

Abstract

In this work we present a new approach to fix the intruder-state problem (ISP) in CASPT2 based on σp regularization. The resulting σp-CASPT2 method is compared to previous techniques, namely, the real and imaginary level shifts, on a theoretical basis and by performing a series of systematic calculations. The analysis is focused on two aspects, the effectiveness of σp-CASPT2 in removing the ISP and the sensitivity of the approach with respect to the input parameter. We found that σp-CASPT2 compares favorably with respect to previous approaches and that different versions, σ1-CASPT2 and σ2-CASPT2, have different potential application domains. This analysis also reveals the unsuitability of the real level shift technique as a general way to avoid the intruder-state problem.
1. Introduction
Among the many options of multireference electron correlation methods,1 approaches based on second-order perturbation theory (PT2) with a multiconfigurational reference function offer an appealing compromise between accuracy and computational complexity. Their popularity is reflected by the large number of available flavors,2−8 which typically differ in the partitioning of the Hamiltonian, the many-electron basis used to express the first-order wave function, or the conditions to obtain the expansion coefficients. One of the most known and used of these multireference perturbation theory (MRPT) approaches is complete active space PT2 (CASPT2),2 whose development has seen some important activity in recent times, with a newly modified zeroth-order Hamiltonian (CASPT2-K),9 new quasi-degenerate variants (XDW-CASPT2 and RMS-CASPT2),10,11 reduced scaling implementations,12−14 and analytic nuclear energy gradients and derivative couplings.15−22 Despite its popularity and general applicability, CASPT2 suffers from an issue common to other MRPT-based approaches, the intruder-state problem (ISP). While this generally does not appear when modeling the ground state of small organic compounds, it is much more common in transition metal complexes and excited-states applications. Two techniques have been introduced during the years to avoid this issue in CASPT2, namely, the real and imaginary level shifts.23,24 In both cases, the idea is to add a uniform shift to the resolvent operator, avoiding the singularity that causes the ISP. As a consequence, these techniques introduce a dependence of the results on a user-defined parameter, which should ideally be minimal, especially in the absence of intruder states. In other words, the results for well-behaved cases should be as insensitive as possible to the value of the input parameter. This is because it is common that, for a molecular system, only a subset of electronic states or conformations is affected by the ISP but all of them are treated with the same intruder-state-removal technique. In principle, the results of well-behaved cases should not change as a function of the parameter, as they did not require any intervention in the first place. This is for instance typical in the calculation of vertical transition energies, where only one or a few states of the excitation manifold are plagued by ISPs, albeit imposing the use of the shift for all states. Furthermore, considering the current wide availability of analytic nuclear energy gradients for CASPT2,15,16,20,21,25,26 exploration of the potential energy surfaces, especially in the excited states,27 has become more common, requiring a robust approach that effectively avoids ISPs and that at the same time does not affect the qualitative description in regions of the PES where these are not present. In this context, the real level shift23 is not an ideal solution, the reason for which the imaginary level shift24 had been developed. This second option is much better, because it selectively corrects large amplitudes. Nevertheless, the results obtained remain susceptible to the choice of input parameter. In an attempt to find a new intruder-state-removal technique that is more insensitive to this, but equally effective in removing the singularities in the first-order wave function, we propose to use σp regularization in CASPT2.28,29 This approach is a simple and effective way to remove the ISP, and that from a theoretical perspective appears to have some advantages over the real and imaginary level shift techniques. In this contribution we introduce the σp-CASPT2 method and critically analyze it in comparison to the established alternatives.
The work is organized as follows. In the next section we present the theoretical foundations of the ISP and the various intruder-state-removal techniques. We then conduct a series of tests to evaluate the effectiveness of σp-CASPT2 in removing the ISPs and study the sensitivity of this new approach with respect to the input parameter. In the last section we summarize our findings and briefly discuss the remaining open issues in this context and possible future directions to solve them.
2. Theoretical Background
In this section we will define the intruder-state problem from a phenomenological perspective, review the theory behind the level shift techniques, and introduce the σp regularization formalism. The discussion is very general and applies, in principle, to any approach based on second-order Rayleigh–Schrödinger perturbation theory (RSPT2). However, in the following, these techniques are presented in the context of CASPT2, as this method is the focus of the present work.
2.1. Elements of Second-Order Perturbation Theory
The starting point is the partitioning of the Hamiltonian into a zeroth-order part and a perturbation operator (also known as fluctuation potential):
| 1 |
The wave function of the reference state Ψ(0) is an eigenfunction of Ĥ(0) by construction (which does not necessarily have to be the ground-state one), with an associated energy eigenvalue E(0). Note that in state-specific CASPT2, this energy does not correspond to the CASSCF one but rather to the expectation value of the generalized Fock operator (this is just as in MP2, where the zeroth-order energy does not correspond to the Hartree–Fock one). The first-order interacting space (FOIS) is spanned by an additional set of M eigenfunctions of Ĥ(0), satisfying the following eigenvalue equation:
| 2 |
In CASPT2, the functions Φi are (linear combinations of) internally contracted configurations generated by the application of excitation operators to the CASSCF reference wave function (eqs 1a–h in ref (2).), while in uncontracted theories such as MRMP2, they are configuration-state functions or Slater determinants.3 In either case, ϵi represents the zeroth-order energy associated with the perturber function Φi. For the remainder of this article we will assume that the eigenfunctions Φi and eigenvalues ϵi are known, unless otherwise stated. This is in practice the case for the diagonal CASPT2 method30 (named CASPT2D in the original publication) but not for the (more conventional) full CASPT2 approach (referred to as CASPT2N by Andersson et al.2). In principle, it is possible to obtain the eigenpairs (Φi, ϵi) in CASPT2 as well, but this would require the diagonalization of Ĥ(0) expressed in the FOIS basis, which is impractical from a computational perspective. Regardless of the particular implementation of RSPT2, the first-order correction to the wave function is expanded as follows,
| 3 |
where the amplitudes Ti are determined by solving the first-order equations:
| 4 |
Owing to eq 2 and the orthogonality of the eigenfunctions, the analytical expression for the amplitudes is simply
| 5 |
where we have introduced a short-hand notation for the right-hand side elements Vi = ⟨Φi|V̂|Ψ(0)⟩ and for the energy denominators Δi = ϵi – E(0). At last, the second-order correction to the energy is obtained either by projection,
| 6 |
or variationally, through the evaluation of the Hylleraas functional,31
| 7 |
Note that the solution of eq 4 is a stationary point of eq 7, hence the variational nature of the Hylleraas expression. Inserting that solution, i.e., eq 5, into either eq 6 or eq 7 results in the same second-order energy correction.
2.2. Intruder-State Problem
The intruder-state problem (ISP) arises when the energy denominator in eq 5 vanishes,
| 8 |
leading to an infinitely large amplitude and a divergent perturbation series. This is caused by a degeneracy between Ψ(0) and a perturber Φi in the zeroth-order approximation. The first obvious solution to this situation would be to include the intruder state Φi in the reference wave function. However, this is not always possible or desired in CASPT2, as it involves changing the active space of the underlying CASSCF optimization and potentially leads to the appearance of new intruder states, the need of additional electronic states in the state-averaging procedure, or exceedingly expensive calculations. The other option is to change the partitioning of the Hamiltonian. If the degeneracy between the intruder state Φi and Ψ(0) is the result of the approximate description provided by the zeroth-order Hamiltonian, while the true energies of these states are different, one could modify the structure of Ĥ(0) to lift this accidental degeneracy. In fact, the potential existence of an ISP is intimately coupled to the form of the zeroth-order Hamiltonian. For instance, an internally contracted approach based on Dyall’s Hamiltonian32 is known to be practically free from intruder states, as in the case of NEVPT2.6,33,34 Thus, it seems a natural choice to focus the efforts for solving the ISP in CASPT2 on modifying Ĥ(0), analogously to the strategy followed by Roos and Andersson23 and Forsberg and Malmqvist24 several years ago, with the introduction of the real and imaginary level shift techniques, respectively. It is important to note that, even though from a formal point of view an intruder state strictly implies an exact degeneracy, in practice, all perturbers Φi associated with small energy denominators are considered intruder states. This is because the corresponding amplitudes will lead to artificially large contributions to the correlation energy. Rigorously speaking, it is possible that higher-order corrections compensate for that, though, for all practical purposes, this is not relevant, because the perturbation expansion is generally considered up to second-order only (for a formal discussion on the matter, we suggest the excellent work by Olsen and Jørgensen35 and references therein).
Before moving on to review the level shift techniques in CASPT2, it is important to introduce a convenient way to identify intruder states in actual calculations. Recognizing that an underlying assumption of perturbation theory is that the fluctuation potential V̂ is a minor modification of the zeroth-order description of the system, it is expected that the first-order correction to the wave function is small compared to Ψ(0). A way to quantify this is to consider the weight of Ψ(0) relative to Ψ(1) in the wave function corrected through first order, Ψ[1] = Ψ(0) + Ψ(1). This gives rise to the diagnostic measure called reference weight, which is given by
| 9 |
Here, intermediate normalization is assumed (this is the case of CASPT2), thus assigning a unit coefficient to the reference wave function. In the trivial case where wref = 1, all amplitudes must be zero and thus the first-order correction does not contribute at all to Ψ[1]. On the other hand, increasingly small values of wref imply larger contributions to the total wave function from Ψ(1); this is the typical situation. The limiting case wref = 0 is formally possible only in the presence of a true intruder state, where the amplitude of the offending state Φi diverges; Ti → ∞. In practice, accidental exact degeneracies almost never occur and small energy denominators are instead the typical cause of troubles, leading to large amplitudes that outweigh the reference wave function. This situation is associated with small values of wref, which we shall recognize as a signature of the intruder-state problem. Clearly, the reference weight is an empirical diagnostic measure, and therefore there is no universal threshold that unambiguously classifies Ψ[1] as affected by intruder states. Typical values of wref for small organic molecules range between 0.7 and 0.9, while transition metal complexes with many open-shell electrons can have lower values due to the dense manifold of electronic states and the presence of a large number of important low-lying configurations.
In any case, wref is expected to decrease with the number of correlated electrons regardless of the type of molecular system considered. Importantly, in most applications the interest is in relative energies, e.g., when computing vertical transition energies or comparing different conformations. For a balanced and consistent account of the electron correlation effects in these situations, it is crucial that the reference weight is commensurate among all structures and states considered. This simple guideline provides a prescription on how to use wref in practice. In calculations involving several electronic states, and in agreement with previous works,9 we consider as a rule of thumb deviations of more than 10% from the state with largest wref, typically the ground state, as potentially damaged by intruder states. At the same time, the reference weight of any electronic state should be a smooth function of the molecular geometry, insomuch as the electronic energy is. This is particularly important in the calculation of potential energy surfaces and curves, such as for molecular dynamics simulations and the dissociation of diatomics.
2.3. Level Shift Techniques
With the realization that the intruder-state problem manifests itself when the energy denominator in eq 5 vanishes, a possible solution would be to add a small value ε > 0 to Δi, such that when Δi → 0, Ti → −Vi/ε. This is the simple idea behind level shift techniques. It can be formally implemented in any perturbation theory approach by modifying the partitioning of the Hamiltonian as follows:
| 10 |
where 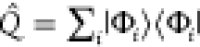 is a projector that shifts only states
orthogonal to the reference one. Solution of the first-order equations
with this modified partitioning leads to amplitudes of the form
is a projector that shifts only states
orthogonal to the reference one. Solution of the first-order equations
with this modified partitioning leads to amplitudes of the form
| 11 |
For a vanishing energy difference, Δi → 0, the amplitude Ti now remains finite owing to the presence of ε in the denominator. This technique is very effective in removing intruder states from ground-state calculations, and it was proposed as a remedy to the ISP in CASPT2 by Roos and Andersson.23 However, because ε uniformly shifts all of the amplitudes of the first-order wave function, it affects also the coefficients of states Φi associated with large denominators, resulting in a second-order energy that strongly depends on it. In an attempt to decrease this dependence, a level shift correction that tries to minimize the impact of the shift on large denominators was proposed. The expression presented in the original contribution23 was derived from manipulating eq 6 and assuming that certain contributions are negligible in the case Δi ≫ ε; however, it was later found24 that this correction actually corresponds to simply evaluating the variational second-order energy, eq 7, using the modified amplitudes (see Section 1.1 in the Supporting Information for a detailed derivation). Unfortunately, the level shift approach with a real parameter does not really remove the singularity but rather moves it at Δi = −ε. While this is not an issue as long as ϵi ≥ E(0), it is possible in CASPT2 that energy denominators take on negative values and accidentally fall near the singularity; that is, Δi ≈ −ε. This is particularly problematic when exploring several conformations, as it can be hard to find a suitable range for ε that avoids all singularities, or when computing excited states and investigating transition metals, where a large a number of small denominators make the results very sensitive to the value of the parameter.36,37
In an attempt at solving the shortcomings of the real level shift, Forsberg and Malmqvist24 proposed to use a purely imaginary parameter instead of a real one. In the imaginary level shift technique, the partitioning is simply modified as
| 12 |
which leads to complex amplitudes of the form
| 13 |
To avoid working with complex arithmetic and considering that the electronic energy is a real number, only the real part of eq 13 is taken, resulting in the following expression for the imaginary-shifted amplitudes:
| 14 |
Interestingly, this is essentially equivalent to the intruder-state avoidance technique developed for MRMP238 and to Tikhonov regularization in linearized coupled cluster.39 In contrast to eq 11, eq 14 does not contain any singularity; hence, the imaginary level shift effectively removes the ISP. This happens by suppressing the contributions associated with small energy denominators: the smaller Δi, the greater the reduction of Ti. Vice versa, when Δi ≫ 0, we have that ε2/Δi → 0 and the amplitudes remain unmodified. Crucially, the ability of the imaginary shift to quench the contributions only when the energy denominators are small comes from its dependence on Δi. As long as the zeroth-order Hamiltonian is diagonal, such as in CASPT2D and MRMP2, the values of these energy denominators are known. However, this is not the case for a nondiagonal Ĥ(0), preventing the use of the true denominators in ε2/Δi. In CASPT2, the imaginary shift is approximated by using only the diagonal entries of the zeroth-order Hamiltonian matrix, which typically yields a reasonable estimate of the exact Δi as long as the off-diagonal couplings are small. It is important to note that the imaginary shift is in practice a real shift too, as can be seen from eq 14. Its formal advantages over the real level shift technique of Roos and Andersson23 are not due to the use of complex algebra but rather to its configuration-specific nature, as opposed to the single uniform parameter appearing in the denominator of eq 11.
At this point, it is convenient for the visual comparison of the two shifts, and for the discussion on the regularizers in the next subsection, to introduce the following general formula for the amplitudes:
| 15 |
where the function f(Δi; ε) embodies the energy denominator and the different approaches that modify it (note the semicolon in eq 15 to highlight that ε is a fixed parameter). For instance, in the case of the real level shift, we have
| 16 |
while for the imaginary shift this is
| 17 |
Note how, in both cases, 1/Δi is recovered for ε = 0. The way in which the amplitudes are affected by the level shift technique is neatly captured by f(Δi; ε), which is plotted in Figure 1 as a function of the energy denominator Δi. As can be seen, f(Δi; ε = 0.2) for the imaginary shift does not contain any pole, while the real one diverges at Δi = −0.2 Eh.
Figure 1.

f(Δi; ε = 0.2) for the real and imaginary level shifts as a function of the energy difference Δi = ϵi – E(0). The gray thick line is the unmodified denominator with the singularity at the origin. The purple line is f(Δi; ε = 0.2) for the real level shift (the dashed vertical line shows the position of the new singularity). The green line is f(Δi; ε = 0.2) for the imaginary level shift.
Finally, note that also, in the case of the imaginary shift, it is possible to reduce the sensitivity of the energy with respect to ε by computing it using the variational expression given in eq 7.
2.4. σp Regularization
An alternative approach to remove the singularity from eq 5 is the σp regularization technique introduced by Head-Gordon and co-workers in the context of (orbital-optimized) second-order Møller–Plesset perturbation theory (MP2).28,29 The theoretical foundation of σp regularization is rooted in the Laplace transform of the energy denominator, which is given by
| 18 |
When Δi = 0, the integrand is a constant function and Ti → ∞. A possibility to avoid such divergence is to change the upper limit of the integral to a value that is directly proportional to the energy difference Δi, such that when Δi = 0 and the integrand is constant, the integral is bound by a finite upper limit. This strategy is implemented by the following expression, where, to accommodate for the possibility for negative denominators, we have introduced the absolute value and sign functions at appropriate places:
| 19 |
In this equation, σ is a non-negative parameter and p is a positive number, which for simplicity we consider to be an integer value. The integration in eq 19 can be carried out analytically, resulting in the σp-regularized amplitude:
| 20 |
For Δi → 0, (1 – e–σ|Δi|p) goes to zero faster than 1/Δi diverges, such that the singularity is effectively suppressed. Similarly to the imaginary shift, the regularized amplitudes depend on the energy denominator appearing in the exponential factor, which in CASPT2 we approximate with the diagonal one as done for the imaginary shift. The integer value p changes the functional form of the regularizer and the way in which the singularity at Δi = 0 is avoided. In this work we consider the values p = 1 and p = 2, as these were found to be the most promising in regularized MP2.29 Interestingly, inserting eq 20 into eq 6 results in the same energy expression as in the driven similarity renormalization group approach.40 The parameter σ is the counterpart of ε in the real and imaginary shifts, and the two can be conveniently related by the following expression (see Section 1.2 in the Supporting Information for more details)
| 21 |
In this way, we can define the regularizer in terms of ε in a manner similar to eqs 16 and 17, that is
| 22 |
The behavior of eq 22 for p = 1 and p = 2 as a function of Δi is shown in Figure 2. In both cases the singularity at the origin is removed. For p = 1 the amplitudes are damped to a finite value, thereby allowing each component of the first-order wave function to contribute to the correlation energy. On the other hand, the case with p = 2 is very similar to the imaginary level shift, where the amplitudes associated with small denominators are completely suppressed. In fact, it is instructive to directly compare f(Δi; ε) for the imaginary shift and the σ2 regularizer, as shown in Figure 3. For matching values of the input parameter, the behavior at Δi → 0 is the same for both techniques. However, the σ2 regularizer follows more closely the unmodified 1/Δi function for larger values of Δi. We thus expect the σ2-regularized energy to be less sensitive to the input parameter ε than the imaginary level shift, which is supported by a Taylor expansion of f(Δi; ε) around Δi → 0 and ε → 0 (see section 1.3 in the Supporting Information for details). Moreover, the energy obtained with σ1-regularized amplitudes is expected to be even less affected by the value of ε, because no contributions to the energy are suppressed; rather they are just damped to a finite value. However, this comes at the price of a discontinuity at Δi = 0. This does not constitute a problem for calculations at a fixed conformation, but a change in the sign of a denominator during a smooth distortion of the molecular geometry will lead to a jump on the potential energy surface. For this reason, the σ1 and σ2 regularizers will likely have different potential applications.
Figure 2.

f(Δi; ε = 0.2) for the σ1 and σ2 regularizers as a function of the energy difference Δi. The light blue line is f(Δi; ε = 0.2) for σ1, while the orange line, for σ2.
Figure 3.

Comparison of f(Δi; ε) for the imaginary level shift and σ2 regularization as a function of the energy difference Δi. A value ε = 0.2 Eh was used for both cases.
2.5. A Note on the IPEA Shift
Another famous shift used in CASPT2 that requires some attention is the IPEA shift.41 While it is technically a shift, its nature is fundamentally different from the techniques discussed in this contribution. The IPEA shift acts on the generalized Fock operator expressed in the molecular orbital basis and was introduced to fix a systematic energy underestimation of open-shell electronic configurations with respect to closed-shell ones. While this has in general a positive effect on the ISP, that is the number of ISPs is generally smaller with the IPEA shift than without, it is not its intended use. The techniques discussed in this work act on the zeroth-order Hamiltonian expressed in the internally contracted basis spanning the FOIS. This directly targets the energies appearing in the denominators of eq 5, whereas the IPEA shift has a more complicated indirect effect on them due to the internal contraction formalism. In other words, the IPEA shift modifies the energies ϵi of eq 5 in nontrivial ways that depend on the generalized Fock eigenvalues. Given this difference, a detailed account of the IPEA shift is omitted from the current work.
3. Results and Discussion
In this section we present a series of calculations to evaluate the effectiveness of σp regularization in solving the intruder-state problem in CASPT2 and compare it to the level shift techniques. We have implemented the σ1 and σ2 regularizers in a development branch of OpenMolcas42,43 and note that they work with any quasi-degenerate variant of CASPT2 currently available in the package. In the following, we shall refer to CASPT2 with σp regularization simply as σp-CASPT2.
3.1. Chromium Dimer
The dissociation of the chromium dimer has proven to be one of the most challenging systems for multireference quantum chemical methods, and it has been used to test a plethora of approaches.9,25,44−55 Due to one 4s and five 3d unpaired electrons in the 7S ground state of each atom, the chromium dimer has a formal sextuple bond, which requires at least an active space of 12 electrons in 12 orbitals for the description of its dissociation. One of the challenges in this molecular system is the large imbalance of the role that dynamic correlation plays at different internuclear distances. Around the equilibrium, there is a significant overlap between the compact 3d orbitals, and the presence of many electrons in such a small space requires an accurate description of short-range correlation effects. This can be achieved by including a large number of Slater determinants in the expansion of the wave function, leading to a sizable contribution of dynamic electron correlation in this region of the potential energy curve (PEC). On the contrary, a crude description of the Coulomb cusp results in a significant overestimation of the repulsion between the electrons, which explains why the CASSCF PEC is strongly repulsive at short internuclear distances (see Figure S1 in the Supporting Information). The situation is qualitatively and quantitatively different during dissociation, where only the more diffuse 4s orbitals overlap, thereby decreasing the importance and contribution of dynamical electron correlation. Another difficulty encountered in the description of the chromium dimer, which is common to transition metal systems, is the presence of a dense manifold of low-lying electronic states. This makes MRPT approaches, and in particular the CASPT2 method, very susceptible to intruder states. As a matter of fact, the Cr2 molecule has been the quintessential test of the intruder-state problem since the early days of CASPT2 and has been used as a representative system to highlight the effectiveness of both the real and imaginary level shift techniques.23,24 In this work we keep this tradition and assess the σp regularizers on the dissociation of Cr2 as well. In particular, we adopt the minimal 12 electrons in 12 orbitals active space in association with the cc-pwCV5Z basis set56 and include scalar relativistic effects through the second-order Douglas–Kroll–Hess Hamiltonian.57,58 The CASPT2 potential energy curve is computed for the lowest singlet state using the modified g1 zeroth-order Hamiltonian,59 which provides a qualitatively correct shape of the PEC. All energies are given relative to twice that of the single atom and no correction for the basis set superposition error was considered, as it does not affect the potential existence of intruder states. The variational energy expression, eq 7, has been used in all cases unless otherwise stated. As a reference, we report the experimental curve of Dattani60 in all of the plots, which predicts a dissociation energy of 1.66 eV. Note that the main focus of these calculations is not to obtain the best possible ab initio results reproducing the experimental data but rather to investigate the effectiveness of the regularizers in suppressing the intruder states.
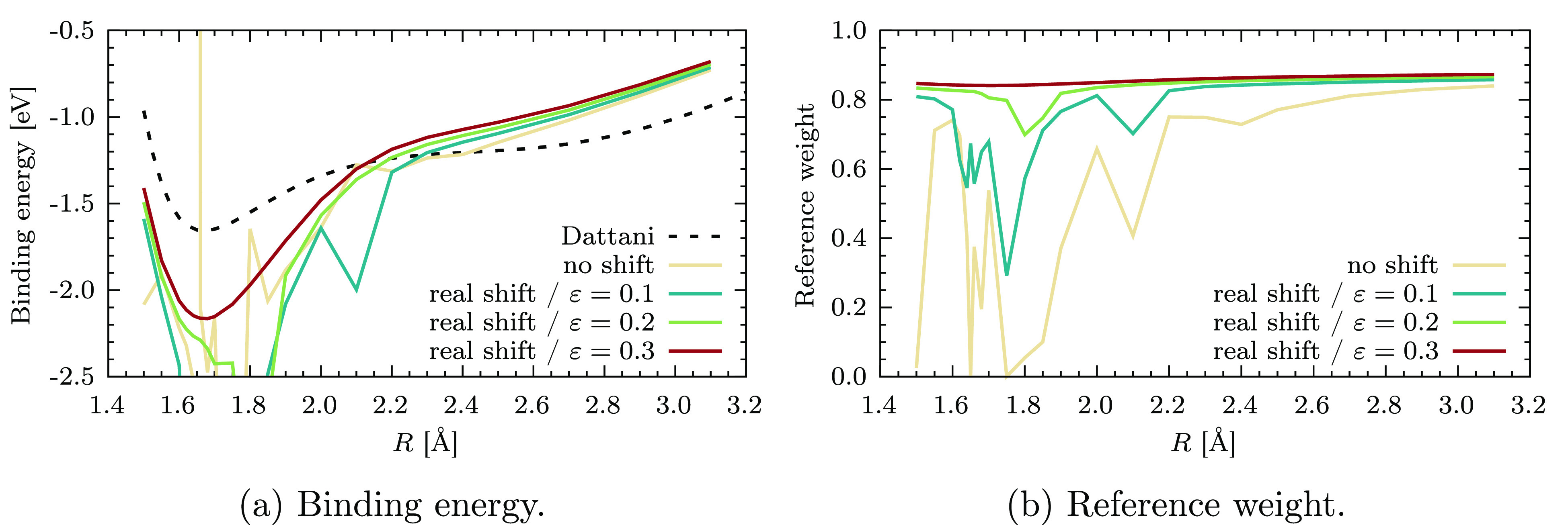
To this end, we start by showing in Figure 4a the CASPT2 PEC obtained with and without the real level shift, for an increasing value of ε. In the unmodified CASPT2 results, there are several places along the curve where the energy is clearly unphysical, and these are all associated with low reference weights, a typical signature of intruder states. This can be clearly seen from Figure 4b, where we plot wref as a function of the internuclear distance R. Due to the presence of negative denominators around Δi ≈ −0.1 Eh, a level shift parameter of ε = 0.1 Eh is not sufficient to avoid all singularities. A value of ε = 0.2 Eh significantly improves the situation, though only at ε = 0.3 Eh the obtained potential energy curve is smooth. For this last case, wref varies only very little as a function of R, meaning that the first-order wave function provides a consistent correction at any point of the dissociation. Repeating the same systematic calculations with σ2-CASPT2 results in a smooth PEC with a much smaller value of ε, as can be seen from Figure 5a. In particular, already at ε = 0.06 Eh the reference weight is approximately constant throughout the dissociation, apart from a minor drop around R = 1.6 Å; see Figure 5b. At ε = 0.1 Eh both the binding energy and wref are extremely smooth functions of the internuclear distance, highlighting the effectiveness of σ2-CASPT2 in removing the intruder states, however, without requiring a large value of the regularization parameter. The curves shown in Figure 5a,b are essentially equivalent to those obtained with the imaginary shift, which we report in the Supporting Information (Figure S2a,b). Additional plots with other values of ε for both approaches confirm the comparable effectiveness of σ2-CASPT2 and the imaginary level shift technique (see Figures S2c–f and S3 in the Supporting Information). We analyze the sensitivity of the results with respect to the input parameter by computing the energy difference ΔE(ε) = E(ε) – Eref throughout the dissociation for increasing values of ε. Considering the results obtained with ε = 0.1 Eh converged (that is, all intruder states are fully removed), we can set Eref = E(ε = 0.1) and compute ΔE(ε = 0.2) and ΔE(ε = 0.3) for both techniques. These energy difference curves are shown in Figure 6. As elucidated by the theoretical discussion in the previous section, the second-order energy with imaginary level shift is more sensitive to the value of ε than the σ2-CASPT2 one. For instance, around R = 1.6 Å, the deviation from Eref is almost double as much for the former approach for both values of ε considered.
Figure 4.
CASPT2 potential energy curves of Cr2 with and without using the real level shift (a) and corresponding reference weight wref (b) as a function of the internuclear distance R.
Figure 5.
σ2-CASPT2 potential energy curves of Cr2 (a) and corresponding reference weights wref (b) as a function of the internuclear distance R.
Figure 6.

Energy differences ΔE(ε) = E(ε) – Eref as a function of the internuclear distance R, for two different values of ε. The reference energy Eref used for each technique was obtained with the value ε = 0.1 Eh.
However, these deviations are less than 1% of the total correlation energy introduced by the second-order correction.
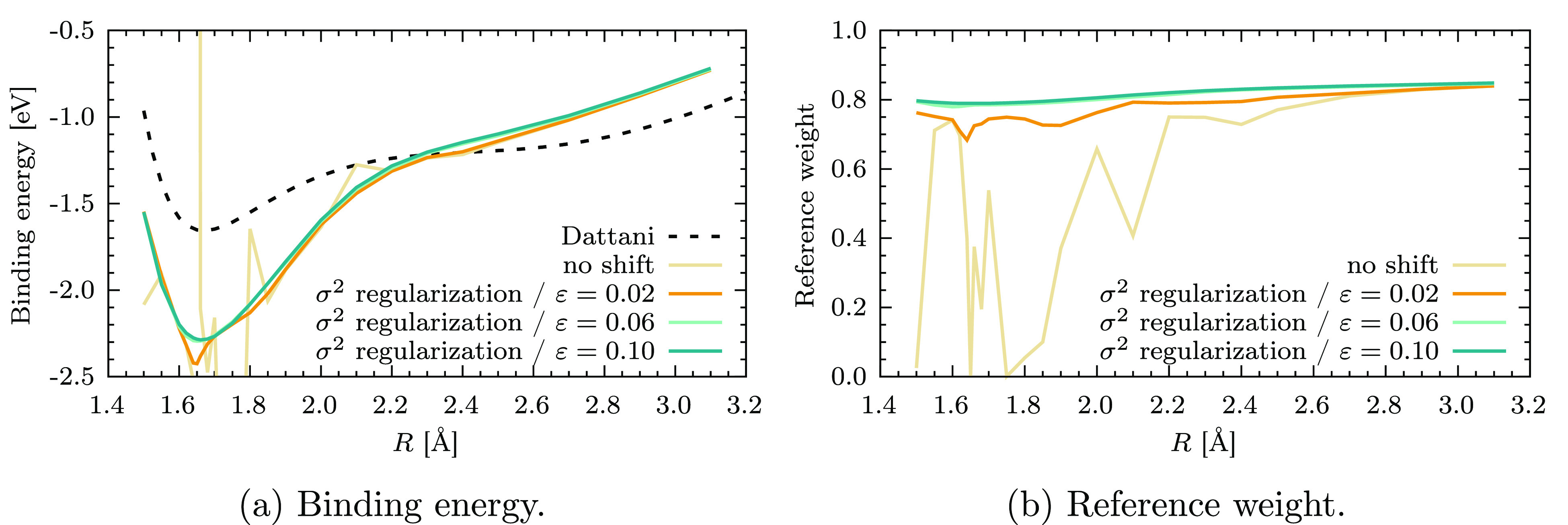
The dissociation of the chromium dimer with σ1-CASPT2 is shown in Figure 7a, and the value of wref as a function of R is reported in Figure 7b. Also, in this case, ε = 0.1 Eh is sufficient to obtain a balanced first-order wave function throughout the dissociation and remove all intruder states. However, a noticeable jump in the energy is present near the equilibrium distance. This discontinuity is due to two perturbers, whose denominators flip sign from R = 1.66 Å to R = 1.68 Å. As a result, a net change of about 0.04 eV in the correlation energy happens between these two points, despite avoiding the singularity at Δi = 0. This is the issue alluded to earlier in the theoretical discussion of the σ1 regularizer, which limits its applicability to situations in which the molecular structure does not change. Note that, despite this potential problem, σ1-CASPT2 effectively removes the ISP and is very insensitive to the value of the regularization parameter ε (see Figures S4 and S5 in the Supporting Information).
Figure 7.
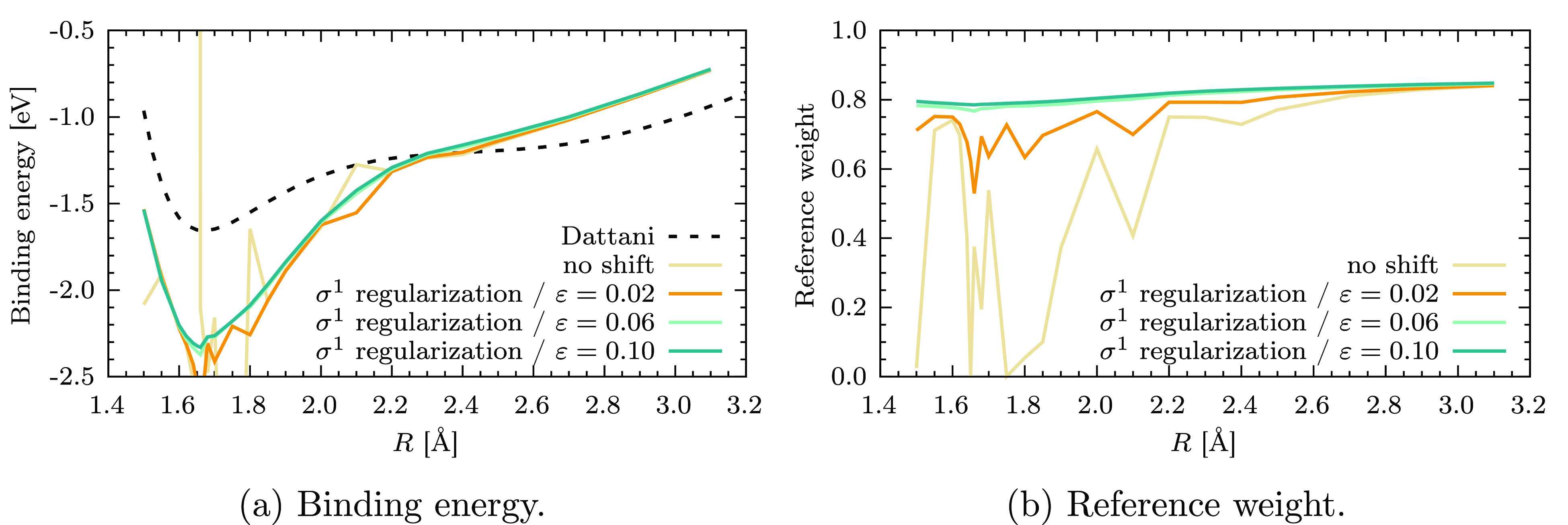
σ1-CASPT2 potential energy curves of Cr2 (a) and corresponding reference weights wref (b) as a function of the internuclear distance R.
Besides showing PECs with the smallest possible value of ε for which the ISPs are removed, we also report in Figure S6 of the Supporting Information the PECs obtained with artificially large values of ε in combination with all intruder-state-removal techniques. This highlights another aspect of these techniques, which is briefly discussed in the Conclusions section.
At last, we shall note that σp-CASPT2 is less dependent on the expression used to obtain the energy, in contrast to the real and imaginary level shifts. For these, the difference between the energy obtained with eq 6 and eq 7—the level shift correction—is about 0.45 eV and 0.2 eV, respectively, for a level shift parameter ε = 0.3 Eh. On the other hand, for σ2-CASPT2 and σ1-CASPT2 this is about 0.08 eV and 0.15 eV, respectively, for the same value of the input parameter ε = 0.3 Eh (see Figures S7–S12 in the Supporting Information).
3.2. Vertical Excitation Energies
The dissociation of the chromium dimer has shown that both the imaginary shift and σ2-CASPT2 are equally effective in removing the intruder-state problem and provide a smooth potential energy curve everywhere. This is the case for the real shift too, albeit with a relatively large value of the shift parameter, which potentially has a negative impact on the overall accuracy. On the other hand, Cr2 was the perfect example to evidence the main shortcoming of σ1-CASPT2—the discontinuity at Δi = 0—which limits its use for this type of applications even though it effectively removed the intruder states. We shall now turn our attention to calculations at a fixed molecular geometry, and in particular to a typical application of the CASPT2 approach: vertical transition energies. In the following, the objective is 2-fold. First, we evaluate the effect of σp regularization and the level shift techniques in cases where no intruder-state problems are present. In particular, we are interested in measuring the sensitivity of the results with respect to the input parameter and quantify to what extent these deviate from the unmodified CASPT2 energies. Ideally, the best technique affects these results the least, as in these situations a modification is not really needed. We call this NOISP analysis. Second, we test the effectiveness of these techniques in fixing the ISP. This case is similar to the chromium dimer dissociation, where removal of singularities was necessary to obtain physical results, though the context is different here, because we consider fixed molecular geometries. To this end, we investigate vertical excitation energies which are affected by intruder-state problems. We call this ISP analysis. To carry out these two analyses, we employ Thiel’s benchmark set61 and systematically investigate a large number of singlet and triplet vertical excitation energies in 28 small organic compounds. The geometries are taken from the original work and correspond to MP2-optimized structures, in combination with the 6-31G*62,63 basis set. The selection of active spaces and the number of states included in the SA-CASSCF optimization follow closely that of the original reference.61 The starting point for both analyses is the calculation of 311 singlet and triplet excitation energies using the MS-CASPT2 method64 (and its diagonal counterpart, MS-CASPT2D), in association with the TZVP basis set65 and the atomic compact Cholesky decomposition of the two-electron integrals66 (using the default threshold value of 10–4Eh). Here, the IPEA shift41 was set to zero and the unmodified generalized Fock operator was used (i.e., no g1 zeroth-order Hamiltonian59). First, we performed these calculations without any shift or regularization, which resulted in 117 states with a reference weight deviating by more than 10% from the ground-state one (109 for MS-CASPT2D). These were considered affected by intruder states and hence excluded from the NOISP analysis. The MS-CASPT2 transition energies of the remaining 194 (202 for MS-CASPT2D) states form the set of reference values for the NOISP analysis, as they do not require any shift or regularization. The full set of 311 states is instead used in the ISP analysis. Please refer to Section S3.1 of the Supporting Information for the detailed account of which states are included in which analysis. In the NOISP analysis, we compare the results obtained with an increasing value of ε to the reference values (ε = 0). This provides a quantitative measure of the sensitivity of the excitation energies with respect to ε. Instead, in the ISP analysis we track the distribution of the reference weights as a function of ε. In particular, for each molecule we compute the relative deviation of the excited states wref (wrefes) with respect to the ground-state one (wref); that is,
| 23 |
(this is the same method used to identify the set of states affected by ISPs). An effective method fixing intruder-state problems should provide a distribution of Δwref that quickly becomes narrow and peaked around zero for increasing values of ε. Instead of plotting the distributions directly, we compute the cumulative distribution function (CDF), which neatly captures how effective the different techniques are. For a given value of Δwref, the CDF essentially counts how many states have a reference weight deviation lower than Δwref in proportion to the total number of states. Mathematically, this can be described as
| 24 |
where p(xi) is the (empirical) probability that there is a reference weight deviation xi in the data set. The signature of an effective intruder-state-removal technique is a CDF function that quickly grows to 1.
From a theoretical perspective, we have seen in the previous section that the imaginary shift and σp regularization depend on the spectrum of the zeroth-order Hamiltonian. For a diagonal zeroth-order Hamiltonian, the use of any of these techniques strictly guarantees that intruder states are progressively removed by increasing the value of the input parameter. This is not the case for CASPT2, where the off-diagonal couplings in Ĥ(0) could accidentally introduce intruder states. To provide an unbiased comparison between the shifts and the regularizers, we perform the NOISP and ISP analyses using the CASPT2D approach first. Nonetheless, we repeat the analyses using CASPT2 as well, as this is the method typically used in practice.
The root-mean-square and maximum absolute deviations (RMSD and MAD, respectively) of all techniques applied to CASPT2D for the NOISP set of energies are shown in Table 1. As it was the case for the chromium dimer, the results for the real level shift highlight its fundamental flaw; that is, the singularity is simply moved at a different place and not really removed. Even though among the 202 states considered here there are no small denominators in the unshifted calculation, increasing the value of ε has catastrophic effects. In a number of cases, negative denominators close to Δi ≈ −0.1 Eh and Δi ≈ −0.2 Eh cause divergences in the first-order wave function and lead to a very large root-mean-square deviation when ε = 0.1 Eh and ε = 0.2 Eh. For instance, of the several hundred thousands of denominators appearing in the wave function expansion of the first 1B1g and 3B1g states of benzoquinone, two of them have a denominator equal to −0.103 Eh and −0.096 Eh, respectively. These did not cause any issue for the reference calculations but led to ISPs for ε = 0.1 Eh. The excitation energy obtained in these two cases is off by more than 8 eV, which is clearly incorrect. It is only with a larger value of ε = 0.3 Eh that no accidental ISPs are introduced by the real level shift. In the remaining lines of Table 1 we see a clear trend for the imaginary shift and the σp regularizers. First, no accidental intruder state appears in any of them, empirically proving the theoretical analysis carried out in the previous section. Importantly, for increasing values of ε, wref monotonically increases (see Figures S13–S15 in the Supporting Information for an example with the second 1B2 state of pyrimidine). Second, for matching values of ε, σp-CASPT2 consistently shows smaller deviations from the reference energies compared to the imaginary level shift. Third, the RMSD and MAD increase at different rates as a function of ε for the three techniques. These results suggest that σp-CASPT2 is less sensitive to the input parameter than CASPT2 with the imaginary shift. In particular, σ1 shows the least deviation, highlighting its potential in this scenario.
Table 1. NOISP Analysis for CASPT2Da.
| technique | ε = 0.1 Eh | ε = 0.2Eh | ε = 0.3Eh |
|---|---|---|---|
| real | 0.916 (9.80) | 3.197 (45.3) | 0.096 (0.39) |
| imaginary | 0.013 (0.07) | 0.029 (0.12) | 0.052 (0.21) |
| σ2-reg | 0.011 (0.07) | 0.021 (0.10) | 0.034 (0.12) |
| σ1-reg | 0.008 (0.04) | 0.017 (0.07) | 0.029 (0.12) |
The values represent root-mean-square deviations (maximum absolute deviations in parentheses) of vertical transition energies obtained with different values of ε > 0 with respect to the reference ones obtained with ε = 0. All values are given in eV.
The situation is slightly different for the ISP analysis. Here we perform the calculation on the entire set that includes 311 excited states and track their reference weight deviations Δwref with respect to the ground-state ones. Recall that, for ε = 0, there are 109 states that have a reference weight deviating by more than 10% from the ground-state one. In Figure 8a we plot the cumulative distribution function of Δwref for ε = 0.1 Eh and ε = 0.3 Eh, respectively (the plot for ε = 0.2 Eh is shown in the Supporting Information, Figure S19). For the unmodified calculation, one-fifth of the excited states obtained with the unmodified CASPT2D method have a Δwref that is larger than 20% (note the CDF function at around 0.8 in the top right part of Figure 8a). Correspondingly, the first series of points shown in the center of Figure 8a—the red pluses—extends beyond the 20% mark (hence, it is not visible in the plot), resulting in a large average deviation. The real level shift improves this situation, producing a significantly more compact distribution of the Δwref and a CDF that grows much faster in the range Δwref < 10%. Nevertheless, several states remain affected by the ISP, which keep the average Δwref relatively large. In contrast, the imaginary shift and σp regularization are much more robust, as evidenced by a CDF curve that reaches the value of 1 for Δwref smaller than 15%. This means that no state of the 311 ones considered has a deviation of the reference weight larger than 15% with respect to the ground-state one. The distributions of the weights are very similar across the three approaches, with the imaginary shift slightly outperforming the regularizers, e.g., displaying a lower mean deviation of about 4% compared to ≈4.5% for σp-CASPT2D. Increasing the value of ε to 0.3 Eh has a dramatic effect for the real level shift, which becomes the most effective way to fix the ISP, as evidenced by the fastest-growing CDF shown in Figure 8b. All intruder states are removed, and the average wref deviation is as low as ≈2%. The imaginary shift also improves significantly, with no deviation of the reference weights above the 10% mark. The smallest difference between ε = 0.1 Eh and ε = 0.3 Eh is obtained with σp regularization. This result is in agreement with the lower sensitivity with respect to the input parameter observed in the NOISP analysis.
Figure 8.
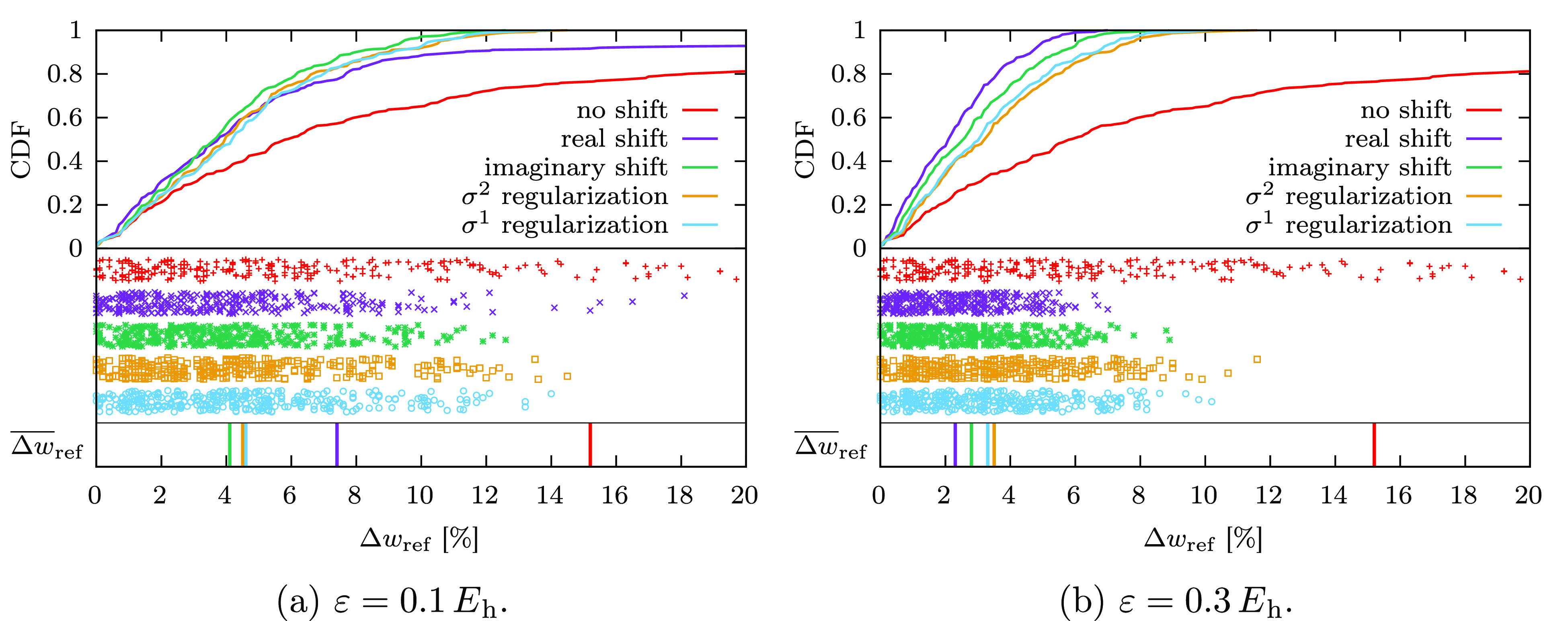
ISP analysis for CASPT2D. The cumulative distribution function is shown in the top half of the plots, while the values Δwref < 20% are shown as points in the center of the plots. In the bottom stripe we show the position of the average reference weight deviation Δwref.
Calculations using CASPT2D served to carry out an unbiased analysis of the various techniques; however, in practice the full CASPT2 approach is the method of choice. Hence, we repeated all of the calculations with the latter, investigating to what extent the off-diagonal couplings in the zeroth-order Hamiltonian affect the results obtained with CASPT2D. In Table 2 we show the RMSD and MAD obtained in the NOISP analysis with the CASPT2 method. The real shift is still plagued by accidental intruder states due to denominators that are close to the negative level shift parameter, and only for ε = 0.3 Eh there are no such cases. For the other three techniques, the results are almost identical to those in Table 1, and thus the same discussion holds. This is the case for the ISP analysis as well, with CASPT2 results virtually equal to CASPT2D ones; hence, we report the plots of the cumulative distribution functions in the Supporting Information (Figures S20–S22). As before, the real shift improves significantly upon increasing ε and shows the most compact distribution compared to the other techniques when ε = 0.3 Eh. The imaginary shift and the σp-CASPT2 differ only slightly and can be considered equally effective. It is interesting to note that, in a few cases, the off-diagonal couplings in the zeroth-order Hamiltonian have led to a decreasingwref for increasing values of ε, as illustrated for the second 1B2 state of pyrimidine in the Supporting Information (Figures S16–S18).
Table 2. NOISP Analysis for CASPT2a.
| technique | ε = 0.1 Eh | ε = 0.2 Eh | ε = 0.3 Eh |
|---|---|---|---|
| real | 0.866 (8.17) | 1.765 (24.4) | 0.095 (0.37) |
| imaginary | 0.013 (0.06) | 0.029 (0.12) | 0.051 (0.21) |
| σ2-reg | 0.010 (0.05) | 0.020 (0.08) | 0.033 (0.12) |
| σ1-reg | 0.008 (0.03) | 0.017 (0.06) | 0.029 (0.12) |
The values represent root-mean-square deviations (maximum absolute deviations in parentheses) of vertical transition energies obtained with different values of ε > 0 with respect to the reference ones obtained with ε = 0. All values are given in eV.
Overall, the NOISP and ISP analyses suggest that the imaginary level shift and σp regularization have a comparable performance. On the one hand, the former is more sensitive to the level shift parameter but appears to be slightly more robust in solving the ISP. On the other hand, σp-CASPT2 provides the smallest deviations from the reference energies in the NOISP analysis, in particular with p = 1. According to this latter result only, it appears that σ1-CASPT2 would be the best choice for calculations at a fixed molecular geometry. However, this comes with the inherent risk that small differences in the structures used (for instance obtained with a different methodology) might significantly affect the results in the unlucky case that one or more denominators change sign. Nevertheless, one can consider that already for ε = 0.1 Eh all ISPs are effectively removed by σp-CASPT2 (and the imaginary shift), even though some states are above the (arbitrary) Δwref = 10% threshold. We can see this by noting, e.g., that the energy differences are not unphysical and agree with values obtained with higher values of ε. After all, the reference weight is one possible measure to identify ISPs, and other criteria may be used as well, such as an analysis in terms of the excitation character of the perturbers with large contributions to the correlation energy. At last, the most striking result is for the real shift. A small value of ε is prone to generate accidental intruder states, but a large value, which is most effective in increasing the reference weight, is associated with the largest RMS deviations in the NOISP analysis. Remarkably, for all methods, no significant difference is observed between the CASPT2D and CASPT2 results.
4. Conclusions
In this work we have implemented the σp-regularization technique in CASPT2. The resulting methodology, σp-CASPT2, compares favorably to previous approaches based on the real and imaginary level shifts in terms of its efficacy in removing the intruder-state problem and the sensitivity of the results with respect to the input parameter. It was found that the two variants considered, σ1-CASPT2 and σ2-CASPT2, are suited for different use-case scenarios. σ1-CASPT2 is the least sensitive approach to the input parameter and effectively removes all intruder states for sufficiently large values of ε. However, due to the discontinuity of the regularized amplitudes at Δi = 0, its application is likely limited to calculations at a fixed molecular geometry, e.g., the determination of vertical transition energies. For calculations involving different molecular geometries, such as the dissociation of diatomic molecules or the exploration of potential energy surfaces, only the imaginary shift and σ2-CASPT2 ensure smooth results. This is because, in these two cases, the regularized amplitudes are a continuous function of the zeroth-order energy denominator. Both approaches remove all singularities equally well; however, σ2-CASPT2 is slightly less sensitive to the input parameter and, hence, our preferred choice. Nonetheless, we should note that the difference between them is probably insignificant compared to the overall accuracy offered by CASPT2 in the first place, such that in practice they both are valid options. Overall, the real level shift is the worst performer and should be avoided, due to its uniform action on all amplitudes. Only large values of the input parameter somewhat ensure no intruder states, but its results are the most sensitive to it, such that they may be considerably different from unshifted CASPT2 ones.
An important aspect related to σp-CASPT2 and the imaginary level shift technique is their dependence on the energy denominator. From a strict theoretical perspective, these techniques are truly intruder-state-free only when used in combination with CASPT2D. In this case, increasing ε always leads to increased reference weights. This is not true for CASPT2, and there are cases in which the off-diagonal couplings of the Fock operator are such that accidental degeneracies appear for ε > 0. The use of the true eigenvalues of the CASPT2 Ĥ(0) remains an open problem, and its solution is rather involved from the computational perspective because it would require the (at least partial) diagonalization of Ĥ(0). On the positive side, the results obtained in this work for excitation energies suggest that this issue is statistically not so relevant.
It is also interesting to note that the level shift technique and σp regularization can be considered from a different point of view than that of intruder-state-removal approaches. In fact, for a given partitioning of the Hamiltonian, a renormalization of the first-order amplitudes can be interpreted as a way to introduce correlation effects from higher-order terms. This has the net impact of decreasing the amount of electron correlation captured by second-order perturbation theory, which is typically overestimated with respect to the infinite-order limit. Within this perspective, one could determine an optimal value for ε by comparing to experiment or high-level computational reference data, essentially defining a new separate methodology that is free of intruder states by design and potentially with a better accuracy. This is for instance the rationale behind regularized MP2 as proposed by Head-Gordon and co-workers,29 and what has been done with the intruder-state-avoidance technique in MRMP2.67 Nevertheless, the simple functional form of the shifts and regularizers cannot compensate the limitations inherent to a second-order perturbation theory framework. For instance, whereas a value of ε = 1.15 Eh for σ2-CASPT2 is such that the PEC of the chromium dimer matches the experimental one around the equilibrium, this comes at the cost of a far worse agreement at longer internuclear distances (see Figure S6 in the Supporting Information). This is because the effect of the regularizer is different at different correlation regimes, and there is no guarantee that a given value of ε is adequate everywhere. In light of this, our take on σp-CASPT2 is for an approach that can be used routinely with a (small) fixed value of ε > 0 which fixes the intruder states, but that provides results as similar as possible to an unmodified version of CASPT2.
At last, we shall note that σp regularization can be used in combination with any variant of CASPT2 and RASPT2 currently implemented in the OpenMolcas package, and that its use does not pose a problem for the implementation of analytic nuclear energy gradients.
Acknowledgments
S.B. acknowledges the Swiss National Science Foundation (SNSF) for the funding received through the Postdoc.Mobility fellowship (Grant No. 199192). R.L. acknowledges the Swedish Research Council (VR, Grant No. 2020-03182). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC). We thank N. Dattani for sharing the Cr2 PEC he deduced from experimental data.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.2c00368.
Additional formal derivations regarding level shifts and regularization; additional results on the chromium dimer; computational details and additional results on the ISP and NOISP analyses (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lischka H.; Nachtigallová D.; Aquino A. J.; Szalay P. G.; Plasser F.; Machado F. B.; Barbatti M. Multireference Approaches for Excited States of Molecules. Chem. Rev. 2018, 118, 7293–7361. 10.1021/acs.chemrev.8b00244. [DOI] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P.-Å.; Roos B. O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. 10.1063/1.462209. [DOI] [Google Scholar]
- Hirao K. Multireference Møller-Plesset method. Chem. Phys. Lett. 1992, 190, 374–380. 10.1016/0009-2614(92)85354-D. [DOI] [Google Scholar]
- Hoffmann M. R. Canonical van Vleck Quasidegenerate Perturbation Theory with Trigonometric Variables. J. Phys. Chem. 1996, 100, 6125–6130. 10.1021/jp952753r. [DOI] [Google Scholar]
- Mahapatra U. S.; Datta B.; Mukherjee D. Development of a size-consistent state-specific multireference perturbation theory with relaxed model-space coefficients. Chem. Phys. Lett. 1999, 299, 42–50. 10.1016/S0009-2614(98)01227-5. [DOI] [PubMed] [Google Scholar]
- Angeli C.; Cimiraglia R.; Evangelisti S.; Leininger T.; Malrieu J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. 10.1063/1.1361246. [DOI] [Google Scholar]
- Fink R. F. The multi-reference retaining the excitation degree perturbation theory: A size-consistent, unitary invariant, and rapidly convergent wavefunction based ab initio approach. Chem. Phys. 2009, 356, 39–46. 10.1016/j.chemphys.2008.10.004. [DOI] [Google Scholar]
- Li C.; Evangelista F. A. Multireference driven similarity renormalization group: A second-order perturbative analysis. J. Chem. Theory Comput. 2015, 11, 2097–2108. 10.1021/acs.jctc.5b00134. [DOI] [PubMed] [Google Scholar]
- Kollmar C.; Sivalingam K.; Neese F. An alternative choice of the zeroth-order Hamiltonian in CASPT2 theory. J. Chem. Phys. 2020, 152, 214110. 10.1063/5.0010019. [DOI] [PubMed] [Google Scholar]
- Battaglia S.; Lindh R. Extended Dynamically Weighted CASPT2: The Best of Two Worlds. J. Chem. Theory Comput. 2020, 16, 1555–1567. 10.1021/acs.jctc.9b01129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia S.; Lindh R. On the role of symmetry in XDW-CASPT2. J. Chem. Phys. 2021, 154, 034102. 10.1063/5.0030944. [DOI] [PubMed] [Google Scholar]
- Song C.; Martínez T. J. Reduced scaling CASPT2 using supporting subspaces and tensor hyper-contraction. J. Chem. Phys. 2018, 149, 044108. 10.1063/1.5037283. [DOI] [PubMed] [Google Scholar]
- Song C.; Martínez T. J. Reduced scaling extended multi-state CASPT2 (XMS-CASPT2) using supporting subspaces and tensor hyper-contraction. J. Chem. Phys. 2020, 152, 234113. 10.1063/5.0007417. [DOI] [PubMed] [Google Scholar]
- Kollmar C.; Sivalingam K.; Guo Y.; Neese F. An efficient implementation of the NEVPT2 and CASPT2 methods avoiding higher-order density matrices. J. Chem. Phys. 2021, 155, 234104. 10.1063/5.0072129. [DOI] [PubMed] [Google Scholar]
- MacLeod M. K.; Shiozaki T. Communication: Automatic code generation enables nuclear gradient computations for fully internally contracted multireference theory. J. Chem. Phys. 2015, 142, 051103. 10.1063/1.4907717. [DOI] [PubMed] [Google Scholar]
- Vlaisavljevich B.; Shiozaki T. Nuclear Energy Gradients for Internally Contracted Complete Active Space Second-Order Perturbation Theory: Multistate Extensions. J. Chem. Theory Comput. 2016, 12, 3781–3787. 10.1021/acs.jctc.6b00572. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Shiozaki T. Analytical Derivative Coupling for Multistate CASPT2 Theory. J. Chem. Theory Comput. 2017, 13, 2561–2570. 10.1021/acs.jctc.7b00018. [DOI] [PubMed] [Google Scholar]
- Park J. W. Single-State Single-Reference and Multistate Multireference Zeroth-Order Hamiltonians in MS-CASPT2 and Conical Intersections. J. Chem. Theory Comput. 2019, 15, 3960–3973. 10.1021/acs.jctc.9b00067. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Al-Saadon R.; Strand N. E.; Shiozaki T. Imaginary Shift in CASPT2 Nuclear Gradient and Derivative Coupling Theory. J. Chem. Theory Comput. 2019, 15, 4088–4098. 10.1021/acs.jctc.9b00368. [DOI] [PubMed] [Google Scholar]
- Song C.; Neaton J. B.; Martínez T. J. Reduced scaling formulation of CASPT2 analytical gradients using the supporting subspace method. J. Chem. Phys. 2021, 154, 014103. 10.1063/5.0035233. [DOI] [PubMed] [Google Scholar]
- Nishimoto Y. Analytic gradients for restricted active space second-order perturbation theory (RASPT2). J. Chem. Phys. 2021, 154, 194103. 10.1063/5.0050074. [DOI] [PubMed] [Google Scholar]
- Nishimoto Y.; Battaglia S.; Lindh R. Analytic First-Order Derivatives of (X)MS, XDW, and RMS Variants of the CASPT2 and RASPT2Methods. J. Chem. Theory Comput. 2022, 18, 4269–4281. 10.1021/acs.jctc.2c00301. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Andersson K. Multiconfigurational perturbation theory with level shift - the Cr2 potential revisited. Chem. Phys. Lett. 1995, 245, 215–223. 10.1016/0009-2614(95)01010-7. [DOI] [Google Scholar]
- Forsberg N.; Malmqvist P.-Å. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196–204. 10.1016/S0009-2614(97)00669-6. [DOI] [Google Scholar]
- Celani P.; Stoll H.; Werner H.; Knowles P. J. The CIPT2 method: Coupling of multi-reference configuration interaction and multi-reference perturbation theory. Application to the chromium dimer. Mol. Phys. 2004, 102, 2369–2379. 10.1080/00268970412331317788. [DOI] [Google Scholar]
- Shiozaki T.; Györffy W.; Celani P.; Werner H.-J. Communication: Extended multi-state complete active space second-order perturbation theory: Energy and nuclear gradients. J. Chem. Phys. 2011, 135, 081106. 10.1063/1.3633329. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Al-Saadon R.; MacLeod M. K.; Shiozaki T.; Vlaisavljevich B. Multireference Electron Correlation Methods: Journeys along Potential Energy Surfaces. Chem. Rev. 2020, 120, 5878–5909. 10.1021/acs.chemrev.9b00496. [DOI] [PubMed] [Google Scholar]
- Lee J.; Head-Gordon M. Regularized Orbital-Optimized Second-Order Møller-Plesset Perturbation Theory: A Reliable Fifth-Order-Scaling Electron Correlation Model with Orbital Energy Dependent Regularizers. J. Chem. Theory Comput. 2018, 14, 5203–5219. 10.1021/acs.jctc.8b00731. [DOI] [PubMed] [Google Scholar]
- Shee J.; Loipersberger M.; Rettig A.; Lee J.; Head-Gordon M. Regularized Second-Order Møller-Plesset Theory: A More Accurate Alternative to Conventional MP2 for Noncovalent Interactions and Transition Metal Thermochemistry for the Same Computational Cost. J. Phys. Chem. Lett. 2021, 12, 12084–12097. 10.1021/acs.jpclett.1c03468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P.-Å.; Roos B. O.; Sadlej A. J.; Wolinski K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. 10.1021/j100377a012. [DOI] [Google Scholar]
- Hylleraas E. A. Über den Grundterm der Zweielektronenprobleme von H–, He, Li+, Be+2 usw. Zeitschrift für Physik 1930, 65, 209–225. 10.1007/BF01397032. [DOI] [Google Scholar]
- Dyall K. G. The choice of a zeroth-order Hamiltonian for second-order perturbation theory with a complete active space self-consistent-field reference function. J. Chem. Phys. 1995, 102, 4909–4918. 10.1063/1.469539. [DOI] [Google Scholar]
- Angeli C.; Cimiraglia R.; Malrieu J.-P. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 117, 9138–9153. 10.1063/1.1515317. [DOI] [Google Scholar]
- Angeli C.; Pastore M.; Cimiraglia R. New perspectives in multireference perturbation theory: The n-electron valence state approach. Theor. Chem. Acc. 2007, 117, 743–754. 10.1007/s00214-006-0207-0. [DOI] [Google Scholar]
- Olsen J.; Jørgensen P. Convergence patterns and rates in two-state perturbation expansions. J. Chem. Phys. 2019, 151, 084108. 10.1063/1.5110554. [DOI] [PubMed] [Google Scholar]
- Camacho C.; Yamamoto S.; Witek H. A. Choosing a proper complete active space in calculations for transition metal dimers: ground state of Mn2 revisited. Phys. Chem. Chem. Phys. 2008, 10, 5128. 10.1039/b805125a. [DOI] [PubMed] [Google Scholar]
- Camacho C.; Witek H. A.; Yamamoto S. Intruder states in multireference perturbation theory: The ground state of manganese dimer. J. Comput. Chem. 2009, 30, 468–478. 10.1002/jcc.21074. [DOI] [PubMed] [Google Scholar]
- Witek H. A.; Choe Y.-K.; Finley J. P.; Hirao K. Intruder state avoidance multireference Møller-Plesset perturbation theory. J. Comput. Chem. 2002, 23, 957–965. 10.1002/jcc.10098. [DOI] [PubMed] [Google Scholar]
- Taube A. G.; Bartlett R. J. Rethinking linearized coupled-cluster theory. J. Chem. Phys. 2009, 130, 144112. 10.1063/1.3115467. [DOI] [PubMed] [Google Scholar]
- Evangelista F. A. A driven similarity renormalization group approach to quantum many-body problems. J. Chem. Phys. 2014, 141, 054109. 10.1063/1.4890660. [DOI] [PubMed] [Google Scholar]
- Ghigo G.; Roos B. O.; Malmqvist P.-Å. A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem. Phys. Lett. 2004, 396, 142–149. 10.1016/j.cplett.2004.08.032. [DOI] [Google Scholar]
- Fdez. Galván I.; Vacher M.; Alavi A.; Angeli C.; Aquilante F.; Autschbach J.; Bao J. J.; Bokarev S. I.; Bogdanov N. A.; Carlson R. K.; Chibotaru L. F.; Creutzberg J.; Dattani N.; Delcey M. G.; Dong S. S.; Dreuw A.; Freitag L.; Frutos L. M.; Gagliardi L.; Gendron F.; Giussani A.; González L.; Grell G.; Guo M.; Hoyer C. E.; Johansson M.; Keller S.; Knecht S.; Kovačević G.; Källman E.; Li Manni G.; Lundberg M.; Ma Y.; Mai S.; Malhado J. P.; Malmqvist P.-Å.; Marquetand P.; Mewes S. A.; Norell J.; Olivucci M.; Oppel M.; Phung Q. M.; Pierloot K.; Plasser F.; Reiher M.; Sand A. M.; Schapiro I.; Sharma P.; Stein C. J.; Sørensen L. K.; Truhlar D. G.; Ugandi M.; Ungur L.; Valentini A.; Vancoillie S.; Veryazov V.; Weser O.; Wesołowski T. A.; Widmark P.-O.; Wouters S.; Zech A.; Zobel J. P.; Lindh R. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. 10.1021/acs.jctc.9b00532. [DOI] [PubMed] [Google Scholar]
- Aquilante F.; Autschbach J.; Baiardi A.; Battaglia S.; Borin V. A.; Chibotaru L. F.; Conti I.; De Vico L.; Delcey M.; Fdez. Galván I.; Ferré N.; Freitag L.; Garavelli M.; Gong X.; Knecht S.; Larsson E. D.; Lindh R.; Lundberg M.; Malmqvist P.-Å.; Nenov A.; Norell J.; Odelius M.; Olivucci M.; Pedersen T. B.; Pedraza-González L.; Phung Q. M.; Pierloot K.; Reiher M.; Schapiro I.; Segarra-Martí J.; Segatta F.; Seijo L.; Sen S.; Sergentu D. C.; Stein C. J.; Ungur L.; Vacher M.; Valentini A.; Veryazov V. Modern quantum chemistry with [Open]Molcas. J. Chem. Phys. 2020, 152, 214117. 10.1063/5.0004835. [DOI] [PubMed] [Google Scholar]
- Dachsel H.; Harrison R. J.; Dixon D. A. Multireference Configuration Interaction Calculations on Cr2: Passing the One Billion Limit in MRCI/MRACPF Calculations. J. Phys. Chem. A 1999, 103, 152–155. 10.1021/jp982648s. [DOI] [Google Scholar]
- Roos B. O. The Ground State Potential for the Chromium Dimer Revisited. Collect. Czechoslov. Chem. Commun. 2003, 68, 265–274. 10.1135/cccc20030265. [DOI] [Google Scholar]
- Angeli C.; Bories B.; Cavallini A.; Cimiraglia R. Third-order multireference perturbation theory: The n-electron valence state perturbation-theory approach. J. Chem. Phys. 2006, 124, 054108. 10.1063/1.2148946. [DOI] [PubMed] [Google Scholar]
- Müller T. Large-Scale Parallel Uncontracted Multireference-Averaged Quadratic Coupled Cluster: The Ground State of the Chromium Dimer Revisited. J. Phys. Chem. A 2009, 113, 12729–12740. 10.1021/jp905254u. [DOI] [PubMed] [Google Scholar]
- Ruipérez F.; Aquilante F.; Ugalde J. M.; Infante I. Complete vs Restricted Active Space Perturbation Theory Calculation of the Cr2 Potential Energy Surface. J. Chem. Theory Comput. 2011, 7, 1640–1646. 10.1021/ct200048z. [DOI] [PubMed] [Google Scholar]
- Kurashige Y.; Yanai T. Second-order perturbation theory with a density matrix renormalization group self-consistent field reference function: Theory and application to the study of chromium dimer. J. Chem. Phys. 2011, 135, 094104. 10.1063/1.3629454. [DOI] [PubMed] [Google Scholar]
- Purwanto W.; Zhang S.; Krakauer H. An auxiliary-field quantum Monte Carlo study of the chromium dimer. J. Chem. Phys. 2015, 142, 064302. 10.1063/1.4906829. [DOI] [PubMed] [Google Scholar]
- Vancoillie S.; Malmqvist P.-Å.; Veryazov V. Potential Energy Surface of the Chromium Dimer Re-re-revisited with Multiconfigurational Perturbation Theory. J. Chem. Theory Comput. 2016, 12, 1647–1655. 10.1021/acs.jctc.6b00034. [DOI] [PubMed] [Google Scholar]
- Sokolov A. Y.; Chan G. K.-L. A time-dependent formulation of multi-reference perturbation theory. J. Chem. Phys. 2016, 144, 064102. 10.1063/1.4941606. [DOI] [PubMed] [Google Scholar]
- Guo S.; Watson M. A.; Hu W.; Sun Q.; Chan G. K.-L. N-Electron Valence State Perturbation Theory Based on a Density Matrix Renormalization Group Reference Function, with Applications to the Chromium Dimer and a Trimer Model of Poly(p-Phenylenevinylene). J. Chem. Theory Comput. 2016, 12, 1583–1591. 10.1021/acs.jctc.5b01225. [DOI] [PubMed] [Google Scholar]
- Li J.; Yao Y.; Holmes A. A.; Otten M.; Sun Q.; Sharma S.; Umrigar C. J. Accurate many-body electronic structure near the basis set limit: Application to the chromium dimer. Phys. Rev. Res. 2020, 2, 012015. 10.1103/PhysRevResearch.2.012015. [DOI] [Google Scholar]
- Larsson H. R.; Zhai H.; Gunst K.; Chan G. K.-L. Matrix Product States with Large Sites. J. Chem. Theory Comput. 2022, 18, 749–762. 10.1021/acs.jctc.1c00957. [DOI] [PubMed] [Google Scholar]
- Balabanov N. B.; Peterson K. A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc-Zn. J. Chem. Phys. 2005, 123, 064107. 10.1063/1.1998907. [DOI] [PubMed] [Google Scholar]
- Douglas M.; Kroll N. M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. (N. Y.) 1974, 82, 89–155. 10.1016/0003-4916(74)90333-9. [DOI] [Google Scholar]
- Hess B. A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742–3748. 10.1103/PhysRevA.33.3742. [DOI] [PubMed] [Google Scholar]
- Andersson K. Different forms of the zeroth-order Hamiltonian in second-order perturbation theory with a complete active space self-consistent field reference function. Theor. Chim. Acta 1995, 91, 31–46. 10.1007/BF01113860. [DOI] [Google Scholar]
- Dattani N. S.; Li Manni G.; Tomza M.. An improved empirical potential for the highly multi-reference sextuply bonded transition metal benchamrk molecule Cr2. 2016 International Symposium on Molecular Spectroscopy,2016. http://hdl.handle.net/2142/91417.
- Schreiber M.; Silva-Junior M. R.; Sauer S. P. A.; Thiel W. Benchmarks for electronically excited states: CASPT2, CC2, CCSD, and CC3. J. Chem. Phys. 2008, 128, 134110. 10.1063/1.2889385. [DOI] [PubMed] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Finley J.; Malmqvist P.-Å.; Roos B. O.; Serrano-Andrés L. The multi-state CASPT2 method. Chem. Phys. Lett. 1998, 288, 299–306. 10.1016/S0009-2614(98)00252-8. [DOI] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Aquilante F.; Lindh R.; Bondo Pedersen T. Unbiased auxiliary basis sets for accurate two-electron integral approximations. J. Chem. Phys. 2007, 127, 114107. 10.1063/1.2777146. [DOI] [PubMed] [Google Scholar]
- Chang S.-W.; Witek H. A. Choice of Optimal Shift Parameter for the Intruder State Removal Techniques in Multireference Perturbation Theory. J. Chem. Theory Comput. 2012, 8, 4053–4061. 10.1021/ct2006924. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.