

Abstract
Genetic modification of the embryo or germline of nonhuman primates is envisioned as a method to develop improved models of human disease, yet the promise of such animal models remains unfulfilled. Here, we discuss current methods and their limitations for producing nonhuman primate genetic models that faithfully genocopy and phenocopy human disease. We reflect on how to ethically maximize the translational relevance of such models in the search for new therapeutic strategies to treat human disease.
Single Sentence Summary:
Here, we discuss current methods to introduce genetic mutations into NHP embryos and reflect on the advances, limitations and translational relevance of such studies.
Introduction
Advances in medical research have improved and extended human life, yet many diseases are still waiting for a cure. There are many reasons for why treatments are lost in translation, including lack of reproducibility among studies which diminishes trust in laboratory results and investment in emerging technologies (1). The mapping of the human genome (https://www.genome.gov/human-genome-project) has accelerated the identification of disease-associated genes. According to the Online Mendelian Inheritance in Man catalogue (https://www.omim.org/statistics/geneMap) 4,211 single gene disorders have been identified (as of 7 February 2022). Interestingly, individual mutations are infrequent even in relatively common diseases, such as the leucine-rich repeat kinase 2 (LRRK2) G2019S mutation in Parkinson’s disease (PD) (Fig. 1).
Figure 1. Prevalence of the PD-associated LRRK2 G2019S mutation.
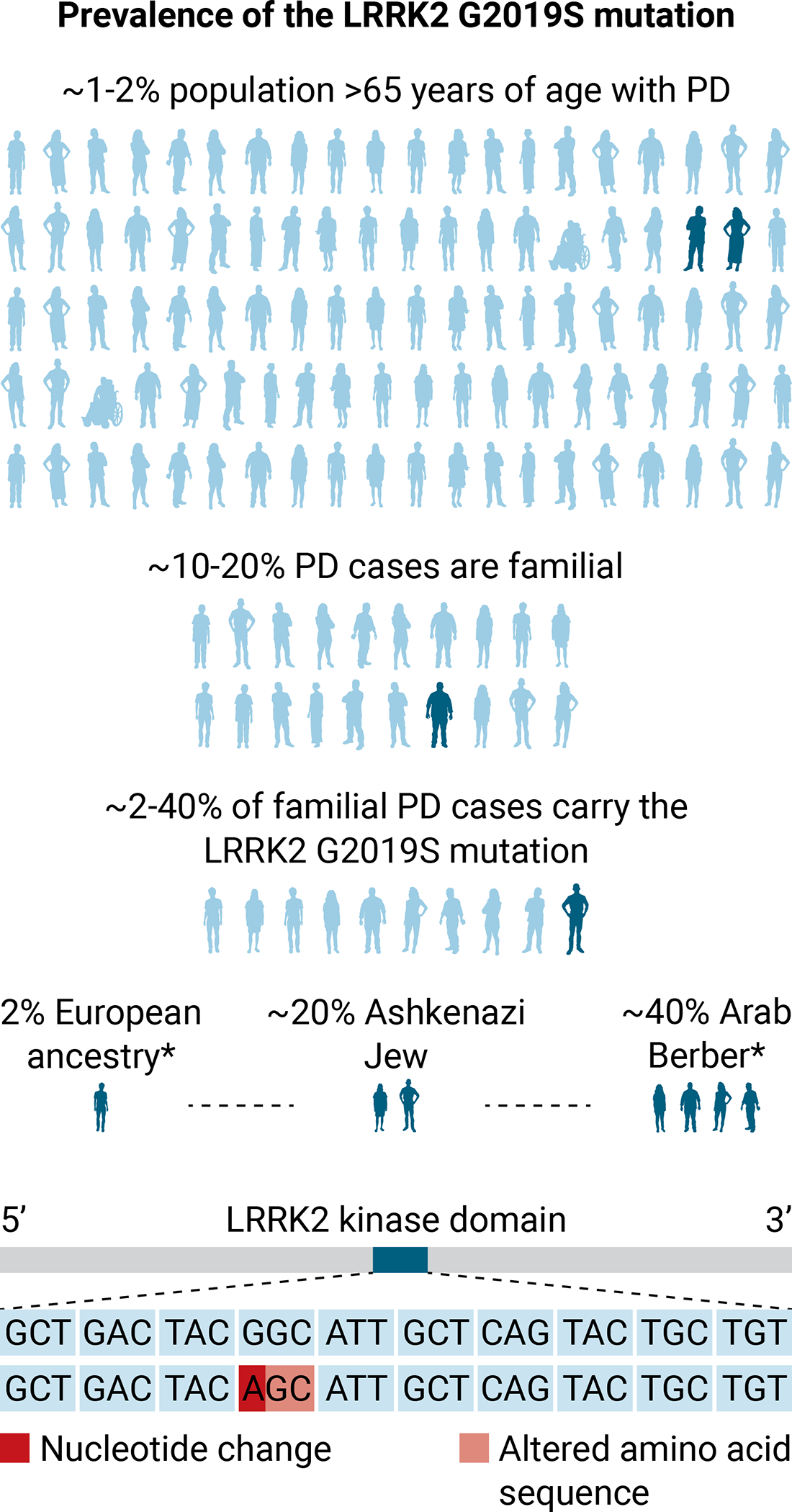
The prevalence of PD in the global population over the age of 65 is 1 to 2%. The G6055A point mutation in the LRRK2 gene results in a change from glutathione to serine at position 2019 (G2019S) in the LRRK2 kinase domain (70). The LRRK2 G2019S mutation is the most frequently found mutation in PD and is associated with both familial and sporadic diseases. Of familial PD cases, 2 to 40% carry the LRRK2 G2019S mutation (2% European ancestry, 20% Ashkenazi Jew, 40% Berber). The genetics and motor behavior of marmosets make this NHP species well suited to model PD caused by the LRRK2 G2019S mutation. Source* (70).
Research using nonhuman primates (NHPs) plays a critical role toward understanding mechanisms of human disease and bringing effective and safe therapies into the clinic. NHPs are our closest relatives according to phylogenetics, anatomy and behavior and have proven critical for translational studies. For example, studies in NHPs helped to decipher the brain’s basal ganglia neural network that led to the development of deep brain stimulation for treating patients with PD who respond to drug treatment but have motor complications (2). NHP studies also prevented a clinical trial for PD when preneoplasia pancreatic lesions were found during the testing of a “Trojan horse technology” based on a monoclonal antibody against the human insulin receptor for trophic factor delivery to the brain (3). Mapping of NHP genomes has highlighted similarities and differences among model species for disease-related research (4). It helped unravel the genetic basis and biological mechanisms of vision and ocular diseases in macaques that resulted in gene therapies for treating inherited and age-related retinal disorders (5). Efforts to identify spontaneous gene mutations in NHPs that are associated with human diseases are currently ongoing [e.g.: (6)], yet this approach is limited by the low incidence of spontaneous mutations in NHPs. Embryonic and germline genome modification has been proposed as a more effective way to create faithful NHP models that genocopy and phenocopy human disease, although its promise remains unfulfilled.
Technologies for generating mutations can be broadly divided into transgenic and genome editing methods. Transgenesis refers to the insertion of a gene or DNA sequence into the genome and is typically achieved by delivery of a viral vector encoding the sequence of interest. Genome editing is the process by which genetic material is added (knocked-in), removed (knocked-out), or altered (point mutation) at a target site in the genome. Zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced palindromic repeats (CRISPR) combined with CRISPR associated protein 9 (Cas9) are molecular methods currently available for genome editing (Fig. 2).
Figure 2. Genome editing of the NHP LRRK2 gene.
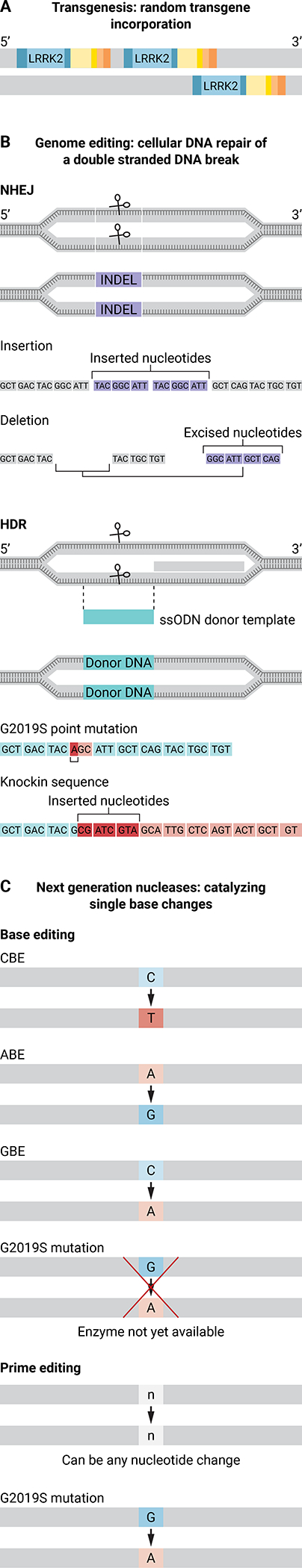
Transgenesis or genome editing using CRISPR-Cas could be used to introduce the G6055A point mutation into the NHP LRRK2 gene. The G6055A point mutation results in a glutathione to serine switch at position 2019 in the LRRK2 kinase domain and is the most common PD-associated mutation. (A) Random incorporation of a mutant LRRK2 transgene into the NHP genome by transgenesis. (B) Genome editing using CRISPR-Cas results in a double-stranded DNA break in the NHP LRRK2 gene that then undergoes cellular DNA repair. DNA repair occurs through one of two processes: non-homologous end joining (NHEJ), which creates insertions or deletions (INDELs) in the LRRK2 gene, or homology directed repair (HDR), which uses a single-stranded oligodeoxynucleotide donor template (ssODN) to introduce a specific nucleotide sequence change. (C) Next-generation nucleases are modified versions of Cas9 that create precise edits in the target gene sequence. Base editor nucleases create a C-to-T nucleotide change [cytosine base editors (CBE)], an A-to-G nucleotide change [adenosine base editors (ABE)], or a C-to-G nucleotide change [guanosine base editors (GBE)]. There is currently no base editor that can introduce a G-A conversion to create the G6055A point mutation in the LRRK2 gene. Prime editors nucleases are able to create any nucleotide change (represented as n-n).
Theoretically, exact genetic modifications can be introduced into NHP gametes or embryos in vitro and upon successful embryo transfer to a female NHP recipient can produce a genetically modified offspring that genocopies and phenocopies the human disease condition. NHP species vary in their reproductive traits, which affects the methods of assisted reproduction needed for generating these animal models of human disease (Table 1). Compared to rodents that have short gestational length (~21 days), large litter sizes (~6 to 10 pups) and multiple pregnancies per year, rhesus or cynomolgus macaques (Old World monkeys) produce one offspring annually after a pregnancy of ~165 days. Across NHPs, common marmosets (New World monkeys) are often viewed as a better choice for genetic modification based on their smaller size, shorter lifespan, shorter time to puberty and ability to produce twins or triplets either annually or biannually. However, the application of assisted reproductive technologies to macaques yields more consistent results across and within research groups compared to marmosets. Differences in genetic, physiological, anatomical and behavioral characteristics define the choice of NHP species. For example, high quality brain imaging is easier in rhesus macaques using standard equipment and techniques, whereas study of vocalizations and social dynamics can be more readily evaluated in common marmosets.
Table 1.
Comparison of reproductive and colony/experimental traits of nonhuman primate species. IVF, in vitro fertilization; ICSI, intracytoplasmic sperm injection; ET, embryo transfer; AI, artificial insemination
| Marmoset (Callithrix jacchus) | African green monkeys/Vervets (Clorocebus aethiops) | Capuchin (Cebus apella) | Squirrel monkey (Saimiri sciureus) | Cynomolgus macaque (Macaca fascicularis) | Rhesus macaque (Macaca mulatta) | Pigtail macaque (Macaca nemestrina) | Olive baboon (Papio anubis) | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Reproductive Traits | ||||||||
|
| ||||||||
| Sexual maturity (years) | 1.5–1.7 | 2.5–4.0 | 4.0 | 2.5–5.0 | 4.0–6.0 | 4.0–5.0 | 3.0 | 4.0–6.0 |
|
| ||||||||
| Female cycle | ovarian | menstrual | menstrual | ovarian | menstrual | menstrual | menstrual | ovarian |
|
| ||||||||
| Breeding | year round | seasonal | year round | seasonal | year round | seasonal | year round | year round |
|
| ||||||||
| Ovulation (days) | 28–30 | 33 | 20.8 | 18–20 | 26–30 | 29 | 32 | 37.5 |
|
| ||||||||
| Gestation (months) | 4.5 | 5.5 | 5.0 | 5.5 | 5.5 | 5.5 | 5.7 | 6.0 |
|
| ||||||||
| Applied reproductive technologies | IVF, ICSI, ET (92–94) | IVF, ICSI, ET (95) | IVF, ICSI (95) | IVF, attempted ET, AI (94) | IVF, ICSI, ET, AI (88,94,97) | IVF, ICSI, ET, AI (90,94,97) | IVF, attempted ET, AI (94, 97) | IVF, attempted |
|
| ||||||||
| Colony/Experimental Traits | ||||||||
|
| ||||||||
| Housing | paired or family group | single, paired or group | single, paired, or group | single, paired, or group | single, paired, or group | single, paired, or group | single or paired | single or group |
|
| ||||||||
| average lifespan (years) | 7–8 | 20 | 25 | 20 | 25–30 | 35 | 26 | 30 |
|
| ||||||||
| Reproductive senescence (years) | >8 | >12 | >18 | >10 | >20 | >20 | >20 | > 20 |
|
| ||||||||
| Weight: male | 0.30–0.50 kg | 4.0–8.0 kg | 1.7–7.36 kg | 0.60–1.25 kg | 3.9–11.4 kg | 5.7–16.8 kg | 6.0–14.0 kg | 23.0–29.0 kg |
| female | 0.30–0.50 kg | 3.0–5.0 kg | 1.8–3.99 kg | 0.60–0.90 kg | 3.0–6.1 kg | 5.0–15.6 kg | 4.5–11.0 kg | 14.0–17.0 kg |
Inefficiencies in assisted reproductive methods, coupled with a high degree of variability in response to ovarian stimulation and restrictions on the number of oocyte retrievals to be performed per animal, necessitate abundant resources to generate live NHP offspring. Many NHP studies report the number of oocytes used rather than the number of oocyte donors that underwent ovarian stimulation, including those that failed to respond to hormonal treatment, thus it is difficult to assess and estimate the true number of females needed for these studies. The need for substantial animal resources in turn affects the reproducibility of the approach and its outcome, a cornerstone of biomedical research. Notably, the efficiency of transferring in vitro produced embryos is very low in NHPs, often resulting in miscarriage or stillbirth and the birth of few live offspring.
Here, we provide an overview of current methods to introduce genetic mutations into NHP embryos and reflect on the limitations and translational relevance of such studies. Scientific and ethical considerations regarding choice of disease model, experimental design, ability to genocopy and phenocopy the human disease and emerging technologies are included to stimulate dialogue on how to prioritize the use of limited animal and human resources. Furthermore, the role of the stakeholders including scientists, veterinarians, funders, and institutions, for selecting the human disease to be modeled is emphasized. The disease to be modeled should be highly impactful to human health and physiologically feasible in NHPs. The stakeholders are also ultimately responsible for recognizing disease outcomes and providing humane treatment of NHPs in response to debilitating phenotypes.
Transgenesis in NHPs: proof of principle
Transgenic NHPs have been generated by microinjection of retroviral vectors into oocytes or embryos for transgene incorporation into host DNA (7). The vector design depends on the transgene of interest and the scientific question driving the research. Commonly used retroviral vectors include gammaretroviruses (e.g. murine Maloney leukemia virus, MMLV) and lentiviruses (e.g. human immunodeficiency virus, HIV) (7). Ubiquitous promoters like human ubiquitin C (UbC), cytomegalovirus (CMV), elongation factor-1 alpha (EF1-α) and chicken ß-actin with a CMV early enhancer (CAG) can drive gene expression in most cell types throughout the body (7). However, heterogeneous distribution across tissues has been detected in transgenic cynomolgus macaques (8). Cell-type-specific promoters (e.g. neural-specific synapsin) attain a restricted transgene expression pattern and confer cell directed phenotypes. The maximum insert size of the promoter and gene sequence into a lentiviral vector is <10 kb (7), and <5 kb has performed optimally for NHP germline transgenesis (9). Larger insert sizes are proportional to decreased viral titers (10), thereby reducing the efficiency of inserting larger DNA sequences.
In proof-of-principle studies, the reporter gene enhanced green fluorescent protein (eGFP) was introduced by transgenesis into macaques and marmosets. A pioneering study by Chan et al. (11) described the delivery of a MMLV vector carrying eGFP into rhesus oocytes. Oocyte microinjection before fertilization resulted in transgene expression in all three germ layers with transgene incorporation and expression in one live male and miscarried twins. In comparison, microinjection into embryos, e.g., into the morula or the blastocoel cavity, restricted transgene expression to the placenta as shown in rhesus macaques by Wolfgang et al. (12) and in marmosets by Sasaki et al. (13). Niu and colleagues (14) reported that viral vector delivery to one- to eight-cell stage rhesus macaque embryos yielded between 33 and 50% transgenic offspring. In contrast, Seita et al. (15) reported that viral vector delivery to cynomolgus macaque oocytes before fertilization resulted in 100% transgenic offspring (two live births). Relatively few offspring (three to eight per study; one to four live births) were generated in each of these preliminary eGFP transgenic studies upon transfer of 22 to 40 embryos to female NHP recipients, resulting in three to seven pregnancies.
Using Transgenesis To Model Human Disease in NHPs
As transgenesis through viral vector microinjection often introduces multiple copies of a gene or nucleotide sequence, this method may be well-suited for NHP modeling of human diseases associated with gene or sequence multiplication. PolyQ diseases, including Huntington’s Disease (HD) and Spinocerebellar ataxia, are caused by an expansion of trinucleotide CAG repeats (Table 2)(16). Yang et al. (17) obtained transgenic HD rhesus macaques by microinjection of oocytes with a lentiviral vector containing exon 1 of the human Huntingtin (HTT) gene with 84 CAG repeats (HTT-84) under the polyubiquitin-C promoter. A transgenic animal with a normal range of CAG repeats (<35) (18) was apparently healthy. Four live transgenic rhesus macaques with expansion of CAG repeats displayed HD-like motor symptoms (e.g. dystonia, chorea) and three of them died early, most likely due to the higher number of CAG repeats (~69 to 88) and mutant HTT copy number. Tomioka et al. (19) introduced a lentiviral vector containing 120 CAG repeats in ataxin 3 (the gene mutated in spinocerebellar ataxia) into marmoset four-cell embryos to morula stage embryos. A high spontaneous abortion rate was observed with miscarried fetuses having 3.7 to 12.9-fold higher transgene expression. Three live transgenic marmosets with two to three transgene integration sites had decreased body weight at birth and developed neurological symptoms at 3 to 4 months of age.
Table 2.
Reported transgenic and genome-edited NHP models of human disease associated with specific gene mutations. Source for human gene information: https://ghr.nlm.nih.gov, unless otherwise specified. bp, base pair; −, deletion; +, insertion; INDEL, insertion or deletion.
| Disease | Gene | Human Mutation | Model Species | NHP Mutation | Type of human disease/mutation |
|---|---|---|---|---|---|
| Transgenesis | |||||
| Huntington’s Disease | Huntingtin (HTT) | CAG trinucleotide repeat expansions of >36 in the first exon of HTT gene (often 36120) | rhesus macaque | HTT gene exon 1 with 84 CAG (Q) repeats (HTT-84Q) under the polyubiquitin-C promoter lentiviral vector. Five transgenic offspring containing 27–88Q (17). | Autosomal, dominant |
| MECP2 Duplication Syndrome | Methyl-CpG binding protein 2 (MECP2) | Duplication of the MECP2 gene on the long arm of X chromosome | cynomolgus macaque | MECP2 gene copy number ranged from 1–8.5; Synapsin-promoter-driven, lentiviral vector. Fourteen MECP2 transgenic offspring (9 live, 5 stillborn) carrying 1–8.5 copies of MECP2(21). | X-linked |
| Parkinson’s Disease | α-Synuclein (SCNA) | SCNA A53T | rhesus macaque | SNCA A53T inserted under human ubiquitin-C promoter lentiviral vector. Eleven transgenic offspring (6 live, 5 aborted) carrying 2–7 copies of SNCA A53T(22). | autosomal, dominant |
| Spinocerebellar ataxia | Ataxin 3 (ATXN3) | CAG trinucleotide repeat expansions of > 50 in ATXN3 gene | common marmoset | Ataxin 3 transgene with 120 CAG (Q) repeats (ataxin 3-120Q); cytomegalovirus-promoterdriven lentiviral vector. Seven live transgenics containing 120Q(19). | autosomal, dominant |
| Genomic Editing | |||||
| ZFNs | |||||
| Severe Combined Immunodeficiency Disorder | Interleukin 2 receptor, gamma (IL2RG) | Any of 300 mutations within the IL2RG gene | common marmoset | Two nucleases (HiFi and eHiFI) introduced these mutations: −21 bp (HiFi-ZFN) in one offspring and a −1 bp, −97 bp and +4 bp, and −20 bp and −86 bp (eHiFi-ZFN) in three offspring(45). | X-linked, recessive |
| TALENs | |||||
| Severe Combined Immunodeficiency Disorder | Interleukin 2 receptor, gamma (IL2RG) | Any of 300 mutations within the IL2RG gene | common marmoset | Exon 2 and 4 targeted resulting in −9 bp (exon 2), −1 bp and −20 bp (exon 4), −9 (exon 2) and −25 bp (exon 2) mutations in three offspring(45). | X-linked, recessive |
| Microcephaly | Microcephalin 1 (MCPH1) | Any of 7 mutations within the MCPH1 gene(98) | cynomolgus macaque | MCPH1 exons 2 and 3 were targeted causing −8 bp and +6 bp as well as −6 bp and +3 bp mutations in one live offspring(51). | autosomal, recessive |
| Rett Syndrome | Methyl-CpG binding protein 2 (MECP2) | Any of 620 mutations within the MECP2 gene | rhesus and cynomolgus macaques | MECP2 exon 3 targeted by two research groups(47–49). Thirteen edited offspring (8 stillborn and 5 live) carrying INDELS and point mutations(47,48). A stillborn monkey carried a −38 bp and +4 bp mutation resulting in a frame-shift(49). | X-linked, dominant Occurs in ~1 in 10,000 girls; the mutation in males is embryonic lethal(50). |
| CRISPR/Cas9 | |||||
| Parkinson’s Disease | PTEN-induced putative kinase 1 (PINK1) | Any of 70 mutations within the PINK1 gene | rhesus macaque | Eight edited-offspring (4 died soon after birth) that carried a −7 kb in PINK1 between exon 2 and exon 4 and/or point mutations and INDELS at each sgRNA target site(71). | autosomal, recessive PINK1 mutations cause 4%–9% of early onset Parkinson’s disease cases(70). |
| Duchenne Muscular Dystrophy | Dystrophin (DMD) | Any of 2000 mutations within the DMD gene. | rhesus macaque | Targeting exons 4 or 46 of the DMD gene to create gene disruption with −2–9 bp or a −2 bp and +20 bp mutations in either exon 4 or 46(59). | X-linked, recessive |
| Adrenal Hypoplasia Congenita and Hypogonadotropic Hypogonadism | Nuclear receptor subfamily 0 group B member 1 (NR0B1) (alias DAX1) | Any of 110 mutations within the NR0B1 gene | cynomolgus macaque | Four mutated aborted fetuses carrying either −8 bp, +15 bp, −4 bp, or −8 bp and −6 bp mutations(66). | X-linked |
| Autism Spectrum Disorder | SH3 and multiple ankyrin repeat domains 3 (SHANK3) | Any of 43 mutations within the SHANK3 gene | cynomolgus macaque | Targeting of exon 21 of SHANK3 led to INDELs including in 5 NHPs including a −1 bp, −12 bp, −38 bp, −40 bp and/or −96 bp mutation(62, 64). Targeting of exon 6 and 12 resulted in 3 edited NHPs carrying: an −11.5 kb, −22 bp and −8 bp; −5 bp and +1 bp; or −2 bp mutations(63). | autosomal, dominant, chromosomal aberrations leading to haploinsufficiency(61); SHANK3 mutations are present in 1–2% of autism spectrum disorder cases |
| Polycystic Kidney Disease | Polycystin 1, transient receptor potential channel interacting (PKD1) | Any of 250 mutations within the PKD1 gene | cynomolgus macaque | Biallelic or heterozygous in-frame and frameshift mutations within exon 2 of 14 edited offspring (4 miscarried/stillborn, 10 live)(73). | autosomal, dominant |
| Hutchinson-Gilford Progeria Syndrome | Lamin A/C (LMNA) | LMNA C1824T, other mutations within the LMNA gene | cynomolgus macaque | Three offspring with homozygous C-T mutation in LMNA and two offspring mosaic for the C-T mutation(76). | autosomal, dominant |
Patients with methyl-CpG binding protein 2 (MECP2) gene duplication overproduce the MECP2 protein leading to disruption of neuronal function (20). Liu et al. (21) introduced the human MECP2 gene driven by the neuronal-specific synapsin promoter into cynomolgus macaque one-cell embryos by microinjection. Transgene expression in a stillborn fetus was restricted to neuronal tissues. In 14 live transgenic births and one stillborn animal, the transgene copy number ranged from 1 to 8.5 with integration and expression from autosomal chromosomes, whereas in humans, the MECP2 gene is located on the X-chromosome.
Genes containing point mutations associated with human disease also have been introduced into NHPs by transgenesis. Niu et al. (22) reported the microinjection of rhesus macaque oocytes with a lentiviral vector carrying the SNCA gene encoding alpha synuclein with the A53T mutation, which is one of 90 independent risk-associated gene variants linked to PD (23). The mutant alpha synuclein protein was expressed in various regions of a stillborn infant’s brain with enriched expression in synaptic terminals (22). In the MECP2 and SNCA A53T NHP studies, the random transgene insertion and multiple integration sites created overexpression of the mutated protein.
Considerations and Caveats for Transgenic NHP Studies
NHP transgenic studies underscore the importance of the timing of viral vector delivery by microinjection relative to the efficiency of transgene propagation. Across studies, viral vectors have been delivered to a considerable number of NHP oocytes (94 to 264 oocytes) or early embryos (29 to 476 embryos), but embryo transfer to recipient females has resulted in low pregnancy rates (11–15, 17, 19, 21, 22, 24, 25). The proportion of transferred embryos that produce a transgenic offspring ranges across studies from 4.8 to 22.6% (11–15, 17, 19, 21, 22, 24, 25), which showcases the need for abundant animal resources to procure a few live transgenic animals. The design of the viral vector construct requires careful consideration. The efficiency of virus-mediated infectivity can result in widespread transgene expression under a ubiquitously expressed promoter. Moreover, multiple transgene copies and insertion sites can contribute to a more severe phenotypic outcome with disease onset at an earlier age than observed in the human disease. Furthermore, transgenic animals express both the mutated and the wild-type gene, which may also affect the resulting phenotype. Future studies should consider the association between copy number of mutated genes, gene expression of mutated and wild-type protein and the severity of disease symptoms in transgenic NHP models.
Germline transmission of the transgene is essential for breeding animal carriers of the mutation, by either natural or assisted reproductive technologies. Sasaki et al. (13) demonstrated that founder (F0) transgenic eGFP marmosets, when crossed with wild-type animals, were capable of producing transgenic offspring. Similarly, germline transmission of mutant HTT has been demonstrated by Putkhao et al. (26) and Moran et al. (27). Sperm cells from the F0 male carried mutant HTT with 71 to 74 CAG repeats, whereas mutant HTT in the infant offspring ranged from 53 to 61 or 72 to 74 CAG repeats, suggesting that the CAG repeats are present but variable in F1 offspring (26, 27). Embryonic stem cells were derived from in vitro produced embryos fertilized by F0 mutant HTT males and were used as an in vitro platform to assess pathogenesis of mutant HTT in NHPs (26–31). These studies demonstrate germline transmission for supporting colony expansion of transgenic NHPs, although the extent to which additional transgenic animals have been generated from founder animals remains unclear.
The Genome-Editing Revolution and its Impact on NHP Research
Targeted genome modification in NHPs for modeling human disease has been made possible by the development of ZFNs, TALENs, and CRISPR-Cas9 genome editing technologies (Table 2 and Fig. 2). Each technology uses a unique recognition system that directs an endonuclease to the target DNA sequence, resulting in the creation of a double-stranded DNA break (32). The ability to harness the nuclease activity along with sequence-specific recognition has revolutionized genetic engineering.
ZFNs and TALENs bind antiparallel to a DNA sequence, and then a double-stranded DNA break is introduced through dimerization of a FokI restriction enzyme. ZFNs were first recognized for their gene editing potential in 1996 by Kim and colleagues (34, 36, 37), and the potential of TALENs emerged in 2010 as an alternative to ZFNs for genome editing (33, 38). A ZFN array is constructed for each DNA strand by assembling three to six ZFNs linked to a FokI restriction enzyme. Each ZFN is engineered to recognize a three-nucleotide sequence that when assembled will recognize the target DNA region spanning 18 to 36 base pairs (bp) (32). TALEN technology also utilizes a FokI restriction enzyme, but applies TAL arrays as the target DNA recognition element. A TAL array is composed of a sequence of TAL effector proteins that bind 1:1 to the DNA nucleotide sequence. When assembled together, the TAL effector proteins target a DNA region spanning 30 to 40 bp (32, 38). Although both ZFNs and TALENs involve protein engineering to create the editing construct, TALENs are more advantageous because of the ease of their monomeric design, high efficiency rate, and expansive targeting range (33, 35).
In 2012, the CRISPR-Cas9 mechanism identified in prokaryotes was exploited as a technology to create double-stranded DNA breaks in eukaryotic cells (32, 39). The CRISPR-Cas9 complex is composed of Cas9 protein and a single guide RNA (sgRNA). Specific sequence recognition occurs when the sgRNA aligns to the target DNA sequence. The DNA is then cleaved 3 bp upstream of the Cas9 recognition sequence, termed the protospacer adjacent motif (PAM) which consists of a 5’-NGG-3’sequence (39, 40). In contrast to ZFNs and TALENs, CRISPR-Cas9 technology does not require intense protein engineering, thus facilitating more rapid design and production of the constructs. Online tools are available for designing sgRNAs at the DNA target site and also provide predictions for on-target and off-target efficiencies. Laboratories can readily synthesize sgRNA and Cas9 mRNA, and Cas9 mRNA and Cas9 protein are commercially available. CRISPR-Cas9 constructs can then be delivered to cells in the form of plasmids, RNA or ribonucleoprotein complexes (RNPs) (39, 41). CRISPR-Cas9 has emerged as a superior technology for gene modification because of its low off-target effects and limited toxicity to cells and embryos compared to other methods (32).
The double-stranded DNA breaks created by the Fok1 or Cas9 nucleases are repaired by the cell’s intrinsic DNA repair mechanisms, through either nonhomologous end-joining (NHEJ) or homology-directed repair (HDR) (32). NHEJ may occur throughout the cell cycle and is error prone, because base substitutions, insertions and deletions may occur (32, 42, 43). Alternatively, DNA repair by HDR requires a donor strand or an endogenous DNA sequence to serve as the repair template (43, 44). A gene knockout can be created by NHEJ through the formation of insertions or deletions (INDELs) leading to missense or frameshift mutations, whereas a gene knockin can be introduced by HDR upon delivery of an exogenous DNA repair template (Fig. 2).
Creating Mutations by ZFNs and TALENs in Marmosets and Macaques
Sato et al. (45) compared targeting by ZFNs and TALENs in the first report of genome editing in marmosets. To model severe combined immunodeficiency (SCID), a variety of ZFN and TALEN constructs were tested to knockout the Interleukin 2 receptor subunit gamma (IL2RG) gene (45). Patients with SCID can carry one of 62 different mutations across all eight exons of the X-linked IL2RG gene (46). An obligate FokI heterodimer ZFN (HiFi-ZFN), a ZFN vector with improved FokI restriction endonuclease specificity (eHiFi-ZFN) (45), or TALENs targeting exons 2 or 4 of the marmoset IL2RG gene were introduced into marmoset embryos. Editing of the IL2RG gene was observed in 37.5% of HiFi-ZFN, 100% of eHiFi-ZFN, and 100% of TALEN-exon 2 and TALEN-exon 4 blastomeres. The eHiFi-ZFNs and TALENs introduced editing events soon after injection, whereas HiFi-ZFN editing occurred later at the four- to eight-cell stage. The IL2RG gene-edited marmosets, like patients with SCID, showed reduced thymic development, specifically a decreased number of T cells and natural killer (NK) cells and an increase in B cells.
TALENs also induced gene disruption in rhesus and cynomolgus macaque embryos. Two separate research groups (47–49) targeted exon 3 of the MECP2 gene, aiming to model the development of human Rett syndrome, an X-linked monogenic neurological disorder. The MECP2 mutation is embryonic lethal in males, but in females there is a wide range of phenotypes due to random X-inactivation (50), which was also observed in the macaque studies. MECP2 gene mutations reduced cellular MeCP2 protein and alterations in neurodevelopment and behaviors were observed in MECP2-edited cynomolgus macaques compared to control animals.
Ke et al. (51) used TALENs to target exons 2 and 3 and disrupt the microcephalin 1 (MCPH1) gene in cynomolgus macaques. MCPH1 protein in biallelically-edited offspring was reduced in both peripheral blood mononuclear cells and dermal fibroblasts compared to controls. Human MCPH1 mutations are associated with a head circumference that is three SDs below that for healthy children of the same age and sex (52). The head circumference of the MCPH1-edited cynomolgus macaque offspring was −4 to −11 SDs below the mean head circumference for age-matched controls in the first year of life. Magnetic resonance imaging (MRI) showed that brain volume was reduced by −8 SDs with hypoplasia of the corpus callosum in the MCPH1-edited macaque offspring.
Using CRISPR-Cas9 in Macaques: Proof of principle
The delivery of CRISPR-Cas9 constructs to NHP embryos was reported by Niu and colleagues (14, 53). Genes encoding nuclear receptor subfamily 0 group B member 1 (NR0B1), peroxisome proliferator-activated receptor gamma (PPARγ) and recombination activating gene 1 (RAG1) were simultaneously targeted in one-cell cynomolgus macaque embryos to create gene disruptions. Gene editing at the NR0B1, PPARγ and RAG1 target sites occurred in ~27, 46 and 60% of embryos, respectively. Live female twin offspring contained mutations in PPARγ and RAG1, with no wildtype sequences for RAG1 in one offspring. A mosaic editing pattern (that is, the presence of more than one edited genotype) was observed in both preimplantation embryos and tissues obtained from the live offspring, with no off-target gene editing observed. A subsequent report showed germline acquisition of CRISPR-Cas9 mutations in both the male and female gonad of the gene edited offspring (54).
The live birth of a cynomolgus macaque with a biallelic mutation of p53 introduced by CRISPR-Cas9 was reported by Wan et al. (55). Microinjection of macaque fertilized oocytes with a higher Cas9 mRNA:sgRNA ratio produced a high rate of biallelic gene editing at the cost of reduced embryonic development. Additional experiments were performed to determine the optimal ratio of Cas9 mRNA:sgRNA that would result in high gene-editing efficiency and low embryotoxicity. Three live births and two miscarried fetuses were produced with INDELs in p53 present in four of the five offspring. No wildtype sequences were present in one live CRISPR-edited animal, suggesting that biallelic editing took place at the one-cell embryo stage.
Delivery of Cas9 mRNA or protein to NHP embryos can produce various on-target editing efficiencies and genetic mosaicism. To minimize genetic mosaicism, Tu et al. (56) engineered Cas9 mRNA containing a ubiquitin-proteasomal degradation signal (Ubi-Cas9) to decrease its half-life. The delivery of the engineered, Ubi-Cas9 nuclease to cynomolgus macaque embryos resulted in similar on-target gene-editing efficiencies (73.8%) compared to wildtype Cas9 mRNA (77.3%), but with less diverse gene-editing events and reduced mosaicism. The Cas9 protein-injected embryos had a ~55% reduction in on-target gene-editing efficiency compared to microinjection of wildtype Cas9 mRNA or Ubi-Cas9 mRNA. In contrast, Midic et al. (57) reported that microinjection of fertilized oocytes with wildtype Cas9 protein resulted in 100% of the blastocysts (n=5) containing an on-target edited gene that likely occurred at the one-cell to two-cell embryo stage. The appropriate ratio of Cas9 protein to sgRNA is crucial to forming RNP complexes (41, 58). Differing ratios of Cas9 and sgRNA between studies plausibly contributed to the different results.
Creating Mutations in Macaques Using CRISPR-Cas9
Modeling human diseases in NHPs using CRISPR-Cas9 has primarily focused on disruption of human disease-associated genes. Duchenne muscular dystrophy (DMD) is an X-linked recessive muscle degenerative disorder caused by mutations within exons 2 to 19 or 45 to 55 of the DMD gene encoding the muscle protein dystrophin. DMD is characterized by progressive muscle degeneration leading to early death in male patients. Chen et al. (59) have reported that the targeting of exons 4 and 46 of the rhesus macaque DMD gene resulted in ~61% of the offspring carrying an edited DMD gene. Two stillborn fetuses had mosaic editing patterns across their tissues. Dystrophin protein was reduced or absent in the muscle tissues of stillborn offspring with a corresponding disruption in muscle fiber architecture compared to control fetuses. At 6 months of age, the nine live DMD-edited offspring did not present phenotypic symptoms of DMD. No off-target editing was observed after whole genome sequencing of cells and tissues from two live DMD-edited rhesus macaques (60).
Gene disruption of the SH3 and multiple ankyrin repeat domains 3 (SHANK3) gene in cynomolgus macaques was explored in order to model human autism spectrum disorders (ASD). SHANK3 encodes a scaffold protein at excitatory synapses of brain neurons, and 43 different mutations have been associated with ASD (Table 2). Haploinsufficiency of SHANK3 is thought to be the mechanism responsible for the developmental disorder Phelan-McDermid syndrome (61). Zhao et al. (62) targeted exons 6 and 12 of SHANK3 by CRISPR-Cas9 in one-cell cynomolgus macaque embryos. These exons are present in all transcript variants of SHANK3, hence creating a disruption in either exon that could alter gene function. A range of edited genotypes was observed in NHP offspring including an 11.5-kb deletion between exon 6 and 12 in a miscarried fetus, a 5-bp deletion and a 1-bp insertion in a stillborn infant, and a 2-bp deletion in a live infant. The brains of two deceased fetuses had reduced SHANK3 protein with near-complete loss of SHANK3 in the prefrontal cortex. Zhou et al. (63) reported a different approach by targeting exon 21, which resulted in two homozygous and three mosaic live infants. A homozygous-edited F0 animal produced an F1 offspring with a mix of wild-type and edited SHANK3 alleles, further demonstrating germline transmission of CRISPR-edited SHANK3 in NHPs.
In a follow up study to the Zhao et al. report (62), Tu et al. (64) compared a cynomolgus macaque offspring carrying a 2-bp SHANK3 deletion in exon 12 to control offspring. The edited NHPs generated by both Zhou et al. (63) and Tu et al. (64) had a greater frequency and duration of stereotypic behaviors reminiscent of human ASD and initiated fewer social interactions with other macaques. Positron emission tomography imaging showed reduced glucose metabolism in multiple brain regions, and MRI revealed reduced gray matter and regional differences in hypo-connectivity and hyper-connectivity in the brains of SHANK3-edited macaques compared to control animals. A result that differed between the two studies was that only Tu et al. (64) found reduced overall growth, delayed vocalizations and impaired social interactions in the one edited offspring compared to age- and gender-matched controls (63). The low embryo transfer efficiency (116 embryos transferred to 37 female recipients resulted in 3 pregnancies; 0.86% of transferred embryos produced live edited offspring) in the study by Tu et al. (64) underscores the need for sufficient animal resources to produce live edited offspring.
The disruption of genes with known roles in human development has been achieved in cynomolgus macaques using CRISPR-Cas9 gene targeting. In humans, the NR0B1 gene, also known as DAX1, plays a critical role in the development of the adrenal gland and the hypothalamic-pituitary-gonadal axis, and if disturbed, results in adrenal insufficiency, hypogonadotropic hypogonadism, and, in some cases cryptorchidism (65). Kang et al. (66) reported the generation of a stillborn cynomolgus macaque offspring with an 8-bp deletion in DAX1 that had an enlarged fetal adrenal cortex zone and disordered neovascularization and abnormal fibrosis in the testis compared to control animals. The in utero developmental defects associated with DAX1 editing observed in the edited stillborn macaque could have affected adrenal and reproductive physiology if the animal had survived. Zhang et al. (67) targeted exons 5 and 7 of the Sirtuin-6 (SIRT6) gene in cynomolgus macaque one-cell embryos to create a gene knockout and to assess its role in development (no disease has yet been linked to mutations in this gene). A lack of SIRT6 protein expression was observed in the brain and muscle of SIRT6-edited offspring that carried biallelic deletions, all of which died shortly after birth. The brain and muscle of SIRT6 mutation carriers were underdeveloped at term compared to wildtype control animals. Interestingly, knockout of SIRT6 was associated with up-regulation of the imprinted gene H19. SIRT6 was proposed to bind to the imprinting control region of H19, which contributes to maturation of the primate brain.
A knockout of the brain and muscle aryl hydrocarbon receptor nuclear translocator-like 1 (BMAL1) gene to disrupt circadian rhythms in cynomolgus macaques was reported by Qiu et al. (68). BMAL1 forms a heterodimeric complex with CLOCK that acts as a transcriptional activator of genes regulating circadian rhythm (69). Five of eight live offspring produced from CRISPR-Cas9 microinjected embryos contained BMAL1 mutations, and three of them were heterozygous for the BMAL1 deletion. Widespread absence of BMAL1 protein was observed by immunoblotting of tissues from a stillborn fetus with a heterozygous BMAL1 deletion. Live mutated offspring displayed increased nocturnal activity by 10 months of age, reduced melatonin concentrations, reduced sleep in one mutant animal, and an increased stress response.
Mutations in the PTEN-induced kinase 1 (PINK1) gene are associated with a rare form of autosomal recessive early-onset (24 to 47 years old) PD. Several missense, nonsense, and frameshift mutations, as well as large deletions of multiple exons have been identified that lead to loss of function of PINK1 protein (70). Yang et al. (71) targeted exon 2 and 4 of PINK1 in rhesus macaques as these exons encode the kinase domain. Eleven rhesus macaque offspring were produced and eight carried PINK1 mutations. Notably, a ~7.2-kb deletion in PINK1 was observed in the brains of two offspring that died shortly after birth. Another PINK1-edited offspring with the ~7.2-kb deletion was euthanized at 3 years of age due to progression of neurodegenerative disease symptoms. Evaluation of the brain of this animal revealed neuronal degeneration in the cortex, substantia nigra, and striatum (72).
Disease-causing mutations can be heterozygous, such as mutations in the polycystic kidney disease 1 (PKD1) gene that cause autosomal dominant polycystic kidney disease. Tsukiyama et al. (73) targeted exon 2 of the PKD1 gene in cynomolgus macaque embryos to create a frameshift mutation. An embryonic editing efficiency of 88.7% was reported, the highest to date in NHP embryos. As embryonic lethality has been noted in Pkd1-null mice, the authors opted to microinject embryos with a lower concentration of Cas9 and sgRNA. The live birth rate after embryo transfer was reduced for PKD1-edited compared to wildtype offspring. This finding highlights that genes targeted by CRISPR-Cas9 may have unknown roles in embryonic development and that their editing may result in reduced embryo or fetal development rates. Genetic analysis of kidney DNA from 12 stillborn or deceased infants showed that eight animals contained biallelic PKD1 mutations, three were heterozygotes, and one could not be genotyped. Heterozygous editing of PKD1 was achieved, which is likely attributable to the low rate of biallelic editing that occurs in NHP embryos. Histopathological analysis of kidney specimens revealed severe to mild cyst formation with a tendency towards greater pathologies observed in macaques with frameshift mutations. Notably, the appearance of small cysts in the perinatal stages of kidney development in the heterozygotes parallels human polycystic kidney disease.
Next-Generation Editing Technologies for NHP Research
Next-generation Cas nucleases such as base editors and prime editors were engineered to overcome limitations in creating point mutations and are promising alternatives for introducing precise gene edits (Fig. 2). These technologies use modified Cas9 variants, such as Cas9 nickase (nCas9) or catalytically deficient Cas9 (dCas9), to nick one strand of DNA and bypass activation of DNA repair mechanisms at both strands thereby mitigating potentially adverse editing outcomes (74, 75). In base editing, nCas9 is fused with a nucleobase deaminase enzyme. Upon DNA recognition, a deamination reaction occurs that facilitates the following nucleotide changes: C to T [cytosine base editors (CBE)], A to G [adenosine base editors (ABE)], or C to G [guanosine base editors (GBE)] (75).
Wang et al. (76) applied base-editing technology to cynomolgus macaque embryos aiming to create a C-T conversion in the lamin A/C (LMNA) gene to mimic the point mutation that causes Hutchinson-Gilford progeria syndrome in humans. Live edited macaques displayed clinical features of this disease including reduced growth, hair loss, skeletal aberrations and expression of progerin protein, a toxic protein formed by truncation of the prelamin A protein.
Multiplexed editing in NHPs would revolutionize disease modeling by enabling creation of point mutations underlying polygenetic disorders. Zhang et al. (77) demonstrated that multiplex base-editing in cynomolgus macaque embryos successfully created precise edits in three targeted genes in 23% of the injected embryos. Testing of four base editors across 11 loci showed that two enzymes, the Staphylococcus aureus Cas9-based SaKKH-BE3 and ABE7.10, consistently created C-T and A-G conversions, respectively. No INDEL formation was observed, but mosaic editing occurred regardless of the base editor used.
Considerations and Caveats of Genome Editing in NHPs
The reports of gene disruption by CRISPR-Cas9 editing in macaque embryos demonstrate the ability to target genes associated with human disease. Such studies have revealed phenotypic outcomes for aberrantly expressed genes in macaques and identified gaps in our knowledge about how to efficiently and accurately perform genome editing in NHPs. Biallelic and heterozygous gene editing were achieved without reports of off-target effects (53, 55, 60, 78), although editing mosaicism occurred across tissues. Precise control of gene editing including propagation of the mutation to all cells of the body remains a challenge. Similar to transgenesis, embryonic genome editing relies on the ability to manipulate embryos followed by the successful establishment and maintenance of a pregnancy after embryo transfer. Across studies of genome editing in NHPs (45, 47–49, 53, 55, 59, 62, 63, 67, 71, 79, 80), the number of embryos transferred ranged from ~21 to 179 and the number of female NHP recipients of CRISPR-Cas9-targeted embryos ranged from ~4 to 113; the percentage of transferred embryos resulting in live edited NHP offspring ranged from 0.87 to 7.1%. These results further exemplify the necessity for a large cohort of both oocyte donors and embryo transfer recipients. The total number of female NHPs that have undergone ovarian stimulation and that serve as a pool of candidate embryo transfer recipients is not well described in many studies, hence the total number of animals required to produce a few offspring is likely to be under estimated. In addition, the low live birth rate after embryo transfer, resulting in one to a few edited offspring that contain diverse mosaic editing patterns, hinders the ability to draw conclusions about genotype to phenotype relationships and the development of reproducible genetic models.
Optimization of the delivered concentration and volume of CRISPR-Cas9 constructs should be considered when assessing editing outcomes relative to the impact on embryo viability. An inverse relationship between editing efficiency and embryonic development has been noted upon microinjection with TALENs (51) and CRISPR-Cas9 constructs (55, 80). A ratio of 100 ng/μL Cas9 10 ng/μL sgRNA produced a high rate of biallelic mutants, but 200 ng/μL:10 ng/μL resulted in reduced embryonic development (55). In addition, Yao et al. (80) described that a microinjection volume of ~4 pL of CRISPR-Cas9 construct resulted in no pregnancies, whereas reducing the volume to ~2 pL increased the pregnancy rate to ~41%. It is possible that the targeted gene may have an unknown role in embryonic or fetal development resulting in lower pregnancy or live birth rates compared to control animals. Although NHP resources are limited, testing multiple concentrations to assess editing efficiencies and embryonic development rates may improve pregnancy rates upon embryo transfer and should be performed with control embryos when feasible to determine whether a mutation contributes to embryonic or fetal lethality.
Introducing a DNA sequence, such as a fluorescent reporter gene, has been successfully achieved using TALENs and CRISPR-Cas9 in NHP embryos (79–82). Chu et al. (83) reported a TALEN-mediated gene knockin of GFP that was introduced into the cynomolgus macaque OCT4 locus. TALEN-mediated GFP expression was observed in ~11% of the injected embryos. The reporter genes mCherry and GFP were introduced into cynomolgus macaque embryos via CRISPR-Cas9 by Yao et al. (80) and Cui et al. (79), respectively. Mosaic editing patterns were noted in both studies. Yoshimatsu et al. (82) described the first knockin marmoset using CRISPR-Cas9 to incorporate a modified PLP exon 2 sequence. A ~10% increase in editing rate was reported for NHP embryos where CRISPR-Cas9 constructs were delivered by electroporation rather than microinjection. Furthermore, Kumita et al. (81) described optimization of the design of the DNA donor repair template and showed that a 36-nucleotide donor template orientated in the sense strand direction more efficiently produced a c-Kit knockin with 30.8% of marmoset embryos containing the precise mutation. Allele-specific gene targeting offers promise in generating heterozygous mutations. Tsukiyama et al. (73) fertilized cynomolgus macaque oocytes containing a single-nucleotide polymorphism (SNP) in exon 4 of the PKD1 gene with wildtype sperm followed by microinjection of CRISPR-Cas9 sgRNAs targeting the wildtype paternal allele; the SNP present in the maternal allele would not confer sgRNA recognition at the target site. This targeting approach reduced mosaicism and was more efficient at generating heterozygote offspring.
Improving the specificity and targeting efficiency of both base-editing and prime-editing technologies is ongoing (75). The ability to create gene edits without the need for HDR makes these Cas9 nucleases more advantageous for use in NHP embryos. The implementation of next-generation nucleases will accelerate the ability to create precise gene edits in NHP embryos. Future NHP studies are needed to assess the degree of mosaicism, INDEL formation and on- and off-target effects with these technologies compared to wildtype Cas9 in NHP embryos. In the cynomolgus macaque study modeling Hutchinson-Gilford progeria syndrome, only two mosaic LMNA-edited animals lived beyond 5 months of age (76), underscoring the difficulty of the task and the need to prioritize efforts towards specific genes and diseases.
The ethical development of NHP genetic models of human disease
The available peer-reviewed studies of genetically modified NHPs showcase the technical challenges of developing these disease models and the importance of ethical and responsible use of NHPs. There are thousands of possible human disease gene targets that could be explored. Stakeholders have the critical and difficult task of prioritizing the gene mutations that should be investigated using limited NHP resources.
Animal research is justified by the potential benefit to humans, other animals and science. The close phylogenetic relationship of NHPs to humans as well as their complex behavior and cognition require application of the highest criteria and welfare standards (84). As developing genetic models of human disease creates risk of harm or discomfort to NHPs (e.g. the severe muscle weakness associated with DMD) the justification of the studies becomes even more critical. We propose that the target gene/human disease should affect a large number of individuals and affect specific traits unique to primates (e.g. frontotemporal dementia requires a highly developed neocortex) and that previous models in rodent or other non-primate species have proven inadequate to understand the pathological process or to evaluate the efficacy of interventions (e.g. Alzheimer’s disease). Yet ultimately the decision to model severely debilitating diseases in NHPs should be carefully considered by multiple stakeholders. Some human diseases may not be advisable or possible to model. For example, generation of NHPs with genetic mutations that impair fertility would not allow for colony expansion, resulting in a reduced number of subjects for meaningful research. Chromosomal disorders, such as Down’s syndrome, cannot be modeled with current technologies, yet even if modeling was possible, the different number of chromosomes between humans (23 pairs) and rhesus macaques (21 pairs) would further complicate the challenge.
For investigators, the well-known principles of the 3Rs (Replacement, Reduction and Refinement) provide a framework for guiding the ethical development of NHP genetic disease models at every step of the process. Several factors need to be considered before targeting the NHP embryonic genome to create models of human disease. First, the choice of NHP species requires careful consideration as the nucleic acid and protein sequences of the disease-associated gene must be highly conserved between humans and the NHP species to be genetically modified. Behavior, anatomy and physiology of the NHP species should also be considered to phenocopy and detect disease traits. For example, common marmosets naturally have a threonine at the 53rd amino acid residue of α-synuclein (85), which makes them an unsuitable species for modeling the α-synuclein A53T point mutation associated with PD. However, their genetics and motor behavior make marmosets well suited for modeling the most frequent PD mutation, the LRRK2 G2019S protein mutation caused by a G6055A point mutation in the LRRK2 gene (Fig. 1). Second, construct design for accurate, impactful disease modeling should aim to introduce the same mutation as occurs in humans. Most genetic diseases are associated with single point mutations that cause amino acid substitutions, such as A53T in α-synuclein or G2019S in LRRK2. Deleting regions of the kinase domain of LRRK2 may provide valuable information about PD pathogenic mechanisms, but will not specifically model PD that is associated with the LRRK2 G2019S mutation. Deletion mutations may be valid for modeling some PD-associated PINK1 mutations, where multiple exons have been deleted (70, 71). Third, selection of highly efficient guide RNAs is needed before injection into embryos. NHP cell culture systems can serve to initially screen guide RNAs and allow for on-target and off-target editing analysis before introducing the genome-editing constructs into NHP embryos (86–88). Moreover, NHP stem cell cultures can be directed toward cellular differentiation after gene targeting, enabling in vitro disease modeling. Vermilyea et al. (88) introduced the precise point mutation G2019S into the PD-associated LRRK2 of marmoset stem cells and then induced these cells to differentiate into neurons. The edited neurons displayed a reduced response to oxidative stress and reduced viability compared to wild-type LRRK2. Fourth, extensive resources are required to generate a sufficient number of live genome-edited NHP offspring. Meaningful research requires studies that are appropriately statistically powered. Similar to transgenesis, embryonic genome editing relies on the ability to manipulate embryos followed by the successful establishment and maintenance of a pregnancy after embryo transfer. Assisted reproductive technologies are well established for macaques and marmosets, yet the limited consistency in yield of oocytes and embryos necessitates a large pool of animals. Fifth, veterinarians and medical scientists with knowledge of disease-specific and species-specific care and evaluation should be ready to provide long-term animal care. These resources are required for years compared to rodent models because of the longer life span of NHP species.
Germline-based editing relies on the reproductive efficiency of assisted reproductive technologies, and improvements in these technologies would aid in reducing the number of animals needed. Without experimental genetic manipulation, in vitro oocyte fertilization rates are typically ~70 to 80%, and of those that are fertilized, ~30 to 40% develop to the blastocyst stage. There is high embryonic and fetal loss after embryo transfer, and the attrition greatly hinders the success rate of producing live genetically-modified NHPs. A large pool of female macaques undergoing regular ovarian cycles is needed to increase the likelihood that a female will be available for embryo transfer, because ovarian stimulation and embryo transfer in this species are initiated relative to the timing of menses onset (89, 90). In addition, rhesus macaques are seasonal breeders with reduced oocyte quality in the non-breeding season, thus restricting the window for optimal embryo development each year (91). Optimization of methods for synchronizing the macaque menstrual cycles of oocyte donors and embryo recipients would help to decrease the number of NHPs required. In comparison, the marmoset’s ovarian cycle can be regulated pharmacologically, which facilitates synchronization (92, 93). Improving cryopreservation strategies for NHP gametes and embryos to ensure post-thaw viability would allow for flexibility in embryo transfer timing and also would allow for preserving genetically-edited animals for future colony expansion. In addition, steps to optimize construct design in cell culture systems, plus incorporation of fluorescent tags and utilization of next-generation editor nucleases may improve selection of edited embryos for transfer. The greatest hurdle is the production of a cohort of genetically modified animals that contain the human disease mutation of interest with phenotypic observations that parallel human disease. Moreover, reproducibility of a model has not yet been proven and often expansion of the genetically modified animal is hindered by symptom progression resulting in humane euthanasia before the animal reaches puberty.
Technological advances will modestly aid in reducing NHP numbers required for human disease modeling in the near future, and alternative strategies should be pursued. Progress in creating such NHP models of human disease will likely not be a one-size fits all approach. Somatic cell delivery of gene-editing constructs by viral vectors or nanoparticles are promising alternatives that have limitations but would mitigate delays in waiting for offspring to be born and develop to adulthood. Another possibility is the screening of NHP colonies for naturally-occurring mutations using whole genome and exome sequencing. Larger primate centers could maximize resources for creating colonies of NHP disease models. Breeding of NHPs with naturally-occurring mutations or genetically-modified NHPs will require careful attention in expanding the mutation while minimizing inbreeding. Disease symptoms in genetically modified NHPs will require extensive monitoring. Some primate centers are already equipped with specialized units for infant and aged NHP care. Both national and international consortia are needed to establish guidelines that are specific for the care and use of genetically-modified NHPs. A collaborative approach with an initiative to establish a shared genome database of naturally-occurring and introduced mutations in NHPs as well as gamete and embryo cryobanks and tissue banks would facilitate greater access to specimens (age-matched, control or mutated) and reduce the number of NHPs required.
Conclusions
Currently, modeling of human genetic diseases in NHPs is limited by existing technologies, animal availability and NHP reproductive biology. Studies have focused on multiplying genes by transgenesis or disrupting genes by genome editing, which may not faithfully model human disease. Advances in genome modification have put pressure on research systems to create NHP models of human disease, yet now more than ever scientific and ethical considerations should guide such research. Stakeholders, including scientists, veterinarians, funders and institutions should consider the overall impact of creating genetically-modified NHPs. National and international collaborations can help to maximize efforts and apply guidelines for the ethically responsible use of NHPs. Safeguarding the overall scientific enterprise requires prioritizing the use of resources and finding suitable alternatives. It would be naïve to think that NHP genetic models will solve all therapeutic shortcomings in human disease, because disease etiologies are complex and are not limited to genetic factors, but NHP genetic models can support the process toward improved medical outcomes and ultimately cures.
Acknowledgements
We thank T. Golos, I. Slukvin, and C. Swanson as well as the reviewers for suggestions.
Funding
This study was supported by NIH grants P51OD011106, UG3NS111688 and R61NS115102 (to M.E.E.), and K99HD099154 (to J.K.S.).
Footnotes
Competing interests: none
References
- 1.Seyhan AA, Lost in translation: the valley of death across preclinical and clinical divide – identification of problems and overcoming obstacles. Translational Medicine Communications 4, 1–19 (2019). [Google Scholar]
- 2.DeLong MR, Benabid AL. Discovery of high-frequency deep brain stimulation for treatment of Parkinson disease: 2014 Lasker Award. JAMA 312, (2014). [DOI] [PubMed] [Google Scholar]
- 3.Ohshima-Hosoyama S, Simmons HA, Goecks N, Joers V, Swanson CR, Bondarenko V, Velotta R, Brunner K, Wood LD, Hruban RH, Emborg ME. A monoclonal antibody-GDNF fusion protein is not neuroprotective and is associated with proliferative pancreatic lesions in parkinsonian monkeys. PloS One 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers J, Gibbs RA, Comparative primate genomics: emerging patterns of genome content and dynamics. Nat. Rev. Genet. 15, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Picaud S, Dalkara D, Marazova K, Goureau O, Roska B, Sahel JA. The primate model for understanding and restoring vision. Proc. Natl. Acad.Sci. U. S. A 116, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peterson SM, McGill TJ, Puthussery T, Stoddard J, Renner L, Lewis AD, Colgin LMA, Gayet J, Wang X, Prongay K, Cullin C, Dozier BL, Ferguson B, Neuringer M, Bardet-Biedl Syndrome in rhesus macaques: A nonhuman primate model of retinitis pigmentosa. Exp. Eye Res. 189, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park F, Lentiviral vectors: are they the future of animal transgenesis? Physiol. Genomics 31, 159–173 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Seita Y, Tsukiyama T, Azami T, Kobayashi K, Iwatani C, Tsuchiya H, Nakaya M, Tanabe H, Hitoshi S, Miyoshi H, Nakamura S, Kawauchi A, Ema M. Comprehensive evaluation of ubiquitous promoters suitable for the generation of transgenic cynomolgus monkeys†. Biol. Reprod. 100, (2019). [DOI] [PubMed] [Google Scholar]
- 9.Sasaki E, in The Common Marmoset in Captivity and Biomedical Research, Marini RP, Wachtman LM, Tardiff SD, Mansfield K, Fox JG, Eds. (Academic Press, United States, 2018), chap. 20, pp. 335–353. [Google Scholar]
- 10.Kumar M, Keller B, Makalou N, Sutton RE. Systematic determination of the packaging limit of lentiviral vectors. Human GeneTherapy 12, (2001). [DOI] [PubMed] [Google Scholar]
- 11.Chan AW, Chong KY, Martinovich C, Simerly C, Schatten G, Transgenic monkeys produced by retroviral gene transfer into mature oocytes. Science 291, 309–312 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Wolfgang MJ, Eisele SG, Browne MA, Schotzko ML, Garthwaite MA, Durning M, Ramezani A, Hawley RG, Thomson JA, Golos TG, Rhesus monkey placental transgene expression after lentiviral gene transfer into preimplantation embryos. Proc. Natl. Acad. Sci. U. S. A. 98, 10728–10732 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki E, Suemizu H, Shimada A, Hanazawa K, Oiwa R, Kamioka M, Tomioka I, Sotomaru Y, Hirakawa R, Eto T, Shiozawa S, Maeda T, Ito M, Ito R, Kito C, Yagihashi C, Kawai K, Miyoshi H, Tanioka Y, Tamaoki N, Habu S, Okano H, Nomura T, Generation of transgenic non-human primates with germline transmission. Nature 459, 523–U550 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Niu Y, Yu Y, Bernat A, Yang S, He X, Guo X, Chen D, Chen Y, Ji S, Si W, Lv Y, Tan T, Wei Q, Wang H, Shi L, Guan J, Zhu X, Afanassieff M, Savatier P, Zhang K, Zhou Q, Ji W, Transgenic rhesus monkeys produced by gene transfer into early-cleavage-stage embryos using a simian immunodeficiency virus-based vector. Proc. Natl. Acad. Sci. U. S. A. 107, 17663–17667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seita Y, Tsukiyama T, Iwatani C, Tsuchiya H, Matsushita J, Azami T, Okahara J, Nakamura S, Hayashi Y, Hitoshi S, Itoh Y, Imamura T, Nishimura M, Tooyama I, Miyoshi H, Saitou M, Ogasawara K, Sasaki E, Ema M, Generation of transgenic cynomolgus monkeys that express green fluorescent protein throughout the whole body. Sci. Rep. 6, 24868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan H-C, Ho L-I, Chi C-S, Chen S-J, Peng G-S, Chan T-M, Lin S-Z, Harn H-J, Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant. 23, 441–458 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Yang S-H, Cheng P-H, Banta H, Piotrowska-Nitsche K, Yang J-J, Cheng ECH, Synder B, Larkin K, Liu J, Orkin J, Fang Z-H, Smith Y, Bachevalier J, Zola SM, Li S-H, Li X-J, Chan AWS, Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 453, 921–924 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC, Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 7, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomioka I, Ishibashi H, Minakawa EN, Motohashi HH, Takayama O, Saito Y, Popiel HA, Puentes S, Owari K, Nakatani T, Nogami N. Yamamoto K, Noguchi S, Yonekawa T, Tanaka Y, Fujita N, Suzuki H, Kikuchi H, Aizawa S, Nagano S, Yamada D, Nishino I, Ichinohe N, Wada K, Kohsaka S, Nagai Y, Seki K, Transgenic Monkey Model of the Polyglutamine Diseases Recapitulating Progressive Neurological Symptoms. eNeuro 4, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang T, Ramocki MB, Neul JL, Lu W, Roberts L, Knight J, Ward CS, Zoghbi HY, Kheradmand F, Corry DB, Overexpression of methyl-CpG binding protein 2 impairs T(H)1 responses. Sci. Transl. Med. 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Li Z, Zhang J-T, Cai Y-J, Cheng T-L, Cheng C, Wang Y, Zhang C-C, Nie Y-H, Chen Z-F, Bian W-J, Zhang L, Xiao J, Lu B, Zhang Y-F, Zhang X-D, Sang X, Wu J-J, Xiu X, Xiong Z-Q, Zhang F. Yu X, Gong N, Zhou W-H, Sun Q, Qiu Z, Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2. Nature 530, 98–102 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Niu Y, Guo X, Chen Y, Wang C-E, Gao J, Yang W, Kang Y, Si W, Wang H, Yang S-H, Li S, Ji W, Li X-J, Early Parkinson’s disease symptoms in alpha-synuclein transgenic monkeys. Hum. Mol. Genet. 24, 2308–2317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blauwendraat C, Nalls MA, Singleton AB, The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JE, Zhang XF, Choi S-H, Okahara J, Sasaki E, Silva AC, Generation of transgenic marmosets expressing genetically encoded calcium indicators. Sci. Rep. 6, 34931 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomioka I, Nogami N, Nakatani T, Owari K, Fujita N, Motohashi H, Takayama O, Takae K, Nagai Y, Seki K, Generation of transgenic marmosets using a tetracyclin-inducible transgene expression system as a neurodegenerative disease model. Biol. Reprod. 97, 772–780 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Putkhao K, Kocerha J, Cho I-K, Yang J, Parnpai R, Chan AWS, Pathogenic cellular phenotypes are germline transmissible in a transgenic primate model of Huntington’s disease. Stem Cells Dev. 22, 1198–1205 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moran SS, Chi T, Prucha MS, Ahn KS, Connor-Stroud F, Jean S, Gould K, Chan AWS, Germline transmission in transgenic Huntington’s disease monkeys. Theriogenology 84, 277–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan AW, Cheng PH, Neumann A, Yang JJ, Reprogramming Huntington monkey skin cells into pluripotent stem cells. Cell Reprogram. 12, 509–517 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laowtammathron C, Cheng EC, Cheng P-H, Synder BR, Yang S-H, Johnson Z, Lorthongpanich C, Kuo H-C, Parnpai R, Chan AWS, Monkey hybrid stem cells develop cellular features of Huntington’s disease. BMC Cell Biol. 11, 12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laowtammathron C, Chan AW, Pluripotent hybrid stem cells from transgenic Huntington’s disease monkey. Methods Mol. Biol. 1010, 61–77 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Carter RL, Chen Y, Kunkanjanawan T, Xu Y, Moran SP, Putkao K, Yang J, Huang AHC, Parnpai R, Chan AWS, Reversal of cellular phenotypes in neural cells derived from Huntington’s disease monkey-induced pluripotent stem cells. Stem Cell Reports 3, 585–593 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LaFountaine JS, Fathe K, Smyth HD, Delivery and therapeutic applications of gene editing technologies ZFNs, TALENs, and CRISPR/Cas9. Int. J. Pharm. 494, 180–194 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186, 757–761 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U. S. A. 93, 1156–1160 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujikawa T, Ishihara H, Leach JE, Tsuyumu S. Suppression of defense response in plants by the avrBs3/pthA gene family of Xanthomonas spp. Mol. Plant Microbe. Interact. 19, 342–349 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Bibikova M, Beumer K, Trautman JK, Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science 300, 764 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Bak RO, Gomez-Ospina N, Porteus MH. Gene Editing on Center Stage. Trends Genet. 34, 600–611 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 39, 359–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jinek M, Chylinski K, Fonfara E, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders C, Niewoehner O, Duerst A, Jinek M, Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kouranova E, Forbes K, Zhao G, Warren J, Bartels A. Wu Y, Cui X, CRISPRs for Optimal Targeting: Delivery of CRISPR Components as DNA, RNA, and Protein into Cultured Cells and Single-Cell Embryos. Hum. Gene Ther. 27, 464–475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis AJ, Chen DJ, DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2, 130–143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kakarougkas A, Jeggo PA, DNA DSB repair pathway choice: an orchestrated handover mechanism. Br. J. Radiol. 87, 20130685 (2014).44. X. Li, W. D. Heyer, Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18, 99–113 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.dummy reference [Google Scholar]
- 45.Sato K, Oiwa R, Kumita W, Henry R, Sakuma T, Ito R, Nozu R, Inoue T, Katano I, Sato K, Okahara N, Okahara J, Shimizu Y, Yamamoto M, Hanazawa K, Kawakami T. Kametani Y, Suzuki R, Takahashi T, Weinstein EJ, Yamamoto T, Sakakibara Y, Habu S, Hata J-I, Okano H, Sasaki E, Generation of a Nonhuman Primate Model of Severe Combined Immunodeficiency Using Highly Efficient Genome Editing. Cell Stem Cell 19, (2016). [DOI] [PubMed] [Google Scholar]
- 46.Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, Conley ME, Fischer RE, Rosenblatt HM, Small TN, Buckley RH, Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood 89, 1968–1977 (1997). [PubMed] [Google Scholar]
- 47.Liu H, Chen Y, Niu Y, Zhang K, Kang Y, Ge W, Liu X, Zhao E, Wang C, Lin S, Jing B, Si C, Lin Q, Chen X, Lin H, Pu X, Wang Y, Qin B, Wang F, Wang H, Si W, Zhou J, Tan T, Li T, Ji S, Xue Z, Luo Y, Cheng L, Zhou Q, Li S, Sun YE, Ji W, TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14, 323–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Yu J, Niu Y, Qin D, Liu H, Li G, Hu Y, Wang J, Lu Y, Kang Y, Jiang Y, Wu K, Li S, Wei J, He J, Wang J, Liu X, Luo Y, Si C, Bai R, Zhang K, Liu J, Huang S, Chen Z, Wang S, Chen X. Bao X, Zhang Q, Li F, Geng R, Liang A, Shen D, Jiang T, Hu X, Ma Y, Ji W, Sun YE, Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell 169, 945–955.e910 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Zhou X, Zhu Y, Chen Z-F, Yu B, Wang Y, Zhang C-C, Nie Y-H, Sang Z, Cai Y-J, Zhang Y-F, Zhang C, Zhou W-H, Sun Q, Qiu Z, Generation of a monkey with MECP2 mutations by TALEN-based gene targeting. Neurosci. Bull. 30, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amir RE, Van den Veyver IV, Wan M, Tran CQ, Francke U, Zoghbi HY, Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics 23, (1999). [DOI] [PubMed] [Google Scholar]
- 51.Ke Q, Li W, Lai X, Chen H, Huang L, Kang Z, Li K, Ren J, Lin X, Zheng H, Huang W, Y Ma, Xu D, Chen Z, Song X, Lin X, Zhuang M, Wang T, Zhuang F, Xi J, Mao FF, Xia H, Lahn BT, Zhou Q, Yang S, Xiang AP, TALEN-based generation of a cynomolgus monkey disease model for human microcephaly. Cell Res. 26, 1048–1061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pirozzi F, Nelson B, Mirzaa G, From microcephaly to megalencephaly: determinants of brain size. Dialogues Clin. Neurosci. 20, 267–282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F. Ji S, Zhou Q, Huang X, Ji W., Sha J, Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Chen Y, Cui Y, Shen B, Niu Y, Zhao X, Wang L, Wang J, Li W, Zhou Q, Ji W, Sha J, Huang X, Germline acquisition of Cas9/RNA-mediated gene modifications in monkeys. Cell Res. 25, 262–265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wan H, Feng C, Teng F, Yang S, Hu B, Niu Y, Xiang AP, Fang W, Ji W, Li W, Zhao X, Zhou Q, One-step generation of p53 gene biallelic mutant cynomolgus monkey via the CRISPR/Cas system. Cell Res. 25, 58–261 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tu Z, Yang W, Yan S, Yin A, Gao J, Liu X, Zheng Y, Zheng J, Li Z, Yang S, Li S, Guo X, Li X-J, Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep. 7, 42081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Midic U, Hung P-H, Vincent KA, Goheen B, Schupp PG, Chen DD, Bauer DE, VandeVoort CA, Latham KE, Quantitative assessment of timing, efficiency, specificity and genetic mosaicism of CRISPR/Cas9-mediated gene editing of hemoglobin beta gene in rhesus monkey embryos. Hum. Mol. Genet. 26, 2678–2689 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacobi AM, Rettig GR, Turk R, Collingwood MA, Zeiner SA, Quadros RM, Harms DW, Bonthuis PJ, Gregg C, Ohtsuka M, Gurumurthy CB, Behlke MA, Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods 121–122, 16–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen Y, Zheng Y, Kang Y, Yang W, Niu Y, Guo X, Tu Z, Si C, Wang H, Xing R, Pu X, Yang S-H, Li S, Ji W, Li X-J,.Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum. Mol. Genet. 24, 3764–3774 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Ren S, Bai R, Xiao P, Zhou Q, Zhou Y, Zhou Z, Niu Y, Ji W, Chen Y, No off-target mutations in functional genome regions of a CRISPR/Cas9-generated monkey model of muscular dystrophy. J. Biol. Chem. 293, 11654–11658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Costales JL, Kolevzon A, Phelan-McDermid Syndrome and SHANK3: Implications for Treatment. Neurotherapeutics 12, 620–630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao H, Tu Z, Xu H, Yan S, Yan H, Zheng Y, Yang W, Zheng J, Li Z, Tian R, Lu Y, Guo X, Jiang Y-H, Li XJ, Zhang YQ, Altered neurogenesis and disrupted expression of synaptic proteins in prefrontal cortex of SHANK3-deficient non-human primate. Cell Res. 27, 1293–1297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Y, Sharma J, Ke Q, Landman R, Yuan J, Chen H, Hayden DS, Fisher JW 3rd, Jian M, Menegas W, Aida T, Yan T, Zou Y, Xu D, Parmar S, Hyman JB, Fanucci-Kiss A, Meisner O, Wang D, Huang Y, Li Y, Bai Y, Ji W, Lai X, Li W, Huang L, Lu Z, Wang L, Anteraper SA, Sur M, Zhou H, Xiang AP, Desimone R, Feng G, Yang S, Atypical behavior and connectivity in SHANK3-mutant macaques. Nature 570, 326–331 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Tu Z, Zhao H, Li B, Yan S, Wang L, Tang Y, Li Z, Bai D, Li C, Lin Y, Li Y, Liu J, Xu H, Guo X, Jiang Y-H, Zhang YQ, Li X-J, CRISPR/Cas9-mediated disruption of SHANK3 in monkey leads to drug-treatable autism-like symptoms. Hum. Mol. Genet. 28, 561–571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Achermann JC, Weiss J, Lee EJ, Jameson JL, Inherited disorders of the gonadotropin hormones. Mol. Cell. Endocrinol. 179, (2001). [DOI] [PubMed] [Google Scholar]
- 66.Kang Y, Zheng B, Shen B, Chen Y, Wang L, Wang J, Niu Y, Cui Y, Zhou J, Wang H, Guo X, Hu B, Zhou Q, Sha J, Ji W, Huang X, CRISPR/Cas9-mediated Dax1 knockout in the monkey recapitulates human AHC-HH. Hum. Mol. Genet. 24, 7255–7264 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Zhang W, Wan H, Feng G, Qu J, Wang J, Jing Y, Ren R, Liu Z, Zhang L, Chen Z, Wang S, Zhao Y, Wang Z, Yuan Y, Zhou Q, Li W, Liu G-H, Hu B, SIRT6 deficiency results in developmental retardation in cynomolgus monkeys. Nature 560, 661–665 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Qiu P, Jiang J, Liu Z, Cai Y, Huang T, Wang Y, Liu Q, Nie Y, Liu F, Cheng J, Li Q, Tang Y-C, Poo M-M, Chang QSH-C, BMAL1 knockout macaque monkeys display reduced sleep and psychiatric disorders. Natl. Sci. Rev. 6, 87–100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gekakis N, Stankis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ, Role of the CLOCK protein in the mammalian circadian mechanism. Science (New York, N.Y.) 280, (1998). [DOI] [PubMed] [Google Scholar]
- 70.Schulte C, Gasser T, Genetic basis of Parkinson’s disease: inheritance, penetrance, and expression. Appl Clin Genet. 4, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang W, Liu Y, Tu Z, Xiao C, Yan S, Ma X, Guo X, Chen X, Yin P, Yang Z, Jiang T, Li S, Qin C, Li X-J, CRISPR/Cas9-mediated PINK1 deletion leads to neurodegeneration in rhesus monkeys. Cell Res. 29, 334–336 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang W, Li S, Li XJ, A CRISPR monkey model unravels a unique function of PINK1 in primate brain. Mol Neurodegener. 14, 17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsukiyama T, Kobayashi K, Nakaya M, Iwatani C, Seita Y, Tsuchiya H, Matsushita J, Izumi H, Itagaki I, Kume S, Maegawa H, Nishinakamura R, Nishio S, Nakamura S, Kawauchi A, Ema M, Monkeys Mutant for PKD1 Recapitulate Human Autosomal Dominant Polycystic Kidney Disease. Nat. Commun. 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeballos C MA., Gaj T. Next-Generation CRISPR Technologies and Their Applications in Gene and Cell Therapy. Trends Biotechnol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang F, Zhang W, Yang Q, Kang Y, Fan Y, Wei J, Liu Z, Dai S, Li H, Li Z, Xu L, Chu C, Qu J, Si C, Ji W, Liu GH, Long C, Niu Y. Generation of a Hutchinson-Gilford progeria syndrome monkey model by base editing. Protein & Cell 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang W, Aida T, Del Rosario RCH, Wilde JJ, Ding C, Zhang X, Baloch Z, Huang Y, Tang Y, Li D, Lu H, Zhou Y, Jiang M, Xu D, Fang Z, Zheng Z, Huang Q, Feng G, Yang S. Multiplex precise base editing in cynomolgus monkeys. Nat. Commun. 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo X, He Y, Zhang C, He X, Yan L, Li M, Hu T, Hu Y, Jiang J, Meng X, Ji W, Zhao X, Zheng P, Xu S, Su B, Trio deep-sequencing does not reveal unexpected off-target and on-target mutations in Cas9-edited rhesus monkeys. Nat. Commun. 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cui Y, Niu Y, Zhou J, Chen Y, Cheng Y, Li S, Ai Z, Chu C, Wang H, Zheng B, Chen X, Sha J, Guo X, Huang X, Ji W, Generation of a precise Oct4-hrGFP knockin cynomolgus monkey model via CRISPR/Cas9-assisted homologous recombination. Cell Res. 28, 383–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yao X, Liu Z, Wang X, Wang Y, Nie Y-H, Lai L, Sun R, Shi L, Sun Q, Yang H, Generation of a knock-in cynomolgus monkey via CRISPR/Cas9 editing. Cell Res. 28, 379–382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumita W, Sato K, Suzuki Y, Kurotaki Y, Harada T, Zhou Y, Kishi N, Sato K, Aiba A, Sakakibara Y, Feng G, Okano H, Sasaki E, Efficient generation of Knock-in/Knock-out marmoset embryo via CRISPR/Cas9 gene editing. Sci. Rep. 9, 12719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshimatsu S, Okahara J, Sone T, Takeda Y, Nakamura M, Sasaki E, Kishi N, Shiozawa S, Okano H, Robust and efficient knock-in in embryonic stem cells and early-stage embryos of the common marmoset using the CRISPR-Cas9 system. Sci. Rep. 9, 1528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chu C, Yang Z, Yang J, Yan L, Si C, Kang Y, Chen Z, Chen Y, Ji W, Niu Y, Homologous recombination-mediated targeted integration in monkey embryos using TALE nucleases. BMC Biotechnol. 19, 7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feng G, Jensen FE, Greely HT, Okano H, Treue S, Roberts AC, Fox JG, Caddick S, Poo M, Newsome WT, Morrison JH, Opportunities and limitations of genetically modified nonhuman primate models for neuroscience research. Proc. Natl. Acad. Sci. U. S. A. 117, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hamilton BA, alpha-Synuclein A53T substitution associated with Parkinson disease also marks the divergence of Old World and New World primates. Genomics 83, (2004). [DOI] [PubMed] [Google Scholar]
- 86.Peterson CW, Wang J, Norman KK, Norgaard ZK, Humbert O, Tse CK, Yan JJ, Trimble RG, Shivak DA, Rebar EJ, Gregory PD, Holmes MC, Kiem H-P, Long-term multilineage engraftment of autologous genome-edited hematopoietic stem cells in nonhuman primates. Blood 127, 2416–2426 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shin TH, Baek EJ, Corat MAF, Chen S, Metais J-Y, AlJanahi AA, Zhou Y, Donahue RE, Yu K-R, Dunbar CE, CRISPR/Cas9 PIG-A gene editing in nonhuman primate model demonstrates no intrinsic clonal expansion of PNH HSPCs. Blood 133, 2542–2545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vermilyea SC, Babinski A, Tran N, To S, Guthrie S, Kluss JH, Schmidt JK, Wiepz GJ, Meyer MG, Murphy ME, Cookson MR, Emborg ME, Golos TG, In Vitro CRISPR/Cas9-Directed Gene Editing to Model LRRK2 G2019S Parkinson’s Disease in Common Marmosets. Sci. Rep. 10, 3447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Curnow E, Hayes E, In Vitro Culture of Embryos From the Cynomolgus Macaque (Macaca fascicularis). Methods Mol. Biol. (Clifton, N. J.) 2006, (2019). [DOI] [PubMed] [Google Scholar]
- 90.Ramsey C, Hanna C, In Vitro Culture of Rhesus Macaque (Macaca mulatta) Embryos. Methods Mol. Biol. (Clifton, N.J.) 2006, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.VandeVoort CA, Mtango NR, Midic U, Latham KE, Disruptions in follicle cell functions in the ovaries of rhesus monkeys during summer. Physiol. Genomics 47, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schmidt JK, Golos TG, In Vitro Culture of Embryos from the Common Marmoset (Callithrix jacchus). Methods Mol. Biol. 2006, 309–319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kropp J, Di Marzo A, Golos T, Assisted reproductive technologies in the common marmoset: an integral species for developing nonhuman primate models of human diseasesdagger. Biol. Reprod. 96, 277–287 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wolf DP, Thomson JA, Zelinski-Wooten MB, Stouffer RL, In Vitro Fertilization-Embryo Transfer in Nonhuman Primates: The Technique and its Applications. Mol. Reprod. Dev. 27, 261–280 (1990). [DOI] [PubMed] [Google Scholar]
- 95.Sparman ML, Ramsey CM, Thomas CM, Mitalipov SM, Fanton JW, Maginnis GM, Stouffer RL, Wolf DP, Evaluation of the Vervet (Clorocebus aethiops) as a Model for the Assisted Reproductive Technologies. Am. J. Primatol. 69, 917–929 (2007). [DOI] [PubMed] [Google Scholar]
- 96.Lima JS, Leao DL, Sampaio RV, Brito AB, Santos RR, Miranda MS, Ohashi OM, Domingues SFS, Embryo production by parthenogenetic activation and fertilization of in vitro matured oocytes from Cebus apella. Zygote (Cambridge, England) 21, (2013). [DOI] [PubMed] [Google Scholar]
- 97.Wolf DP, Artificial insemination and the assisted reproductive technologies in non-human primates. Theriogenology 71, 123–129 (2009). [DOI] [PubMed] [Google Scholar]
- 98.Naveed M, Kazmi SK, Amin M, Asif Z, Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genet. Res (Camb.) 100, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]