

Abstract
Numerous brain disorders demonstrate structural brain abnormalities, which are thought to arise from molecular perturbations or connectome miswiring. The unique and shared contributions of these molecular and connectomic vulnerabilities to brain disorders remain unknown, and has yet to be studied in a single multi-disorder framework. Using MRI morphometry from the ENIGMA consortium, we construct maps of cortical abnormalities for thirteen neurodevelopmental, neurological, and psychiatric disorders from N = 21,000 participants and N = 26,000 controls, collected using a harmonised processing protocol. We systematically compare cortical maps to multiple micro-architectural measures, including gene expression, neurotransmitter density, metabolism, and myelination (molecular vulnerability), as well as global connectomic measures including number of connections, centrality, and connection diversity (connectomic vulnerability). We find a relationship between molecular vulnerability and white-matter architecture that drives cortical disorder profiles. Local attributes, particularly neurotransmitter receptor profiles, constitute the best predictors of both disorder-specific cortical morphology and cross-disorder similarity. Finally, we find that cross-disorder abnormalities are consistently subtended by a small subset of network epicentres in bilateral sensory-motor, inferior temporal lobe, precuneus, and superior parietal cortex. Collectively, our results highlight how local molecular attributes and global connectivity jointly shape cross-disorder cortical abnormalities.
Subject terms: Network models, Diseases of the nervous system, Neurological disorders
Changes to structural and functional connectivity can give rise to neurodegeneration and neurodevelopmental diseases. Here the authors investigate molecular and connectomic patterns in 13 different neurological, psychiatric and neurodevelopmental diseases from the ENIGMA consortium.
Introduction
The brain is a network with intricate connection patterns among individual neurons, neuronal populations, and large-scale brain regions. The wiring of the network supports propagation of electrical signals, as well as molecules needed for growth and repair. This complex system is vulnerable to multiple neurological, psychiatric and neurodevelopmental disorders. Pathological perturbations—including altered cellular morphology, cell death, aberrant synaptic pruning and miswiring—disrupt inter-regional communication and manifest as overlapping groups of sensory, motor, cognitive and affective symptoms. How different disorders are shaped by local and global vulnerability is unknown.
Indeed, several studies have demonstrated cross-disorder connectomic vulnerability, where regions and white-matter pathways are targeted non-randomly. In particular, regions that are highly connected and potentially important for communication tend to be disproportionately affected by disease1,2. A similar phenomenon is observed for connections that support multiple communication pathways3. In neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases, emerging evidence suggests pathological misfolded proteins spread trans-synaptically, such that the connectivity of the brain shapes the course and expression of these diseases4–11. Recent evidence also suggests that patterns of tissue volume loss in schizophrenia are circumscribed by structural and functional connection patterns12,13. Collectively, these studies demonstrate that both neurodevelopmental and neurodegenerative brain diseases are influenced by network connectivity14,15.
The effects of disease can also be driven by local cellular and molecular vulnerability. Namely, local patterns of gene expression16–18, neurotransmitter receptor profiles19, cellular composition20, and metabolism21–24 may predispose individual regions to stress and, ultimately, pathology. Importantly, local and global vulnerability are not necessarily mutually exclusive; some diseases may originate from local pathologies that spread selectively along the network to other vulnerable regions. How local attributes and global connectivity shape cross-disorder pathology remains an open question.
Here, we map local molecular attributes ("molecular vulnerability”) and global network connectivity ("connectomic vulnerability”) to case versus control cortical thickness abnormalities of thirteen different neurological, psychiatric, and neurodevelopmental diseases and disorders from the ENIGMA consortium25. We consistently find that disorder-specific cortical abnormality is shaped more by the local molecular fingerprints of brain regions than network embedding. Interestingly, for disorders that are better predicted by molecular attributes, we find that the spatial patterning of cortical abnormalities reflects the underlying network architecture, suggesting that the joint contribution of local molecular and global connectomic mechanisms is greater than their individual contribution. Next, we study cross-disorder similarity and find that regions with similar molecular make-up tend to be similarly affected across disorders. Collectively, the present report highlights how local and global factors interact to shape cross-disorder cortical morphology.
Results
We collected thirteen spatial maps of cortical abnormalities from the ENIGMA consortium for the following diseases, disorders, and conditions: 22q11.2 deletion syndrome26, attention-deficit/hyperactivity disorder (ADHD)27, autism spectrum disorder (ASD)28, idiopathic generalised epilepsy29, right temporal lobe epilepsy29, left temporal lobe epilepsy29, depression30, obsessive-compulsive disorder (OCD)31, schizophrenia32, bipolar disorder33, obesity34, schizotypy35, and Parkinson’s disease36. For simplicity, we refer to diseases, disorders, and conditions as “disorders” throughout the text. While most disorders show decreases in cortical thickness, some (e.g., 22q11.2 deletion syndrome, ASD, schizotypy) also show regional increases in cortical thickness. We therefore refer to the cortical measure as “cortical abnormality”. All cortical abnormality maps were collected from adult participants (except ASD which included younger participants), following identical processing protocols, for a total of over 21,000 scanned participants against almost 26,000 controls. To assess the extent to which each abnormality pattern is informed by molecular attributes and network connectivity, we defined a molecular and connectivity fingerprint at each brain region. The molecular fingerprint of a region was defined using the gene expression gradient (a potential proxy for cell type distribution16,20,37–39), neurotransmitter receptor gradient, excitatory-inhibitory receptor density ratio, glycolytic index, glucose metabolism, synapse density, and myelination (Fig. 1a). Likewise, we defined the connectivity fingerprint of a region by calculating the strength, betweenness centrality, closeness centrality, mean Euclidean distance, participation coefficient, clustering coefficient, and mean first passage time of a weighted structural connectivity matrix from 70 healthy adults (Fig. 1b; see Methods for details). Collectively, these graph measures aim to capture the connectedness, centrality, and connection diversity of regions in the network. All analyses were conducted using the 68-region Desikan-Killiany parcellation40,41, as this is the native and only available representation of ENIGMA datasets.
Fig. 1. Molecular and connectomic cortical profiles.

a, b Brain surfaces show the z-scored molecular (a) and connectomic (b) predictors used in the multilinear regression models. Heatmaps on the right show Pearson's correlation coefficients between pairs of features. See Methods for details on how each feature was derived. Molecular predictors: gene PC1 = first component of 11 560 genes' expression; receptor PC1 = first component of 18 PET-derived receptor/transporter density; E:I ratio = excitatory:inhibitory receptor density ratio; glycolytic index = amount of aerobic glycolysis; glucose metabolism -labelled fluorodeoxyglucose (FDG) PET image; synapse density = synaptic vesicle glycoprotein 2A (SV2A)-binding UCB-J PET tracer; myelination = T1w/T2w ratio. Connectivity predictors: strength = sum of weighted connections; betweenness = fraction of all shortest paths traversing region i; closeness = mean shortest path length between region i and all other regions; Euclidean distance = mean Euclidean distance between region i and all other regions; participation coefficient = diversity of connections from region i to the seven Yeo-Krienen resting-state networks164; clustering = fraction of triangles including region i; mean first passage time = average time for a random walker to travel from region i to any other region.
Local and global contributions to disorder-specific cortical morphology
To assess the extent to which cortical abnormalities of all thirteen disorders are informed by molecular gradients versus measures of network connectivity, we fit a multilinear model between molecular or connectivity predictors and abnormality maps for each disorder separately, for a total of 13 × 2 = 26 model fits (Fig. 2a; for results when molecular and connectomic predictors are combined, see Supplementary Fig. 1). Next, we conducted a dominance analysis for each multilinear model42–45. Dominance analysis distributes the across input variables as a measure of contribution ("dominance”) that each input variable has on the cortical abnormality pattern (Fig. 2b). Each model was cross-validated in a distance-dependent manner (Supplementary Fig. 239).
Fig. 2. Local and global contributions to disorder-specific cortical morphology.

a A total of twenty-six multilinear models were fit between local molecular and global connectome predictors to cortical abnormality maps of thirteen different disorders (surface plots, left). Adjusted R2 is shown in the bar plot (orange: molecular; blue: connectivity). b Dominance analysis was applied to assess the contribution of each input variable (done separately for molecular (orange) and connectivity (blue) predictors) to the fit of the model.
We find that the fit between molecular predictors and cortical abnormality is greater than that between connectivity predictors and cortical abnormality for most disorders (Fig. 3). Notably, the variance in cortical thickness of schizotypy (a possible precursor of schizophrenia that is poorly defined in the brain46) and idiopathic generalised epilepsy (a form of epilepsy that is thought to be informed by genetics instead of brain structural abnormalities47,48) are poorly explained by both biological gradients and network measures of the brain. On the other hand, ADHD, ASD, OCD, Parkinson’s, and depression are better predicted by molecular predictors, whereas schizophrenia, 22q11.2 deletion syndrome, and bipolar disorder are better predicted by connectivity predictors (Fig. 3).
Fig. 3. Comparing molecular and connectomic contributions to disorder-specific cortical differences.
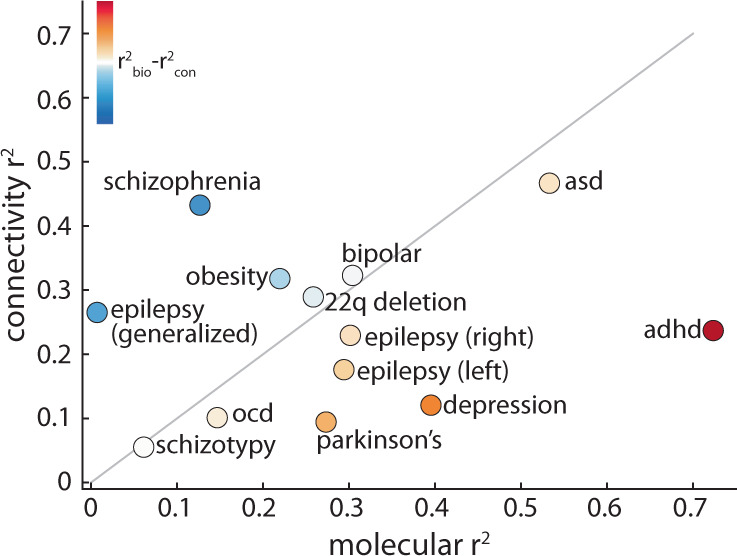
The local molecular of each disorder is plotted against the global connectivity . The grey line indicates the identity line and circle colour represents the difference between molecular and connectomic , such that warm colours represent disorders that are better predicted by molecular predictors, and cool colours represent disorders that are better predicted by connectomic predictors.
From the dominance analysis, we find that certain predictors are consistently unimportant. Indeed, synapse density and myelination demonstrate less dominance than microscale gradients such as the gene expression gradient (a potential proxy for cell type distribution16,20,37–39), neurotransmitter receptor gradient, and metabolic gradients. Connectivity predictors, particularly measures of centrality, demonstrate less dominance than more fundamental measures of connectivity such as number of connections (strength), distance, and connection diversity (participation coefficient). For completeness, we tested a third family of predictors related to temporal dynamics: magnetoencephalography (MEG)-derived power spectral densities for six canonical frequency bands (Supplementary Fig. 3).
One important consideration with this analysis is that disorder-specific pathology and symptom presentation are heterogeneous over time. The analysis in Fig. 2 is limited to adults and encompasses multiple stages of disease progression. We therefore sought to investigate changes across different ages (paediatric, adolescent, and adult) and different disease severities. First, we tracked the model fit () of regression models that fit molecular/connectomic features to paediatric, adolescent, and adult cortical abnormality profiles for the four available disorders with this data (ADHD, bipolar disorder, depression, and OCD; Supplementary Fig. 4a). We find that model fit is greatest in adulthood, except for OCD, which shows little change for connectomic predictors and a lower model fit in adulthood for molecular predictors. Model fit significantly improves when molecular features are used to predict cortical abnormality patterns of ADHD and depression (F > Fcritical, one-sided). Next, focusing on disease severity, we show how model fit changes across four levels of Parkinson’s disease severity (Hoehn and Yahr (HY) stages49; Supplementary Fig. 4b). Interestingly, we find that from stage HY2, molecular predictors perform worse with disease severity whereas connectomic predictors perform better (although note the changes in model fit are not statistically significant), supporting the notion that Parkinson’s pathology is influenced by the spread of misfolded proteins on the structural connectome10,50,51. Altogether, these analyses provide a more nuanced and transdiagnostic representation of molecular and connectomic contributions to cortical disorder vulnerability.
Interactions between local and global vulnerability
The previous section separately addresses molecular and connectomic contributions to disease-specific cortical abnormalities. However, molecular attributes likely interact with network connectivity to shape disease pathology. These molecular mechanisms include gene expression, neurotransmitter expression, and metabolic pathways in the cell. In neurodegenerative diseases, this interaction may result in synaptic pruning and cortical atrophy whereas in neurodevelopmental disorders, the pathology may manifest as perturbations in network wiring during development52. We hypothesised that abnormalities in such molecular mechanisms at the regional level may spread trans-synaptically between connected regions, resulting in connectome-informed changes in cortical morphology that reflect an interplay between local vulnerability and network structure. For instance, two regions may both participate in many connections (have high degree), but one may be connected to more regions with local vulnerability. Thus, despite the fact that their connectomic profiles are similar, one region may have greater disease exposure than the other10,53.
To test the hypothesis that a region’s cortical thickness is driven by "exposure” to abnormalities of connected regions, we measured the extent to which disorders demonstrate network-spreading patterns of cortical morphology9,13,54. The extent to which a disorder displays network-informed cortical changes is defined as the correlation between regional abnormality and mean abnormality of structurally connected neighbours (Fig. 4a). Importantly, significance was assessed using the spin-test to control for the effect of spatial autocorrelation on cortical abnormality patterns. We also test the hypothesis that this network-spreading effect is functionally informed, whereby the cortical thickness of structurally connected neighbours is weighted by the functional connectivity between regions when calculating the mean (Fig. 4b; see Methods for details and Supplementary Figs. 5 and 6 for scatter plots of regional abnormality versus mean neighbour abnormality across all thirteen disorders). We find that multiple disorders display a significant correlation between regional abnormality and abnormality of connected neighbours (0.23 < r < 0.80), suggesting that spatial patterning of disorders reflects the connection patterns between brain regions, above and beyond the effect of spatial autocorrelation (Supplementary Fig. 5).
Fig. 4. Interactions between molecular and connectomic vulnerability.

a Left: schematic of structural connectivity informing disorder-related cortical changes. The correlation between SC-weighted mean neighbour abnormality and region abnormality represents the extent to which a disorder demonstrates network-spreading disorder-specific cortical morphology. Right: this correlation coefficient was then correlated (Pearson’s r, two-sided) to both local molecular (left) and global connectivity (right) . Yellow points refer to disorders where the correlation between region abnormality and SC-weighted mean neighbour abnormality is significant (pspin < 0.05). b Left: likewise, mean neighbour abnormality can be additionally weighted by functional connectivity between regions. Right: correlation (Pearson’s r, two-sided) between the extent to which a disorder demonstrates SC- and FC-informed network-spreading cortical morphology and local molecular (left) and global connectivity (right) . Yellow points refer to disorders where the correlation between region abnormality and SC- and FC-weighted mean neighbour abnormality is significant (pspin < 0.05). c Left: a region with high abnormality that is also connected to regions with high abnormality is considered a likely disorder epicentre. Middle: epicentre likelihood was calculated as the mean rank of region and neighbour abnormality (see Supplementary Fig. 7 for individual epicentre likelihoods). Right: median epicentre likelihood was calculated for the disorders that show a significant correlation between regional and neighbour abnormality. To limit biasing cross-disorder epicentre likelihood towards epilepsy epicentre likelihood, left and right temporal lobe epilepsy epicentre likelihood was merged into a single mean epicentre likelihood map, prior to calculating the median. For completeness, Supplementary Fig. 8 shows cross-disorder epicentre likelihood when calculated using alternative statistics.
Does molecular or connectomic predictability of a disorder pattern (Fig. 2a) relate to network spreading? Interestingly, the extent to which a disorder can be predicted from molecular attributes (i.e., yellow R2 in Fig. 2a) is positively correlated with the extent to which a disorder displays evidence of network spreading (r(11) = 0.61, p = 0.03, CI = [0.10, 0.87] when weighted by SC only as shown in Fig. 4a; r(11) = 0.75, p = 0.003, CI = [0.33, 0.92] when weighted by FC and SC as shown in Fig. 4b). Notably, we do not observe this relationship with the extent to which a disorder can be predicted from global connectivity (i.e., blue R2 in Fig. 2a; r(11) = 0.24, p = 0.42, CI = [ − 0.36, 0.70] when weighted by SC only, Fig. 4a; r(11) = 0.06, p = 0.84, CI = [ − 0.50, 0.59] when weighted by FC and SC, Fig. 4b). In other words, for disorders with cortical morphologies that more strongly depend on molecular attributes, we also observe a greater effect of disorder exposure. Although we previously found that the cortical patterning of a disorder is less influenced by network embedding per se (e.g., centrality or connection diversity), here we show that it is instead more influenced by network-driven exposure to regions with local vulnerability. This finding is significant because it suggests that the combined effect of local vulnerability and connectome architecture is greater than their individual contribution.
Brain regions with high abnormality and high neighbour abnormality are likely to act as an epicentre of the network-spreading disorder pattern, since the region is both heavily affected and facilitates the spread of atypical morphology13,53,55. We calculated epicentre likelihood of each brain region as the mean rank of regional and neighbour abnormality, such that regions with high node and neighbour abnormality would be labelled as likely epicentres (Fig. 4c). The measure identifies "disorder hubs”—regions that are both vulnerable to disorder-specific changes but also embedded in a highly atypical network cluster. Epicentre likelihood was only calculated for brain maps with significant correlation between their node and neighbour abnormality (network-spreading disorders), and we did not find evidence for epicentre likelihood being driven by distance (Supplementary Fig. 14a. This list comprised of: 22q11.1 deletion syndrome, ADHD, ASD, right and left temporal lobe epilepsy, bipolar disorder, and schizotypy (Supplementary Fig. 7). Next, we aimed to construct a single cross-disorder epicentre likelihood map (Fig. 4c). To avoid having left and right temporal lobe epilepsy—which demonstrate similar epicentre likelihood maps—bias the cross-disorder likelihood map, we combined left and right epilepsy epicentre likelihood into a single average map. We calculated the median epicentre likelihood across these six disorders and find that cross-disorder epicentre likelihood is highest in bilateral sensory-motor cortex, angular gyrus, inferior temporal lobe, precuneus, and superior parietal cortex. In Supplementary Fig. 8 we show mean epicentre likelihood as well as a map that shows the frequency with which a brain region is in the top 50% of most likely epicentres across the six disorders. Across all three methods (mean, median, frequency), cross-disorder epicentre likelihood is consistent.
Brain regions with similar molecular annotations are similarly affected across disorders
In the previous sections, we mapped molecular annotations and network measures to each disorder separately. Here, we focused on disorder similarity. For every region we constructed a 13-element vector of abnormality values, where each element corresponds to cortical change (i.e., cortical abnormality) in that region in one disorder. We then correlated regional vectors with each other to estimate how similarly two regions are affected across the thirteen disorders (Fig. 5a). Disorder similarity is analogous to other measures of inter-regional attribute similarity including anatomical covariance56–58, morphometric similarity59, correlated gene expression60–62, receptor similarity63, temporal profile similarity44, and microstructural similarity64.
Fig. 5. Brain regions with similar molecular annotations are similarly affected across disorders.

a Disorder similarity was computed as the pairwise correlation of regional cortical abnormality across all thirteen disorders such that pairs of regions with high disorder similarity are similarly affected across disorders. b A histogram depicting the upper triangle of the disorder similarity matrix. c Disorder similarity is significantly correlated to molecular attribute similarity (Pearson's r(2276) = 0.45, pspin = 0.0001, CI = [0.42, 0.49], two-tailed). d Disorder similarity is not significantly correlated with connectomic similarity (Pearson's r(2276) = 0.25, pspin = 0.063, CI = [0.21, 0.29], two-tailed). e Disorder similarity is significantly correlated to neurotransmitter receptor similarity (Pearson's r(2276) = 0.41, pspin = 0.001, CI = [0.38, 0.45], two-tailed). f Left hemisphere disorder similarity is significantly correlated to correlated gene expression (Pearson's r(559) = 0.46, pspin = 0.0001, CI = [0.40, 0.53], two-tailed). g Disorder similarity is significantly greater within intrinsic functional networks than between networks, against the spin-test (p = 0.01; bottom). Disorder similarity is non-significantly greater between structurally connected regions than regions that are not connected, against a degree- and edge-length-preserving null model (p = 0.028101). Bounds of the box represent the 1st (25%) and 3rd (75%) quartiles, the centre line represents the median, and whiskers represent the minima and maxima of the distribution. Nconnected = 592 edges, Nnotconnected = 1686. Nwithin = 388, Nbetween = 1890. h Disorder similarity is significantly correlated to functional connectivity (Pearson's r(2276) = 0.36, pspin = 0.004, CI = [0.33, 0.40], two-tailed).
We first asked whether brain regions with similar molecular versus connectivity fingerprints show greater disorder similarity. Molecular similarity was likewise computed as the pairwise regional correlation of molecular predictors, and vice versa for connectivity. We find that disorder similarity is significantly correlated with molecular similarity (r(2276) = 0.45, pspin = 0.0001, CI = [0.42, 0.49]; Fig. 5c). On the other hand, the correlation between distance-regressed connectivity similarity and disorder similarity is smaller and non-significant (r(2276) = 0.25, pspin = 0.063, CI = [0.21, 0.29]; Fig. 5d).
Two of the molecular predictors included in the present report are summary measures of much more expansive molecular annotations: the gene expression gradient and the neurotransmitter receptor gradient. We therefore asked whether inter-regional similarity of these molecular attributes confers similar predisposition to disease. We computed correlated gene expression and neurotransmitter receptor similarity matrices63,65, and correlated these matrices with disorder similarity (Fig. 5e, f). We find a significant correlation between disorder similarity and neurotransmitter receptor similarity (r(2276) = 0.41, pspin = 0.001, CI = [0.38, 0.45]), as well as correlated gene expression (r(559) = 0.46, pspin = 0.0001, CI = [0.40, 0.53])60,63. These results hold when distance-regression is applied to the similarity networks instead of spin-tests (Supplementary Fig. 9). Altogether, our results indicate that regions with similar molecular composition are similarly affected across disorders.
We finally ask whether disorder similarity might analogously be informed by structural and functional connectivity between regions. We compared the disorder similarity matrix to weighted structural and functional connectomes. First, we find that brain regions that are structurally connected are more likely to change similarly across disorders than regions that are not structurally connected, although this result is non-significant against a degree and edge-length preserving null model (Fig. 5g66; see Null models). Second, we find that brain regions that are within the same intrinsic functional network are more likely to change similarly than regions between functional networks, against the spin-test (Fig. 5g, pspin = 0.01). Finally, we find a positive significant correlation between disorder similarity and functional connectivity (r(2276) = 0.36, pspin = 0.004, CI = [0.33, 0.40]; Fig. 5h). Consistent with the previous subsection, these results collectively suggest that areas that share molecular attributes and connections are similarly affected across disorders.
Sensitivity and robustness analyses
To ensure the results are not driven by choice of dataset, acquisition parameters and processing methodology, we repeated the analyses using structural and functional networks from the Human Connectome Project (N = 326), for which acquisition parameters and processing methodologies differ. The connectomic predictors from the Lausanne dataset used in the main text are highly correlated with the same metrics calculated using HCP data (Supplementary Fig. 10). As a result, the regression models and dominance analyses show consistent results (Supplementary Fig. 11). We also repeat the analysis in Fig. 2 using connectomic predictors calculated based on the binary structural connectome and the functional connectome from the Lausanne dataset (Supplementary Fig. 12).
Next, since the Desikan-Killiany atlas parcellates the brain into unequally sized parcels, we tested the effect of parcel size on disorder abnormality maps. Parcel size was defined as the number of voxels assigned to the parcel using the MNI-152 volumetric parcellation. Across all thirteen disorder maps, we do not find a significant correlation between parcel size and cortical abnormality (Supplementary Fig. 13). Likewise, we compare effects of parcel size on epicentre likelihood maps (Supplementary Fig. 14b). We find no significant correlations except between parcel size and bipolar epicentre likelihood (r = 0.44, pspin = 0.03). Finally, since epicentre likelihood is calculated using the structural connectome, we also assessed the relationship between epicentre likelihood and distance. Specifically, we correlate epicentre likelihood with the average distance between a brain region and all other brain regions (Supplementary Fig. 14a). We do not find any significant correlations between epicentre likelihood and distance.
Discussion
In the present report, we comprehensively map local molecular attributes and global measures of connectivity to the cortical morphology of thirteen different neurological, psychiatric, and neurodevelopmental disorders. We consistently find that local attributes govern both disorder-specific abnormalities and cross-disorder similarity more than global connectivity. In addition, we find that molecular mechanisms interact with the structural and functional architectures of the brain to guide cross-disorder abnormality patterns. Altogether, our results highlight how molecular and connectomic vulnerability shape cross-disorder cortical abnormalities.
This work builds on a growing literature on cross-disorder effects, and how shared vulnerability may potentially transcend traditional diagnostic boundaries3,67–69. It is becoming increasingly clear that pathology is governed by layers of abnormal processes, at the molecular and cellular level, to neural dynamics, to large-scale brain networks. Aligning high-quality maps of disorder-specific cortical changes to a common reference frame of local and global attributes allows us to systematically relate the effect of disease to multiple scales of organisation. By taking a cross-modal and cross-disorder approach we reveal that, despite different clinical presentation and label, there exists some commonality across diseases including predictors that are ubiquitously important as well as interplay between local vulnerability and network structure.
Interestingly, we find that the principal gradient of receptor distribution is particularly dominant towards disease-specific cortical morphology. This receptor gradient represents the maximal variance of density distributions from fourteen receptors and four transporters across nine different neurotransmitter systems, and therefore captures how brain regions may integrate exogenous signals differently63,70. This gradient is a powerful predictor of ADHD, left temporal lobe epilepsy, depression, and Parkinson’s disease. Indeed, neurotransmitter dysfunction is thought to underlie multiple disorders, including dopamine release in Parkinson’s and schizophrenia or serotonin reuptake in depression. Modern therapeutics are designed to selectively manipulate neurotransmitter function for the purpose of alleviating behavioural symptoms, as opposed to altering brain structure. Our findings confirm the fundamental contribution of neurotransmitters to a wide spectrum of diseases, but they also highlight an important link between the spatial patterning of neurotransmitter receptors and cortical disorder morphology itself63.
We generally find that cortical abnormality is better predicted by local vulnerability compared to global connectomic vulnerability. One possible reason for the relatively poorer performance of connectivity predictors is that they are generic measures of a region’s embedding in a network (number of connections, centrality, connection diversion) but do not consider how this embedding exposes regions to pathology elsewhere in the network. Indeed, we find that disorders whose cortical morphology is better reflected by local vulnerability also bear a prominent signature of network architecture (e.g., ASD, ADHD, 22q11.2 deletion syndrome, temporal lobe epilepsy, schizotypy, bipolar disorder). This suggests a network-spreading phenomenon where focal pathology or perturbation propagates to connected regions, resulting in cortical abnormality that is correlated with the underlying connection patterns14. This interaction between local vulnerability and connectomic vulnerability has previously been reported in neurodegenerative syndromes where the trans-synaptic spreading of misfolded proteins appears to be guided and amplified by local gene expression10,71–74. In other words, the poorer performance of connectivity predictors does not suggest that the white-matter architecture is less relevant to disease progression. Indeed, pathogenesis of multiple diseases is thought to originate in the white matter of the brain14,75,76. A promising future direction for studying cross-disorder brain abnormalities is to focus on disruptions in white-matter pathways instead of cortical thickness3,77.
The interaction between molecular vulnerability and network structure naturally raises the question of what are the network epicentres of cortical disorder maps. We find epicentres—regions with high abnormality that are also strongly connected with other regions with high abnormality—in primarily transmodal regions (e.g., inferior temporal lobe, angular gyrus, precuneus, superior parietal cortex), although the motor cortex also appears as an epicentre. That the sensory-motor cortex is an epicentre is consistent with recent reports that multiple psychiatric and neurological disorders are accompanied by sensory deficits and reduced motor control78–80. Indeed, the sensory-motor cortex has been previously established as a functional hub in temporal lobe epilepsies and across multiple psychiatric disorders68,81. Interestingly, both the bilateral precuneus and superior parietal cortex are members of the brain’s putative rich club—densely inter-connected regions that are thought to support the integration and broadcasting of signals82. Rich club regions undergo changes in connectivity patterns in multiple diseases such as schizophrenia, Alzheimer’s, and Huntington’s1,3,15,83,84. We complement this work by showing that the precuneus and superior parietal cortex are both vulnerable to cortical abnormality and, by virtue of their network embedding, increase disease exposure to connected regions. Conversely, although the anterior cingulate cortex (ACC) is implicated across multiple psychiatric disorders13,85, we do not find that the ACC is an epicentre of cross-disorder cortical morphology. This suggests that although the ACC demonstrates considerable local vulnerability in a subset of brain disorders, it is not consistently involved across the seven disorders included in the epicentre analyses. Altogether, despite variable cortical morphology patterns across the thirteen disorders, when looked at through the lens of network connectivity, we see a more consistent and compact subset of potential epicentres, suggesting greater commonality among diseases than previously appreciated.
One strength of the ENIGMA consortium is that the datasets are pooled over thousands of individuals. However, such large-scale analyses obscure the important inter-subject variability that exists within all disorders. We conduct supplementary analyses in which cortical disorder profiles are stratified by age and disease severity (Supplementary Fig. 4) and find that molecular and connectomic contributions vary. For example, we find that molecular and connectomic influences on Parkinson’s disease differ with disease severity: molecular predictors become less powerful predictors and connectomic predictors become more powerful predictors as the disease progresses. This complements previous work that suggests that atrophy in Parkinson’s is the result of network-mediated spread of alpha-synuclein10. Furthermore, we find a similar trend of increased connectomic influence for severity of psychotic symptoms: namely, schizotypy and schizophrenia. Although schizotypy is not an earlier stage of schizophrenia (indeed, schizotypy is not a disorder per se but rather a multidimensional continuum of traits related to psychosis), individuals with schizotypy exhibit similar, albeit attenuated, characteristics as schizophrenia patients35. We find that the connectomic influence is considerably greater in schizophrenia compared to schizotypy, which may suggest that the structural network gradually becomes more implicated in disease progression. One key factor that we were not able to study in more depth is that of biological sex. Since ENIGMA datasets are all sex-corrected, we are unable to make conclusions about how molecular and connectomic contributions may differ between the sexes. Multiple disorders show well-established sex-differences, including schizophrenia86, autism87, and depression88. Designing effective clinical interventions will require more nuanced studies that consider the many heterogeneities that exist within each disease.
This work considers multimodal molecular and connectomic contributions to disorders but does not make conclusions about two important features of disease: cognitive phenotypes and genetics. An exciting future direction is to explore whether molecular and connectomic contributions to disease can be related to phenotypic or genetic similarity. Lee et al.89 compare single-nucleotide polymorphism data across eight psychiatric disorders and find that schizophrenia and bipolar disorder show greatest genetic similarity. This complements our finding that schizophrenia and bipolar disorder have consistent connectomic profiles. Lee et al.89 also find a clique among the disorders that we find are best predicted by molecular features: ADHD, autism, and major depressive disorder. On the other hand, a comprehensive battery of cognitive and behavioural tests was not uniformly applied to all the disease groups in the ENIGMA datasets. As a result, robust cross-disorder phenotypic profiles are less well-established. Our findings potentially suggest an executive function (anchored by schizophrenia) versus attention (anchored by ADHD and ASD) cognitive axis that separates connectomic versus molecularly informed disorder profiles, but more work is needed to standardise cognitive testing and assess how cognitive/behavioural phenotypes may be related to brain structure. Altogether, future work is necessary to explore how overlapping genetic and neurocognitive disturbances correspond to molecular and connectomic contributions to disease.
The present work should be considered along some important methodological considerations. First, although the ENIGMA consortium standardises pre-processing pipelines and provides large N datasets, allowing for robust results and meaningful comparison between disorder-specific cortical abnormality maps, working with ENIGMA data also has caveats: (1) the measures of cortical abnormality are effect sizes between patients and controls and do not represent tissue volume loss/gain, (2) some of the patient populations included have co-morbidities and patients may be undergoing treatment, including treatment that may have an effect on cortical thickness90, and (3) all analyses were conducted at the level of 68 cortical brain areas, limiting regional specificity and precluding analyses of the subcortex and cerebellum. Second, despite the fact that structural connectomes were reconstructed from high-resolution diffusion spectrum imaging, diffusion tractography is still prone to false-positives and false-negatives91–93. Third, both local molecular and global connectivity predictors are derived from state-of-the-art open-access datasets in healthy participants, but they do not capture individual variability or changes across the lifespan—both of which are key factors in neurological, psychiatric, and neurodevelopmental disorders. Additionally, the molecular predictors are limited by imaging modality (in particular myelination, for which the T1w/T2w ratio is an indirect estimate94,95), and, in the case of the gene and receptor gradients, by the subset of genes and receptors included in the data decomposition. Fourth, we assessed contribution of multiple predictors to disorder maps using simple but robust linear models that are not sensitive to nonlinear contributions or higher-order interactions among the predictors. In addition to this, the correlational structure of the predictor subsets affects predictive power, which limits our ability to compare molecular and connectomic model fits (Supplementary Fig. 15). Fifth, the linear models used in the present analyses assume independence between observations, which is not the case in the brain; we therefore employ spatial-autocorrelation-preserving null models to account for the spatial dependencies between regions throughout the report. Finally, although the present report spans a wide range of neurological, psychiatric, and neurodevelopmental disorders, results are only valid for this subset of disorders. Future work is needed to map local and global vulnerabilities to the many more brain diseases and disorders that exist.
In summary, we find that molecular and connectomic vulnerability jointly shape cross-disorder cortical abnormalities. Cross-disorder regional vulnerability is largely driven by molecular fingerprints, including neurotransmitter receptor densities and gene expression, while connection patterns among vulnerable regions further amplify this vulnerability. Our results highlight how an integrative, multimodal approach can illuminate the contributions of local biology and connectome architecture to brain disease.
Methods
Cortical disorder maps
Patterns of cortical thickness were collected for the available thirteen neurological, neurodevelopmental, and psychiatric disorders from the ENIGMA consortium and the enigma toolbox (https://github.com/MICA-MNI/ENIGMA96), including: 22q11.2 deletion syndrome (N = 474 participants, N = 315 controls)26, attention-deficit/hyperactivity disorder (ADHD; N = 733 participants, N = 539 controls)27, autism spectrum disorder (ASD; N = 1571 participants, N = 1651 controls)28, idiopathic generalised (N = 367 participants), right temporal lobe (N = 339 participants), and left temporal lobe (N = 415 participants) epilepsies (N = 1727 controls)29, depression (N = 2148 participants, N = 7957 controls)30, obsessive-compulsive disorder (OCD; N = 1905 participants, N = 1760 controls)31, schizophrenia (N = 4474 participants, N = 5098 controls)32, bipolar disorder (N = 1837 participants, N = 2582 controls)33, obesity (N = 1223 participants, N = 2917 controls)34, schizotypy (N = 3004 participants)35, and Parkinson’s disease (N = 2367 participants, N = 1183 controls)36. The ENIGMA (Enhancing Neuroimaging Genetics through Meta-Analysis) Consortium is a data-sharing initiative that relies on standardised processing and analysis pipelines, such that disorder maps are comparable25. Altogether, over 21,000 participants were scanned across the thirteen disorders, against almost 26,000 controls. The analysis was limited to adults in all cases except ASD where the cortical abnormality map is only available aggregated across all ages (2–64 years). The values for each map are z-scored effect sizes (Cohen’s d) of cortical thickness in patient populations versus healthy controls. Imaging and processing protocols can be found at http://enigma.ini.usc.edu/protocols/, and detailed demographic information can be found in the supplement of each accompanying article. Local review boards and ethics committees approved each individual study separately, and written informed consent was provided according to local requirements.
Structural and functional networks
Lausanne dataset
Structural and functional data were collected at the Department of Radiology, University Hospital Center and University of Lausanne, on n = 70 healthy young adults (16 females, 25.3 ± 4.9 years)97. Informed consent was obtained from all participants and the protocol was approved by the Ethics Committee of Clinical Research of the Faculty of Biology and Medicine, University of Lausanne. The scans were performed in a 3-T MRI scanner (Trio; Siemens Medical), using a 32-channel head coil. The protocol included (1) a magnetisation-prepared rapid acquisition gradient echo (MPRAGE) sequence sensitive to white/grey matter contrast (1 mm in-plane resplution, 1.2 mm slice thickness), (2) a DSI sequence (128 diffusion-weighted volumes and a single b0 volume, maximum b-value 8000 s/mm2, 2.2 × 2.2 × 3.0 mm voxel size), and (3) a gradient echo-planar imaging (EPI) sequence sensitive to blood-oxygen-level-dependent (BOLD) contrast (3.3 mm in-plane resolution and slice thickness with a 0.3 mm gap, TR 1920 ms, resulting in 280 images per participant). Participants were not subject to any overt task demands during the fMRI scan. The Lausanne dataset is available at https://zenodo.org/record/2872624#.XOJqE99fhmMand has been used in other work98,99.
Grey matter was parcellated according to the 68-region Desikan-Killiany cortical atlas40. Structural connectivity was estimated for individual participants using deterministic streamline tractography. The procedure was implemented in the Connectome Mapping Toolkit100, initiating 32 streamline propagations per diffusion direction for each which matter voxel. Collating each individual’s structural connectome was done using a group-consensus approach that seeks to preserve the density and edge-length distributions of the individual connectomes (see Group-consensus structural network101). The binary density for the final whole-brain structural connectome was 24.6%. For the weighted structural connectome, edges were weighted by the average log-transform of non-zero streamline density, scaled to values between 0 and 1.
Functional MRI data were pre-processed using procedures designed to facilitate subsequent network exploration102. fMRI volumes were corrected for physiological variables, including regression of white matter, cerebrospinal fluid, and motion (3 translations and 3 rotations, estimated by rigid body coregistration). BOLD time-series were then subjected to a low-pass filter (temporal Gaussian filter with full width at half maximum equal to 1.92 s). The first four time points were excluded from subsequent analysis to allow the time-series to stabilise. Motion scrubbing was performed as described by ref. 102. The data were parcellated according to the same 68-region Desikan-Killiany atlas used for the structural network. Individual functional connectivity matrices were defined as zero-lag Pearson correlation among the fMRI BOLD time-series. A group-consensus functional connectivity matrix was estimated as the mean connectivity of pairwise connections across individuals. Note that one individual did not undergo an fMRI scan and therefore the functional connectome was composed of n = 69 participants.
Human Connectome Project
Following the procedure described in de Wael et al.103, we obtained structural and functional magnetic resonance imaging (MRI) data for 326 unrelated participants (age range 22–35 years, 145 males) from the Human Connectome Project (HCP; S900 release104; informed consent obtained). All four resting-state fMRI scans (two scans (R/L and L/R phase encoding directions) on day 1 and two scans (R/L and L/R phase encoding directions) on day 2, each about 15 min long; TR = 720 ms), as well as diffusion-weighted imaging (DWI) data were available for all participants. All the structural and functional MRI data were pre-processed using HCP minimal pre-processing pipelines104,105. We provide a brief description of data pre-processing below, while detailed information regarding data acquisition and pre-processing is available elsewhere104,105.
DWI data was pre-processed using the MRtrix3 package106 (https://www.mrtrix.org/). More specifically, fibre orientation distributions were generated using the multi-shell multi-tissue constrained spherical deconvolution algorithm from MRtrix107,108. White-matter edges were then reconstructed using probabilistic streamline tractography based on the generated fibre orientation distributions109. The tract weights were then optimised by estimating an appropriate cross-section multiplier for each streamline following the procedure proposed by Smith et al.110 and a connectivity matrix was built for each participant using the 68-region Deskian-Killiany parcellation40,41. Collating each individual’s structural connectome was done using a group-consensus approach that seeks to preserve the density and edge-length distributions of the individual connectomes (see Group-consensus structural network101). The binary density for the final whole-brain structural connectome was 31.2%. For the weighted structural connectome, edges were weighted by the average log-transform of non-zero streamline density, scaled to values between 0 and 1.
All 3T functional MRI time-series were corrected for gradient nonlinearity, head motion using a rigid body transformation, and geometric distortions using scan pairs with opposite phase encoding directions (R/L, L/R)103. Further pre-processing steps include coregistration of the corrected images to the T1w structural MR images, brain extraction, normalisation of whole-brain intensity, high-pass filtering (>2000s FWHM; to correct for scanner drifts), and removing additional noise using the ICA-FIX process103,111. The pre-processed time-series were then parcellated to 68 cortical brain regions according to the Desikan-Killinay atlas40,41. The parcellated time-series were used to construct functional connectivity matrices as a Pearson correlation coefficient between pairs of regional time-series for each of the four scans of each participant. A group-average functional connectivity matrix was constructed as the mean functional connectivity across all individuals and scans.
Group-consensus structural network
To construct a representative group-level connectome, we used a consensus approach that seeks to preserve the density and edge-length distributions of the individual connectomes (first applied in Mišić et al.112 and presented formally in Betzel et al.101). This procedure better captures important organisational features of subject-level networks compared to other consensus methods (i.e., thresholding based on whether an edge is observed in a fraction of subjects)101. The procedure for generating the consensus network is as follows. First, existing edges across participants were binned according to length. The number of bins was determined heuristically as the square root of the mean binary density across participants. Within each bin, the k most frequently occurring edges across participants were retained. k was set as the average across the number of edges each participant has in the bin. To ensure that interhemispheric edges are not underrepresented, we carried out this procedure separately for inter- and intrahemispheric edges.
Molecular predictors
A total of seven local molecular predictors were used in the multilinear model to represent the influence that local molecular attributes have on disorder-specific cortical morphology.
Gene expression gradient
The first principal component of gene expression (“gene gradient”) was used to represent the variation in gene expression levels across the left cortex. This gradient has been previously related to cell type distributions and cell-specific gene expression, which suggests the gradient is related to the cellular architecture of the brain16,20,37–39. Gene expression data was collected by the Allen Human Brain Atlas as described in Hawrylycz et al.37 and processed by abagen, an open-source Python toolbox113. A total of 11,560 genes with differential stability greater than 0.1 were retained in the region by gene matrix114. The left gene gradient was mirrored in the right hemisphere. A detailed account of the specific processing choices made can be found in Hansen et al.39.
Receptor gradient
The first principal component of receptor density (“receptor gradient”) was used to represent the variation in receptor densities across the cortex. Receptor densities were estimated using PET tracer studies for a total of 18 receptors and transporters, across 9 neurotransmitter systems. These include dopamine (D1115, D2116–119, DAT120), norepinephrine (NET121–124), serotonin (5-HT1A125, 5-HT1B125–132, 5-HT2A133, 5-HT4133, 5-HT6134,135, 5-HTT133), acetylcholine (α4β2132,136, M1137, VAChT138,139), glutamate (mGluR5140,141), GABA (GABAA142), histamine (H3143), cannabinoid (CB1144–147), and opioid (MOR148). Volumetric PET images were registered to the MNI-ICBM 152 nonlinear 2009 (version c, asymmetric) template, averaged across participants within each study, then parcellated to 68 cortical regions. Parcellated PET maps were then z-scored before compiling all receptors/transporters into a region × receptor matrix of relative densities. Data were originally presented as an atlas in Hansen et al.63.
Excitatory-inhibitory ratio
The excitatory-inhibitory ratio was computed as the ratio of z-scored PET-derived excitatory to inhibitory neurotransmitter receptor densities in the cortex, using the same dataset that was used to compute the receptor gradient. Excitatory neurotransmitter receptors included are: 5-HT2A, 5-HT4, 5-HT6, D1, mGluR5, α4β2, and M1. Inhibitory neurotransmitter receptors included are: 5-HT1A, 5-HT1B, CB1, D2, GABAA, H3, and MOR.
Glycolytic index
Aerobic glycolysis is the process of converting glucose to lactate in the presence of oxygen. It is traditionally calculated as the ratio of oxygen metabolism to glucose metabolism. Here, we use glycolytic index, a measure of aerobic glycolysis that mitigates certain limitations of using the traditional ratio149. Glycolytic index is defined as the residual after fitting glucose metabolism to oxygen metabolism in a linear regression model. Larger values indicate more aerobic glycolysis. Note that glycolytic index and the traditional ratio are highly correlated (see Vaishnavi et al.149). Data were collected, calculated, and made available by Vaishnavi et al.149. Glucose metabolism was obtained as described in the section below, and oxygen metabolism was collected in the same participants by administering -labelled water, carbon monoxide, and oxygen. All experiments were approved by the Human Research Protection Office and the Radioactive Drug Research Committee at Washington University in St. Louis. Written informed consent was provided by all participants.
Glucose metabolism
Glucose metabolism in the cortex was measured in 33 healthy adults (19 female, mean age 25.4 ± 2.6 years) by administering -labelled fluorodeoxyglucose (FDG) for a PET scan, as described in detail in Vaishnavi et al.149. All experiments were approved by the Human Research Protection Office and the Radioactive Drug Research Committee at Washington University in St. Louis. Written informed consent was provided by all participants.
Synapse density
Synapse density in the cortex was measured in 76 healthy adults (45 males, 48.9 ± 18.4 years of age) by administering UCB-J, a PET tracer that binds to the synaptic vesicle glycoprotein 2A (SV2A)150–161. Data were collected on an HRRT PET camera for 90 min post injection. Non-displaceable binding potential (BPND) was modelled using SRTM2, with the centrum semiovale as reference and fixed to 0.027 (population value). Each study was performed under a protocol approved by the Yale University Human Investigation Committee and the Yale New Haven Hospital Radiation Safety Committee, and written informed consent was obtained from all participants.
Myelination
Data from the Human Connectome Project (HCP, S1200 release)104,105 was used for measures of T1w/T2w ratios—a proxy for intracortical myelin—for 417 unrelated participants (age range 22–37 years, 193 males), as approved by the WU-Minn HCP Consortium. Images were acquired on a Siemens Skyra 3T scanner, and included a T1-weighted MPRAGE sequence at an isotropic resolution of 0.7 mm, and a T2-weighted SPACE also at an isotropic resolution of 0.7 mm. Details on imaging protocols and procedures are available at http://protocols.humanconnectome.org/HCP/3T/imaging-protocols.html. Image processing includes correcting for gradient distortion caused by non-linearities, correcting for bias field distortions, and registering the images to a standard reference space. T1w/T2w ratios for each participant was made available in the surface-based CIFTI file format and parcellated into 68 cortical regions according to the Lausanne anatomical atlas41. Note that the T1w/T2w ratio is an MRI-based estimate of myelin content that has not yet been validated against myelin histology94. Other MRI-based proxies may be more suitable alternatives, such as magnetisation transfer or simultaneous tissue relaxometry of R1 and R2 relaxation rates and proton density (SyMRI), which have been validated using myelin histology and are closely correlated to one another94,95. Additionally, PET imaging may be a promising avenue for mapping myelin content in the brain162,163.
Connectivity predictors
A total of nine global connectome predictors were used in the multilinear model to represent the influence that global connectivity has on disorder-specific cortical morphology. In the main text, connectome measures were computed on the weighted structural connectome. Analyses were repeated using a binary structural connectome and an absolute functional connectome (Supplementary Fig. 12). All connectivity measures were computed using the Python-equivalent of the Brain Connectivity Toolbox, bctpy.
Strength
The strength of region i is the sum of the edges connected to region i. For a binary structural connectome, the strength is equivalent to the degree, which is the number of links connected to region i.
Betweenness centrality
Betweenness centrality of region i is the fraction of all shortest paths between any two regions that traverse region i.
Closeness centrality
Closeness centrality is equivalent to the mean shortest path distance from region i to every other region in the network.
Euclidean distance
Mean Euclidean distance of a region to all other regions in the network represents how spatially close one region is to all other regions.
Participation coefficient
Participation coefficient was computed using the putative intrinsic functional networks of the brain164. Participation coefficient represents the connection diversity of a region. A region with high participation coefficient is well connected to several different networks, whereas a region with low participation coefficient primarily makes local (within-network) connections.
Clustering coefficient
The clustering coefficient of region i is the fraction of all triangles that are around region i. Equivalently, it is the fraction of all of region i’s neighbours that are also neighbours with each other. In the case of the weighted structural connectome, clustering coefficient is the average geometric mean of all triangles associated with the region.
Mean first passage time
The mean first passage time from region i to j is the expected amount of time it takes a random walker to reach region j from i for the first time. For each region, mean first passage time was averaged across regions, resulting in a mean mean first passage time representing the average amount of time it takes a random walker to travel from region i to any other region in the network for the first time.
Temporal predictors
Six-minute resting-state eyes-open magenetoencephalography (MEG) time-series were acquired from the Human Connectome Project (HCP, S1200 release) for 33 unrelated subjects (age range 22–35, 17 males)104,105. Complete MEG acquisition protocols can be found in the HCP S1200 Release Manual. For each subject, we computed the power of the run at the vertex level across six different frequency bands: delta (2–4 Hz), theta (5–7 Hz), alpha (8–12 Hz), beta (15–29 Hz), low gamma (30–59 Hz), and high gamma (60–90 Hz), using the open-source software, Brainstorm165. Each power band was then parcellated into 68 cortical regions41.
Dominance analysis
Dominance analysis seeks to determine the relative contribution (“dominance”) of each input variable to the overall fit (adjusted R2) of the multiple linear regression model (https://github.com/dominance-analysis/dominance-analysis42,43). This is done by fitting the same regression model on every combination of input variables (2p − 1 submodels for a model with p input variables). Total dominance is defined as the average of the relative increase in R2 when adding a single input variable of interest to a submodel, across all 2p − 1 submodels. The sum of the dominance of all input variables is equal to the total adjusted R2 of the complete model, making total dominance an intuitive measure of contribution. Note that significance testing is not applied to the individual dominances because the contributions of input variables are relative to other predictors in the model and input variables do not act in isolation.
Each multilinear model was cross-validated using a distance-dependent method proposed by Hansen et al.39. Briefly, for each of 1000 iterations, the 75% of regions closest in Euclidean distance to a randomly chosen source node were selected as the training set, and the remaining 25% of regions as the test set. Predicted values in the test set were then correlated to true abnormality patterns, and the correlations are shown in Supplementary Fig. 2.
Network spreading
Network spreading was computed as first introduced in Shafiei et al.13 and later adopted in Chopra et al.54, Shafiei et al.9. Briefly, regional abnormality was defined as the normalised effect size used in all ENIGMA brain maps. For each region i, its neighbours are those with which region i is connected via a structural connection, as defined by the structural connectivity matrix. Mean neighbour abnormality of region i (Di) is the average abnormality of region i’s neighbours, where dj represents the abnormality of neighbour j. Notably, this method normalises neighbour abnormality by the number of connections made by region i (Ni).
| 1 |
when neighbour abnormality is weighted by functional connectivity, each neighbour’s abnormality are weighted by the functional connection to node i (FCij).
| 2 |
Each brain region was assigned a rank in terms of their node abnormality and their mean neighbour abnormality. The average of node and neighbour abnormality ranks was defined as the epicentre likelihood of the node, where nodes with high abnormality and whose neighbours are also highly atypical are more likely to be an epicentre of the disorder.
Disorder similarity
For every brain region, we constructed a 13-element vector of disorder abnormality, where each element represents a disorder’s cortical abnormality at the region. For every pair of brain regions, we correlated the abnormality vectors to quantify how similarly two brain regions are affected across disorders. This results in a region-by-region matrix of “disorder similarity” (Fig. 5a). We verified that no single disorder pattern was driving the disorder similarity matrix by recalculating the disorder similarity when a single disorder is excluded. We then correlated the leave-one-out disorder similarity matrix with the original disorder similarity matrix. The minimum correlation was r = 0.95 (Supplementary Fig. 16a). Finally, influence on the disorder similarity matrix by a disorder i was quantified as
| 3 |
where D is the original disorder similarity matrix and Di is the disorder similarity matrix constructed when disorder i is excluded (Supplementary Fig. 16b).
Null models
Spatial-autocorrelation-preserving permutation tests were used to assess statistical significance of associations across brain regions, termed “spin tests”166–168. We created a surface-based representation of the parcellation on the FreeSurfer fsaverage surface, via files from the Connectome Mapper toolkit (https://github.com/LTS5/cmp). We used the spherical projection of the fsaverage surface to define spatial coordinates for each parcel by selecting the coordinates of the vertex closest to the center of the mass of each parcel98. These parcel coordinates were then randomly rotated, and original parcels were reassigned the value of the closest rotated parcel (1000 repetitions). Parcels for which the medial wall was closest were assigned the value of the next most proximal parcel instead. The procedure was performed at the parcel resolution rather than the vertex resolution to avoid upsampling the data, and to each hemisphere separately. This spin-permuted null model involves conflating and collapsing the brain surface to and from a sphere. The geometry of the cortical surface is therefore not retained in the spinning process, which may result in null distributions that are too wide. Other methods for constructing spatial null models exist, such as generative models169 and 2D spatial wavestrapping170.
A second null model was used to test whether disorder similarity is greater in connected regions than unconnected regions. This model generates a null structural connectome that preserves the density, edge length, and degree distributions of the empirical structural connectome66,171,172. Briefly, edges were binned according to Euclidean distance. Within each bin, pairs of edges were selected at random and swapped. This procedure was then repeated 10,000 times. To compute a p-value, the mean disorder similarity of unconnected edges was subtracted from the mean disorder similarity of connected edges, and this difference was compared to a null distribution of differences computed on the rewired networks.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
This research was undertaken thanks in part to funding from the Canada First Research Excellence Fund, awarded to McGill University for the Healthy Brains for Healthy Lives initiative. B.M. acknowledges support from the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant RGPIN #017-04265), the Canada Research Chairs Programme, the Brain Canada Future Leaders Fund and the Healthy Brains for Healthy Lives initiative. J.Y.H. acknowledges support from the Helmholtz International BigBrain Analytics & Learning Laboratory, the Natural Sciences and Engineering Research Council of Canada, and the Fonds de reserches de Québec. The research studies produced by the ENIGMA Working Groups would not be possible without the contributions of many researchers across the globe and the authors of this work thank all scientists who contribute to making this work possible. A full list of ENIGMA Consortium current and past members can be found here http://enigma.ini.usc.edu/ongoing/members/. The authors acknowledge the NIH Big Data to Knowledge (BD2K) award for foundational support and consortium development (U54 EB020403 to P.M.T.) and support from NIMH R01MH116147 (P.M.T.), NIMH R01MH116147 (T.G.M.v.E.), NIMH R01 MH117601 (N.J., L.S.), NIMH R01MH085953 (C.E.B.), NIMH R21MH116473 (C.E.B.), NIMH 1U01MH119736 (C.E.B.). For a complete list of ENIGMA-related grant support please see here: http://enigma.ini.usc.edu/about-2/funding. J.B. has been supported by the EU-AIMS (European Autism Interventions) and AIMS-2-TRIALS programmes, which receive support from Innovative Medicines Initiative Joint Undertaking Grant No. 115300 and 777394, the resources of which are composed of financial contributions from the European Union’s FP7 and Horizon2020 Programmes, and from the European Federation of Pharmaceutical Industries and Associations (EFPIA) companies’ in-kind contributions, and AUTISM SPEAKS, Autistica and SFARI; and by the Horizon2020 supported programme CANDY Grant No. 847818). B.F. is supported by the European Community’s Horizon 2020 Programme (H2020/2014-2020) under grant agreements no. 667302 (CoCA), no. 728018 (Eat2beNICE), and no. 847879 (PRIME). This work was supported by a personal Veni grant to M.H. from the Netherlands Organisation for Scientific Research (NWO, grant number 91619115). C.R.M. is supported by NIH R01 NS065838; R21 NS107739. D.J.S. is supported by South African Medical Research Council. G.M. is funded by a Wellcome Trust & The Royal Society Sir Henry Dale Fellowship [202397/Z/16/Z]. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
J.Y.H. and B.M. conceived the study and wrote the manuscript, with valuable revision from all authors. J.Y.H. performed the formal analysis, with contribution from G.S. J.Y.H. interpreted the results with contribution from G.S., J.W.V., A.D., and B.M. K.S., C.E.B., M.H., B.F., D.V., J.B., C.R.M., S.M.S., L.S., D.J.V., O.A.V., D.J.S., T.G.M.v.E, C.R.K.C., O.A.A., T.H., N.O., G.M., A.A., Y.V., N.J., S.I.T., P.M.T., and R.E.C. provided data. B.M. was the project administrator.
Peer review
Peer review information
Nature Communications thanks Michael Breakspear, Elika Garg, Rafael Romero-Garcia and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
All data used in the present report is openly available at https://github.com/netneurolab/hansen_crossdisorder_vulnerability. More specifically, ENIGMA datasets are available through the ENIGMA consortium and the ENIGMA toolbox (https://github.com/MICA-MNI/ENIGMA96). The Lausanne dataset is available at https://zenodo.org/record/2872624#.XOJqE99fhmM97. The HCP dataset is available at https://db.humanconnectome.org/. Molecular predictors are available as volumetric images in the neuromaps toolbox (https://netneurolab.github.io/neuromaps/173). The Allen Human Brain Atlas is available at https://human.brain-map.org/.
Code availability
All code used to perform the analyses can be found at https://github.com/netneurolab/hansen_crossdisorder_vulnerabilityand on Zenodo (10.5281/zenodo.6795748).
Competing interests
C.R.K.C., N.J., P.M.T. received partial research support from Biogen, Inc., for research unrelated to this manuscript. J.B. has been in the past 3 years a consultant to/member of advisory board of/and/or speaker for Takeda/Shire, Roche, Medice, Angelini, Janssen, and Servier. He is not an employee of any of these companies, and not a stock shareholder of any of these companies. He has no other financial or material support, including expert testimony, patents, royalties. B.F. has received educational speaking fees from Medice GmbH. D.J.S. has received research grants and/or consultancy honoraria from Lundbeck and Sun. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-32420-y.
References
- 1.Crossley NA, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain. 2014;137:2382–2395. doi: 10.1093/brain/awu132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Heuvel MP, Sporns O. Network hubs in the human brain. Trend. Cogn. Sci. 2013;17:683–696. doi: 10.1016/j.tics.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 3.de Lange SC, et al. Shared vulnerability for connectome alterations across psychiatric and neurological brain disorders. Nat. Human Behav. 2019;3:988–998. doi: 10.1038/s41562-019-0659-6. [DOI] [PubMed] [Google Scholar]
- 4.Warren JD, et al. Molecular nexopathies: a new paradigm of neurodegenerative disease. Trend. Neurosci. 2013;36:561–569. doi: 10.1016/j.tins.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73:1204–1215. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj A, et al. Network diffusion model of progression predicts longitudinal patterns of atrophy and metabolism in alzheimer’s disease. Cell Rep. 2015;10:359–369. doi: 10.1016/j.celrep.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iturria-Medina Y, Sotero RC, Toussaint PJ, Evans AC, Initiative ADN. Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput. Biol. 2014;10:e1003956. doi: 10.1371/journal.pcbi.1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pievani M, Filippini N, Van Den Heuvel MP, Cappa SF, Frisoni GB. Brain connectivity in neurodegenerative diseases-from phenotype to proteinopathy. Nat. Rev. Neurol. 2014;10:620–633. doi: 10.1038/nrneurol.2014.178. [DOI] [PubMed] [Google Scholar]
- 9.Shafiei, G. et al. Network structure and transcriptomic vulnerability shape atrophy in frontotemporal dementia. Brain. 10.1093/brain/awac069 (2022). [DOI] [PMC free article] [PubMed]
- 10.Zheng Y-Q, et al. Local vulnerability and global connectivity jointly shape neurodegenerative disease propagation. PLoS Biol. 2019;17:e3000495. doi: 10.1371/journal.pbio.3000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yau Y, et al. Network connectivity determines cortical thinning in early parkinson’s disease progression. Nat. Commun. 2018;9:1–10. doi: 10.1038/s41467-017-02416-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wannan CM, et al. Evidence for network-based cortical thickness reductions in schizophrenia. Am. J. Psychiatry. 2019;176:552–563. doi: 10.1176/appi.ajp.2019.18040380. [DOI] [PubMed] [Google Scholar]
- 13.Shafiei G, et al. Spatial patterning of tissue volume loss in schizophrenia reflects brain network architecture. Biol. Psychiatry. 2020;87:727–735. doi: 10.1016/j.biopsych.2019.09.031. [DOI] [PubMed] [Google Scholar]
- 14.Fornito A, Zalesky A, Breakspear M. The connectomics of brain disorders. Nat. Rev. Neurosci. 2015;16:159–172. doi: 10.1038/nrn3901. [DOI] [PubMed] [Google Scholar]
- 15.van den Heuvel MP, Sporns O. A cross-disorder connectome landscape of brain dysconnectivity. Nat. Rev. Neurosci. 2019;20:435–446. doi: 10.1038/s41583-019-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burt JB, et al. Hierarchy of transcriptomic specialization across human cortex captured by structural neuroimaging topography. Nat. Neurosci. 2018;21:1251–1259. doi: 10.1038/s41593-018-0195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan SE, et al. Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc. Natl Acad. Sci. USA. 2019;116:9604–9609. doi: 10.1073/pnas.1820754116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnatkeviciute, A., Fulcher, B. D., Bellgrove, M. A. & Fornito, A. (2021b). Imaging transcriptomics of brain disorders. Biol. Psychiatry Glob. Open Sci.Phenotype. 10.1016/j.bpsgos.2021.10.002 (2021b). [DOI] [PMC free article] [PubMed]
- 19.Hoftman GD, et al. Altered gradients of glutamate and gamma-aminobutyric acid transcripts in the cortical visuospatial working memory network in schizophrenia. Biol. Psychiatry. 2018;83:670–679. doi: 10.1016/j.biopsych.2017.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seidlitz, J. et al. Transcriptomic and cellular decoding of regional brain vulnerability to neurodevelopmental disorders. Nat. Commun.11, 3358 (2020). [DOI] [PMC free article] [PubMed]
- 21.Buckner RL, et al. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to alzheimer’s disease. J. Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buckner RL, et al. Molecular, structural, and functional characterization of alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J. Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vlassenko AG, et al. Aerobic glycolysis and tau deposition in preclinical alzheimer’s disease. Neurobiol. Aging. 2018;67:95–98. doi: 10.1016/j.neurobiolaging.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vlassenko AG, et al. Spatial correlation between brain aerobic glycolysis and amyloid-β (aβ) deposition. Proc. Natl Acad. Sci. USA. 2010;107:17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson PM, et al. Enigma and global neuroscience: a decade of large-scale studies of the brain in health and disease across more than 40 countries. Transl. Psychiatry. 2020;10:1–28. doi: 10.1038/s41398-020-0705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun D, et al. Large-scale mapping of cortical alterations in 22q11. 2 deletion syndrome: convergence with idiopathic psychosis and effects of deletion size. Mol. Psychiatry. 2020;25:1822–1834. doi: 10.1038/s41380-018-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoogman M, et al. Brain imaging of the cortex in adhd: a coordinated analysis of large-scale clinical and population-based samples. Am. J. Psychiatry. 2019;176:531–542. doi: 10.1176/appi.ajp.2019.18091033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Rooij D, et al. Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: results from the enigma asd working group. Am. J. Psychiatry. 2018;175:359–369. doi: 10.1176/appi.ajp.2017.17010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whelan CD, et al. Structural brain abnormalities in the common epilepsies assessed in a worldwide enigma study. Brain. 2018;141:391–408. doi: 10.1093/brain/awx341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmaal L, et al. Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the enigma major depressive disorder working group. Mol. Psychiatry. 2017;22:900–909. doi: 10.1038/mp.2016.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boedhoe PS, et al. Cortical abnormalities associated with pediatric and adult obsessive-compulsive disorder: findings from the enigma obsessive-compulsive disorder working group. Am. J. Psychiatry. 2018;175:453–462. doi: 10.1176/appi.ajp.2017.17050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Erp TG, et al. Cortical brain abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the enhancing neuro imaging genetics through meta analysis (enigma) consortium. Biol. Psychiatry. 2018;84:644–654. doi: 10.1016/j.biopsych.2018.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hibar D, et al. Cortical abnormalities in bipolar disorder: an mri analysis of 6503 individuals from the enigma bipolar disorder working group. Mol. Psychiatry. 2018;23:932–942. doi: 10.1038/mp.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opel N, et al. Brain structural abnormalities in obesity: relation to age, genetic risk, and common psychiatric disorders. Mol. Psychiatry. 2020;26:1–14. doi: 10.1038/s41380-021-01191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirschner, M. et al. Cortical and subcortical neuroanatomical signatures of schizotypy in 3004 individuals assessed in a worldwide enigma study. Mol. Psychiatry27, 1167–1176 (2021). [DOI] [PMC free article] [PubMed]
- 36.Laansma, M. A. et al. (2021). International multicenter analysis of brain structure across clinical stages of parkinson’s disease. Mov. Disord.36, 2583–2594 (2021). [DOI] [PMC free article] [PubMed]
- 37.Hawrylycz MJ, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson KM, et al. Convergent molecular, cellular, and cortical neuroimaging signatures of major depressive disorder. Proc. Natl Acad. Sci. USA. 2020;117:25138–25149. doi: 10.1073/pnas.2008004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen JY, et al. Mapping gene transcription and neurocognition across human neocortex. Nat. Human Behav. 2021;5:1240–1250. doi: 10.1038/s41562-021-01082-z. [DOI] [PubMed] [Google Scholar]
- 40.Desikan RS, et al. An automated labeling system for subdividing the human cerebral cortex on mri scans into gyral based regions of interest. NeuroImage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Cammoun L, et al. Mapping the human connectome at multiple scales with diffusion spectrum mri. J. Neurosci. Meth. 2012;203:386–397. doi: 10.1016/j.jneumeth.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 42.Budescu DV. Dominance analysis: a new approach to the problem of relative importance of predictors in multiple regression. Psychol. Bull. 1993;114:542. doi: 10.1037/0033-2909.114.3.542. [DOI] [Google Scholar]
- 43.Azen R, Budescu DV. The dominance analysis approach for comparing predictors in multiple regression. Psychol. Methods. 2003;8:129. doi: 10.1037/1082-989X.8.2.129. [DOI] [PubMed] [Google Scholar]
- 44.Shafiei G, et al. Topographic gradients of intrinsic dynamics across neocortex. Elife. 2020;9:e62116. doi: 10.7554/eLife.62116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Z-Q, et al. Time-resolved structure-function coupling in brain networks. Commun. Biol. 2022;5:1–10. doi: 10.1038/s42003-021-02997-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lenzenweger MF. Schizotaxia, schizotypy, and schizophrenia: Paul e. meehl’s blueprint for the experimental psychopathology and genetics of schizophrenia. J. Abnormal Psychol. 2006;115:195. doi: 10.1037/0021-843X.115.2.195. [DOI] [PubMed] [Google Scholar]
- 47.Feng Y-CA, et al. Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am. J. Hum. Genet. 2019;105:267–282. doi: 10.1016/j.ajhg.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consortium, T. I. L. A. E. et al. (2018). Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun.9, 5269 (2018). [DOI] [PMC free article] [PubMed]
- 49.Hoehn M, Yahr M. Parkinsonism: onset, progression and mortality. Neurology. 1967;17:427–442. doi: 10.1212/WNL.17.5.427. [DOI] [PubMed] [Google Scholar]
- 50.Luk KC, et al. Pathological α-synuclein transmission initiates parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Henderson MX, Trojanowski JQ, Lee VM-Y. α-synuclein pathology in parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 2019;709:134316. doi: 10.1016/j.neulet.2019.134316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Martino A, et al. Unraveling the miswired connectome: a developmental perspective. Neuron. 2014;83:1335–1353. doi: 10.1016/j.neuron.2014.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown JA, et al. Patient-tailored, connectivity-based forecasts of spreading brain atrophy. Neuron. 2019;104:856–868. doi: 10.1016/j.neuron.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chopra, S. et al. Network constraints on longitudinal grey matter changes in first episode psychosis. Preprint at medRxiv10.1101/2022.01.11.22268989 (2022).
- 55.Zeighami Y, et al. Network structure of brain atrophy in de novo parkinson’s disease. Elife. 2015;4:e08440. doi: 10.7554/eLife.08440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Romero-Garcia R, et al. Structural covariance networks are coupled to expression of genes enriched in supragranular layers of the human cortex. NeuroImage. 2018;171:256–267. doi: 10.1016/j.neuroimage.2017.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans AC. Networks of anatomical covariance. Neuroimage. 2013;80:489–504. doi: 10.1016/j.neuroimage.2013.05.054. [DOI] [PubMed] [Google Scholar]
- 58.Váša F, et al. Adolescent tuning of association cortex in human structural brain networks. Cereb. Cort. 2018;28:281–294. doi: 10.1093/cercor/bhx249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seidlitz J, et al. Morphometric similarity networks detect microscale cortical organization and predict inter-individual cognitive variation. Neuron. 2018;97:231–247. doi: 10.1016/j.neuron.2017.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnatkeviciute, A., Fulcher, B., Bellgrove, M. & Fornito, A. Where the genome meets the connectome: understanding how genes shape human brain connectivity. Neuroimage244, 118570 (2021a). [DOI] [PubMed]
- 61.Fulcher BD, Fornito A. A transcriptional signature of hub connectivity in the mouse connectome. Proc. Natl Acad. Sci. USA. 2016;113:1435–1440. doi: 10.1073/pnas.1513302113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richiardi J, et al. Correlated gene expression supports synchronous activity in brain networks. Science. 2015;348:1241–1244. doi: 10.1126/science.1255905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hansen, J. Y. et al. Mapping neurotransmitter systems to the structural and functional organization of the human neocortex. Preprint at bioRxiv10.1101/2021.10.28.466336 (2121b). [DOI] [PMC free article] [PubMed]
- 64.Paquola C, et al. Microstructural and functional gradients are increasingly dissociated in transmodal cortices. PLoS Biol. 2019;17:e3000284. doi: 10.1371/journal.pbio.3000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fornito A, Arnatkevičiūtė A, Fulcher BD. Bridging the gap between connectome and transcriptome. Trend. Cogn. Sci. 2019;23:34–50. doi: 10.1016/j.tics.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 66.Betzel RF, Bassett DS. Specificity and robustness of long-distance connections in weighted, interareal connectomes. Proc. Natl Acad. Sci. USA. 2018;115:E4880–E4889. doi: 10.1073/pnas.1720186115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yee CM, Javitt DC, Miller GA. Replacing dsm categorical analyses with dimensional analyses in psychiatry research: the research domain criteria initiative. JAMA Psychiatry. 2015;72:1159–1160. doi: 10.1001/jamapsychiatry.2015.1900. [DOI] [PubMed] [Google Scholar]
- 68.Kebets V, et al. Somatosensory-motor dysconnectivity spans multiple transdiagnostic dimensions of psychopathology. Biol. Psychiatry. 2019;86:779–791. doi: 10.1016/j.biopsych.2019.06.013. [DOI] [PubMed] [Google Scholar]
- 69.Vanasse TJ, et al. Brain pathology recapitulates physiology: A network meta-analysis. Commun. Biol. 2021;4:1–11. doi: 10.1038/s42003-021-01832-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shine JM, et al. Computational models link cellular mechanisms of neuromodulation to large-scale neural dynamics. Nat. Neurosci. 2021;24:765–776. doi: 10.1038/s41593-021-00824-6. [DOI] [PubMed] [Google Scholar]
- 71.Cornblath EJ, et al. Computational modeling of tau pathology spread reveals patterns of regional vulnerability and the impact of a genetic risk factor. Sci. Adv. 2021;7:eabg6677. doi: 10.1126/sciadv.abg6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henderson MX, et al. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 2019;22:1248–1257. doi: 10.1038/s41593-019-0457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raj A, Powell F. Network model of pathology spread recapitulates neurodegeneration and selective vulnerability in huntington’s disease. NeuroImage. 2021;235:118008. doi: 10.1016/j.neuroimage.2021.118008. [DOI] [PubMed] [Google Scholar]
- 74.Shafiei, G. et al. (2022) Network structure and transcriptomic vulnerability shape atrophy in frontotemporal dementia. Brain10.1093/brain/awac069 (2022). [DOI] [PMC free article] [PubMed]
- 75.Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging. 2011;32:1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 77.Binette AP, et al. Bundle-specific associations between white matter microstructure and aβ and tau pathology in preclinical alzheimer’s disease. Elife. 2021;10:e62929. doi: 10.7554/eLife.62929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marco EJ, Hinkley LB, Hill SS, Nagarajan SS. Sensory processing in autism: a review of neurophysiologic findings. Pediat. Res. 2011;69:48–54. doi: 10.1203/PDR.0b013e3182130c54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Javitt DC, Freedman R. Sensory processing dysfunction in the personal experience and neuronal machinery of schizophrenia. Am. J. Psychiatry. 2015;172:17–31. doi: 10.1176/appi.ajp.2014.13121691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bernard JA, Mittal VA. Updating the research domain criteria: the utility of a motor dimension. Psychol. Med. 2015;45:2685–2689. doi: 10.1017/S0033291715000872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Larivière S, et al. Network-based atrophy modeling in the common epilepsies: a worldwide enigma study. Sci. Adv. 2020;6:eabc6457. doi: 10.1126/sciadv.abc6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Den Heuvel MP, Sporns O. Rich-club organization of the human connectome. J. Neurosci. 2011;31:15775–15786. doi: 10.1523/JNEUROSCI.3539-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Van Den Heuvel MP, et al. Abnormal rich club organization and functional brain dynamics in schizophrenia. JAMA Psychiatry. 2013;70:783–792. doi: 10.1001/jamapsychiatry.2013.1328. [DOI] [PubMed] [Google Scholar]
- 84.McColgan P, et al. Selective vulnerability of rich club brain regions is an organizational principle of structural connectivity loss in huntington’s disease. Brain. 2015;138:3327–3344. doi: 10.1093/brain/awv259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goodkind M, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72:305–315. doi: 10.1001/jamapsychiatry.2014.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abel KM, Drake R, Goldstein JM. Sex differences in schizophrenia. Int. Rev. Psychiatry. 2010;22:417–428. doi: 10.3109/09540261.2010.515205. [DOI] [PubMed] [Google Scholar]
- 87.Werling DM, Geschwind DH. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013;26:146. doi: 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Altemus M, Sarvaiya N, Epperson CN. Sex differences in anxiety and depression clinical perspectives. Front. Neuroendocrinol. 2014;35:320–330. doi: 10.1016/j.yfrne.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee PH, et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–1482. doi: 10.1016/j.cell.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Voineskos AN, et al. Effects of antipsychotic medication on brain structure in patients with major depressive disorder and psychotic features: neuroimaging findings in the context of a randomized placebo-controlled clinical trial. JAMA Psychiatry. 2020;77:674–683. doi: 10.1001/jamapsychiatry.2020.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maier-Hein KH, et al. The challenge of mapping the human connectome based on diffusion tractography. Nat. Commun. 2017;8:1–13. doi: 10.1038/s41467-017-01285-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones DK, Knösche TR, Turner R. White matter integrity, fiber count, and other fallacies: the do’s and don’ts of diffusion mri. Neuroimage. 2013;73:239–254. doi: 10.1016/j.neuroimage.2012.06.081. [DOI] [PubMed] [Google Scholar]
- 93.Zalesky A, et al. Connectome sensitivity or specificity: which is more important? Neuroimage. 2016;142:407–420. doi: 10.1016/j.neuroimage.2016.06.035. [DOI] [PubMed] [Google Scholar]
- 94.van der Weijden CW, et al. Myelin quantification with mri: A systematic review of accuracy and reproducibility. NeuroImage. 2021;226:117561. doi: 10.1016/j.neuroimage.2020.117561. [DOI] [PubMed] [Google Scholar]
- 95.Hagiwara A, et al. Myelin measurement: comparison between simultaneous tissue relaxometry, magnetization transfer saturation index, and t1w/t2w ratio methods. Sci. Rep. 2018;8:1–12. doi: 10.1038/s41598-018-28852-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Larivière S, et al. The enigma toolbox: multiscale neural contextualization of multisite neuroimaging datasets. Nat. Methods. 2021;18:698–700. doi: 10.1038/s41592-021-01186-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Griffa, A., Alemán-Gómez, Y. & Hagmann, P. Structural and functional connectome from 70 young healthy adults [data set]. Zenodohttps://zenodo.org/record/2872624 (2019).
- 98.Vázquez-Rodríguez B, et al. Gradients of structure–function tethering across neocortex. Proc. Natl Acad. Sci. USA. 2019;116:21219–21227. doi: 10.1073/pnas.1903403116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bazinet V, de Wael RV, Hagmann P, Bernhardt BC, Misic B. Multiscale communication in cortico-cortical networks. NeuroImage. 2021;243:118546. doi: 10.1016/j.neuroimage.2021.118546. [DOI] [PubMed] [Google Scholar]
- 100.Daducci A, et al. The connectome mapper: an open-source processing pipeline to map connectomes with MRI. PLoS ONE. 2012;7:e48121. doi: 10.1371/journal.pone.0048121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Betzel RF, Griffa A, Hagmann P, Mišić B. Distance-dependent consensus thresholds for generating group-representative structural brain networks. Netw. Neurosci. 2019;3:475–496. doi: 10.1162/netn_a_00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity mri networks arise from subject motion. Neuroimage. 2012;59:2142–2154. doi: 10.1016/j.neuroimage.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Wael RV, et al. Anatomical and microstructural determinants of hippocampal subfield functional connectome embedding. Proc. Natl Acad. Sci. USA. 2018;115:10154–10159. doi: 10.1073/pnas.1803667115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Van Essen DC, et al. The wu-minn human connectome project: an overview. Neuroimage. 2013;80:62–79. doi: 10.1016/j.neuroimage.2013.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Glasser MF, et al. The minimal preprocessing pipelines for the human connectome project. Neuroimage. 2013;80:105–124. doi: 10.1016/j.neuroimage.2013.04.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tournier J-D, et al. Mrtrix3: A fast, flexible and open software framework for medical image processing and visualisation. Neuroimage. 2019;202:116137. doi: 10.1016/j.neuroimage.2019.116137. [DOI] [PubMed] [Google Scholar]
- 107.Dhollander, T., Raffelt, D. & Connelly, A. Unsupervised 3-tissue response function estimation from single-shell or multi-shell diffusion mr data without a co-registered t1 image. In: ISMRM Workshop on Breaking the Barriers of Diffusion MRI, Vol. 5. (ISMRM, 2016).
- 108.Jeurissen B, Tournier J-D, Dhollander T, Connelly A, Sijbers J. Multi-tissue constrained spherical deconvolution for improved analysis of multi-shell diffusion mri data. NeuroImage. 2014;103:411–426. doi: 10.1016/j.neuroimage.2014.07.061. [DOI] [PubMed] [Google Scholar]
- 109.Tournier, J. D. et al. Improved probabilistic streamlines tractography by 2nd order integration over fibre orientation distributions. In: Proc. International Society for Magnetic Resonance in Medicine, Vol. 1670 (John Wiley & Sons, Inc. New Jersey, USA, 2010).
- 110.Smith RE, Tournier J-D, Calamante F, Connelly A. Sift2: Enabling dense quantitative assessment of brain white matter connectivity using streamlines tractography. Neuroimage. 2015;119:338–351. doi: 10.1016/j.neuroimage.2015.06.092. [DOI] [PubMed] [Google Scholar]
- 111.Salimi-Khorshidi G, et al. Automatic denoising of functional mri data: combining independent component analysis and hierarchical fusion of classifiers. Neuroimage. 2014;90:449–468. doi: 10.1016/j.neuroimage.2013.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mišić B, et al. Cooperative and competitive spreading dynamics on the human connectome. Neuron. 2015;86:1518–1529. doi: 10.1016/j.neuron.2015.05.035. [DOI] [PubMed] [Google Scholar]
- 113.Markello RD, et al. Standardizing workflows in imaging transcriptomics with the abagen toolbox. eLife. 2021;10:e72129. doi: 10.7554/eLife.72129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hawrylycz M, et al. Canonical genetic signatures of the adult human brain. Nat. Neurosci. 2015;18:1832. doi: 10.1038/nn.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaller S, et al. Test–retest measurements of dopamine d 1-type receptors using simultaneous pet/mri imaging. Eur. J. Nucl. Med. Mol. Imag. 2017;44:1025–1032. doi: 10.1007/s00259-017-3645-0. [DOI] [PubMed] [Google Scholar]
- 116.Smith CT, et al. Partial-volume correction increases estimated dopamine d2-like receptor binding potential and reduces adult age differences. J. Cereb. Blood Flow Metab. 2019;39:822–833. doi: 10.1177/0271678X17737693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sandiego CM, et al. Reference region modeling approaches for amphetamine challenge studies with [11c] flb 457 and pet. J. Cereb. Blood Flow Metab. 2015;35:623–629. doi: 10.1038/jcbfm.2014.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zakiniaeiz Y, et al. Sex differences in amphetamine-induced dopamine release in the dorsolateral prefrontal cortex of tobacco smokers. Neuropsychopharmacology. 2019;44:2205–2211. doi: 10.1038/s41386-019-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Slifstein M, et al. Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry. 2015;72:316–324. doi: 10.1001/jamapsychiatry.2014.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dukart J, et al. Cerebral blood flow predicts differential neurotransmitter activity. Sci. Rep. 2018;8:1–11. doi: 10.1038/s41598-018-22444-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ding Y-S, et al. Pet imaging of the effects of age and cocaine on the norepinephrine transporter in the human brain using (s, s)-[11c] o-methylreboxetine and hrrt. Synapse. 2010;64:30–38. doi: 10.1002/syn.20696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chiang-shan RL, et al. Decreased norepinephrine transporter availability in obesity: positron emission tomography imaging with (s, s)-[11c] o-methylreboxetine. Neuroimage. 2014;86:306–310. doi: 10.1016/j.neuroimage.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sanchez-Rangel E, et al. Norepinephrine transporter availability in brown fat is reduced in obesity: a human pet study with [11 c] mrb. Int. J. Obesity. 2020;44:964–967. doi: 10.1038/s41366-019-0471-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Belfort-DeAguiar R, et al. Noradrenergic activity in the human brain: a mechanism supporting the defense against hypoglycemia. J. Clin. Endocrinol. Metab. 2018;103:2244–2252. doi: 10.1210/jc.2017-02717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Savli M, et al. Normative database of the serotonergic system in healthy subjects using multi-tracer pet. Neuroimage. 2012;63:447–459. doi: 10.1016/j.neuroimage.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 126.Gallezot J-D, et al. Kinetic modeling of the serotonin 5-ht1b receptor radioligand [11c] p943 in humans. J. Cereb. Blood Flow Metab. 2010;30:196–210. doi: 10.1038/jcbfm.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murrough JW, et al. The effect of early trauma exposure on serotonin type 1b receptor expression revealed by reduced selective radioligand binding. Arch. Gen. Psychiatry. 2011;68:892–900. doi: 10.1001/archgenpsychiatry.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Murrough JW, et al. Reduced ventral striatal/ventral pallidal serotonin 1b receptor binding potential in major depressive disorder. Psychopharmacology. 2011;213:547–553. doi: 10.1007/s00213-010-1881-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matuskey D, et al. Reductions in brain 5-ht1b receptor availability in primarily cocaine-dependent humans. Biol. Psychiatry. 2014;76:816–822. doi: 10.1016/j.biopsych.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pittenger C, et al. Ocd is associated with an altered association between sensorimotor gating and cortical and subcortical 5-ht1b receptor binding. J. Affect. Disorder. 2016;196:87–96. doi: 10.1016/j.jad.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Saricicek A, et al. Test–retest reliability of the novel 5-ht 1b receptor pet radioligand [11 c] p943. Eur. J. Nucl. Med. Mol. Imag. 2015;42:468–477. doi: 10.1007/s00259-014-2958-5. [DOI] [PubMed] [Google Scholar]
- 132.Baldassarri SR, et al. Use of electronic cigarettes leads to significant beta2-nicotinic acetylcholine receptor occupancy: evidence from a pet imaging study. Nicotine Tob. Res. 2018;20:425–433. doi: 10.1093/ntr/ntx091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Beliveau V, et al. A high-resolution in vivo atlas of the human brain’s serotonin system. J. Neurosci. 2017;37:120–128. doi: 10.1523/JNEUROSCI.2830-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Radhakrishnan R, et al. Age-related change in 5-ht6 receptor availability in healthy male volunteers measured with 11c-gsk215083 pet. J. Nucl. Med. 2018;59:1445–1450. doi: 10.2967/jnumed.117.206516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Radhakrishnan R, et al. In vivo 5-ht6 and 5-ht2a receptor availability in antipsychotic treated schizophrenia patients vs. unmedicated healthy humans measured with [11c] gsk215083 pet. Psychiatry Res. Neuroimag. 2020;295:111007. doi: 10.1016/j.pscychresns.2019.111007. [DOI] [PubMed] [Google Scholar]
- 136.Hillmer AT, et al. Imaging of cerebral α4β2* nicotinic acetylcholine receptors with (-)-[18f] flubatine pet: implementation of bolus plus constant infusion and sensitivity to acetylcholine in human brain. Neuroimage. 2016;141:71–80. doi: 10.1016/j.neuroimage.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Naganawa M, et al. First-in-human assessment of 11c-lsn3172176, an m1 muscarinic acetylcholine receptor pet radiotracer. J. Nucl. Med. 2021;62:553–560. doi: 10.2967/jnumed.120.246967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Aghourian M, et al. Quantification of brain cholinergic denervation in alzheimer’s disease using pet imaging with [18 f]-feobv. Mol. Psychiatry. 2017;22:1531–1538. doi: 10.1038/mp.2017.183. [DOI] [PubMed] [Google Scholar]
- 139.Bedard M-A, et al. Brain cholinergic alterations in idiopathic rem sleep behaviour disorder: a pet imaging study with 18f-feobv. Sleep Med. 2019;58:35–41. doi: 10.1016/j.sleep.2018.12.020. [DOI] [PubMed] [Google Scholar]
- 140.Smart K, et al. Sex differences in [11 c] abp688 binding: a positron emission tomography study of mglu5 receptors. Eur. J. Nucl. Med. Mol. Imaging. 2019;46:1179–1183. doi: 10.1007/s00259-018-4252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.DuBois JM, et al. Characterization of age/sex and the regional distribution of mglur5 availability in the healthy human brain measured by high-resolution [11 c] abp688 pet. Eur. J. Nucl. Med. Mol. Imaging. 2016;43:152–162. doi: 10.1007/s00259-015-3167-6. [DOI] [PubMed] [Google Scholar]
- 142.Nørgaard M, et al. A high-resolution in vivo atlas of the human brain’s benzodiazepine binding site of gabaa receptors. NeuroImage. 2021;232:117878. doi: 10.1016/j.neuroimage.2021.117878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gallezot J-D, et al. Determination of receptor occupancy in the presence of mass dose:[11c] gsk189254 pet imaging of histamine h3 receptor occupancy by pf-03654746. J. Cereb. Blood Flow Metab. 2017;37:1095–1107. doi: 10.1177/0271678X16650697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Normandin MD, et al. Imaging the cannabinoid cb1 receptor in humans with [11c] omar: assessment of kinetic analysis methods, test–retest reproducibility, and gender differences. J. Cereb. Blood Flow Metab. 2015;35:1313–1322. doi: 10.1038/jcbfm.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.D’Souza DC, et al. Rapid changes in cannabinoid 1 receptor availability in cannabis-dependent male subjects after abstinence from cannabis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging. 2016;1:60–67. doi: 10.1016/j.bpsc.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 146.Ranganathan M, et al. Reduced brain cannabinoid receptor availability in schizophrenia. Biol. Psychiatry. 2016;79:997–1005. doi: 10.1016/j.biopsych.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Neumeister A, et al. Positron emission tomography shows elevated cannabinoid cb 1 receptor binding in men with alcohol dependence. Alcohol. Clin. Exp. Res. 2012;36:2104–2109. doi: 10.1111/j.1530-0277.2012.01815.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 148.Kantonen T, et al. Interindividual variability and lateralization of μ-opioid receptors in the human brain. NeuroImage. 2020;217:116922. doi: 10.1016/j.neuroimage.2020.116922. [DOI] [PubMed] [Google Scholar]
- 149.Vaishnavi SN, et al. Regional aerobic glycolysis in the human brain. Proc. Natl Acad. Sci. USA. 2010;107:17757–17762. doi: 10.1073/pnas.1010459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Finnema SJ, et al. Kinetic evaluation and test–retest reproducibility of [11c] ucb-j, a novel radioligand for positron emission tomography imaging of synaptic vesicle glycoprotein 2a in humans. J. Cereb. Blood Flow Metab. 2018;38:2041–2052. doi: 10.1177/0271678X17724947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen, M.-K. et al. Comparison of [11c] ucb-j and [18f] fdg pet in alzheimer’s disease: a tracer kinetic modeling study. J. Cereb. Blood Flow Metab.41, 2395–2409 (2021). [DOI] [PMC free article] [PubMed]
- 152.O’Dell RS, et al. Association of aβ deposition and regional synaptic density in early Alzheimer’s disease: a pet imaging study with [11 c] ucb-j. Alzheimer’s Res. Ther. 2021;13:1–12. doi: 10.1186/s13195-020-00736-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smart K, et al. Binding of the synaptic vesicle radiotracer [11c] ucb-j is unchanged during functional brain activation using a visual stimulation task. J. Cereb. Blood Flow Metab. 2021;41:1067–1079. doi: 10.1177/0271678X20946198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weiss JJ, et al. Preliminary in vivo evidence of reduced synaptic density in human immunodeficiency virus (hiv) despite antiretroviral therapy. Clin. Infect. Dis. 2021;73:1404–1411. doi: 10.1093/cid/ciab484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Radhakrishnan R, et al. In vivo evidence of lower synaptic vesicle density in schizophrenia. Mol. Psychiatry. 2021;26:7690–7698. doi: 10.1038/s41380-021-01184-0. [DOI] [PubMed] [Google Scholar]
- 156.Finnema SJ, et al. Reduced synaptic vesicle protein 2a binding in temporal lobe epilepsy: a [11c] ucb-j positron emission tomography study. Epilepsia. 2020;61:2183–2193. doi: 10.1111/epi.16653. [DOI] [PubMed] [Google Scholar]
- 157.Bini J, et al. Human adult and adolescent biodistribution and dosimetry of the synaptic vesicle glycoprotein 2a radioligand 11 c-ucb-j. EJNMMI Res. 2020;10:1–8. doi: 10.1186/s13550-020-00670-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mecca AP, et al. In vivo measurement of widespread synaptic loss in alzheimer’s disease with sv2a pet. Alzheimer’s Dement. 2020;16:974–982. doi: 10.1002/alz.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Finnema SJ, et al. A single-center, open-label positron emission tomography study to evaluate brivaracetam and levetiracetam synaptic vesicle glycoprotein 2a binding in healthy volunteers. Epilepsia. 2019;60:958–967. doi: 10.1111/epi.14701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Holmes SE, et al. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019;10:1–10. doi: 10.1038/s41467-019-09562-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chen M-K, et al. Assessing synaptic density in alzheimer disease with synaptic vesicle glycoprotein 2a positron emission tomographic imaging. JAMA Neurol. 2018;75:1215–1224. doi: 10.1001/jamaneurol.2018.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Auvity S, et al. Repurposing radiotracers for myelin imaging: a study comparing 18f-florbetaben, 18f-florbetapir, 18f-flutemetamol, 11c-medas, and 11c-pib. Eur. J. Nucl. Med. Mol. Imaging. 2020;47:490–501. doi: 10.1007/s00259-019-04516-z. [DOI] [PubMed] [Google Scholar]
- 163.Zeydan B, et al. Pittsburgh compound-b pet white matter imaging and cognitive function in late multiple sclerosis. Mult. Scler. J. 2018;24:739–749. doi: 10.1177/1352458517707346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yeo B, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 2011;106:1125–1165. doi: 10.1152/jn.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tadel F, Baillet S, Mosher JC, Pantazis D, Leahy RM. Brainstorm: a user-friendly application for meg/eeg analysis. Comput. Intell. Neurosci. 2011;2011:879716. doi: 10.1155/2011/879716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Alexander-Bloch AF, et al. On testing for spatial correspondence between maps of human brain structure and function. NeuroImage. 2018;178:540–551. doi: 10.1016/j.neuroimage.2018.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Markello RD, Misic B. Comparing spatial null models for brain maps. NeuroImage. 2021;236:118052. doi: 10.1016/j.neuroimage.2021.118052. [DOI] [PubMed] [Google Scholar]
- 168.Vasa, F. & Misic, B. (2022). Null models in network neuroscience. Nat. Rev. Neurosci. 23, 493–504 (2022). [DOI] [PubMed]
- 169.Burt, J. B., Helmer, M., Shinn, M., Anticevic, A. & Murray, J. D. Generative modeling of brain maps with spatial autocorrelation. NeuroImage220, 117038 (2020). [DOI] [PubMed]
- 170.Breakspear M, Brammer MJ, Bullmore ET, Das P, Williams LM. Spatiotemporal wavelet resampling for functional neuroimaging data. Hum. Brain Mapp. 2004;23:1–25. doi: 10.1002/hbm.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Roberts JA, et al. The contribution of geometry to the human connectome. Neuroimage. 2016;124:379–393. doi: 10.1016/j.neuroimage.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 172.Gollo LL, et al. Fragility and volatility of structural hubs in the human connectome. Nat. Neurosci. 2018;21:1107–1116. doi: 10.1038/s41593-018-0188-z. [DOI] [PubMed] [Google Scholar]
- 173.Markello, R. D. et al. Neuromaps: structural and functional interpretation of brain maps. Preprint at bioRxiv10.1101/2022.01.06.475081 (2022). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in the present report is openly available at https://github.com/netneurolab/hansen_crossdisorder_vulnerability. More specifically, ENIGMA datasets are available through the ENIGMA consortium and the ENIGMA toolbox (https://github.com/MICA-MNI/ENIGMA96). The Lausanne dataset is available at https://zenodo.org/record/2872624#.XOJqE99fhmM97. The HCP dataset is available at https://db.humanconnectome.org/. Molecular predictors are available as volumetric images in the neuromaps toolbox (https://netneurolab.github.io/neuromaps/173). The Allen Human Brain Atlas is available at https://human.brain-map.org/.
All code used to perform the analyses can be found at https://github.com/netneurolab/hansen_crossdisorder_vulnerabilityand on Zenodo (10.5281/zenodo.6795748).