

Abstract
Future energy demands for green hydrogen have fueled intensive research on proton‐exchange membrane water electrolyzers (PEMWE). However, the sluggish oxygen evolution reaction (OER) and highly corrosive environment on the anode side narrow the catalysts to be expensive Ir‐based materials. It is very challenging to develop cheap and effective OER catalysts. Herein, Co‐hexamethylenetetramine metal–organic framework (Co‐HMT) as the precursor and a fast‐quenching method is employed to synthesize RuO2 nanorods loaded on antimony‐tin oxide (ATO). Physical characterizations and theoretical calculations indicate that the ATO can increase the electrochemical surface areas of the catalysts, while the tensile strains incorporated by quenching can alter the electronic state of RuO2. The optimized catalyst exhibits a small overpotential of 198 mV at 10 mA cm−2 for OER, and keeps almost unchanged after 150 h chronopotentiometry. When applied in a real PEMWE assembly, only 1.51 V is needed for the catalyst to reach a current density of 1 A cm−2.
Keywords: electrochemical catalysis, oxygen evolution reaction, proton‐exchange membrane water electrolyzer, RuO2 , water electrolysis
The tensile strained RuO2 nanorod catalyst loaded on antimony‐tin oxide (ATO) substrate is prepared by a fast‐quenching method. With multiple characterizations, it is proved that the tensile strain and ATO substrate are beneficial for the oxygen evolution reaction (OER). This work has demonstrated the OER enhancements in RuO2 when tensile strains are incorporated.
1. Introduction
Hydrogen energy can offer a promising way to reduce fossil energy usage and alleviate carbon dioxide emissions.[ 1 ] Water electrolysis, consisting of two half‐reactions of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), is regarded as one of the best solutions to hydrogen production.[ 2 ] The HER is known to be more favorable in kinetics and mass transport,[ 3 ] so it is more compulsory to develop OER catalysts for coupling industrial applications. In alkaline electrolytes, layered double hydroxides are active and stable enough to drive the sluggish OER.[ 4 ] However, most of the reported catalysts suitable for alkaline OER are thermodynamically unstable when operated in acid electrolytes. For now, it is commonly accepted that proton‐exchange membrane water electrolyzers (PEMWE) can deliver much higher current densities than alkaline electrolyzers due to the intrinsic high ionic conductivity of the proton membrane.[ 5 ] Therefore, PEMWE devices are more practicable for industrial usage. Nevertheless, the acidic media and high operating voltages in PEMWE have limited the anodic catalysts to be expensive Ir‐based materials.[ 6 ] Beyond Ir‐based materials, much cheaper Ru‐based materials can also be employed for OER in acid media.[ 7 ] Furthermore, Nørskov's volcano curve has indicated that the activity of RuO2 toward OER is located near the peak position.[ 8 ] Though with relatively higher OER activity than IrO2, RuO2 cannot operate for a long time due to the formation of volatile RuO4, which tends to dissolve in the electrolyte.[ 9 ]
In previous reports, doping of foreign atoms is the mainstream for boosting the OER activity of RuO2.[ 10 ] Indeed, Sargent et al. had done high‐throughput density functional theory (DFT) screenings and experimental trials to indicate that the Ru‐Ir‐Sr ternary oxide can achieve exceptionally high OER activity and stability.[ 7b ] However, the selective leaching of unstable elements (dopants) is essential for obtaining high OER activity.[ 11 ] In real PEMWE devices, transition metal ions leached from the anode catalysts would irreversibly poison the proton membranes, thus degrading the electrolyzer performances.[ 12 ] So alternative methods must be proposed to modify RuO2 without highlighting the doping of unstable atoms. To this end, strains can be introduced as positive enhancements for electrolysis.[ 13 ] For instance, the self‐generated strained Pt skins outside the Pt‐M (transition metals) alloy can offer a much higher mass activity than the pristine Pt in ORR.[ 14 ] While for the OER, Yang et al. prepared a strained iridium oxide catalyst based on lattice mismatches, and the strained sample exhibited a mass activity of 3.7 times higher than that of bare iridium oxide.[ 15 ] Strains would cause electron structure modifications.[ 16 ] Since the electron structure is closely involved in the binding with oxygen intermediates (*OH, *O, and *OOH), changes of it would tune the catalytic activity.[ 16 , 17 ] However, studies related to strain‐influenced RuO2 activity toward OER are still missing due to the complex preparation procedures like epitaxial growths of thin films.[ 18 ] So easy methods to induce strains in RuO2 and systematic investigations on it are urgently needed.
Here, we report tensile strained RuO2 nanorods growing on antimony‐tin oxide (ATO) particles using the Co‐hexamethylenetetramine metal‐organic framework (Co‐HMT) and fast‐quenching treatment. The tensile strains were introduced on RuO2 nanorods due to the fast‐quenching process instead of lattice mismatches between ATO and RuO2. As a result, the modified electronic states promote the OER activity with an overpotential as low as 198 mV at 10 mA cm−2. Moreover, the catalyst shows no apparent degradations after 12 h chronopotentiometry on the carbon electrode or 150 h chronopotentiometry on the Ti felt electrode. When applied in PEMWE devices, only 1.51 V (IR‐free) is needed to drive a current density of 1 A cm−2. Our work has demonstrated that tensile strains can be employed as a positive enhancement for OER of RuO2, thus would inspire future OER catalyst designs.
2. Results and Discussion
As illustrated in Figure 1a, the tensile‐strained RuO2 loaded on ATO was prepared via ion substitutions, pyrolysis, and fast quenching. The ATO was prepared by the method presented in the previous report.[ 19 ] The Co‐HMT was first formed in ATO dispersions by adding the Co salt and hexamethylenetetramine, and the self‐assembled product was noted as Co‐HMT/ATO. From the scanning electron microscope (SEM) image shown in Figure S1 (Supporting Information), it is found that the Co‐HMT exists as spindle‐like crystals together with irregular ATO nanoparticles. The as‐prepared Co‐HMT/ATO composites were then subscribed to Ru3+ substitutions, where the RuCl3•3H2O/Co‐HMT mass ratios are fixed to be around 1:1 to ensure sufficient ion replacements. After Ru3+ substitutions, no separated Co‐HMT can be observed (Figure S2, Supporting Information), indicating the destruction of Co‐HMT structures during the substitution process (noted as “Ru‐Co‐HMT/ATO”). The XRD patterns in Figure 1b also prove that no pure Co‐HMT residuals are retained in Ru‐Co‐HMT/ATO. Besides, the transmission electron microscopy (TEM) and corresponding elementals mapping images (Figure S3, Supporting Information) show that the Ru elements tend to reside on the ATO surfaces, implying that the Co‐HMT might undergo dissolution and redeposition in the Ru3+ substitution process. In the last step, final pyrolysis was conducted to transform the Ru species into RuO2. In order to incorporate tensile strains into the RuO2, a fast‐quenching process was performed.[ 20 ] In detail, the powder product was immediately taken out from a hot furnace and quenched in liquid N2 to preserve the residual stresses. After quenching, the final sample was collected and named s‐RuO2/ATO (“s” refers to “strained”).
Figure 1.
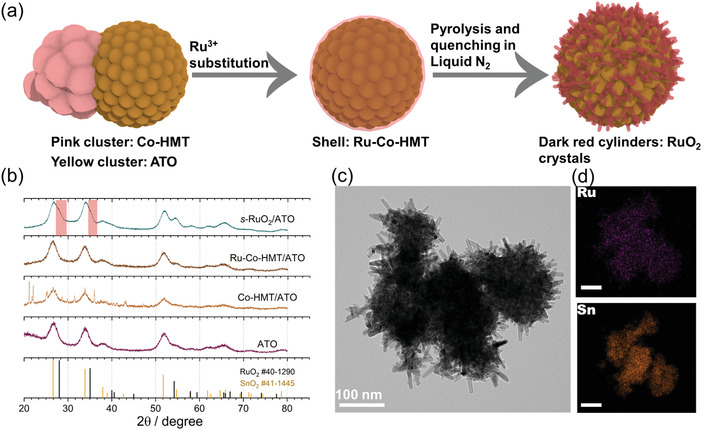
a) The schematic illustration of the synthesis procedures. b) The XRD patterns of ATO, Co‐HMT/ATO, Ru‐Co‐HMT/ATO, and s‐RuO2/ATO. The red areas inserted indicate the bumps induced by the existence of RuO2. c) The TEM image of s‐RuO2/ATO. d) The elements mapping images of s‐RuO2/ATO. The scale bars inserted are 100 nm.
In the X‐ray diffraction (XRD) patterns of s‐RuO2/ATO, no prominent diffraction peaks attributed to RuO2 are presented. Conversely, only small bumps near the ATO peaks (SnO2) can be identified (Figure 1b). As shown in the TEM images (Figure 1c), s‐RuO2/ATO exhibits a nanorod morphology, totally different from the initial ATO (Figures S4 and S5, Supporting Information). Similar morphology has been reported in Co‐doped RuO2.[ 10a ] Furthermore, elementals mapping images (Figure 1d) show that Ru elements are more prone to distribute on the outside while Sn elements are on the inside, proving that the nanorods outside are RuO2 crystals instead of ATO. The nanorod structures of s‐RuO2/ATO can favor the maximum exposure of OER active sites.
Apart from s‐RuO2/ATO, another three samples including n‐RuO2/ATO (without quenching process, “n” refers to “normal”), s‐RuO2 (without ATO substrate), and n‐RuO2 (without quenching process and ATO substrate) were prepared as counterpart references. As shown in Figure S6 (Supporting Information), n‐RuO2/ATO shows identical morphology to s‐RuO2/ATO, implying that the quenching process does not change the overall morphology. Similar morphologies can also be observed between s‐RuO2 and n‐RuO2 (Figure S7, Supporting Information). However, samples without ATO substrates show thicker and broader nanorods. Some of the nanorods are even broader than 30 nm, much bigger than samples with ATO substrates (Figure S8, Supporting Information), indicating that ATO substrates can reduce the sizes of RuO2 crystals. It is easy to conjecture that thin‐layer precursors containing Ru elements covered on ATO surfaces have fewer sources to contact and grow, so relatively smaller RuO2 nanorods can grow on the ATO substrates. The Ru and Co contents in s‐RuO2/ATO checked by inductively coupled plasma mass spectrometry (ICP‐MS) are 19.5 and 2.4 wt%, and similar results are also obtained from n‐RuO2/ATO provided by the same precursors (Table S1, Supporting Information). Here, it should be noted that small fractions of Co residuals are inevitable due to Co‐HMT precursors. Since several reports have clarified that Co dopants positively influence the OER,[ 10 , 21 ] we have tentatively kept the Ru/Co ratios during the substitution processes for a rational comparison.
The electrochemical OER performances were first assessed on a three‐electrode rotating ring electrode configuration in 0.1 m HClO4 solution. For comparison, the load masses of Ru for all samples keep at 0.19 mg cm−2. In Figure 2a; and Figure S9 (Supporting Information), the linear sweep voltammetry (LSV) curves are plotted in the range of 1.3–1.6 V (vs reversible hydrogen electrode (RHE)). As can be seen, the optimized s‐RuO2/ATO exhibits an overpotential of 198 mV at the current density of 10 mA cm−2 (η 10), which is much lower than that of n‐RuO2/ATO (η 10 = 224 mV), indicating that the quenching process is beneficial for boosting the OER activity. Here, the reported η 10 of s‐RuO2/ATO is lower than most previous works (Table S2, Supporting Information). Additionally, the sample without the ATO substrate (s‐RuO2) exhibits lower activity (η 10 = 241 mV) than n‐RuO2/ATO, demonstrating that the ATO substrates are also vital for enhancing the OER activity. Identical to the above assumption, the n‐RuO2 without quenching process and ATO substrate show the worst OER activity (η 10 = 276 mV).
Figure 2.
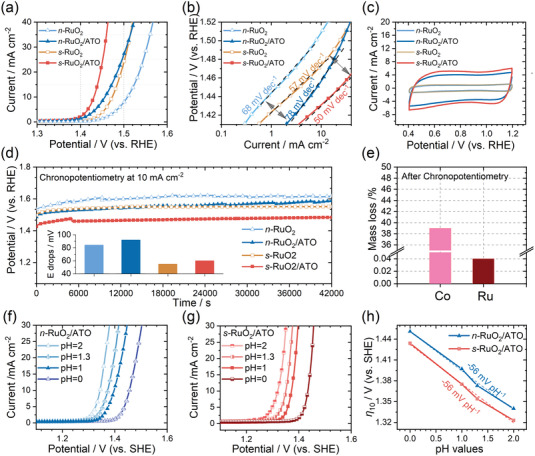
a) The LSV curves, b) The Tafel slopes, c) The CV curves, and d) The chronopotentiometry curves of n‐RuO2, n‐RuO2/ATO, s‐RuO2, and s‐RuO2/ATO. e) The mass loss percentages evaluated from ICP‐MS results in the electrolyte after 12 h chronopotentiometry of s‐RuO2/ATO. f) The LSV curves of n‐RuO2/ATO recorded at pH = 2, 1.3, 1, and 0. g) The LSV curves of s‐RuO2/ATO recorded at pH = 2, 1.3, 1, and 0. h) The η 10 from f,g) recorded at different pH values for n‐RuO2/ATO and s‐RuO2/ATO.
The LSV curves were further plotted on the logarithm axis. As shown in Figure 2b, the Tafel slopes for s‐RuO2/ATO, n‐RuO2/ATO, s‐RuO2, n‐RuO2 are 50, 78, 57, and 68 mV dec−1, respectively. The trend summarized from the obtained Tafel slopes is that the quenching process could reduce the Tafel slope. Besides, the quenched samples seem to exhibit similar Tafel slopes, and so do the unquenched samples. Since the Tafel slopes reflect the intrinsic microkinetic in electrochemical processes, we suggest that the quenching process could alter the catalyst's binding strengths with the OER intermediates, thus rendering different coverages of oxygen intermediates.[ 22 ]
In order to investigate the intrinsic activity of the samples, we estimated the electrochemical surface areas (ECSA) by calculating the double‐layer capacitances based on the cyclic voltammetry (CV) measurements between 0.4 and 1.2 V (vs RHE) under continuous N2 bubbling. In Figure 2c; and Figure S10 (Supporting Information), it is observed that the ECSAs of s‐RuO2/ATO and n‐RuO2/ATO are nearly four times larger than those of s‐RuO2 and n‐RuO2. Though the total load masses for samples with the ATO substrates are much higher, the recorded CV curve of bare ATO at the load mass equal to the quantity in s‐RuO2/ATO indicates that the ATO is not the main factor for the enhanced ECSA (Figure S11, Supporting Information). As discussed above, the ATO can decrease the crystal sizes of RuO2, which might be the primary reason for the enhanced ECSA after using ATO as substrates. Besides, we also observe that the quenching process also improves ECSAs from the differences between quenched and unquenched samples. To clarify if the enhanced OER activity originated from the enhanced ECSAs, we normalized the LSV curves by ECSA capacitances in Figure S12 (Supporting Information). As can be seen, these differences between samples with and without ATO substrates are diminished. Nevertheless, for the quenched and unquenched counterparts, a significant difference still exists.
As mentioned above, the Co dopants are inevitable. Since the Ru/Co ratios are similar in samples, it can be deduced that the above conclusions about the positive effects of ATO and quenching are reasonable. To extend the quenching effect, we have also prepared RuO2 from the fast‐quenching process with no Co involvement (see detailed information in the Experimental Section). As shown in Figure S13 (Supporting Information), the quenching step enhances the OER performances of RuO2, proving its transferability to undoped RuO2.
The long term‐stability tests were performed at 10 mA cm−2 for 12 h and presented in Figure 2d. As can be seen, after 12 h chronopotentiometry, these samples show increased overpotentials at different degrees. The increased overpotentials were plotted in the inserted image of Figure 2d. Intriguingly, these samples with ATO as substrates show no significant differences compared to their counterparts, evidencing that the ATO has limited effects on improving the stability of RuO2. However, previous studies have presented that the ATO substrate can improve the stability of IrO2.[ 19 , 23 ] Given the similarities in rutile structures between RuO2 and IrO2, we believe that the insufficient contacts between RuO2 and ATO may result in the limited effects of ATO in our experiment. Nevertheless, samples with quenching processes exhibit improved stability compared to their counterparts, proving that quenching can be used as a positive factor to enhance the stability of RuO2. Combining with the Tafel slopes and ECSAs analyzed above, we tentatively suggest that the ATO substrate can increase the exposed active sites, and the quenching process can alter the intrinsic characteristics of Ru sites. In addition, the CV curve of s‐RuO2/ATO after chronopotentiometry shows a 13% increment in ECSA. (Figure S14, Supporting Information). As evidenced by the ICP‐MS results (Figure 2e), we attribute the increased ECSA to the leaching of Co atoms.
The glass carbon electrode is not suitable for more extended chronopotentiometry tests due to the detachment of active materials and oxidation of the carbon electrode. Further evaluations of s‐RuO2/ATO's stability were performed on Ti felt electrode. There were no apparent attenuations of the potential after a continuous 150 h chronopotentiometry test (Figure S15, Supporting Information).
The LSV curves recorded at different pH values are presented in Figure 2f,g on the standard hydrogen electrode (SHE) scales. As can be seen, the LSV curves gradually move to higher potentials under lower pH values. In Figure 2h, we observed a clear pH‐dependent η 10 with the same slopes of −56 mV pH−1 (concerted proton‐electron transfer: −59 mV pH−1), demonstrating the absence of the lattice oxygen mechanism (LOM) pathway. The LOM pathway is not beneficial for the structural stability of the catalysts due to the collapse of the metal–oxygen framework. The absence of the LOM pathway can partly explain the excellent stability of s‐RuO2/ATO.[ 24 ] The above results demonstrate the synergic positive effects of the ATO substrate and quenching process toward OER, but pertinent proofs are still needed to unravel the mechanism.
The XRD is sensitive to subtle differences in crystal structures. To carefully screen the differences between samples, prolonged scans with 0.5° min−1 were used. As shown in Figure 3a, s‐RuO2/ATO and n‐RuO2/ATO show two sets of diffraction peaks, which can be assigned to SnO2 (PDF No: 41‐1445) and RuO2 (PDF No: 40‐1290). The peaks of RuO2 in s‐RuO2/ATO and n‐RuO2/ATO are heavily overlapped with peaks from SnO2, and only small bumps at higher degrees can be distinguished. Additionally, the diffraction peaks of s‐RuO2 and n‐RuO2 concord very well with the RuO2. It can be seen that the diffraction peaks of RuO2 in s‐RuO2 and n‐RuO2 are much more pronounced and narrower than s‐RuO2/ATO and n‐RuO2/ATO (Figures S16 and S17, Supporting Information), confirming that the crystal sizes in samples without the ATO substrates are larger, and agreeing well with the above discussions. When closely inspecting the peak positions (Figure 3b), the corresponding peaks are negatively shifted for s‐RuO2 compared to n‐RuO2. Based on the diffraction equation, lower diffraction angles mean expanded lattice parameters, so it can be deduced that tensile strains are incorporated in s‐RuO2.[ 25 ] The XRD patterns after Rietveld refinements indicate that the lattice parameters of s‐RuO2 are slightly larger than n‐RuO2 (Figure 3c; and Table S3, Supporting Information). The samples with ATO substrates are not suitable for Rietveld refinements due to the very weak diffraction peaks of the RuO2 phase. However, the boarding of lattice parameters in s‐RuO2 can hint that the induced tensile strains are related to the quenching process. Besides, when s‐RuO2 was further annealed at 350 °C and cooled down to room temperature naturally, these characteristic peaks of RuO2 moved back to positions of n‐RuO2 due to the release of residual strains (Figure S18, Supporting Information).
Figure 3.
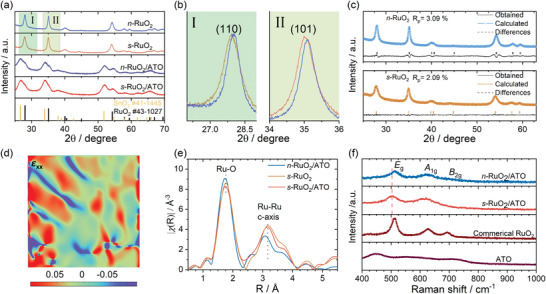
a) The XRD patterns of n‐RuO2, n‐RuO2/ATO, s‐RuO2, and s‐RuO2/ATO. b) The magnified regions XRD patterns of n‐RuO2 and s‐RuO2. c) Rietveld refinements of the XRD patterns for n‐RuO2 and s‐RuO2. d) The GPA image of s‐RuO2/ATO for axial strain (ɛ xx). e) The Ru K‐edge FT‐EXAFS spectra of n‐RuO2/ATO, s‐RuO2, and s‐RuO2/ATO after phase corrections. f) The Raman spectra of n‐RuO2/ATO, s‐RuO2/ATO, Commercial RuO2, and ATO.
High‐resolution TEM images can provide direct evidence for the differences existing in crystal structures. As shown in Figures S19 and S20 (Supporting Information), the interplanar spacings of (110) were obtained by averaging 8 accumulations. Similar to Rietveld refinements (Table S3, Supporting Information), the interplanar spacings of (110) of rutile‐structured RuO2 are 3.20, 3.23, and 3.23 Å for n‐RuO2/ATO, s‐RuO2/ATO, and s‐RuO2, respectively. Nevertheless, a large number of grain boundaries and stacking faults are also frequently observed in s‐RuO2/ATO and s‐RuO2 (Figure S21, Supporting Information), which could be related to the fast quenching process.[ 26 ] Geometric phase analysis (GPA) was also performed near these defective regions.[ 27 ] As shown in Figure 3d; and Figures S22 and S23 (Supporting Information), s‐RuO2/ATO and s‐RuO2 process high distortions in these regions. In terms of effects from ATO, no evidence of its influences on crystal structures can be identified. As s‐RuO2/ATO exhibits excellent long‐term stability, post‐OER (On glass carbon electrode) TEM characterization is also critical to assess the structural integrity. Figure S24 (Supporting Information) shows that tensile strains are preserved after 12 h chronopotentiometry, demonstrating the excellent stability of s‐RuO2/ATO.
X‐ray absorption spectroscopy (XAS) and Raman spectrum were also employed to investigate the incorporated tensile strains presented in s‐RuO2/ATO. In the Fourier transformed extended X‐ray absorption fine structure (FT‐EXAFS), the first and second shells of s‐RuO2/ATO and s‐RuO2 have longer radial distances than n‐RuO2/ATO (Figure 3e). The longer radial distances observed in FT‐EXAFS indicate the existence of tensile strains.[ 26 ] It should be mentioned that the scattering paths in FT‐EXAFS do not identically correspond to the real atomic distances due to multiple scattering.[ 28 ] Hence, further discussions using lattice parameters are based on the results from TEM and XRD.
According to the previous reports, the E g peak tends to redshift for the tensile‐strained RuO2 in the Raman spectrum.[ 29 ] As expected, the E g peaks of s‐RuO2/ATO and s‐RuO2 are shifted to a lower wavenumber than n‐RuO2/ATO (Figure 3f; and Figure S25, Supporting Information). In conclusion, the diffractometry and spectroscopy methods can testify that the quenching process could induce tensile strains in the RuO2. However, we do not observe enough evidence that ATO could influence the crystal structures of RuO2 in the above characterizations, possibly due to the insufficient contacts between RuO2 nanorods and ATO substrates.
The chemical environments were characterized by the X‐ray absorption near‐edge structure (XANES) and X‐ray photoelectron spectroscopy (XPS). In the XANES spectra (Figure 4a), the samples with tensile strains (s‐RuO2 and s‐RuO2/ATO) show slightly lower absorption edges than n‐RuO2/ATO, indicating the lower valence states induced by tensile strains. Similar negative shifts are also observed in the XPS Ru 3d regions (Figure 4b; and Figure S26, Supporting Information), supporting the results from XANES. The Ru site in high chemical valence states tends to be leached out, compromising the stability of crystal structures.[ 9 ] So, we tentatively attribute that the stability of s‐RuO2/ATO benefits from the lower valence states of Ru sites. Besides, there is no sufficient evidence of the interactions between ATO and RuO2 that can be identified due to the minor difference in s‐RuO2 and s‐RuO2/ATO (XANES and XPS results), suggesting that tensile strains are the leading causes of the modified electronic states.
Figure 4.
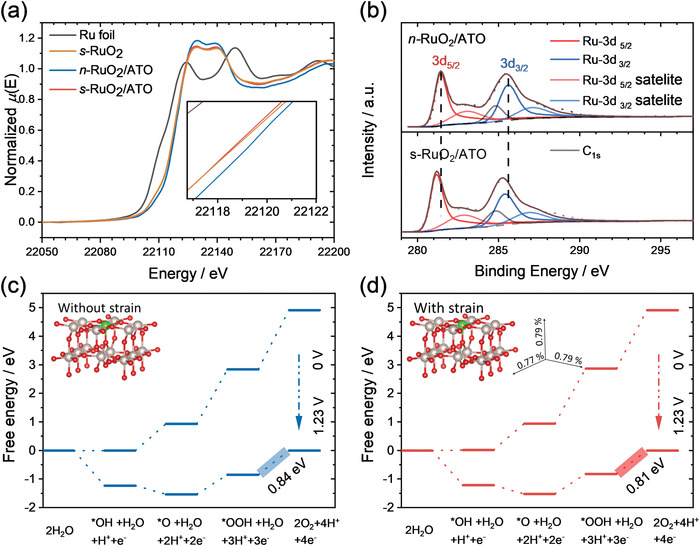
a) The XANES spectra of Ru foil, n‐RuO2/ATO, s‐RuO2, and s‐RuO2/ATO. b) The XPS‐Ru3d spectra of n‐RuO2/ATO and s‐RuO2/ATO. The peaks used for Ru 3d fitting are asymmetric, while the carbon C1s peaks are symmetric.[ 30 ] The same parameters were used to fit all the Ru 3d peaks. c,d) The free energy diagrams of RuO2 and strained RuO2 calculated on the coordinatively unsaturated sites (CUS, colored with green). The inserted axis gives the strain values in different directions of the strained RuO2.
DFT was employed to investigate the effect of tensile strains. Two relatively simple models with no consideration of cobalt dopants and involvements of linear relation breaking sites (Insets in Figure 4c,d). Four elemental steps of OER are considered in the calculation
| (1) |
| (2) |
| (3) |
| (4) |
As shown in Figure 4c,d, the barrier of the limiting step of OER decreases after the introduction of tensile strains into RuO2. Besides, the absorption energies of oxygen intermediates are weakening on the tensile‐strained RuO2. According to Nørskov's results, relatively lower binding strengths of oxygen intermediates could improve the OER in RuO2.[ 8a ] When considering the bridge‐O coverages on the surfaces of RuO2 in DFT calculations, a similar trend with decreased limiting step after applying the tensile strains is also observed (Figures S27 and S28, Supporting Information). Notably, the limiting step on the strained RuO2 with bridge‐O coverage is smaller and much closer to the experimental results.
As a model catalyst, s‐RuO2/ATO, incorporated with the ATO substrate and tensile strains, was used as the anode material in PEMWE. Detailed configurations of the PEMWE assembly can be found in Figure S29 (Supporting Information). The PEMWEs were tested at 80 °C with a water flow of 100 mL min−1 to the anode only. Figure 5a shows the recorded LSV curves. As shown, the s‐RuO2/ATO has displayed a remarkably high activity with the potential of only 1.51 V at 1 A cm−2 (IR‐free), far surpassing the commercial RuO2. Furthermore, the assembled PEMWEs were operated at 0.5 A cm−2. As shown in Figure 5b, the PEMWE using commercial RuO2 as the anode cannot work anymore after 5 h. The PEMWE using s‐RuO2/ATO as the anode shows no significant decline even after 40 h continuous operation. As discussed above, the high efficiency and robustness of the PEMWE using s‐RuO2/ATO as the anode can be ascribed to the incorporations of ATO substrate and tensile strains.
Figure 5.
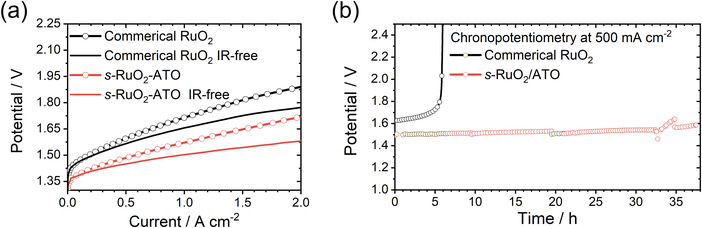
a) The LSV curves of s‐RuO2/ATO and commercial RuO2 in PEMWEs. b) The chronopotentiometry curves (without IR corrections) at 500 mA cm−2 of s‐RuO2/ATO and commercial RuO2 in PEMWEs.
3. Conclusions
In summary, we have successfully prepared a catalyst with strained RuO2 nanorods loaded on the ATO substrate by employing the ATO as substrate and the quenching process to induce tensile strains. The ATO substrate enhances the ECSA of RuO2, while tensile strains induced by quenching can modify the electronic states of RuO2. Synergetic effects from the ATO and tensile strains enable s‐RuO2/ATO to be one of the best OER catalysts with only 198 mV overpotential at 10 mA cm−2, and it is kept almost unchanged after 150 h chronopotentiometry. In addition, the PEMWE assembled by using s‐RuO2/ATO as anode also displays remarkably high efficiency and stability. This work demonstrated that tensile strains in RuO2 would positively influence the OER activity, and present a new perspective toward future catalyst designs.
4. Experimental Section
Preparations of ATO
The ATO was synthesized following previous reports with slight modifications.[ 31 ] First, 1.28 g dodecylamine (Adamas) was mixed with 65 mL ethanol and 160 mL deionized water, then stirred for 3 h to form an emulsion. At the same time, antimony triacetate (0.48 g, Macklin) and SnCl4 (2.15 mL, Macklin) were added into 20 mL ethanol. Then, 20 mL of the metal contained ethanol were slowly added into the above emulsion, followed by stirring for 1 h. After that, the above solution was then slowly added to 200 mL of 1.5 × 10−3 m NH3H2O solution and kept stirred for 1 h. The prepared solution was then transformed into an oil bath at 80 °C for 72 h. Then the precipitate was collected by centrifugation (5000 rpm, 10 min) and washed with water five times. The obtained wet product was dispersed in 60 mL water and transformed into a 100 mL autoclave at 120 °C for 24 h. The as‐prepared product was collected by centrifugation (5000 rpm, 10 min) and washed with water five times. Finally, the gray powder was calcinated in air at 400 °C for 3 h.
Preparations of Co‐HMT/ATO
First, 200 mg of the prepared ATO was dispersed in 50 mL of isopropanol by sonication for 4 h. Then, 197 mg cobalt nitrate hexahydrate (Macklin) was dissolved into the ATO dispersion. The hexamethylenetetramine (HMT) (Sinopharm) solution was prepared by adding 950 mg HMT into a mixture of 2 mL water and 5 mL ethanol. Then, the HMT solution was dropwise added into ATO dispersion to initialize the formation of the Co‐HMT metal–organic framework. After reaction for 30 min, the sample was collected by centrifugation (5000 rpm, 5 min) and washed with isopropanol five times. Then, the sample was dried in a vacuum condition.
Preparations of Ru‐Co‐HMT/ATO
The prepared 100 mg Co‐HMT/ATO was dispersed in 5 mL tetrahydrofuran by sonication for 10 min. Then 60 mg of the RuCl3• 3H2O (Adamas) was added, followed by a 24 h vigorous stir. The final sample was collected by centrifugation (10 000 rpm, 10 min) and washed with tetrahydrofuran two times. Then, the sample was dried in a vacuum condition.
Preparations of s‐RuO2/ATO
The prepared Ru‐Co‐HMT/ATO was annealed in air at 350 °C for 2 h in a muffle furnace. After 2 h annealing, the sample was quickly taken out and immersed in liquid N2 (−196 °C) to finish the quenching process.
Preparations of n‐RuO2/ATO
Sample of n‐RuO2/ATO were prepared similar to s‐RuO2/ATO except without the last quenching step. In detail, the sample was cooled down to room temperature inside the muffle furnace in nature.
Preparations of s‐RuO2
The sample of s‐RuO2 was prepared similar to s‐RuO2/ATO except without the addition of ATO. To achieve a similar substitution degree of Co2+ by Ru3+, the mass ratio of Co‐HMT to RuCl3•3H2O is setted to be 1:1.
Preparations of n‐RuO2
Sample of s‐RuO2 was prepared similar to s‐RuO2/ATO except without the addition of ATO and quenching step.
Preparations of EG‐s‐RuO2 and EG‐n‐RuO2
The Ru nanoparticles were first synthesized by oil bathing the mixture of 60 mg RuCl3 and 30 mL ethylene glycol at 190 °C under stirring for 30 min. Then, the obtained Ru nanoparticles were collected by centrifugation and washed with ethanol and water several times. Later, the Ru nanoparticles were annealed (with a quenching step for EG‐s‐RuO2) in the same condition as s‐RuO2 and n‐RuO2.
Preparations of s‐RuO2/C and n‐RuO2/C
30 mg Ketjen Black were mixed with 100 mg RuCl3• 3H2O in 15 mL H2O by simple sonication for 1 h. After that, the mixture was free‐dried. Then the same annealing process mentioned above was used to synthesize s‐RuO2/C and n‐RuO2/C.
Physical Characterizations
X‐ray powder diffraction spectroscopy was recorded on Miniflex600 (Rigaku) with Cu K α X‐ray (λ = 0.154 nm). Scanning electron microscope images were obtained in a Su‐8010 electron microscope. Transmission electron microscope (TEM) images were obtained in JEOL 2100F (JEOL). XAS spectra were collected at Shanghai Synchrotron Radiation Facility (SSRF) and analyzed by Athena software. The Raman spectra were recorded in the Horiba Jobin Yvon LabRAM ARAMIS system equipped with 633 nm light. Before recording the Raman spectra, the system was calibrated using a Si plate. XPS spectra were measured on ESCALAB 250Xi (ThermoFisher) and calibrated to adventitious C1s peak at 284.8 eV.
Theoretical Calculations
All spin‐polarized DFT calculations for periodic material systems were performed with the Vienna Ab initio simulation package (VASP)[ 32 ] with the projector‐augmented wave (PAW) method. The exchange‐correlation function was handled using the generalized gradient approximation (GGA) formulated by the Revised Perdew–Burke–Ernzerhof (RPBE). The van der Waals (vdW) interactions are described with the DFT‐D3 method in Grimme's scheme. The interaction between the atomic core and electrons was described by the projector augmented wave method. The plane‐wave basis set energy cutoff was set to 500 eV. The Brillouin zone was sampled with a 3 × 3 × 1 grid centered at the gamma (Γ) point for geometry relaxation. The perfect RuO2 bulk was fully optimized using 5 × 5 × 5 k‐point Gamma (Γ) centered Monkhorst‐Pack mesh sampling. The RuO2 facets were modeled using a four atomic layer 1 × 2 supercell slab with 6.22 × 6.39 Å in x and y directions, respectively. The RuO2 facets with strain were modeled with 6.27 × 6.44 Å in x and y directions, respectively. A 15 Å vacuum region, ensuring negligible lateral interaction of adsorbates. The bottom two atomic layers were kept frozen at the lattice position. All structures with a dynamic magnetic moment were fully relaxed to optimize without any restriction until their total energies were converged to < 1×10−6 eV, and the average residual forces were < 0.02 eV Å−1. Moreover, the Gibbs free energy calculation is operated with the computational hydrogen electrode (CHE) model.[ 33 ]
Electrochemical Tests
A three‐electrode configuration including a rotating disk electrode (RDE, working electrode), Ag/AgCl electrode (reference electrode), and graphite electrode (counter electrode) was used to record the linear sweep voltammetry (LSV), cyclic voltammetry (CV), and chronopotentiometry curves. The electrochemical workstation used was CHI 760E (CH Instruments). The ink was prepared by dispersing 5 mg sample into a mixture of 970 µL isopropanol and 30 µL Nafion (5 wt%), and sonicated for at least 1 h. The load masses on the RDE for all the samples are based on the Ru contents (ICP results), which were set to be 0.19 mg cm−2. The LSV and CV curves were recorded under N2 bubbling. Before the LSV and CV tests, at least 50 CV curves (0.4–1.2 V) were carried out to stabilize the catalyst. The LSV curves were recorded at 5 mV s−1, while the CV curves were at 100 mV s−1. Here, it should be mentioned that all the LSV curves were IR corrected automatically in CHI software.
Electrolytes of different pH were prepared from concentrated HClO4 (Sigma) by diluting. To reduce high ionic resistances in electrolytes with pH 1.3 and 2, certain K2SO4 (Sigma) was added to make the total ionic concentrations 0.1 m.
Proton‐exchange membrane water electrolyzer (PEMWE) was tested on Gamry (Gamry Instruments) Interface 5000E. The Membrane Electrode Assembly (MEA) was prepared using Nafion 212 by the Catalyst Coated Membrane (CCM) method with a geometric area of 2 cm2. The catalyst inks were prepared at the same I:C ratio (Nafion: Catalyst, mass ratio) used for three‐electrode measurements. In addition, the total load mass for the anode (s‐RuO2/ATO) was weighted to be 3.3 mg cm−2, while it was 0.6 mg cm−2 for the cathode (Pt/C, 20 wt%). It has to note that the PEMWE using commercial RuO2 also employed an anodic load mass of 3.3 mg cm−2. During the data recording, both the anode and cathode plate were heated to 80 °C. Besides, a water flow preheated to 80 °C at 100 mL min−1 was supplied to the anode side.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 22171266), the FJIRSM&IUE Joint Research Fund (No. RHZX‐2019‐002), and the STS project (No. KFJ‐STS‐QYZD‐2021‐09‐002). This work was also supported by CAS‐Shanghai Science Research Center and user Experiment Assist System of SSRF. The authors thank the 11B beamline staff at the Shanghai Synchrotron Radiation Facility (SSRF) for data collection.
Huang B., Xu H., Jiang N., Wang M., Huang J., Guan L., Tensile‐Strained RuO2 Loaded on Antimony‐Tin Oxide by Fast Quenching for Proton‐Exchange Membrane Water Electrolyzer. Adv. Sci. 2022, 9, 2201654. 10.1002/advs.202201654
Contributor Information
Hengyue Xu, Email: xuhy21@mails.tsinghua.edu.cn.
Lunhui Guan, Email: guanlh@fjirsm.ac.cn.
Data Availability Statement
Research Data are not shared.
References
- 1. Meng X., Gu A., Wu X., Zhou L., Zhou J., Liu B., Mao Z., Int. J. Hydrog. Energy 2021, 46, 28887. [Google Scholar]
- 2.a) Ursua A., Gandia L. M., Sanchis P., Proc. IEEE 2012, 100, 410; [Google Scholar]; b) Liang H., Cao Z., Xia C., Ming F., Zhang W., Emwas A.‐H., Cavallo L., Alshareef H. N., CCS Chem. 2021, 3, 1553. [Google Scholar]
- 3. Chatterjee S., Peng X., Intikhab S., Zeng G., Kariuki N. N., Myers D. J., Danilovic N., Snyder J., Adv. Energy Mater. 2021, 11, 2101438. [Google Scholar]
- 4. Oliver‐Tolentino M. A., Vázquez‐Samperio J., Manzo‐Robledo A., González‐Huerta R. d. G., Flores‐Moreno J. L., Ramírez‐Rosales D., Guzmán‐Vargas A., J. Phys. Chem. C 2014, 118, 22432. [Google Scholar]
- 5. Vincent I., Bessarabov D., Renewable Sustainable Energy Rev. 2018, 81, 1690. [Google Scholar]
- 6.a) Ayers K., Curr. Opin. Chem. Eng. 2021, 33, 100719; [Google Scholar]; b) Daiane Ferreira da Silva C., Claudel F., Martin V., Chattot R., Abbou S., Kumar K., Jiménez‐Morales I., Cavaliere S., Jones D., Rozière J., Solà‐Hernandez L., Beauger C., Faustini M., Peron J., Gilles B., Encinas T., Piccolo L., Barros de Lima F. H., Dubau L., Maillard F., ACS Catal. 2021, 11, 4107; [Google Scholar]; c) Shan J., Ye C., Chen S., Sun T., Jiao Y., Liu L., Zhu C., Song L., Han Y., Jaroniec M., Zhu Y., Zheng Y., Qiao S. Z., J. Am. Chem. Soc. 2021, 143, 5201. [DOI] [PubMed] [Google Scholar]
- 7.a) Lin C., Li J.‐L., Li X., Yang S., Luo W., Zhang Y., Kim S.‐H., Kim D.‐H., Shinde S. S., Li Y.‐F., Liu Z.‐P., Jiang Z., Lee J.‐H., Nat. Catal. 2021, 4, 1012; [Google Scholar]; b) Wen Y., Chen P., Wang L., Li S., Wang Z., Abed J., Mao X., Min Y., Dinh C. T., Luna P., Huang R., Zhang L., Wang L., Wang L., Nielsen R. J., Li H., Zhuang T., Ke C., Voznyy O., Hu Y., Li Y., Goddard W. A. III, Zhang B., Peng H., Sargent E. H., J. Am. Chem. Soc. 2021, 143, 6482; [DOI] [PubMed] [Google Scholar]; c) Briquet L. G. V., Sarwar M., Mugo J., Jones G., Calle‐Vallejo F., ChemCatChem 2017, 9, 1261. [Google Scholar]
- 8.a) Rossmeisl J., Qu Z. W., Zhu H., Kroes G. J., Nørskov J. K., J. Electroanal. Chem. 2007, 607, 83; [Google Scholar]; b) Man I. C., Su H. Y., Calle‐Vallejo F., Hansen H. A., Martínez J. I., Inoglu N. G., Kitchin J., Jaramillo T. F., Nørskov J. K., Rossmeisl J., ChemCatChem 2011, 3, 1159. [Google Scholar]
- 9.a) Klyukin K., Zagalskaya A., Alexandrov V., J. Phys. Chem. C 2019, 123, 22151; [DOI] [PubMed] [Google Scholar]; b) Dickens C. F., Nørskov J. K., J. Phys. Chem. C 2017, 121, 18516. [Google Scholar]
- 10.a) Tian Y., Wang S., Velasco E., Yang Y., Cao L., Zhang L., Li X., Lin Y., Zhang Q., Chen L., iScience 2020, 23, 100756; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou C., Chen X., Liu S., Han Y., Meng H., Jiang Q., Zhao S., Wei F., Sun J., Tan T., Zhang R., J. Am. Chem. Soc. 2022, 144, 2694. [DOI] [PubMed] [Google Scholar]
- 11.a) Chen Y., Sun Y., Wang M., Wang J., Li H., Xi S., Wei C., Xi P., Sterbinsky G. E., Freeland J. W., Fisher A. C., Ager J. W., Feng Z., Xu Z. J., Sci. Adv. 2021, 7, eabk1788; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ji M., Yang X., Chang S., Chen W., Wang J., He D., Hu Y., Deng Q., Sun Y., Li B., Xi J., Yamada T., Zhang J., Xiao H., Zhu C., Li J., Li Y., Nano Res. 2022, 15, 1959. [Google Scholar]
- 12.a) Stiber S., Sata N., Morawietz T., Ansar S. A., Jahnke T., Lee J. K., Bazylak A., Fallisch A., Gago A. S., Friedrich K. A., Energy Environ. Sci. 2022, 15, 109; [Google Scholar]; b) Gao J., Tao H., Liu B., Adv. Mater. 2021, 33, 2003786. [DOI] [PubMed] [Google Scholar]
- 13.a) Meng G., Sun W., Mon A. A., Wu X., Xia L., Han A., Wang Y., Zhuang Z., Liu J., Wang D., Li Y., Adv. Mater. 2019, 31, 1903616; [DOI] [PubMed] [Google Scholar]; b) Yu J., He Q., Yang G., Zhou W., Shao Z., Ni M., ACS Catal. 2019, 9, 9973; [Google Scholar]; c) Wang Z., Xiao B., Lin Z., Shen S., Xu A., Du Z., Chen Y., Zhong W., J. Energy Chem. 2021, 54, 510. [Google Scholar]
- 14. Yang C.‐L., Wang L.‐N., Yin P., Liu J., Chen M.‐X., Yan Q.‐Q., Wang Z.‐S., Xu S.‐L., Chu S.‐Q., Cui C., Ju H., Zhu J., Lin Y., Shui J., Liang H.‐W., Science 2021, 374, 459. [DOI] [PubMed] [Google Scholar]
- 15. Sun W., Zhou Z., Zaman W. Q., Cao L. M., Yang J., ACS Appl. Mater. Interfaces 2017, 9, 41855. [DOI] [PubMed] [Google Scholar]
- 16. You B., Tang M. T., Tsai C., Abild‐Pedersen F., Zheng X., Li H., Adv. Mater. 2019, 31, 1807001. [DOI] [PubMed] [Google Scholar]
- 17. Sun S., Zhou X., Cong B., Hong W., Chen G., ACS Catal. 2020, 10, 9086. [Google Scholar]
- 18. Kim H., Charipar N. A., Figueroa J., Bingham N. S., Piqué A., AIP Adv. 2019, 9, 015302. [Google Scholar]
- 19. Oh H. S., Nong H. N., Reier T., Bergmann A., Gliech M., Ferreira de Araujo J., Willinger E., Schlogl R., Teschner D., Strasser P., J. Am. Chem. Soc. 2016, 138, 12552. [DOI] [PubMed] [Google Scholar]
- 20. Kumar A., Liu X., Lee J., Debnath B., Jadhav A. R., Shao X., Bui V. Q., Hwang Y., Liu Y., Kim M. G., Lee H., Energy Environ. Sci. 2021, 14, 6494. [Google Scholar]
- 21. Halck N. B., Petrykin V., Krtil P., Rossmeisl J., Phys. Chem. Chem. Phys. 2014, 16, 13682. [DOI] [PubMed] [Google Scholar]
- 22.a) Shinagawa T., Garcia‐Esparza A. T., Takanabe K., Sci. Rep. 2015, 5, 13801; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yuan S., Peng J., Cai B., Huang Z., Garcia‐Esparza A. T., Sokaras D., Zhang Y., Giordano L., Akkiraju K., Zhu Y. G., Hubner R., Zou X., Roman‐Leshkov Y., Shao‐Horn Y., Nat. Mater. 2022, 21, 673. [DOI] [PubMed] [Google Scholar]
- 23. Böhm D., Beetz M., Schuster M., Peters K., Hufnagel A. G., Döblinger M., Böller B., Bein T., Fattakhova‐Rohlfing D., Adv. Funct. Mater. 2020, 30, 1906670. [Google Scholar]
- 24.a) Stoerzinger K. A., Diaz‐Morales O., Kolb M., Rao R. R., Frydendal R., Qiao L., Wang X. R., Halck N. B., Rossmeisl J., Hansen H. A., Vegge T., Stephens I. E. L., Koper M. T. M., Shao‐Horn Y., ACS Energy Lett. 2017, 2, 876; [Google Scholar]; b) An L., Wei C., Lu M., Liu H., Chen Y., Scherer G. G., Fisher A. C., Xi P., Xu Z. J., Yan C. H., Adv. Mater. 2021, 33, 2006328. [DOI] [PubMed] [Google Scholar]
- 25. Liu T., Yang S., Guan J., Niu J., Zhang Z., Wang F., Small Methods 2022, 6, 2101156. [DOI] [PubMed] [Google Scholar]
- 26. Hao S., Sheng H., Liu M., Huang J., Zheng G., Zhang F., Liu X., Su Z., Hu J., Qian Y., Zhou L., He Y., Song B., Lei L., Zhang X., Jin S., Nat. Nanotechnol. 2021, 16, 1371. [DOI] [PubMed] [Google Scholar]
- 27. Hÿch M. J., Potez L., Philos. Mag. A 1997, 76, 1119. [Google Scholar]
- 28. Wang M., Feng Z., Curr. Opin. Electrochem. 2021, 30, 100803. [Google Scholar]
- 29. Meng L.‐J., dos Santos M. P., Thin Solid Films 2000, 375, 29. [Google Scholar]
- 30. Rochefort D., Dabo P., Guay D., Sherwood P. M. A., Electrochim. Acta 2003, 48, 4245. [Google Scholar]
- 31. Oh H. S., Nong H. N., Reier T., Gliech M., Strasser P., Chem. Sci. 2015, 6, 3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.a) Kresse G., Furthmüller J., Comput. Mater. Sci. 1996, 6, 15; [Google Scholar]; b) Kresse G., Furthmüller J., Phys. Rev. B 1996, 54, 11169. [DOI] [PubMed] [Google Scholar]
- 33. Nørskov J. K., Rossmeisl J., Logadottir A., Lindqvist L., Kitchin J. R., Bligaard T., Jonsson H., J. Phys. Chem. B 2004, 108, 17886. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
Research Data are not shared.