

Abstract
Glioblastoma (GBM) is the most frequent and aggressive primary tumor type in the central nervous system in adults. Resistance to chemotherapy remains one of the major obstacles in GBM treatment. Identifying and overcoming the mechanisms of therapy resistance is instrumental to develop novel therapeutic approaches for patients with GBM. To determine the major drivers of temozolomide (TMZ) sensitivity, we performed shRNA screenings in GBM lines with different O6-methylguanine-DNA methyl-transferase (MGMT) status. We then evaluated dianhydrogalactitol (Val-083), a small alkylating molecule that induces interstrand DNA crosslinking, as a potential treatment to bypass TMZ-resistance mechanisms. We found that loss of mismatch repair (MMR) components and MGMT expression are mutually exclusive mechanisms driving TMZ resistance in vitro. Treatment of established GBM cells and tumorsphere lines with Val-083 induces DNA damage and cell-cycle arrest in G2–M phase, independently of MGMT or MMR status, thus circumventing conventional resistance mechanisms to TMZ. Combination of TMZ and Val-083 shows a synergic cytotoxic effect in tumor cells in vitro, ex vivo, and in vivo. We propose this combinatorial treatment as a potential approach for patients with GBM.
Introduction
Gliomas comprise a large group of brain cancers, with glioblastoma (GBM), a WHO grade IV malignant astrocytoma, being the most common and lethal primary central nervous system tumor in adults (1). Standard therapy for GBM begins with maximal resection of the tumor mass, followed by concurrent radiotherapy and chemotherapy (2–4). Despite significant advances in the treatment of other solid tumors, an effective therapy for GBM is still elusive. The median survival has remained nearly unchanged over the last 50 years, averaging 15 months (2). One of the main pitfalls of current GBM therapies is that they quickly develop resistance, which renders the treatment futile (5). It remains unclear whether the resistance is a consequence of tumor progression or if it is associated with the genetic events that lead to tumorigenesis. Defining the pathways that determine this poor response to treatment is instrumental for the development of new therapeutic approaches.
Temozolomide (TMZ) is an oral alkylating agent used for treating GBM. TMZ methylates the DNA at positions N-7 and O-6 of guanine and position N-3 of adenine (6). Despite being the less frequent, methylation at O-6 exerts the highest cytotoxic effect. However, it can be removed by the O6-methylguanine-DNA methyl-transferase (MGMT), thus abrogating the cytotoxic effect (7). The O6-methyl-guanine pairs with thymine in subsequent replication rounds and causes a mismatch, which is recognized by the mismatch repair (MMR) system and leads to cell-cycle arrest and cell death (6). Mutations in MMR components are associated with hypermutations and microsatellite instability and mediate chemotherapy resistance in GBM, among other cancer types (6, 8, 9).
The bi-alkylating agent dianhydrogalactitol, also known as Val-083, is a first-in-class molecule that has been shown to exert antineoplastic effects (10). Val-083 induces interstrand-crosslinks at N7-guanine (11), which leads to persistent DNA double-strand breaks (DSB) and cell-cycle arrest in a p53-dependent or p53-independent manner (12). It is able to cross the blood–brain barrier, has a long half-life in the brain compartment, and it acts preferentially in tumor cells (13). As of today, there is not any known cross-resistance of Val-083 with other conventional chemotherapeutic agents (12). Currently, it is been tested in phase II clinical trials, in combination with radiotherapy in patients with newly diagnosed GBM with MGMT-unmethylated (NCT03050736) and for patients with MGMT-unmethylated, bevacizumab-naïve GBM (NCT02717962). Furthermore, its use has been expanded to patients with relapsed/refractory GBM that are not eligible for the trials (NCT03138629).
Val-083 has a small therapeutic window, displaying toxicity at relatively low doses when administered in vivo (14, 15). However, a recent halfway report of the phase II clinical trial NCT03050736 has shown that after dose escalation, Val-083 in combination with radiotherapy was generally safe and well tolerated (16).
Here, we present evidence that Val-083 overcomes the main mechanisms of TMZ resistance, MGMT expression, and MMR defects. Moreover, we show that the combination of low doses of TMZ and Val-083 efficiently targets and kills GBM cells with various genetic backgrounds and therefore it might decrease the emergence of chemotherapy-resistant tumors.
Materials and Methods
Cell lines
Standard cell lines U-251 MG, T98G, U-87 MG, LN-18, SF268, and GP2 were cultured in DMEM media (Sigma-Aldrich) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). DNA fingerprinting was performed for authentication of the cell lines (data available upon request). Human GBM tumorspheres H543, H516, and H676 were cultured in human NeuroCult NS-A Proliferation Kit (Stem Cell Technologies, Catalog No. 05751) and supplemented with 10 ng/mL recombinant human EGF (Gibco, Catalog No. PHG0313), 20 ng/mL basic-FGF (Sigma-Aldrich, Catalog No. F0291–25UG), 1 mg/mL heparin (Stem Cell Technologies, Catalog No. 07980), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were incubated at 37°C, 5% CO2 with humidity. Both established and GBM tumorsphere lines were kindly provided by Eric Holland. All the cell lines were routinely checked for Mycoplasma contamination by PCR analysis.
shRNA library generation and pooled shRNA screenings
A custom shRNA library of a total of 3,790 shRNA targeting 643 DDR genes (GO: DNA damage stimulus/DNA damage checkpoint/DNA repair) was synthesized using mir30-adapted DSIR predictions refined with “sensor” rules (17) and constructed by PCR-cloning a pool of oligonucleotides synthesized on a custom array (Agilent Technologies) into the MLP-mir-E vector (LTR-miR-E-PGK-PuroR-IRESGFP; ref. 18). The library was designed to have at least five shRNAs targeting a specific transcript and it was divided in three pools, each containing approximately 1,500 shRNAs, targeting 200 to 220 transcripts (Supplementary Table S1). The pools were transduced into U-251 MG and T98G cells using viral concentrations that led to 10% to 20% of GFP-positive cells (MOI < 0.25) and represent each shRNA in calculated number >1,000 cells. Transduced cells were selected with puromycin (U-251 MG, 3 μg/mL and T98G 1 μg/mL) for 7 days. Aliquots of 5 million cells were then frozen to preserve library representation throughout the experiments.
To perform the screening, cells were thawed and maintained for one passage before starting the drug treatments: U-251 MG and T98G cells were treated with 100 μmol/L TMZ or 1 μmol/L Val-083 (U-251 MG) and 200 μmol/L TMZ or 2 μmol/L Val-083 (T98G). Genomic DNA from control and treated samples (7 days for TMZ and 5 days for Val-083) was isolated by two rounds of phenol extraction using PhaseLock tubes (5 Prime) followed by isopropanol precipitation. Deep-sequencing template libraries were generated by PCR amplification of shRNA guide strands as described previously (18, 19). Briefly, templates for deep-sequencing were generated by PCR amplification of shRNA guide strands using primers that tag the product with standard Illumina adapters (p7+loop, CAAGCAGAAGACGGCATACGA [INDEX]TAGTGAAGCCACAGATGTA; p5+miRE AATGATACGGCGACCACCGAGAaTTCtagccccttgaaGtc). For each sample, DNA was amplified in 12 parallel 50-μL PCR reactions using 3 μg of genomic DNA as PCR template. PCR products were combined for each sample, precipitated and purified on a 2% agarose gel. Samples were analyzed on an Illumina High Seq and sequenced using a primer that reads in reverse into the guide strand (TAGCCCCTTGAAGTCCGAGGCAGTAGGCA).
Bioinformatics analysis of shRNA screenings
Raw sequencing reads were trimmed using Trimmomatic software (v.0.32; ref. 20) with default parameters, by removing the adaptor from the miR30 construct from the raw sequencing reads. Only processed reads with at least 17 nucleotide lengths were retrieved. Processed reads were aligned to the shRNA library reference using Bowtie2 (v.2.1.0; ref. 21), using the options for a very sensitive alignment with no mismatches following manual instructions (-D 20 -R 3 -N 0 -L 17 -i S, 1, 0.50). The read counts per hairpin were obtained with a custom Perl script that processes the Sequence Alignment/Map (SAM) file. Within-sample normalization was performed by calculating the read counts per million per hairpin for each independent pool from the shRNA library design. Contrasts of hairpin representation upon treatment were obtained by the average log2(fold-change) between treated and untreated cell lines from three replicates. We tested for differentially represented sets of RNAi hairpins, which target the same gene, using gene set enrichment methods implemented in limma R package (v.3.24.15; ref. 22). We defined two ad-hoc scores for target gene prioritization, the sensitizer score (sScore) and RNAi score (RiScore; Supplementary Tables SII–SV). The sScore and RiScore were obtained using the ROMER and ROAST methods (22, 23), respectively. The scores were calculated by transforming the resulting P value via log10-transformation with the sign of the direction of the enrichment yield by the statistical test. The two methods differ in the type of enrichment test and the alternative hypothesis tested. We used ROMER, a competitive enrichment test, with the alternative hypothesis of overrepresentation of depleted hairpin sets to rank candidate target genes to sensitize cells to the treatment. Thus, the sScore indicates whether there is an overrepresentation towards depletion of the set of hairpins which share the same target gene. The more negative is the value, the stronger is the evidence for depletion. We used ROAST, a self-contained enrichment test with the alternative hypothesis of mixed directional enrichment to depict individual enrichments of a subset of the hairpin-set in any direction. The RiScore represents whether there is any difference between two groups (e.g., treated vs. control) across the hairpins for a given gene. The larger is the value, the better is the score for the tendency.
Plasmids and cell transfection
shRNAs were cloned into the MLP-mir-E vector (LTR-miR-E-PGK-PuroR-IRESGFP) as described previously (18). Sequences are listed in Supplementary Table S1. In brief, GP2 cells (ATCC) were cotransfected with VSGg and the MLP-mir-E vector. After 1 day, conditioned media was incubated with U-251 MG or T98G cells for infection. Cells were selected with puromycin. In addition, U-251 MG and T98G parental cells were transfected with LoxP1-His-H2B-Cherry-2A plasmid, expressing histone H2B fused with mCherry (Addgene No. 99616) with Lipofectamine (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were selected with G418 and sorted by FACS and the top 10% brightest cells were kept to expand and use in downstream experiments. The U-251 MG cells harboring the SAR1A-MGMT fusion were previously generated in our lab as described (24).
Immunofluorescence
A total of 2 × 103 U-251 MG cells per well were plated in 96 wells mClear black plates (Grenier). Different concentrations of TMZ (Merck, Catalog No. T2577), Val-083 (Adooq Bioscience, Catalog No. A15269), or DMSO (Sigma-Aldrich) were added. At the specified timepoints, media was removed and cells were fixed with 4% PFA for 15 minutes at room temperature (RT). Cells were permeabilized with 0.2% BSA, 0.1% Triton X-100 in PBS for 15 minutes at RT. Cells were blocked by incubating with 3% BSA in PBS for 45 minutes. The primary antibody (anti-phospho-H2AX, Merck, 05-636, 1 μg/mL) was incubated at 4°C overnight. After washing with PBS, cells were incubated with an anti-mouse IgG antibody conjugated with Alexa Fluor 488 (Thermo Fisher Scientific) for 1 hour. DNA was labeled with 1 μg/mL DAPI for 5 minutes. The Opera High Content Screening System (Perkin Elmer) was used to acquire and analyze the images. In brief, nuclei labelled with DAPI were selected, and mean intensity of phospho-H2AX staining was quantified. Each experimental condition was normalized to its DMSO control and expressed as fold increase.
MTT assays
A total of 103 cells (U-251 MG, T98G, and LN-18), 1.5 × 103 cells (U-87 MG and SF268), or 104 cells (human GBM tumorspheres) per well were plated in 96-well plates. TMZ and Val-083 were dissolved in DMSO to generate a stock of 100 and 10 mmol/L respectively, which were aliquoted and frozen at −20°C. After addition of the indicated concentrations of TMZ, Val-083, a combination of both, or vehicle, cells were incubated for 5 days (established cell lines) or 7 days (human GBM tumorspheres). Ten microliters of MTT reagent (Sigma, 5 mg/mL in PBS) were added to the media, and incubated for 4 hours. After adding 100 μL of a 1% SDS, 4 mmol/L HCl solution, absorbance at 595 nm was recorded with a plate reader.
Synergy studies
Cells were treated with a combination of different doses of TMZ and Val-083, as specified in the text, and analyzed by MTT. The measurements were processed with the SynergyFinder R package (25). Synergy scores were obtained for each combination, with the highest single agent (HSA) model (26).
Competition assay
U-251 MG or T98G cells expressing either H2B-mCherry or eGFP alongside the indicated shRNA were mixed in a one-to-one ratio to reach a final number of 103 cells per well in 96 wells μClear black plates, and TMZ, Val-083, or DMSO were added. After 5 days, cells were washed with PBS and incubated for 15 minutes with Hoechst 33342 diluted in DMEM without phenol red. Cells were immediately imaged using the Opera High Content Screening System. The number of eGFP+ or mCherry+ cells was quantified with ImageJ.
Colony-forming assay
A total of 2 × 103 U-251 MG or T98G cells expressing different shRNAs were plated per well in 12-well plates, and incubated with the specified concentrations of DMSO, TMZ, Val-083, and/or O6-benzylguanine (O6-BG). After 9 days, cells were washed, stained with crystal violet, and imaged using an office scanner.
Flow cytometry
105 U-251 MG or T98G cells were plated in 10-cm plates and incubated in the presence of the specified concentrations of TMZ, Val-083, or DMSO. After 48 hours, cells were harvested, washed in DPBS, and fixed by overnight incubation at 4°C in 70% ethanol. After two washes in DPBS, cells were stained with propidium iodide for at least 1 hour at 4° C. Data acquisition was performed with BD FACSCanto II (BD Biosciences), and analysis was performed with FlowJo v10 (FlowJo, LLC).
Ex vivo treatments
Five-to 10-week-old Foxn1-null mice (Jackson) were intracranially injected with 5 × 105 luciferase-expressing U-251 MG cells (27). Mice were anesthetized by inhalation of 2.5% isoflurane, and placed in a stereotactic device to immobilize the head. After disinfection of the area, a 1-cm incision was performed to expose the skull. A 1- to 2-mm hole was drilled in the lower right corner below the bregma. Afterwards, 1 μL of cell suspension was injected with a microliter syringe (Hamilton). Mice were intraperitoneally injected with buprenorphine 10 minutes prior to surgery, followed by two more injections every 24 hours afterward.
Three weeks after implantation, mice were sacrificed by CO2 inhalation, and the brain was dissected in HBSS supplemented (HBSSS) with HEPES (pH 7.4, 2.5 mmol/L), D-glucose (30 mmol/L), CaCl2 (1 mmol/L), MgCl2 (1 mmol/L), and NaHCO3 (4 mmol/L), and embedded in low-melting agarose (Lonza). Embedded brains were sectioned into 250 μmol/L slices using a vibratome (Leica). The slices were placed on top of a 13-mm polycarbonate membrane (0.8 μmol/L pore size, Whatman), floating in DMEM media supplemented with HBSSs, FBS 5%, l-glutamine (1 mmol/L), 100 IU/mL penicillin, and 100 mg/mL streptomycin. Brain slices were imaged to confirm the presence of tumors with the IVIS system (Perkin Elmer). Drug treatment was added to the media and the slices were cultured for 7 days, with renewal of media and treatment at day 4. Upon addition of luciferin to the media, photons emitted by the tumor-bearing slices were measured, compared with those obtained at the beginning of the experiment, and normalized to those cultured with DMSO (100%).
In vivo treatments
A total of 2 × 105 luciferase-expressing U-251 MG or 5 × 103 H676 cells were surgically implanted into Foxn1-null mice, both male and female mice, as for the ex vivo experiments. Ten or 3 days after the intervention, respectively, mice were randomized and treatments were administered intraperitoneally. TMZ was dissolved in DMSO to a concentration of 15 mg/mL, and then diluted in saline. Val-083 was dissolved in saline to a concentration of 1 mg/mL, and then diluted in saline. Mice were injected three times per week for 3 weeks, and monitored until the end of the experiment. Presence of tumors was confirmed by H&E staining of paraffin-embedded tissue sections.
All animal procedures have been approved by the “Research Ethics and Animal Welfare” from the “Instituto de Salud Carlos III” and from the “Área de Protección Animal” committees (CBA 31_2019-v2 and PROEX 250/19).
Results
TMZ-mediated toxicity is dependent on MMR status and abrogated by MGMT expression
The standard therapies for patients with GBM, ionizing radiation (IR) and TMZ, create DSBs, the most deleterious form of DNA damage. DSBs lead to initiation of the DNA damage response (DDR) and consequently the activation of DNA repair pathways and cell-cycle checkpoints. We have previously presented evidence that alterations in key DNA repair and checkpoint proteins can modulate GBM treatment response (27–29). The intricacy of the DDR signaling pathways makes it susceptible to multiple concomitant alterations of its components in a given tumor patient, offering both challenges and opportunities from a treatment perspective. Loss of components of a specific DNA repair pathway might be compensated by the increased activity of other components or pathways. Upregulated DNA repair pathways can lead to resistance to radiotherapy and/or DNA-damaging chemotherapy. Therefore, inhibitors of these pathways can potentially increase the sensitivity of the tumor cells to those therapies. In contrast, lost pathways represent weaknesses in the DNA-repairing ability of the tumor cell. These weaknesses can be exploited by choosing a suitable chemotherapy to induce unrepairable DNA damage.
Here, we explored which DDR components contribute to TMZ-induced toxicity in the presence or absence of MGMT, the key DNA repair enzyme which removes the alkylation introduced by TMZ. We initially measured the response to TMZ of a group of well-established GBM cell lines. We observed that the cell lines expressing MGMT (T98G and LN18) were more resistant to TMZ as compared with those not expressing MGMT (U-251 MG, SF268, and U-87 MG; Fig. 1A). We selected U-251 MG cells (MGMT−) and T98G cells (MGMT+) for further analysis.
Figure 1.
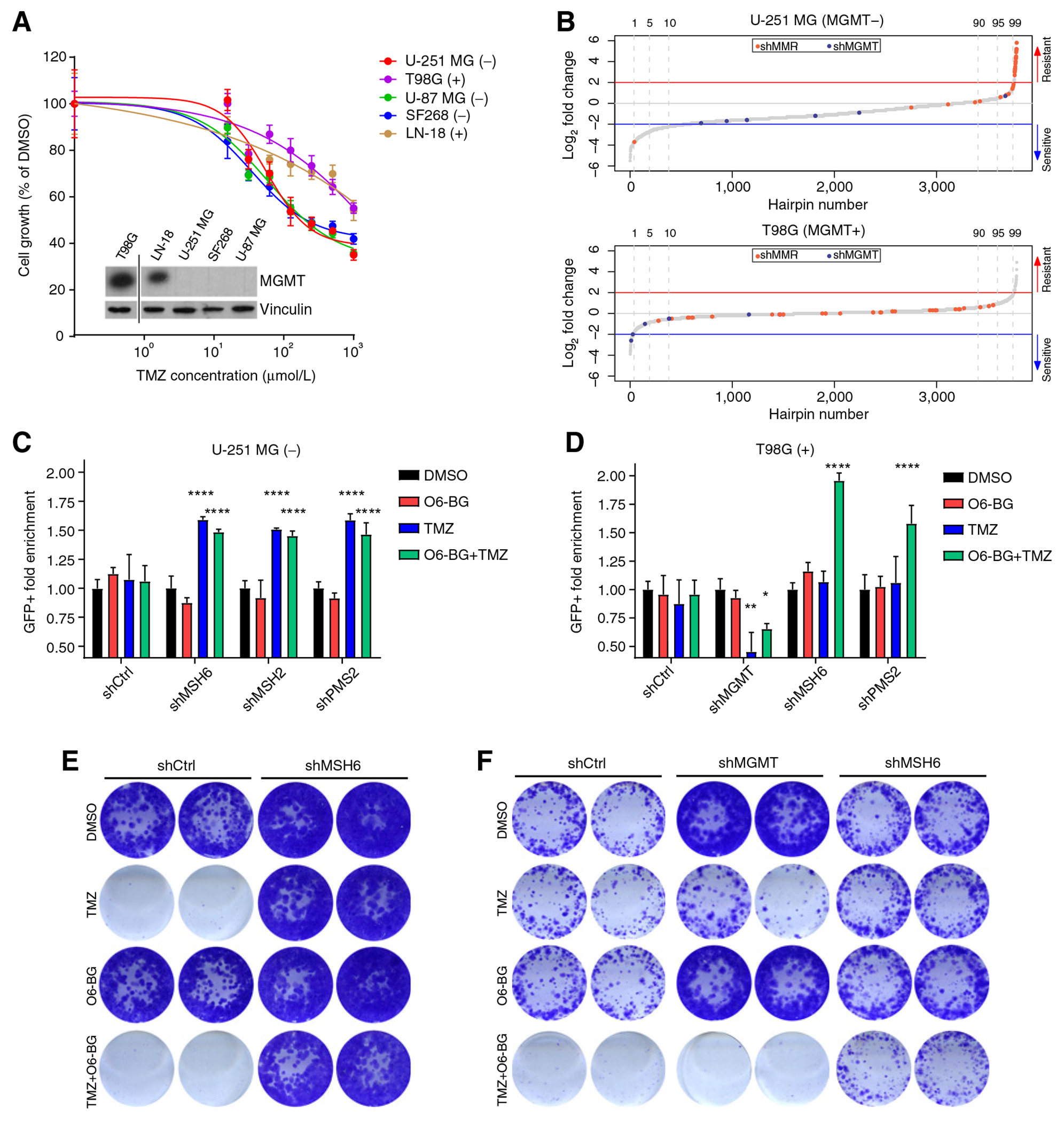
TMZ cytotoxicity depends on MMR functionality and MGMT expression. A, Viability of parental U-251 MG, T98G, U-87 MG, LN-18, and SF268 cell lines upon exposure to different concentrations of TMZ measured by MTT. Values are represented as percentage of cell viability compared with DMSO control. The inlay is a Western blot analysis of these cell lines showing the expression of MGMT (top) as compared with the loading control (vinculin, bottom). MGMT expression for each cell lines is also indicated in the legend: (−) = MGMT negative and (+) = MGMT positive. B, HiSeq shRNA screening results of U-251 MG and T98G cells transduced with the DDR library and treated with TMZ (U-251 MG 100 μmol/L and T98G 200 μmol/L). shRNAs targeting the components of the MMR machinery and MGMT are highlighted in red and green, respectively. C and D, Competition assay of U-251 MG (C) and T98G (D) cells expressing shRNAs (shRen.660, shMGMT.430, shMSH6.3908, shPMS2.946, and shMSH2.357) targeting different components of the MMR machinery after treatment with TMZ (100 μmol/L) and/or the MGMT inhibitor O6-BG (200 μmol/L). The represented value is the fold enrichment of the shRNA-expressing cells (eGFP+) compared with the control cells (mCherry+) and normalized to the DMSO control. Statistical test: Two-way ANOVA (P values: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001, compared with DMSO treated). E and F, CFA of U-251 MG (E) and T98G (F) cells expressing control shRNA (shRen.660), shMSH6.3908, or shMGMT.430 after treatment with TMZ and/or O6-BG for 9 days.
To explore potential sensitivity and resistance mechanisms due to alterations in different DNA repair pathways, we performed a set of shRNA screenings with a custom-generated shRNA library targeting different components of DDR pathways. The library was designed to target 643 DDR genes with at least five shRNAs per transcript (see Materials and Methods for details). After transducing the library into U-251 MG and T98G, the infected cells were treated with TMZ and analyzed by high-throughput sequencing (HiSeq). Similarly to what has been recently reported in a genome-wide CRISPR/Cas9 gRNA screen performed on GBM stem cell cultures (30), we observed a strong enrichment of multiple shRNAs targeting various components of the MMR pathway (MSH2, PMS2, MSH6, and MLH1) in U-251 MG cells (Fig. 1B, top; Supplementary Table S2). Because of the lack of MGMT expression, shRNAs targeting MGMT were not significantly lost. In contrast, the library-infected T98G cells (MGMT+) exposed to TMZ had a loss of representation of cells expressing shRNAs targeting MGMT, whereas those against MMR components were not enriched (Fig. 1B, bottom; Supplementary Table S3).
The differences between U-251 MG and T98G cells might be explained by MGMT activity, which readily repairs the TMZ-induced damage, avoiding the intervention of the MMR machinery. Therefore, MGMT-expressing cells do not rely on MMR alterations for their survival in the presence of an alkylating agent. To corroborate this hypothesis and to validate the screening results, we generated cell lines expressing shRNAs from our library targeting MGMT or MMR components (Supplementary Figs. S1A and S1B). We performed competition assays by co-incubating control cells with cells expressing shRNAs for MGMT or for MMR components (Supplementary Fig. S1C). Upon TMZ treatment, U-251 MG cells deficient in different MMR components were significantly enriched, independently of the presence of the MGMT inhibitor O6-BG (Fig. 1C). However, enrichment of MMR-deficient T98G cells was only evident in those cotreated with O6-BG (Fig. 1D). In addition, the ratio of T98G cells expressing shMGMT was significantly reduced as compared with the control cells after TMZ exposure.
We further confirmed these results in short- and long-term experiments. CFAs (Fig. 1E) and dose–response assessment (Supplementary Fig. S2A) reproduced the strong phenotype of TMZ resistance associated with MSH6 silencing in U-251 MG cells. Overexpression of MGMT in U-251 MG cells, mediated by the SAR1A–MGMT fusion gene (24), abrogated the toxic effect of TMZ (Supplementary Fig. S2B). On the other hand, MGMT knockdown increased TMZ sensitivity in T98G cells, which was further enhanced by blocking the residual MGMT activity with O6-BG (Fig. 1F; Supplementary Fig. S2C). Finally, MSH6 knockdown in T98G cells, although it did not alter cell viability in the presence of TMZ alone (Fig. 1F; Supplementary Fig. S2D), it clearly protected them from TMZ-induced toxicity when the cells were cotreated with TMZ and O6-BG (Fig. 1F).
In summary, our data confirmed that MGMT expression and/or loss of MMR components act as major mechanisms of TMZ resistance in vitro. Most importantly, we showed that MGMT inhibition in MGMT+ cells makes them susceptible to develop resistance mediated by the loss of MMR components.
Val-083-mediated cytotoxicity is independent of MGMT expression or MMR deficiency
The high frequency of resistance to TMZ in patients with GBM pre- and posttreatment (6) urges to find new chemotherapeutics. In this line, we aimed to explore potential alternative treatments. Val-083 is known to act as a bi-alkylating agent, and the main mechanism of cytotoxicity is thought to be DNA crosslinking (11).
We incubated GBM cell lines with Val-083 to test its effect in the GBM context. Unlike treatment with TMZ (Fig. 1A), Val-083 did not differentially affect cell proliferation based on MGMT expression (Fig. 2A). Cell-cycle profile of cells exposed to Val-083 showed a clear dose-dependent arrest in G2–M phase, both in U-251 MG (MGMT−) and T98G (MGMT+) cells. TMZ lead to cell-cycle arrest only in the MGMT− cells (Fig. 2B). These results were validated in human GBM tumorspheres (Supplementary Figs. S3A and S3B). In line with the treatment of the established GBM lines, Val-083 efficiently reduced cell proliferation independently of MGMT expression.
Figure 2.
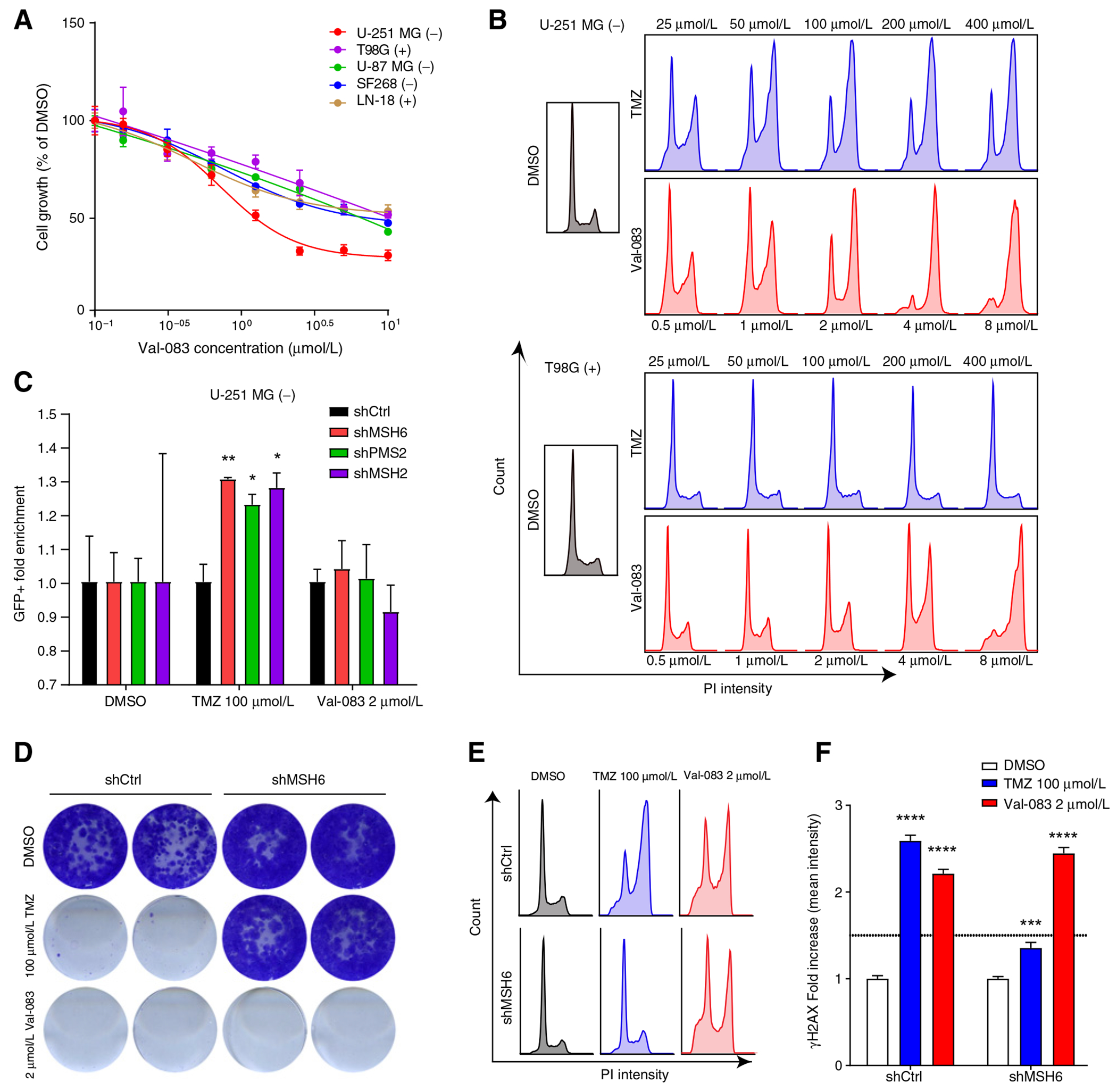
Val-083 cytotoxicity is exerted independently of MMR or MGMT expression. A, Viability of parental U-251 MG, T98G, U-87 MG, LN-18, and SF268 cell lines upon exposure to different concentrations of Val-083 measured by MTT. Values are represented as percentage of cell viability compared with DMSO control. B, Cell-cycle profile of U-251 MG or T98G cells treated with different doses of TMZ or Val-083 for 48 hours. C, Competition assay of U-251 MG cells expressing shRNAs targeting MSH6 (shMSH6.3908), PMS2 (shPMS2.946), and MSH2 (shMSH2.357) after treatment with TMZ or VAL-083. The plotted value is the fold enrichment of the shRNA-expressing cells (eGFP+) compared with the control cells (mCherry+) and normalized to the DMSO control. Statistical test: Two-way ANOVA (P values: *P < 0.05, **P < 0.01, compared with DMSO treated). D, CFA of U-251 MG cells expressing control shRNA or shMSH6 after treatment with TMZ or Val-083 for 9 days. E, Cell-cycle profile of U-251 MG cells expressing control shRNA (shRen.660) or shMSH6.3908 after treatment with TMZ or Val-083 for 48 hours. F, Quantification of mean γH2AX fluorescence in nuclei of cells treated with vehicle, TMZ, or Val-083 for 48 hours. Values are normalized to the DMSO control, and fold increase in H2AX signal is plotted. Statistical test: Two-way ANOVA (P values: ***, P < 0.001; ****, P < 0.0001, compared with DMSO treated).
We then investigated whether Val-083 cytotoxicity was mediated by the activity of the MMR pathway. In contrast to what we observed with TMZ exposure, silencing of MMR in U-251 MG cells did not cause any selective advantage upon treatment with Val-083 by competition assay (Fig. 2C). CFAs, cell-cycle profiling, and dose–response assessments further confirmed that Val-083 efficiently prevented cell proliferation independently of MGMT expression or MMR deficiency (Fig. 2D and E; Supplementary Figs. S2E–S2H).
TMZ and Val-083 lead to DSBs as an indirect consequence of their DNA-damaging mechanism. In addition to the cell-cycle arrest (Fig. 2E), we observed an increase in DNA damage in U-251 MG cells upon incubation with TMZ or Val-083, as evidenced by the increase in phosphorylation of H2AX (γH2AX; Fig. 2F; Supplementary Fig. S1D).
To corroborate the MGMT- and MMR-independent action of Val-083, and to potentially uncover DNA repair pathways involved in resistance or sensitivity to this drug, we performed a shRNA screening with our DDR library. In contrast to our observations in the TMZ screenings (Fig. 1B), there were not significative changes in the distribution of shRNAs against MMR components or MGMT in the HiSeq analysis of U-251 MG and T98G cells treated with Val-083 (Supplementary Figs. S4A and S4B; Supplementary Tables S4 and S5). Moreover, we did not observe any consistent enrichment or loss of shRNAs targeting any other DNA repair genes.
Overall, our data demonstrate that Val-083 could represent an effective treatment that overcomes multiple known mechanisms of TMZ resistance, with potentially less signaling pathways that could mediate its resistance.
TMZ and Val-083 have synergistic effect on tumor cell proliferation inhibition
Combination therapy is a well-known approach for treating a wide range of diseases, including cancer. A variety of molecules targeting multiple DDR components (e.g., PARP, ATM, and ATR, among others) have shown to potentiate TMZ efficacy in cells with different genetic background (31–34). Our data clearly indicate that TMZ and Val-083 toxicity is mediated by independent mechanisms. We speculated that both drugs could synergize in decreasing GBM cell proliferation, thus also reducing the likelihood of resistance.
We analyzed the growth of GBM cells exposed to combinations of increasing doses of TMZ and Val-083. We found that some of these combinations synergistically reduced cell viability in treated U-251 MG cells as compared with sham-treated cells (Fig. 3A and B). Importantly, this synergy was observed in both MGMT-deficient and MGMT-proficient cells, albeit higher doses of TMZ were required in the second group (Fig. 3C; Supplementary Fig. S5). Similar results were obtained in GBM tumorspheres (Fig. 3C; Supplementary Fig. S5).
Figure 3.
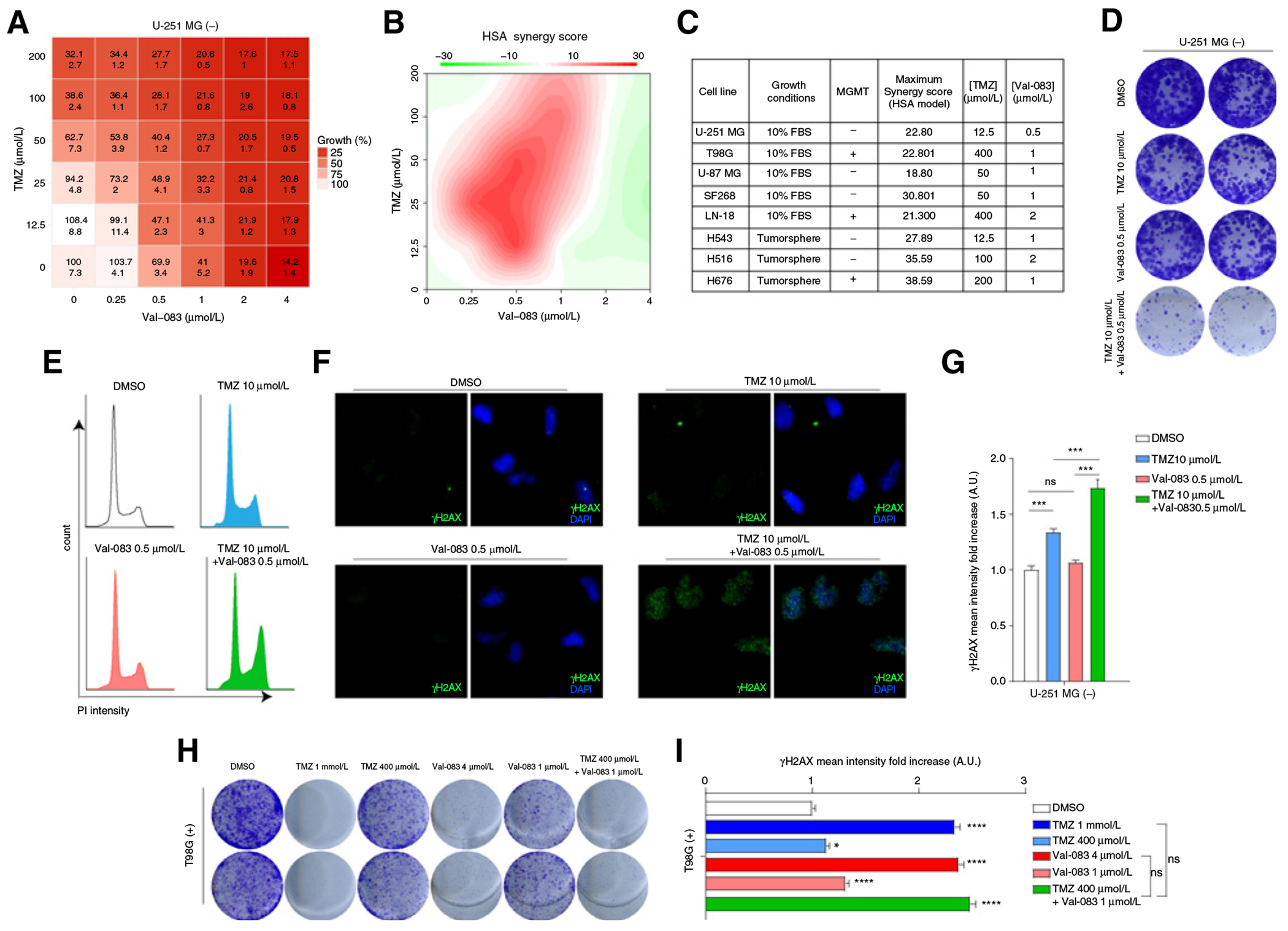
TMZ and Val-083 combination treatment show synergistic cytotoxic effect. A, Table showing the percentage of cell viability (as compared with DMSO, top number in each cell) and SD (bottom number). B, Plot of synergy score calculated with the HSA method of U-251 MG cells treated with different doses of TMZ and Val-083. C, Table showing the maximum synergy scores of different adherent cell lines (U-251 MG, T98G, U-87 MG, SF268, LN-18) and tumorspheres grown in suspension (H543, H516, H676), as well as the doses of TMZ and Val-083, which rendered said score. MGMT expression is also indicated. D, CFA of U-251 MG cells incubated with low doses of TMZ, Val-083, or the combination for 9 days. E, Cell-cycle profile of U-251 MG cells incubated for 48 hours with TMZ, Val-083, or the combination of both. F, Representative pictures of γH2AX staining of U-251 MG cells treated for 48 hours with DMSO, TMZ, Val-083, or the combination of the last two. G, Quantification of γH2AX signal intensity in nuclei of U-251 MG cells treated for 48 hours with TMZ, Val-083, or the combination of both. Statistical test (P values: ***, P < 0.001). H, CFA of T98G cells incubated with high and low doses of TMZ, Val-083, or the combination for 9 days. I, Quantification of γH2AX signal intensity in nuclei of T98G cells treated for 48 hours with TMZ, Val-083, or the combination of both. Statistical test: t test (P values: *, P < 0.05; ***, P < 0.001).
Next, we tested the combination of concentrations of TMZ and Val-083, which showed the highest degree of synergism in U-251 MG and T98G cells. Notably, these combinations used concentrations lower than the IC50 of each drug. Reduction in colony-forming ability (CFA) (Fig. 3D and H), cell-cycle arrest (Fig. 3E), and accumulation of DNA damage (Fig. 3F, G, and I) were clearly potentiated in cells treated with the combination of TMZ and Val-083, as compared with the single drugs in low concentration.
Cotreatment with TMZ and Val-083 increases survival in a mouse xenograft model
We further validated the synergy between TMZ and Val-083 by analyzing its efficacy ex vivo with organotypic cultures. We obtained brain slices from mice bearing U-251 MG-derived tumors, and treated them with TMZ and/or Val-083. As in the in vitro scenario, combination of low doses of TMZ (10 μmol/L) and Val-083 (0.5 μmol/L) caused a notable decrease in tumor cell proliferation, when compared with those treated with DMSO. Notably, this reduction was similar as that achieved with treatment with higher doses of TMZ (25 μmol/L; Fig. 4A).
Figure 4.
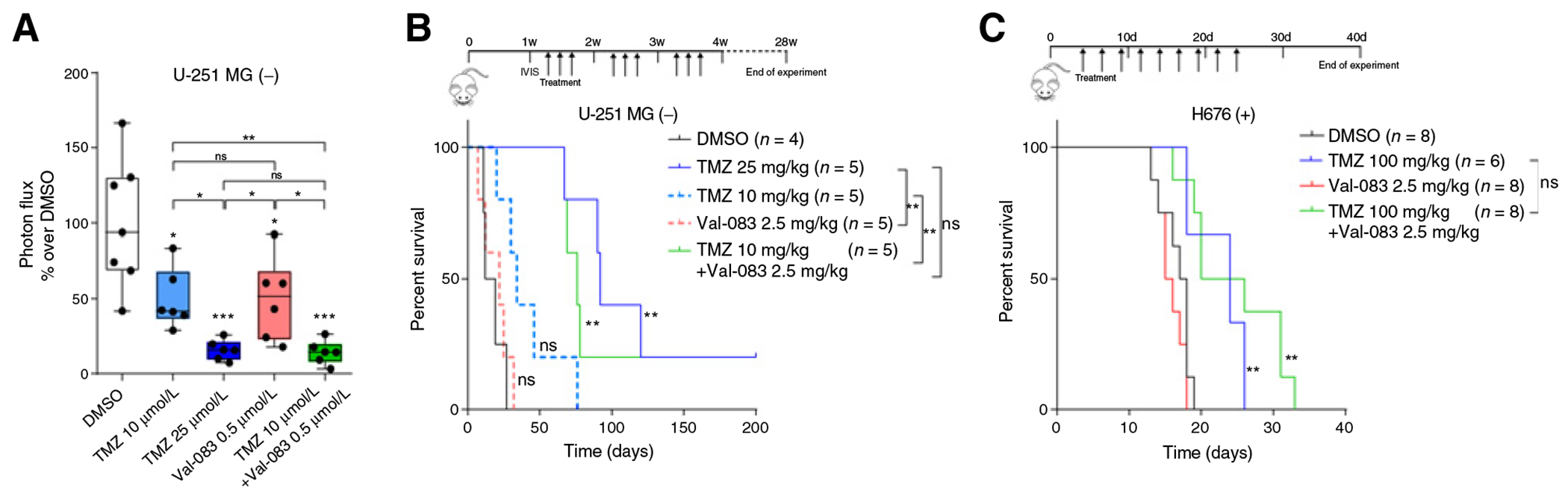
Treatment with combination of TMZ and Val-083 increases survival in mice. A, Quantification of the ratio of luminescence emission by U251-MG in ex vivo-treated brain slices after 7 days as compared with the initial. Plots represent the fold change compared with the DMSO control. Statistical test: t test (P values: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, nonsignficant); P values on top of the boxes are comparison over the DMSO control. B, Kaplan–Meier curve showing the survival proportions of mice transplanted with U-251 MG cells and treated with TMZ, Val-083, or the combination of both. Log-rank P values: **, P < 0.01; ns, nonsignificant). C, Kaplan–Meier curve showing the survival proportions of mice transplanted with H676 cells and treated with TMZ, Val-083, or the combination of both. Log-rank P values: **, P < 0.01; ns, nonsignificant).
The ability of Val-083 and TMZ to cross the brain–blood barrier makes them ideal partners for a potential combination therapy for patients with GBM. To validate the efficacy of the combination in vivo, we treated mice bearing U-251 MG-derived intracranial tumors TMZ and/or Val-083 (Fig. 4B). As expected, both vehicle-treated and mice administered with the lower dose of TMZ (10 mg/kg) were the first to develop signs of brain tumors, which was later confirmed by histology (Supplementary Fig. S6). Most importantly, similarly to what observed in the ex vivo approach, mice treated with high doses of TMZ (25 mg/kg) and with the combination of TMZ (10 mg/kg) plus Val-083 (2.5 mg/kg) displayed similar survival kinetics, significantly expanding the life span of these mice. No significant differences between both longer-surviving groups were found.
To further validate our results in the context of MGMT expressing tumors, we tested the combination treatment in mice transplanted with the H676 human neurosphere line (Supplementary Fig. S3A). We did observe a significant increase in survival of mice treated with the combination of TMZ and Val-083, when compared with the DMSO- or Val-083-treated mice. However, this increase was similar to that of the TMZ-treated mice. Of note, the combination-treated mice developed pale skin and petechiae, probably due to the described toxic effect of both drugs over the hematopoietic system (12, 35).
Discussion
TMZ has been used as the standard chemotherapy for patients with GBM for the last 15 years. Its mechanism of action has been extensively characterized, and comprises alkylation in the DNA, which if unrepaired, causes mismatches in following rounds of replication. These mismatches are recognized by the MMR system, which attempts and fails to remove them, leading to the generation of DSBs and subsequent cell death. MGMT expression, induced either by promoter hypomethylation or MGMT genomic rearrangements (24), and alterations in MMR components have been identified as the main mechanisms driving TMZ resistance in patients with GBM. Epigenetic silencing by methylation of the promoter of the MGMT gene prevents its synthesis. As a consequence, there is an increase in the sensitivity of tumors to TMZ. Hypermethylation of the MGMT promoter is currently the only known predictive biomarker for TMZ response in patients with GBM. Our shRNA screening results, with the caveat of having being performed on only one MGMT-proficient and one MGMT-deficient cell line, provide a strong genetic basis of the mutually exclusivity between MGMT expression and defects in MMR observed in patients with GBM at recurrence (24, 36).
Strategies for MGMT inhibition are under investigation to potentiate TMZ activity in patients with GBM (37). MGMT inhibitors, such as the O6-BG and O6-(4-bromothenyl) guanine (O6-BTG) are very effective in increasing TMZ cytotoxicity in vitro. However, when combined systemically with TMZ, these drugs have failed in clinical trials due to toxicity, mainly myelosuppression. Hematopoietic cells rely on MGMT to be protected from the cytotoxic action of TMZ (37, 38). We argue that blocking MGMT activity would ultimately result in the dependence on an intact MMR system for induction of cell death (Fig. 5). Our results suggest that MGMT inhibitors could lead to a positive selective pressure to inactivate MMR components to acquire TMZ resistance, thus rendering MGMT inhibition a much less attractive treatment strategy.
Figure 5.
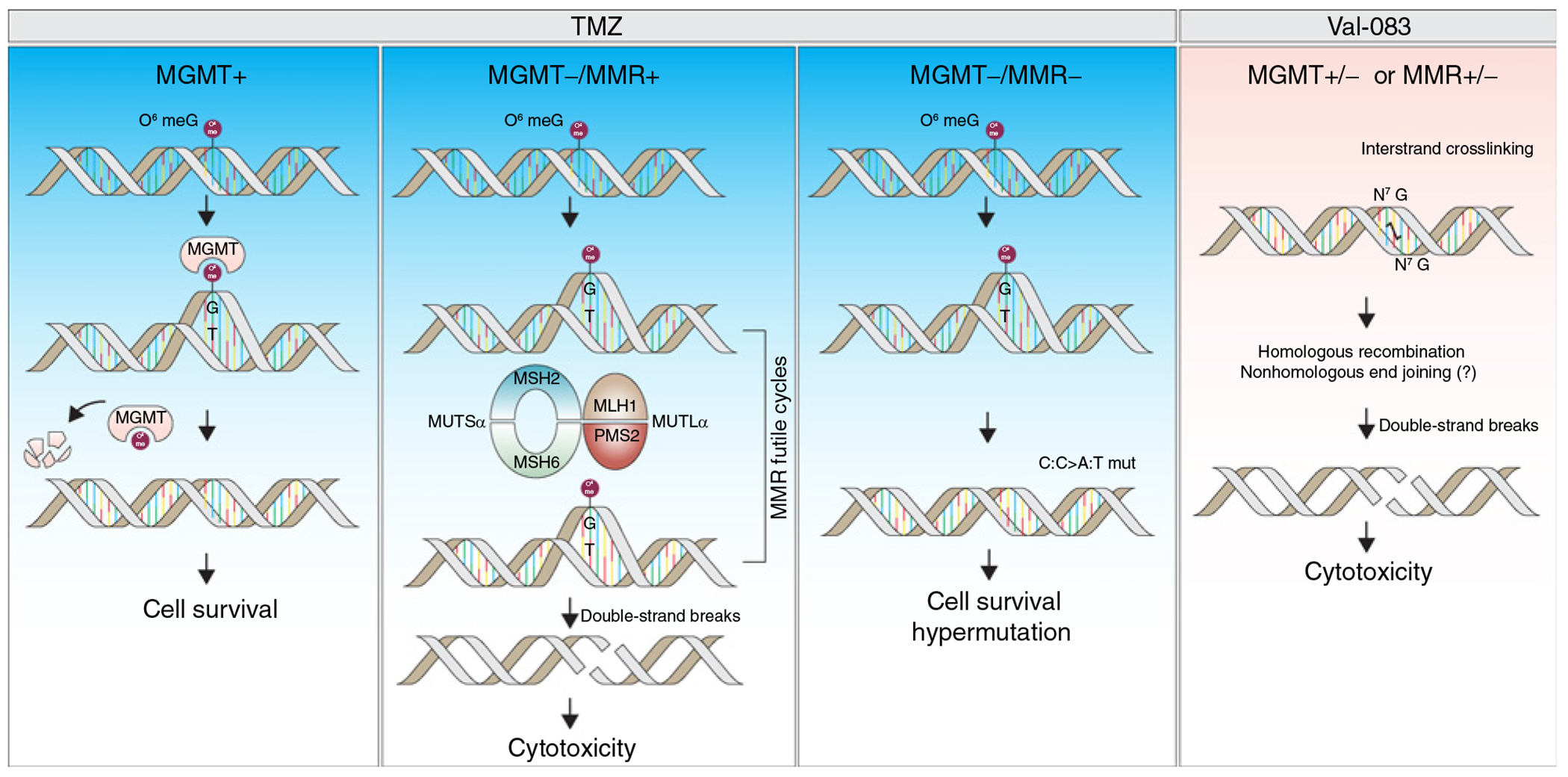
Mechanisms of action of TMZ and Val-083. Expression of MGMT effectively removes the O6-guanine methylation induced by TMZ exposure, which allows cell survival. Lack of MGMT allows the formation of mismatches in the next replication round, which leads to the generation of double DNA-strand breaks mediated by the MMR machinery. The accumulation of double-strand breaks ultimately lead to cell death. However, defects in the MMR pathway paves the way for the accumulation of unresolved mismatches, which allows cell survival and a hypermutation phenotype. In the case of Val-083, there is a formation of interstrand adducts, that are not resolved by MGMT, and lead to cytotoxicity independently of MMR status.
Dianhydrogalactitol, also known as Val-083, is able to cross the blood-brain barrier, and it was considered to use it as a chemotherapeutic in brain tumors (39). It was recently granted orphan drug designation by the U.S. FDA for the treatment of glioma, medulloblastoma and ovarian cancers, as well as a fast-track status for the treatment of recurrent GBM. Recent data on newly diagnosed GBM showed Val-083 safety in combination with radiotherapy (16). Moreover, it has been recently shown to be more effective than TMZ at similar doses in patient-derived organoids, and consistent with our data, the effect was independent on their MGMT promoter methylation status (40). However, evidence of its efficacy in prolonging patient survival has not yet been demonstrated.
The clinical use in oncology of a variety of DNA crosslinking drugs (e.g., cisplatin, mitomycin C, and nitrogen mustards) is well established (41). Val-083 induces interstrand-crosslinks at N7-guanine (11), which are possibly repaired by the homologous recombination (HR) pathway (Fig. 5; ref. 12). However, its exact mechanism of action is not yet fully understood. Although it was shown that silencing of the HR component BRCA2 increased Val-083 cytoxicity in lung cancer cells (12), our shRNA screening did not point to specific DNA repair pathways as key players in its sensitivity, nor resistance. Extending the analysis to cell lines with more diverse genetic backgrounds and using additional shRNA libraries targeting other signaling pathways might point to Val-083-specific sensitivity and/or resistance mechanisms. Nevertheless, we believe that we could definitely rule out any implication of the MMR pathway or MGMT in Val-083 sensitivity or resistance.
Targeting multiple cellular pathways is the base of many combination treatments for tumors, and often result in synergic therapeutic effects. In GBM, the use of concomitant treatments along TMZ and IR, such as carmustine (42), tumor-treating fields (43) or bevacizumab (44), has been approved. However, the increase of patient survival is still very limited. Our data show a significant synergistic cytotoxic effect upon combination of Val-083 and TMZ in MGMT-proficient and MGMT-deficient cells. Thus, addition of Val-083 to the repertoire of concomitant therapies used along TMZ and IR might prove beneficial in patients with both methylated and unmethylated MGMT promoter.
In conclusion, we provide mechanistic insights into the cytotoxic effect of TMZ and Val-083 and propose that the combinatorial treatment of the two drugs warrant further investigation as a potential therapeutic approach for patients with GBM.
Supplementary Material
Acknowledgments
This research was supported by funds from the Seve Ballesteros Foundation and the Asociación Española Contra el Cáncer (AECC; LABAE16015SQUA) to M. Squatrito. M. Jiménez-Alcázar is a recipient of an EMBO Long Term Fellowship (ALTF 1199–2018). A. Curiel-García is a recipient of a Severo-Ochoa PhD fellowship. P. Nogales is a recipient of an AECC fellowship (PPL2013). A. Schuhmacher is a recipient of a Ramón y Cajal contract from the Spanish Ministry for the Economy, Industry and Competitiveness (RYC-2015-17622). L. Zhu is a La Caixa-Severo Ochoa International PhD fellow (LCF/BQ/SO16/52270014). We are grateful to Eusebio Manchado for his help with the shRNA library and screening design. We would like to thank Orlando Domínguez from the CNIO Genomic Unit for his help in designing the primers for the HiSeq and the CNIO Confocal Microscopy Unit for their support. Finally, we would like to acknowledge Dennis Brown and DelMar pharmaceuticals for kindly providing the Val-083 for the in vivo experiments.
Authors’ Disclosures
P. Nogales reports grants from Asociación Española Contra el Cáncer during the conduct of the study. M. Galán-Ganga reports personal fees from Fundación Científica de la Asociación Española Contra el Cáncer (AECC) during the conduct of the study; grants from Roche Farma S.A.; and personal fees from Roche Farma S.A. outside the submitted work. L. Zhu reports personal fees from Fundación La Caixa during the conduct of the study. S.W. Lowe reports personal fees from Oric Pharmaceuticals, Blueprint Medicines, PMV Pharmaceuticals, and Constellation Pharmaceuticals outside the submitted work. M. Squatrito reports grants from Seve Ballesteros Foundation and Asociación Española Contra el Cáncer (AECC) during the conduct of the study. No disclosures were reported by the other authors.
Footnotes
Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
References
- 1.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016;131:803–20. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 3.Mischel PS, Cloughesy TF. Targeted molecular therapy of GBM. Brain Pathol 2006;13:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro Oncol 2010;12:304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirimanoff RO, Gorlia T, Mason W, Van den Bent MJ, Kortmann R-D, Fisher B, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: Recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol 2006;24:2563–9. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Stevens MFG, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol 2012;5:102–14. [DOI] [PubMed] [Google Scholar]
- 7.Kaina B, Christmann M, Naumann S, Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst) 2007;6:1079–99. [DOI] [PubMed] [Google Scholar]
- 8.Felsberg J, Thon N, Eigenbrod S, Hentschel B, Sabel MC, Westphal M, et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int J Cancer 2011;129:659–70. [DOI] [PubMed] [Google Scholar]
- 9.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 2007;13:2038–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nemeth L, Institoris L, Somfai S, Gál F, Pályi I, Sugár J, et al. Pharmacologic and antitumor effects of 1,2:5,6-dianhydrogalactitol (NSC-132313). Cancer Chemother Rep 1972;56:593–602. [PubMed] [Google Scholar]
- 11.Institoris E, Tamas J. Alkylation by 1,2:5,6-dianhydrogalactitol of deoxyribonucleic acid and guanosine. Biochem J 1980;185:659–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhai B, Steinø A, Bacha J, Brown D, Daugaard M. Dianhydrogalactitol induces replication-dependent DNA damage in tumor cells preferentially resolved by homologous recombination. Cell Death Dis 2018;9:1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levin VA, Freeman-Dove MA, Maroten CE. Dianhydrogalactitol (NSC-132313): pharmacokinetics in normal and tumor-bearing rat brain and antitumor activity against three intracerebral rodent tumors. J Natl Cancer Inst 1976;56:535–9. [DOI] [PubMed] [Google Scholar]
- 14.Elson LA, Jarman M, Ross WC. Toxicity, haematological effects and anti-tumour activity of epoxides derived from disubstituted hexitols. mode of action of mannitol myleran and dibromomannitol. Eur J Cancer 1968;4:617–25. [DOI] [PubMed] [Google Scholar]
- 15.Kralovanszky J, Prajda N, Kerpel-Fronius S, Gal F, Szentirmay Z. Effect of a single high dose and repeated small doses of dianhydrogalactitol (DAG; NSC-132313) on rat intestinal mucosa. Cancer Chemother Pharmacol 1983;11:167–71. [DOI] [PubMed] [Google Scholar]
- 16.Guo C,Yang Q, Li J, Wu S, Deng M, Du X, et al. Phase 2 clinicaltrial ofVAL-083 as first-line treatment in newly-diagnosed MGMT-unmethylated glioblastoma multiforme (GBM): halfway report. Glioma 2019;2:167. [Google Scholar]
- 17.Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell 2011;41:733–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, et al. An optimized microRNA backbone for effective single-Copy RNAi. Cell Rep 2013;5:1704–13. [DOI] [PubMed] [Google Scholar]
- 19.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol 2011;29:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, et al. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood 2010;116:731–9. [DOI] [PubMed] [Google Scholar]
- 23.Wu D, Lim E, Vaillant F, Asselin-Labat ML, Visvader JE, Smyth GK. ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 2010;26:2176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oldrini B, Vaquero-Siguero N, Mu Q, Kroon P, Zhang Y, Galán-Ganga M, et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat Commun 2020;11:3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, et al. Methods for high-throughput drug combination screening and synergy scoring. Methods Mol Biol 2018;1711:351−98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berenbaum MC. What is synergy? Pharmacol Rev 1989;41:93–141. [PubMed] [Google Scholar]
- 27.Squatrito M, Vanoli F, Schultz N, Jasin M, Holland EC. 53BP1 is a haploinsufficient tumor suppressor and protects cells from radiation response in glioma. Cancer Res 2012;72:5250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell 2010;18:619–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res 2011;71:5945–9. [DOI] [PubMed] [Google Scholar]
- 30.MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, et al. Genome-wide CRISPR-Cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep 2019;27:971–86. [DOI] [PubMed] [Google Scholar]
- 31.Higuchi F, Nagashima H, Ning J, Koerner MVA, Wakimoto H, Cahill DP. Restoration of temozolomide sensitivity by Poly(ADP-Ribose) polymerase inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin Cancer Res 2020;26:1690–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jackson CB, Noorbakhsh SI, Sundaram RK, Kalathil AN, Ganesa S, Jia L, et al. Temozolomide sensitizes MGMT-deficient tumor cells to ATR inhibitors. Cancer Res 2019;79:4331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta SK, Smith EJ, Mladek AC, Tian S, Decker PA, Kizilbash SH, et al. PARP inhibitors for sensitization of alkylation chemotherapy in glioblastoma: impact of blood-brain barrier and molecular heterogeneity. Front Oncol 2019;8:670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadkarni A, Shrivastav M, Mladek AC, Schwingler PM, Grogan PT, Chen J, et al. ATM inhibitor KU-55933 increases the TMZ responsiveness of only inherently TMZ sensitive GBM cells. J Neurooncol 2012;110:349–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kourelis T, Buckner JC, Gangat N, Patnaik MM. Temozolomide induced bone marrow suppression—a single institution outcome analysis and review of literature. Blood 2014;124:1602. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Cazzato E, Ladewig E, Frattini V, Rosenbloom DIS, Zairis S, et al. Clonal evolution of glioblastoma under therapy. Nat Genet 2016;48:768–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaina B, Margison GP, Christmann M. Targeting O 6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell Mol Life Sci 2010;67:3663–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J Clin Oncol 2009;27:1262–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eagan RT, Childs DS Jr, Layton DD Jr, Laws ER Jr, Bisel HF, Holbrook MA, et al. Dianhydrogalactitol and radiation therapy. Treatment of supratentorial glioma. JAMA 1979;241:2046–50. [PubMed] [Google Scholar]
- 40.Golebiewska A, Hau A-C, Oudin A, Stieber D, Yabo YA, Baus V, et al. Patient-derived organoids and orthotopic xenografts of primary and recurrent gliomas represent relevant patient avatars for precision oncology. Acta Neuropathol 2020;140:919–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011;11:467–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jungk C, Chatziaslanidou D, Ahmadi R, Capper D, Bermejo JL, Exner J, et al. Chemotherapy with BCNU in recurrent glioma: analysis of clinical outcome and side effects in chemotherapy-naïve patients. BMC Cancer 2016;16:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trusheim J, Dunbar E, Battiste J, Iwamoto F, Mohile N, Damek D, et al. A state-of-the-art review and guidelines for tumor treating fields treatment planning and patient follow-up in glioblastoma. CNS Oncol 2017;6:29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diaz RJ, Ali S, Qadir MG, De La Fuente MI, Ivan ME, Komotar RJ. The role of bevacizumab in the treatment of glioblastoma. J Neurooncol 2017;133:455–67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.