

ABSTRACT
In formin-family proteins, actin filament nucleation and elongation activities reside in the formin homology 1 (FH1) and FH2 domains, with reaction rates that vary by at least 20-fold between formins. Each cell expresses distinct formins that assemble one or several actin structures, raising the question of what confers each formin its specificity. Here, using the formin Fus1 in Schizosaccharomyces pombe, we systematically probed the importance of formin nucleation and elongation rates in vivo. Fus1 assembles the actin fusion focus, necessary for gamete fusion to form the zygote during sexual reproduction. By constructing chimeric formins with combinations of FH1 and FH2 domains previously characterized in vitro, we establish that changes in formin nucleation and elongation rates have direct consequences on fusion focus architecture, and that Fus1 native high nucleation and low elongation rates are optimal for fusion focus assembly. We further describe a point mutant in Fus1 FH2 that preserves native nucleation and elongation rates in vitro but alters function in vivo, indicating an additional FH2 domain property. Thus, rates of actin assembly are tailored for assembly of specific actin structures.
KEY WORDS: Actin cytoskeleton, Cell–cell fusion, Fission yeast Schizosaccharomyces pombe, Formin
Summary: Systematic alteration of formin nucleation and elongation rates shows that the actin assembly properties of formin Fus1 are tailored to its function in vivo and identifies an additional function for the Fus1 FH2 domain.
INTRODUCTION
Formins are a large family of conserved proteins that act as both nucleators and elongators of linear actin filaments (Breitsprecher and Goode, 2013; Courtemanche, 2018). They are involved in the assembly of many actin structures that underlie cellular processes such as polarization, motility, or division (Bohnert et al., 2013b; Goode and Eck, 2007; Pollard and O'Shaughnessy, 2019; Skau and Waterman, 2015). Most cells and organisms express different formin genes, which contribute to the assembly of distinct actin structures. For example, mammals have 15 formins (Schönichen and Geyer, 2010) and Arabidopsis thaliana has at least 21 (Blanchoin and Staiger, 2010), while the yeast Saccharomyces cerevisiae possesses only two (Breitsprecher and Goode, 2013) and Schizosaccharomyces pombe (S. pombe) three that organize distinct actin networks (Kovar et al., 2011). In this organism, formin 3 (For3) supports the formation of polarizing actin cables (Feierbach and Chang, 2001), cell division control protein 12 (Cdc12) assembles the contractile cytokinetic ring (Chang et al., 1997), and Fus1 supports the formation of the fusion focus (Dudin et al., 2015; Petersen et al., 1995), an aster of actin filaments necessary for the fusion of gametes to form the diploid zygote. One important question is what defines the functional specificity of each formin to assemble a specific actin structure.
The defining feature of formins is their highly conserved formin homology 2 (FH2) domain, which forms a dimer that nucleates new actin filaments and processively binds the elongating actin filament barbed end, protecting it from growth arrest by capping proteins (Courtemanche, 2018). The FH2 domain is flanked by a FH1 domain, which extrudes from the FH2 dimer as variable numbers of flexible proline-rich tracks that bind profilin-actin to feed it to the elongating filament. The number and quality of the tracks, but also their spacing in relationship to the FH2 dimer, dictate filament elongation speed (Courtemanche and Pollard, 2012; Paul and Pollard, 2008; Scott et al., 2011). For example, the fission yeast formin Fus1 possesses a single proline-rich track, compared to the two and four tracks in Cdc12 and For3 FH1 domains respectively, making Fus1 a slower elongator than Cdc12 and For3 in vitro (Scott et al., 2011). However, because Cdc12 and For3 have very similar elongation speeds, properties other than the number of proline-rich tracks also regulate elongation speed. Furthermore, exchanging the FH1 domains between Fus1 and Cdc12 is not sufficient to convert their elongation speeds, indicating that the FH2 domain also contributes in setting elongation speed. The contribution of the FH2 domains depends on the time the domain spends in the open state (Aydin et al., 2018; Zweifel and Courtemanche, 2020). Elongation speed can also be influenced by tensile and compressive forces (Zimmermann and Kovar, 2019). In addition to F-actin nucleation and elongation, some formins exhibit additional non-canonical actin regulatory properties, such as actin filament bundling or severing (Courtemanche, 2018). For instance, both Fus1 and Cdc12 were shown to bundle filaments, through the FH1-FH2 domain for Fus1 (Scott et al., 2011) and through the long C-terminal tail for Cdc12 (Bohnert et al., 2013a).
The functional specificity of diverse formins in the same cytosol is controlled in part by regulation of their localization and activation (Breitsprecher and Goode, 2013; Chesarone et al., 2010; Yonetani et al., 2008). Indeed, the FH1-FH2 domains are flanked by less conserved N- and C-terminal regulatory regions that control formin localization, activation, nucleation and processivity, often through interaction with additional proteins (Breitsprecher and Goode, 2013). For instance, in Diaphanous-related formins including S. pombe For3, N- to C-terminal binding mediates autoinhibition, relieved by small GTPase binding (Kühn and Geyer, 2014; Martin et al., 2007). However, many formins, such as Cdc12 or Fus1, are thought not to be autoinhibited by a canonical N- to C-terminus interaction (Scott et al., 2011; Yonetani et al., 2008). There is less information on how the specific actin assembly and regulatory properties of formins – which vary significantly between formins – participate in functional specificity in vivo. Recent findings by Homa et al., have shed some light on the importance of Cdc12's specific actin assembly properties in supporting cytokinesis (Homa et al., 2021) but these findings have not been generalized. The FH1-FH2 domains of all three S. pombe formins have been characterized in vitro and their specific actin assembly rates are known (Scott et al., 2011), which makes S. pombe a good model organism to understand the actin assembly specificities of each formin necessary to organize its own actin network. Interestingly for this work, Fus1 was shown to be a potent nucleator (one filament per two formin dimers) but a modest elongator (5 subunits s−1 µM−1).
Cell–cell fusion is a fundamental process that relies on actin assembly (Martin, 2016). In S. pombe, cell fusion takes place between haploid cells to form the diploid zygote during sexual reproduction, which is initiated by nitrogen starvation. Yeast cells are encased in a cell wall that protects them from external insult and internal turgor pressure. During cell fusion, the cell wall is locally digested precisely at the site of partner cell contact to allow plasma membrane merging without risk of cell lysis. The process is coordinated by the fusion focus, a Fus1-assembled actin structure (Dudin et al., 2015). From light and electron microscopy data, the fusion focus is best described as an aster-like arrangement of actin filaments, which concentrates glucanase-loaded secretory vesicles transported by the type V myosin Myo52 and promotes local cell wall digestion (Dudin et al., 2015; Muriel et al., 2021). The fusion focus shares characteristics with other formin-assembled structures: it requires profilin (Petersen et al., 1998a) and tropomyosin (Dudin et al., 2017; Kurahashi et al., 2002), and Fus1 competes with capping protein (Billault-Chaumartin and Martin, 2019). Importantly, deletion of fus1 completely blocks cell fusion (Petersen et al., 1995). As this formin is only expressed during sexual differentiation and plays no role during mitotic growth (Petersen et al., 1995), this allows to change Fus1 properties at will and to study the resulting effect without impact on viability.
In this work, we address the importance of Fus1 actin assembly properties. By systematically changing the nucleation and elongation rates through chimeras of the FH1 and FH2 domains previously characterized in vitro (Scott et al., 2011), we demonstrate that changes in actin assembly have direct, visible consequences on the assembly of the fusion focus and that Fus1 actin assembly properties are tailored to its function. We further establish that the Fus1 FH2 domain contains an additional, non-canonical function that contributes to fusion focus assembly.
RESULTS
Fus1 has essential properties for fusion not contained in the other pombe formins
To investigate whether Fus1 formin contains any unique property, we tested the ability of the two other pombe formins, Cdc12 and For3, to rescue the fusion defect of fus1Δ cells (Fig. 1A-C and Fig. S1). Whereas a construct expressing fus1 at the ura4 locus under the fus1 promoter in a fus1Δ strain was able to sustain cell fusion in a manner indistinguishable from the WT strain, constructs expressing either for3 or cdc12 were completely fusion deficient (Fig. 1B,C). The inability of For3 and Cdc12 to complement fus1Δ may be due to their different N-terminal regulatory regions, which mediate localization. However, lack of localization is not the sole reason for lack of function as For3, like Fus1, localizes to the region of cell–cell contact (Fig. 1B).
Fig. 1.

Fus1 has essential properties for cell fusion not contained in the other S. pombe formins. (A) Scheme of the constructs used in panels (B-C). All are tagged C-terminally with sfGFP and expressed from the fus1 promoter at the ura4 locus. (B) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of homothallic WT strains expressing Fus1-sfGFP at the endogenous locus or homothallic fus1Δ strains expressing the formins listed in A. (C) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in B. (D) Scheme of the constructs used in panels E and F. Formins are cut between the N-terminal regulatory region and FH1 domain with a BamHI restriction site, tagged C-terminally with sfGFP and expressed from the fus1 promoter. (E) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of homothallic fus1Δ strains expressing the formin chimeras described in D at the ura4 locus. (F) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in E compared to the WT. Error bars are standard deviations. All P-values are relative to WT. Bars are 5 µm.
Nevertheless, to alleviate any problem due to improper localization or lack of other regulatory elements, we constructed a set of chimeric formins that keep Fus1N constant while varying the C-terminal part, similarly expressed at the ura4 locus under the fus1 promoter in a fus1Δ strain (Fig. 1D-F and Fig. S1). The control Fus1N-Fus1C was able to support fusion in a manner indistinguishable from the WT (Fig. 1E,F). The other chimeric formins instead supported cell fusion to varying reduced degrees (Fig. 1E,F). Fus1N-For3C performed very poorly, with only 12% of the cells fused 24 h post nitrogen starvation, and exhibited a hazy localization at the region of contact between the two cells. Fus1N-Cdc12C performed relatively well, with a fusion efficiency >75%, and showed a sharper localization to the region of cell contact. However, in cell pairs that failed to fuse, Fus1N-Cdc12C formed two distinct foci that failed to come together, indicating a position more distant from the membrane than that of WT Fus1 (Movie 1) (Dudin et al., 2015). This localization was caused by the Cdc12 long C-terminal extension, suggesting it is due to its described function in oligomerization and actin bundling (Bohnert et al., 2013a), as truncation after Cdc12 FH2 domain led to loss of the double-focus localization and alteration of fusion efficiency (Fig. 1E,F). Because neither For3C nor Cdc12C were able to completely replace Fus1C, these findings suggest a key role of Fus1 actin assembly properties to nucleate the fusion focus. The observation that these chimeras performed better than full-length For3 and Cdc12 also indicates a role of Fus1 N-terminal regulatory region, which we address elsewhere (Billault-Chaumartin et al., 2022 preprint). Here we focus on the specificities contained within the formin C-terminal half.
Importance of Fus1 expression levels and leucine prototrophy for cell fusion
In the process of repeating the experiments described above with chimeras integrated at the native fus1 locus, we obtained initially puzzling results (Fig. 2 and Fig. S1). Indeed, these constructs appeared to be more strongly expressed (compare Fig. 2Ai with Fig. 1E) but exhibited reduced fusion efficiency (<10% of fused cell pairs 24 h post starvation for both Fus1N-For3C and Fus1N-Cdc12C). Through systematic analysis we resolved that these discrepancies are accounted for by two variables, i.e. the leucine auxotrophy status of the strain and the formin expression level.
Fig. 2.

Leucine auxotrophy and formin expression levels have an impact on cell fusion. (A) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of fus1Δ homothallic strains expressing the formin chimeras described in Fig. 1D, either (i) at the fus1 locus in leu1− cells, (ii) at the fus1 locus in leu+ cells, (iii) at the ura4 locus in leu1− cells, or (iv) at both ura4 and fus1 loci in leu1− cells. The same chimeras expressed at the ura4 locus in leu+ cells are shown in Fig. 1E. (B) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in Ai-ii compared to the WT. P-values between bars compare leu1− and leu+ backgrounds. (C) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in Aiii and Fig. 1E (same data as Fig. 1F shown for comparison here) compared to the WT. P-values between bars compare leu1− and leu+ backgrounds. (D) Boxplot of fusion times in strains expressing Fus1N-Fus1C as indicated. (E) Boxplot of the normalized mean total fluorescence intensity at fusion time in indicated strains. (F) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in Aiv compared to the WT. All strains are leu1−. (G) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of fus1Δ homothallic strains expressing Fus1-sfGFP from the ste11 or the pak2 mating-specific promoter at the ura4 locus. All strains are leu1−. (H) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in G compared to the WT. All P-values are relative to WT unless indicated otherwise. Bars are 5 µm.
The main contributor was the leucine status. Indeed, after systematically creating strains with constructs integrated at the fus1 or the ura4 locus in a leu+ or leu1− background, we found that all leu1− strains fused strikingly less well than their leu+ counterparts (Fig. 2A-C). Even the Fus1N-Fus1C control exhibited an increase in fusion time in the leu1− background compared to its leu+ counterpart, independently of the site of insertion (Fig. 2D). Although we do not currently understand the reason why leucine auxotrophy is detrimental for cell fusion (uracil and adenine auxotrophies did not show the same effect), these results stress the importance of using strains with identical auxotrophies, which is what we did for this work.
Independently of the auxotrophy effect, we also found that constructs inserted at the ura4 locus generally exhibited reduced fusion efficiencies than constructs inserted at the native fus1 locus (Fig. 2A-C). In fact, the otherwise fully functional Fus1N-Fus1C control was not able to completely support fusion when expressed from the ura4 locus in a leu1− background (Fig. 2A,C). Quantification of Fus1N-Fus1C-sfGFP total fluorescence levels at the time of cell fusion, when it is maximal (Dudin et al., 2015), showed a 0.7-fold lower expression from the ura4 than the fus1 locus, independently of auxotrophy (Fig. 2E). Thus, the genomic context influences the expression of the inserted 1kb-fus1 promoter and Fus1 expression levels matter in fusion.
To further probe how expression levels influence cell fusion, we constructed a set of strains with varied Fus1 expression levels in the more sensitized leu1− background. First, we inserted the Fus1N-forminC constructs at the ura4 and fus1 loci (Fig. 2Aiv). This yielded a 1.3-fold increased expression, as measured on Fus1N-Fus1C, compared to insertion at the fus1 locus alone (Fig. 2E), but had no significant effect on fusion efficiency (Fig. 2A,F). We then expressed full-length Fus1 under ste11 and pak2 mating-specific promoters at the ura4 locus in fus1Δ cells (Fig. 2G), which, respectively, gave similar and 2.1-fold higher expression than expression from the endogenous locus (Fig. 2E). Unexpectedly, expression from the ste11 promoter did not allow for a fully successful fusion efficiency, although it was superior to that observed upon lower-level expression under the fus1 promoter at the same genomic locus (Fig. 2H). We hypothesize that lower levels are expressed by cell pairs that fail to fuse and that our measures overestimate the expression level because – to ensure quantification is done at the same functional timepoint – we only quantified fluorescence in successfully fusing pairs. Expression from the pak2 promoter allowed for a much better fusion efficiency just slightly, but significantly, inferior to the endogenous and the dually expressed Fus1 (94%, Fig. 2H). These results indicate that Fus1 function in cell fusion is more robust to increase than reduction in levels of Fus1 protein, and suggest that native Fus1 levels are close to those minimally required for function.
To control for these variables in all subsequent experiments, we systematically used leu1− strains with constructs inserted at the native fus1 locus.
The actin assembly properties of Fus1 are tailored to its function
The Fus1N-forminC chimera experiments described above indicate an important role of the formin C-terminal half in achieving cell fusion. The formin C-terminal halves contain FH1 and FH2 domains that are required for actin assembly but also a C-terminal regulatory region. In Cdc12, the C-terminal extension is long and contains an oligomerization domain (Bohnert et al., 2013a). In For3, the C-terminal tail bears the diaphanous-autoregulatory domain (DAD) region for autoinhibition (Martin et al., 2007). The C-terminal regulatory region of Fus1 is short and does not display any detectable DAD domains. To test whether this region of Fus1 is important for cell fusion, we constructed the mutant form Fus1ΔCter, which lacks the C-terminal regulatory extension (Fig. 3 and Fig. S1). Whereas Fus1ΔCter localized to the region of contact between the two cells, it distributed more widely along the contact zone than full-length Fus1 (Fig. 3A-C). However, Fus1ΔCter did not cause a significant increase in fusion time (Fig. 3D), had only minor effects on fusion efficiency (Fig. 3A,E) and showed very mild post-fusion morphogenetic phenotypes (Fig. 3A, arrowheads). Because the reduction in fusion efficiency of fus1ΔCter cells is mild but replacement of the entire Fus1C with either For3C or Cdc12C leads to dramatic loss of function in the same conditions, this suggests specific aspects of Fus1 FH1 and FH2 domains are crucial for cell–cell fusion.
Fig. 3.

Deletion of Fus1 C-terminal tail leads to spreading of Fus1 localization but has only minor impact on fusion efficiency. (A) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of strains expressing sfGFP-tagged Fus1 or Fus1ΔCter from the endogenous fus1 locus. Fus1ΔCter is cut just after the FH2 domain. Black arrowheads point to cell wall remnants in fused cells. (B) Profiles of Fus1-sfGFP bleach-corrected fluorescence intensities perpendicular to the cell pair long axis at fusion time in strains as in A. P-values are calculated at the curve maximum. (C) Boxplot of the width at half maximum of the fluorescence profiles shown in B. (D) Boxplot of fusion times in strains as in A. (E) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in A. All P-values are relative to WT. Bars are 5 µm.
To examine the specificity in the actin assembly properties of Fus1 FH1-FH2 domains to build the fusion focus, we constructed a set of formin chimeras in which Fus1 N and C-terminal regulatory regions remained constant, and only the FH1 and FH2 domains were exchanged. We built upon the extensive in vitro characterization of Fus1, Cdc12 and For3 FH1-FH2 fragments (Scott et al., 2011), which allowed us to control for actin assembly parameters. We first assessed a set of constructs where we kept the FH2 domain of Fus1 and varied the FH1 domain to increase Fus1 elongation speed, while keeping the other actin assembly properties constant (Fig. 4A and Fig. S1). These constructs localized to the fusion site normally (Fig. 4B). Doubling the Fus1 FH1 domain or replacing it with half of the Cdc12 FH1 domain has previously been shown to double actin filament elongation rate in vitro, whereas replacing it with two copies of Cdc12 FH1 was shown to triple it (Scott et al., 2011). This increase in elongation speed was reflected in vivo by a larger actin fusion focus, as labeled by Lifeact, which associates with F-actin, or tropomyosin mNeonGreen-Cdc8 (mNG-Cdc8), a highly sensitive probe that associates specifically with all formin-assembled actin filaments and allows to distinguish the fusion focus linear filaments from those assembled by Arp2/3 at neighboring actin patches (Christensen et al., 2017; Hatano et al., 2022 preprint) (Fig. 4C,F). These findings agree with a higher actin filament assembly capacity for the faster formin chimeras. Interestingly, although none of the constructs significantly reduced fusion efficiency at 24 h post starvation (Fig. 4B,D), they induced a delay in the fusion process. Indeed, fusion duration, measured from fusion focus formation to cytosolic mixing, showed an apparent correlation with the formin elongation speed (Fig. 4E). We conclude that increasing Fus1 elongation speed is detrimental to its function.
Fig. 4.

An increase in Fus1 actin elongation rate delays cell fusion. (A) Scheme of the constructs used in the figure. All constructs were constructed seamlessly and integrated at the endogenous fus1 locus. As they keep their N- and C-terminal regulatory parts and their FH2 domain constant, they are referred to only by their variable FH1 domain. Indicative actin filament elongation rates as measured in vitro on FH1-FH2 fragments by Scott et al. (2011) are shown on the right, as multiple of Fus1 elongation rate. (B) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of homothallic strains expressing formin chimeras as in A, C-terminally tagged with sfGFP. (C) Merge and GFP fluorescence images ∼8 h post starvation of Myo52-tdTomato (magenta) and either (left) LifeAct-sfGFP or (right) mNG-Cdc8 in homothallic strains expressing untagged formin chimeras with Fus1FH1 or Cdc122FH1. (D) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in B. (E) Boxplot of fusion times in strains as in B. (F) Boxplot of mNG-Cdc8 intensity normalized to the WT at the cell contact site at fusion time in strains as in C. All P-values are relative to WT. Bars are 5 µm.
We then set to examine chimeras with different FH2 domains (Fig. 5A and Fig. S1). Using the Cdc12 FH2 domain and varying FH1 domains, we were able to express formin constructs with a wider range of in vitro measured elongation rates than those obtained with Fus1 FH2. In vitro studies have shown that Cdc12 has a nucleation rate very close to that of Fus1 (Scott et al., 2011), suggesting it is adequate in these constructs and permitting us to test for an optimum elongation speed in fusion (Fig. 5A and Fig. S1). The Cdc12FH1-Cdc12FH2 chimera, with twice the Fus1 elongation speed, supported fusion in only 33% of cell pairs (Fig. 5B,C). Importantly, reducing the elongation speed to that of native Fus1 by halving Cdc12 FH1 domain increased the fusion efficiency to 78% of cell pairs (Fig. 5B,C). By contrast, further decrease (Fus1FH1-Cdc12FH2) or increase (Cdc122FH1-Cdc12FH2) of the elongation rate compromised cell fusion efficiency to 17% or 15%, respectively, demonstrating an optimum at the native elongation rate of Fus1. The architecture of the fusion focus reflected the elongation rates measured in vitro. Indeed, all constructs localized to the site of cell–cell contact (Fig. 5B) but the worst elongator was barely able to form an actin aster, showing minimal tropomyosin localization (Fig. 5D,E). In contrast, the faster elongators formed a F-actin-rich focus from which emanated prominent mNG-Cdc8-decorated actin cables, an organization not observed with native Fus1 (compare Fig. 5D,E, with Fig. 4C). Quantification of tropomyosin mNG-Cdc8 signal intensity further showed that the amount of actin incorporated at the cell contact site largely correlates with the in vitro elongation rate (Fig. 5E,F). These results demonstrate that the native Fus1 elongation rate is best adapted to assemble the fusion focus.
Fig. 5.

Low elongation and nucleation rates are detrimental for cell fusion and Fus1 contains an additional property within its FH2 domain absent from Cdc12 FH2 domain. (A) Scheme of the constructs used in the figure. All constructs were constructed seamlessly and integrated at the endogenous fus1 locus. As they keep their N- and C-terminal regulatory parts constant, they are referred to only by their variable FH1 and FH2 domains. Indicative actin filament elongation rates as measured in vitro on FH1-FH2 fragments by Scott et al. (2011) are shown on the right, as multiple of Fus1 elongation rate. Note that For3 exhibits ∼80-fold lower nucleation rates than Fus1 (not shown). (B) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of homothallic strains expressing the formin chimeras shown in A, C-terminally tagged with sfGFP. (C) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in B. P-values are relative to the most efficient chimera, Cdc12½FH1-Cdc12FH2. (D) Merge and GFP fluorescence images ∼8 h post starvation of Myo52-tdTomato and LifeAct-sfGFP in homothallic strains expressing untagged formin chimeras shown in A. (E) Merge and GFP fluorescence images ∼8 h post starvation of Myo52-tdTomato and mNG-Cdc8 in homothallic strains expressing untagged formin chimeras shown in A. (F) Boxplot of mNG-Cdc8 intensity normalized to the WT (shown in Fig. 4E) at the cell contact site at fusion time in strains as in E. P-values are relative to WT. Bars are 5 µm.
In vitro studies showed that Fus1 is an efficient nucleator (Scott et al., 2011). To test for the importance of actin filament nucleation, we used the For3 FH2 domain that, in vitro, exhibits a nucleation efficiency over 80-fold lower than Fus1. A chimera with For3 FH1-FH2 domains (Fig. 5A) performed poorly, with barely more than 10% of the mating pairs fusing (Fig. 5B,C). For3 elongates actin filaments at about twice the rate of Fus1, and we observed a perturbed actin focus with stronger LifeAct and tropomyosin signal (Fig. 5D-F). This stronger signal suggests that the For3 FH1-FH2 chimera either efficiently elongates pre-existing filaments or is a better nucleator in vivo than in vitro. Some of the inability of For3 FH1-FH2 to support cell fusion might stem from its faster elongation rate. However, compared to the constructs with matched elongation speed described above (Fus12FH1-Fus1FH2 and Cdc121/2FH1-Fus1FH2 in Fig. 4, or Cdc12FH1-Cdc12FH2 in Fig. 5), the poorer performance of the For3 chimera suggests that a high nucleation rate is beneficial for the assembly of the fusion focus.
Together, these results show that known changes in the in vitro actin assembly properties of Fus1 induce clear alterations in the architecture of the fusion focus in vivo and an associated loss of function. This demonstrates that the native actin assembly properties of Fus1, i.e. a relatively low elongation rate (5 subunits s−1 µM−1) and high nucleation rate (one filament per two dimers), are tailored to its function in fusion focus assembly.
Identification of a mutation blocking an additional function in Fus1 FH2 domain
We noticed that, even for matching elongation rates, chimeras containing the Fus1 FH2 domain systematically performed better than those containing the For3 or Cdc12 FH2 domain. Whereas for the For3 chimera this can be rationalized to the importance of efficient nucleation, comparisons with Cdc12 FH2 chimeras – where both elongation and nucleation rates are matched – suggested to us that Fus1 FH2 contains a specific property that is crucial for cell fusion. The clearest demonstration of this lies in the comparison of Cdc122FH1-Fus1FH2 with Cdc122FH1-Cdc12FH2, as both constructs differ only through their FH2 domains but perform at 97% and 15%, respectively (Figs 4D and 5C, respectively). The mNG-Cdc8 labeling of linear actin structure also reveals dramatic differences between constructs bearing the Fus1 or Cdc12 FH2 domain (compare Fig. 4C and Fig. 5E).
With the objective to identify amino acids required for Fus1 FH2 specific property, we first tested whether it was conserved in Fus1 orthologues. To this aim, we replaced the S. pombe Fus1 FH2 domain with the corresponding FH2 domains in Schizosaccharomyces octosporus (S. octosporus) and the more-distant Schizosaccharomyces japonicus (S. japonicus). We performed this replacement in two distinct construct backgrounds (Figs S2A and S1). First, we used the construct carrying Cdc122FH1 because it is the only FH1 domain to yield identical elongation rates in combination with either Fus1 FH2 or Cdc12 FH2 domain, allowing to control for this variable. As noted above, this background also resulted in the most striking difference in cell fusion efficiency between Cdc12 and Fus1 FH2 domains, affording better sensitivity to our assay. Second, we used the S. pombe WT Fus1 background as a control to ensure that FH2 exchanges as well as further mutations (described below) would not disturb other actin assembly properties. Both the S. octosporus and S. japonicus FH2 domains were able to functionally replace S. pombe FH2 in combination with either Fus1FH1 or Cdc122FH2, and localizations and fusion efficiencies were equivalent to those conferred by the S. pombe Fus1 FH2 domain (Fig. S2B,C). Thus, the characteristic we are looking for must be conserved within the Schizosaccharomyces clade.
We used sequence alignments of Schizosaccharomyces Fus1 and Cdc12 FH2 domains, and homology modeling of S. pombe Fus1 and Cdc12 FH2 domains, to identify residues conserved in Fus1 but different in Cdc12 and regions with local charge difference (Fig. 6A and Fig. S2D). The homology models helped to eliminate residues likely to affect actin binding, homodimerization or overall formin structure (Fig. 6A). Our comparisons identified two amino acid residues, L959 and R1054, and three poorly conserved regions located in flexible loops, 935KEYTG939, 1006EEVMEV1011 and 1182NHK1184 (numbering and residues refer to S. pombe Fus1).
Fig. 6.

The R1054E mutation in Fus1 FH2 domain partly recapitulates the cell fusion deficiencies observed with Cdc12 FH2. (A) (Left) Overlay of Fus1 and Cdc12 FH2 domain structures, which were constructed by homology modeling with murine FMNL3. (Right) Dimeric Fus1 FH2 homology model with mutated residues shown in turquoise. Residues were selected in regions that were unlikely to disrupt the formin FH2 dimerization or actin binding, were surface-exposed and had a charge difference between Cdc12 and Fus1 or were in variable loops. (B) Scheme of the constructs used in the figure. All constructs were constructed seamlessly and integrated at the endogenous fus1 locus. The set of 5 mutations as shown in A were introduced in (left) Fus1 or (right) chimeras with Cdc122FH1. The latter shows identical elongation rate when combined with either Fus1FH2 or Cdc12FH2 [as indicated on the right from measurements in vitro on FH1-FH2 fragments by Scott et al. (2011)]. (C) DIC images ∼16 h post starvation and GFP fluorescence images ∼8 h post starvation of homothallic strains expressing either (left) mutant Fus1-sfGFP or (right) mutant sfGFP-tagged Cdc122FH1-Fus1FH2 formin chimeras. (D) Percentage of cell pair fusion and lysis 24 h after nitrogen removal in strains as in B compared to the non-mutated controls. P-values are relative to the non-mutated Fus1 controls. Chimeras with Cdc12FH2 are shown for information but note that Fus1FH1-Cdc12FH2 (left) cannot be used for direct comparison due to its lower elongation rate. (E) Merge and GFP fluorescence images ∼8 h post starvation of Myo52-tdTomato and either LifeAct-sfGFP or mNG-Cdc8 in homothallic strains expressing the untagged mutant formins Fus1R1054E (left) or Cdc122FH1-Fus1R1054EFH2 (right). Note the extended actin network, compared to non-mutated equivalents in Fig. 4C and Fig. 5E. Bars are 5 µm.
We mutated these residues in the Fus1FH1 and Cdc122FH1 construct backgrounds described above, replacing them with the corresponding amino acids found in the S. pombe Cdc12 sequence (Fig. 6B and Fig. S1). All formins with mutant Fus1 FH2 localized correctly to the site of cell–cell contact (Fig. 6C). In the WT background, none of these mutations had any significant impact on fusion efficiency (Fig. 6C,D, left), suggesting preservation of dimerization, actin binding and assembly function. In the Cdc122FH1 background, most mutations did not have a significant effect on fusion efficiencies. However, the R1054E mutation in Fus1 FH2 showed a strikingly similar phenotype to that of Cdc12 FH2 (Fig. 6C,D, right), i.e. severely reduced fusion efficiency and largely increased cell lysis. These cells also showed altered fusion focus architecture, very similar to that caused by cdc122FH1-cdc12FH2, with cable-like structures originating from the fusion focus (Fig. 6E, right), which are absent from the cdc122FH1-fus1FH2 strain (see Fig. 4C). The R1054E mutation in Fus1 FH2 also caused the formation of visible, although weaker, mN-Cdc8-labeled cables in an otherwise WT background (Fig. 6E, left). This suggests that the Fus1R1054E FH2 has lost the Fus1-specific property and behaves similar to Cdc12 FH2, both in terms of fusion focus architecture and fusion/lysis efficiency.
Encouraged by these results, we introduced the complementary mutation in Cdc12 FH2, replacing E1168 by the R found in Fus1 (Figs S3 and S1). This mutation had no marked effect on formin localization or fusion focus architecture of cdc122FH1-cdc12FH2-expressing cells (Fig. S3A,B) but led to a slight rescue of the fusion efficiency (Fig. S3A,C), albeit lower than that in WT. This is not very surprising and suggests that Cdc12 requires additional mutations – perhaps some of the changes tested above that caused mild phenotypes – to acquire the Fus1-specfic property. Put together, these results suggest that R1054 is one of the amino acids needed to render Fus1 so well equipped to support cell fusion.
The R1054E mutation does not alter the biochemical properties of Fus1 FH2 on actin in vitro
One hypothesis to explain our in vivo results is that a particular biochemical property is significantly altered by the R1054E mutation. The reported actin bundling activity of the Fus1 actin assembly domains (Scott et al., 2011) appeared as a possible Fus1-specific characteristic not shared with Cdc12 FH2 domain. We, therefore, expressed and purified Cdc122FH1-Fus1FH2 protein constructs that do or do not contain the R1054E mutation (Fig. 7A and Fig. S4C), and tested their actin filament bundling, assembly and disassembly activities in vitro (Fig. 7 and Fig. S4). Although we were unable to reproduce the bundling activity to the extent described for Fus1 FH1-FH2 with low-speed sedimentation or fluorescence microscopy assays (Scott et al., 2011), both Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2 resulted in significantly more pelleted actin than the control actin-only samples, indicating moderate bundling activity (Fig. 7B,C). However, there was no significant difference in the bundling activity between the two Cdc122FH1-Fus1FH2 constructs.
Fig. 7.

The R1054E mutation does not alter the biochemical properties of Cdc122FH1-Fus1FH2 in vitro. (A) Cdc122FH1-Fus1FH2 constructs with and without the R1054E mutation utilized for in vitro actin biochemistry assays. (B-C) Low-speed sedimentation of 3 µM preassembled Mg-ATP actin filaments incubated with 1.5 µM of Cdc122FH1-Fus1FH2, Cdc122FH1-Fus1R1054EFH2, or the F-actin bundler Fim1. Samples were spun at 10,000 g for 20 min. (B) Coomassie Blue-stained gel of pellets. (C) Graph of the density of actin in the pellets of the assay in B. Error bars indicate standard errors. ** indicates P<0.01; ***P<0.001; ****P<0.0001. (D-E) Spontaneous assembly of 2.5 µM Mg-ATP-actin (10% pyrene-labeled) in the absence and presence of increasing concentrations of Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2. (D) Representative curves in the absence (gray) and presence of 25 nM formin (light blue and light green) or 100 nM formin (dark blue and dark green). (E) Representative graph from one of three independent trials of the dependence of the initial assembly slopes (linear phase) on formin concentration. Average effective concentration at half maximal assembly rate (EC50) is reported for each formin±s.e. (P=0.18). (F-G) Barbed end disassembly of preassembled actin filaments (50% pyrene-labeled) upon dilution to 0.1 µM in the absence and presence of increasing concentrations of Cdc122FH1Fus1FH2 or Cdc122FH1-Fus1R1054EFH2. (F) Representative curves in the absence (gray) and presence of 0.5 nM formin (light blue and light green) or 50 nM formin (dark blue and dark green). (G) Representative graph from one of three independent trials of the dependence of the initial disassembly slopes on formin concentration. Average dissociation constant (KD) for the barbed end is reported for each formin±s.e. (P=0.1).
We next tested whether these constructs differed in their actin assembly properties, by performing spontaneous pyrene actin assembly assays in the presence of varying concentrations of the formin (Fig. 7D). By plotting the dependence of the initial actin assembly rates on the concentration of formin, we calculated the concentrations at which the formins achieve half-maximum activity (EC50) (Fig. 7E). For the wild-type Fus1 chimera, the average EC50 from three trials was 33.9±3.9 nM, and for the mutant Fus1 chimera, the average EC50 from three trials was 52.7±10.9 nM (n.s., P=0.18) (Fig. 7E), indicating that the overall actin assembly properties of these constructs are similar.
To assess the affinity of these formins for actin filament barbed ends, we performed pyrene actin disassembly assays in the presence of varying concentrations of formin (Fig. 7F). Pre-assembled actin filaments were diluted to the critical concentration of 0.1 μM, and the rate of barbed end disassembly was followed. Both Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2 associated with and reduced disassembly from the barbed end to similar extents (Fig. 7F). Furthermore, we plotted the initial disassembly rates against the concentrations of formin, fitted the data by using a nonlinear least squares regression analysis and calculated the dissociation constant (KD) (Fig. 7G). We calculated the KD of Cdc122FH1-Fus1FH2 as 1.76±0.33 nM, and that of Cdc122FH1-Fus1R1054EFH2 as 0.98±0.16 nM (n.s., P=0.1), revealing that wild-type and mutant formin chimeras have similar affinities for actin filament barbed ends.
Last, we measured actin filament elongation rates by using TIRF microscopy to directly observe individual formin-assembled filaments (Fig. S4A,B). With both Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2 two populations of filaments were produced in the absence of profilin, i.e. control filaments that elongate at ∼10 to 13 subunits s−1 and formin-dependent filaments that elongate significantly slower. Cdc122FH1-Fus1FH2-associated filaments elongate at 1.1 subunits s−1, which is consistent with previous results (Scott et al., 2011) (Fig. S4B). Cdc122FH1-Fus1R1054EFH2-associated filaments elongate at 1.0 subunits s−1. In the presence of profilin Cdc3 the elongation rates of control and formin-associated filaments were indistinguishable but, again, similar for both Cdc122FH1-Fus1FH2 (10.2 subunits s−1) and Cdc122FH1-Fus1R1054EFH2 (12.8 subunits s−1) (Fig. S4B). Furthermore, given that both Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2-dependent filaments elongate at almost identical rates (Fig. S4) and have similar ‘bulk’ pyrene actin assembly rates (Fig. 7), their actin filament nucleation rates must be similar. These in vitro experiments provide strong confirmation that the reduced functionality of the R1054E mutation in vivo is due to changes other than the intrinsic ability to nucleate, elongate or bundle actin filaments.
DISCUSSION
How cells can assemble functionally diverse actin structures from a common cytosolic actin pool is a complex question. Actin nucleators confer part of the identity of a structure. Naturally, the different mode of actin nucleation of the Arp2/3 complex and formins yield vastly differing branched and linear networks. However, even formin-nucleated actin networks exist in a wide variety of sizes and shapes, which depend on the specific formin nucleator. Regulation of the formin localization and activity is one well-established way to control its time and place of action. Here, we focused on how the intrinsic actin assembly properties of formin contribute to the specificity of the assembled actin network architecture. We exploited the simplicity of the fission yeast formin assortment, in which only three formins assemble a specific actin network each, and the prior knowledge on their in vitro actin assembly characteristics (Scott et al., 2011) to systematically test the requirement of Fus1 FH1-FH2 domains in assembly of the fusion focus. Our results show that (1) Fus1 actin assembly properties are tailored to its function as decrease and increase in elongation rates or reduction in nucleation rates are detrimental, and (2) Fus1 FH2 also comprises an additional specific property that helps confer the fusion focus its specific architecture.
Fus1 C-terminal region is largely dispensable for cell fusion
The C-terminal tail of formins, like their N-terminal half, is highly variable and often contains determinants that regulate the formin activity. In Diaphanous-related formins, this region harbors the DAD, which binds the N-terminal diaphanous inhibitory domain (DID) to keep the protein in an inactive state (Alberts, 2001). In S. pombe, For3 is regulated by DAD–DID interaction (Martin et al., 2007) but the Fus1 C-terminal sequence does not show a recognizable DAD motif. The C-terminus of budding yeast formins interact with Aip5 and Bud6, which promote nucleation (Graziano et al., 2011; Xie et al., 2022); the For3 C-terminus also binds Bud6 (Martin et al., 2007) and the Cdc12 C-terminus contains an oligomerization domain that promotes actin bundling (Bohnert et al., 2013a). In our initial experiments (Fig. 1), we found that this Cdc12 tail boosts Cdc12 FH1-FH2 performance in replacement of Fus1. However, it is unlikely that the Fus1 C-terminus has a similar function. Indeed, by truncating it, we found that this region is largely dispensable for Fus1 function, as the Fus1ΔCter exhibits cell fusion activity that is indistinguishable from wild type. In fact, Fus1 does self-assemble but this is mediated by a disordered region in the Fus1 N-terminus, which is essential for function (Billault-Chaumartin et al., 2022 preprint). The broader localization of Fus1ΔCter is surprising, as changes in Fus1 distribution typically correlate with changes in cell fusion/lysis activity (Billault-Chaumartin and Martin, 2019; Billault-Chaumartin et al., 2022 preprint; Dudin et al., 2015), and suggests that the Fus1 C-terminus contributes weakly to the regulation of Fus1.
Fus1 actin assembly properties are tailored for the assembly of the fusion focus
With 5 subunits s−1 µM−1 in vitro, Fus1 exhibits a relatively low elongation rate (Scott et al., 2011). Our experiments showed that this rate is optimal for fusion focus architecture. Indeed, increasing it by doubling the Fus1 FH1 domain or providing that of Cdc12 – as in the chimeras comprising the Fus1FH2 domain (Fig. 4) – led to the formation of a larger fusion focus and slowed down the fusion process proportionally to the rate of elongation. Similarly, we found that in chimeras with a Cdc12FH2 domain (Fig. 5), the construct whose elongation rate was matched to that of Fus1 in vitro provided the highest fusion efficiency. By contrast, Cdc12FH2-based constructs with faster elongation rates formed actin structures with an increase in linear actin and reduced fusion efficiency, again in a manner proportional to elongation rate. These fast-elongating formin chimeras also showed a high percentage of lysed mating pairs. In the context of cells attempting cell fusion, we interpret this as inappropriate cell wall digestion due to loss of spatial precision. The Cdc12FH2-based chimeras also allowed us to test the function of a construct with a reduced elongation rate when using the Fus1FH1 domain. This slow-elongating formin also did not support function in cell fusion but showed very little cell lysis, probably because it leads to filaments that are too short to produce a functional actin structure, unable to capture myosin V-associated vesicles for cell wall digestion. Collectively, these experiments demonstrate that the Fus1 endogenous elongation rate, which for a cellular concentration of actin of ∼20 µM (Wu and Pollard, 2005) corresponds to a filament elongation of ∼270 nm s−1, is tailored to its function in fusion focus assembly.
Fus1 is a very potent nucleator, with every other dimer able to initiate a new actin filament in vitro (Scott et al., 2011). The observation that For3 FH1-FH2, which has an 80-fold lower nucleation rate in vitro, is unable to replace Fus1 and performs worse than Cdc12 with matched elongation rate (compare, i.e. For3FH1-For3FH2 and Cdc12FH1-Cdc12FH2 in Fig. 5) suggests that high nucleation rate is beneficial for fusion focus assembly. However, we noticed that the For3FH1-For3FH2 chimera efficiently assembled F-actin at the cell–cell contact site, yielding a structure with long actin cables. Owing to the contribution of additional factors – e.g. budding yeast nucleation promoting factor Bud6 interacts with the DAD region of formin Bni1 to enhance nucleation by recruiting actin monomers (Graziano et al., 2011) – we cannot exclude that the nucleation rate is higher in vivo than in vitro, but the For3 DAD region that also binds Bud6 (Martin et al., 2007) is absent in these constructs. The For3 chimera might also trigger this actin organization by elongating pre-existing filaments, e.g. at actin patches. The importance of Fus1 nucleation rate should also be viewed in relation with the importance of Fus1 expression levels, reduction of which reduces fusion efficiency, at least in the leu1− background. Indeed, combination of both the nucleation rate and the number of formin molecules will dictate the number of filaments that assemble at the fusion focus. Together, these experiments suggest that high nucleation capacity is required for formation of the fusion focus.
In this study, we have not specifically controlled for variations in formin dissociation constants from the filament barbed end, which have also been measured in vitro. This is because, whereas the rates were measured on ‘purified reactions’ containing only actin and formins, actin structures are regulated at much faster rates in vivo by other actin-interacting proteins that either sever the actin filaments or actively unload the formin (Shekhar et al., 2016). Indeed, Fus1 remains bound to an actin filament for over 25 min and assembles a >400 µm-long filament according to the dissociation constant measured in vitro (Scott et al., 2011), but Fus1-assembled filaments in vivo are at most a few micrometers long. In particular, this difference is due to capping protein, which competes with formins for the barbed end of the actin filament by forming a ternary complex that decreases the affinity of both proteins for the barbed end (Bombardier et al., 2015; Shekhar et al., 2015), and which we have shown competes with Fus1 (Billault-Chaumartin and Martin, 2019).
During our experiments, when initially observing inconsistent phenotypes between strains that express the same construct, we discovered that leucine auxotrophy influences cell fusion. Indeed, strains prototroph for leucine fuse considerably faster and better than their leu1− counterparts. The underlying reason is currently unknown and awaits further investigation, but these findings underscore the importance of using identical strain backgrounds, especially during mating.
The Fus1 FH2 domain supports an additional function important for cell fusion
The poorer performance of Cdc12 FH2-based constructs for matched nucleation and elongation rates, suggested to us that the FH2 domain of Fus1 requires an additional, not-yet attributed property conserved within the Schizosaccharomyces clade, to be fully competent in assembling the fusion focus. The absence of this function in Cdc12 or For3 correlates with the formation of long actin cables emanating from the region of contact between the two cells. Through sequence alignment, homology modeling and mutational analysis, we were able to identify R1054 in Fus1 as being crucial for this function. The corresponding residue is an aspartic acid in both Cdc12 and For3. The Fus1 FH2 R1054E mutation has no effect in vitro on either elongation or nucleation rates, consistent with the position of this residue on the external face of the FH2 coiled-coil region, not predicted to contact actin. However, it produces phenotypes similar to those observed with Cdc12 or For3 FH2 domains, namely the presence of long cables emanating from the fusion focus. In the context of a fast-elongating construct (with Cdc122FH1), it is also unable to support cell fusion and, instead, leads to strong cell lysis phenotypes. However, in an otherwise unmodified Fus1, this mutation does not decrease fusion efficiency. Collectively, these data suggest that R1054 contributes to the functional specificity of Fus1 FH2, independently of nucleation and elongation rates, but that additional mutations are necessary to completely abrogate this specific Fus1 role.
One important question is what this role might be. We initially thought that bundling activity would be a good candidate. However, the R1054E mutation did not alter the weak bundling activity of the Cdc122FH1-Fus1FH2 fragment in vitro, indicating that the mutation does not change the intrinsic bundling activity of the Fus1 FH2 domain (Scott et al., 2011). Two arguments support the idea that the mutation may still reduce filament bundling or crosslinking in vivo. First, our initial chimeras using the entire C-terminal half of formins (Fig. 1), indicate that, under the conditions tested, the C-terminal tail of Cdc12, which contains an oligomerization domain that promotes actin bundling (Bohnert et al., 2013a), enhances Cdc12 FH1-FH2 function. Second, the extended cables systematically observed for all constructs not containing wild-type Fus1 FH2 are consistent with a lack of actin filament crosslinking, yielding filaments able to extend out of the fusion focus nucleation zone. While further work will be required to test this hypothesis, a likely scenario, given the in vitro results, is that any bundling activity is indirect and requires additional proteins, potentially binding Fus1 on an interface containing R1054 and absent in our in vitro reactions.
The finding that formins are tailored to their cellular function makes intuitive sense in the light of evolution, where selection of the most-adapted parameters will have been refined through generations. However, a recent review concluding with open questions on formins stated that “perhaps most challenging of all: how does possessing particular actin polymerization activities render a formin isoform most suitable to fulfil its cellular role?” (Courtemanche, 2018). Our work, along with other recent work on S. pombe Cdc12 (Homa et al., 2021) and Physcomitrella patens formin For2 (Vidali et al., 2009), contributes to establishing how the formin actin assembly activities are customized to build specific, functional actin structures.
MATERIALS AND METHODS
Strain construction
Strains were constructed using standard genetic manipulation of S. pombe, either by tetrad dissection or transformation, and can be found in Table S1. Oligonucleotides and plasmids used can be found in Tables S2 and S3, respectively, with details on how the plasmids were constructed.
The tags myo52-tdTomato:natMX and fus1-sfGFP:kanMX were constructed by PCR-based gene targeting of a fragment from a template pFA6a plasmid containing the appropriate tag and resistance cassette, amplified with primers carrying 5′ extensions corresponding to the last 78 coding nucleotides of the ORF and the first 78 nucleotides of the 3′UTR, which was transformed and integrated in the genome by homologous recombination, as previously described (Bähler et al., 1998). Similarly, fus1Δ::hphMX was constructed by PCR-based gene targeting of a fragment from a template pFA6a plasmid containing the appropriate resistance cassette, amplified with primers carrying 5′ extensions corresponding to the last 78 nucleotides of the 5′UTR and the first 78 nucleotides of the 3′UTR, which was transformed and integrated in the genome by homologous recombination. fus1Δ1278-1372-sfGFP:kanMX was constructed by PCR-based gene targeting of a fragment from a template pFA6a plasmid with primers carrying 5′ extensions corresponding to the 78 nucleotides upstream of the deleted region and the first 78 nucleotides of the 3′UTR, which was transformed and integrated in the genome by homologous recombination.
Construction of the strains expressing formin constructs from the fus1 promoter at the ura4 locus as a multicopy integration (ura4-294:pfus1:fus1-sfGFP:ura4+, ura4-294:pfus1:cdc12-sfGFP:ura4+, ura4-294:pfus1:for3-sfGFP:ura4+ in Fig. 1A-C) was done by homologous recombination of a transformed ura4EndORF-ura43′UTR-pfus1-ForminConstruct-sfGFP-ura4StartORF-ura45′UTR fragment, obtained from StuI digestion of a pRIP based plasmid (pSM1656, pSM1658, pSM1657). Such recombination recreates a new integration site that was shown to be unstable and can lead to multiple insertion (Vještica et al., 2020). This is why we switched to single integration vectors for the rest of the study.
Construction of the strains expressing Fus1N-Fus1C, Fus1N-Cdc12C, Fus1N-For3C and Fus1N-Cdc12CΔC chimeras from the fus1 promoter at the ura4 locus as a single integration (ura4+:pfus1:fus1N1-792-fus1C793-1372-sfGFP, ura4+:pfus1:fus1N1-792-cdc12C888-1841-sfGFP, ura4+:pfus1:fus1N1-792-for3C715-1461-sfGFP, ura4+:pfus1:fus1N1-792-cdc12CΔolig888-1451-sfGFP in Figs 1D-F and 2) was done by homologous recombination of a transformed ura45′UTR-ura4ORF-ura43′UTR-pfus1-ForminConstruct-sfGFP-ura43″ fragment, obtained from PmeI digestion of a pUra4PmeI based plasmid (pSM2594, pSM2595, pSM2604 and pSM2596, respectively). This leads to a stable single integration at the ura4 locus (Vještica et al., 2020).
Construction of the strains expressing fus1 under either ste11 or pak2 promoter at the ura4 locus as a single integration (ura4+:pste11:fus1-sfGFP and ura4+:ppak2:fus1-sfGFP in Fig. 2) was done by homologous recombination of a transformed ura45′UTR-ura4ORF-ura43′UTR-pste11 or pak2-Fus1-sfGFP-ura43″ fragment, obtained from PmeI digestion of a pUra4PmeI based plasmid (pSM2828 and pSM2829, respectively).
Construction of the strains expressing tagged formin constructs from the endogenous locus (fus1N1-792-fus1C793-1372-sfGFP:kanMX, fus1N1-792-cdc12C888-1841-sfGFP:kanMX, fus1N1-792-for3C715-1461-sfGFP:kanMX, fus11-868-fus1792-1372-sfGFP:kanMX, fus11-791-cdc12928-972-fus1869-1372-sfGFP:kanMX, fus11-791-cdc12882-972-cdc12882-972-fus1869-1372-sfGFP:kanMX, fus11-791-for3718-1265-fus11278-1372-sfGFP:kanMX, fus11-868-cdc12973-1390-fus11278-1372-sfGFP:kanMX, fus11-791-cdc12928-1390-fus11278-1372-sfGFP:kanMX, fus11-791-cdc12882-1390-fus11278-1372-sfGFP:kanMX, fus11-791-cdc12882-972-cdc12882-1390-fus11278-1372-sfGFP:kanMX, fus11-868-Sjfus1908-1317-fus11278-1372-sfGFP:kanMX, fus11-868-Sofus1857-1265-fus11278-1372-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-Sjfus1908-1317-fus11278-1372-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-Sofus1857-1265-fus11278-1372-sfGFP:kanMX, fus1(KEYTG935-939CARTD)-sfGFP:kanMX, fus1(L959 K)-sfGFP:kanMX, fus1(EEVMEV1006-1011NGGDLVNS)-sfGFP:kanMX, fus1(R1054E)-sfGFP:kanMX, fus1(NHK1182-1184DPT)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(KEYTG935-939CARTD)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(L959 K)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(EEVMEV1006-1011NGGDLVNS)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(R1054E)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(NHK1182-1184DPT)-sfGFP:kanMX, fus11-792-cdc12882-972-cdc12882-1390(E1168R)-fus11278-1372-sfGFP:kanMX in Figs 2-6 and Fig. S2-S3) was done by homologous recombination of a transformed fus15′UTR-ForminConstruct-sfGFP-kanMX-fus13′UTR fragment, obtained from a gel purified, SalI and SacII or AatII and SacII (Cdc12 FH2 contains an endogenous SalI site) digested pFA6a based plasmid (pSM2690, pSM2691, pSM2692, pSM2621, pSM2702, pSM2700, pSM2631, pSM2701, pSM2632, pSM2622, pSM2699, pSM2855, pSM2857, pSM2854, pSM2856, pSM2849, pSM2850, pSM2851, pSM2852, pSM2853, pSM2844, pSM2845, pSM2846, pSM2847, pSM2848 and pSM2924, respectively).
Similarly, construction of the strains expressing untagged formin constructs from the endogenous locus (fus1:kanMX, fus11-791-cdc12882-972-cdc12882-972-fus1869-1372:kanMX, fus11-791-for3718-1265-fus11278-1372:kanMX, fus11-868-cdc12973-1390-fus11278-1372:kanMX, fus11-791-cdc12928-1390-fus11278-1372:kanMX, fus11-792-cdc12882-972-cdc12882-1390-fus11278-1372:kanMX, fus1(R1054E):kanMX, fus11-792-cdc12882-972-cdc12882-972-fus1869-1372(R1054E):kanMX, fus11-792-cdc12882-972-cdc12882-1390(E1168R-fus11278-1372):kanMX in Figs 4-6 and Fig. S3) was done by homologous recombination of a transformed fus15′UTR-ForminConstruct-kanMX-fus13′UTR fragment, obtained from a gel-purified SalI- and SacII- or AatII- and SacII-digested pFA6a-based plasmid (pSM2913, pSM2914, pSM2918, pSM2915, pSM2916, pSM2917, pSM2961, pSM2962 and pSM2963, respectively).
Constructs leu1-32:pcdc8:mNeonGreen-cdc8:termcdc8:termScADH1:leu1+ (Hatano et al., 2022 preprint), fus1Δ::LEU2+ (Petersen et al., 1998b) and ade6+:pact1:LifeAct-sfGFP:termScADH1:bsdMX (Vještica et al., 2020) trace back to the aforementioned papers, and are kind gifts from the aforementioned labs.
Growth conditions
For mating experiments, homothallic (h90) strains able to switch mating types were used, where cells were grown in liquid or agar minimum sporulation media (MSL), with or without nitrogen (+N or −N, respectively) (Egel et al., 1994; Vjestica et al., 2016).
Live imaging of S. pombe mating cells protocol was adapted from (Vjestica et al., 2016). Briefly, cells were first pre-cultured overnight in MSL+N at 25°C, then diluted to OD600=0.05 into MSL+N at 25°C for 20 h. Exponentially growing cells were then pelleted, washed in MSL−N by three rounds of centrifugation, and resuspended in MSL−N to an OD600 of 1.5. Cells were then grown 3 h at 30°C to allow mating in liquid, added on 2% agarose MSL−N pads, and sealed with VALAP. We allowed the pads to rest either for 30 min at 30°C before overnight imaging or for 21 h at 25°C for snapshot imaging 24 h post-starvation to measure fusion efficiencies.
Live cell imaging microscopy
Images presented in Figs 1-6, and Figs S2,S3 were obtained using a DeltaVision platform (Applied Precision) composed of a customized inverted microscope (IX-71; Olympus), a UPlan Apochromat 100×/1.4 NA oil objective, a camera (CoolSNAP HQ2; Photometrics or 4.2Mpx PrimeBSI sCMOS camera; Photometrics), and a color combined unit illuminator (Insight SSI 7; Social Science Insights). Images were acquired using softWoRx v4.1.2 software (Applied Precision). Images were acquired every 5 min during 9 h-15 h. To limit photobleaching, overnight videos were captured by optical axis integration (OAI) imaging of a 4.6-μm z-section, which is essentially a real-time z-sweep.
Quantification and statistical analysis of live imaging data
Percentages of cell fusion and lysis as in Figs 1C,F, 2B,C,F,H, 3E, 4D, 5C, 6D, Fig. S2C and S3C were calculated as described by Dudin et al. (2015). Briefly, 24 h post starvation, fused cell pairs, lysed pairs and the total number of cell pairs were quantified using the ImageJ Plugin ObjectJ; percentages were calculated using the following equations:
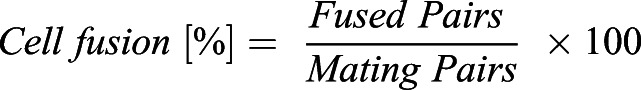 |
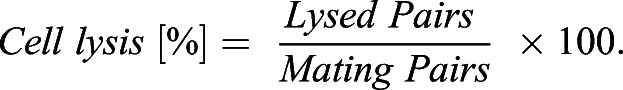 |
Fusion Times as in Figs 2D, 3D and 4E were calculated in overnight time lapse movies at 5-min intervals using the 2-dot Myo52-tdTomato stage (Dudin et al., 2015) as a marker for the beginning of the fusion process and either the entry of GFP expressed under control of the P-cell-specific pmap3 promoter into the h− partner, or the maximum intensity of the Myo52-tdTomato dot, the two of which perfectly correlate (Dudin et al., 2015), as a marker for the end of the process.
Fusion Focus intensities at fusion time as in Fig. 3B, 4F and 5F were obtained in overnight time lapse movies at 5-min intervals using either the entry of GFP into the h− partner, or the maximum intensity of the Myo52-tdTomato dot to determine the moment of fusion. At that time frame, a fluorescence profile across the fusion focus perpendicular to the long axis of the mating pair was recorded. Profiles were background-subtracted and corrected for bleaching as follows: First, the cell fluorescence intensity was recorded over time in a square of about 7×7 pixels in 12 control (non-mating) cells. These fluorescence profiles were averaged, and the mean was fitted to a double exponential as it was describing our data better (Vicente et al., 2007):
We then used this fit to correct the fluorescence profiles across the fusion focus for photobleaching. After subtracting background signal, the value at each timepoint was divided by the photo-bleaching correction signal:
 |
Corrected profiles were then averaged and directly plotted as in Fig. 3B, or further normalized to the WT and only the central point was plotted as a boxplot as in Figs 4F and 5F. Widths at Half maximum (D50) as in Fig. 3C were then calculated using these fluorescence profiles. This was done for each cell and then plotted as a boxplot.
Total fluorescence intensities as in Fig. 2E in mating pairs at fusion time were obtained using 5-min time lapse overnight movies where fusion time was assessed for each mating cell, by outlining the mating pairs and recording the mean fluorescence intensity for each of them at the determined time point. The signal was bleaching-corrected as described above.
All plots, fittings, corrections and normalizations in Figs 1-6, and Figs S2,S3 were made using MATLAB home-made scripts. For boxplots, the central line indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considering outliers. For bar plots, error bars represent the standard deviation. Statistical P-values were obtained using a two-sided t-test, after normal distribution had been visually checked using a simple histogram. No further verification was made to ascertain that the data met assumptions of the statistical approach. All values below 0.05 are mentioned in the figures, including sample size. Sample size was not pre-defined. No samples were excluded from analysis. No randomization or blinding was used.
Homology modeling
The suitable templates for Fus1 and Cdc12 structure modeling in Fig. 6A were found using the HHpred tool (Zimmermann et al., 2018). The models of Fus1 protein were calculated based on the template of diaphanous protein from mice, stored under the 3OBV code in the Protein Data Bank (Otomo et al., 2010). The sequence identity between these proteins is 20%. The yeast S. cerevisiae Bni1 protein structure, stored under the 5UX1 code in Protein Data Bank, served as a template for Cdc12 protein (Xu et al., 2004). Both sequences share 34% of sequence identity. For the modeling of Fus1 and Cdc12 protein dimers we used the dimeric structure of FMNL3 protein bound to actin, stored in the PDB under the 4EAH code (Thompson et al., 2013). Fus1 shared around 23% and Cdc12 around 25% of sequence identity with this template, respectively. The models' structures were calculated using Modeller 9v18 program (Šali and Blundell, 1993). The models of Fus1 and Cdc12 proteins were aligned with UCSF Chimera visualization program (Pettersen et al., 2004). The alignments points, where opposite charge residues were present in both sequences or where a group of charged amino acid appeared in only one of the sequences, were identified. We took into account the fragments that were exposed on the surface of the models – and consequently accessible for interactions with other proteins – and not interfering with dimerization of the domain or with actin elongation.
Protein expression and purification
Actin was purified from rabbit skeletal-muscle acetone powder (Spudich and Watt, 1971) and labeled on Cys374 with pyrenyliodoacetamide (Pollard and Cooper, 1984), or lysines with Alexa488-succinimidylester (Isambert et al., 1995). Fission yeast profilin Cdc3 was overexpressed and purified from Escherichia coli (Lu and Pollard, 2001). Fimbrin Fim1 was expressed in E. coli and purified via His-tag affinity to Talon Metal Affinity Resin (Clontech, Mountain View, CA) (Skau and Kovar, 2010). SNAP-tagged Cdc122FH1-Fus1FH2 and Cdc122FH1-Fus1R1054EFH2 constructs (including the C-terminal tail) were expressed in E. coli strain BL21-Codon Plus (DE3) RP (Agilent Technologies, Santa Clara, CA) with 0.5 mM isopropyl β-D-1-thiogalactopyranoside for 16 h at 16°C. Cells were lysed with sonication in extraction buffer [50 mM NaH2PO4 (anhydrous), 500 mM NaCl, 10% glycerol, 10 mM imidazole, 10 mM BME, pH 8] with EDTA-free Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and were clarified by centrifugation. The extract was incubated for 1 h at 4°C with Talon resin (Clontech, Mountain View, CA), loaded onto a column, washed with extraction buffer, and the protein was eluted with 250 mM imidazole. Formin proteins were dialyzed into SNAP buffer [20 mM HEPES (pH 7.4), 200 mM KCl, 0.01% NaN3, 10% glycerol, and 1 mM DTT] and filtered on a Superdex 200 10/300 GL column (GE Healthcare, Little Chalfont, UK). Aliquots of the protein were flash-frozen in liquid nitrogen and stored at −80°C.
Low-speed sedimentation
F-actin bundling activity was determined from low-speed sedimentation assays. Mg-ATP-actin (15 μM) was preassembled for 1 h at 25°C, then 3.0 μM was aliquoted into Eppendorf tubes and incubated with 1.5 μM of Cdc122FH1-Fus1FH2, Cdc122FH1-Fus1R1054EFH2, or fission yeast fimbrin (Fim1) for 20 min at 25°C. Samples were spun at 10,000 g for 20 min at 25°C. Pellets were resuspended in sample buffer and separated by 12.5% SDS–PAGE and stained with Coomassie Blue (Fig. 7B). The density of protein bands as in Fig. 7C was analysed with ImageJ.
Pyrene assembly and disassembly assays
Assembly and disassembly of actin filaments were measured by observing changes in pyrene fluorescence over time. Fluorescence of 10%-labeled pyrene actin (excitation 364 nm, emission 407 nm) was measured with m200Pro (Tecan) fluorescent plate reader. Final protein concentrations are indicated in the figure legends. For spontaneous assembly assays, 15 μM 10% pyrene-labeled Mg-ATP-actin with 100× anti-foam 204 (0.005%; Sigma) was added to the upper row of a 96-well nonbinding black plate (Corning, Corning, NY). A range of concentrations of formin, plus 10× KMEI [500 mM KCl, 10 mM MgCl2, 10 mM EGTA, 100 mM imidazole (pH 7.0)] and Mg-buffer G [2 mM Tris (pH 8.0), 0.2 mM ATP, 0.1 mM MgCl2, 1 mM NaN3, 0.5 mM DTT] was added to the lower row of the plate. Reactions were initiated by transferring the contents of the lower wells to the upper wells with a 12-channel pipet, and fluorescence was read in the upper wells.
For depolymerization assays, a 5.0 μM mixture of 50% pyrene-labeled Mg-ATP-actin monomers was preassembled for 1 h in the upper row of a 96-well nonbinding black plate. Protein, 10× KMEI, and SNAP buffer [20 mM HEPES (pH 7.4), 200 mM KCl, 0.01% NaN3, 10% glycerol, and 1 mM DTT] were placed in a lower row of the plate. Reactions were initiated by mixing the contents of the lower wells with the pre-polymerized filaments, which diluted the actin to 0.1 μM.
Analysis of pyrene data
Fluorescence data as in Fig. 7D and F was plotted in Rstudio (https://www.rstudio.com/). Initial assembly rates as in Fig. 7E were calculated by finding the slope of a linear regression to the linear portions of the graph from 0-150 s. Initial depolymerization rates as in Fig. 7G were calculated by finding the slope of a linear regression to the linear portions of the graphs from 0-500 s.
Dissociation constants (KD) were calculated by fitting the depolymerization data using the nonlinear least squares function, and solving the equation:
 |
Vi is the observed elongation or depolymerization rate, Vif is the elongation or depolymerization rate when barbed ends are free, Vib is the elongation or depolymerization rate when barbed ends are bound, [ends] and [formin] are barbed-end and formin concentrations.
TIRF microscopy
Mg-ATP-Actin (10% Alexa88-labeled) polymerization was triggered in the presence of 10 mM imidazole (pH 7.0), 50 mM KCl, 1 mM MgCl2, 1 mM EGTA, 50 mM DTT, 0.2 mM ATP, 50 mM CaCl2, 15 mM glucose, 20 mg/ml catalase, 100 mg/ml glucose oxidase, and 0.5% (400 centipoise) methylcellulose. This polymerization mix was complemented with proteins of interest as indicated in the figure legends. TIRF-microscopy images as in Fig. S3A were acquired using an Olympus IX-71 microscope through TIRF illumination, recorded with a iXon EMCCD camera (Andor Technology), and a cellTIRF 4Line system (Olympus). Actin filament elongation rates as in Fig. S3B were measured using the ImageJ software (NIH, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2015). To compare Cdc122FH1-Fus1FH2 elongation rates to previously reported rates (Scott et al., 2011), we used the same normalization method where rates were adjusted based on normalization of internal control filaments to 10.0 subunits s−1 μM−1.
Supplementary Material
Acknowledgements
We thank Mohan Balasubramanian, Daniel Mulvihill, Iain Hagan and Aleksander Vještica for sharing strains as well as Boris Sieber for careful reading of the manuscript. I.B.-C. and S.G.M. conceived the project. S.G.M. performed the experiments in Fig. 1B. C.A.A., S.E.Y. and C.S. performed the experiments in Fig. 7 and Fig. S4. J.I. and V.Z. performed the molecular modeling part of the project. I.B.-C. performed all other experiments with technical assistance from L.M. S.G.M. and D.R.K. acquired funding. S.G.M. coordinated the project. I.B.-C. and S.G.M. wrote the first draft of the manuscript, which was revised by all authors.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: I.B.-C., S.G.M.; Methodology: I.B.-C., S.G.M.; Formal analysis: I.B.-C.; Investigation: I.B.-C., L.M., C.A.A., S.E.Y., C.S., J.I., V.Z., D.R.K., S.G.M.; Writing - original draft: I.B.-C, S.G.M.; Writing - review & editing: I.B.-C., L.M., C.A.A., S.E.Y., C.S., J.I., V.Z., D.R.K., S.G.M.; Supervision: D.R.K., S.G.M.; Funding acquisition: D.R.K., S.G.M.
Funding
This work was funded by grants from the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (310030B_176396 and 310030_191990) and the European Research Council (CoG CellFusion) to S.G.M., National Institutes of Health's Molecular and Cellular Biology Training Grant T32 GM007183 to S.E.Y., and the National Institutes of Health grant R01 GM079265 to D.R.K. Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/article-lookup/doi/10.1242/jcs.260289.
References
- Alberts, A. S. (2001). Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J. Biol. Chem. 276, 2824-2830. 10.1074/jbc.M006205200 [DOI] [PubMed] [Google Scholar]
- Aydin, F., Courtemanche, N., Pollard, T. D. and Voth, G. A. (2018). Gating mechanisms during actin filament elongation by formins. Elife 7, e37342. 10.7554/eLife.37342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler, J., Wu, J. Q., Longtine, M. S., Shah, N. G., McKenzie, A., 3rd, Steever, A. B., Wach, A., Philippsen, P. and Pringle, J. R. (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943-951. [DOI] [PubMed] [Google Scholar]
- Billault-Chaumartin, I. and Martin, S. G. (2019). Capping protein insulates Arp2/3-assembled actin patches from formins. Curr. Biol. 29, 3165-3176.e6. 10.1016/j.cub.2019.07.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billault-Chaumartin, I., Muriel, O., Michon, L. and Martin, S. G. (2022). Condensation of the fusion focus by the intrinsically disordered region of the formin Fus1 is essential for cell-cell fusion. bioRxiv, 2022.05.05.490810. 10.1101/2022.05.05.490810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchoin, L. and Staiger, C. J. (2010). Plant formins: diverse isoforms and unique molecular mechanism. Biochim. Biophys. Acta 1803, 201-206. 10.1016/j.bbamcr.2008.09.015 [DOI] [PubMed] [Google Scholar]
- Bohnert, K. A., Grzegorzewska, A. P., Willet, A. H., Vander Kooi, C. W., Kovar, D. R. and Gould, K. L. (2013a). SIN-dependent phosphoinhibition of formin multimerization controls fission yeast cytokinesis. Genes Dev. 27, 2164-2177. 10.1101/gad.224154.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert, K. A., Willet, A. H., Kovar, D. R. and Gould, K. L. (2013b). Formin-based control of the actin cytoskeleton during cytokinesis. Biochem. Soc. Trans. 41, 1750-1754. 10.1042/BST20130208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombardier, J. P., Eskin, J. A., Jaiswal, R., Corrêa, I. R., Jr., Xu, M.-Q., Goode, B. L. and Gelles, J. (2015). Single-molecule visualization of a formin-capping protein ‘decision complex’ at the actin filament barbed end. Nat. Commun. 6, 8707. 10.1038/ncomms9707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitsprecher, D. and Goode, B. L. (2013). Formins at a glance. J. Cell Sci. 126, 1-7. 10.1242/jcs.107250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, F., Drubin, D. and Nurse, P. (1997). cdc12p, a protein required for cytokinesis in fission yeast, is a component of the cell division ring and interacts with profilin. J. Cell Biol. 137, 169-182. 10.1083/jcb.137.1.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesarone, M. A., DuPage, A. G. and Goode, B. L. (2010). Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62-74. 10.1038/nrm2816 [DOI] [PubMed] [Google Scholar]
- Christensen, J. R., Hocky, G. M., Homa, K. E., Morganthaler, A. N., Hitchcock-DeGregori, S. E., Voth, G. A. and Kovar, D. R. (2017). Competition between Tropomyosin, Fimbrin, and ADF/Cofilin drives their sorting to distinct actin filament networks. Elife 6, e23152. 10.7554/eLife.23152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche, N. (2018). Mechanisms of formin-mediated actin assembly and dynamics. Biophys. Rev. 10, 1553-1569. 10.1007/s12551-018-0468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtemanche, N. and Pollard, T. D. (2012). Determinants of Formin Homology 1 (FH1) domain function in actin filament elongation by formins. J. Biol. Chem. 287, 7812-7820. 10.1074/jbc.M111.322958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudin, O., Bendezú, F. O., Groux, R., Laroche, T., Seitz, A. and Martin, S. G. (2015). A formin-nucleated actin aster concentrates cell wall hydrolases for cell fusion in fission yeast. J. Cell Biol. 208, 897-911. 10.1083/jcb.201411124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudin, O., Merlini, L., Bendezú, F. O., Groux, R., Vincenzetti, V. and Martin, S. G. (2017). A systematic screen for morphological abnormalities during fission yeast sexual reproduction identifies a mechanism of actin aster formation for cell fusion. PLoS Genet. 13, e1006721. 10.1371/journal.pgen.1006721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egel, R., Willer, M., Kjærulff, S., Davey, J. and Nielsen, O. (1994). Assessment of pheromone production and response in fission yeast by a halo test of induced sporulation. Yeast 10, 1347-1354. 10.1002/yea.320101012 [DOI] [PubMed] [Google Scholar]
- Feierbach, B. and Chang, F. (2001). Roles of the fission yeast formin for3p in cell polarity, actin cable formation and symmetric cell division. Curr. Biol. 11, 1656-1665. 10.1016/S0960-9822(01)00525-5 [DOI] [PubMed] [Google Scholar]
- Goode, B. L. and Eck, M. J. (2007). Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 76, 593-627. 10.1146/annurev.biochem.75.103004.142647 [DOI] [PubMed] [Google Scholar]
- Graziano, B. R., DuPage, A. G., Michelot, A., Breitsprecher, D., Moseley, J. B., Sagot, I., Blanchoin, L. and Goode, B. L. (2011). Mechanism and cellular function of Bud6 as an actin nucleation-promoting factor. Mol. Biol. Cell 22, 4016-4028. 10.1091/mbc.e11-05-0404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano, T., Lim, T. C., Billault-Chaumartin, I., Dhar, A., Gu, Y., Massam-Wu, T., Adishesha, S., Springall, L., Sivashanmugam, L., Scott, W.et al. (2022). mNeonGreen-tagged fusion proteins and nanobodies reveal localization of tropomyosin to patches, cables, and contractile actomyosin rings in live yeast cells. bioRxiv, 2022.05.19.492673. 10.1101/2022.05.19.492673 [DOI] [Google Scholar]
- Homa, K. E., Zsolnay, V., Anderson, C. A., O'Connell, M. E., Neidt, E. M., Voth, G. A., Bidone, T. C. and Kovar, D. R. (2021). Formin Cdc12's specific actin assembly properties are tailored for cytokinesis in fission yeast. Biophys. J. 120, 2984-2997. 10.1016/j.bpj.2021.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isambert, H., Venier, P., Maggs, A. C., Fattoum, A., Kassab, R., Pantaloni, D. and Carlier, M. F. (1995). Flexibility of actin filaments derived from thermal fluctuations. Effect of bound nucleotide, phalloidin, and muscle regulatory proteins. J. Biol. Chem. 270, 11437-11444. 10.1074/jbc.270.19.11437 [DOI] [PubMed] [Google Scholar]
- Kovar, D. R., Sirotkin, V. and Lord, M. (2011). Three's company: the fission yeast actin cytoskeleton. Trends Cell Biol. 21, 177-187. 10.1016/j.tcb.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn, S. and Geyer, M. (2014). Formins as effector proteins of Rho GTPases. Small GTPases 5, e29513. 10.4161/sgtp.29513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi, H., Imai, Y. and Yamamoto, M. (2002). Tropomyosin is required for the cell fusion process during conjugation in fission yeast. Genes Cells 7, 375-384. 10.1046/j.1365-2443.2002.00526.x [DOI] [PubMed] [Google Scholar]
- Lu, J. and Pollard, T. D. (2001). Profilin binding to poly-L-proline and actin monomers along with ability to catalyze actin nucleotide exchange is required for viability of fission yeast. Mol. Biol. Cell 12, 1161-1175. 10.1091/mbc.12.4.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, S. G. (2016). Role and organization of the actin cytoskeleton during cell-cell fusion. Semin. Cell Dev. Biol. 60, 121-126. 10.1016/j.semcdb.2016.07.025 [DOI] [PubMed] [Google Scholar]
- Martin, S. G., Rincón, S. A., Basu, R., Pérez, P. and Chang, F. (2007). Regulation of the formin for3p by cdc42p and bud6p. Mol. Biol. Cell 18, 4155-4167. 10.1091/mbc.e07-02-0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muriel, O., Michon, L., Kukulski, W. and Martin, S. G. (2021). Ultrastructural plasma membrane asymmetries in tension and curvature promote yeast cell fusion. J. Cell Biol. 220, e202103142. 10.1083/jcb.202103142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo, T., Tomchick, D. R., Otomo, C., Machius, M. and Rosen, M. K. (2010). Crystal structure of the Formin mDia1 in autoinhibited conformation. PLoS One 5, e12896. 10.1371/journal.pone.0012896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, A. S. and Pollard, T. D. (2008). The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9-19. 10.1016/j.cub.2007.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, J., Weilguny, D., Egel, R. and Nielsen, O. (1995). Characterization of fus1 of Schizosaccharomyces pombe: a developmentally controlled function needed for conjugation. Mol. Cell. Biol. 15, 3697-3707. 10.1128/MCB.15.7.3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, J., Nielsen, O., Egel, R. and Hagan, I. M. (1998a). F-actin distribution and function during sexual differentiation in Schizosaccharomyces pombe. J. Cell Sci. 111, 867-876. 10.1242/jcs.111.7.867 [DOI] [PubMed] [Google Scholar]
- Petersen, J., Nielsen, O., Egel, R. and Hagan, I. M. (1998b). FH3, a domain found in formins, targets the fission yeast formin Fus1 to the projection tip during conjugation. J. Cell Biol. 141, 1217-1228. 10.1083/jcb.141.5.1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C. and Ferrin, T. E. (2004). UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605-1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pollard, T. D. and Cooper, J. A. (1984). Quantitative analysis of the effect of Acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry 23, 6631-6641. 10.1021/bi00321a054 [DOI] [PubMed] [Google Scholar]
- Pollard, T. D. and O'Shaughnessy, B. (2019). Molecular Mechanism of Cytokinesis. Annu. Rev. Biochem. 88, 661-689. 10.1146/annurev-biochem-062917-012530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali, A. and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779-815. 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Schönichen, A. and Geyer, M. (2010). Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim. Biophys. Acta 1803, 152-163. 10.1016/j.bbamcr.2010.01.014 [DOI] [PubMed] [Google Scholar]
- Scott, B. J., Neidt, E. M. and Kovar, D. R. (2011). The functionally distinct fission yeast formins have specific actin-assembly properties. Mol. Biol. Cell 22, 3826-3839. 10.1091/mbc.e11-06-0492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar, S., Kerleau, M., Kühn, S., Pernier, J., Romet-Lemonne, G., Jégou, A. and Carlier, M. F. (2015). Formin and capping protein together embrace the actin filament in a ménage à trois. Nat. Commun. 6, 8730. 10.1038/ncomms9730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar, S., Pernier, J. and Carlier, M. F. (2016). Regulators of actin filament barbed ends at a glance. J. Cell Sci. 129, 1085-1091. 10.1242/jcs.179994 [DOI] [PubMed] [Google Scholar]
- Skau, C. T. and Kovar, D. R. (2010). Fimbrin and tropomyosin competition regulates endocytosis and cytokinesis kinetics in fission yeast. Curr. Biol. 20, 1415-1422. 10.1016/j.cub.2010.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skau, C. T. and Waterman, C. M. (2015). Specification of Architecture and Function of Actin Structures by Actin Nucleation Factors. Annu. Rev. Biophys. 44, 285-310. 10.1146/annurev-biophys-060414-034308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich, J. A. and Watt, S. (1971). The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866-4871. 10.1016/S0021-9258(18)62016-2 [DOI] [PubMed] [Google Scholar]
- Thompson, M. E., Heimsath, E. G., Gauvin, T. J., Higgs, H. N. and Kull, F. J. (2013). FMNL3 FH2-actin structure gives insight into formin-mediated actin nucleation and elongation. Nat. Struct. Mol. Biol. 20, 111-118. 10.1038/nsmb.2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente, N. B., Zamboni, J. E. D., Adur, J. F., Paravani, E. V. and Casco, V. H. (2007). Photobleaching correction in fluorescence microscopy images. J. Phys., Conf. Ser. 90, 012068. 10.1088/1742-6596/90/1/012068 [DOI] [Google Scholar]
- Vidali, L., van Gisbergen, P. A., Guérin, C., Franco, P., Li, M., Burkart, G. M., Augustine, R. C., Blanchoin, L. and Bezanilla, M. (2009). Rapid formin-mediated actin-filament elongation is essential for polarized plant cell growth. Proc. Natl. Acad. Sci. USA 106, 13341-13346. 10.1073/pnas.0901170106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vjestica, A., Merlini, L., Dudin, O., Bendezu, F. O. and Martin, S. G. (2016). Microscopy of fission yeast sexual lifecycle. J. Vis. Exp. 109, 53801. 10.3791/53801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vještica, A., Marek, M., Nkosi, P. J., Merlini, L., Liu, G., Bérard, M., Billault-Chaumartin, I. and Martin, S. G. (2020). A toolbox of stable integration vectors in the fission yeast Schizosaccharomyces pombe. J. Cell Sci. 133, jcs240754. 10.1242/jcs.240754 [DOI] [PubMed] [Google Scholar]
- Wu, J.-Q. and Pollard, T. D. (2005). Counting cytokinesis proteins globally and locally in fission yeast. Science 310, 310-314. 10.1126/science.1113230 [DOI] [PubMed] [Google Scholar]
- Xie, Y., Zhou, F., Ma, Q., Lu, L. and Miao, Y. (2022). A teamwork promotion of formin-mediated actin nucleation by Bud6 and Aip5 in Saccharomyces cerevisiae. Mol. Biol. Cell 33, ar19. 10.1091/mbc.E21-06-0285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y., Moseley, J. B., Sagot, I., Poy, F., Pellman, D., Goode, B. L. and Eck, M. J. (2004). Crystal structures of a Formin Homology-2 domain reveal a tethered dimer architecture. Cell 116, 711-723. 10.1016/S0092-8674(04)00210-7 [DOI] [PubMed] [Google Scholar]
- Yonetani, A., Lustig, R. J., Moseley, J. B., Takeda, T., Goode, B. L. and Chang, F. (2008). Regulation and targeting of the fission yeast formin cdc12p in cytokinesis. Mol. Biol. Cell 19, 2208-2219. 10.1091/mbc.e07-07-0731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann, D. and Kovar, D. R. (2019). Feeling the force: formin's role in mechanotransduction. Curr. Opin. Cell Biol. 56, 130-140. 10.1016/j.ceb.2018.12.008 [DOI] [PubMed] [Google Scholar]
- Zimmermann, L., Stephens, A., Nam, S.-Z., Rau, D., Kübler, J., Lozajic, M., Gabler, F., Söding, J., Lupas, A. N. and Alva, V. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237-2243. 10.1016/j.jmb.2017.12.007 [DOI] [PubMed] [Google Scholar]
- Zweifel, M. E. and Courtemanche, N. (2020). Competition for delivery of profilin-actin to barbed ends limits the rate of formin-mediated actin filament elongation. J. Biol. Chem. 295, 4513-4525. 10.1074/jbc.RA119.012000 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.