

SUMMARY
Intracellular lipopolysaccharide (LPS) sensing by the noncanonical inflammasome comprising caspase-4 or −11 governs antibacterial host defense. How LPS gains intracellular access in vivo is largely unknown. Here, we show that CD14—an LPS-binding protein with a well-documented role in TLR4 activation—plays a vital role in intracellular LPS sensing in vivo. By generating Cd14−/− and Casp11−/− mice strains on a Tlr4−/− background, we dissociate CD14’s known role in TLR4 signaling from its role in caspase-11 activation and show a TLR4-independent role for CD14 in GSDMD activation, pyroptosis, alarmin release, and the lethality driven by cytosolic LPS. Mechanistically, CD14 enables caspase-11 activation by mediating cytosolic localization of LPS in a TLR4-independent manner. Overall, our findings attribute a critical role for CD14 in noncanonical inflammasome sensing of LPS in vivo and establish—together with previous literature—CD14 as an essential proximal component of both TLR4-based extracellular and caspase-11-based intracellular LPS surveillance.
Graphical abstract
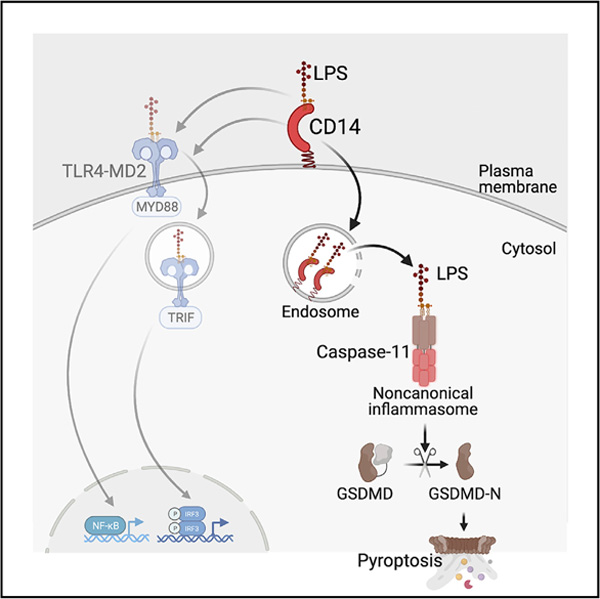
In brief
How LPS attains cytosolic access in vivo is unclear. Vasudevan et al. define a TLR4-independent role for CD14 in the cytosolic localization of LPS, triggering noncanonical inflammasome activation and pyroptosis in vivo. This finding positions CD14 as an integral component of both extracellular and intracellular LPS surveillance pathways.
INTRODUCTION
Innate immune defense, a frontline host defense strategy, relies on sensing microbial products and activities through an array of germline-encoded pattern recognition receptors (PRRs) (Kawai and Akira, 2010). Upon recognizing microbes, the PRRs initiate signaling cascades involving proteins with scaffolding and enzymatic activities that trigger a wide range of inflammatory and antimicrobial responses contingent in part on the nature of microbial signals and the subcellular sites of their detection. The PRR sensing of microbial products and activities in the cytosol assembles a specialized supramolecular organizing center called inflammasomes (Fitzgerald and Kagan, 2020). The canonical forms of inflammasomes have a receptor such as a nucleotide-binding domain and leucine-rich-repeat-containing (NLR) protein or an absent in melanoma (AIM2)-like receptor (ALR) protein, an adaptor protein (apoptosis-associated speck-like protein containing a CARD, ASC), and an effector molecule, procaspase-1. Inflammasomes convert procaspase-1 into biologically active caspase-1, which subsequently cleaves and activates proinflammatory cytokines IL-1β and IL-18 (Mathur et al., 2017). Caspase-1 also activates a pore-forming protein called gasdermin D (GSDMD) by liberating its N-terminal domain from the inhibitory C-terminal domain leading to a lytic form of cell death, pyroptosis (Chen et al., 2016; Kayagaki et al., 2015; Liu et al., 2016; Shi et al., 2015).
In contrast to the receptor-adaptor-caspase-1 organization in canonical inflammasomes, a recently discovered noncanonical inflammasome has inflammatory caspases (caspase-11 in rodents and caspase-4 and −5 in humans) functioning as PRRs for intracellular lipopolysaccharide (LPS), a major constituent of Gram-negative bacterial outer membrane (Hagar et al., 2013; Kayagaki et al., 2013; Shi et al., 2014). Direct binding of lipid A of LPS triggers oligomerization and activation of caspase-11, −4, and −5, leading to GSDMD processing and pyroptosis. Furthermore, the membrane disruption by GSDMD’s N-terminal fragment (GSDMD-N) leads to the activation of the NLRP3 inflammasome and the maturation of caspase-1, IL-1β, and IL-18 (Kayagaki et al., 2015; Rühl and Broz, 2015; Schmid-Burgk et al., 2015). The pyroptotic cell death and the concomitant release of proinflammatory mediators, including IL-1 cytokines and alarmins, are considered to play important roles in Gram-negative bacterial infections and septic shock (Kayagaki et al., 2011; Rathinam et al., 2019; Russo et al., 2021; Wang et al., 1999).
A poorly understood aspect of the noncanonical inflammasome pathway is the molecular basis of cytosolic access of LPS. Purified LPS lacks the ability to cross biological membranes to reach the cytosol of cells in vitro (Vanaja et al., 2016). However, during infections, LPS associated with bacteria or outer membrane vesicles (OMVs) secreted by bacteria gains access to the host cell cytosol (Vanaja et al., 2016). In contrast, free LPS shed from bacteria or even purified LPS can access the cytosol and activate caspase-11 uniquely in vivo but not in vitro (Kayagaki et al., 2013; Vanaja et al., 2016). HMGB1, a damage-associated molecular pattern (DAMP) with a capacity to bind LPS, has been shown to be involved in the intracellular transport of LPS (Deng et al., 2018; Kim and Kim, 2018). Nonetheless, the mechanisms by which free LPS translocates into the cytosol and activates the noncanonical inflammasome in vivo mostly remain elusive.
CD14 is a glycophosphatidylinositol-anchored plasma membrane protein and has a long-established role in LPS-mediated TLR4 activation; CD14 binds and transfers LPS to the TLR4-MD2 complex initiating the myddosome assembly and signal transduction (Dagvadorj et al., 2015; Ferrero et al., 1993; Gutsmann et al., 2001; Haziot et al., 1995, 1996; Latz et al., 2002; Moore et al., 2000; Perera et al., 1997; Schromm et al., 1996; Tan et al., 2015; Wright et al., 1990; Youn et al., 2008). Furthermore, CD14 promotes MD2-dependent TLR4 internalization into endosomes, where TLR4 engages TRIF-TRAF3 signaling to stimulate IRF3-dependent type I interferon expression (Kagan et al., 2008; Zanoni et al., 2011). Whereas the role of CD14 in TLR4-mediated host responses to LPS is well characterized, its role in noncanonical inflammasome sensing of cytosolic LPS is unknown. By generating Cd14−/− mice on a Tlr4-deficient background, we reveal that CD14 plays an essential TLR4-independent role in caspase-11 activation in vivo by delivering LPS intracellularly. Thus, we demonstrate CD14 as an integral component of the noncanonical inflammasome in vivo.
RESULTS
Noncanonical inflammasome activation by LPS in vivo is CD14-dependent
To investigate the role of CD14 in noncanonical inflammasome activation, we injected wild-type and Cd14−/− mice with LPS and measured plasma IL-1β and IL-18. CD14 is necessary for TLR4 activation of TRIF signaling and type I interferons (Zanoni et al., 2011), which prime the noncanonical inflammasome by upregulating caspase-11 expression (Rathinam et al., 2012). Therefore, to ensure that the priming step is not compromised in Cd14−/− mice, mice were first injected with IFN-γ. IFN-γ priming obviates TLR4-dependent priming of the noncanonical inflammasome, and as a result, caspase-11-mediated responses such as GSDMD activation and IL-18 secretion to cytosolic LPS are intact in IFN-γ-primed Tlr4−/− mice (Figures 1A and 1B). IL-1β release was, however, reduced in IFN-γ-primed and LPS-injected Tlr4−/− mice (Figure S1A), which is most likely due to impaired proIL-1β transcription. Importantly, all these noncanonical inflammasome responses, but not TNF, were abolished in IFN-γ-primed Casp11−/− mice, reinforcing the TLR4-independent intracellular sensing of LPS by caspase-11 (Figures 1A, 1B, and S1B).
Figure 1. Noncanonical inflammasome activation by LPS in vivo is CD14-dependent.
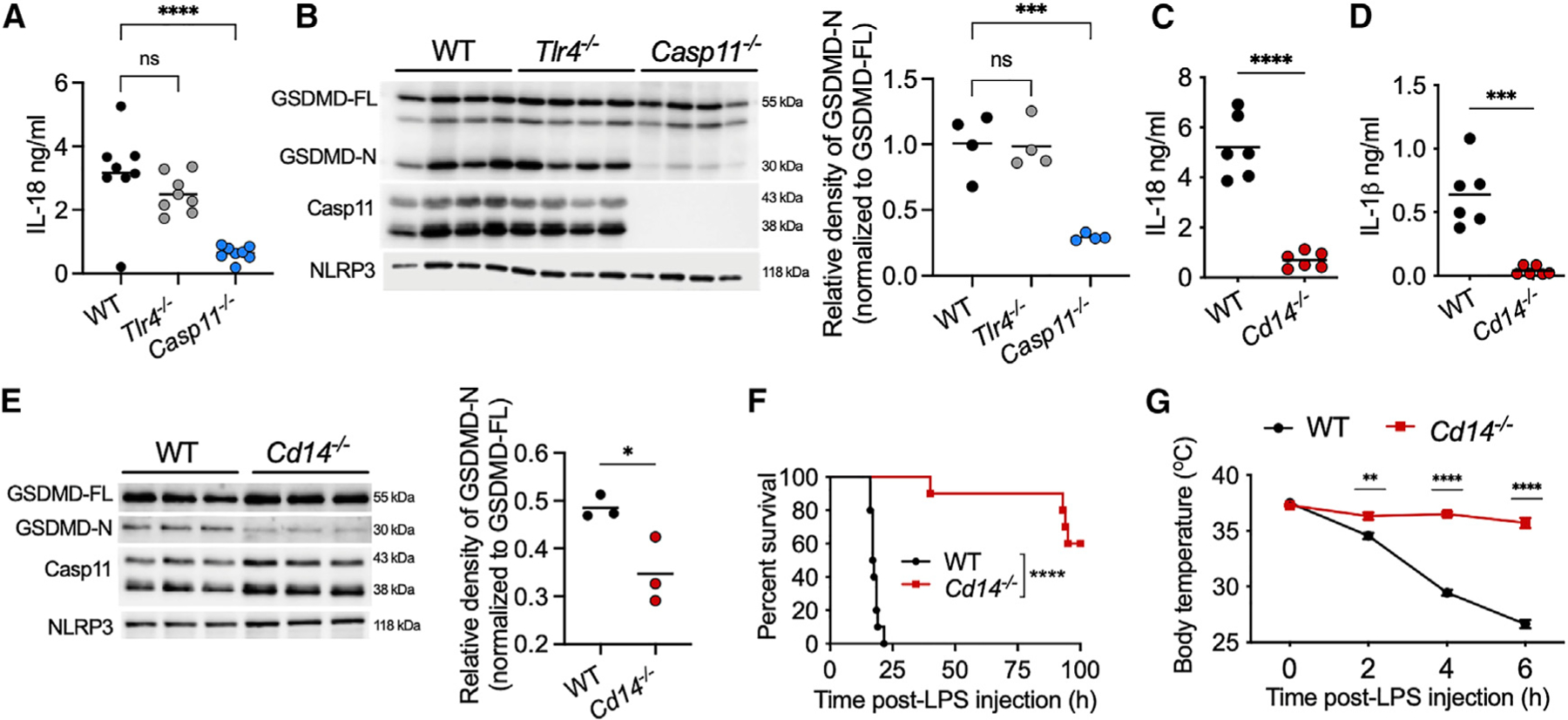
(A, C, and D) Plasma IL-18 (A and C) and IL-1β (D) in IFN-γ (1 μg; 3 h)-primed mice 6 h post-LPS (20 mg/kg) injection.
(B and E) Full-length and cleaved GSDMD (GSDMD-FL and GSDMD-N, respectively), caspase-11, and NLRP3 in spleen homogenates from indicated mice treated as above.
(F and G) Survival (F; n = 10 for each genotype) and body temperature (G; n = 10 for each genotype) of indicated mice primed with IFN-γ (0.25 μg; 3 h) and injected with LPS (10 mg/kg).
Data from two experiments (A, F, and G) or one experiment representative of two (B–E) are shown. Each circle represents a mouse, and horizontal lines represent mean (A–E). Data are presented as the mean ± SEM (G). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns: non-significant; one-way ANOVA/Dunnett’s test (A and B), unpaired two-tailed t test (C–E), Mantel-Cox test (F), or two-way ANOVA/Sidak’s test (G).
See also Figure S1.
Notably, IFN-γ-primed Cd14−/− mice displayed tremendously impaired noncanonical inflammasome responses to LPS characterized by a minimal secretion of IL-18 and IL-1β into the plasma (Figures 1C and 1D). Furthermore, cytosolic LPS-induced GSDMD activation was also significantly reduced in the spleens of IFN-γ-primed Cd14−/− mice compared with IFN-γ-primed wild-type mice (Figure 1E). Thus, Cd14−/− mice phenocopied Casp11−/− mice with respect to host responses to intracellular LPS. Moreover, IFN-γ-primed Cd14−/− mice were highly resistant to endotoxic shock (Figures 1F and 1G). Splenic caspase-11, GSDMD, and NLRP3 expression was comparable between IFN-γ-primed wild-type and Cd14−/− mice (Figure 1E). Taken together, these data indicate that CD14 is necessary for mediating noncanonical inflammasome responses to LPS in vivo and that poor noncanonical inflammasome responses in Cd14−/− mice are not due to an inherent priming issue but rather due to impaired caspase-11 activation.
CD14 functions independent of TLR4 to activate the noncanonical inflammasome
Given CD14’s significant roles in TLR4 signaling, we next sought to determine if CD14 functions independent of TLR4 in noncanonical inflammasome activation. To examine CD14’s role in a TLR4-deficient experimental system, we generated Tlr4−/− Cd14−/− mice by crossing Tlr4−/− mice and Cd14−/− mice. Additionally, we made Tlr4−/− Casp11−/− mice for comparison purposes by crossing Tlr4−/− mice and Casp11−/− mice. Tlr4−/−, Tlr4−/− Cd14−/−, and Tlr4−/− Casp11−/− mice were primed with IFN-γ and administered with LPS; Tlr4−/− mice displayed a robust noncanonical inflammasome activation evident from GSDMD processing in the spleen and IL-1β and IL-18 secretion into the plasma (Figures 2A–2F), confirming TLR4-independent activation of the noncanonical inflammasome. In contrast, Tlr4−/− Cd14−/− mice, like Tlr4−/− Casp11−/− mice, failed to mount IL-1β and IL-18 responses to LPS (Figures 2A, 2B, 2D, and 2E). In addition, GSDMD cleavage was also significantly reduced in Tlr4−/− Cd14−/− and Tlr4−/− Casp11−/− mice compared with Tlr4−/− mice (Figures 2C and 2F), whereas splenic caspase-11, GSDMD, and NLRP3 expression was comparable. Similarly, cytosolic LPS-induced release of DAMPs such as IL-1α and the recently identified galectin-1 (Russo et al., 2021) was also reduced in Tlr4−/− Cd14−/− mice compared with Tlr4−/− mice (Figures 2G, 2H, and 2I).
Figure 2. CD14 functions independent of TLR4 to activate the noncanonical inflammasome.
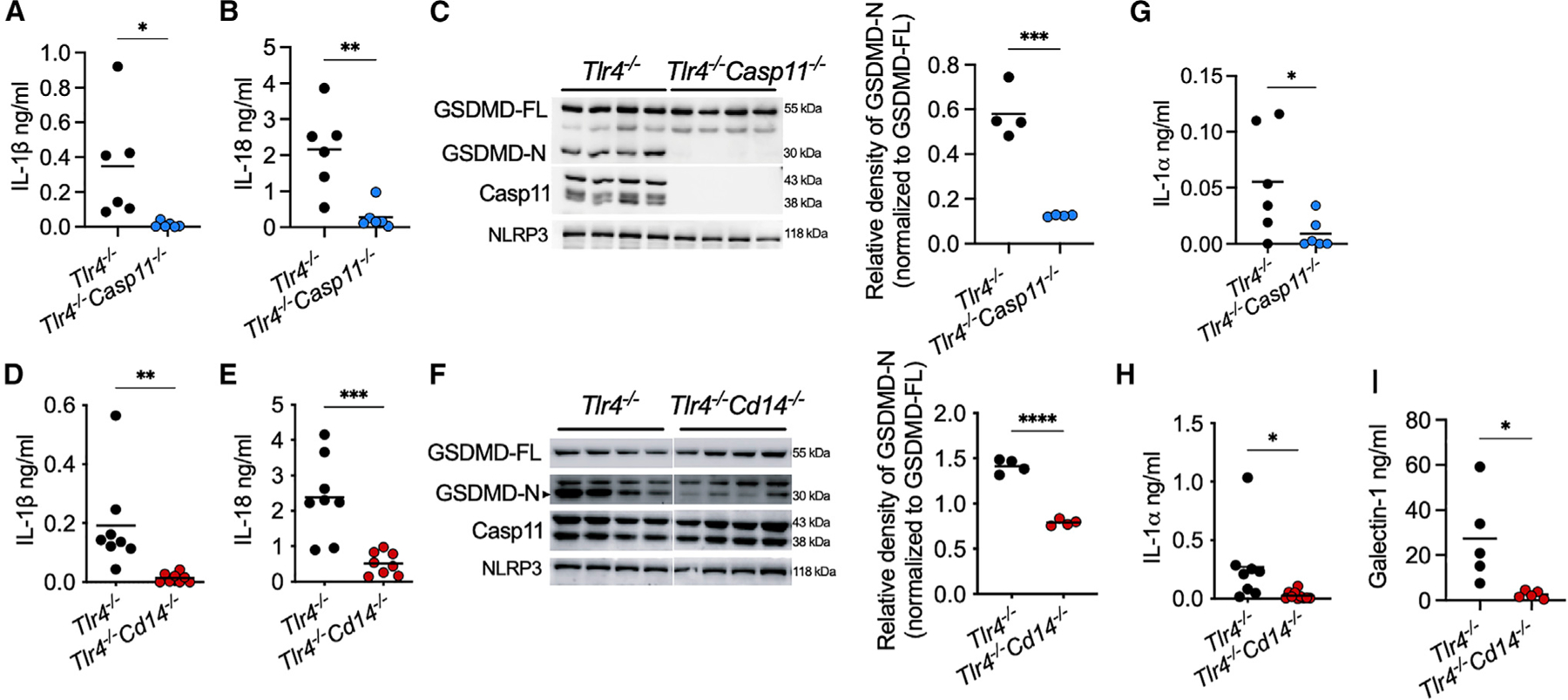
(A, B, D, E, G, H, and I) Plasma IL-1β (A and D), IL-18 (B and E), IL-1α (G and H), and galectin-1 (I) in IFN-γ-primed mice 6 h post-LPS injection.
(C and F) GSDMD, caspase-11, and NLRP3 in spleen homogenates from indicated mice treated as above.
Data from two experiments (A, B, D, E, G, and H) or one experiment representative of two (C, F, and I) are shown. Each circle represents a mouse, and horizontal lines represent mean. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; unpaired two-tailed t test.
Furthermore, IFN-γ-primed Tlr4−/− mice succumbed to the LPS challenge, whereas IFN-γ-primed Tlr4−/− Cd14−/− and Tlr4−/− Casp11−/− mice were almost completely resistant (Figures 3A and 3B). In line with the improved survival, Tlr4−/− Cd14−/− mice were also significantly protected from hypothermia (Figure 3C) and vascular coagulopathy, which is evident from their lower plasma levels of tissue factor-3 and prothrombin time (Figures 3D and 3E). It is clear from these data that the TLR4-independent function of CD14 is necessary for cytosolic LPS-elicited caspase-11 activation and pathophysiological manifestations. Notably, data from Tlr4−/− Cd14−/− mice provide solid genetic evidence that CD14’s roles in TLR4 and caspase-11 pathways are separable.
Figure 3. The contribution of CD14 to caspase-11-mediated lethal responses is TLR4-independent.
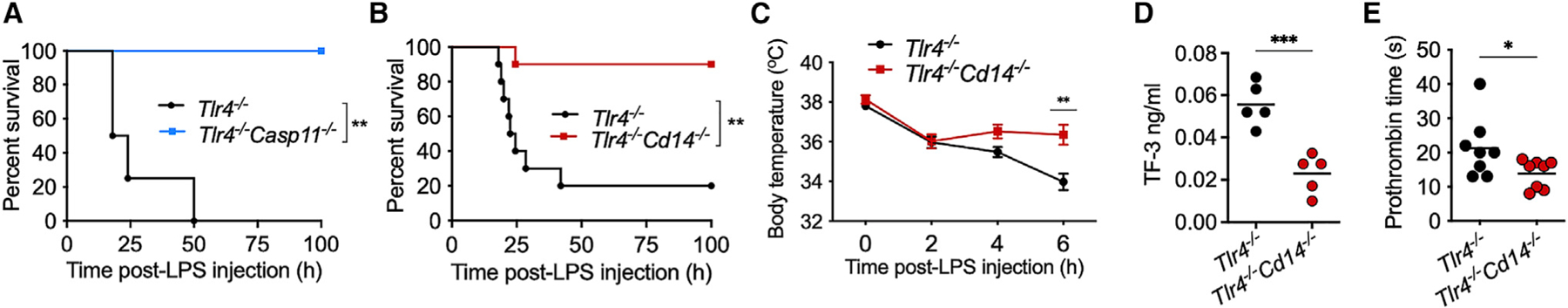
(A and B) Survival of IFN-γ-primed LPS-injected Tlr4−/− (n = 5) and Tlr4−/− Casp11−/− (n = 5) (A) and Tlr4−/− (n = 10) and Tlr4−/− Cd14−/− (n = 10) (B) mice.
(C–E) Body temperature (n = 10) (C), plasma tissue factor-3 (TF-3) (D), and prothrombin clotting time (E) in Tlr4−/− and Tlr4−/− Cd14−/− mice treated as above. Data from two experiments (B–E) or one experiment (A) are shown. Data are presented as the mean ± SEM (C). Each circle represents a mouse, and horizontal lines represent mean (D and E). *p < 0.05; **p < 0.01; ***p < 0.001; Mantel-Cox test (A and B), two-way ANOVA/Sidak’s test (C), or unpaired two-tailed t test (D and E).
CD14 enables caspase-11 activation by facilitating intracellular LPS localization in a TLR4-independent manner
Having established the TLR4-independent role of CD14 in caspase-11-mediated endotoxic shock, we next sought to identify the mechanism underlying CD14’s role in noncanonical inflammasome activation. Considering that CD14 is known to bind LPS and endogenous lipids such as oxPAPC and induce their internalization as well as that of TLR4 (Dagvadorj et al., 2015; Kitchens and Munford, 1998; Luchi and Munford, 1993; Zanoni et al., 2011, 2017), we hypothesized that CD14 mediates caspase-11 activation by mediating cytosolic translocation of LPS. To examine this, we injected mice with LPS and measured LPS quantity in the cytosol of splenic myeloid cells—which we identified as a primary cell type involved in cytosolic LPS sensing in vivo (Kumari et al., 2021)—using the HEK-Blue TLR4 reporter cell and the standard limulus amebocyte lysate (LAL) assays. The cytosol was extracted from CD11b+ myeloid cells, sorted from the spleen, using a digitonin-based fractionation method that we optimized previously to assess cytosolic LPS (Vanaja et al., 2016). We found that cytosolic LPS levels, as revealed by the HEK-Blue TLR4 reporter and LAL assays, were comparable between wild-type and Tlr4−/− myeloid cells (Figures 4A and 4B). In contrast, the cytosol of splenic myeloid cells from Tlr4−/− Cd14−/− mice contained little LPS (Figures 4A and 4B).
Figure 4. CD14 enables caspase-11 activation by facilitating intracellular LPS localization in vivo.
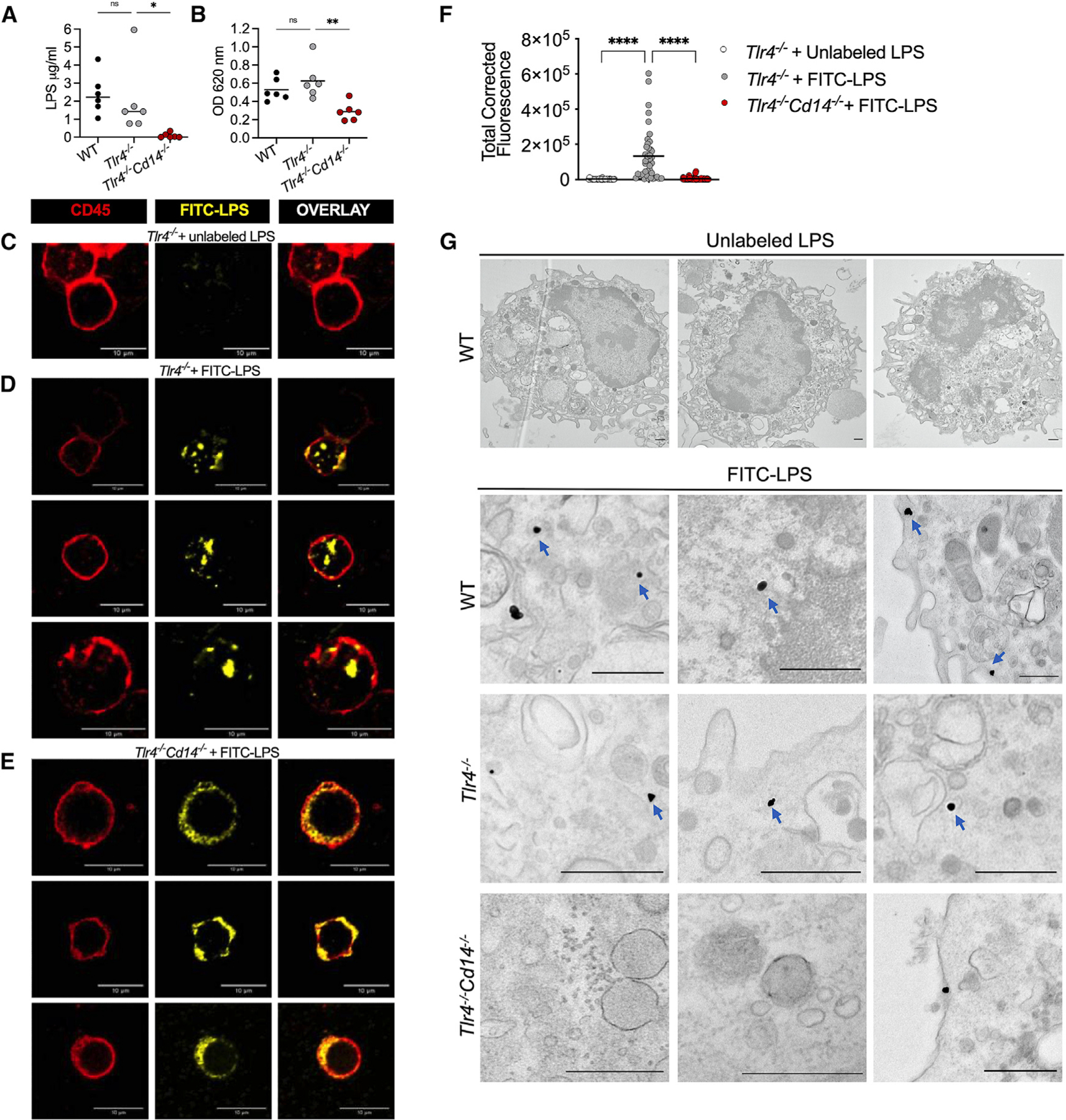
(A and B) LPS levels in the splenic myeloid cell cytosol of WT, Tlr4−/−, and Tlr4−/− Cd14−/− mice 5 h post-LPS injection assessed by the LAL (A) and HEK-TLR4 reporter cell (B) assays. Each circle represents a mouse, and horizontal lines represent mean.
(C–F) Confocal images of CD11b+ splenocytes sorted from unlabeled LPS-injected Tlr4−/− mice (C) or FITC-LPS-injected Tlr4−/− (D) and Tlr4−/− Cd14−/− (E) mice and stained with anti-FITC and anti-CD45 antibodies and the quantification of intracellular FITC-LPS intensity (F) (each circle represents a cell and horizontal lines represent mean).
(G) EM images of peritoneal lavage cells from indicated mice injected with unlabeled or FITC-labeled LPS and stained with anti-FITC gold particles. Blue arrows indicate cytosolic localization of LPS.
Data from five experiments (A and B), representative images (C–E) from four experiments, combined data (F) from four experiments, and representative images from three experiments (G) are shown. *p < 0.05; **p < 0.01; ****p < 0.0001; ns: non-significant; one-way ANOVA/Dunnett’s test. Scale bar: 10 μM (C–E) or 0.5 μM (G).
See also Figures S2, S3, and S4.
To further test our hypothesis, we used confocal microscopy to visualize intracellular LPS. Control and mutant mice were injected with FITC-LPS, or unlabeled LPS as control, and the LPS localization was tracked in CD11b+ myeloid cells sorted from the spleen. FITC-LPS colocalized with CD14 on the cell surface and in the intracellular space (Figure S2A). Furthermore, FITC-LPS was visualized in early endosomes, lysosomes, and nonlysosomal compartments (Figure S2B). Importantly, intracellular FITC-LPS was visualized in Tlr4−/− cells but not in Tlr4−/− Cd14−/− cells (Figures 4C, 4D, and 4E). Quantification of intracellular FITC intensity confirmed that Tlr4−/− Cd14−/− cells have significantly reduced intracellular LPS (Figure 4F). We next used electron microscopy (EM) to better visualize cytosolic localization of LPS; peritoneal lavage cells from FITC-LPS-injected or unlabeled LPS-injected (as control) mice were subjected to staining with gold-conjugated anti-FITC antibody followed by EM imaging. The cytosolic localization of LPS—indicated by intracellular LPS staining outside organelle structures— was visible in cells from FITC-LPS-injected wild-type and Tlr4−/− mice but not Tlr4−/− Cd14−/− mice (Figure 4G). These data reinforce the role of CD14 in the cytosolic translocation of LPS. These data collectively indicate that CD14 mediates LPS internalization into the cytosol in vivo independently of TLR4. In line with this finding, the introduction of LPS into the cytosol of BMDMs by transfection bypasses the requirement for CD14 and activates pyroptosis and IL-1β secretion in a CD14-independent manner (Figure S2C).
Since HMGB1 has been shown to mediate LPS uptake and release into the cytosol, we tested if CD14 plays a role in this process. Whereas the BMDM uptake of LPS was poor, which could be attributed to the low dose of LPS used, we found that the BMDM internalization of LPS complexed with HMGB1 occurred in a CD14-independent but receptor for advanced glycation end-products (RAGE)-dependent manner (Figure S3). Finally, to understand how LPS internalized via CD14 eventually translocates into the cytosol, we tested if CD14-mediated uptake of LPS results in early endosomal disruption, which perhaps is sufficient for LPS to gain access to the cytosol. For this, we assessed the early endosomal localization of galectin-3 (a sensor of endomembrane damage) as a proxy for the loss of endosomal integrity (Canton et al., 2020; Paz et al., 2010). We observed galectin-3 recruitment to Rab5+ early endosomes in splenic myeloid cells from LPS-injected wild-type and Tlr4−/− mice but not Tlr4−/− Cd14−/− mice (Figure S4). These data lend evidence to the idea that the endolysosomal membrane permeabilization occurring upon CD14-dependent LPS uptake may underlie the cytosolic access of LPS as it has been proposed for HMGB1-mediated delivery of LPS (Deng et al., 2018).
DISCUSSION
Intracellular sensing of microbial products dictates critical host defense responses during infections (Wright et al., 2021). The noncanonical inflammasome—via caspase-11 in mice and caspase-4 in humans—senses bacterial LPS that gains access to the cytosol and triggers inflammatory responses characterized by pyroptosis (Hagar et al., 2013; Kayagaki et al., 2013; Shi et al., 2014). Here, we identify CD14 as an essential component of the intracellular LPS sensing pathway in vivo. We found that CD14 facilitates the intracellular localization of LPS and caspase-11 activation in vivo. Using Tlr4−/− Cd14−/− mice, we show that the role of CD14 in intracellular LPS sensing is separable from its known role in TLR4 signaling. Given that pathological inflammation mediated by caspase-11 is linked to sepsis in murine models (Cheng et al., 2017; Kayagaki et al., 2011; Kumari et al., 2021; Russo et al., 2021), the specific role of CD14 in facilitating intracellular LPS sensing and noncanonical inflammasome activation reported in this study likely contributes to CD14’s detrimental effects in endotoxemia.
Though CD14 has been known to mediate LPS internalization (Dagvadorj et al., 2015; Kitchens and Munford, 1998; Luchi and Munford, 1993), its biological relevance—particularly in the context of caspase-11-dependent intracellular LPS sensing—is not known. Our data show that LPS internalization via CD14 is a consequential event that enables cytosolic translocation of LPS, licensing pyroptotic and IL-1 responses from the host. Furthermore, it has been shown that CD14 captures endogenous lipid DAMPs, such as oxPAPC, in the extracellular space and transports them into endosomes, leading to inflammasome activation and hyperactivation of dendritic cells (Zanoni et al., 2017). Thus, the capacity to directly capture lipids of host and microbial origins and trigger their internalization (Dagvadorj et al., 2015; Kitchens and Munford, 1998; Luchi and Munford, 1993; Zanoni et al., 2011, 2017) makes CD14 highly relevant in the context of cytosolic immune surveillance.
Following CD14-mediated internalization, LPS is visualized in EEA1+ early endocytic compartments in this study. We have previously shown that LPS associated with bacterial OMVs traffics to early endosomes from where it gains access to the cytosol (Vanaja et al., 2016). Considering that LPS trafficking to early endosomes favors its cytosolic translocation (Vanaja et al., 2016), CD14-mediated internalization likely traffics LPS into an early endosomal compartment that permits LPS translocation into the cytosol in vivo. Furthermore, the early endosomal membrane disruption, evident from early endosomal accumulation of galectin-3, occurring following CD14-mediated internalization of LPS is perhaps sufficient for its luminal contents such as LPS to be exposed to the cytosol. Supporting this possibility, it has been recently shown that following RAGE-mediated internalization, HMGB1-complexed LPS becomes cytosolic owing to the destabilization of lysosomes by HMBG1 (Deng et al., 2018; Kim and Kim, 2018).
Limitations of the study
While this study has revealed the TLR4-independent contribution of CD14 to caspase-11 activation, it has a few limitations. First, CD14’s role is likely to be limited to transporting LPS to early endosomes, and the precise mechanism underlying how LPS translocates from early endosomes to the cytosol following CD14-mediated uptake was not identified in this study. While this study observed CD14-dependent endosomal membrane disruption following LPS internalization, whether this is responsible for the endosomal escape of LPS was not examined. Furthermore, while our experiments show no role for CD14 in HMGB1-mediated internalization of LPS in vitro, this work did not determine whether this is also the case in vivo. Moreover, the CD14-mediated cytosolic delivery of LPS demonstrated in this study occurs only in vivo but not in BMDMs in vitro despite the expression of CD14 by BMDMs, which implicates the involvement of a yet-to-be-identifi in vivo-specific factor in this event. Another limitation is that our studies are done in the murine model, and we have not evaluated the role of CD14 in promoting LPS internalization and noncanonical inflammasome activation in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests regarding resources and reagents can be directed to Vijay Rathinam (rathinam@uchc.edu).
Materials availability
Mouse lines generated in this study are available from the lead contact upon request (subject to their availability at the time of request).
Data and code availability
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J, Cd14−/−, and Tlr4−/− mice from the Jackson Laboratory (Bar Harbor, ME) and Casp11−/− mice (a kind gift of Dr. Vishva Dixit) were bred and maintained in specific pathogen-free conditions in the animal facility of UConn Health. Tlr4−/− Casp11−/− and Tlr4−/− Cd14−/− mice were generated by breeding the respective single KO mice with Tlr4−/− mice. Genetic ablation of Tlr4, Cd14, and Casp11 was confirmed by PCR analysis of tail genomic DNA. Eight–24-week-old male and female mice were used. All experiments were carried out in accordance with the guidelines set forth by the UConn Health Institutional Animal Care and Use Committee.
Cell culture
Primary BMDMs were generated as described previously (Russo et al., 2021). TLR4-expressing HEK293 cells (HEK-Blue hTLR4; InvivoGen) were cultured in complete DMEM supplemented with 10% FBS and HEK-Blue selection (InvivoGen).
METHOD DETAILS
In vivo LPS challenge and cytokine analysis
Age- and sex-matched C57BL/6J, Cd14−/−, Tlr4−/−, Casp11−/−, Tlr4−/− Casp11−/−, and Tlr4−/− Cd14−/− mice were injected intraperitoneally (i.p.) with recombinant mouse IFN-γ (1 μg; R&D Systems) for 3 h, followed by 20 or 25 mg/kg of E. coli O 111:B4 LPS (Sigma) unless otherwise indicated. Body temperature and survival were monitored for indicated periods. Plasma was collected at 6 h or 18 h post-LPS injection for cytokine analysis. IL-1β, IL-1α, and TNF levels in the plasma were assessed by using appropriate ELISA kits (ThermoFisher). IL-18 was measured by ELISA as previously described (Kumari et al., 2021). Galectin-1 levels were assessed with the DuoSet ELISA kit (R&D Systems) according to the manufacturer’s protocol.
Immunoblotting
Spleen lysates were prepared by homogenizing spleen in PBS with protease inhibitor cocktail (Invitrogen). BCA assay was performed to measure protein concentration. Protein (25 μg) was mixed with NuPAGE sample buffer and separated by SDS-PAGE. Samples were transferred to nitrocellulose membrane using Trans-Blot Turbo Transfer System (Bio-Rad) and blocked with 2.5% milk. The membrane was probed with antibodies for mouse GSDMD (clone EPR19828; Abcam catalog no. ab209845), mouse caspase-11 (clone 17D9; Cell Signaling Technology catalog no. 14340) and NLRP3 (clone D4D8T; Cell Signaling Technology catalog no. 15101).
Tissue Factor-III assay and prothrombin time (PT)
Tissue Factor-III in the plasma was assessed by DuoSet Mouse Coagulation Factor III/TF ELISA kit (R&D systems). Prothrombin time was measured using Pacific Hemostasis Thromboplastin-D (Thermo Scientific) according to the manufacturer’s instructions (Wu et al., 2019; Yang et al., 2019). Briefly, blood was collected by cardiac puncture in 3.8% trisodium citrate (1:10 ratio) as an anticoagulant. Plasma was collected by centrifugation and prothrombin time was measured manually by rapidly adding pre-warmed thromboplastin-D to the plasma.
Cytosol fractionation and LPS assessment
Cytosolic fraction was isolated as previously described (Vanaja et al., 2016). Spleens were collected 5 h post-LPS injection and crushed through 100 μm nylon-mesh strainer. RBCs were lysed using ACK lysis buffer (Gibco). Splenocytes were incubated with Fc block, stained with mouse CD11b antibody (Clone M1/70; BioLegend catalog no. 101212) and sorted on BD FACS Aria II. Sorted CD11b + cells were resuspended in 0.001% digitonin buffer for 8 min and supernatant containing the cytosolic content was collected. Cytosolic LPS was quantified using the Limulus Amebocyte Lysate (LAL) assay (Associates of Cape Cod) according to the manufacturer’s instructions. HEK293 cells expressing hTLR4 (Invivogen) were cultured with HEK-Blue Detection and HEK-Blue Selection (Invivogen) in a 96-well plate according to the manufacturer’s protocol. Cytosol from CD11b+ splenocytes was added to the cell culture medium and SEAP activity was assessed by measuring absorbance at 620–655 nm.
Confocal microscopy
Mice were injected with FITC-conjugated or unlabeled LPS (25 mg/kg) from E. coli O111:B4 (Sigma). Spleen was dissected 5 h post-LPS injection and splenocytes were isolated by collagenase digestion with DNase I in BSS at 37°C for 20 min. Splenocytes were crushed through 100 μm nylon-mesh strainer and RBCs were lysed using ACK lysis buffer. Cytospins of CD11b+ splenocytes were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X. Slides were blocked with 10% goat serum and stained with fluorophore-conjugated antibodies against FITC (Clone 1F8.1E4; Biotium catalog no. 20210) and mouse CD45 (Clone 104; Invitrogen catalog no. 17–0454-82). Staining with an anti-FITC antibody was performed to enhance FITC signal, which otherwise might be difficult to detect in vivo. Furthermore, immunofluorescence staining was also done as indicated with anti-EEA1 (Clone C45B10; Cell signaling catalog no. 3288S), anti-Lamp1 (Clone eBio1D4B; eBioscience catalog no. 14–1071-85), APC-labeled anti-mouse CD14 (Clone Sa2–8; Invitrogen catalog no. 17–0141-81), anti-galectin-3 (Clone D4I2R; Cell Signaling catalog no. 12733) and anti-Rab5A (Clone E6N8S; Cell Signaling catalog no. 46449S) antibodies and the respective fluorophore-conjugated secondary antibodies. Cells were visualized using Zeiss LSM880 microscope and the intracellular FITC fluorescence was quantified by measuring integrated density of FITC signal normalized to background signal using ImageJ (Parry and Hemstreet, 1988).
BMDM stimulation and staining
To assess cell death and IL-1β secretion, cells were primed with IFN-γ (10 ng/mL; R&D Systems) and Pam3CSK4 (0.5 μg/mL; InvivoGen) for 3 h and stimulated with 1 μg LPS or transfected with LPS (1 μg)-Lipofectamine 2000 (Invitrogen). Supernatants were collected 16 h post-stimulation and cell death was measured by LDH release. IL-1β levels in the supernatant was measured by ELISA. To visualize LPS internalization, BMDMs were stimulated with FITC-LPS (100 ng/mL) or FITC-LPS pre-incubated with recombinant HMGB1 (400 ng/mL) for 20 min at room temperature as described previously (Deng et al., 2018). RAGE-mediated FITC-LPS internalization was assessed by treating BMDMs with DMSO or RAGE inhibitors (1 μM FPS-ZM1 (Cayman), or 10 μM RAGE antagonist peptide (RAP; Millipore) (Deng et al., 2018; Senatus et al., 2020)) 1 h before LPS treatment. Cells were washed with PBS 2 h later, fixed, permeabilized, and blocked as described above and stained with antibodies against FITC and CD45.
Transmission electron microscopy
Peritoneal lavage cells were collected 5 h post FITC-conjugated or unlabeled LPS (25 mg/kg) injection. Cells were plated on 35 mm confocal dish and fixed with 4% paraformaldehyde, followed by blocking and permeabilization with 0.1% saponin in 5% goat serum. To visualize cytosolic LPS, anti-FITC antibody conjugated with nanogold (Electron microscopy science catalog no. 25581) was used. After fixation with 2% glutaraldehyde and the gold particle size was augmented using GoldEnhance EM kit (Nanoprobes) to enhance visualization of gold labelled FITC particle. To obtain a high contrast and image organelles distinctly, cells were stained with 1% osmium tetroxide for 30 min, followed by 1% uranyl acetate staining for 30 min. The samples were then dehydrated and embedded on resin. Ultrathin sections were cut and imaged on a Hitachi H-7650 Transmission Electron Microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed on GraphPad Prism using unpaired two-tailed t test, one-way ANOVA, two-way ANOVA, or Mantel-cox test as indicated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-human/mouse NLRP3 (Clone D4D8T) | Cell Signaling Technology | Cat# 15101; RRID:AB_2722591 |
| Rabbit monoclonal anti-mouse GSDMD (Clone EPR 19828) | Abcam | Cat# Ab209845; RRID:AB_2783550 |
| Rat monoclonal anti-mouse Caspase 11 (Clone 17D9) | Cell Signaling Technology | Cat# 14340S; RRID:AB_2728693 |
| Anti-Rat IgG HRP | Jackson ImmunoResearch Labs | Cat# 712-035-150; RRID:AB_2340638 |
| Anti-Rabbit IgG HRP | Jackson ImmunoResearch Labs | Cat# 711-035-152; RRID:AB_10015282 |
| Rat monoclonal anti-IL-18 antibody (Clone 74; ELISA capture antibody) | MBL International | Cat# D047-3; RRID:AB_592016 |
| Rat monoclonal anti-IL-18 antibody Biotin (Clone 93-10C; ELISA detection antibody) | MBL International | Cat# D048-6; RRID:AB_592012 |
| APC-anti-mouse/human CD11b antibody (Clone M1/70) | BioLegend | Cat# 101212; RRID:AB_312795 |
| FITC-anti-mouse/human CD11b antibody (Clone M1/70) | BioLegend | Cat# 101206; RRID:AB_312789 |
| Monoclonal mouse anti-fluorescein (FITC) antibody (Clone 1F8.1E4) | Biotium | Cat# 20210 |
| APC-anti-mouse CD45.2 antibody (Clone 104) | Invitrogen | Cat# 17-0454-82 |
| Rabbit monoclonal anti-human/mouse/rat EEA1 (Clone 45B10) | Cell Signaling Technology | Cat# 3288S; RRID:AB_2096811 |
| Rat monoclonal anti-mouse CD107a (Lamp1) (Clone eBio1D4B) | eBioscience | Cat# 14-1071-85; RRID:AB_657533 |
| APC-anti-mouse CD14 (Clone Sa2-8) | Invitrogen | Cat# 17-0141-81; RRID:AB_469352 |
| Rabbit polyclonal anti-human/mouse/rat Galectin-3 (Clone D4I2R) | Cell Signaling Technology | Cat# 12733; RRID:AB_2798009 |
| Mouse monoclonal Rab5A (Clone E6N8S) | Cell Signaling Technology | Cat# 46449S; RRID:AB_2799303 |
| CF405M-Goat anti-rat IgG (H+L) | Biotium | Cat# 20374 |
| CF405M-Goat anti-rabbit IgG (H+L) | Biotium | Cat# 20373 |
| CF647-Goat anti-rabbit IgG (H+L) | Biotium | Cat# 20282; RRID:AB_10853478 |
| Mouse monoclonal anti-FITC gold conjugate | Electron Microscopy Sciences | Cat# 25581 |
|
Chemicals, peptides, and recombinant proteins | ||
| LPS E. coli O111:B4 | Sigma | Cat# L3024-25MG |
| FITC-conjugated LPS E. coli O111:B4 | Sigma | Cat# F3665-10MG |
| Recombinant mouse IFN-γ | R&D Systems | Cat# 485-MI-100 |
| Collagenase | Sigma Aldrich | Cat# C5138-5G |
| DNase I | Sigma Aldrich | Cat# DN25-1G |
| Recombinant mouse IL-18 (ELISA standard) | MBL | Cat# B002-5 |
| Pam3CSK4 | InvivoGen | Cat# tlrI-pms |
| Recombinant HMGB1 | HMGBiotech | Cat# HM-114 |
| RAGE inhibitor FPS-ZM1 | Cayman | Cat# 11909 |
| RAGE Antagonist Peptide (RAP) | Millipore | Cat# 553031 |
|
Critical commercial assays | ||
| Mouse IL-1β ELISA kit | Thermo Fisher Scientific | Cat# 50-171-85 |
| Mouse TNF ELISA kit | Thermo Fisher Scientific | Cat# 88-7324 |
| Mouse IL-1α ELISA kit | Thermo Fisher Scientific | Cat# 88-5019 |
| Mouse Galectin-1 ELISA kit | R&D Systems | Cat# DY1245 |
| Pierce™ BCA Protein Assay kit | Thermo Fisher Scientific | Cat# 23227 |
| Halt™ Protease Inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat# 1861279 |
| NuPAGE LDS sample buffer (4X) | Invitrogen | Cat# NP0007 |
| Trans-Blot Turbo Transfer System | Bio-Rad | Cat# 1704271 |
| Clarity ECL HRP Substrate | Bio-Rad | Cat# 170-5060S |
| Mouse Coagulation Factor III/TF ELISA kit | R&D systems | Cat# DY3178-05 |
| Pacific Hemostasis Thromboplastin-D | Thermo Scientific | Cat# 100356TS |
| ACK Lysis buffer | Gibco | Cat# A1049201 |
| Chromo-LAL | Associates of Cape Cod | Cat# C0031-5 |
| Glucashield® (1→3)-ß-D-Glucan Inhibiting Buffer | Associates of Cape Cod | Cat# GB051-5 |
| HEK-Blue Detection | Invivogen | Cat# hb-det2 |
| HEK-Blue Selection | Invivogen | Cat# hb-sel |
| Lipofectamine 2000 | Invitrogen | Cat# 11668027 |
| GoldEnhance EM kit | Nanoprobes | Cat# 2113-8ML |
|
Experimental Models: Cell lines | ||
| HEK-Blue™ hTLR4 | InvivoGen | Cat# hkb-htlr4 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Casp11−/− | Kayagaki et al., Nature 2011 Genentech | N/A |
| Mouse: Tlr4−/− | The Jackson Laboratory | RRID:IMSR_JAX:007227 |
| Mouse: Cd14−/− | The Jackson Laboratory | RRID:IMSR_JAX:003726 |
| Mouse: Tlr4−/−Cd14−/− | Generated in this study | N/A |
| Mouse: Tlr4−/−Casp11−/− | Generated in this study | N/A |
|
Oligonucleotides | ||
| Casp11−/− genotyping Primer 1: CCCTGGAAAAATCGATGACT | Kayagaki et al., Nature 2011 & Integrated DNA Technologies | Mouse listed above |
| Casp11−/− genotyping Primer 2: TGAAATGCATGTACTGAGAGCAAGG | Kayagaki et al., Nature 2011 & Integrated DNA Technologies | Mouse listed above |
| Casp11−/− genotyping Primer 3: CAATTGACTTGGGGATTCTGG | Kayagaki et al., Nature 2011 & Integrated DNA Technologies | Mouse listed above |
| Tlr4−/− genotyping Primer 1 oIMR8365: GCAAGTTTCTATATGCATTCTC | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Tlr4−/− genotyping Primer 2 oIMR8366: CCTCCATTTCCAATAGGTAG | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Tlr4−/− genotyping Primer 3 oIMR8367: TATGCATGATCAACACCACAG | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Tlr4−/− genotyping Primer 4 oIMR8368: TTTCCATTGCTGCCCTATAG | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Cd14−/− genotyping Primer 1 oIMR0662: CCGCTTCCATTGCTCAGCGG | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Cd14−/− genotyping Primer 2 oIMR1314: CCAAGTTTTAGCGCTGCGTAAC | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
| Cd14−/− genotyping Primer 3 oIMR1315: GCCAGCCAAGGATACATAGCC | The Jackson Laboratory & Integrated DNA Technologies | Mouse listed above |
|
Software and Algorithms | ||
| GraphPad Prism 9.3 | GraphPad Software | N/A |
| Fiji/ImageJ (version 10.2) | https://imagej.net/Fiji | N/A |
Highlights.
CD14 is vital for cytosolic LPS sensing by the noncanonical inflammasome in vivo
CD14 enables caspase-11 activation by facilitating the cytosolic transfer of LPS
The role of CD14 in the cytosolic sensing of LPS is TLR4-independent
ACKNOWLEDGMENTS
We thank V. Dixit and K. Fitzgerald for Casp11−/− mice and N. Frank for the HEK-Blue TLR4 reporter cell line. This work was supported by the NIH (AI119015 and AI148491 to V.R. and AI132850 to S.K.V.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110755.
DECLARATION OF INTERESTS
Authors declare no competing interests.
REFERENCES
- Canton J, Blees H, Henry CM, Buck MD, Schulz O, Rogers NC, Childs E, Zelenay S, Rhys H, Domart M-C, et al. (2020). The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat. Immunol, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, He W-T, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, and Han J (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26, 1007.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. (2017). Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest 127, 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, Wawrowsky KA, Crother TR, and Arditi M (2015). Lipopolysaccharide induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to interleukin-1a release. Immunity 42, 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. (2018). The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity 49, 740–753.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero E, Jiao D, Tsuberi BZ, Tesio L, Rong GW, Haziot A, and Goyert SM (1993). Transgenic mice expressing human CD14 Are hypersensitive to lipopolysaccharide. Proc. Nat. Acad. Sci. U S A, 2380–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, and Kagan JC (2020). Toll-like receptors and the control of immunity. Cell 180, 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutsmann T, Müller M, Carroll SF, MacKenzie RC, Wiese A, and Seydel U (2001). Dual role of lipopolysaccharide (LPS)-Binding protein in neutralization of LPS and enhancement of LPS-induced activation of mononuclear cells. Infect. Immun 69, 6942–6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar JA, Powell DA, Aachoui Y, Ernst RK, and Miao EA (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haziot A, Rong GW, Lin XY, Silver J, and Goyert SM (1995). Recombinant soluble CD14 prevents mortality in mice treated with endotoxin (lipopolysaccharide). J. Immunol. Baltim Md 1950 154, 6529–6532. [PubMed] [Google Scholar]
- Haziot A, Ferrero E, Köntgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, and Goyert SM (1996). Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity 4, 407–414. [DOI] [PubMed] [Google Scholar]
- Kagan JC, Su T, Horng T, Chow A, Akira S, and Medzhitov R (2008). TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-b. Nat. Immunol 9, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kim HM, and Kim Y-M (2018). HMGB1: LPS delivery vehicle for caspase-11-mediated pyroptosis. Immunity 49, 582–584. [DOI] [PubMed] [Google Scholar]
- Kitchens RL, and Munford RS (1998). CD14-dependent internalization of bacterial lipopolysaccharide (LPS) is strongly influenced by LPS aggregation but not by cellular responses to LPS. J. Immunol 160, 1920–1928. [PubMed] [Google Scholar]
- Kumari P, Russo AJ, Wright SS, Muthupalani S, and Rathinam VA (2021). Hierarchical cell-type-specific functions of caspase-11 in LPS shock and antibacterial host defense. Cell Rep 35, 109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, and Espevik T (2002). Lipopolysaccharide rapidly traffics to and from the golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J. Biol. Chem 277, 47834–47843. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchi M, and Munford RS (1993). Binding, internalization, and deacylation of bacterial lipopolysaccharide by human neutrophils. J. Immunol 151, 959–969. [PubMed] [Google Scholar]
- Mathur A, Hayward JA, and Man SM (2017). Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol 103, 233–257. [DOI] [PubMed] [Google Scholar]
- Moore KJ, Andersson LP, Ingalls RR, Monks BG, Li R, Arnaout MA, Golenbock DT, and Freeman MW (2000). Divergent response to LPS and bacteria in CD14-deficient murine macrophages. J. Immunol 165, 4272–4280. [DOI] [PubMed] [Google Scholar]
- Parry WL, and Hemstreet GP (1988). Cancer detection by quantitative fluorescence image analysis. J. Urol 139, 270–274. [DOI] [PubMed] [Google Scholar]
- Paz I, Sachse M, Dupont N, Mounier J, Cederfur C, Enninga J, Leffler H, Poirier F, Prevost M, Lafont F, et al. (2010). Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell Microbiol 12, 530–544. [DOI] [PubMed] [Google Scholar]
- Perera PY, Vogel SN, Detore GR, Haziot A, and Goyert SM (1997). CD14-dependent and CD14-independent signaling pathways in murine macrophages from normal and CD14 knockout mice stimulated with lipopolysaccharide or taxol. J. Immunol 158, 4422–4429. [PubMed] [Google Scholar]
- Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, and Fitzgerald KA (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VAK, Zhao Y, and Shao F (2019). Innate immunity to intracellular LPS. Nat. Immunol 20, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rühl S, and Broz P (2015). Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol 45, 2927–2936. [DOI] [PubMed] [Google Scholar]
- Russo AJ, Vasudevan SO, Méndez-Huergo SP, Kumari P, Menoret A, Duduskar S, Wang C, Sáez JMP, Fettis MM, Li C, et al. (2021). Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat. Immunol 22, 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, and Hornung V (2015). Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur. J. Immunol 45, 2911–2917. [DOI] [PubMed] [Google Scholar]
- Schromm AB, Brandenburg K, Rietschel E.Th., Flad H-D, Carroll SF, and Seydel U (1996). Lipopolysaccharide-binding protein mediates CD14-independent intercalation of lipopolysaccharide into phospholipid membranes. FEBS Lett 399, 267–271. [DOI] [PubMed] [Google Scholar]
- Senatus L, López-Díez R, Egaña-Gorroño L, Liu J, Hu J, Daffu G, Li Q, Rahman K, Vengrenyuk Y, Barrett TJ, et al. (2020). RAGE impairs murine diabetic atherosclerosis regression and implicates IRF7 in macrophage inflammation and cholesterol metabolism. Jci Insight 5, e137289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Tan Y, Zanoni I, Cullen TW, Goodman AL, and Kagan JC (2015). Mechanisms of toll-like receptor 4 endocytosis reveal a common immune-evasion strategy used by pathogenic and commensal bacteria. Immunity 43, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, and Rathinam VAK (2016). Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165, 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. [DOI] [PubMed] [Google Scholar]
- Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, and Mathison JC (1990). CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431–1433. [DOI] [PubMed] [Google Scholar]
- Wright SS, Vasudevan SO, and Rathinam VA (2021). Mechanisms and consequences of noncanonical inflammasome-mediated pyroptosis. J. Mol. Biol 434, 167245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, Grover SP, Zhang X, Li L, Xiang B, et al. (2019). Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 50, 1401–1411.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, Wu J, Wang Z, Liu Y, Chen F, et al. (2019). Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity 51, 983–996.e6. [DOI] [PubMed] [Google Scholar]
- Youn JH, Oh YJ, Kim ES, Choi JE, and Shin J-S (2008). High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-α production in human monocytes. J. Immunol 180, 5067–5074. [DOI] [PubMed] [Google Scholar]
- Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, Granucci F, and Kagan JC (2011). CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Gioia MD, Springstead JR, and Kagan JC (2017). By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity 47, 697–709.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.