

Summary
We here investigate the impact of antiviral treatments such as remdesivir on intra-host genomic diversity and emergence of SARS-CoV2 variants in patients with a prolonged course of infection. Sequencing and variant analysis performed in 112 longitudinal respiratory samples from 14 SARS-CoV2-infected patients with severe disease progression show that major frequency variants do not generally arise during prolonged infection. However, remdesivir treatment can increase intra-host genomic diversity and result in the emergence of novel major variant species harboring fixed mutations. This is particularly evident in a patient with B cell depletion who rapidly developed mutations in the RNA-dependent RNA polymerase gene following remdesivir treatment. Remdesivir treatment-associated emergence of novel variants is of great interest in light of current treatment guidelines for hospitalized patients suffering from severe SARS-CoV2 disease, as well as the potential use of remdesivir to preventively treat non-hospitalized patients at high risk for severe disease progression.
Keywords: SARS-CoV-2 variants, remdesivir treatment, prolonged SARS-CoV-2 infection, escape mutations, intra-host evolution
Graphical abstract
Highlights
-
•
Analysis of SARS-CoV2 intra-host diversity in longitudinal samples from 14 patients
-
•
Prolonged infection does not generally result in fundamentally increased diversity
-
•
Remdesivir treatment can result in rapid fixation of newly acquired mutations
-
•
Treatment-associated evolutionary bottlenecks promote emergence of novel variants
Heyer et al. investigate SARS-CoV2 intra-host genomic diversity in longitudinal samples from 14 patients suffering from prolonged infection. Whereas viral populations are surprisingly stable overall, novel variant species can rapidly emerge in remdesivir-treated patients, suggesting that antiviral treatment can create evolutionary bottlenecks, which promote emergence of SARS-CoV2 variants.
Introduction
Both cell-mediated and humoral immunity are required to control SARS-CoV2 infection.1, 2, 3, 4, 5 Recently, several case reports have reported prolonged respiratory tract-associated or systemic infection in patients with pre-existing conditions, sometimes with positive viral detection over several months.1, 2, 3, 4, 5, 6, 7, 8, 9 Prolonged infection was primarily detected in immunodeficient patients suffering from conditions such as chronic lymphocytic leukemia, follicular lymphoma, or B cell immunodeficiency. Prolonged infections in such patients have been repeatedly discussed as major contributors to viral evolution. It is thought that decreased immune restriction may result in a broad increase of viral intra-host diversity, thus favoring the emergence of novel variants, especially if antiviral treatments such as remdesivir or convalescent plasma exert selection pressure for the acquisition of escape mutations.2,3 Most recently, the emergence of the Omicron variant of concern (VOC) has refueled interest in the mechanisms contributing to variant emergence.
In addition to patients suffering from primary or secondary immune deficiencies such as HIV-1,10 prolonged infections are mainly observed in patients on B cell depleting therapies or in patients on CAR T cell therapy who have failed to clear the virus efficiently.4, 5, 6, 7, 8,11 In these patients, the presence of novel mutations was observed particularly in the orf1ab and spike region, as well as deletions in the spike region and in orf7 and orf8, which are thought to have an immunoregulatory function.12 While there are also a few reports of prolonged infections in patients without known impairment of the humoral and cellular immune response, these are typically patients suffering from underlying conditions such as diabetes or cardiovascular disease, known risk factors for a severe COVID-19 progression.13,14
Treatment of COVID-19 has changed throughout the pandemic with limited therapeutic options, including antiviral therapy with remdesivir, administration of convalescent plasma, or anti-inflammatory therapy through the use of dexamethasone, hydrocortisone, JAK inhibitors, and tocilizumab. While current ICU guidelines in Germany do not generally recommend remdesivir or convalescent plasma treatment, remdesivir can be given to hospitalized patients on oxygen supplementation.15 Similarly, NIH guidelines give a moderate recommendation for remdesivir treatment for patients who require supplemental oxygen, or remdesivir together with dexamethasone for patients who additionally require high-flow oxygen therapy or invasive ventilation.16 Furthermore, several agencies have recommended remdesivir administration as an option in treatment guidelines for high-risk patients who have not been hospitalized and are within the 5–7 day window following symptom onset.17, 18, 19, 20
Considering the use of antiviral treatments and increasing reports on prolonged SARS-CoV2 infection, it is of high interest to investigate whether patients with prolonged diseases in general display fundamentally increased intra-host evolution that may allow more rapid emergence of SARS-CoV2 variants, or whether specific treatment regimens foster the emergence of novel variant species.
Results
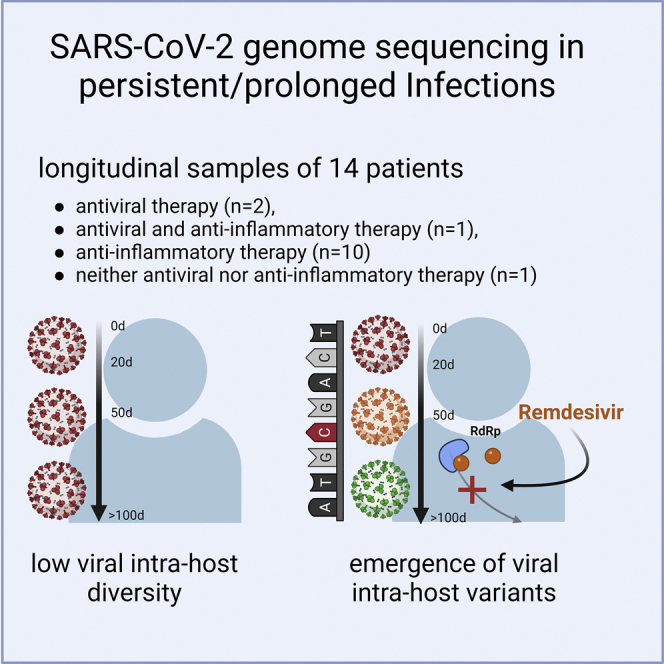
We addressed this by investigating intra-host genomic diversity in longitudinal samples from 14 patients with prolonged viral persistence (30–146 days) during severe COVID-19 disease, including immune-compromised and immune-competent patients with or without antiviral treatment to evaluate the emergence of mutations with and without selection pressure. We tested respiratory samples of patients for SARS-CoV-2 by qRT-PCR. Viral loads measured by cycle threshold (Ct) values ranged from 20 to 38, and only samples with Ct value < 33 were included in viral sequencing. Fourteen patients, in total 112 respiratory samples, with severe COVID-19 disease and prolonged detection of SARS-CoV-2 in the respiratory tract were included (Table 1). The infection time, defined as time with detectable SARS-CoV-2 RNA in the respiratory tract, ranged from 30 to 146 days. The mean age of the patients was 62.2 years (±12.2 years), and 46% of the patients were female. All patients suffered from underlying diseases, such as diabetes, renal insufficiency, obesity, and arterial hypertension (Table 1). Few patients showed pre-existing conditions due to chronic obstructive pulmonary disease (COPD) (P10, P16, and P17) or rheumatoid arthritis (P13). Two patients were immunosuppressed due to follicular lymphoma (P01) and liver transplantation (P14), and two patients received mild immunoregulatory treatment against rheumatic arthritis (P13, P17). All patients except one, P13, who died of multi-organ failure, were eventually discharged from the hospital. In the majority without symptoms, two patients presented symptoms such as atrial fibrillation or polyneuropathy, P07 and P08, respectively (Table 1). Patients were classified based on their treatments as individuals receiving (1) antiviral therapy (n = 2) including remdesivir (R), (2) antiviral (R) and anti-inflammatory therapy (dexamethasone) (n = 1), (3) anti-inflammatory therapy (dexamethasone, hydrocortisone, or both) (n = 10), and (4) neither antiviral nor anti-inflammatory therapy (n = 1).
Table 1.
Patient data
| ID | Age/gender | Infection timea | Pango lineage | Seq. interval | Comorbidities | Symptoms | Immunosuppressive medicationb | Complications | ICU | Ventilationc | Treatment (R/D/P)d | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P01 | 53 F | 90 | B.1.8 | 0–83 days | Follicular lymphoma (in remission) | Dyspnea, cough | Obinutuzumab (3 months prior to admission) | Pneumonia | No | – | R (d63-d72/d91-95), P (d95; d97, d99) |
Discharged with no symptoms on day 138 |
| P03 | 34 M | 30 | A | 0–28 days | Diabetes, renal failure, adiposity | Cough, fever | – | – | No | LFOT | R (d9/d12) | Discharged with no symptoms on day 33 |
| P05 | 61 M | 65 | B.1.36.1 | 0–65 days | Diabetes | Hypoxia with accompanying syncope | – | Severe ARDS, septic shock, acute renal failure | Yes | Invasive | D (continuously), Hydrocortisone (d4-12) | Discharged with no symptoms on day 87 |
| P06 | 43 M | 47 | B.1.177.81 | 0–46 days | Pre-diabetes, adiposity | Dyspnea, cough | – | Severe ARDS, acute renal failure, septic shock | Yes | Invasive | D (d2-d18), Hydrocortisone | Transfer to rehabilitation center on day 35 |
| P07 | 75 F | 57 | B.1.1 | 0–47 days | Renal failure, granulomatosis with polyangiitis, hypertension | Dyspnea, cough | Cyclophosphamide (1 month prior to admission) | Severe ARDS | Yes | LFOT/HFOT/invasive | D (d35-d41) | Discharged with atrial fibrillation and dyspnea on day 43 |
| P08 | 50 F | 38 | B.1.1 | 0–24 days | Diabetes, adiposity, hypertension | Pyrexia, dyspnea, nausea, cephalalgia | – | Severe ARDS, acute renal failure, asystole with successful cardiopulmonary resuscitation | Yes | Invasive | D (before admission and d12-d16) | Discharged with Dyspnea and on day 41 |
| P09 | 62 M | 39 | B.1.177.81 | 0–12 days | Diabetes, adiposity, hypertension | Pyrexia, dyspnea, fatigue | – | Severe ARDS, acute renal failure, pneumogenic sepsis | Yes | LFOT/HFOT/invasive | D (before admission and d1-d9) | Discharged with no symptoms on day 54 |
| P10 | 75 M | 55 | A | 0–24 days | COPD, adiposity, hypertension | Progressive pulmonary failure | – | Moderate ARDS, acute renal failure, sepsis, circulatory failure with successful cardiopulmonary resuscitation | Yes | Invasive | – | Discharged with no symptoms on day 56 |
| P13 | 60 F | 57 | A | 0–57 days | Rheumatoid arthritis | – | Methotrexate (weekly) Rituximab (most recent administration unknown) |
Severe ARDS, acute renal failure, sepsis | Yes | LFOT/invasive | R (before admission and d9-15), D (before admission and d9 –d39) | Deceased 41 days after admission, septic shock with multi-organ failure |
| P14 | 59 M | 57 | B.1.1.277 | 0–29 days | Liver transplant recipient, hypertension | Dyspnea | Tacrolimus (on admission) Basiliximab (most recent administration unknown) |
Pneumonia | Yes | LFOT/HFOT/invasive | D (d20 – d24) | Discharged with no symptoms on day 67 |
| P15 | 70 F | 63 | AC.1 | 0–33 days | Diabetes, adiposity, hypertension | Dyspnea, cough | – | Severe ARDS, heparin induced thrombocytopenia | Yes | Invasive | D (d4 - d6) | Discharged with no symptoms on day 66 |
| P16 | 75 M | 26 | B.1 | 0–25 days | COPD, hypertension | Pyrexia, dyspnea, fatigue, progressive pulmonary failure | – | ARDS, acute atrial fibrillation, acute renal failure, intrahepatic cholestasis | Yes | Invasive | Prednisolone | Discharged with no symptoms on day 92 |
| P17 | 77 M | 52 | B.1.1 | 0–13 days | COPD, rheumatoid arthritis, hypertension, diabetes, renal failure | Dyspnea, pyrexia, diarrhea | Methotrexate (weekly) | Severe ARDS, acute renal failure, multiple septic episodes, critical illness polyneuropathy, and myopathy | Yes | LFOT/invasive | Prednisolone, Hydrocortisone continuously |
Discharged with no respiratory symptoms on day 205 |
| P18 | 66 F | 146 | A.9 | 0–117 days | Dementia, hypertension, adiposity, renal failure, Goodpasture syndrome | Dyspnea, altered vigilance | Single dose rituximab, cyclophosphamide (8–10 months prior to admission) | ARDS, AV block I°, sepsis, cardiac decompensation (NYHA IV) | Yes | HFOT/invasive | Hydrocortisone for 3days, Prednisolone continuously |
Discharged with no symptoms on day 160 |
Interval between admission to the hospital and last qRT-PCR positive SARS-CoV-2 report in the respiratory tract.
Immunosuppressive medication upon admission.
LFOT: low-flow oxygen therapy; HFOT: high-flow oxygen therapy.
D: dexamethasone; R: remdesivir; P: convalescent plasma.
All patients had been hospitalized between April 2020 and January 2021, prior to the spread of the Alpha variant or other VOCs in the Hamburg metropolitan area. Accordingly, whole-genome SARS-CoV-2 sequencing from the respiratory samples confirmed that no SARS-CoV-2 VOC contributed to the infections at any time (Table 1 and Figures S2, S3, S4, and S5).
For all patients included, the overall viral load after an initial decrease remained at a low level with Ct values of 30–33, indicating insufficient immune control to clear the infection. Except for patient P10, all patients were considered viremic due to low viral loads detectable in the blood in the first days of disease progression. However, blood viral loads were too low to perform whole viral genome sequencing and variant profiling.
We investigated changes in genomic diversity by amplicon sequencing of whole SARS-CoV-2 genomes in longitudinal respiratory samples that were collected over time periods of up to 117 days (mean 43 days; see Figure 1A for an overview of overall sampling periods, as well as individual sample collection intervals for each patient). In each sample, we identified nucleotide variants (NVs) and evaluated changes in NV abundance and frequency over time. Figures 1B and 1C show a summary analysis of all patients and samples, in which the distance of each sample from the first investigated time point is represented by changes in the total count (A) or frequency (B) across all detected NVs. We furthermore employed a statistical model to identify transition events (defined as events whenever an NV transits between minor and major frequency states with abundance values below or above 50%, respectively), and we calculated p values for the hypothesis that the total number of transition events in a particular sample is higher than the expected average across all samples (see STAR Methods for calculation details and Figure S1). Figures 1D and 1E show the total number of transitions and associated p values for each sample, respectively. Heat maps with individual NV frequencies, along with corresponding major or minor calls are shown in Figure 2B for patient P01, and Figures S2 through S5 for the remaining patients.
Figure 1.

Cross-comparison of NV diversity in longitudinal patient samples
(A) Overview of sequenced samples with each sample sequenced once, and sample collection time points, relative to the collection date of the first investigated sample. Consecutive longitudinal samples are shown as stacked columns marked in alternating shades of gray, with the height of each segment denoting the time span between sampling dates. Samples exhibiting NV transition counts significantly above the mean expectation value (see below) are shown in orange or red for significance levels of 0.01 or 0.001, respectively, and are additionally labeled with the corresponding sample collection time point.
(B–E) Summary analysis of patient samples. Individual samples are denoted by filled circles, and mean values for each patient are shown as short horizontal lines. (B) and (C) show the aggregated NV distance relative to the first investigated sample, as measured by either the relative number of SNVs with an estimated frequency of at least 1% (B) or the sum of frequency alterations across all SNVs (C). (D) shows the total number of transitions (i.e., events in which SNVs transition from a non-significant or minor to a major state, or vice versa) between consecutive longitudinal samples. (E) shows p values for the hypothesis that the number of transitions observed in a given sample is greater than that expected from the estimated average transition frequency across all samples (pT; see STAR Methods for calculation details). The 0.05 significance cutoff is shown as a horizontal line. Circles filled in blue, orange, or red across all panels denote samples for which this value reaches significance levels of 0.05, 0.01, or 0.001, respectively.
Figure 2.

Summary of key clinical parameters, treatment regimen, and sequencing results in patient 1
(A) Temporal representation of the infection interval indicating dates when respiratory samples were collected and whether they tested positive for SARS-CoV-2, along with remdesivir treatment dates (light gray boxes) and inflammatory markers (CRP and IL-6). CRP values are shown in green and IL-6 in turquoise. Viral loads are shown for respiratory samples (green diamonds) and blood samples (gray triangles). Filled symbols depict samples subjected for sequencing.
(B) Upper panel: Heatmap depicting the frequency of nucleotide variants (NVs) in longitudinal samples from patient 1. Each longitudinal sample was sequenced once. Nucleotide variants (relative to reference sequence NC_045512.2) and potentially resulting amino acid mutations are indicated at the bottom or top of the map, respectively. Nucleotide variant C28628T only occurred in the background of C28629T, thus resulting in an A > F amino acid exchange in a fraction of C28629T mutations that alone led to an A > V exchange. The corresponding amino acid exchanges are marked with an asterisk. Variant frequency is indicated by heatmap colors, ranging from gray (reference) over yellow to dark blue, as indicated in the legend shown to the right. Ct values and treatment regimen are shown to the left. None of the investigated samples exhibited coverage levels below 10 (as labeled with a blue dot in Figures S2, S3, S4, and S5). Lower panel: Color code map indicating classification of nucleotide variants according to the observed coverage/frequency values, as further described in the STAR Methods section and illustrated in Figure S1. Classification calls corresponding to individual colors are shown in the legend to the right of the map (n.s.: not significant).
Most patients exhibited surprisingly stable viral populations, with a relatively low number of transition events observed even over long infection periods (Figure 1D). Statistical analysis suggested a total of four samples with a significantly higher number of transitions (Figure 1E) when compared to expected mean values. One of these was from patient P01, a remdesivir-treated patient who also exhibited the overall highest relative genomic diversity among all patients (Figures 1B and 1C). The remaining three were collected from P07 on day 38 (relative to the initial sampling date) and from patient P14 on days 20 and 23. Transitioning NVs in these samples include silent mutations in nsp13, ORF8, and the 3′-UTR in patient P07, as well as silent and non-silent nsp3 mutations in addition to deletions in the spike gene in patient 14 (see Figure S3, P07: nsp13:F422, ORF8:F120, and G29751A, and Figure S4, P14: nsp3:T725I, nsp3:N1369). Interestingly, both patients also exhibit deletions spanning amino acids 141–144 or 141–145 of the spike gene. This segment in the N-terminal region of the spike protein has been described as a recurrent deleted region, and deletions with this region have been shown to influence antigenicity.9 However, as with other mutations in patients receiving anti-inflammatory treatment only, these deletions did not become fixed (i.e., did not reach near-100% allelic frequency), indicating insufficient selection advantage to allow displacement of the original genotype(s).
We made substantially different observations in patient P01, a patient suffering from prolonged COVID-19 disease and SARS-CoV-2 viremia that required two rounds of remdesivir treatment (the second one combined with convalescent plasma therapy) to achieve a sustained response. A detailed report of this patient’s clinical course has been published before4; key parameters are recapitulated in Figure 2A. As shown in Figure 2B, longitudinal samples exhibited a total of nine newly acquired NVs that reached fixation within the investigated time period. Strikingly, six of these underwent transition at day 64, shortly after initiation of the first round of remdesivir treatment. Another two transitioned at day 67, and the ninth mutation (nsp12:V166L) registered 6 days later (day 73). All nine mutations had reached 100% allelic frequency by day 79. Except for a silent mutation in the nsp7-coding region, all mutations result in amino acid exchanges within the envelope, nucleocapsid (N), spike (S), nsp4, nsp8, nsp12, and nsp14 gene products. The two mutations at positions 166 and 536 of nsp 12, the gene encoding the catalytic subunit of the RNA-dependent RNA polymerase (RdRp), are of particular interest (Table 2 and Figures 2B, 3A, and 3B): nsp12:536:I:V resides in a region responsible for nsp8 binding, (another structural component of the active multi-protein RdRp complex), while nsp12:166:V:L maps to the nidovirus RdRp-associated nucleotidyl transferase (NiRAN) domain. Figure 3B shows a structural view of this residue’s location relative to a remdesivir molecule (distance: 19.28Å) bound at the catalytically active polymerization site of nsp12.21 Four additional variants in nsp3 and nsp4 (a protein contributing to the binding to the endoplasmic reticulum and subsequent formation of double-membrane vesicles involved in viral replication) were registered on day 73, but they did not become fixed and remained at frequency levels below 50% (Figure 1B, nsp4:S209P and A457V).
Table 2.
Mutations arising under remdesivir treatment
| Patient | Treatment | Location of samplingc | 100% allele frequency aa changes | <100% allele frequency aa changes | Transient aa changes |
|---|---|---|---|---|---|
| P01 | Ra: d63–d72; R: d91–95 | LRT |
d83 nsp4:457:A:V nsp7:S61d nsp8:20:E:K nsp12:166:V:L nsp12:536:I:V S:50:S:L S:260:A:D S:490:F:S E:30:T:I |
d83 nsp3:1614:R:K nsp4:209:S:P nsp4:272:I:L nsp4:336:S:L |
d64 orf8:1614:R:KQ91d N:119:A:F N:119:A:F/V N:119:A:G |
| P03 | R: d9; R: d12–d15 | URT | – |
d28 NC:29,742:G:A |
– |
| P13 | R: before admission and d38–36; Db:d28–d36 | LRT | – | – |
d42 nsp10:138:L:F E:30:T:I S:850:I:M orf8:84:L:S orf8:92:E:K |
Mutations observed in multiple patients are shown in bold.
R: remdesivir.
D: dexamethasone.
LRT: lower respiratory tract, URT: upper respiratory tract.
Silent mutation.
Figure 3.

Schematic representation of the mutations identified in patient 1
(A) Overview of the location of mutations in the SARS-CoV-2 genome emerging over the infection interval 0–83 days. Open reading frames (ORFs) in which mutations emerge are shown in blue, whereas unaffected ORFs are shown in gray. Non-synonymous mutations that establish during infection are shown in black, synonymous mutations in gray, and mutations that are gradually lost during infection are shown in green.
(B) Representation of mutation 166:V:L within the nsp12 structure (PDB:7BV2). Nsp12 structural elements are depicted in green, the RNA structure in magenta, and remdesivir in pink.
Besides patient P01, two other patients had received remdesivir treatment, either together with or without anti-inflammatory drugs (P13 and P03; see Table 1 for treatment regimen). Patient P03 received single-dose remdesivir treatments on d9 and d12. We did not observe emergence of variants passing our stringency filters (Figure S2A). However, our ability to detect such variants may have been hampered by a limited number of available post-treatment samples and low viral loads (Ct 33) in the last investigated sample (d28). In patient 13, a mutation in the envelope protein (E), E:30:T:I, emerged on day 28, shortly after remdesivir treatment (Figure S2B). The mutation reached a maximum allelic frequency of 95% during the following 2 weeks but eventually was lost on day 57, a total of 28 days after cessation of antiviral treatment. A mutation in the nsp10 region (nsp10:138:L:F) showed similar patterns but generally remained below frequencies of 50%. Similar to transiently increased intra-host diversity observed in patients with anti-inflammatory treatment, emergence of these mutations coincided with an increase in viral replication, as signified by a decrease of Ct values from 32 to 24 between days 29 and 34 (Figure S2B).
Discussion
Thus far, the appearance of novel SARS-CoV-2 mutations resulting from intra-host evolution has been described mainly in case reports with small patient numbers or few longitudinal samples from immunocompromised individuals with prolonged viral infection.1, 2, 3, 4, 5, 6,8,11,22,23 Our study extends these reports by a systematic side-by-side analysis of intra-host diversity and emerging mutations in prolonged SARS-CoV-2 infection in patients with severe COVID-19 under different therapeutic regimens. We demonstrate marked viral intra-host diversification, with emerging mutations that reach frequency levels of 100%, in a patient with prolonged SARS-CoV-2 infection and antiviral remdesivir treatment. In contrast, in patients receiving exclusively anti-inflammatory treatment, we only sporadically observed the emergence of novel variants. Associated mutations often appeared only transiently, and their allelic frequency generally remained below 50%. Hence, our data suggest that prolonged infection per se is unlikely to promote the emergence of novel variant species. Our observation differs from some previous studies, for example, a study that examined two time points of SARS-CoV-2-positive individuals with mild symptoms in 13 cases with an infection interval of 14 days and generally observed a detectable increase in intra-host diversity.24 In addition to the increased number of serial samples included in our study, we suspect that the stringent variant calling (variants are only included in the analysis if they are present in at least two consecutive longitudinal samples, with an allelic frequency of minimally 20%) may contribute to these differences. While this method does not allow us to observe mutations that may be present at very low frequency, it efficiently eliminates noise, including sequencing errors as well as technical artifacts introduced during reverse transcription and PCR amplification.
Interestingly, Lythgoe and colleagues examined intra-host diversity in temporal samples of infected individuals in households in a 20-day time window and observed little consistency in the frequency of minor variants between time points within individuals.25 The authors concluded that these within-host dynamics suggest inherent stochasticity associated with low viral load samples and that within-host emergence of vaccine- and therapeutic-escape mutations are likely to be relatively rare when viral loads are high. The authors furthermore suggest that potentially beneficial adaptive mutations might be present even in the absence of immunological or therapeutic selection pressure and could then become established under therapy.25 We tend to side with this view, especially considering the fact that our study preferentially registered changes in intra-host diversity following a low-to-high switch in viral replication levels. This is in line with previous observations24 and suggests that a given low-frequency variant may rapidly achieve dominance after a severe replication bottleneck is encountered. Our study strongly supports the notion that antiviral treatments such as remdesivir represent such a bottleneck. However, whether the resulting dominant species emerge in a stochastic fashion or whether they may represent escape mutants is another issue. In our study, we observed emergence of variants in nsp10 or the envelope gene, regions of the viral genome that are unlikely to be associated with a potential increase in remdesivir resistance, in one of the remdesivir-treated patients. In contrast, the pattern of mutations observed after the first round of remdesivir treatment in patient P1, and the fact that a viral rebound in this patient could only be controlled by another combinatorial remdesivir and convalescent plasma treatment suggest an at least partial escape to the initial treatment regimen. A prime candidate for a potential resistance mutation was V166L. Interestingly, while we did not reverse-engineer variant genotypes, an independent study26 performing an experimental remdesivir resistance screen observed selection for another mutation (V166A) at the exact same position of nsp12. We thus opine that our findings suggest a critical role for V166L in providing increased remdesivir resistance, although proof of this hypothesis will ultimately require experimental testing in appropriate in vitro systems.
Taken together, our work demonstrates that, while not generally observed in long-lasting infections, novel viral intra-host species may rapidly emerge in the context of antiviral treatments such as remdesivir therapy. Differentiating between true escape and stochastically arising variants in such patients, however, represents a particular challenge that will greatly benefit from additional data.
Our findings are significant in light of recent discussions regarding the use of remdesivir to treat non-hospitalized patients at high risk for disease progression,19 as well as potentially new antiviral therapeutics that are about to be incorporated into patient treatment strategies (e.g., protease inhibitors such as Paxlovid or RdRp-inhibiting compounds such as molnupiravir27,28). Unlike remdesivir, for which patient reports and in vitro studies on resistance mutations are available,2,3,5,8,11,23,26 similar data for Paxlovid or mulnopiravir treatment regimen are still lacking.
Therefore, we suggest monitoring patients with long-lasting viral infections by frequent testing of respiratory samples with whole viral genome analysis and variant determination and extending this monitoring to new directly acting therapeutics that may impose a different form of selection pressure when used in these patient populations.
Limitations of the study
The present study was performed in a relatively small number of patients with prolonged disease courses due to limited numbers of patients with sustained infection, comparable concomitant diseases and treatments during the pandemic. Some of the longitudinal samples exhibited low viral loads, and only one replicate per time point was sequenced. Despite the measures described in the STAR Methods section, there thus is the potential risk of potential false positive as well as false negative calls, which may have resulted from amplification biases or stringent filtering procedures implemented to avoid erroneous calls. Our primary conclusion—that remdesivir treatment increases diversity within the host and selects for mutations in the RNA polymerase—has to be validated in a larger number of samples. In addition, our study does not include direct experimental validation to confirm a direct linkage between observed mutations such as V166L and remdesivir resistance.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| CleanPlex SARS-CoV-2 Flex Panel | Paragon Genomics, CA, USA | Cat.# 918015 |
| Mid output NextSeq500 | Illumina, CA, USA | Cat.# 20024905 |
| CleanPlex plated Unique Dual-Indexed PCR Primers for Illumina Set C |
Paragon Genomics, CA, USA | Cat.# 716037 |
| CleanPlex plated Unique Dual-Indexed PCR Primers for Illumina Set D |
Paragon Genomics, CA, USA | Cat.# 716038 |
| CleanPlex plated Unique Dual-Indexed PCR Primers for Illumina Set E |
Paragon Genomics, CA, USA | Cat.# 716039 |
| CleanPlex plated Unique Dual-Indexed PCR Primers for Illumina Set F |
Paragon Genomics, CA, USA | Cat.# 716040 |
| cobas® SARS-CoV-2 | Roche | Cat.# 09175431190 |
| cobas® SARS-CoV-2 Control Kit | Roche | Cat.# 09175440190 |
| cobas® 6800/8800 Buffer Negative Control Kit | Roche | Cat.# 07002238190 |
| MagNA Pure 96 DNA and Viral Nucleic Acid Small Volume Kit | Roche | Cat.# 06543588001 |
| Deposited data | ||
| All SARS-CoV2 sequences are available at | ENA | PRJEB50471 |
| Software and algorithms | ||
| FastQC | Andrews S. (2010). | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Cutadapt | Martin M. (2011). | https://cutadapt.readthedocs.io/en/stable/ |
| Vsearch | Rognes T. (2016). | https://github.com/torognes/vsearch |
| FASTX-Toolkit | Hannon, G.J. (2010) | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Samtools | Danecek P. (2021). | https://github.com/samtools/samtools |
| Sambamba | Tarasov A. (2015). | https://lomereiter.github.io/sambamba/ |
| Samclip | N/A | https://github.com/tseemann/samclip |
| bamaddrg | N/A | https://github.com/ekg/bamaddrg |
| seqtk | N/A | https://github.com/lh3/seqtk |
| iVAR | Grubaugh, N.D. (2019). | https://github.com/andersen-lab/ivar |
| bedtools | Aaron R. Quinlan (2010). | https://bedtools.readthedocs.io/en/latest/ |
| Frrebayes | Garrison E. (2012). | https://github.com/freebayes/freebayes |
| bcftools | Danecek P. (2021). | https://samtools.github.io/bcftools/bcftools.html |
| vcflib | Garrison E. | https://github.com/vcflib/vcflib |
| Pangolin | O’Toole A. (2021). | https://github.com/stevenlovegrove/Pangolin |
| Nexstrain | Aksamentov, I. (2021). | https://clades.nextstrain.org/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Nicole Fischer (nfischer@uke.de).
Material availability
This study did not generate new unique reagents.
Experimental model and subject details
Fourteen patients with severe COVID-19 and persistent viral detection in the respiratory tract were included in this study. Patients were admitted to the University Medical Center Hamburg-Eppendorf between April 2020 and January 2021. RNA was extracted from routine diagnostic respiratory samples, and subjected to RT-qPCR. SARS-CoV-2 infection was considered prolonged if the infection period, defined as the period from hospital admission to the last PCR positive result in the respiratory tract, exceeded 14 days. Longitudinal sampling of respiratory samples of ICU patients was conducted every three to four days. Clinical data included patients' preexisting conditions, ventilation, and therapies. Additional parameters including inflammatory markers, such as CRP, and IL-6 levels were documented. The study was approved by the local ethics committee (PV7306).
Method details
SARS-CoV-2 qRT-PCR
SARS-CoV-2 loads were determined by qRT-PCR in a certified diagnostic environment as described previously.29,30
SARS-CoV-2 amplicon sequencing and variant calling
Whole genome amplicon sequencing and downstream bioinformatic analysis was performed as described.31,32 Sequencing libraries were generated using the CleanPlex SARS-CoV-2 Panel (Paragon Genomics, CA, USA). Libraries were multiplexed and sequenced on a Mid output NextSeq500 (Illumina). Complete or partially remaining sequencing adapters sequences are trimmed from the reads pairs extremities using cutadapt (-u -1 -U -1 -e 0.1 -O 9).33 Mate reads pairs are then merged with vsearch,34 allowing merging of staggered reads, and filtered for a minimum base quality of 15 (--fastq_qminout 15 –fastq_allowmergestagger). Unmerged reads are kept for downstream processing. A more stringent quality step in applied on merged reads using FASTX-Toolkit35 where only merged reads with a minimum base quality of 20 on at least 95% of the reads length are kept (fastq_quality_filter -q 20 -p 95). Merged reads are aligned to NC_045512.2 using minimap2 32 with default settings for short read alignment (-ax sr). Reads clipped from a minimum of 1 base are filtered out using samclip (https://github.com/tseemann/samclip) for downstream processing. Previously identified unmerged reads are aligned to NC_045512.2 using minimap2 32 with default settings for short read alignment (-ax sr) and all reads clipped from a minimum of 1 base are filtered out using samclip (https://github.com/tseemann/samclip) to be use further in the analysis stream. All original template of clipped reads from merged and unmerged pairs are recovered from filtered fastq files using seqtk (https://github.com/lh3/seqtk) to be aligned with STAR 33 for detection of larger deletions. The four obtained alignment files (merged reads not-clipped aligned with minimap2, unmerged reads not-clipped aligned with minimap2, merged reads clipped during minimap2 alignment and realigned with STAR, unmerged reads clipped during minimap2 alignment and realigned with STAR) are merged in a singled BAM file per sample. The final BAM file is then analyzed by iVAR36 to remove forward and reversed multiplex primers sequences at the edges of the reads. Coverage table are calculated using genomecov from BEDTools.37
Major and minor allele frequency variants within the samples of individual patients (≥1% allelic frequency) were called using FreeBayes Bayesian haplotype caller v1.3.138 with ploidy and haplotype independent detection parameters to generate frequency-based calls for all variants supported by a minimum coverage of 10 (-K -F 0.05 -min-coverage 9) in at least one data point per patient. The resulting variants were annotated using ANNOVAR 34 and consensus sequences based on major alleles (>90% AF) were generated using in-house scripts. Pangolin lineage assignment of consensus sequences was performed using the pangolin package.39
Variant filtering and classification
Our study aims at identifying nucleotide variant frequency changes across longitudinal patient samples. In this context, variable viral loads (and consequently coverage levels) pose a particular challenge to sample cross-comparison. To mitigate this problem, we employ a statistical model to estimate the significance of observed frequency changes. In this model, for each NV and sampling point, we assume that the frequency distribution at a given coverage c is equivalent to a random draw of c reads from a virtually unlimited pool of amplicons with a composition that approximates the authentic distribution of positive and negative parental genomes (note that this model does not consider potential PCR biases, and therefore must be considered optimistic). If so, we can calculate the minimum coverage required to conclude that, within a given error margin e, the observed frequency value fo does not substantially deviate from the unknown authentic NV frequency as:
where fo is the observed NV frequency (0 ≥ fo ≥ 1), z is the z score at the chosen confidence interval, and e is the acceptable error margin (for example, for fo = 0.3 and e = 0.05, the authentic frequency may fall between 0.25 and 0.35). Accordingly, we can test if the coverage value co at which a given NV was observed is sufficient to reasonably conclude that the authentic NV frequency is above or below a specific threshold ft. If so, error intervals centered on fo and ft must be smaller than the absolute distance between fo and ft:
The minimum coverage required to differentiate observed frequency values from a given threshold can then be calculated as:
In our longitudinal analysis, we test observed frequency and coverage values against a set of threshold values to filter datapoints and classify SNVs. Figure S1 shows a graphical depiction of the filtering and classification scheme. Firstly, to exclude spurious hits, we generally require the coverage to be minimally 10 and, depending on the observed frequency, sufficiently high to conclude that the authentic frequency is greater than 1% (ft > 0.01 in Figure S1). Secondly, for any given NV, at least one datapoint must exhibit coverage and frequency values that allow the conclusion that the authentic NV frequency is significantly above 20% (ft > 0.2 in Figure S1). We assume that there is little evidence for a biological selection advantage (or disadvantage) associated with a given NV if frequency values do not reach 20% in any of the investigated samples. Lastly, to principally allow us to establish whether a NV may undergo a minor-to-major or major-to-minor transition, values from at least two sampling points (which may or may not include the data point meeting the 20% frequency cutoff) must allow the reasonable classification of a NV as a major or minor variant. To meet this condition, coverage values must be sufficient to differentiate observed frequency values from the midpoint value (i.e, ft < 0.5 or ft > 0.5, for observed frequency value below or above 0.5, respectively).
SNVs with a sufficient number of qualifying data points are then subjected to classification as not significant, minor or major according to observed frequencies and coverage levels across all samples. Hybrid calls are being made in cases where a NV cannot be placed in a distinct category (see Figure S1).
Calling of transition events and calculation of transition p-values
To detect changes in variant frequencies that may reflect parental genotypes which acquire or loose dominance, we identify time points in which NV states transition from not significant or minor and major (or vice versa) when compared to the closest preceding time point meeting the selection criteria. Data points having ambiguous classification calls (4 and 5 in Figure S1), or coverage values below 10 do not qualify for this analysis. For each variant, we then calculate a transition probability corresponding to the mean of transition events divided by the number of intervening days across all qualifying samples. This value provides a lower estimate for the probability that a variant may undergo a transition per day. We then calculate an average variant transition probability across all samples by calculating the mean of individual transition probabilities.
We calculate p-values for the hypothesis that, in a given sample, the total number of observed transitions is greater than that expected from the average variant transition probability. To do so, for each variant which meets the above selection criteria (i.e., sufficient coverage levels such that a transition event could theoretically be observed), we first calculate an expected value by multiplying the total number of days between the time point in question and that of the closest preceding sample in which the variant meets selection criteria. The expected sample mean then corresponds to the sum of values from all variants:
where n is the number of variants v meeting selection criteria, d is the time period between the considered time point and the closest preceding sample in which v meets selection criteria, and is the average variant transition probability.
The p-value for above hypothesis then corresponds to the cumulative Poisson probability that the number of transition events is between the observed value and the total number of variants meeting selection criteria. Note that this value does not provide an absolute measure; it only indicates an unexpectedly high number of transitions under the null hypothesis that transition events occur with equal likelihood across all patient samples.
Acknowledgments
We thank Svenja Reucher, Christina Herrde, Kerstin Reumann, Anthea Spier, and Kathrin Cermann for excellent technical support. The work was supported by the Deutsche Forschungsgemeinschaft (DFG., German Research Foundation) (grant numbers AE 11/10-1 given to M.A., FI 782/6-1 given to N.F., and GR 3318/4-1 given to A.G.). J.S.zW. is funded by the DZIF and the DFG [SFB841 A6, SFB 1328 A12].
Author contributions
Conceptualization, J.S.zW., M.L., T.G., D.J., S.K., M.M.A., M.A., A.G., and N.F. Methodology, A.G. and N.F. Software, A.G. and A.R. Formal Analysis, A.G., A.R., and T.G. Investigation, T.H., A.R., T.G., and M.L. Resources T.H., J.S.zW., S.K., D.J., M.M.A., N.F., M.L., and M.A. Data curation, A.G., A.R., and T.G. Writing, N.F., J.S.zW., and A.G. Visualization, A.G., A.R., and T.G. Supervision, N.F. and A.G.
Declaration of interests
S.K. received research support from Cytosorbents and Daiichi Sankyo. He also received lecture fees from Astra, Bard, Baxter, Biotest, Cytosorbents, Daiichi Sankyo, Fresenius Medical Care, Gilead, Mitsubishi Tanabe Pharma, MSD, Pfizer, Philips, and Zoll. He received consultant fees from Fresenius, Gilead, MSD, and Pfizer. J.S.zW. received consultant and lecture fees from Gilead.
Published: September 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100735.
Contributor Information
Nicole Fischer, Email: nfischer@uke.de.
Adam Grundhoff, Email: adam.grundhof@leibniz-liv.de.
Supplemental information
Data and code availability
Whole genome amplicon sequencing data have been deposited at ENA and are publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Avanzato V.A., Matson M.J., Seifert S.N., Pryce R., Williamson B.N., Anzick S.L., Barbian K., Judson S.D., Fischer E.R., Martens C., et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell. 2020;183:1901–1912.e9. doi: 10.1016/j.cell.2020.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi B., Choudhary M.C., Regan J., Sparks J.A., Padera R.F., Qiu X., Solomon I.H., Kuo H.H., Boucau J., Bowman K., et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 2020;383:2291–2293. doi: 10.1056/NEJMc2031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kemp S.A., Collier D.A., Datir R.P., Ferreira I.A.T.M., Gayed S., Jahun A., Hosmillo M., Rees-Spear C., Mlcochova P., Lumb I.U., et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592:277–282. doi: 10.1038/s41586-021-03291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malsy J., Veletzky L., Heide J., Hennigs A., Gil-Ibanez I., Stein A., Lütgehetmann M., Rosien U., Jasper D., Peine S., et al. Sustained response after remdesivir and convalescent plasma therapy in a B-cell depleted patient with protracted COVID-19. Clin. Infect. Dis. 2020;73:e4020–e4024. doi: 10.1093/cid/ciaa1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nussenblatt V., Roder A.E., Das S., de Wit E., Youn J.H., Banakis S., Mushegian A., Mederos C., Wang W., Chung M., et al. Year-long COVID-19 infection reveals within-host evolution of SARS-CoV-2 in a patient with B cell depletion. Preprint at medRxiv. 2021 doi: 10.1101/2021.10.02.21264267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borges V., Isidro J., Cunha M., Cochicho D., Martins L., Banha L., Figueiredo M., Rebelo L., Trindade M.C., Duarte S., et al. Long-term evolution of SARS-CoV-2 in an immunocompromised patient with non-Hodgkin lymphoma. mSphere. 2021;6:e0024421. doi: 10.1128/mSphere.00244-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hensley M.K., Bain W.G., Jacobs J., Nambulli S., Parikh U., Cillo A., Staines B., Heaps A., Sobolewski M.D., Rennick L.J., et al. Intractable coronavirus disease 2019 (COVID-19) and prolonged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication in a chimeric antigen receptor-modified T-cell therapy recipient: a case study. Clin. Infect. Dis. 2021;73:e815–e821. doi: 10.1093/cid/ciab072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinot M., Jary A., Fafi-Kremer S., Leducq V., Delagreverie H., Garnier M., Pacanowski J., Mékinian A., Pirenne F., Tiberghien P., et al. Emerging RNA-dependent RNA polymerase mutation in a remdesivir-treated B-cell immunodeficient patient with protracted coronavirus disease 2019. Clin. Infect. Dis. 2021;73:e1762–e1765. doi: 10.1093/cid/ciaa1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy K.R., Rennick L.J., Nambulli S., Robinson-McCarthy L.R., Bain W.G., Haidar G., Duprex W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science. 2021;371:1139–1142. doi: 10.1126/science.abf6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cunha M.D.P., Vilela A.P.P., Molina C.V., Acuña S.M., Muxel S.M., Barroso V.d.M., Baroni S., Gomes de Oliveira L., Angelo Y.d.S., Peron J.P.S., et al. Atypical prolonged viral shedding with intra-host SARS-CoV-2 evolution in a mildly affected symptomatic patient. Front. Med. 2021;8:760170. doi: 10.3389/fmed.2021.760170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi S., Klein J., Robertson A., Peña-Hernández M.A., Lin M.J., Roychoudhury P., Lu P., Fournier J., Ferguson D., Mohamed Bakhash S.A., et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Preprint at medRxiv. 2021 doi: 10.1101/2021.11.08.21266069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan A.T., Linster M., Tan C.W., Le Bert N., Chia W.N., Kunasegaran K., Zhuang Y., Tham C.Y.L., Chia A., Smith G.J.D., et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021;34:108728. doi: 10.1016/j.celrep.2021.108728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu B., Guo H., Zhou P., Shi Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021;19:141–154. doi: 10.1038/s41579-020-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang B., Fan J., Huang J., Guo E., Fu Y., Liu S., Xiao R., Liu C., Lu F., Qin T., et al. Clinical and molecular characteristics of COVID-19 patients with persistent SARS-CoV-2 infection. Nat. Commun. 2021;12:3501. doi: 10.1038/s41467-021-23621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malin J.J., Spinner C.D., Janssens U., Welte T., Weber-Carstens S., Schalte G., Gastmeier P., Langer F., Wepler M., Westhoff M., et al. Key summary of German national treatment guidance for hospitalized COVID-19 patients : key pharmacologic recommendations from a national German living guideline using an Evidence to Decision Framework. Infection. 2021;50:93–106. doi: 10.1007/s15010-021-01645-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., et al. Remdesivir for the treatment of covid-19 - final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ansems K., Grundeis F., Dahms K., Mikolajewska A., Thieme V., Piechotta V., Metzendorf M.I., Stegemann M., Benstoem C., Fichtner F. Remdesivir for the treatment of COVID-19. Cochrane Database Syst. Rev. 2021;8:CD014962. doi: 10.1002/14651858.CD014962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gandhi R.T., Malani P.N., del Rio C. COVID-19 therapeutics for nonhospitalized patients. JAMA. 2022;327:617–618. doi: 10.1001/jama.2022.0335. [DOI] [PubMed] [Google Scholar]
- 19.Gottlieb R.L., Vaca C.E., Paredes R., Mera J., Webb B.J., Perez G., Oguchi G., Ryan P., Nielsen B.U., Brown M., et al. Early remdesivir to prevent progression to severe covid-19 in outpatients. N. Engl. J. Med. 2021;386:305–315. doi: 10.1056/NEJMoa2116846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spinner C.D., Gottlieb R.L., Criner G.J., Arribas López J.R., Cattelan A.M., Soriano Viladomiu A., Ogbuagu O., Malhotra P., Mullane K.M., Castagna A., et al. Effect of remdesivir vs standard Care on clinical status at 11 Days in patients with moderate COVID-19: a randomized clinical trial. JAMA. 2020;324:1048–1057. doi: 10.1001/jama.2020.16349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bravo J.P.K., Dangerfield T.L., Taylor D.W., Johnson K.A. Remdesivir is a delayed translocation inhibitor of SARS-CoV-2 replication. Mol. Cell. 2021;81:1548–1552.e4. doi: 10.1016/j.molcel.2021.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., COVID-19 Genomics UK COG-UK Consortium. Robertson D.L. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szemiel A.M., Merits A., Orton R.J., MacLean O.A., Pinto R.M., Wickenhagen A., Lieber G., Turnbull M.L., Wang S., Furnon W., et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021;17:e1009929. doi: 10.1371/journal.ppat.1009929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voloch C.M., da Silva Francisco R., Jr., de Almeida L.G.P., Brustolini O.J., Cardoso C.C., Gerber A.L., Guimarães A.P.d.C., Leitão I.d.C., Mariani D., Ota V.A., et al. Intra-host evolution during SARS-CoV-2 prolonged infection. Virus Evol. 2021;7:veab078. doi: 10.1093/ve/veab078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lythgoe K.A., Hall M., Ferretti L., de Cesare M., MacIntyre-Cockett G., Trebes A., Andersson M., Otecko N., Wise E.L., Moore N., et al. SARS-CoV-2 within-host diversity and transmission. Science. 2021;372:eabg0821. doi: 10.1126/science.abg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stevens L.J., Pruijssers A.J., Lee H.W., Gordon C.J., Tchesnokov E.P., Gribble J., George A.S., Hughes T.M., Lu X., Li J., et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022;14:eabo0718. doi: 10.1126/scitranslmed.abo0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021;12:6055. doi: 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabinger F., Stiller C., Schmitzová J., Dienemann C., Kokic G., Hillen H.S., Höbartner C., Cramer P. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 2021;28:740–746. doi: 10.1038/s41594-021-00651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nörz D., Fischer N., Schultze A., Kluge S., Mayer-Runge U., Aepfelbacher M., Pfefferle S., Lütgehetmann M. Clinical evaluation of a SARS-CoV-2 RT-PCR assay on a fully automated system for rapid on-demand testing in the hospital setting. J. Clin. Virol. 2020;128:104390. doi: 10.1016/j.jcv.2020.104390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfefferle S., Reucher S., Nörz D., Lütgehetmann M. Evaluation of a quantitative RT-PCR assay for the detection of the emerging coronavirus SARS-CoV-2 using a high throughput system. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.9.2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Günther T., Czech-Sioli M., Indenbirken D., Robitaille A., Tenhaken P., Exner M., Ottinger M., Fischer N., Grundhoff A., Brinkmann M.M. SARS-CoV-2 outbreak investigation in a German meat processing plant. EMBO Mol. Med. 2020;12:e13296. doi: 10.15252/emmm.202013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pfefferle S., Günther T., Kobbe R., Czech-Sioli M., Nörz D., Santer R., Oh J., Kluge S., Oestereich L., Peldschus K., et al. SARS Coronavirus-2 variant tracing within the first Coronavirus Disease 19 clusters in northern Germany. Clin. Microbiol. Infect. 2021;27:130.e5. doi: 10.1016/j.cmi.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:3. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 34.Rognes T., Flouri T., Nichols B., Quince C., Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hannon G.J. 2011. FASTX-Toolkit. [Google Scholar]
- 36.Grubaugh N.D., Gangavarapu K., Quick J., Matteson N.L., De Jesus J.G., Main B.J., Tan A.L., Paul L.M., Brackney D.E., Grewal S., et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019;20:8. doi: 10.1186/s13059-018-1618-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrison E., Marth G. Haplotype-based variant detection from short-read sequencing. Preprint at arXiv. 2012 doi: 10.48550/arxiv.1207.3907. [DOI] [Google Scholar]
- 39.O'Toole Á., Scher E., Underwood A., Jackson B., Hill V., McCrone J.T., Colquhoun R., Ruis C., Abu-Dahab K., Taylor B., et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021;7:veab064. doi: 10.1093/ve/veab064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome amplicon sequencing data have been deposited at ENA and are publicly available as of the date of publication. Accession number is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.