

Abstract
Meningiomas are the most common primary intracranial tumors. There are no effective medical therapies for meningioma patients, and new treatments have been encumbered by limited understanding of meningioma biology. Here we use DNA methylation profiling on 565 meningiomas integrated with genetic, transcriptomic, biochemical, proteomic, and single-cell approaches to show meningiomas are comprised of 3 DNA methylation groups with distinct clinical outcomes, biological drivers, and therapeutic vulnerabilities. Merlin-intact meningiomas (34%) have the best outcomes and are distinguished by NF2/Merlin regulation of susceptibility to cytotoxic therapy. Immune-enriched meningiomas (38%) have intermediate outcomes and are distinguished by immune infiltration, HLA expression, and lymphatic vessels. Hypermitotic meningiomas (28%) have the worst outcomes and are distinguished by convergent genetic and epigenetic mechanisms driving the cell cycle and resistance to cytotoxic therapy. To translate these findings into clinical practice, we show cytostatic cell cycle inhibitors attenuate meningioma growth in cell culture, organoids, xenografts, and patients.
DNA methylation profiling was performed on 565 meningiomas from patients with comprehensive clinical follow-up who were treated at 2 independent institutions from 1991 to 2019 (Supplementary table 1). Consistent with typical meningioma outcomes, local freedom from recurrence (LFFR) and overall survival (OS) were worse with higher World Health Organization (WHO) grade, recurrent presentation, or subtotal resection (Supplementary figure 1). Meningiomas were stratified into a 200-sample discovery cohort from the University of California San Francisco (median follow-up 6.3 years), and a consecutive 365-sample validation cohort from The University of Hong Kong (median follow-up 5.3 years) (Supplementary table 1). Meningiomas have an abundance of genomic copy number variants (CNVs)1,2 (Figure 1a), and standard bioinformatic approaches can report inaccurate β methylation values at genomic loci with CNVs3. We hypothesized that controlling for the influence of CNVs on β methylation values, coupled with mechanistic and functional studies, may reveal insights into meningioma biology and inform new treatments for meningioma patients. Prior meningioma DNA methylation studies have not accounted for the influence of CNVs on β methylation values, and have reported variable groups of tumors2,4–8, sometimes with overlapping clinical outcomes4. Thus, questions remain regarding the biological drivers and therapeutic vulnerabilities across meningioma DNA methylation groups.
Figure 1. Meningiomas are comprised of 3 DNA methylation groups with distinct outcomes.
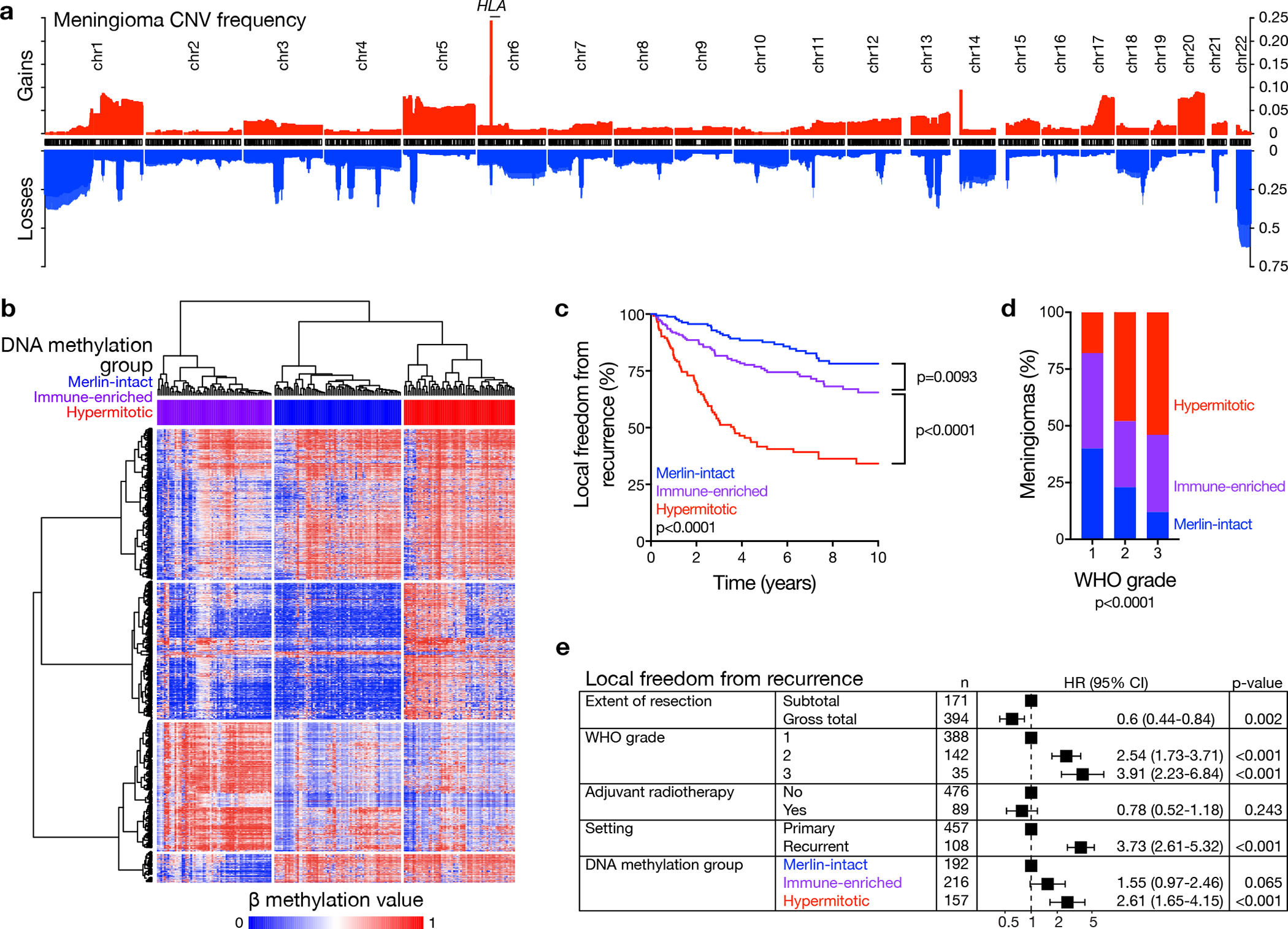
a, Frequency of copy number losses (blue) and gains (red) across the discovery and validation cohorts (n=565). b, Unsupervised hierarchical clustering of meningiomas from the discovery cohort (n=200) using 2000 differentially methylated DNA probes. c, Kaplan-Meier curves for meningioma local freedom from recurrence from the discovery and validation cohorts (n=565) across DNA methylation groups (Log-rank test). d, Meningioma WHO grades (n=565) across DNA methylation groups (Chi-squared test, two-sided). e, Multivariable regression hazard ratio (HR) forest plots for local freedom from recurrence using meningioma clinical variables and DNA methylation groups (n=565, Cox proportional hazards model, Wald test, two-sided, no adjustment for multiple comparisons). Boxes represent means, and error bars represent 95% confidence intervals (CI).
The 565 meningioma DNA methylation profiles comprising our discovery and validation cohorts were analyzed using the SeSAMe preprocessing pipeline (Extended data figure 1a–e), which controls for the influence of homozygous or heterozygous CNVs on β methylation values3. K-means consensus clustering, continuous distribution functions, and unsupervised hierarchical clustering revealed 3 meningioma DNA methylation groups in the discovery cohort (Figure 1b and Extended data figure 1a–e), which was validated as the optimal number of groups in the validation cohort (Extended data figure 2a). A multi-class support vector machine classifier with 97.9% accuracy (95% CI 89.2–99.9%, p<2.2×10−16) was constructed to assign meningiomas from the validation cohort into DNA methylation groups (Extended data figure 2b). Kaplan-Meier analyses showed DNA methylation groups were distinguished by differences in LFFR and OS (Figure 1c and Supplementary figure 2a), and correlated with WHO grade, sex, prior radiotherapy, and location (Figure 1d and Supplementary figure 2b). DNA methylation groups were independently prognostic for LFFR on Kaplan-Meier analysis across WHO grades ( Supplementary figure 2c, d), and on multivariable regression (Figure 1e and Supplementary figure 2e).
Meningioma DNA methylation groups had different CNV landscapes in aggregate (Figure 1a and Supplementary figure 4a, 4b), but individual CNVs or combinations of CNVs were insufficient to define all meningiomas in each DNA methylation group (Supplementary figure 4c). Reanalysis using the minfi preprocessing pipeline9, which does not control for the influence of CNVs on β methylation values, reassigned 21% of meningiomas across an unclear number of DNA methylation groups with overlapping clinical outcomes (Extended data figure 3). Thus, controlling for the influence of CNVs on β methylation values improves meningioma DNA methylation grouping and discrimination of clinical outcomes.
NF2/Merlin drives meningioma apoptosis
Meningiomas are common in patients with neurofibromatosis type 2, a complex autosomal syndrome caused by loss of NF2 on chromosome 22q, which encodes the tumor suppressor protein Merlin10. NF2 is also the most recurrently mutated gene in sporadic and radiation-induced meningiomas11–15. CNVs defined using DNA methylation profiles revealed chromosome 22q copy number deletions of any size containing the entire NF2 locus in 86% of syndromic (n=18) or radiation-induced meningiomas (n=34), which were predominantly found in DNA methylation groups with intermediate or poor clinical outcomes (96%). Only 17% of meningiomas in the DNA methylation group with the best outcomes had chromosome 22q copy number deletions of any size containing the entire NF2 locus (n=32 of 192), compared to 76% (n=165 of 216) or 98% (n=154 of 157) of meningiomas in the DNA methylation groups with intermediate or poor outcomes, respectively (Figure 2a and Supplementary table 2). RNA sequencing of 200 meningiomas from the discovery cohort confirmed higher NF2 expression in the DNA methylation group with the best outcomes compared to other groups (Figure 2b). The combined distribution of NF2 copy number deletions and somatic short variants from DNA amplicon sequencing of 65 meningiomas showed 89% of tumors in the DNA methylation group with the best outcomes encoded at least 1 wildtype copy of NF2 (Extended data figure 4a and Supplementary table 3). An orthogonal comparison of meningiomas with deletion of at least 1 copy of NF2 revealed Merlin protein was only expressed in the DNA methylation group with the best outcomes (Figure 2c).
Figure 2. NF2/Merlin drives meningioma apoptosis and susceptibility to cytotoxic therapy.
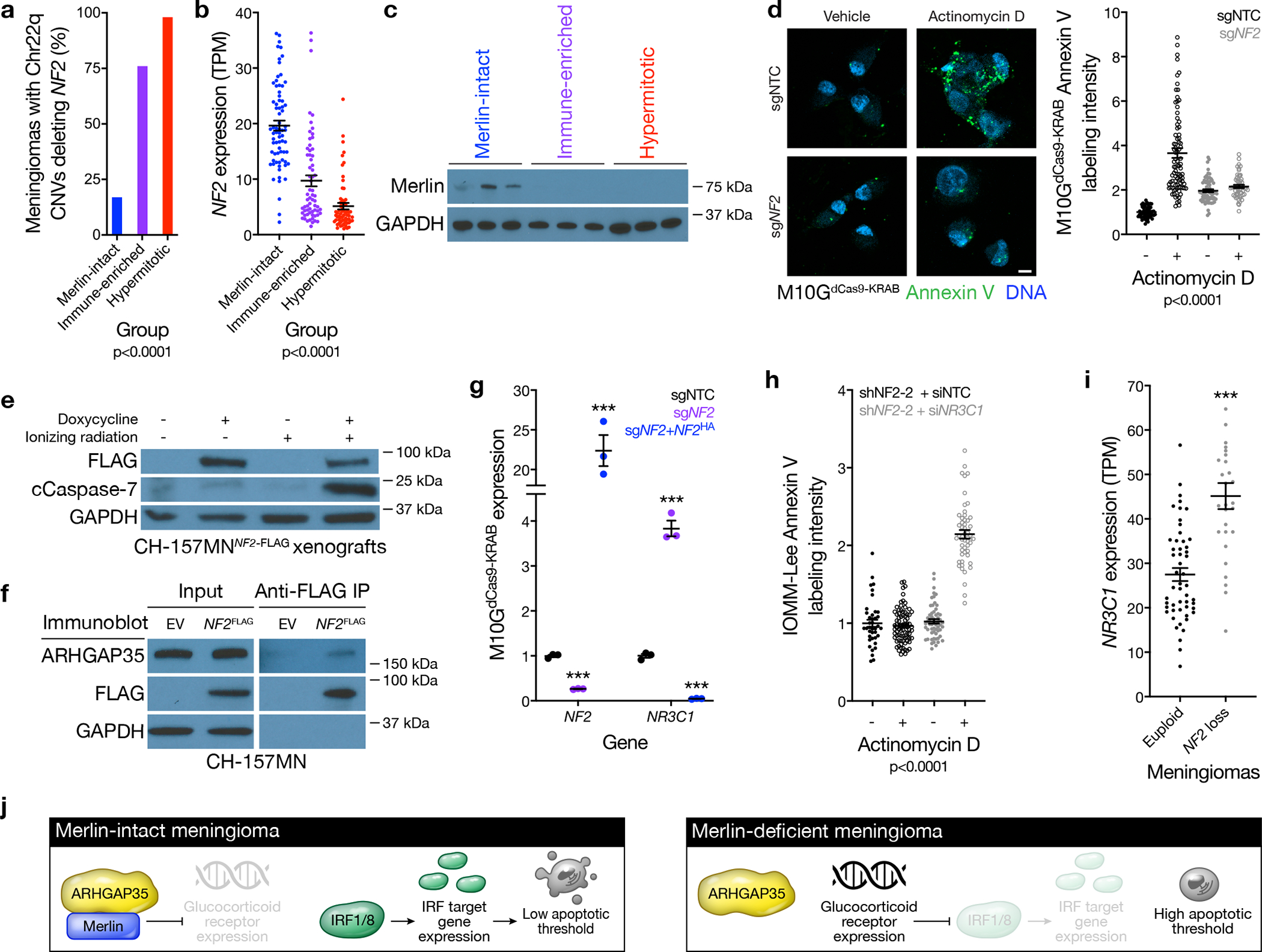
a, Meningioma DNA methylation analysis of chromosome 22q segment copy number deletions of any size containing the entire NF2 locus across Merlin-intact (n=32 of 192 meningiomas, 17%), Immune-enriched (n=165 of 216 meningiomas, 76%), and Hypermitotic (n=154 of 157 meningiomas, 98%) DNA methylation groups (n=565, Chi-squared test, two-sided). b, Meningioma NF2 transcripts per million (TPM) expression across Merlin-intact (n=72), Immune-enriched (n=65), and Hypermitotic (n=63) DNA methylation groups (n=200, ANOVA, one-sided). c, Immunoblot for Merlin or GAPDH in 3 meningiomas with loss of at least one copy of the NF2 locus from each meningioma DNA methylation group. d, Confocal microscopy and quantification of Annexin V in M10GdCas9-KRAB cells stably expressing a non-targeting control single-guide RNA (sgNTC) or a single-guide RNA suppressing NF2 (sgNF2) after 24 hours of actinomycin D or vehicle control treatment. DNA is marked with DAPI. Scale bar 10 μM. From left to right, 53, 88, 69, or 56 cells are shown (ANOVA, one-sided). e, Immunoblot for FLAG, cleaved Caspase-7 (cCaspase-7), or GAPDH from CH-157MN xenografts stably expressing doxycycline-inducible Merlin encoding a FLAG tag (NF2-FLAG) in NU/NU mice after 7 days of doxycycline or vehicle treatment, and 24 hours after 4 Gy ionizing radiation or control treatment. f, Immunoblot for ARHGAP35 or FLAG after FLAG immunoprecipitation from CH-157MN cells stably expressing Merlin encoding a FLAG tag (NF2FLAG). EV, empty vector. g, QPCR for NF2 or NR3C1 in M10GdCas9-KRAB cells stably expressing sgNTC, sgNF2, or sgNF2 with NF2 rescue (sgNF2+NF2HA). 3 biological replicates per condition (Student’s t test, one-sided). h, Quantification of Annexin V confocal microscopy in IOMM-Lee cells stably expressing a short-hairpin RNA suppressing NF2 (sgNF2-2) and transiently expressing a non-targeting control siRNA (siNTC) or siRNAs suppressing NR3C1 (siNR3C1). Cells were treated as in d. From left to right, 39, 80, 58, or 52 cells are shown (ANOVA, one-sided). i, NR3C1 TPM expression in euploid meningiomas (n=52) or meningiomas with loss of NF2 as the only CNV (n=28) (Student’s t test, one-sided). j, Model of Merlin pro-apoptotic tumor suppressor function in meningioma cells. Lines represent means, and error bars represent standard error of the means. ***p≤0.0001.
NF2 variants are mutually exclusive from TRAF7 somatic short variants in meningiomas11,13,16, and TRAF7 variants were enriched in Merlin-intact meningiomas compared to other groups (Supplementary table 4). Many Merlin-intact meningiomas did not encode TRAF7 variants (79%), suggesting the DNA methylation group with the best outcomes may not be unified by a single genetic driver. Indeed, meningioma histologic subtypes associated with AKT1E17K variants were enriched in Merlin-intact meningiomas compared to tumors from other groups17 (Supplementary table 4). Further, analysis of matched exome sequencing and DNA methylation profiling on 53 meningiomas revealed no solitary TRAF7, AKT1, KLF4, or other somatic short variants associated with favorable outcomes4,13,17,18 in DNA methylation groups with intermediate or poor outcomes (Supplementary table 5).
Merlin has myriad tumor suppressor functions in schwannoma cells19–21, but Merlin tumor suppressor functions in meningiomas are incompletely understood. M10G and IOMM-Lee meningioma cells express Merlin22,23, which inhibits cell proliferation in these cells (Extended data figure 4b–f). To identify gene expression programs underlying Merlin tumor suppressor functions in meningioma cells, RNA sequencing was performed on triplicate M10G cultures stably expressing the CRISPR interference (CRISPRi) components dCas9-KRAB24,25 and either non-targeting control sgRNA (sgNTC), sgRNA suppressing NF2 (sgNF2), or sgNF2 with NF2 rescue (Extended data figure 4c). Differential expression and ontology analyses revealed Merlin induced pro-apoptotic interferon regulatory factor (IRF) pathways (Extended data figure 4g, h and Supplementary table 6). Merlin suppression blocked IRF target gene expression in MSC1 cells22 (Extended data figure 4i, j), whose DNA methylation profiles were assigned as Merlin-intact using the multi-class support vector machine classifier. Thus, to determine if Merlin regulates meningioma cell apoptosis, MSC1, M10GdCas9-KRAB, and IOMM-Lee cultures were treated with the chemotherapy agent actinomycin D, revealing that Merlin suppression reduced apoptosis (Figure 2d and Extended data figure 5a–c). To define the relevance of this mechanism in vivo, CH-157MN meningioma cells, which do not express Merlin26, were grown as xenografts harboring an inducible Merlin construct. Merlin rescue in CH-157MN xenografts increased apoptosis in response to ionizing radiation compared to meningiomas lacking Merlin (Figure 2e and Extended data figure 5d).
Merlin regulates protein degradation to control schwannoma cell proliferation21, but Merlin suppression did not alter IRF stability or subcellular localization in meningioma cells (Extended data figure 5e). To determine if Merlin instead regulates IRF activity, Merlin constructs encoding ascorbic acid peroxidase (APEX) tags were expressed in meningioma cells for proximity-labeling proteomic mass spectrometry27 (Supplementary table 7). ARHGAP35, a DNA binding factor that inhibits glucocorticoid receptor expression28,29, was detected in proximity to wild-type Merlin but not MerlinL46R, a missense variant associated with neurofibromatosis type 221 (Extended data figure 5f). Glucocorticoid signaling inhibits IRF activity to prevent apoptosis30,31, and immunoprecipitation of Merlin from meningioma cells validated Merlin interaction with ARHGAP35 (Figure 2f). IRF proteins were not detected in proximity to MerlinAPEX constructs (Supplementary table 7), or in Merlin immunoprecipitates (Extended data figure 5g), suggesting Merlin indirectly regulates IRF activity through ARHGAP35. In support of this hypothesis, NF2 suppression in meningioma cells induced glucocorticoid receptor expression, which was inhibited by NF2 rescue (Figure 2g). Further, glucocorticoid receptor suppression rescued meningioma cell apoptosis in the absence of Merlin (Figure 2h and Extended data figure 5h), and glucocorticoid receptor expression was increased in human meningiomas with NF2 loss compared to euploid tumors (Figure 2i). In sum, these data shed light on a pro-apoptotic tumor suppressor function of Merlin regulating glucocorticoid signaling and susceptibility to cytotoxic therapy in meningiomas (Figure 2j).
Convergent mechanisms underlie meningioma immune enrichment
Meningiomas in the DNA methylation group with intermediate outcomes had fewer CNVs than other groups (Supplementary figure 3b), suggesting bulk bioinformatic analyses of meningiomas in this group may be influenced by non-tumor cells in the meningioma microenvironment (Supplementary table 8). SeSAMe cell-type deconvolution of DNA methylation profiles showed immune cell enrichment in the meningioma DNA methylation group with intermediate outcomes compared to other groups (Figure 3a). xCell RNA sequencing deconvolution and PAMES tumor purity analysis validated these findings32,33 (Supplementary figure 4a–d). Further, differential expression and gene ontology analyses showed enrichment of immune genes in the meningioma DNA methylation group with intermediate outcomes (Supplementary figure 4e and Supplementary table 9), and immunohistochemistry revealed T cell enrichment in this group compared to tumors from other groups (Figure 3b). Differential expression and gene ontology analyses across minfi groups failed to distinguish meningiomas by immune or inflammatory gene expression programs (Supplementary figure 4f).
Figure 3. HLA expression and meningeal lymphatics underlie meningioma immune enrichment.
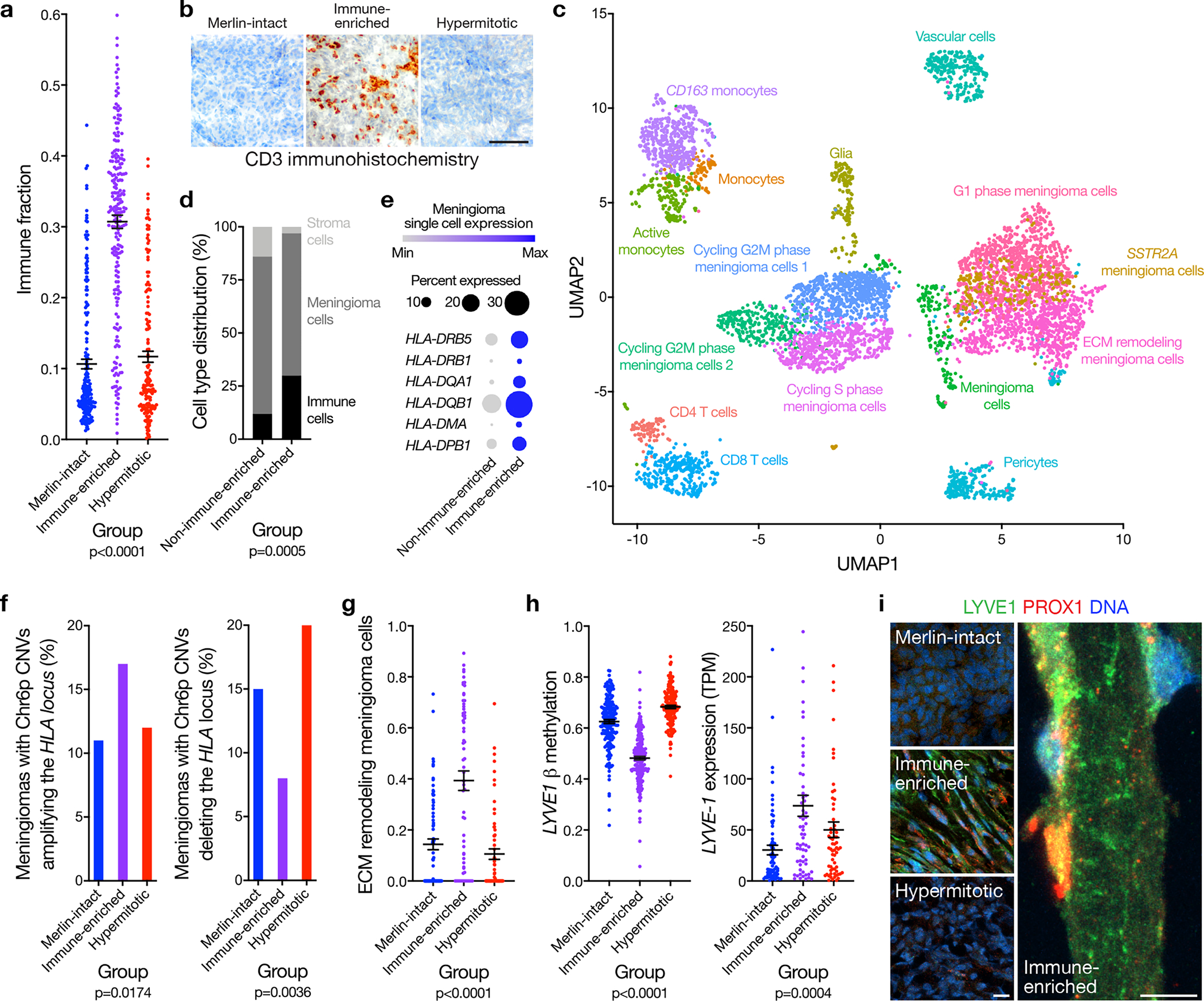
a, Meningioma DNA methylation leukocyte fractions (n=565) across DNA methylation groups (ANOVA, one-sided). b, Representative images of T cell immunohistochemistry across meningioma DNA methylation groups (n=87, p<0.0001, Chi-squared test, two-sided). Scale bar 100 μM. c, UMAP of single-cell RNA sequencing transcriptomes of 57,114 cells from 8 human meningioma samples and 2 human dura samples, colored by assignments from Louvain clustering. d, Quantification of single-cell types from c in Immune-enriched (n=5) and non-Immune-enriched (n=3) meningioma samples (Chi-squared test, two-sided). e, Single-cell RNA sequencing relative expression of HLA genes in meningioma cells across Immune-enriched (n=5) and non-Immune-enriched (n=3) meningioma samples. Circle size denotes percentage of cells. Shading denotes average expression. f, Meningioma DNA methylation analysis of chromosome 6p segment CNVs containing the entire polymorphic HLA locus encompassing HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1 across Merlin-intact (n=192 meningiomas, 21 gains, 28 losses), Immune-enriched (n=216 meningiomas, 37 gains, 18 losses), and Hypermitotic (n=157 meningiomas, 12 gains, 32 losses) DNA methylation groups (Chi-squared test, two-sided). g, Fraction of meningioma samples (n=200) classified as extracellular matrix (ECM) remodeling meningioma cells across DNA methylation groups, based on single-cell reference transcriptomes from c (ANOVA, one-sided). h, Meningioma DNA methylation (n=565) of LYVE1 (cg26455970) and transcripts per million (TPM) expression (n=200) of LYVE-1 across DNA methylation groups (ANOVA, one-sided). i, Representative images of meningioma LYVE1 and PROX1 immunofluorescence microscopy across DNA methylation groups (n=12). DNA is marked with Hoechst 33342. Scale bars 10 μM. Lines represent means, and error bars represent standard error of the means.
To define the cellular architecture of meningiomas, single-cell RNA sequencing was performed on 57,114 cells from 8 meningioma samples with each DNA methylation group represented (Figure 3c and Supplementary figure 5). Reduced dimensionality clusters of meningioma and non-meningioma cells were distinguished by chromosome 22q loss using CONICSmat34 (Supplementary figure 6a). Non-meningioma cell clusters with intact chromosome 22q were classified by expression of immune, neural, or vascular marker genes (Supplementary figure 6b, c and Supplementary table 10). Meningioma cell clusters with chromosome 22q loss were distinguished by differentially expressed cellular pathways or meningioma marker genes (Supplementary figure 5c, 6b, 6c and Supplementary table 10). Single-cell transcriptomes revealed more immune cells in Immune-enriched meningiomas compared to tumors from other groups (Figure 3d). Further, analysis of DNA methylation profiles on 86 spatially-distinct samples from 13 meningiomas revealed 92% of samples classified in concordance with the consensus DNA methylation group of each tumor22 (Supplementary figure 7a), suggesting meningioma DNA methylation grouping is not significantly confounded by intratumor heterogeneity or spatial sampling bias.
Single-cell transcriptomes demonstrated increased expression of HLA-DRB5, HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DMA, and HLA-DPB1 in Immune-enriched meningioma cells compared to meningioma cells from other groups (Figure 3e). HLA loss on chromosome 6p can decrease immune infiltration in cancer35, and there was a polymorphic locus on chromosome 6p encompassing HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1, with recurrent gains and losses across meningioma DNA methylation groups (Figure 1a, Supplementary figure 3a, and Supplementary table 2). Copy number amplifications of any size containing the entire polymorphic HLA locus were more frequent in Immune-enriched meningiomas (17%, n=37 of 216) compared to Merlin-intact (11%, n=21 of 192) or Hypermitotic meningiomas (12%, n=12 of 157) (p=0.0174, Chi-squared test) (Figure 3f). Conversely, copy number deletions of any size containing the entire polymorphic HLA locus were less frequent in Immune-enriched meningiomas (8%, n=18 of 216) compared to Merlin-intact (15%, n=28 of 192) or Hypermitotic meningiomas (20%, n=32 of 157) (p=0.0036, Chi-squared test) (Figure 3f). Analysis of matched whole exome sequencing and DNA methylation profiling revealed no instances of HLA loss of heterozygosity in Immune-enriched meningiomas2 (Supplementary figure 7b). Expression of HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1 correlated with CNVs amplifying or deleting these genes, and the expression of HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1 was enriched in Immune-enriched meningiomas compared to other groups (Supplementary figure 7c). HLA-DMA and HLA-DPB1, located outside the polymorphic locus on chromosome 6p, had increased expression in single-cell transcriptomes from Immune-enriched meningioma cells (Figure 3e), and were hypomethylated with increased expression in Immune-enriched meningiomas compared to other groups (Supplementary figure 7d–f). In sum, these data reveal convergent genetic and epigenetic mechanisms underlying meningioma HLA expression.
To identify additional mechanisms underlying Immune-enriched meningiomas, reference transcriptomic signatures of meningioma single-cell clusters were generated using CIBERSORTx36, and cell type proportions across meningioma DNA methylation groups were deconvolved from RNA sequencing of the discovery cohort (Extended data figure 6a). Changes in the extracellular matrix can create a permissive microenvironment for lymphatic vessel formation37–40. Accordingly, we observed meningioma cells expressing extracellular matrix remodeling genes were enriched in Immune-enriched meningiomas compared to tumors from other groups (Figure 3g). Consistently, DNA methylation probes that were hypomethylated in Immune-enriched meningiomas compared to other groups were concentrated at genes involved in vasculature, vessel, and circulatory system development (Supplementary table 8). Meningeal lymphatics near dural venous sinuses are necessary for central nervous system immune surveillance41–45, but preoperative magnetic resonance imaging showed meningiomas from multiple DNA methylation groups were likely to involve dural venous sinuses (Extended data figure 6b, c), suggesting meningioma location may be necessary but not sufficient for immune infiltration. Compared to tumors from other DNA methylation groups, Immune-enriched meningiomas had hypomethylation and increased expression of meningeal lymphatic genes such as LYVE1, CCL21, and CD3E44,46–48 (Figure 3h and Extended data figure 6d, e). Immunofluorescence confirmed lymphatic enrichment in Immune-enriched meningiomas compared to tumors from other groups (Figure 3i), and also in CH-157MN xenografts (Extended data figure 6f), whose DNA methylation profiles were assigned as Immune-enriched using the multi-class support vector machine classifier. Thus, HLA expression and lymphatic vessels are associated with meningioma immune infiltration.
Convergent mechanisms misactivate the meningioma cell cycle
High-grade meningiomas are defined by brisk cell proliferation leading to local recurrence and death in 50–90% of patients49,50, and accordingly, cell proliferation was highest in the meningioma DNA methylation group with the worst clinical outcomes (Figure 4a). To elucidate mechanisms driving cell proliferation in Hypermitotic meningiomas, RNA sequencing, gene ontology analysis, and immunohistochemistry revealed FOXM1 and the FOXM1 gene expression program were enriched in Hypermitotic meningiomas compared to tumors from other groups (Figure 4b, Supplementary figure 8a, and Supplementary table 9), correlating with meningioma cell proliferation ( Supplementary figure 8b, c). FOXM1 is a biomarker for meningioma recurrence and drives meningioma cell proliferation, and putative FOXM1 target genes are accessible in the chromatin of meningioma DNA methylation groups with adverse clinical outcomes2,7,51,52. To define the specificity of FOXM1 signaling across meningioma DNA methylation groups, differential expression and ontology analyses were compared between Hypermitotic meningiomas with elevated Ki-67 labeling index and non-Hypermitotic meningiomas with elevated Ki-67 labeling index, revealing the FOXM1 gene expression program was specifically enriched in Hypermitotic meningiomas (Supplementary figure 8d). Although many FOXM1 targets are induced by other cell cycle regulators, such as E2F153, FOXM1 targets that were nonoverlapping with E2F1 targets were enriched in Hypermitotic meningiomas compared to tumors from other groups (Supplementary figure 8e), and E2F1 targets that were nonoverlapping with FOXM1 targets did not distinguish meningioma DNA methylation groups (Supplementary figure 9). To elucidate the function of FOXM1 target genes in Hypermitotic meningiomas, differentially expressed genes with FOXM1 binding motifs were analyzed across 25 meningiomas with matched RNA sequencing, H3K27ac ChIP sequencing, and DNA methylation profiling54. FOXM1 targets in Hypermitotic meningiomas regulated the cell cycle, tumor metabolism, and the DNA damage response (Extended data figure 7a), suggesting FOXM1 may underlie Hypermitotic meningioma resistance to cytotoxic therapy. In support of this hypothesis, FOXM1 protein increased in meningioma cells treated with actinomycin D (Extended data figure 7b), and over-expression of FOXM1 increased meningioma cell resistance to cytotoxic chemotherapy (Extended data figure 7c, d).
Figure 4. Convergent genetic and epigenetic mechanisms misactivate the cell cycle in meningioma.
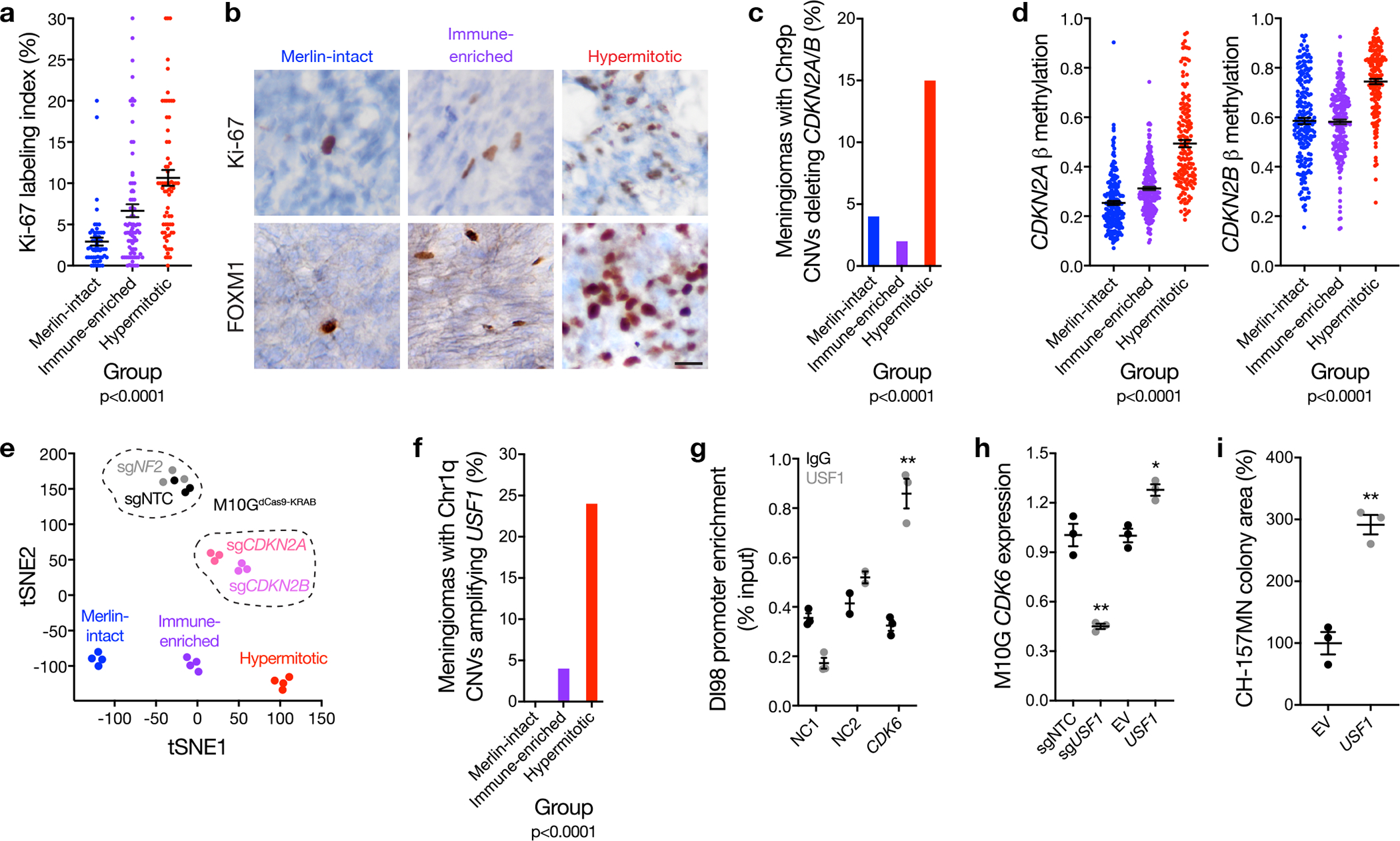
a, Ki-67 labeling index from meningioma clinical pathology reports (n=206) across DNA methylation groups (ANOVA, one-sided). b, Representative images of meningioma Ki-67 and FOXM1 immunohistochemistry (n=92) across meningioma DNA methylation groups. Scale bar 10 μM. c, Meningioma DNA methylation analysis of chromosome 9p segment copy number deletions of any size containing the entire CDKN2A/B locus across Merlin-intact (n=8 of 192 meningiomas, 4%), Immune-enriched (n=5 of 216 meningiomas, 2%), and Hypermitotic (n=24 of 157 meningiomas, 15%) DNA methylation groups (n=565, Chi-squared test, two-sided). d, Meningioma DNA methylation (n=565) of CDKN2A (cg26349275) or CDKN2B (cg08390209) across DNA methylation groups (ANOVA, one-sided). e, tSNE plot of meningioma and meningioma cell line DNA methylation profiles. Four representative meningiomas from each DNA methylation group are shown. Triplicate meningioma M10GdCas9-KRAB cultures stably expressing a non-targeting control single-guide RNA (sgNTC) or single-guide RNAs suppressing NF2 (sgNF2), CDKN2A (sgCDKN2A), or CDKN2B (sgCDKN2B) are shown. Differences in DNA methylation groups are captured in tSNE1, and a positive shift from Immune-enriched meningiomas to Hypermitotic meningiomas mimics the shift from M10GdCas9-KRAB-sgNTC and M10GdCas9-KRAB-sgNF2 cells to M10GdCas9-KRAB-sgCDKN2A and M10GdCas9-KRAB-sgCDKN2B cells. Differences between tumors and cell lines, such as the tumor microenvironment, are captured in tSNE2. f, Meningioma DNA methylation analysis of chromosome 1p segment copy number amplifications of any size containing the entire USF1 locus across Merlin-intact (n=0 of 192 meningiomas, 0%), Immune-enriched (n=2 of 216 meningiomas, 4%), and Hypermitotic (n=38 of 157 meningiomas, 24%) DNA methylation groups (n=565, Chi-squared test, two-sided). g, USF1 ChIP-QPCR in DI98 meningioma cells for the CDK6 promoter compared to negative control primers targeting a gene desert (NC1) or a gene not predicted to be bound by USF1 (NC2) from ChIP sequencing. **p=0.001 (Student’s t test, one-sided, no adjustment for multiple comparisons). h, QPCR for CDK6 in M10GdCas9-KRAB cells expressing sgNTC or a single-guide RNA suppressing USF1 (sgUSF1), or M10G cells over-expressing USF1 or empty vector (EV). *p=0.003, **p=0.001 (Student’s t test, one-sided, no adjustment for multiple comparisons). i, Relative colony area of CH-157MN cells stably over-expressing USF1 or EV after 10 days of clonogenic growth. **p=0.001 (Student’s t test, one-sided, no adjustment for multiple comparisons). Lines represent means, and error bars represent standard error of the means.
H3K27ac ChIP sequencing analysis of enhancer and super-enhancer availability showed Hypermitotic meningiomas were dominated by epigenetic regulators and transcription factors that are impractical pharmacologic targets (Supplementary figure 10). More broadly, druggable somatic short variants in meningiomas are rare, and are not associated with adverse clinical outcomes1,10–14,16,17,51,55–58, with infrequent exceptions59–62. There were more CNVs in Hypermitotic meningiomas compared to other groups (Supplementary figure 3a, b), and chromosome instability drives cancer evolution and response to therapy63–65. Thus, we hypothesized CNVs contributing to cell cycle misactivation may harbor therapeutic vulnerabilities informing new treatments for meningioma patients.
Loss of the endogenous CDK4/6 inhibitor CDKN2A/B on chromosome 9p is a rare biomarker for meningioma recurrence62,66. Copy number deletions of any size containing the entire CDKN2A/B locus were identified in 6.5% of meningiomas (n=37 of 565), but were enriched in Hypermitotic meningiomas (15%, n=24 of 157) compared to Merlin-intact (4%, n=8 of 192) or Immune-enriched meningiomas (2%, n=5 of 216) (Figure 4c and Supplementary table 2), and were associated with worse LFFR (Supplementary figure 11a). CDKN2A/B hypermethylation, an alternate mechanism of cell cycle misactivation in cancer67,68, was also more frequent in Hypermitotic meningiomas compared to other groups (Figure 4d). Stable suppression of CDKN2A or CDKN2B increased M10GdCas9-KRAB cell proliferation (Supplementary figure 11b, c), and re-assigned M10GdCas9-KRAB DNA methylation profiles from Immune-enriched to Hypermitotic using the multi-class support vector machine classifier. tSNE visualization of M10GdCas9-KRAB DNA methylation profiles in the context of Merlin-intact, Immune-enriched, and Hypermitotic meningiomas also revealed a shift in the DNA methylation profiles of M10GdCas9-KRAB cells from Immune-enriched to Hypermitotic meningiomas (Figure 4e).
Most Hypermitotic meningiomas did not have CNVs deleting CDKN2A/B (Figure 4c) or misactivation of the FOXM1 gene expression program (Supplementary figure 8e), and clustering of meningioma transcriptomes was unable to identify a unifying mechanism underlying Hypermitotic meningiomas (Supplementary figure 12). Thus, to identify additional mechanisms driving the cell cycle in meningiomas, transcription factor binding sites were mapped to regions of open chromatin defined by H3K27ac ChIP sequencing, revealing a binding site for the poorly understood transcription factor USF1 in the CDK6 promoter in meningiomas (Supplementary figure 13a and Supplementary table 11). Chromosome 1q copy number amplifications of any size containing the entire USF1 locus were enriched in Hypermitotic meningiomas (24%, n=38 of 157) compared to Merlin-intact (0%, n=0 of 192) or Immune-enriched meningiomas (2%, n=4 of 216) (Figure 4f, Supplementary table 2), and were associated with worse LFFR (Supplementary figure 13b). Mechanistic studies showed USF1 bound and activated the CDK6 promoter in Hypermitotic meningioma cells (Figure 4g, h and Supplementary figure 13c, d), and USF1 over-expression increased non-Hypermitotic meningioma cell proliferation (Figure 4i and Supplementary figure 13e).
To define the distribution of genetic and epigenetic mechanisms misactivating the cell cycle in Hypermitotic meningiomas, CDKN2A/B methylation (Figure 4d) or FOXM1 expression (Extended data figure 7 and Supplementary figure 8) in the top quartile of meningiomas were analyzed alongside CNVs of any size deleting the entire CDKN2A/B locus (Figure 4c) or amplifying the entire USF1 locus (Figure 4f). Among the 63 Hypermitotic meningiomas in the discovery cohort (Supplementary table 1), there were 13 tumors with CNVs deleting CDKN2A/B, 14 tumors with CNVs amplifying USF1, 37 tumors with CDKN2A hypermethylation, 32 tumors with CDKN2B hypermethylation, and 26 tumors with increased FOXM1 expression (Supplementary table 12). Removing duplicates, 52 of 63 Hypermitotic meningiomas in the discovery cohort had CDKN2A/B deletion, USF1 amplification, CDKN2A/B hypermethylation, or increased expression of FOXM1 (83%). Multiple genetic, epigenetic, or transcriptomic mechanisms misactivating the cell cycle were identified in 40 of 63 Hypermitotic meningiomas (63%).
Clinical translation of meningioma DNA methylation groups
Merlin regulation of glucocorticoid signaling drives meningioma apoptosis (Figure 2j), and Merlin-intact meningiomas have the best clinical outcomes with current therapies (Figure 1c and Supplementary figure 2a). In contrast, Immune-enriched and Hypermitotic meningiomas have adverse outcomes (Figure 1c and Supplementary figure 2a) and elevated cell proliferation (Figure 4a), and are resistant to cytotoxic therapies due to loss of Merlin (Figure 2j) or misactivation of FOXM1 (Extended data figure 7). The convergence of mechanisms driving cytotoxic resistance and cell cycle misactivation in Immune-enriched and Hypermitotic meningiomas suggests cytostatic cell cycle inhibitors may be effective treatments for meningiomas from these DNA methylation groups. To test this, the CDK4/6 inhibitors abemaciclib, palbociclib, and ribociclib were studied in cell culture, organoids, and xenografts using Immune-enriched and Hypermitotic meningioma cells. CDK4/6 inhibitors blocked clonogenic growth of meningioma cells without cell cycle modifications (Extended data figure 8a), but suppression of CDKN2A/B or over-expression of USF1 increased treatment sensitivity (Extended data figure 8b, c). To test this therapeutic strategy in the context of a tumor microenvironment, meningioma cells were co-cultured with cerebral organoids comprised of pre-differentiated human pluripotent stem-cell derived astrocytes. This model recapitulates intratumor heterogeneity in meningioma cells22, and intratumor heterogeneity drives resistance to therapy69, but CDK4/6 inhibition nevertheless attenuated meningioma cell growth in organoid co-culture (Extended data figure 8d). To define the pharmacodynamics and efficacy of this strategy for meningiomas in vivo, CH-157MN xenografts were treated with CDK4/6 inhibitors, which decreased RB phosphorylation (Extended data figure 8e), inhibited cell proliferation (Extended data figure 8f), attenuated xenograft growth (Figure 5a), and prolonged survival (Figure 5b).
Figure 5. Clinical translation of meningioma DNA methylation groups.
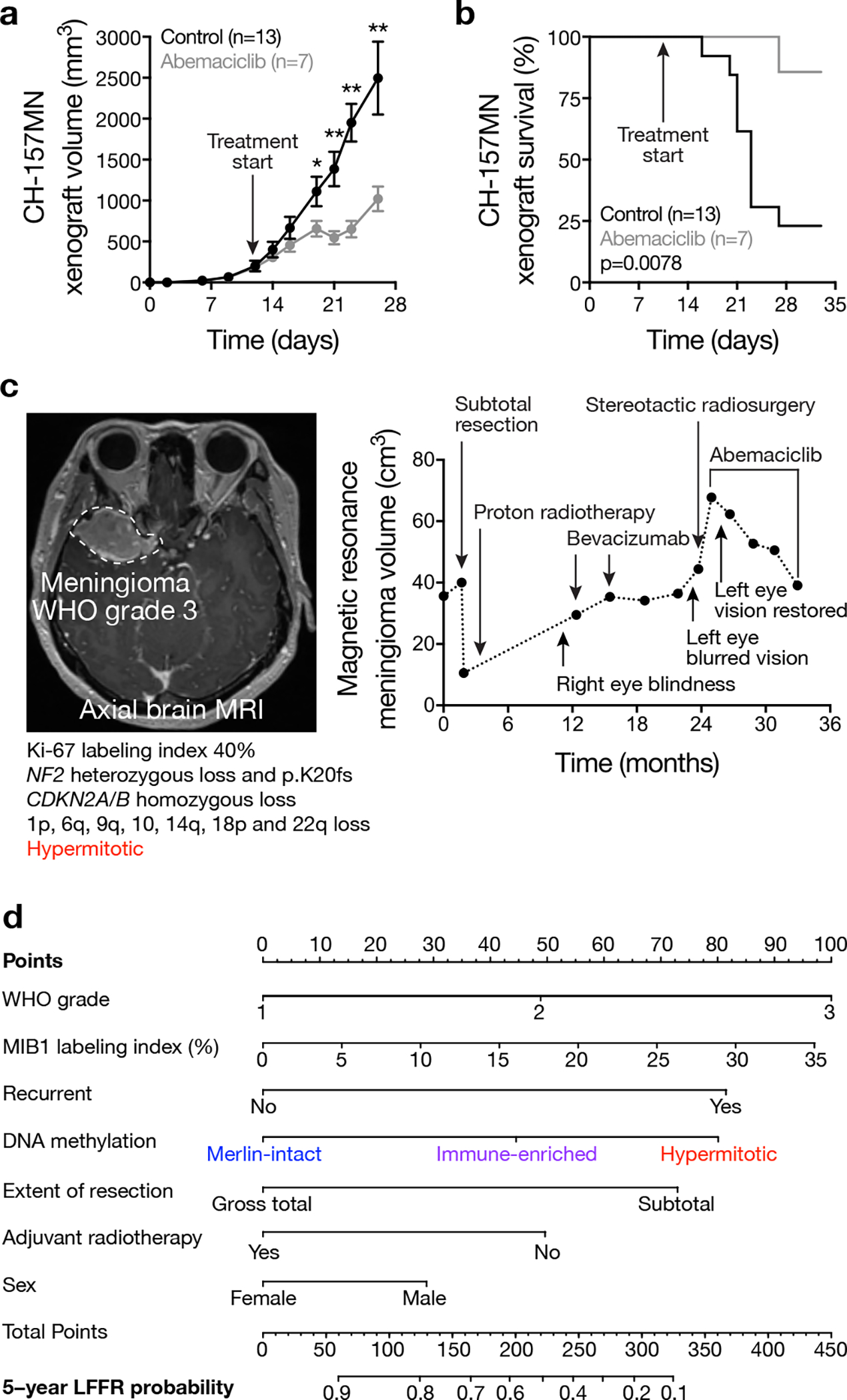
a, Subcutaneous CH-157MN xenograft measurements in NU/NU mice treated with abemaciclib (100 μg/g) by daily oral gavage with versus control. Lines represent means, and error bars represent standard error of the means. *p≤0.05, **p≤0.01 (Student’s t tests, one-sided). b, Kaplan-Meier curve for CH-157MN xenograft overall survival in NU/NU mice treated as in c (Log-rank test). c, Magnetic resonance imaging and molecular features of a representative human meningioma (left) that was resistant to cytotoxic therapies but responded to cytostatic cell cycle inhibition (right). d, Nomogram for meningioma local freedom from recurrence (LFFR, n=201) integrating clinical features and DNA methylation groups. Variables contribute points (top row), which estimate the probably of 5-year LFFR (bottom rows) (https://william-c-chen.shinyapps.io/RaleighLab_MethylationSubgroupNomogram/).
These data provide preclinical rationale to treat patients with Immune-enriched or Hypermitotic meningiomas with cell cycle inhibitors, which achieve therapeutic doses in human meningiomas70. In support of these preclinical investigations, we have observed encouraging early results with compassionate use of CDK4/6 inhibitors in patients with Hypermitotic meningiomas that are resistant to surgery and radiotherapy (Figure 5c and Supplementary figure 14). Clinical trials to establish the efficacy of this and other molecular therapies for meningiomas will require rigorous patient selection and biological stratification. In anticipation, we developed nomograms to compare models based on meningioma DNA methylation groups to models based on CNVs or clinical variables (Figure 5d and Supplementary figure 15, 16).
Discussion
DNA methylation profiling is a powerful tool for biological discovery, but clinical adoption of this approach has been encumbered by a lack of medical indications. The data presented here demonstrate a need for DNA methylation profiling to stratify meningioma patients for molecular therapies. Integrating genetic, epigenetic, transcriptomic, biochemical, proteomic, and single-cell approaches, we find meningiomas are comprised of 3 DNA methylation groups with distinct clinical outcomes and biological drivers. We validate our results using mechanistic and functional studies in cells, organoids, xenografts, and patients to elucidate mechanisms underlying therapeutic resistance and susceptibility in the most common primary intracranial tumor.
Our study tests the hypothesis that controlling for the influence of CNVs on β methylation values may reveal insights into meningioma biology. In support of this approach, meningioma DNA methylation analysis uncontrolled for the influence of CNVs on β methylation values could not identify an optimal number of meningioma groups (Extended data figure 3b, c), or groups with nonoverlapping differences in clinical outcomes, NF2 loss, immune enrichment, cell proliferation, and chromosome instability (Extended data figure 3d, 9). Quantifying the signal-to-noise ratio (SNR) of NF2 loss across meningioma DNA methylation groups, we found an SNR of 5.57 for 3 SeSAMe groups compared to 2.25 for 3 minfi groups.
Integrating epigenetic or gene expression features alongside genetic alterations may be important for understanding the biology of meningioma DNA methylation groups (Figure 6). We identified CNVs of any size deleting the entire CDKN2A/B locus or amplifying the entire USF1 locus in 54 of 157 Hypermitotic meningiomas (35%) (Supplementary table 2), but when integrated with CDKN2A/B hypermethylation and FOXM1 expression, the genetic, epigenetic, and transcriptomic alterations underlying meningioma cell proliferation we report were found in 83% of Hypermitotic tumors (Supplementary table 12). We used the same approach to integrate CNVs of any size amplifying the entire polymorphic HLA locus with hypomethylation of HLA-DMA, HLA-DPB1, or meningeal lymphatic genes (LYVE1, CCL21, CD3E) (Supplementary table 13). Removing duplicates, the epigenetic and genetic alterations underlying meningioma immune infiltration we report were found in 166 of 216 Immune-enriched meningiomas (77%). Multiple genetic or epigenetic mechanisms underlying immune infiltration were identified in 157 of 216 Immune-enriched meningiomas (73%).
Figure 6. Genetic, epigenetic, and transcriptomic mechanisms distinguishing meningioma DNA methylation groups.
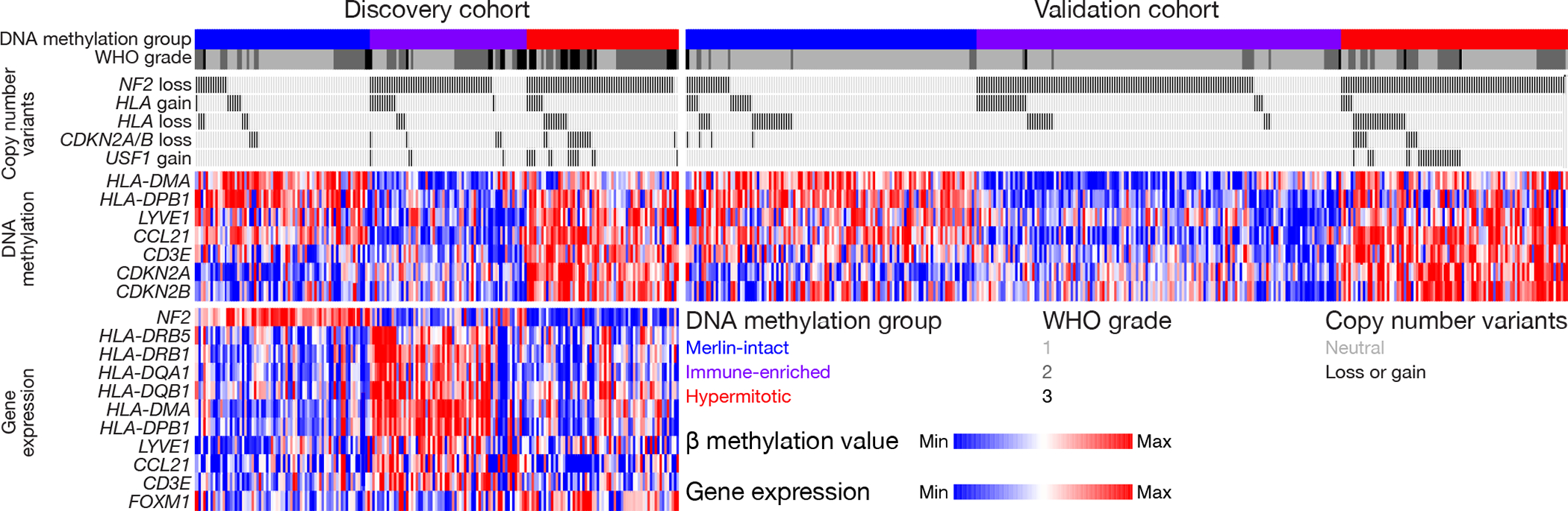
Oncoprint comprised of the 565 meningiomas in this study. CNVs of any size deleting or amplifying entire genes, scaled β methylation values, and scaled transcripts per million (TPM) are shown. The focal versus broad nature of CNVs are described in the Copy number analysis section of the Methods. HLA gain/loss shows the polymorphic locus encompassing HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1. β methylation values and TPM are scaled from the bottom 10th percentile to the top 90th percentile of each row. RNA sequencing was performed on the discovery cohort (n=200) but not on the validation cohort.
Our mechanistic and functional studies validate a biomarker-based treatment for meningiomas with adverse clinical outcomes. We find Immune-enriched meningiomas display multiple markers of immunoediting, and T cells in the meningioma microenvironment display multiple markers of exhaustion (Extended data figure 10). These data suggest immune checkpoint inhibition may be ineffective for Immune-enriched meningiomas, and shed light on why meningioma immune infiltration does not correlate with improved clinical outcomes71, as it does in other cancers72. Thus, we encourage careful consideration of meningioma DNA methylation groups in the context of available preclinical data, and WHO grade, when stratifying meningioma patients for new treatments. Indeed, our data demonstrate DNA methylation grouping does not obviate the importance of meningioma grading (Figure 1e and Supplementary figure 2e). These complementary systems provide independent information about meningioma outcomes, but unlike WHO grade, the meningioma DNA methylation groups we report reveal biological drivers and therapeutic vulnerabilities informing new treatments for meningioma patients (Figure 7).
Figure 7. Molecular, cellular, and clinical features distinguishing meningioma DNA methylation groups.
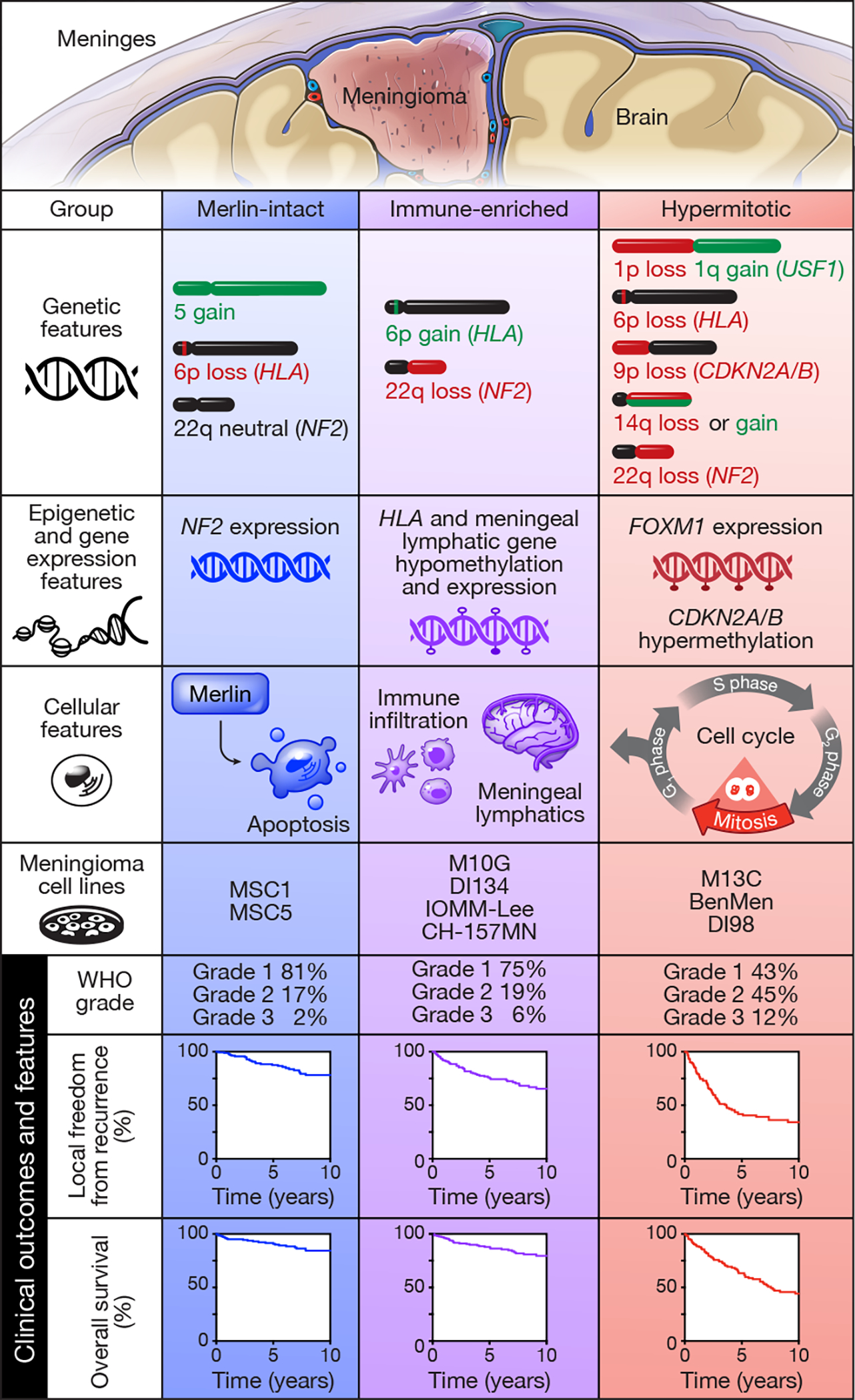
DNA methylation profiling was performed on 565 meningiomas and integrated with genetic, transcriptomic, biochemical, proteomic, and single-cell approaches to show meningiomas are comprised of 3 DNA methylation groups with distinct clinical outcomes, biological drivers, and therapeutic vulnerabilities. DNA methylation profiling was also performed on 9 meningioma cell lines to define reagents to study biological drivers and therapeutic vulnerabilities underlying meningioma DNA methylation groups.
Methods
This study complied with all relevant ethical regulations and was approved by the UCSF Institutional Review Board (IRB #13–12587, #17–22324, #17–23196, and #18–24633), and by the HKU Institutional Review Board (UW 07–273 and UW 21–112). As part of routine clinical practice at both institutions, all patients who were included in this study signed a waiver of informed consent to contribute de-identified data to research projects. Meningiomas and de-identified clinical information were transferred from HKU to UCSF in 2019 for analysis under protection of a Material Transfer Agreement that was certified by both institutions.
Meningiomas and clinical data
Meningioma samples for the discovery cohort were selected from the UCSF Brain Tumor Center Biorepository and Pathology Core in 2017, with an emphasis on high-grade meningiomas and low-grade meningiomas with long clinical follow-up. All WHO grade 2 and grade 3 meningiomas with available frozen samples were included. For WHO grade 1 meningiomas, frozen samples in the tissue bank were cross-referenced for clinical follow-up data from a retrospective institutional meningioma clinical outcomes database, and all cases with available frozen tissue and clinical follow-up greater than 10 years (n=40) were included. To achieve a discovery cohort of 200 cases, additional WHO grade 1 meningiomas with available frozen tissue and the longest possible clinical follow-up (albeit less than 10 years, n=47) were included. The electronic medical record was reviewed for all patients in late 2018, and paper charts were reviewed in early 2019 for patients treated prior to the advent of the electronic medical record. All available clinical pathology material was reviewed for diagnostic accuracy by a board-certified neuropathologist (D.A.S.). WHO grading was performed using contemporary criteria outlined in the WHO classification of tumors of the central nervous system73. Cases for which other tumors remained in the differential diagnosis (such as schwannoma or solitary fibrous tumor/hemangiopericytoma) were excluded. The validation cohort was comprised of 365 consecutive meningiomas from patients who were treated at The University of Hong Kong (HKU) from 2000 to 2019 that had frozen tissue suitable for DNA methylation profiling. The medical record was reviewed for all patients in late 2019. For the discovery and validation cohorts, meningioma recurrence was defined as new radiographic tumor on magnetic resonance imaging after gross total resection, or enlargement/progression/growth of residual tumor on magnetic resonance imaging after subtotal resection. All magnetic resonance imaging studies in the discovery cohort were reviewed for accuracy and meningioma location by a board-certified radiologist with a Certificate of Added Qualification in Neuroradiology (J.E.V-M.) (Supplementary note). Nomograms integrating clinical and molecular features influencing meningioma outcomes were developed to guide clinical translation of meningioma DNA methylation groups (Supplementary note).
DNA methylation profiling and analysis
DNA was extracted from all 565 meningiomas included in this study (Supplementary note). Genomic DNA was processed on the Illumina Methylation EPIC Beadchip (#WG-317–1003, Illumina) according to manufacturer’s instructions at the Molecular Genomics Core at the University of Southern California. Downstream analysis was performed in R (v3.5.3 and v3.6.1) with SeSAMe (Bioconductor v3.10)3. Probes were filtered and analyzed using the standard SeSAMe preprocessing pipeline, including normal-exponential out-of-band background correction, nonlinear dye bias correction, p-value with out-of-band array hybridization masking, and β value calculation (β = methylated/[methylated+unmethylated]). A total of 272,041 probes were masked in at least one sample by the SeSAMe preprocessing pipeline, and 593,877 were retained for subsequent analysis.
Pre-processing and β value calculation were repeated using the minfi R package for comparison (Bioconductor v3.10)9, using functional normalization74. Probes were filtered based on the following criteria: (i) removal of probes containing common single nucleotide polymorphisms (SNPs) within the targeted CpG sites or on an adjacent base pair (n=30,435), (ii) removal of probes targeting the X and Y chromosomes (n=19,298), (iii) removal of cross-reactive probes previously reported in the literature75 (n=39,605), and (iv) removal of probes with a detection p>0.05 in any samples (n=12,572). A total of 763,949 probes were retained for analysis after minfi pre-processing, representing 31.4% more probes than were included for analysis using the SeSAMe preprocessing pipeline.
Principal component analysis was performed independently on the β methylation values from both the SeSAMe and minfi pre-processing pipelines in R using the base command ‘prcomp’ with the parameters ‘center = TRUE, scale. = FALSE’. Variable probes were identified from the first 3 principal components (PCs), which were chosen for analysis in both pre-processing pipelines (SeSAMe and minfi). The elbow method identified 3–4 PCs as the optimal number, but PC4 was excluded from analysis as it contributed to <5% of β value variance in both pipelines. In contrast, PCs 1–3 contributed to >5% of β value variance in both pipelines. The top 700 probes from PC 1–3 (2,100 total probes) were selected for analysis by ranking the absolute gene loading score values within PCs. A cutoff of 700 probes for each PC was chosen based on the distribution of loading scores in an effort to balance signal and noise from probes minimally contributing to β value variance. Using 500, 1000, or 1500 probes only re-grouped 1–4% of meningiomas, suggesting the precise number of probes across the top 3 PCs did not affect unsupervised hierarchical clustering results. Duplicate probes were removed, and probes with the lowest gene loading scores were culled until 2,000 variable probes remained, which were used for unsupervised hierarchical clustering (Pearson correlation distance, Ward’s method). Using all 2094 unique probes did not affect unsupervised hierarchical clustering results, and using as few as 1900 probes only re-clustered 1% of meningiomas, suggesting the precise number of probes from the union set across the top 3 PCs did not affect unsupervised hierarchical clustering results.
Sampling distributions of DNA methylation group proportions were generated via bootstrapping, group number was investigated across principal components and DNA methylation pre-processing pipelines, and cell type deconvolution was performed (Supplementary note). In brief, the number of groups identified was independent of the number of principal components used for probe selection. SeSAMe consensus clustering clearly identified 3 clusters as the optimal number, while minfi consensus clustering was unable to discriminate between 3 and 4 clusters. Even within 3 groups, minfi re-assigned 21% of meningiomas to different DNA methylation groups compared to the SeSAMe preprocessing pipeline.
Copy number variant analysis
CNV profiles from DNA methylation data were generated with the ‘cnSegmentation’ command within the SeSAMe R package76, using the ‘EPIC.5.normal’ dataset from the sesameData package as a copy-number-normal control. CNV profiles were generated independent of meningioma DNA methylation groups, and sample-level DNA methylation group identities were unblinded for integrated analyses in the context of other genetic data only after CNVs were defined. CNV intensity value distributions were manually inspected for local minima and maxima, and nadirs separating copy number losses, gains, and neutral events were identified. Segments with mean intensity values less than -0.1 were defined as copy number losses, segments with mean intensity values greater than 0.15 were defined as copy number gains, and segments with intensity values between -0.1 and 0.15 were defined as neutral copy number events. Chromosome arms with at least 80% of their length meeting these criteria were considered losses or gains of the chromosomal arm, respectively. This analysis excluded sex chromosomes and the p arms of acrocentric chromosomes, which had insufficient methylation probes for robust CNV quantification (13p, 14p, 15p, 21p, and 22p). The percentage of the genome with copy number variation was determined by calculating the average number of segments per sample with mean intensity values less than -0.1 or greater than 0.15, weighted by segment length. Genome-wide CNV plots were generated using the R package karyoplotR (Bioconductor v3.10). CNVs of biological interest were verified using IGV. CNV definitions were validated using exome sequencing, the interdependence of meningioma CNVs and meningioma DNA methylation groups was tested, and the size and specificity of CNVs across meningioma DNA methylation groups was investigated (Supplementary note).
RNA sequencing and analysis
RNA was extracted from the 200 meningioma discovery cohort (Supplementary note). Library preparation was performed using the TruSeq RNA Library Prep Kit v2 (#RS-122–2001, Illumina) and 50 bp single-end reads were sequenced on an Illumina HiSeq 4000 to a mean of 42 million reads per sample at the UCSF IHG Genomics Core. Quality control of FASTQ files was performed with FASTQC (v0.11.9). Reads were trimmed with Trimmomatic to remove Illumina adapter sequences77, leading and trailing bases with quality scores below 20, and any bases that did not have an average quality score of 20 within a sliding window of 4 bases. Any reads shorter than 36 bases after trimming were removed. Reads were mapped to the human reference genome GRCh3878 using HISAT279 (v2.1.0) with default parameters. For downstream expression analysis, exon level count data were extracted from the mapped HISAT2 output using featureCounts (v2.0.1)80. Differential expression analysis and cell type deconvolution were performed, hierarchical clustering using gene expression data was attempted, and the specificity of FOXM1 signaling in Hypermitotic meningiomas was investigated (Supplementary note).
Somatic short variant sequencing and analysis
A custom amplicon DNA sequencing panel was designed with 100% coverage of all coding exons of NF2, somatic short variants in TRAF7 were identified from the RNA sequencing data, whole exome sequencing data were analyzed, CLIA-certified exome sequencing was performed and analyzed, and MHC class I genotypes and mutant neoepitope peptides were inferred (Supplementary note).
Immunoblotting, fractionation, and immunoprecipitation
Immunoblots were used to assess protein expression in tissues, whole cell lysates, subcellular fractions, or immunoprecipitated samples (Supplementary note). Immunoblot cell line samples were prepared by lysis in radioimmunoprecipitation assay (RIPA) buffer containing Complete-Mini EDTA-free protease inhibitor (#11836170001, Sigma-Aldrich) and PhosSTOP phosphatase inhibitor (#04906837001, Sigma-Aldrich), followed by boiling in Laemmli reducing buffer. Immunoblot meningioma samples were first mechanically lysed using a TissueLyser II (QIAGEN) according to the manufacturer’s instructions then processed identically to cell line samples. Samples were separated on 4–15% gradient TGX precast gels (#4561086, Bio-Rad), and transferred onto nitrocellulose membranes (#1620094, Biorad). Membranes were blocked in 5% TBST-milk, incubated in primary antibody, washed, and incubated in secondary antibodies. Membranes were subjected to immunoblot analysis using Pierce ECL (#32209, Thermo Fischer Scientific) or SuperSignal West Femto (#34095, Thermo Fischer Scientific) substrates. Primary antibodies were used against Merlin (#ab88957, clone AF1G4, Abcam, 1:2000), GAPDH (#MA515738, clone GA1R, Thermo Fischer Scientific, 1:2000), Caspase-7 (#9492, Cell Signaling, 1:500), IRF8 (#5628S, clone D20D8, Cell Signaling, 1:500), Tubulin (#T5168, clone B-5–1-2, Sigma, 1:5000), HH3 (#702023, clone 17H2L9, Thermo Fischer Scientific, 1:1000), FLAG (#F1804, clone F1804, Sigma, 1:1000), ARHGAP35 (#2860, clone C59F7, Cell Signaling, 1:1000), FOXM1 (#sc-376471, clone G-5, Santa Cruz, 1:500), pRB-S780 (#8180P, clone D59B7, Cell Signaling, 1:1000), and pRB-S807/811 (#8516P, clone D20B12, Cell Signaling, 1:1000). Secondary antibodies used were mouse (#7076, Cell Signaling, 1:2000) or rabbit (#7074, Cell Signaling, 1:2000).
Cell culture and organoids
HEK293T (#CRL-3216, ATCC), BenMen81, IOMM-Lee23, and CH157-MN26 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (#11960069, Life Technologies), supplemented with 10% fetal bovine serum (FBS) (#16141, Life Technologies), 1X GlutaMAX (#35050–061, Thermo Fischer Scientific), and 1X Penicillin/Streptomycin (#15140122, Life Technologies). DI-98 and DI-134 cells54 and cultured in DMEM supplemented with 7% FBS and 1X Penicillin/Streptomycin. M10G cells22 were cultured in a 1:1 ratio of DMEM/F12 media (#10565, Life Technologies) and Neurobasal media (#21103, Life Technologies), supplemented with 5% FBS, B-27 supplement without vitamin A (#12587, Life Technologies), N-2 supplement (#17502, Life Technologies), 1X GlutaMAX (#35050, Life Technologies), 1mM NEAA (#11140, Life Technologies), 100U/mL Anti-Anti (#15240, Life Technologies), 20 ng/mL EGF (#AF-100–15, Peprotech), and 20 ng/mL FGF (#AF-100–18B, Peprotech). MSC1 cells were cultured in the same conditions as M10G cells, but supplemented with 15% FBS22. Meningioma cell lines were authenticated using DNA methylation profiling and CNV analyses to confirm concordance to tumors of origin, most recently in 2021. Non-meningioma cell lines purchased from reputable commercial suppliers (HEK293T cells from ATCC) were not authenticated. Cell proliferation or apoptosis was investigated (Supplementary note). Human cerebral organoids were created from astrocytes induced from pluripotent human stem cells and co-cultured with meningioma cells22.
CRISPRi gene suppression
Lentiviral particles containing pMH000182 (UCOE-SFFV-dCas9-BFP-KRAB, #85969, Addgene) were produced by transfecting HEK293T cells with standard packaging vectors using the TransIT-Lenti Transfection Reagent (#6605, Mirus). M10G cells were stably transduced with lentiviral particles to generate M10GdCas9-KRAB cells. Successfully transduced cells were isolated through selection of BFP positive cells using fluorescence activated cell sorting on a Sony SH800.
Single-guide RNA (sgRNA) protospacer sequences were individually cloned into the pCRISPRia-v2 vector83 (#84832, Addgene), between the BstXI and BlpI sites, by ligation. Each vector was verified by Sanger sequencing of the protospacer (Supplementary table 14). Lentivirus was generated as described for each sgRNA expression vector. M10GdCas9-KRAB cells were independently transduced with lentivirus from each sgRNA expression vector and selected to purity using 20 μg/mL puromycin over 7 days.
shRNA gene suppression
Lentiviral particles containing pLKO.1 shRNA targeting control (RHS6848, Dharmacon) or NF2 (RHS3979–201768826 or RHS3979–201768830) were generated by transfecting HEK293T cells with standard packaging vectors (psPAX2 and pMD2.9) and shRNA plasmids using TransIT-Lenti Transfection Reagent. After 48 hours of virus production, viral particles were sterilized through a 0.45 μM filter and added to meningioma cells with polybrene 10 μg/ml (TR-1003, MerkMillipore). A polyclonal population of shRNA positive cells was selected using puromycin 2 μg/ml.
siRNA gene suppression
Small interfering RNA (siRNA) targeting the glucocorticoid receptor, NR3C1 (#J-003424–10-0002), or control (#D-001810–01-05), were purchased from Dharmacon. siRNA transfection was performed using Lipofectamine RNAiMax reagent (#13778, Thermo Fischer Scientific). In brief, 2 cocktails containing either siRNA (25 nM) or Lipofectamine RNAiMax (3 μl) in 150 μl OptiMEM were made per transfection. Cocktails were incubated for 5 minutes prior to combination of the 2 solutions, followed by incubated for an additional 10 minutes before adding to cells for 15 hours. The siRNA transfection was repeated after 48 hours and expanded. Cells were harvested 92 hours after first siRNA transfection.
RNA extraction, cDNA synthesis, and QPCR
RNA was extracted from cultured cells using the RNeasy Mini Kit (#74106, QIAGEN) according to manufacturer’s instructions. cDNA was synthesized from extracted RNA using the iScript cDNA Synthesis Kit (#1708891, Bio-Rad). Real-time qPCR was performed using PowerUp SYBR Green Master Mix (#A25918, Thermo Fisher Scientific) on a QuantStudio 6 Flex Real Time PCR system (Life Technologies) using forward and reverse primers (Supplementary table 14). Real-time qPCR data were analyzed using the ΔΔCt method relative to GAPDH expression.
Immunofluorescence
Immunofluorescence of primary meningioma cells was performed on glass coverslips. Cells were fixed in 4% paraformaldehyde (#15710, Electron Microscopy Sciences), blocked in 2.5% BSA (#BP1600, Fischer Scientific) and 0.1% Triton X-100 (#X100, Sigma) in Phosphate Buffered Saline (PBS) for 30 min at room temperature (#14190, Gibco), and labeled with Ki-67 (#ab15580, Abcam) primary antibodies at room temperature for 1 h. Cells were labeled with Alexa Fluor secondary antibodies and either Hoechst 33342 (#H3570, Thermo Fisher Scientific) or DAPI (#D3571, Thermo Fisher Scientific) to mark DNA for 1 h at room temperature, and were mounted in ProLong Diamond Antifade Mountant (#P36970, Thermo Fisher Scientific). For apoptosis assays, cells were washed in Annexin V binding buffer, stained with Annexin V for 15 min (#550911, BD Bioscience), washed, labeled with DAPI to mark DNA for 1 h at room temperature, and mounted in ProLong Diamond Antifade Mountant.
Immunofluorescence of human and xenograft meningiomas was performed on 10 μm cryosections of frozen tissue embedded in OCT Compound (Thermo Fisher Scientific). Slides with tissue were fixed in cold acetone for 10 min, air dried, washed in PBS, permeabilized with 0.3% Triton-X 100 in PBS, and washed again in PBS. Sections were blocked (2% BSA, 1% donkey serum, and 0.1% Triton-X 100 in PBS) for 30 min. Sections were labeled with either LYVE-1 (#ab14917, Abcam, 1:1000) or PROX-1 (#AF2727, R&D Systems, 1:1000) primary antibodies at room temperature for 1 h. Slides were subsequently labeled with rabbit (#A21206, Thermo Fischer Scientific, 1:1000) and goat (#A21469, Thermo Fischer Scientific 1:1000) Alexa Fluor secondary antibodies and Hoechst 33342 to mark DNA for 1 h at room temperature and were mounted in ProLong Diamond Antifade Mountant.
Dual immunofluorescence of human meningiomas for FOXM1 and Ki-67 was performed on 5 μm formalin-fixed, paraffin-embedded (FFPE) human meningioma sections. Following antigen retrieval using CC1 for 32 min (#950–124, Roche Diagnostics), sections were incubated and detected sequentially with rabbit monoclonal Ki-67 (#790–4286, clone 30–9, Ventana, 1:4) and rabbit monoclonal FOXM1 primary antibodies (#ab207298, clone EPR17379, Abcam, 1:600). Each primary antibody incubation was 32 min and single stained controls were used to verify specificity. FOXM1 labeling index was quantified based on the total amount of nuclei with strong immunoreactivity for FOXM1 within a 200x field. Ki-67 labeling index was quantified based on the total amount of nuclei with strong immunoreactivity for Ki-67 within a 200x field. The labeling index for both FOXM1 and Ki-67 was averaged across 2 200x fields for each meningioma.
Histology and immunohistochemistry
Deparaffinization and rehydration of 5 μm FFPE human and mouse meningioma tissue sections and hematoxylin and eosin staining were performed using standard procedures. Immunostaining was performed on an automated Ventana Discovery Ultra staining system. Immunohistochemistry was performed on 5μm FFPE meningioma xenograft sections using rabbit monoclonal Ki-67 (#790–4286, clone 30–9, Ventana, 1:6) with primary antibody incubation for 16 min following CC1 antigen retrieval for 8 min, rabbit monoclonal cleaved Caspase-3 (#9664, clone 5A1E, Cell Signaling, 1:2000) with primary antibody incubation for 32 min at 37°C following CC1 antigen retrieval for 8 min, or rabbit polyclonal CD3 (#A0452, Agilent Technologies, 1:200) with primary antibody incubation for 32 min at 37°C following CC1 antigen retrieval for 8 min. Tumors were scored CD3 positive if multiple aggregates of CD3-positive lymphocytes were identified and were otherwise scored as CD3 negative.
Microscopy
Fluorescence microscopy was performed on a LSM 800 confocal laser scanning microscope with a PlanApo 20X air objective (Zeiss). Images were processed and quantified from representative regions of each tumor or coverslip using ImageJ (v.2.1.0). Histologic and immunohistochemical features were evaluated using light microscopy on an BX43 microscope with standard objectives (Olympus). Images were obtained and analyzed using the Olympus cellSens Standard Imaging Software package.
Proteomic proximity-labeling mass spectrometry
M10G cells encoding pLV.APEX2-Merlin wild type and L46R-mutant constructs were seeded onto 5 × 15 cm plates. For APEX labelling, cells were treated with 0.5 uM Biotin-phenol (#BT1015, Berry & Associates) and returned to 37°C, 5% CO2 for 30 minutes. After 24 hours, Merlin protein expression was induced with 0.1μg/ml doxycycline. Biotin treated cells were subject to free radical formation by adding media containing 1mM H2O2 to cells for exactly 30 seconds, on ice. Immediately, H2O2 media was aspirated and the reaction was quenched (10mM Sodium Ascorbate, 1mM Azide, 5mM Trolox), and cells were pelleted for biotin/streptavidin precipitation, reversed-phase gradient extraction, and mass spectrometry (Supplementary note).
Gene over-expression
For transient over-expression of FOXM1 or USF1, M10G cells were transfected with pCMV6-FOXM1 (#RC202246, Origene) or pCMV6-USF1 (#RC204915, Origene) plasmids at a ratio of 1 μg of DNA to 2 μl of FuGENE transfection reagent (#E2311, Promega). Cells were harvested 48 hours after transfection for RNA extraction. For stable over-expression, USF1 was cloned from pCMV6-USF1 into the pLVX-IRES-puro vector using restriction digest and ligation. Lentivirus was generated and introduced onto CH-157MN cells and stable polyclonal cell lines were generated using antibiotic selection as described above.
Mice
This study was approved by the UCSF Institutional Animal Care and Use Committee (AN174769), and all experiments complied with relevant ethical regulations. Xenograft experiments were performed by implanting 3 million CH-157MN cells into the flank of 5–6-week-old female NU/NU mice (Harlan Sprague Dawley Inc.). To induce Merlin expression in meningiomas in vivo, mice harboring CH-157MN cells encoding pLV.APEX2-Merlin were treated with doxycycline 200 μg/ml (n=3) (#D9891, Sigma) or vehicle (n=3) 14 days post implantation. After 7 days of treatment, 2 Gy of ionizing radiation per day was delivered using a Precision X-RAD 320 Cabinet Irradiation, with normal operating settings, on each of 2 successive days. Tumors were collected 24 hours after the second dose of ionizing radiation for immunoblotting and immunohistochemistry. For preclinical pharmacologic experiments, animals in the treatment arm (n=7) were gavaged with 100 μg/g abemaciclib in 0.5% methylcellulose vehicle daily starting 12 days after injection, until protocol-defined endpoints. Animals in the vehicle arm (n=13) were gavaged at the same frequency for the same duration with 0.5% methylcellulose. For Kaplan-Meier survival analysis, events were recorded when tumors reached the protocol-defined size of 2000 mm3.
Single cell isolation and RNA sequencing
Fresh human meningioma (n=8) and dura (n=2) samples were acquired from the operating room and transported to the laboratory in PBS at 4°C. These samples were not part of the discovery or validation cohorts, but all patients signed a waiver of informed consent to contribute de-identified data to research projects in compliance with all relevant ethical regulations under the supervision of the UCSF Institutional Review Board, as described above. Tissue samples were minced with sterile #10 scalpels (#4–410, Integra LifeSciences) and incubated at 37°C in a Collagenase Type 7 solution (#LS005332, Worthington) until digested (30–60 minutes). Collagenase was used at a concentration of 0.1 mg/mL for tumor and brain-tumor interface samples, and at a concentration of 0.2 mg/mL for dura samples. Samples were incubated in Trypsin-EDTA 0.25% (#25200056, Thermo Fisher Scientific) at 37°C for 5 minutes, and in 1X RBC lysis buffer (#00–4300-54, eBioscience) at room temperature for 10 minutes. Finally, samples were sequentially filtered through 70 μM and 40 μM cell strainers (#352350 and #352340, Corning) to generate single-cell suspensions.
Single cell suspensions were processed for single cell RNA sequencing using a 10X Chromium controller, and libraries were generated using the Chromium Single Cell 3’ Library & Gel Bead Kit v3 on a 10X Chromium controller using the manufacturer recommended default protocol and settings (#1000121, 10X Genomics), at a target cell recovery of 5,000 cells per sample. Libraries were sequenced on an Illumina NovaSeq 6000, targeting >50,000 reads/cell, at the UCSF Center for Advanced Technology.
Single cell RNA sequencing analysis
Library demultiplexing, read alignment to the GRCh38 human reference genome, identification of empty droplets, and UMI quantification was performed using CellRanger version 3.0.2 (https://github/com/10xGenomics/cellranger). Cells with greater than 500 unique genes, less than 10,000 unique genes, and less than 20% of reads attributed to mitochondrial transcripts were retained. Single cell UMI count data were preprocessed in R 1 with the Seurat84,85 package (v3.0.1) using the sctransform86 workflow, with scaling based on regression of UMI count and percentage of reads attributed to mitochondrial genes per cell. Dimensionality reduction was performed using principal component analysis, and principal components were corrected for batch effects using Harmony (v0.1)87. Uniform Manifold Approximation and Projection (UMAP) was performed on the reduction data with a minimum distance metric of 0.2 and Louvain clustering was performed using a resolution of 0.4. Marker identification and differential gene expression was performed in Seurat using a minimum fraction of detection of 0.75 and a minimum log-fold change of 0.5. CNVs were defined in single-cells, cell types were defined using marker genes and cell cycle phases, and reference transcriptomic signatures were defined (Supplementary note).
ChIP sequencing and enhancer/super-enhancer analysis
H3K27ac ChIP sequencing data were derived from 25 previously reported meningiomas54 (3 Merlin-intact, 7 Immune-enriched, and 15 Hypermitotic), and enhancer and super enhancer analyses were performed (Supplementary note). USF1 enrichment at the CDK6 promoter was investigated using ChIP QPCR (Supplementary note).
Statistics
All experiments were performed with independent biological replicates, repeated, and statistics were derived from biological replicates. Biological replicates are indicated in each panel or figure legend. No statistical methods were used to predetermine sample sizes, but our discovery and validation cohort sizes are similar or larger to those reported in previous publications4,5,8. Data distribution was assumed to be normal, but this was not formally tested. Investigators were blinded to conditions during clinical data collection and analysis of mechanistic or functional studies. Bioinformatic analyses were performed blind to clinical features, outcomes, or molecular characteristics. The clinical samples used in this study were retrospective and non-randomized with no intervention, and all samples were interrogated equally. Thus, controlling for covariates among clinical samples is not relevant. Cells, organoids, and animals were randomized to experimental conditions. No clinical, molecular, cellular, or animal data points were excluded from the analyses. Unless specified otherwise, lines represent means, and error bars represent standard error of the means. Results were compared using log-rank tests, Student’s t tests, ANOVA, Chi-squared tests, Fischer’s exact tests, and Wald tests, which are indicated in the text, methods, and figure legends alongside approaches used to adjust for multiple comparisons. Results comparisons in Supplementary tables were performed using Wald tests (Supplementary table 6, 9, 11, two-sided, Benjamin-Hochberg adjustment for multiple comparisons) or Wilcoxon rank sum tests (Supplementary table 10, two-sided, Bonferroni adjustment for multiple comparisons). In general, statistical significance is shown by *p≤0.05, **p≤0.01, or ***p≤0.0001, but exact p-values are provided in the figure legends.
Data availability
DNA methylation (n=565), RNA sequencing (n=185), and single-cell RNA sequencing data (n=8 meningioma samples, n=2 dura samples) of new samples reported in this manuscript have been deposited in the NCBI Gene Expression Omnibus under the accession GSE183656 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183656). Additional RNA sequencing data from previously reported meningiomas (n=15) from the discovery cohort are available under the accession GSE101638 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101638). Whole exome sequencing, ChIP sequencing, and additional DNA methylation profiling data incorporated into this study were derived from previously reported and deposited meningiomas in GSE101638 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101638), GSE139652 (https://www.ncbi.xyz/geo/query/acc.cgi?acc=GSE139652). The publicly available GRCh38 (hg38, https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.39/) and CRCh37.p13 datasets (hg19, https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.25/) were used in this study.
Code availability
The open-source software, tools, and packages used for data analysis in this study, as well as the version of each program, were ImageJ (v2.1.0), R (v3.5.3 and v3.6.1), FASTQC (v0.11.9), HISAT2 (v2.1.0), featureCounts (v2.0.1), Seurat R package (v3.0.1), Harmony R package (v0.1), caret R package (v6.0–90), Rtsne R package (v0.15), PAMES R package (v2.6.2), CONICSmat R package (v1.0), DeepTools (v3.1.2), survival R package (v3.2–13), survAUC R package (v1.0–5), rms R package (v6.2–0), rpart R package (v4.1.16), DynNom R package (v5.0.1), DESeq2 (Bioconductor v3.10), SeSAMe (Bioconductor v3.10), minfi (Bioconductor v3.10), karyoplotR (Bioconductor v3.10), ConsensusClusterPlus (Bioconductor v3.10), and DiffBind (Bioconductor v3.10). No software was used for data collection. A methylation profile multi-class support vector machine (SVM) classifier was generated using the caret R package88, and was deposited in the github repository abrarc/meningioma-svm (DOI:10.5281/zenodo.6353877)89. In brief, a linear kernel SVM was constructed using training data comprising 75% of randomly selected samples from the discovery cohort (n=150) with 10-fold cross validation. 2,000 probes from each pre-processing pipeline were used as variables. The remaining 25% of samples from the discover cohort (n=50) were used to test the model, which performed with 97.9% accuracy when classifying samples into 3 SeSAMe groups (95% CI 89.2–99.9%, p<2.2×10−16). SVM classifiers for 3, 4, 5, or 6 minfi groups were generated using the same approach and performed with 91.8% (95% CI 80.4%–97.7%, p=4.69×10−9), 91.8% (95% CI 80.4%–97.7%, p=9.58×10−16), 93.8% (95% CI 82.8%–98.7%, p=2.98×10−16), and 93.6% (95% CI 82.5%–98.7%, p<2.2×10−16) accuracy, respectively. SVM classification and K-means consensus clustering of the validation cohort was performed with the same parameters as for the discovery cohort using the same probes in the validation cohort that were identified from the discovery cohort.
Extended Data
Extended Data Fig. 1. DNA methylation analysis using SeSAMe to control for the influence of CNVs on β values identifies 3 groups of meningiomas.
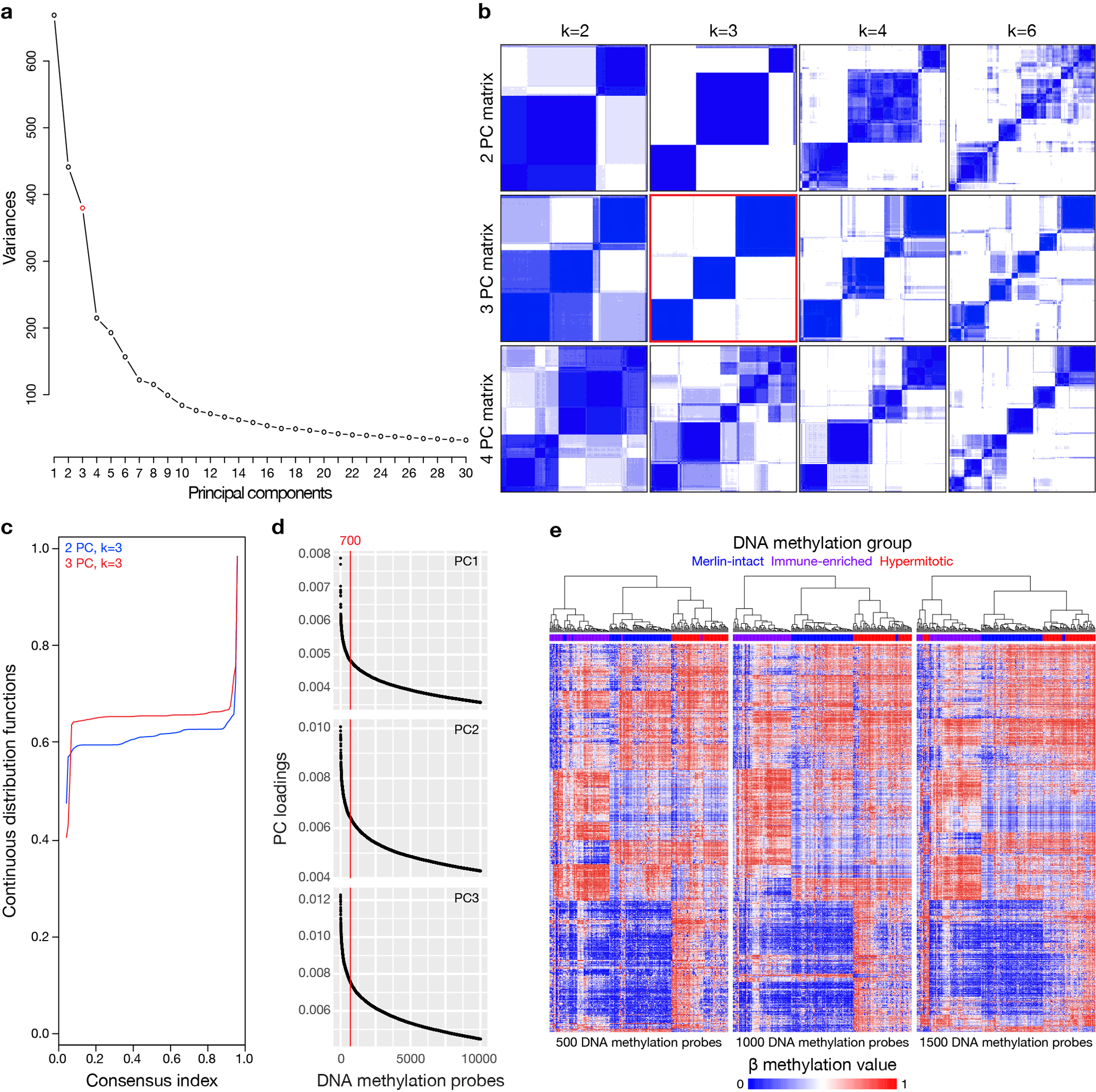
a, Scree/elbow plot of principal component analysis (PCA) of meningioma DNA methylation profiles suggesting differentially methylated DNA probes from the top 3 to 4 principal components (PCs) provide the most information in the discovery cohort (n=200). b, K-means consensus clustering of meningioma DNA methylation profiles from the discovery cohort (n=200) using differentially methylated DNA probes from the top 2–4 PCs across k=2–7 groups, suggesting 3 PCs and k=3 groups are optimal. c, Continuous distribution functions from K-means consensus clustering of meningiomas from the discovery cohort (n=200) using differentially methylated DNA probes from the top 2 or 3 PCs across k=3 groups, validating 3 PCs as the optimal number (p<2.2×10−16, Kolmogorov-Smirnov test). d, Distribution of absolute DNA methylation probe loadings across the top 3 PCs from the discovery cohort (n=200) for the top 10,000 probes for each PC. Loading distribution plots for each PC were similar, and the top 700 probes for each PC were selected using the elbow method for meningioma clustering. e, Unsupervised hierarchical clustering of meningiomas from the discovery cohort (n=200) using 500, 1000, or 1500 differentially methylated DNA probes from each PC demonstrating the precise number of probes from each PC does not significantly influence meningioma DNA methylation grouping. In comparison to Figure 1b, altering the number of probes for meningioma DNA methylation grouping only altered assignments for 3–9 meningiomas (1–4%). Merlin-intact (blue), Immune-enriched (purple), and Hypermitotic (red) DNA methylation group assignments are from Figure 1b.
Extended Data Fig. 2. Independent validation of 3 meningioma DNA methylation groups.
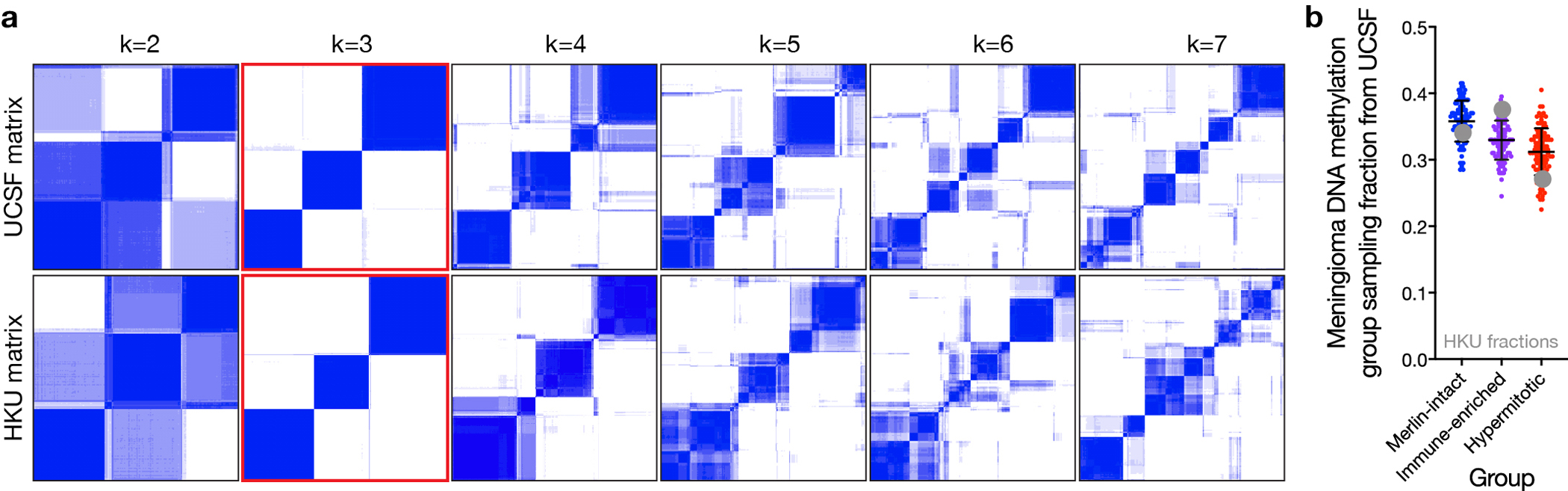
a, K-means consensus clustering of meningioma DNA methylation profiles from the discovery (n=200, UCSF) and validation (n=365, HKU) cohorts. b, Sampling distributions of DNA methylation group fractions from the discovery cohort (n=100 per DNA methylation group), with the observed DNA methylation group fractions from the validation cohort denoted in grey. Lines represent means, and error bars represent standard deviations. The observed fractions of each DNA methylation group from the validation cohort fall within the sampling distributions from the discovery cohort.
Extended Data Fig. 3. Meningioma DNA methylation grouping using SeSAMe to control for the influence of CNVs on β values compared to approaches that do not control for the influence of CNVs on β values.
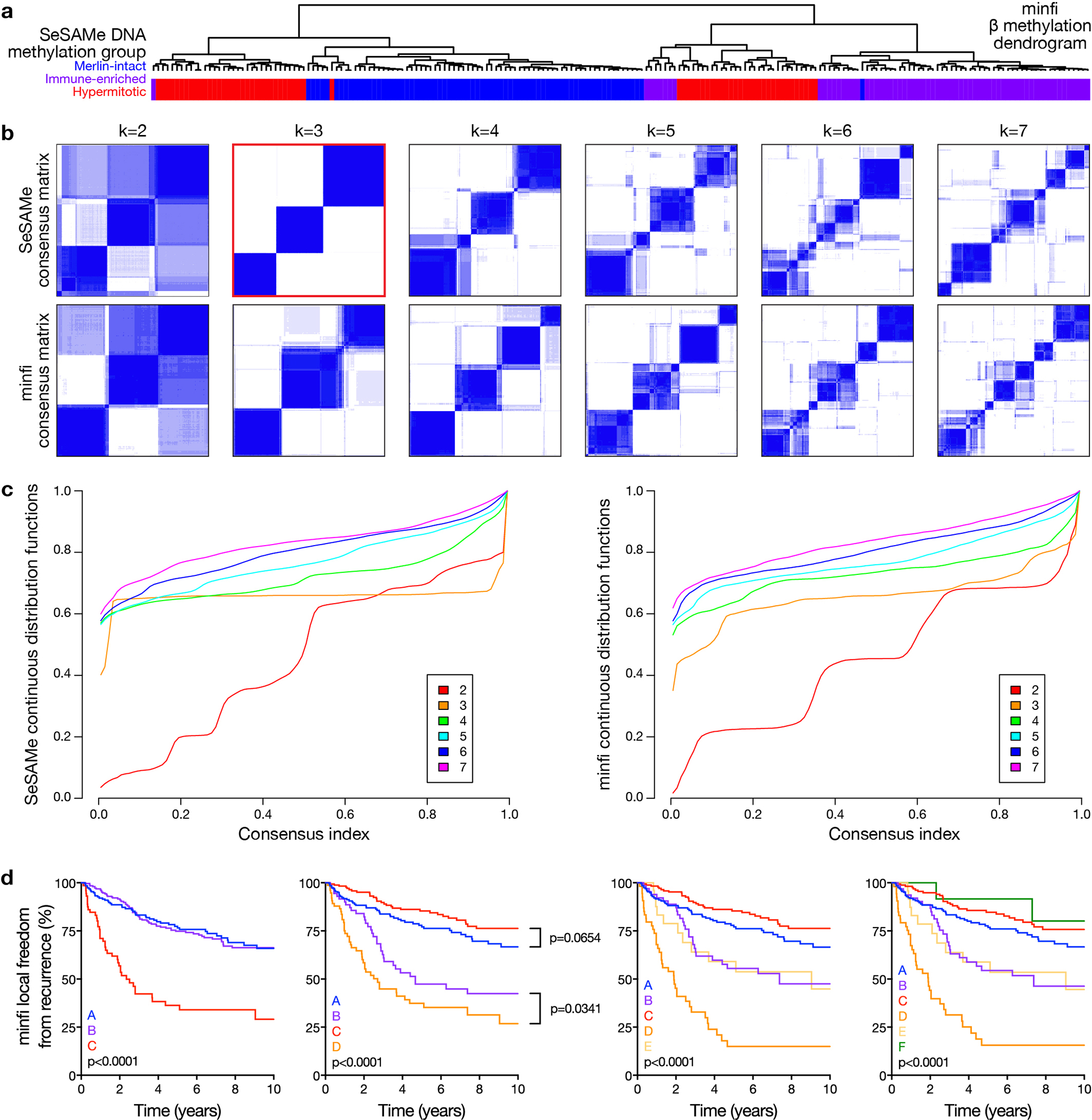
a, Unsupervised hierarchical clustering of meningiomas from the discovery cohort (n=200) using 2,000 differentially methylated DNA probes from the minfi pre-processing pipeline, which does not control for the influence of CNVs on β values. SeSAME meningioma DNA methylation groups (21% altered by minfi) are shown beneath the vertical dendrogram. b, K-means consensus clustering of meningiomas from the discovery and validation cohorts (n=565) using differentially methylated DNA probes and β values from SeSAMe or minfi. SeSAMe consensus clustering identifies 3 groups as the optimal number, but minfi consensus clustering is unable to discriminate between 3 and 4 clusters. c, Continuous distribution functions (CDFs) from K-means consensus clustering of meningiomas from the discovery and validation cohorts (n=565) using differentially methylated DNA probes and β values from SeSAMe or minfi. SeSAMe CDFs validated 3 groups as the optimal number, which was quantitatively different from 3 minfi groups (p=1.341×10−4) or 4 minfi groups (p<2.2×10−6) (Kolmogorov-Smirnov test). d, Kaplan-Meier curves for meningioma local freedom from recurrence (n=565) across minfi DNA methylation groups fails to identify a grouping scheme with non-redundant differences in clinical outcomes, in contrast to SeSAMe DNA methylation groups (Figure 1c) (Log-rank tests). minfi meningioma DNA methylation grouping schemed comprised of 3, 4, 5, or 6 groups are designated by letters A-C, A-D, A-E, or A-F, respectively.
Extended Data Fig. 4. Mechanisms of NF2/Merlin tumor suppression in meningioma cells.
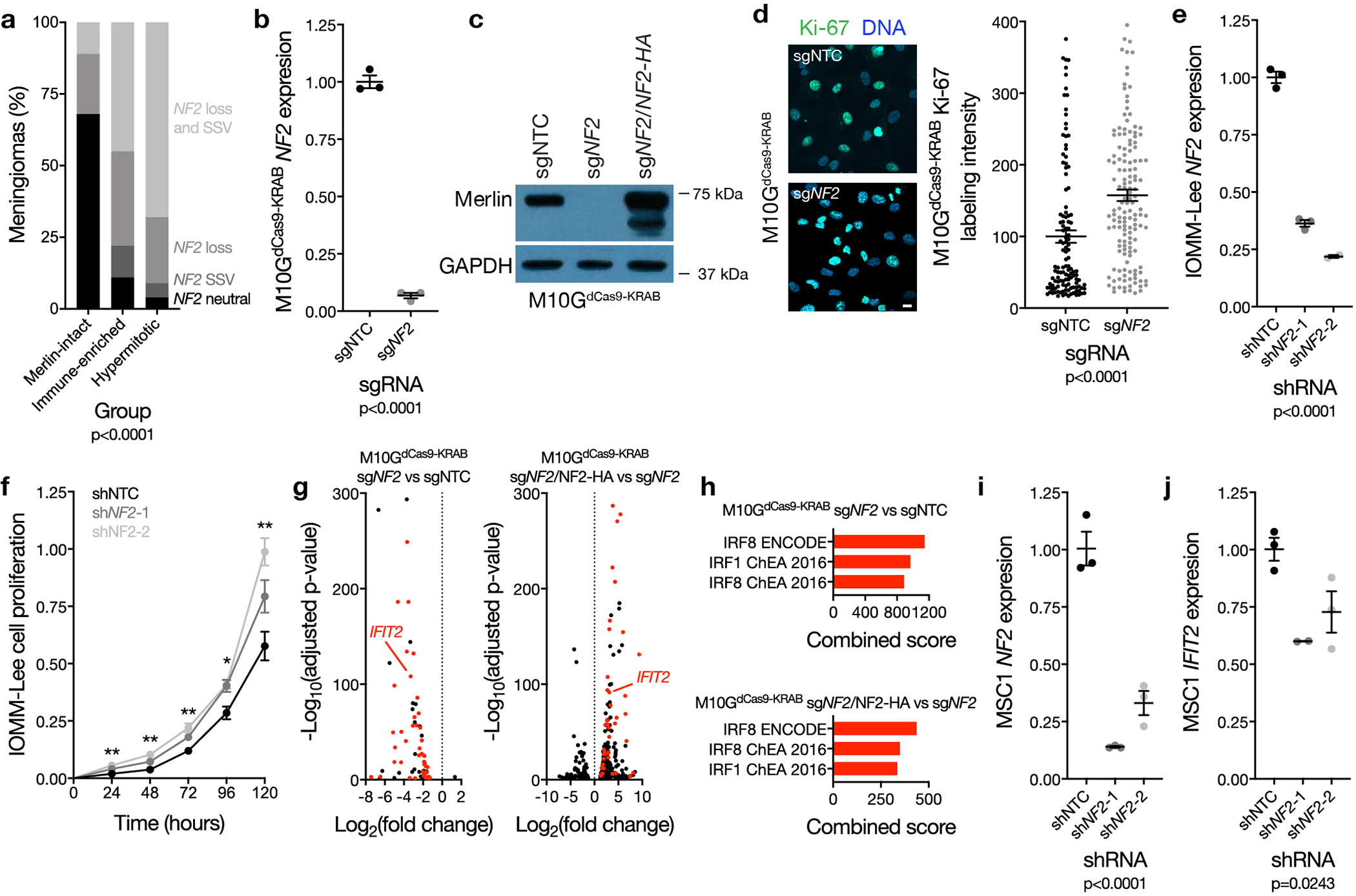
a, Meningioma NF2 copy number deletions containing the entire locus and targeted sequencing of somatic short variants (SSV, n=65) across DNA methylation groups (Chi-squared test, two-sided). b, QPCR for NF2 in M10GdCas9-KRAB cells expressing a non-targeting control single-guide RNA (sgNTC) or a single-guide RNA suppressing NF2 (sgNF2). 3 biological replicates per condition (Student’s t test, one-sided). c, Immunoblot for Merlin or GAPDH in M10GdCas9-KRAB cells expressing sgNTC, sgNF2, or sgNF2 with NF2 rescue (sgNF2+NF2HA). d, Confocal immunofluorescence microscopy and quantification of Ki-67 in M10GdCas9-KRAB cells from b. DNA is marked with Hoechst 33342. Scale bar 10 μM. From left to right, 123 or 145 cells are shown (Student’s t test, one-sided). e, QPCR for NF2 in IOMM-Lee cells stably expressing a non-targeting control shRNA (shNTC) or shRNAs suppressing NF2 (shNF2-1 or shNF2-2). From left to right, 3, 3, or 2 biological replicates are shown (ANOVA, one-sided). f, MTT cell proliferation of IOMM-Lee cells from e, normalized to shNTC at 120 hours. 4 biological replicates per condition per timepoint. *p=0.0101, **p≤0.01 (ANOVA, one-sided). g, Volcano plots of relative gene expression from RNA sequencing of M10GdCas9-KRAB cells in c. Interferon-regulated genes (including IFIT2, validated in j) are marked in red. h, Gene ontology analysis of differentially expressed genes from RNA sequencing of M10GdCas9-KRAB cells in g. i, QPCR for NF2 in MSC1 cells stably expressing shNTC, shNF2-1, or shNF2-2. 3 biological replicates per condition (ANOVA, one-sided). j, QPCR for the IRF target gene IFIT2 in MSC1 cells from i. From left to right, 3, 2, or 3 biological replicates are shown (ANOVA, one-sided). Lines represent means, and error bars represent standard error of the means.
Extended Data Fig. 5. NF2/Merlin drives meningioma apoptosis.
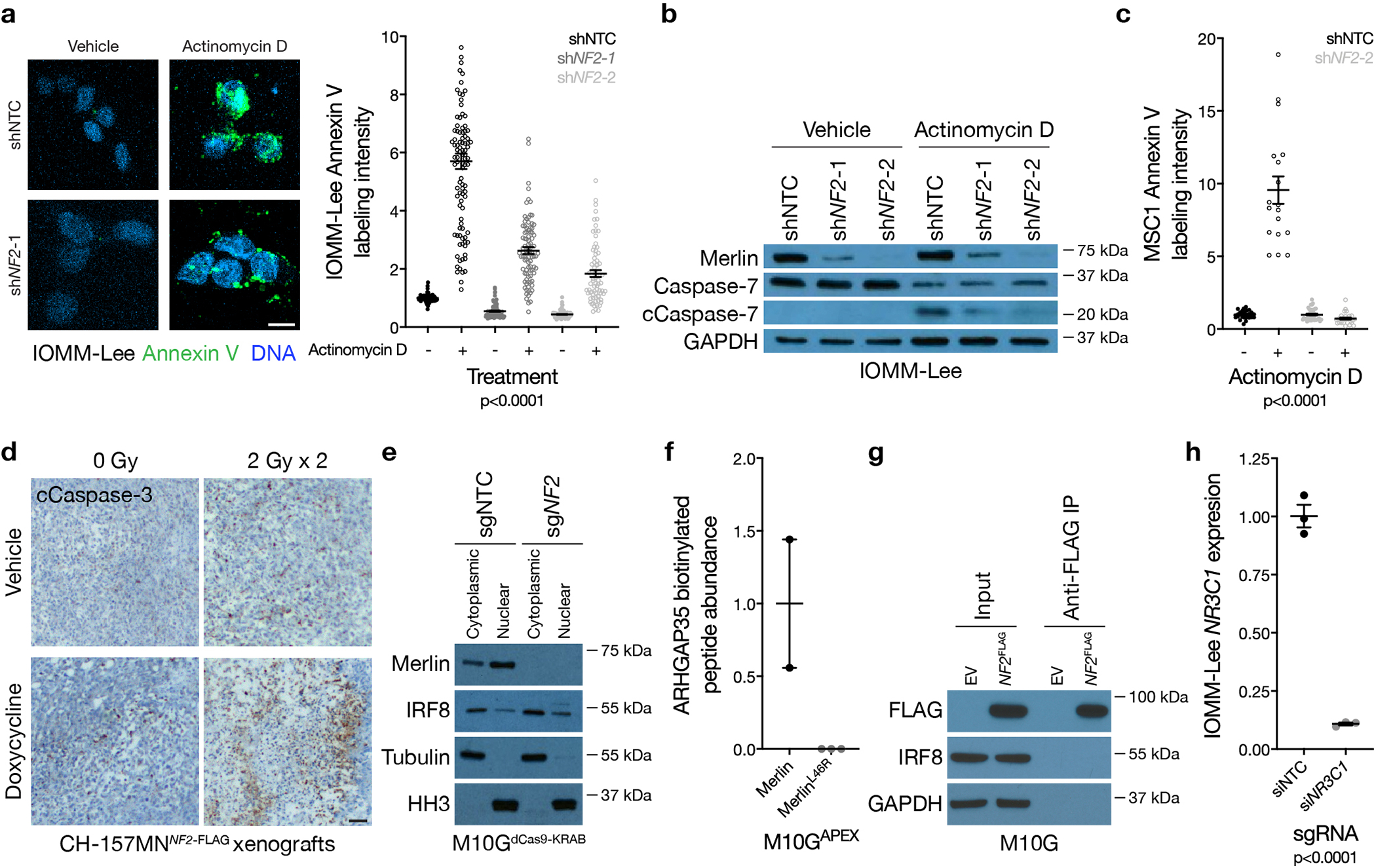
a, Confocal microscopy and quantification of Annexin V in IOMM-Lee cells from Extended data figure 4e treated with actinomycin D or vehicle control for 24 hours. DNA is marked with DAPI. Scale bar 10 μM. From left to right, 96, 101, 95, 90, 98, or 75 cells are shown (ANOVA, one-sided). b, Immunoblot for Merlin, Caspase-7, cleaved Caspase-7 (cCaspase-7), or GAPDH in IOMM-Lee cells from a. c, Quantification of Annexin V confocal microscopy in MSC1 cells stably expressing sgNTC or sgNF2-2. Cells were treated as in a. From left to right, 29, 19, 40, or 30 cells are shown (ANOVA, one-sided). d, Representative images of cleaved Caspase-3 (cCaspase-3) immunohistochemistry from CH-157MN xenografts stably expressing doxycycline-inducible Merlin encoding a FLAG tag (NF2-FLAG) in NU/NU mice after 7 days of doxycycline (n=6) or vehicle treatment (n=6), and 24 hours after 4 Gy ionizing radiation (n=6) or control treatment (n=6). Scale bar 100 μM. e, Immunoblot for Merlin, IRF8, Tubulin, or Histone H3 (HH3) in cytoplasmic or nuclear fractions of M10GdCas9-KRAB cells from Extended data figure 4b. f, Normalized proteomic proximity-labeling mass spectrometry from M10G cells stably expressing Merlin constructs with APEX tags. From left to right, 2 or 3 biological replicates are shown. g, Immunoblot for IRF8 or FLAG after FLAG immunoprecipitation from M10G cells stably expressing Merlin encoding a FLAG tag (NF2FLAG). EV, empty vector. h, QPCR for the glucocorticoid receptor (NR3C1) in IOMM-Lee cells expressing a non-targeting control siRNA (siNTC) or siRNAs suppressing NR3C1 (siNR3C1). 3 biological replicates per condition (Student’s t test, one-sided). Lines represent means, and error bars represent standard error of the means.
Extended Data Fig. 6. Lymphatic vessels underlie meningioma immune infiltration.
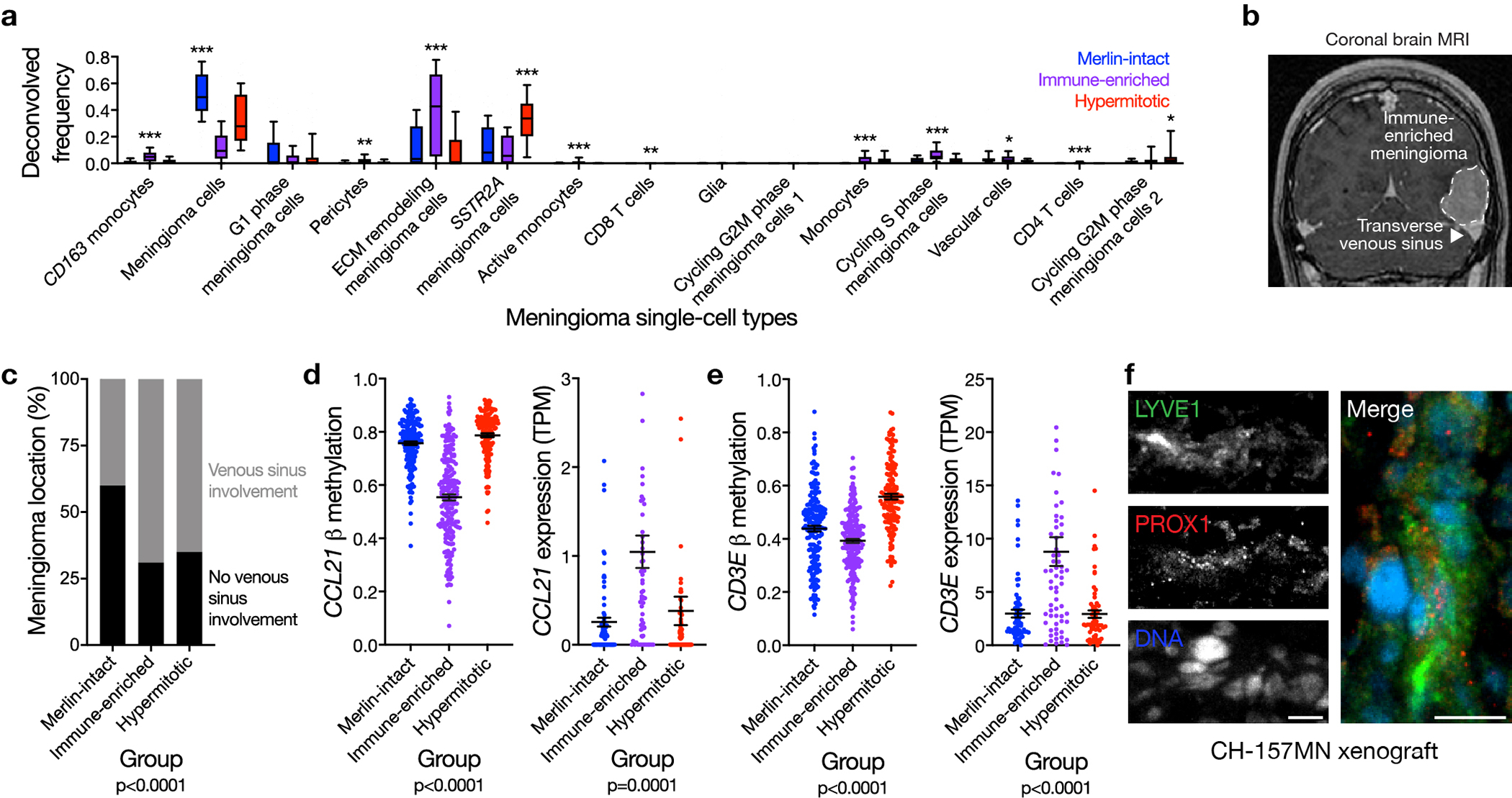
a, Fraction of meningioma samples (n=200) classified meningioma single-cell types across DNA methylation groups, based on single-cell reference transcriptomes. Lines represent means, boxes represent inner quartile ranges, and error bars represent 10th-90th percentiles (ANOVA, one-sided). b, c, Meningioma location on preoperative magnetic resonance imaging (n=169) across DNA methylation groups (Chi-squared test, two-sided). Representative magnetic resonance image shown. d, Meningioma DNA methylation (n=565) of CCL21 (cg27443224) and TPM expression (n=200) of CCL21 across DNA methylation groups (ANOVA, one-sided). e, Meningioma DNA methylation (n=565) of CD3E (cg08956138) and TPM expression (n=200) of CD3E across DNA methylation groups (ANOVA, one-sided). f, Representative image of LYVE1 and PROX1 confocal immunofluorescence microscopy in CH157-MN xenografts in NU/NU mice (n=3). DNA is marked with Hoechst 33342. Scale bars 10 μM. Lines represent means, and error bars represent standard error of the means. *p≤0.05, **p≤0.01, ***p≤0.0001.
Extended Data Fig. 7. FOXM1 target gene functions in meningiomas and meningioma cells.
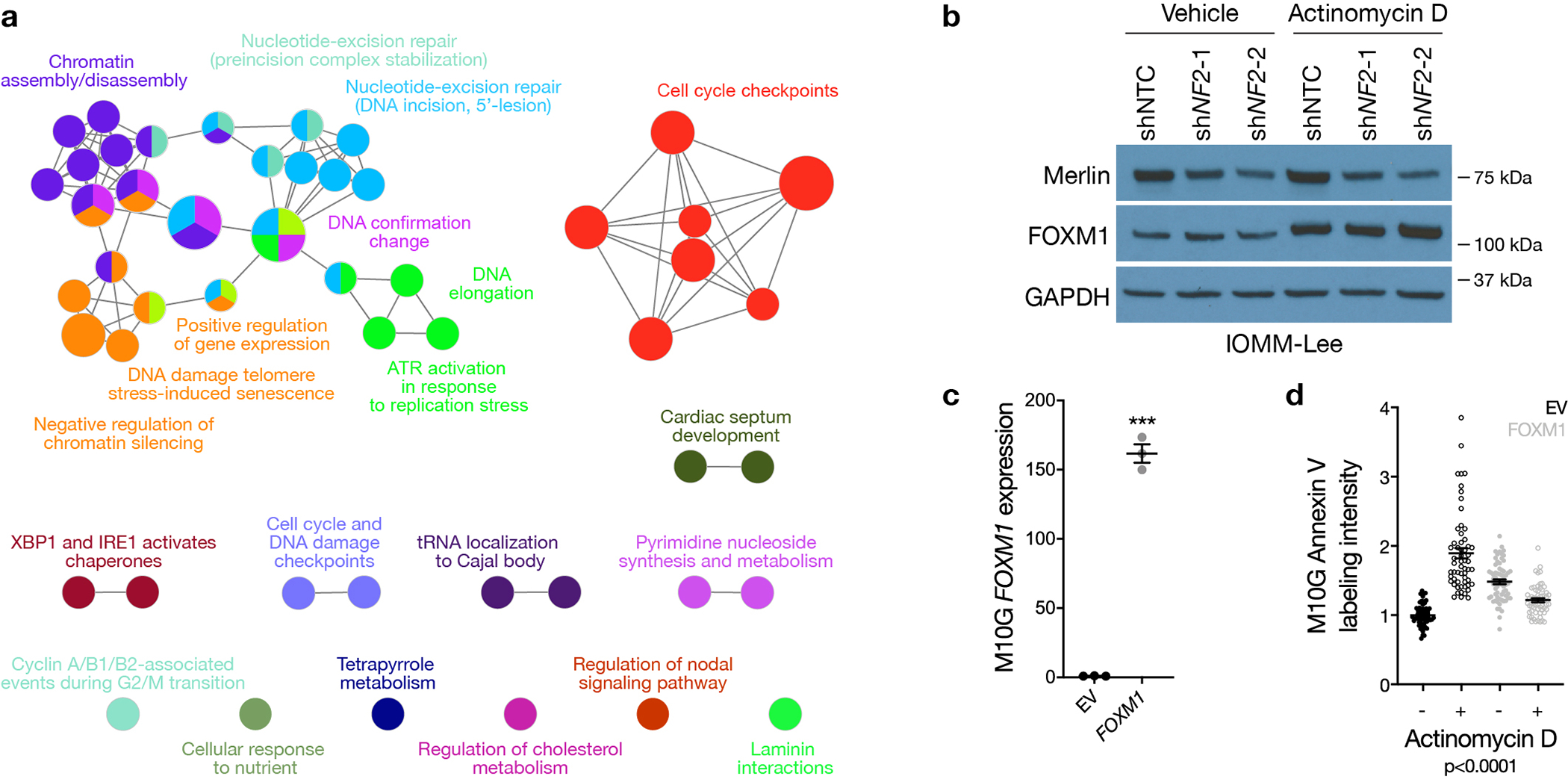
a, Predicted network of FOXM1-regulated pathways in Hypermitotic meningiomas based on H3K27ac ChIP sequencing of 25 meningiomas with matched RNA sequencing and DNA methylation profiling (15 Hypermitotic, 10 non-Hypermitotic). b, Immunoblot for Merlin, FOXM1, or GAPDH in IOMM-Lee meningioma cells stably expressing a non-targeting ontrol shRNA (shNTC) or shRNAs suppressing NF2 (shNF2-1 or shNF2-2), after treatment with actinomycin D or vehicle control for 24 hours. c, QPCR for FOXM1 in M10G meningioma cells over-expressing FOXM1 or empty vector (EV). 3 biological replicates per condition. ***p≤0.0001 (Student’s t test, one-sided). d, Quantification of Annexin V confocal microscopy in M10G cells over-expressing FOXM1 or EV after treatment with actinomycin D or vehicle control for 24 hours. From left to right, 57, 58, 65, or 60 cells are shown (ANOVA, one-sided). Lines represent means, and error bars represent standard error of the means.
Extended Data Fig. 8. Cell cycle inhibition blocks meningioma growth in cells, organoids, and xenografts.
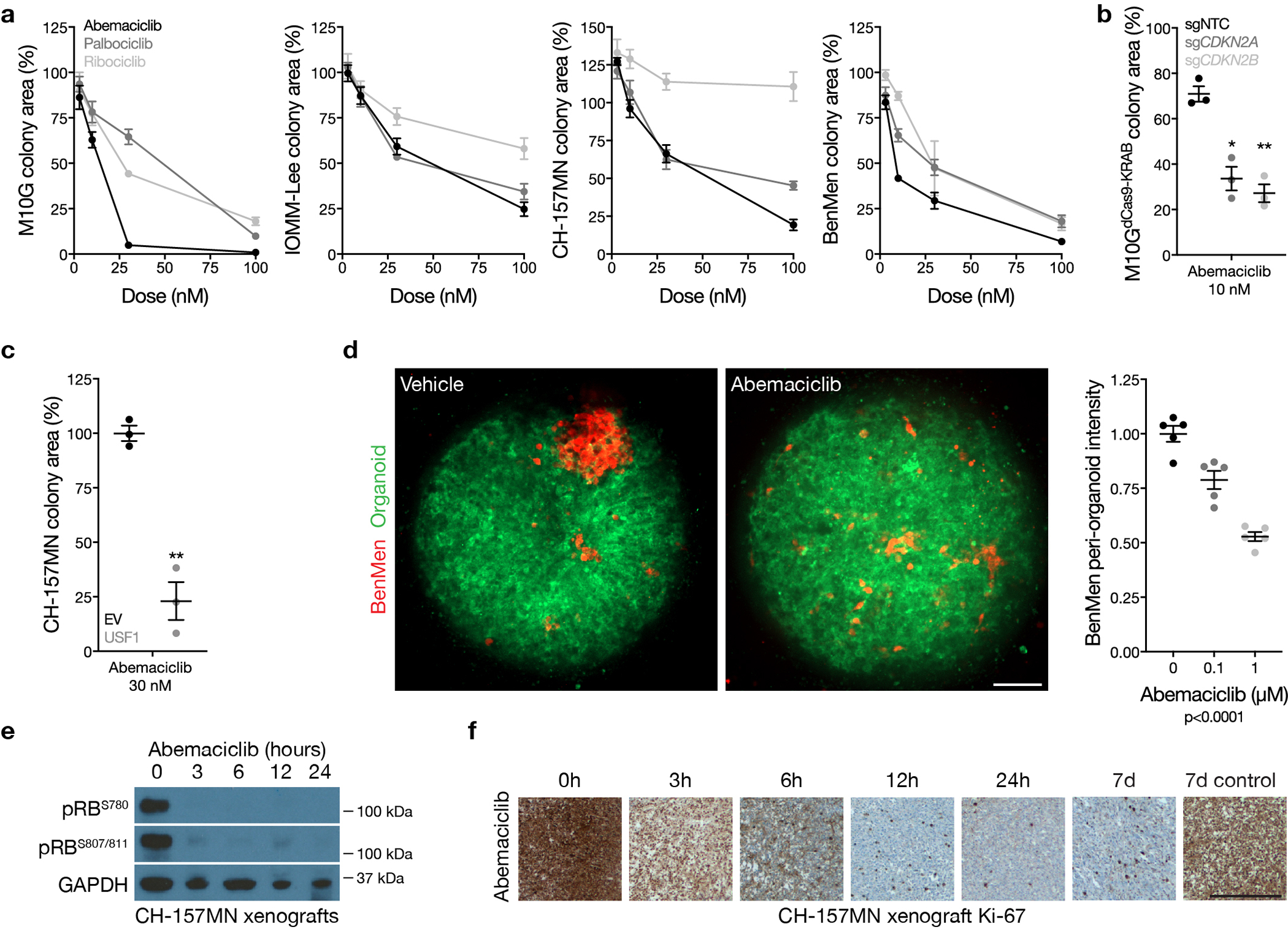
a, Relative colony area of M10G, BenMen, CH-157MN, or IOMM-Lee meningioma cells after 10 days of clonogenic growth and treatment with abemaciclib, ribociclib, or palbociclib. 3 biological replicates per condition per timepoint. b, Relative colony area of M10GdCas9-KRAB cells expressing sgNTC, sgCDKN2A, or sgCDKN2B after 10 days of clonogenic growth and treatment with abemaciclib. 3 biological replicates per condition. *p=0.002, **p=0.001 (Student’s t test, one-sided). Data are normalized to growth with vehicle treatment of each cell lines. c, Relative colony area of CH-157MN cells stably over-expressing USF or empty vector (EV) after 10 days of clonogenic growth and treatment with abemaciclib. 3 biological replicates per condition. **p=0.001 (Student’s t test, one-sided). Data are normalized to growth with vehicle treatment of each cell lines. d, Quantification of BenMen peri-organoid intensity after 10 days of growth and treatment with abemaciclib or vehicle control Representative images of meningioma (red) and organoid (green) cells are shown. Scale bar 100 μM. 5 biological replicates per condition (ANOVA, one-sided). e, Representative immunoblots from CH-157MN xenografts in NU/NU mice (left) harvested at intervals after a single treatment of abemaciclib (100 μg/g) via oral gavage (right). f, Representative images of CH-157MN xenograft Ki-67 immunohistochemistry after a daily treatment of abemaciclib or control. Scale bar 1 mm. Lines represent means, and error bars represent standard error of the means.
Extended Data Fig. 9. Meningioma DNA methylation grouping schemes uncontrolled for the influence of CNVs on β values.
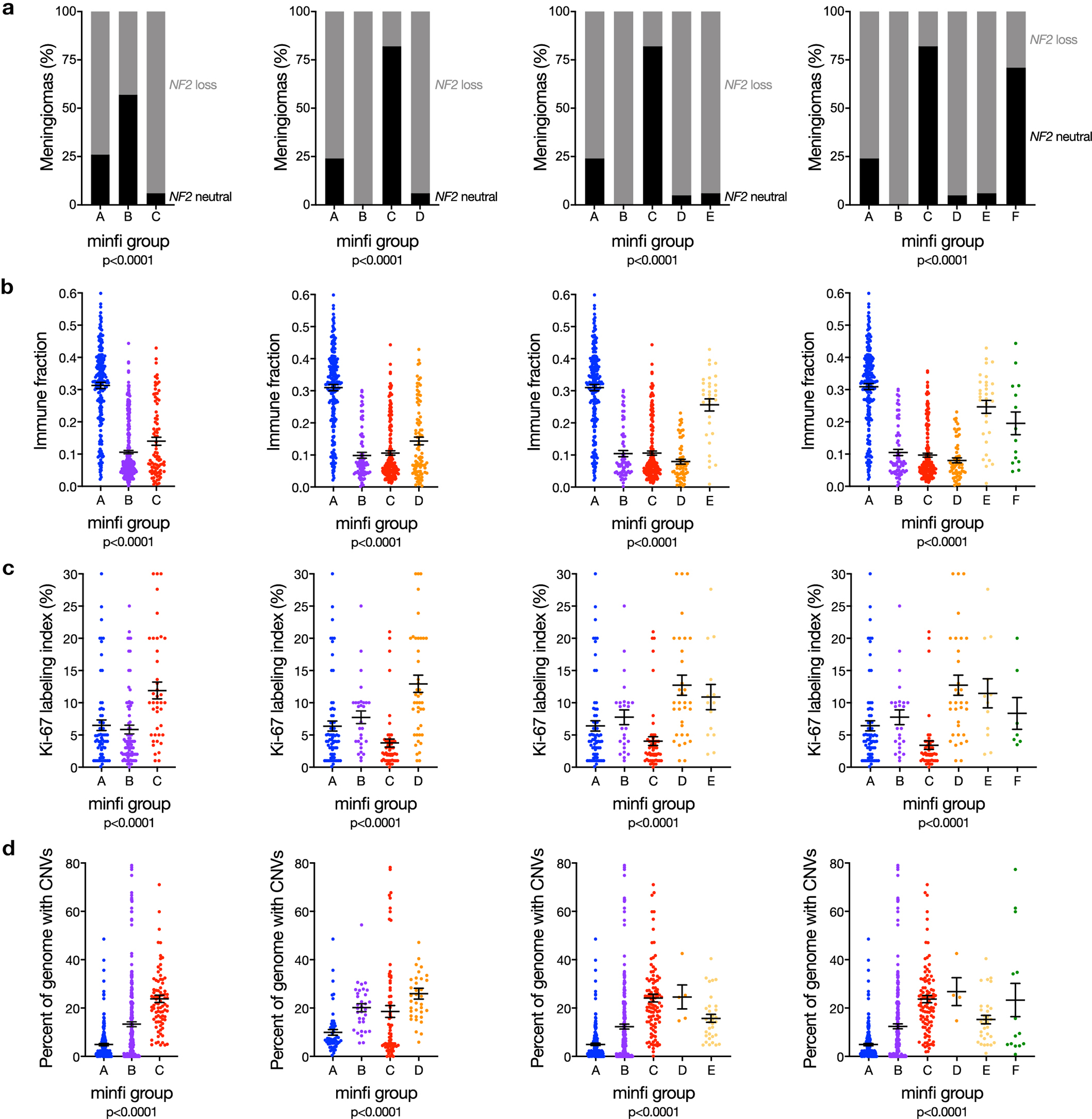
a, Meningioma DNA methylation analysis of copy number loss at the NF2 locus (n=565) across different numbers of DNA methylation groups determined by the minfi pre-processing pipeline (Chi-squared tests, two-sided). b, Meningioma DNA methylation estimation of leukocyte fraction (n=565) across different numbers of DNA methylation groups determined by the minfi pre-processing pipeline (ANOVA, one-sided). c, Ki-67 labeling index from meningioma clinical pathology reports (n=206) across different numbers of DNA methylation groups determined by the minfi pre-processing pipeline (ANOVA, one-sided). b, Meningioma genomes (n=565) with copy number variations (CNVs) across DNA methylation groups determined by the minfi pre-processing pipeline (ANOVA, one-sided). Regardless of the number of groups, meningioma DNA methylation analysis uncontrolled for the influence of CNVs on β values cannot identify a grouping scheme with non-redundant differences in clinical outcomes (Extended data figure 3d), NF2 loss, immune enrichment, cell proliferation, and chromosome instability. Lines represent means, and error bars represent standard error of the means. minfi meningioma DNA methylation grouping schemed comprised of 3, 4, 5, or 6 groups are designated by letters A-C, A-D, A-E, or A-F, respectively.
Extended Data Fig. 10. Immune-enriched meningiomas display markers of T cell exhaustion and immunoediting.
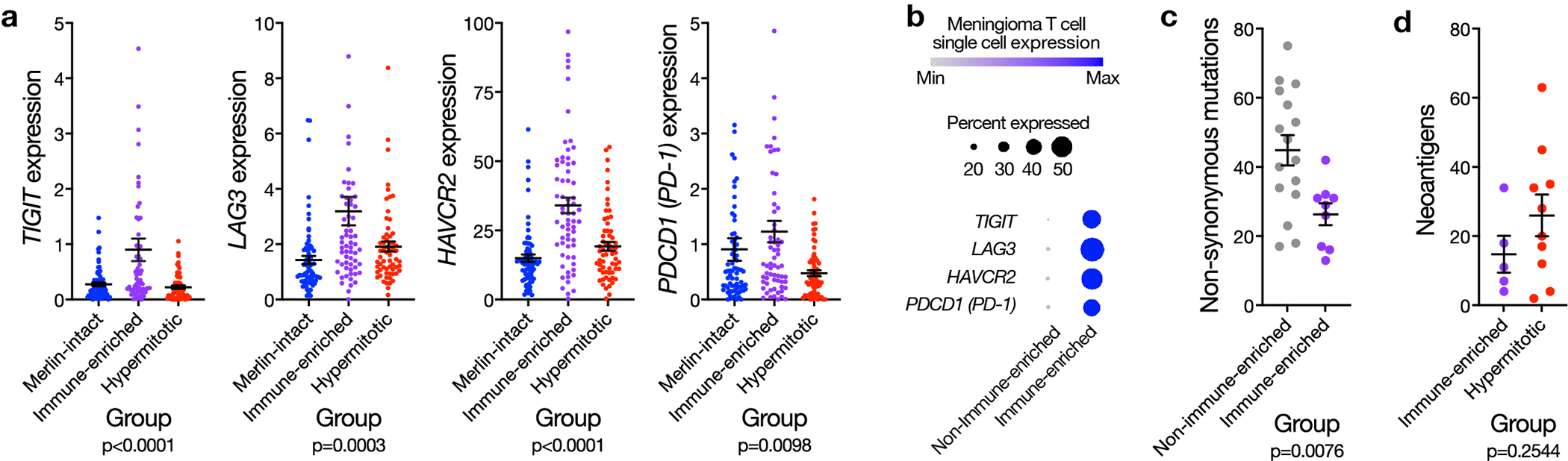
a, Meningioma transcripts per million (TPM) expression of TIGIT, LAG3, HAVCR2, or PDCD1 (n=200) T cell exhaustion markers across DNA methylation groups. Lines represent means, and error bars represent standard error of the means (ANOVA, one-sided). b, Single-cell RNA sequencing relative expression of immune exhaustion genes in T cells across Immune-enriched (n=5) and non-Immune-enriched (n=3) meningioma samples. Circle size denotes percentage of cells. Circle shading denotes average expression. c, Non-synonymous mutations from whole-exome sequencing of Immune-enriched (n=9) and non-Immune-enriched (n=16) and meningiomas, with paired normal samples, overlapping with the discovery cohort. Lines represent means, and error bars represent standard error of the means (Student’s t test, one-sided). d, Neoantigen prediction from whole-exome sequencing of Immune-enriched (n=5) and Hypermitotic (n=9) meningiomas, with paired normal samples, overlapping with the discovery cohort. Lines represent means, and error bars represent standard error of the means (Student’s t test, one-sided).
Supplementary Material
Acknowledgements
The authors thank Adam Abate, Aparna Bhaduri, Aaron Tward, and Bryan Tomlin for comments and reagents, Ken Probst and Noel Sirivansanti for illustrations, Anny Shai and the staff of the University of California San Francisco (UCSF) Brain Tumor Center Biorepository and Pathology Core, Tomoko Ozawa and the staff of the UCSF Brain Tumor Center Preclinical Therapeutics Core, and Eric Chow and the staff of the UCSF Center for Advanced Technology. This study was supported by the UCSF Wolfe Meningioma Program Project and NIH grants F30 CA246808 and T32 GM007618 to A.C.; NIH grant P50 CA097257 to J.J.P.; the UCSF Wolfe Meningioma Program Project and NIH grant F32 CA213944 to S.T.M.; the UCSF Wolfe Meningioma Program Project to C.D.E., J.E.V-M., H.N.V., S.E.B., N.A.O.B., J.S., and N.B.; NIH grant U54 CA209891 to N.J.K; and the UCSF Physician Scientist Scholar Program, the UCSF Wolfe Meningioma Program Project, and NIH grant R01 CA262311 to D.R.R.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Bi WL et al. Genomic landscape of intracranial meningiomas. Journal of Neurosurgery 125, 525–535 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Vasudevan HN et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Reports 22, 3672–3683 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou W, Triche TJ, Laird PW & Shen H SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res 46, e123–e123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahm F et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. The Lancet Oncology 18, 682–694 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Olar A et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol 133, 431–444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nassiri F et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol (2019) doi: 10.1093/neuonc/noz061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harmancı AS et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nature Communications 8, 14433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassiri F et al. A clinically applicable integrative molecular classification of meningiomas. Nature 597, 119–125 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aryee MJ et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans Dg. R. Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis 4, 1–11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark VE et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339, 1077–1080 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brastianos PK et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 45, 285–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youngblood MW et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. Journal of Neurosurgery 1, 1–10 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Agnihotri S et al. Therapeutic radiation for childhood cancer drives structural aberrations of NF2 in meningiomas. Nature Communications 8, 186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sahm F et al. Meningiomas induced by low-dose radiation carry structural variants of NF2 and a distinct mutational signature. Acta Neuropathol 134, 155–158 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Reuss DE et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 125, 351–358 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Sahm F et al. AKT1E17K mutations cluster with meningothelial and transitional meningiomas and can be detected by SFRP1 immunohistochemistry. Acta Neuropathol 126, 757–762 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Findakly S et al. Meningioma cells express primary cilia but do not transduce ciliary Hedgehog signals. Acta Neuropathologica Communications 8, 114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curto M, Cole BK, Lallemand D, Liu C-H & McClatchey AI Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol 177, 893–903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chiasson-MacKenzie C et al. NF2/Merlin mediates contact-dependent inhibition of EGFR mobility and internalization via cortical actomyosin. J Cell Biol 211, 391–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W et al. Merlin/NF2 Suppresses Tumorigenesis by Inhibiting the E3 Ubiquitin Ligase CRL4DCAF1 in the Nucleus. Cell 140, 477–490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magill ST et al. Multiplatform genomic profiling and magnetic resonance imaging identify mechanisms underlying intratumor heterogeneity in meningioma. Nature Communications 11, 4803 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee WH Characterization of a newly established malignant meningioma cell line of the human brain: IOMM-Lee. Neurosurgery 27, 389–395; discussion 396 (1990). [PubMed] [Google Scholar]
- 24.Gilbert LA et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeo NC et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods 15, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai J-C, Goldman CK & Gillespie GY Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB, and bFGF. Journal of Neurosurgery 82, 864–873 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Lobingier BT et al. An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 169, 350–360.e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeClerc S, Palaniswami R, Xie BX & Govindan MV Molecular cloning and characterization of a factor that binds the human glucocorticoid receptor gene and represses its expression. J Biol Chem 266, 17333–17340 (1991). [PubMed] [Google Scholar]
- 29.Leclerc S, Xie BX, Roy R & Govindan MV Purification of a human glucocorticoid receptor gene promoter-binding protein. Production of polyclonal antibodies against the purified factor. J Biol Chem 266, 8711–8719 (1991). [PubMed] [Google Scholar]
- 30.Flammer JR et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol 30, 4564–4574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballegeer M et al. Glucocorticoid receptor dimers control intestinal STAT1 and TNF-induced inflammation in mice. J Clin Invest 128, 3265–3279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aran D, Hu Z & Butte AJ xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biology 18, 220 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benelli M, Romagnoli D & Demichelis F Tumor purity quantification by clonal DNA methylation signatures. Bioinformatics 34, 1642–1649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Müller S, Cho A, Liu SJ, Lim DA & Diaz A CONICS integrates scRNA-seq with DNA sequencing to map gene expression to tumor sub-clones. Bioinformatics 34, 3217–3219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGranahan N et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nature Biotechnology 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frye M et al. Matrix stiffness controls lymphatic vessel formation through regulation of a GATA2-dependent transcriptional program. Nat Commun 9, 1511 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Groopman JE & Wang JF Extracellular matrix regulates endothelial functions through interaction of VEGFR-3 and integrin alpha5beta1. J Cell Physiol 202, 205–214 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Vaahtomeri K, Karaman S, Mäkinen T & Alitalo K Lymphangiogenesis guidance by paracrine and pericellular factors. Genes Dev 31, 1615–1634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiig H, Keskin D & Kalluri R Interaction between the extracellular matrix and lymphatics: consequences for lymphangiogenesis and lymphatic function. Matrix Biol 29, 645–656 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louveau A et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Antila S et al. Development and plasticity of meningeal lymphatic vessels. J Exp Med 214, 3645–3667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahn JH et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 572, 62–66 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Hu X et al. Meningeal lymphatic vessels regulate brain tumor drainage and immunity. Cell Research 30, 229–243 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rustenhoven J et al. Functional characterization of the dural sinuses as a neuroimmune interface. Cell 0, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banerji S et al. LYVE-1, a New Homologue of the CD44 Glycoprotein, Is a Lymph-specific Receptor for Hyaluronan. J Cell Biol 144, 789–801 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noor S & Wilson EH Role of C-C chemokine receptor type 7 and its ligands during neuroinflammation. J Neuroinflammation 9, 77 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wigle JT et al. An essential role for Prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J 21, 1505–1513 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis DN et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131, 803–820 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Rogers L et al. Meningiomas: knowledge base, treatment outcomes, and uncertainties. A RANO review. Journal of Neurosurgery 122, 4–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paramasivam N et al. Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol 138, 295–308 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Chen WC et al. A Prognostic Gene-Expression Signature and Risk Score for Meningioma Recurrence After Resection. Neurosurgery (2020) doi: 10.1093/neuros/nyaa355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Olano N et al. The p38 MAPK-MK2 axis regulates E2F1 and FOXM1 expression after epirubicin treatment. Mol Cancer Res 10, 1189–1202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prager BC et al. The Meningioma Enhancer Landscape Delineates Novel Subgroups and Drives Druggable Dependencies. Cancer Discov (2020) doi: 10.1158/2159-8290.CD-20-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark VE et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet 48, 1253–1259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sievers P et al. YAP1-fusions in pediatric NF2-wildtype meningioma. Acta Neuropathol 139, 215–218 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Williams EA et al. Distinct genomic subclasses of high-grade/progressive meningiomas: NF2-associated, NF2-exclusive, and NF2-agnostic. Acta Neuropathol Commun 8, 171 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Youngblood MW et al. Associations of Meningioma Molecular Subgroup and Tumor Recurrence. Neuro Oncol (2020) doi: 10.1093/neuonc/noaa226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sahm F et al. TERT Promoter Mutations and Risk of Recurrence in Meningioma. J Natl Cancer Inst 108, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spiegl-Kreinecker S et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro-Oncology 20, 1584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shankar GM & Santagata S BAP1 mutations in high-grade meningioma: implications for patient care. Neuro-Oncology 19, 1447 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guyot A et al. Analysis of CDKN2A gene alterations in recurrent and non-recurrent meningioma. J. Neurooncol. 145, 449–459 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Shoshani O et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 1–5 (2020) doi: 10.1038/s41586-020-03064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen-Sharir Y et al. Aneuploidy renders cancer cells vulnerable to mitotic checkpoint inhibition. Nature 590, 486–491 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Quinton RJ et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 590, 492–497 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sievers P et al. CDKN2A/B homozygous deletion is associated with early recurrence in meningiomas. Acta Neuropathol 140, 409–413 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merlo A et al. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nature Medicine 1, 686–692 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Herman JG et al. Distinct Patterns of Inactivation of p15INK4B and p16INK4A Characterize the Major Types of Hematological Malignancies. Cancer Res 57, 837–841 (1997). [PubMed] [Google Scholar]
- 69.Levine AJ, Jenkins NA & Copeland NG The Roles of Initiating Truncal Mutations in Human Cancers: The Order of Mutations and Tumor Cell Type Matters. Cancer Cell 35, 10–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tien A-C et al. MNGI-01. A PHASE 0 TRIAL OF RIBOCICLIB IN AGGRESSIVE MENINGIOMA PATIENTS INCORPORATING A TUMOR PHARMACODYNAMIC- AND PHARMACOKINETIC-GUIDED EXPANSION COHORT. Neuro Oncol 21, vi139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bi WL et al. Activity of PD-1 blockade with Nivolumab among patients with recurrent atypical/anaplastic meningioma: Phase II trial results. Neuro Oncol noab118 (2021) doi: 10.1093/neuonc/noab118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pagès F et al. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29, 1093–1102 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Louis D, Ohgaki H, Wiestler O & Cavenee W WHO Classification of Tumours of the Central Nervous System. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fortin J-P et al. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol 15, 1–17 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pidsley R et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol 17, 1–17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Capper D et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider VA et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Püttmann S et al. Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Laboratory Investigation 85, 1163 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Adamson B et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horlbeck MA et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stuart T et al. Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hafemeister C & Satija R Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.James G, Witten D, Hastie T & Tibshirani R An introduction to statistical learning: with applications in R. (2021). [Google Scholar]
- 89.Choudhury A meningioma-svm. (2022). doi: 10.5281/zenodo.6353877. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA methylation (n=565), RNA sequencing (n=185), and single-cell RNA sequencing data (n=8 meningioma samples, n=2 dura samples) of new samples reported in this manuscript have been deposited in the NCBI Gene Expression Omnibus under the accession GSE183656 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183656). Additional RNA sequencing data from previously reported meningiomas (n=15) from the discovery cohort are available under the accession GSE101638 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101638). Whole exome sequencing, ChIP sequencing, and additional DNA methylation profiling data incorporated into this study were derived from previously reported and deposited meningiomas in GSE101638 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101638), GSE139652 (https://www.ncbi.xyz/geo/query/acc.cgi?acc=GSE139652). The publicly available GRCh38 (hg38, https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.39/) and CRCh37.p13 datasets (hg19, https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.25/) were used in this study.