signaling pathway (PI3K/AKT/mTOR) and integrin signaling (18–20). PSMA provides a growth advantage to cells by generating monoglutamated folates and glutamate, both critical to nucleotide metabolism and DNA synthesis and repair (21–23). We have previously demonstrated that mCRPC with DNA damage repair (DDR) defects, including deleterious alterations in BRCA2 and ATM, have significantly higher PSMA expression (15). Interestingly, preliminary studies suggest that BRCA2 CRISPR knockout and alpha emitting radioisotopes that result in DNA damage may upregulate PSMA (24, 25). Overall, we hypothesized that these data support a functional role for PSMA in nucleotide pool maintenance and that the expression of PSMA is dynamic, not static in prostate cancer cells, with DNA damage–inducing agents increasing PSMA expression. If this hypothesis is correct, this would support the pursuit of pharmacologic strategies that induce PSMA expression by damaging DNA, to enhance the antitumor activity of PSMA-targeting strategies.
Materials and Methods
Cell lines
Prostate cancer cell lines (LNCaP and 22Rv1) and prostate epithelial lines (WPE-1 and RWPE-1) were purchased from ATCC and grown in conditions recommended by the supplier. Briefly, LNCaP and 22Rv1 cells were grown in RPMI-1640 (Thermo Fisher) supplemented with 10% FBS (Biosera). RWPE-1 and WPE-1 lines were grown in complete keratinocyte serum-free media supplemented with human recombinant epidermal growth factor (rEGF) and bovine pituitary extract (Thermo Fisher). The castrate-resistant LNCaP subline LNCaP95 was a generous gift by Drs. Alan K. Meeker and Jun Luo (Johns Hopkins University, Baltimore, MD) and have been cultured in phenol red-free RPMI media (Thermo Fisher) supplemented with 10% charcoal-stripped serum (Biosera) to ensure hormone-free conditions (26). Cell lines were regularly STR profiled and mycoplasma tested.
CellTiter-Glo 2D cell viability assay
Cells were plated in triplicate and allowed to adhere overnight. Media were subsequently removed, and cells were treated with the appropriate vehicle or drug in the respective media at 50 μL final volume. On the final day of growth 50 μL of CellTiter-Glo 2.0 (Promega) was added to each well and incubated at room temperature for 10 minutes on a laboratory rocker, in the dark. Plates were subsequently read on the Biotek Synergy HTX plate using the luminescence setting associated with Gen5 Software. The viability of treated wells was normalized to control wells.
Drug screen
The NCI/NIH FDA-approved anticancer “Oncology Set” was obtained from the NCI/NIH DTP Open Chemical Repository. For the screen, LNCaP95 cells were plated in 24-well plates and left to adhere overnight. Plates had 4 wells allocated to vehicle (DMSO, 0.01% v/v) and 20 wells for drug treatment, resulting in 9 plates. The screen included 147 different compounds, delivered in DMSO at 10 mmol/L and reduced to 1 mmol/L in PBS (Supplementary Table S1). One well was allocated to each drug (n = 1). Cells were treated with 1 μmol/L or 100 nmol/L of each drug for 48 hours, washed with PBS, and prepared for western blot analysis. Protein was quantified through BCA analysis, and an equal amount of protein from each sample lysate was loaded into the electrophoresis gel. Changes in PSMA protein were subsequently investigated through western blot and densitometric analysis of bands, with samples normalized to the housekeeping control vinculin.
Repeat irradiation of cell lines
Cell lines were plated in T75 flasks at a confluency of 15% and delivered doses of 1 Gy or 2 Gy (160 keV, 11.3 mA) using an RS160 cabinet (XStrahl) once every 48 hours, with 3 or 6 repeats. Media were replaced once per week. Cell lysates and pellets were harvested 24 or 72 hours after the last fraction of radiotherapy delivery.
Topoisomerase inhibitor treatment
For short-term exposure, cell lines were plated in 6-well plates at a confluency of 70%. Cell lines were treated with 100 nmol/L of mitoxantrone dihydrochloride (Sigma-Aldrich), 100 nmol/L of daunorubicin dihydrochloride (Sigma-Aldrich), or vehicle (DMSO 0.1% v/v). Cell lysates or cell pellets were harvested after 48 hours of treatment for analysis by western blot and IHC.
Patient-derived xenograft in vivo work
The patient-derived xenograft (PDX) CP286 was derived from a metastatic CRPC patient biopsy with a mismatch-repair defect and deleterious alterations in AR, PTEN, and BRAF taken from a patient treated with androgen deprivation, next-generation hormonal treatment, and taxane chemotherapy. For the drug treatment pilot study, CP286 PDX tumors were implanted bilaterally into the flanks of male NSG mice (n = 10, total) and allowed to grow to 400 mm3. Mice were then randomized into 5 treatment arms (cumulative treatment mg/kg): arm 1 = vehicle (PBS), arm 2 = mitoxantrone (0.5 mg/kg), arm 3 = mitoxantrone (1 mg/kg), arm 4 = daunorubicin (0.5 mg/kg), and arm 5 = daunorubicin (1 mg/kg). Treatment was delivered intraperitoneally twice over 8 days on days 1 and 4. For the repeat irradiation study, CP286 PDX tumors were implanted bilaterally into the flanks of male NSG mice (n = 6) and allowed to grow to 400 mm3. Mice were randomized into 3 treatment arms (cumulative dose/fractions): no treatment (mock), 6 Gy/6# or 12 Gy/6#. Tumors were irradiated on days 1, 3, 5, 7, 9, and 11. Mice were culled 24 hours after the final treatment dose and either 2 or 3 independent tumor cores of bilateral tumors taken for protein, IHC, and RNA analysis. All animal work was performed under an animal license granted by the UK home office authorities in accordance with UK law.
Immunohistochemistry (IHC)
IHC for PSMA was performed on 3- to 5-μm sections of formalin-fixed paraffin-embedded (FFPE) cell pellets and tissue sections run with reagents and methods previously published (15). Briefly, antigen retrieval was achieved for slides designated for PSMA analysis through microwaving in retrieval buffer for 18 minutes at 800 W. Primary antibodies were then applied for 1 hour, and samples were processed via the semiautomated BioGenex (i6000) immunostainer as previously described (15). Quantification of PSMA protein expression was undertaken by an experienced pathologist (B. Gurel) utilizing automated, colorimetric, HALO image analysis software.
Western blot analysis
Cell line and PDX tissue lysates were analyzed via western blot. Whole-cell lysates were generated using RIPA lysis buffer (Thermo Fisher), Halt Protease (Thermo Fisher), and Phosphatase Inhibitor Cocktail 100× (Thermo Fisher). PDX tissue was homogenized using a Qiagen tissue homogenizer with 7-mm stainless steel beads, followed by centrifugation through a QiaShredder column, sonification, and centrifugation (30 minutes, 1500 RPM, 4°C). The protein concentration of the cleared lysate was normalized after BCA analysis and loaded on a 4%–12% Bis-Tris Nupage gel (Thermo Fisher). Blots were blocked with 1% BSA in TBS-T (0.05% Tween in TBS) for 1 hour, and the proteins of interest were detected with primary (overnight at 4°C) and secondary antibodies (1 hour at room temperature), with three washes between the primary and secondary antibodies. Primary and secondary antibodies used, and their dilutions, are listed in Supplementary Table S2. Protein bands were visualized as per the manufacturer's protocol using Pierce ECL Western Blotting Substrate (Thermo Fisher) and analyzed using the ChemiDoc MP Imaging System (Bio-Rad).
Quantitative PCR analysis of mRNA
RNA was extracted from pelleted cells and PDX samples via RNeasy Plus Mini Kits (Qiagen), following the manufacturer's instructions. PDX tissue was homogenized using Qiagen tissue homogenizer with 7-mm stainless steel beads, followed by centrifugation through a QiaShredder column prior to RNA extraction. cDNA was generated using RevertAid First-Strand cDNA Synthesis Kit (Thermo Fisher) from total sample RNA on the Mastercycler Nexus Gradient GSX1 Thermal Cycler (Eppendorf). Quantitative RT-PCR was performed on cDNA samples on a ViiA 7 real-time PCR system (Applied Biosystems) for gene expression with the specified TaqMan probes for FOLH1, TOP2A, GAPDH, and RPLP0 outlined in Supplementary Table S3 using TaqMan Fast Advanced Master Mix (Thermo Fisher). Relative expression levels were compared through the Delta CT method and normalized to GAPDH and RPLP0 expression.
Statistical analysis
Gene and protein expression in quantitative PCR and western blot, respectively, was normalized to housekeeping genes and analyzed through Prism 9 software (GraphPad). Statistical comparisons of gene or protein expression in control versus treatment were carried out with a two-tailed, unpaired student's t-test, assuming equal variance and normal distribution. Mean and standard deviation were presented unless specified otherwise. Significance in experimental data was defined as P < 0.05, with figures presented as the sample mean ± 1 standard deviation of the mean (SD). IHC of PDX tumors treated with topoisomerase-2 inhibitors was analyzed with HALO image software. Tumor cores (n = 3) from each mouse (n = 2) were investigated for changes in the percentage of PSMA cell positivity (total, membranous, and cytoplasmic) and changes in optical density (intensity). Tumor cores (n = 2) from each mouse (n = 2) were investigated for changes in FOLH1 and TOP2A, normalizing to the housekeeping genes GAPDH and RPLP0.
Data availability
Data were generated by the authors and are included in this article.
Results
Topoisomerase-2 inhibition induces PSMA expression in prostate cancer cells
Androgen deprivation–resistant LNCaP95 prostate cancer cells were treated with 147 different FDA-approved compounds (NCI/NIH FDA-approved anticancer “Oncology Set”), including DNA-damaging agents, for 48 hours at 100 nmol/L or 1 μmol/L concentrations (Supplementary Table S1). PSMA protein expression was then evaluated and compared with vehicle controls (DMSO, 0.1% v/v).
Consistent with our previous results demonstrating that high PSMA protein expression is correlated to DDR defects, we found that treatment with topoisomerase-2 inhibitors, in particular daunorubicin, doxorubicin, and mitoxantrone, led to the most robust and significant upregulation of PSMA protein levels (Fig. 1A; Supplementary Figs. S1 and S2; Supplementary Table S4), closely followed by topoisomerase-1 inhibition with topotecan, and interestingly EGFR inhibition with gefitinib. Daunorubicin (1 μmol/L) induced the largest fold change (3.78-fold) compared with the vehicle controls (DMSO) (Fig. 1A–D).
Figure 1.

Topoisomerase-2 inhibitors upregulate PSMA in prostate cancer cells. LNCaP95 cells were treated individually for 48 hours with 1 μmol/L of 147 compounds available in the NIH FDA-approved anticancer drug. Lysates were then probed for PSMA and vinculin (VINC) expression, the latter used to normalize PSMA expression per sample. Densitometry of PSMA blots, normalized to VINC are expressed in A with fold change of PSMA in samples centered at DMSO = 0 (red in the heatmap). Topoisomerase-2 inhibitors (TopII; green), androgen receptor signaling inhibitors (ARSi; black), PI3K inhibitors (PI3K; blue), and mTOR inhibitors (yellow) are highlighted for reference in the heatmap. Every other compound is labeled in the heat map beginning from daunorubicin due to space. An example plot demonstrating PSMA upregulation in response to daunorubicin (B). Topoisomerase-2 inhibitors made up 3 of the top 10 PSMA inducing compounds tested at 1 μmol/L, and the respective changes in the 100 nmol/L screen are noted in the right column (C). Comparison of fold change of PSMA at 100 nmol/L and 1 μmol/L identified daunorubicin as the most consistent inducer of PSMA expression in the screen (D). DMSO is demarcated by a dashed line and a red circle at the intersection.
Enzalutamide and abiraterone, which inhibit androgen signaling and are used in mCRPC, reduced PSMA expression in the androgen-deprivation resistant LNCaP95 cells (Fig. 1B), although this reduction was small (−0.14 and −0.25, respectively). Similarly, although the PI3K/AKT/mTOR signaling axis has been implicated in PSMA function in prostate cancer cells (18), neither the PI3K inhibitors (duvelisib, copanlisib, and idelalisib) nor the mTOR inhibitors (temsirolimus, sirolimus, and everolimus) included in this screen significantly affected PSMA fold expression. Therefore, neither ARSI nor PI3K/AKT/mTOR signaling inhibitors were chosen for further validation.
Overall, however, these data indicated that PSMA expression levels are dynamic and suggested that PSMA protein expression can be modulated through eliciting DNA damage.
Daunorubicin and mitoxantrone induce PSMA expression in castration-sensitive and -resistant prostate cancer cells
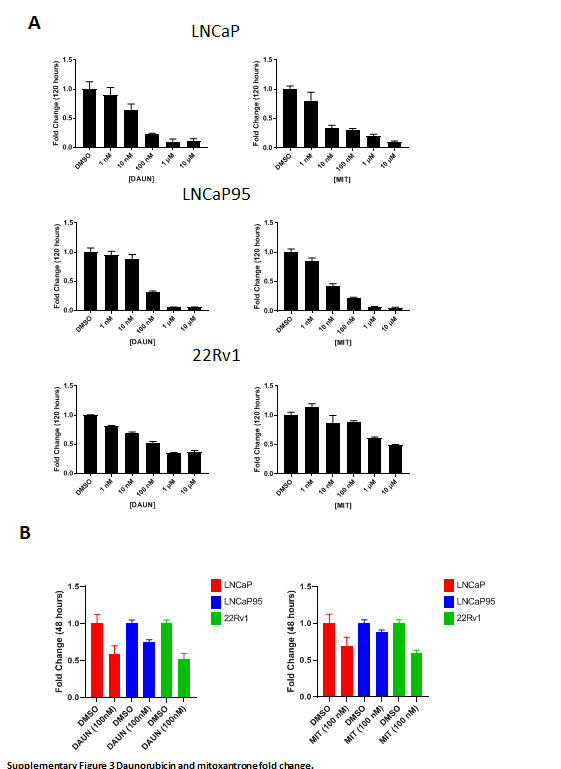
To validate the results of this screen, we next evaluated the effect of daunorubicin and mitoxantrone on PSMA protein expression in the castration-sensitive prostate cancer cell line LNCaP and the castration-resistant prostate cancer cell lines 22Rv1 and LNCaP95. As 1 μmol/L of both mitoxantrone and daunorubicin induced >50% reduction in cell viability after 5 days in most cell lines (Supplementary Fig. S3), treatment with 100 nmol/L of daunorubicin was chosen for validation studies. This resulted in higher cell viability after 48 hours of treatment across LNCaP, LNcaP95, and 22Rv1 cells, respectively as compared with 1 μmol/L daunorubicin (Supplementary Fig. S3). Both daunorubicin and mitoxantrone induced PSMA protein expression and phosphorylation of the Ser-139 residue of the histone variant H2AX (pγH2AX), though the magnitude of PSMA increase was higher in the castration-resistant LNCaP95 and 22Rv1 cell lines (Fig. 2A–C). Densitometric analyses of western blots demonstrated changes in PSMA protein expression to be statistically significant for both inhibitors across all cell lines, though this was most clear in castration-resistant lines (Fig. 2D). Daunorubicin induced a 1.3-, 2.4-, and 7.0-fold increase in LNCaP, LNCaP95, and 22Rv1 cells (P = 0.009, P < 0.0001, P < 0.0001), respectively. Mitoxantrone induced a 1.3-, 1.5-, and 2.0-fold increase in LNCaP, LNCaP95, and 22Rv1 cells (P = 0.014, P = 0.006, P = 0.012), respectively. RNA expression of the FOLH1 gene also increased with increasing doses of mitoxantrone and daunorubicin, however not to the same extent seen at the protein level. Increases in FOLH1 mRNA expression were significant only for mitoxantrone despite this inducing smaller changes in protein expression (Fig. 2E). Overall, these data confirmed our initial screen findings across numerous prostate cancer cell lines suggesting that topoisomerase-2 inhibitor treatment could increase PSMA expression to impact the efficacy of PSMA-targeting.
Figure 2.

Daunorubicin and mitoxantrone induce PSMA protein expression in castration-sensitive and -resistant prostate cancer cells. LNCaP, LNCaP95, and 22Rv1 cell lines were treated with 100 nmol/L of either daunorubicin or mitoxantrone for 48 hours and PSMA expression was quantified by western blot (A–D), qPCR (E), and IHC (F). Western blots of vehicle (DMSO), daunorubicin, or mitoxantrone for each cell line: LNCaP (A), LNCaP95 (B), and 22Rv1 (C) were also subject to the densitometric analysis of PSMA change (n = 3), and analysis of fold change in PSMA is reported for each cell line (D). Daunorubicin-induced upregulation in castration-resistant and sensitive lines was further investigated through IHC, demonstrating increases in membranous and cytoplasmic PSMA. Representative images at 10× and 20× are displayed in F. PSMA protein and RNA expression in treatment versus vehicle groups were subjected to two-sided, unpaired student's t-tests to detect significance. Mean and standard deviation are shown. Normality was assumed. Statistical significance was assumed at P < 0.05, with *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Black scale bars, 100 mm (top row) and 40 mm (bottom row).
Topoisomerase-2 inhibitors increase membranous PSMA expression
The antitumor activity of PSMA-targeting agents, including radionuclides and ADCs, is dependent on their ability to bind to the extracellular portion of PSMA (membranous PSMA). Given daunorubicin (100 nmol/L) induced the greatest fold change in total PSMA protein levels in LNCaP, LNCaP95, and 22Rv1 prostate cancer cell lines, we next evaluated these cells by IHC to study how daunorubicin increases in PSMA is distributed through the cell. Interestingly, although 22Rv1 and LNCaP prostate cancer cells demonstrated homogeneous, moderate, or high PSMA protein in DMSO-treated controls, respectively, PSMA protein levels in LNCaP95 prostate cancer cells were noticeably lower (Fig. 2F). Daunorubicin treatment increased both cytoplasmic and membranous PSMA protein levels (Fig. 2F). Critically, in addition to confirming the findings of our previous western blot experiments, these findings indicate that daunorubicin upregulates membranous PSMA protein levels, thereby potentially modulating the antitumor activity of PSMA-directed therapies.
Ionizing radiation induces membranous PSMA protein in castration-resistant prostate cancer cells
We next studied whether ionizing radiation-induced DNA double-strand breaks (DSB) affected PSMA expression in a comparable manner to topoisomerase-2 inhibitors in the castration-sensitive and -resistant prostate cancer cell lines LNCaP, 22Rv1, and LNCaP95. Cells were irradiated with 3, 6, or 12 Gy of cumulative radiation doses delivered over 3 (3#) or 6 (6#) fractions with 48-hour intervals over a period of 1 to 2 weeks (Fig. 3A). Samples were taken 24 hours after the final dose at week 1 (after 3 fractions), and 24 or 72 hours after the final dose of week 2 (after 6 fractions) and were analyzed for PSMA protein change by western blot. Total PSMA protein expression was upregulated after both 1 and 2 weeks of fractionated radiation across all the cell lines tested in a dose-dependent manner (Fig. 3B). PSMA protein levels increased across all cell lines studied after 12 Gy/6# treatment (LNCaP, P = 0.007; LNCaP95, P = 0.003; 22Rv1, P = 0.017). Interestingly, the androgen deprivation–sensitive cell line LNCaP and its castration-resistant subline LNCaP95 showed similar fold increases of 2.0 and 1.8, respectively, whereas the castration-resistant 22Rv1 prostate cancer cells demonstrated a 10-fold increase in PSMA in response to the same treatment dose. Both LNCaP and LNCaP95 cells also showed significant increases in PSMA protein expression after 6 Gy/6# treatment (LNCaP, P = 0.017; LNCaP95, P = 0.007), with 1.8- and 1.7-fold increases, respectively (Fig. 3C), although the same dose was not sufficient to induce significant increases in 22Rv1. Interestingly, however, qPCR analysis of lysates did not demonstrate an equivalent upregulation of PSMA mRNA at the time points evaluated (Supplementary Fig. S4), suggesting that the accumulation of PSMA on the membranes of treated prostate cancer cells was not transcriptionally driven. Upregulation of PSMA protein was sustained for up to 72 hours after the final dose of both 6 Gy/6# and 12 Gy/6# fractionation schedules in all evaluated prostate cancer cell lines (Fig. 3D); conversely, PSMA upregulation was not observed in nonmalignant prostatic epithelial cell lines even after 6 doses of radiation (Fig. 3E). Similar to our observations after treatment with the topoisomerase-2 inhibitors, mitoxantrone and daunorubicin, PSMA IHC analysis of cell pellets from radiation-treated samples reported upregulated membranous (Fig. 3E) PSMA, confirming our western blot results in Fig. 3B.
Figure 3.

Repeated radiation induces PSMA protein expression in prostate cancer cells. LNCaP, LNCaP95, and 22Rv1 cell lines were subjected to cumulative radiation doses of 3, 6, or 12 Gy in 3 or 6 fractions (3 Gy/3#, 6 Gy/3#, 6 Gy/6#, and 12 Gy/6#). Samples were taken for analysis 24 hours after 3 or 6 fractions and 72 hours after 6 fractions (A). Western blot analysis evaluated PSMA protein change after 1 or 2 weeks of fractionated radiation (B). Densitometric analysis (n = 3) of blots showed significant increases in PSMA protein across all lines at 12-Gy doses delivered in 6 fractions (LNCaP, P = 0.007; LNCaP95, P = 0.003; 22Rv1, P = 0.017) and in LNCaP95 and LNCaP cells at 6-Gy doses delivered in 6 fractions (LNCaP, P = 0.017; LNCaP95, P = 0.007; C). PSMA upregulation was sustained for 72 hours after the final dose of ionizing radiation of either 6 Gy/6# or 12 Gy/6# (D15) across all cell lines (D). FFPE cell pellets treated with 3 Gy/3# underwent IHC analysis for cytoplasmic and membranous PSMA; representative images are shown for each cell line (E). PSMA protein change in treatment versus mock groups was subjected to student's t-tests to detect significance. Mean and standard deviation are shown. Normality was assumed. Black scale bars, 100 mm (top row) and 40 mm (bottom row). Statistical significance was considered at P < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Topoisomerase-2 inhibition and repeat radiation in PDXs induce increased PSMA expression in vivo
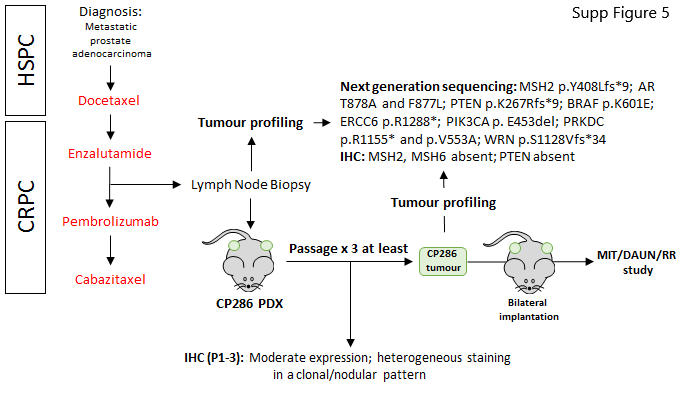
To further support the utility of topoisomerase-2 inhibitors to enhance the expression of PSMA membranous protein, an mCRPC-derived PDX model (CP286) with relatively low and heterogeneous PSMA expression was treated with both daunorubicin and mitoxantrone (Fig. 4). CP286 tumors were also repeat irradiated to demonstrate PSMA upregulation in vivo in response to external beam, ionizing radiation (Fig. 5), as observed in vitro (Fig. 3). Molecular characterization of this model revealed F877L and T878A mutations of the androgen receptor, a BRAF K601E mutation, as well as deleterious mutations of PTEN and MSH2 and MSH6 absence on IHC (Supplementary Table S5). NSG mice were implanted bilaterally with CP286 PDX tumors, heterogeneous for PSMA expression (Supplementary Fig. S5). Once tumors had grown to 400 mm3, mice were randomized and treated. Topoisomerase-2 treatment arms included vehicle (PBS), or 0.5 mg/kg or 1 mg/kg of daunorubicin or mitoxantrone, having confirmed appropriate drug concentrations to be administered from a preliminary dose tolerance study (Fig. 4A; Supplementary Fig. S6). Irradiation arms included mock irradiation (mock) or repeated irradiations with cumulative doses of 6 Gy (6 Gy/6#) or 12 Gy (12 Gy/6#); this was equivalent to in vitro dosing. Tumor samples were harvested on day 8 or day 12 for topoisomerase-2 or irradiation studies, respectively, and investigated for change in PSMA expression through IHC, qPCR, and western blot analyses.
Figure 4.

Topoisomerase-2 inhibition induces significant increases in PSMA protein expression and reduces the heterogeneity of PSMA expression in PDX tumors. CP286 PDX tumor pieces were implanted into NSG mice and grown to 400 mm3 before randomization and treatment with either vehicle (PBS), or 0.5 or 1 mg/kg of daunorubicin (DAUN) or mitoxantrone (MIT; Supplementary Fig. S5; A). Tumor samples were subsequently taken on day 8 and analyzed for PSMA expression through IHC and western blot analysis. Three independent tumor cores from each treatment group were fixed and underwent PSMA IHC. Representative images of daunorubicin-treated tumors at 10× and 20× are displayed. Black scale bars, 100 μm (top row) and 40 μm (bottom row; B). IHC of tumor cores was scanned and analyzed by HALO image analysis software. Percentage cells, percentage cytoplasmic, and percentage membranous (C) positivity for PSMA were compared between treatment groups. Average optical density (OD), a read-out for intensity, was also investigated for changes in cytoplasmic and membranous PSMA (D). Lysates of tumor cores were also analyzed by western blot (E), and densitometry (F) was undertaken to measure change in PSMA. PSMA was normalized to the housekeeping gene vinculin (VINC). The highest treatment group displayed significantly higher PSMA protein in western blot (P = 0.004). Changes in various measures of PSMA in treatment versus vehicle groups were subjected to student's t-test to detect significance. Median and 95% confidence intervals are shown. Normality was assumed. Statistical significance was considered at P < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Figure 5.

Repeat radiation induces significant increases in PSMA protein expression and reduces the heterogeneity of PSMA expression in PDX tumors. CP286 PDX tumor pieces were implanted into NSG mice and grown to 400 mm3 before randomization and treatment with either mock or repeated radiation (RR); RR included 6 repeated doses of 1 Gy (cumulative 6 Gy) or 2 Gy (cumulative 12 Gy) of ionizing external beam irradiation (A). Tumor samples were subsequently taken on day 12 and analyzed for PSMA expression through IHC analysis. Four tumors per treatment group were fixed and underwent PSMA IHC. Representative images of mock or irradiated tumors at 10× and 20× are displayed. Black scale bars represent 100 μm (top row) and 50 μm (bottom row; B). As CP286 mock-treated tumors were heterogeneous for PSMA expression, an example of PSMA-positive and -negative tumor is shown. IHC of tumor cores was scanned and analyzed by HALO image analysis software. Percentage cells positive for PSMA were compared between treatment groups (C). Average optical density (OD), a read-out for intensity, was also investigated for changes in cytoplasmic and membranous PSMA (C). Changes in various measures of PSMA in treatment versus vehicle groups were subjected to student's t-test to detect significance. Median and 95% confidence intervals are shown. Normality was assumed. Statistical significance was considered at P < 0.05 (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
These studies revealed that daunorubicin (1 mg/kg) treatment significantly upregulated membranous and cytoplasmic PSMA expression (Fig. 4B–D). Furthermore, the median percentage of total PSMA-positive cells and cytoplasmic PSMA-positive cells was both 1.67-fold higher in tumors treated with 1 mg/kg of daunorubicin (P = 0.005, P = 0.005). Overall, the median percentage of membranous PSMA-positive cells increased by 4.37-fold (P = 0.02). Median cytoplasmic (P = 0.004) and membranous (P = 0.033) PSMA optical density (OD) also increased 1.93- and 5.26-fold, respectively, demonstrating an increase in staining intensity alongside the decrease in the percentage of PSMA-negative cells 50% (vehicle) vs. 16% (1 mg/kg daunorubicin). These findings were confirmed by an 8-fold increase in PSMA protein (P = 0.004) determined by western blotting of tumor lysates previously treated with 1 mg/kg of daunorubicin (Fig. 4E and F). Similarly, 1 and 0.5 mg/kg doses of mitoxantrone also increased PSMA protein levels, although this was not statistically significant (Supplementary Fig. S6). Interestingly, mRNA expression of PSMA did not change across any treatment group (Supplementary Fig. S7).
Similarly, repeated irradiation induced a significant increase in both membranous and cytoplasmic PSMA OD in CP286 tumors after 12 Gy (P = 0.004, P = 0.013). A 2.9-fold and 1.7-fold increase was observed after a cumulative dose of 12 Gy in membranous and cytoplasmic PSMA, respectively (Fig. 5A–C). Though a repeat cumulative dose increased the percentage of PSMA-positive cells, and cumulative doses of 6 Gy increased membranous and cytoplasmic PSMA OD, none reached statistical significance.
Taken together, these data confirm in vivo that PSMA protein levels are dynamic and can be upregulated by treatment with topoisomerase-2 inhibition or repeated radiation, suggesting the former to be a candidate for combination therapy with PSMA-directed therapies currently in development (Supplementary Fig. S8).
Discussion
Clinical trials evaluating PSMA-directed therapies in patients with mCRPC, such as 177Lu–PSMA-617, have reported significant improvements in overall survival and symptomatic benefit compared with cabazitaxel chemotherapy or best supportive care (1, 2). However, outcomes in patients with low PSMA expression, who are often not eligible for clinical trials evaluating PSMA-directed therapies, remain unsatisfactory (8). Despite the rapid utilization of PSMA-directed therapies in the clinic, their function and regulation remain poorly understood. The results presented herein are the first, to our knowledge, to demonstrate PSMA upregulation in response to DNA damage by ionizing radiation and topoisomerase-2 inhibitors. Furthermore, we report that topoisomerase-2 inhibition and repeat irradiation both increase PSMA expression and reduce the heterogeneity of its expression in a PDX derived from a CRPC patient, suggesting that this could have a direct impact on the clinical utility of PSMA-directed therapies. These studies not only describe a novel way to modulate PSMA expression, through DNA damage induction, but may also provide insight into the underlying function of PSMA in prostate cancer in DNA damage responses.
The topoisomerase-2 proteins, TOP2A and β (TOP2B), generate transient DSBs to reduce the supercoiling of DNA (27). Most topoisomerase-2 inhibitors, such as daunorubicin, inhibit both TOP2A and TOP2B (28). By stabilizing the covalently bound topoisomerase complexes onto DNA, DSBs cannot be repaired leading to irrecoverable DNA damage. Specifically, TOP2A is critical for cell replication and is highest in G2–M phases, whereas TOP2B is expressed throughout the cell cycle (29, 30). Therefore, TOP2A inhibition and DNA DSBs likely account for most of the cytotoxic impact of topoisomerase-2 inhibitors. Our results demonstrate that these effects of topoisomerase-2 inhibition drive PSMA expression. This is in keeping with results from other groups reporting that BRCA2 CRISPR knockout and treatment with PSMA-TCC, an alpha particle emitting RLT toward PSMA, induce PSMA expression (24, 25). Interestingly, our screen also identified that toposisomerase-1 inhibition and EGFR inhibition upregulate PSMA; as topoisomerase-1 inhibitors can also cause DNA single- and double-strand breaks this is perhaps not surprising (31). Moreover, multiple reports indicate that EGFR signaling regulates DNA repair, impacting the expression for example of RAD51 (32). EGFR blockade will therefore likely decrease DNA repair and increase DNA damage, which we now hypothesize may explain why PSMA increases after EGFR blockade.
Ionizing radiation similarly causes cytotoxicity by inducing DSBs during G2–M phase and, like topoisomerase-2 inhibition, also induced PSMA expression. Interestingly, the fractionation of radiation doses can exacerbate DSBs in tumor cells, particularly in cells with DDR aberrations (33). Repeated radiation doses are reported to enrich tumor cells in the G2 phase of the cell cycle, increasing the efficacy of subsequent doses (34, 35). We now hypothesize that this induction of DNA DSBs results in PSMA upregulation, probably to support cell survival through increased AKT signaling and nucleotide pool generation to facilitate DNA repair, through glutamate and folate metabolism.
Cells challenged by low folate conditions can be advantaged by PSMA overexpression in the presence of polyglutamated folates (22) due to the generation of monoglutamated folate that can be utilized intracellularly. Reports indicate that enzymatic inhibition of PSMA in mice bearing ovarian serous adenocarcinoma xenografts results in significantly lower tumor-specific glutamate concentrations and reduced tumor growth (23). Glutamate and folate are critical to the biogenesis of nucleotides and the maintenance of DNA replication (36–38). The relationship of PSMA to the generation of glutamate, monoglutamated folate, and to nucleotide pool maintenance in prostate cancer and other tumor cells merits further study. This requires the in vitro use, however, of folate-deprived media that more accurately reflect cancer cells in patients. Untangling the mechanism whereby DSBs in DNA result in PSMA upregulation may help further elucidate the functions of PSMA in prostate cancer.
Interestingly, across all experiments, PSMA mRNA was not reflective of PSMA protein change. This suggests that PSMA expression may be posttranslationally stabilized in response to DSBs, rather than transcriptionally upregulated. Further work into the posttranslational regulation of PSMA expression, an area poorly understood in the context of PSMA biology, is therefore needed, for example, studying the ubiquitination and recycling of the PSMA protein in response to DNA damage (16, 39, 40). The transcriptional regulation of PSMA has been studied to some extent; AR signaling inhibition is reported to drive PSMA expression suggesting a regulatory mechanism, although we were unable to confirm this from the ARSI compounds investigated in the screen (41–43). The enhancer region of the PSMA gene, which is shown to be responsible for gene repression by androgens, possesses an AR response element half site (44). However, PSMA expression is downregulated with neuroendocrine differentiation and loss of AR signaling in prostate cancer, and there is little evidence of the direct binding of the AR to the FOLH1 promoter to date (12). Other transcription factors are reported to bind FOLH1 directly, including SOX7 and NFATc1, though their impact on PSMA expression appears to be small (45, 46).
In conclusion, our studies are of immediate translational relevance and support the clinical study of PSMA expression by PET scans before and after topoisomerase-2 inhibition to gather data supporting the use of these agents in combination with PSMA-targeting therapeutic strategies. We hypothesize that such a rational combination could reduce the heterogeneity of PSMA expression and increase the efficacy of PSMA-directed treatments, which are changing survival outcomes from lethal prostate cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Work in the AS laboratory is supported by research funding from the Wellcome Trust, the Prostate Cancer Foundation, and the NIHR Biomedical Research Centre. Work in the JdB laboratory is supported by research funding from Prostate Cancer UK, the Movember Foundation through the London Movember Centre of Excellence (CEO13_2-002), the Prostate Cancer Foundation, Cancer Research UK (Centre Programme grant), Experimental Cancer Medicine Centre grant funding from Cancer Research UK and the Department of Health, and Biomedical Research Centre funding to the Royal Marsden. J. de Bono is a National Institute for Health Research (NIHR) Senior Investigator. B. Sheehan was supported by a PhD studentship from the Institute of Cancer Research. J. Jiménez Vacas is supported by a Prostate Cancer UK Travelling Prize Fellowship. The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
A. Paschalis reports a patent for JMJD6 as an actionable therapeutic target in lethal prostate cancer pending. A. Sharp reports nonfinancial support from Sanofi and Roche-Genentech; personal fees from Astellas Pharma; and other support from DE Shaw Research outside the submitted work. In addition, A. Sharp has been the CI/PI of industry-sponsored clinical trials (no PSMA-targeted therapies). J. de Bono has served on advisory boards and received fees from many companies including Amgen, AstraZeneca, Astellas, Bayer, Bioxcel Therapeutics, Boehringer Ingelheim, Cellcentric, Daiichi, Eisai, Genentech/Roche, Genmab, GSK, Harpoon, ImCheck Therapeutics, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Pfizer, Qiagen, Sanofi Aventis, Sierra Oncology, Taiho, Terumo, and Vertex Pharmaceuticals. In addition, J. de Bono is an employee of The ICR, which has received funding or other support for de Bono's research work from AstraZeneca, Astellas, Bayer, Cellcentric, Daiichi, Genentech, Genmab, GSK, Janssen, Merck Serono, MSD, Menarini/Silicon Biosystems, Orion, Sanofi Aventis, Sierra Oncology, Taiho, Pfizer, and Vertex, and which has a commercial interest in abiraterone, PARP inhibition in DNA repair defective cancers and PI3K/AKT pathway inhibitors (no personal income). Finally, J. de Bono was named as an inventor, with no financial interest for patent 8,822,438, submitted by Janssen that covers the use of abiraterone acetate with corticosteroids; J. de Bono has been the CI/PI of many industry-sponsored clinical trials. No disclosures were reported by the other authors.
Authors' Contributions
B. Sheehan: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Neeb: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing. L. Buroni: Investigation, writing–original draft, writing–review and editing. A. Paschalis: Writing–original draft, writing–review and editing. R. Riisnaes: Validation, visualization, writing–review and editing. B. Gurel: Formal analysis, validation, visualization, writing–review and editing. V. Gil: Methodology, writing–review and editing. S. Miranda: Writing–review and editing. M. Crespo: Validation, writing–review and editing. C. Guo: Writing–review and editing. J. Jiménez Vacas: Investigation, writing–review and editing. I. Figueiredo: Validation, visualization, writing–review and editing. A. Ferreira: Validation, visualization, writing–review and editing. J. Welti: Supervision, writing–review and editing. W. Yuan: Data curation, software, writing–review and editing. S. Carreira: Data curation, software, investigation, writing–review and editing. A. Sharp: Supervision, writing–review and editing. J. de Bono: Conceptualization, resources, formal analysis, supervision, funding acquisition, methodology, project administration, writing–review and editing.
References
- 1. Hofman MS, Emmett L, Sandhu S, Iravani A, Joshua AM, Goh JC, et al. [(177)Lu]Lu-PSMA-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (TheraP): a randomised, open-label, phase 2 trial. Lancet 2021;397:797–804. [DOI] [PubMed] [Google Scholar]
- 2. Sartor O, De Bono J, Chi KN, Fizazi K, Herrmann K, Rahbar K, et al. Lutetium-177–PSMA-617 for metastatic castration-resistant prostate cancer. N Engl J Med 2021;385:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sheehan B, Guo C, Neeb A, Paschalis A, Sandhu S, De Bono JS. Prostate-specific membrane antigen biology in lethal prostate cancer and its therapeutic implications. Eur Urol Focus 2021;S2405-4569(21)00168-1. [DOI] [PubMed] [Google Scholar]
- 4. Lawal IO, Bruchertseifer F, Vorster M, Morgenstern A, Sathekge MM. Prostate-specific membrane antigen-targeted endoradiotherapy in metastatic prostate cancer. Curr Opin Urol 2020;30:98–105. [DOI] [PubMed] [Google Scholar]
- 5. Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies: the next big thing in solid tumor therapeutics. Mol Med 2018;24:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emmett L, Crumbaker M, Ho B, Willowson K, Eu P, Ratnayake L, et al. Results of a prospective phase 2 pilot trial of Lu-177-PSMA-617 therapy for metastatic castration-resistant prostate cancer including imaging predictors of treatment response and patterns of progression. Clin Genitourin Cancer 2019;17:15–22. [DOI] [PubMed] [Google Scholar]
- 7. Emmett L, Willowson K, Violet J, Shin J, Blanksby A, Lee J. Lutetium 177 PSMA radionuclide therapy for men with prostate cancer: a review of the current literature and discussion of practical aspects of therapy. J Med Radiat Sci 2017;64:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thang SP, Violet J, Sandhu S, Iravani A, Akhurst T, Kong G, et al. Poor outcomes for patients with metastatic castration-resistant prostate cancer with low prostate-specific membrane antigen (PSMA) expression deemed ineligible for (177)Lu-labelled PSMA radioligand therapy. Eur Urol Oncol 2019;2:670–6. [DOI] [PubMed] [Google Scholar]
- 9. Silver DA, Pellicer I, Fair WR, Heston WD, Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin Cancer Res 1997;3:81–5. [PubMed] [Google Scholar]
- 10. Mannweiler S, Amersdorfer P, Trajanoski S, Terrett JA, King D, Mehes G. Heterogeneity of prostate-specific membrane antigen (PSMA) expression in prostate carcinoma with distant metastasis. Pathol Oncol Res 2009;15:167–72. [DOI] [PubMed] [Google Scholar]
- 11. Mhawech-Fauceglia P, Zhang S, Terracciano L, Sauter G, Chadhuri A, Herrmann FR, et al. Prostate-specific membrane antigen (PSMA) protein expression in normal and neoplastic tissues and its sensitivity and specificity in prostate adenocarcinoma: an immunohistochemical study using mutiple tumour tissue microarray technique. Histopathology 2007;50:472–83. [DOI] [PubMed] [Google Scholar]
- 12. Bakht MK, Derecichei I, Li Y, Ferraiuolo R-M, Dunning M, Oh SW, et al. Neuroendocrine differentiation of prostate cancer leads to PSMA suppression. Endocr Relat Cancer 2018;26:131–46. [DOI] [PubMed] [Google Scholar]
- 13. Emmett L, Yin C, Crumbaker M, Hruby G, Kneebone A, Epstein R, et al. Rapid modulation of PSMA expression by androgen deprivation: serial (68)Ga-PSMA-11 PET in men with hormone-sensitive and castrate-resistant prostate cancer commencing androgen blockade. J Nucl Med 2019;60:950–4. [DOI] [PubMed] [Google Scholar]
- 14. Kratochwil C, Giesel FL, Heussel C-P, Kazdal D, Endris V, Nientiedt C, et al. Patients resistant against PSMA-targeting alpha-radiation therapy often harbor mutations in DNA damage-repair-associated genes. J Nucl Med 2020;61:683–8. [DOI] [PubMed] [Google Scholar]
- 15. Paschalis A, Sheehan B, Riisnaes R, Rodrigues DN, Gurel B, Bertan C, et al. Prostate-specific membrane antigen heterogeneity and DNA repair defects in prostate cancer. Eur Urol 2019;76:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, et al. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J 2006;25:1375–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci U S A 2005;102:5981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaittanis C, Andreou C, Hieronymus H, Mao N, Foss CA, Eiber M, et al. Prostate-specific membrane antigen cleavage of vitamin B9 stimulates oncogenic signaling through metabotropic glutamate receptors. J Exp Med 2018;215:159–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Conway RE, Petrovic N, Li Z, Heston W, Wu D, Shapiro LH. Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol Cell Biol 2006;26:5310–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Conway RE, Rojas C, Alt J, Nováková Z, Richardson SM, Rodrick TC, et al. Prostate-specific membrane antigen (PSMA)-mediated laminin proteolysis generates a pro-angiogenic peptide. Angiogenesis 2016;19:487–500. [DOI] [PubMed] [Google Scholar]
- 21. Yao V, Berkman CE, Choi JK, O'keefe DS, Bacich DJ. Expression of prostate-specific membrane antigen (PSMA), increases cell folate uptake and proliferation and suggests a novel role for PSMA in the uptake of the non-polyglutamated folate, folic acid. Prostate 2010;70:305–16. [DOI] [PubMed] [Google Scholar]
- 22. Yao V, Bacich DJ. Prostate-specific membrane antigen (PSMA) expression gives prostate cancer cells a growth advantage in a physiologically relevant folate environment in vitro. Prostate 2006;66:867–75. [DOI] [PubMed] [Google Scholar]
- 23. Nguyen T, Kirsch BJ, Asaka R, Nabi K, Quinones A, Tan J, et al. Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Rep 2019;27:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakraborty G, Armenia J, Mazzu YZ, Nandakumar S, Stopsack KH, Atiq MO, et al. Significance of BRCA2 and RB1 co-loss in aggressive prostate cancer progression. Clin Cancer Res 2020;26:2047–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hammer S, Schlicker A, Zitzmann-Kolbe S, Baumgart S, Hagemann UB, Scholz A, et al. Darolutamide potentiates the antitumor efficacy of a PSMA-targeted thorium-227 conjugate by a dual mode of action in prostate cancer models. Clin Cancer Res 2021;27:4367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banerjee SR, Foss CA, Castanares M, Mease RC, Byun Y, Fox JJ, et al. Synthesis and evaluation of technetium-99m- and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J Med Chem 2008;51:4504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vos SM, Tretter EM, Schmidt BH, Berger JM. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat Rev Mol Cell Biol 2011;12:827–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Skok Ž, Zidar N, Kikelj D, Ilaš J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J Med Chem 2020;63:884–904. [DOI] [PubMed] [Google Scholar]
- 29. Nielsen CF, Zhang T, Barisic M, Kalitsis P, Hudson DF. Topoisomerase IIα is essential for maintenance of mitotic chromosome structure. Proc Natl Acad Sci U S A 2020;117:12131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Earnshaw WC, Halligan B, Cooke CA, Heck MM, Liu LF. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J Cell Biol 1985;100:1706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Coussy F, El-Botty R, Château-Joubert S, Dahmani A, Montaudon E, Leboucher S, et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci Transl Med 2020;12:eaax2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res 2011;71:1103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin L, Marples B, Coffey M, Lawler M, Hollywood D, Marignol L. Recognition of O6MeG lesions by MGMT and mismatch repair proficiency may be a prerequisite for low-dose radiation hypersensitivity. Radiat Res 2009;172:405–13. [DOI] [PubMed] [Google Scholar]
- 34. Todd P. Fractionated heavy ion irradiation of cultured human cells. Radiat Res 1968;34:378–89. [PubMed] [Google Scholar]
- 35. Ngo FQH, Blakely EA, Tobias CA, Chang PY, Lommel L. Sequential exposures of mammalian cells to low- and high-LET radiations. II. As a function of cell-cycle stages. Radiat Res 1988;115:54–69. [PubMed] [Google Scholar]
- 36. Blount BC, Mack MM, Wehr CM, Macgregor JT, Hiatt RA, Wang G, et al. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc Natl Acad Sci U S A 1997;94:3290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015;162:552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B, et al. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep 2019;28:1136–43. [DOI] [PubMed] [Google Scholar]
- 39. Liu T, Mendes DE, Berkman CE. Functional prostate-specific membrane antigen is enriched in exosomes from prostate cancer cells. Int J Oncol 2014;44:918–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meighan MA, Dickerson MT, Glinskii O, Glinsky VV, Wright GL Jr, Deutscher SL. Recombinant glutamate carboxypeptidase II (prostate specific membrane antigen–PSMA)–cellular localization and bioactivity analyses. J Protein Chem 2003;22:317–26. [DOI] [PubMed] [Google Scholar]
- 41. Evans MJ, Smith-Jones PM, Wongvipat J, Navarro V, Kim S, Bander NH, et al. Noninvasive measurement of androgen receptor signaling with a positron-emitting radiopharmaceutical that targets prostate-specific membrane antigen. Proc Natl Acad Sci U S A 2011;108:9578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kranzbühler B, Salemi S, Umbricht CA, Müller C, Burger IA, Sulser T, et al. Pharmacological upregulation of prostate-specific membrane antigen (PSMA) expression in prostate cancer cells. Prostate 2018;78:758–65. [DOI] [PubMed] [Google Scholar]
- 43. Murga JD, Moorji SM, Han AQ, Magargal WW, Dipippo VA, Olson WC. Synergistic co-targeting of prostate-specific membrane antigen and androgen receptor in prostate cancer. Prostate 2015;75:242–54. [DOI] [PubMed] [Google Scholar]
- 44. Watt F, Martorana A, Brookes DE, Ho T, Kingsley E, O'Keefe DS, et al. A tissue-specific enhancer of the prostate-specific membrane antigen gene, FOLH1. Genomics 2001;73:243–54. [DOI] [PubMed] [Google Scholar]
- 45. Peng W, Guo L, Tang R, Liu X, Jin R, Dong J-T, et al. Sox7 negatively regulates prostate-specific membrane antigen (PSMA) expression through PSMA-enhancer. Prostate 2019;79:370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee S-J, Lee K, Yang X, Jung C, Gardner T, Kim H-S, et al. NFATc1 with AP-3 site binding specificity mediates gene expression of prostate-specific-membrane-antigen. J Mol Biol 2003;330:749–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were generated by the authors and are included in this article.