

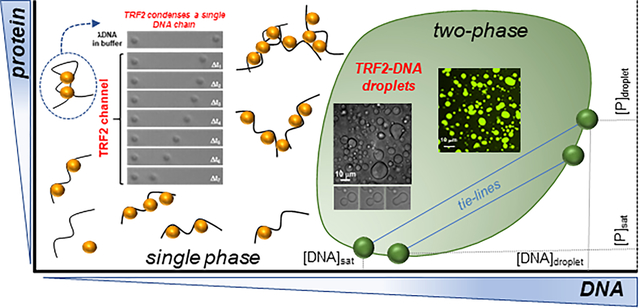
Abstract
Telomeres are nucleoprotein complexes that protect the ends of chromosomes and are essential for chromosome stability in Eukaryotes. In cells, individual telomeres form distinct globules of finite size that appear to be smaller than expected for bare DNA. Moreover, telomeres can cluster together, form telomere-induced-foci or co-localize with promyelocytic leukemia (PML) nuclear bodies. The physical basis for collapse of individual telomeres and coalescence of multiple ones remains unclear, as does the relationship between these two phenomena. By combining single-molecule force spectroscopy measurements, optical microscopy, turbidity assays, and simulations, we show that the telomere scaffolding protein TRF2 can condense individual DNA chains and drives coalescence of multiple DNA molecules, leading to phase separation and the formation of liquid-like droplets. Addition of the TRF2 binding protein hRap1 modulates phase boundaries and tunes the specificity of solution demixing while simultaneously altering the degree of DNA compaction. Our results suggest that the condensation of single telomeres and formation of biomolecular condensates containing multiple telomeres are two different outcomes driven by the same set of molecular interactions. Moreover, binding partners, such as other telomere components, can alter those interactions to promote single-chain DNA compaction over multiple-chain phase separation.
Keywords: TRF2, hRap1, phase separation, DNA condensation, telomeres
Graphical Abstract
Introduction
Biomolecular condensates are non-stochiometric assemblies of biomacromolecules that underlie a variety of cellular processes including the stress response, ribosomal assembly, germ-granule formation and chromatin organization [1–8]. These assemblies are commonly enriched in multivalent DNA/RNA binding proteins and nucleic acids, with multivalency being a critical feature that facilitates formation of a set of reversible inter-molecular interactions [9–13]. As a result of this multivalency, many of the proteins that localize in condensates have been shown to undergo phase separation in vitro and in vivo, lending support to a model in which intracellular phase separation plays a key role in cellular organization and compartmentalization [14–17].
The emergent property of biomolecular condensates parallels previously observed characteristics of ligand-induced condensation of nucleic acids, in which multivalent interactions provided by DNA/RNA binding proteins can promote either the compaction of single nucleic acid chains or the condensation of multiple ones. Indeed, as proposed by Post and Zimm [18], from a polymer physics standpoint, the condensation of a single nucleic acid chain by a ligand and the phase separation of mixtures of nucleic acids and the same ligands can be rationalized as two distinct outcomes of the same set of interactions. Advancements in single-molecule methods and their application have paved the way to a direct investigation of protein-mediated nucleic acid condensation [19–24]. While the relative concentration regimes and molecular architecture of the components play key roles, the physical basis that determines whether protein-nucleic acids interactions lead to intra-molecular loops and chain compaction or to inter-chain interactions and solution demixing remains to be fully elucidated. An important implication of this duality is that, under appropriate conditions, many proteins that can induce the condensation of single nucleic-acid chains will instead promote phase separation. Though rooted into a similar theoretical framework [18, 25], this is a distinct phenomenon from the compaction and phase separation observed for a single protein [26–28], since it entails the interaction between a nucleic acid (often significantly long) and its interaction with ligands. Identifying how these two possible outcomes are encoded in the architecture of proteins and their interaction with nucleic acids having specific and nonspecific sequences is fundamental to our understanding of how the genetic material may be organized and regulated in vivo. To address this question directly we focused on the telomeric protein TRF2 and sought to explore the symmetry between nucleic acid compaction and phase separation.
Telomeres are nucleoprotein complexes that protect the ends of linear chromosomes. Human telomeres are formed by the six shelterin subunits TRF1, TRF2, Rap1, TIN2, TPP2 and POT1 that co-assemble at the end of chromosomes [29] (Figure 1A), with the four-protein core complex TRF1-TIN2-TRF2-Rap1 forming the major scaffold bound to the terminal double-stranded DNA containing T2AG3 repeats. Homodimers of TRF1 and TRF2 bind specifically to the telomeric repeats [30–32]. Rap1 interacts with TRF2 with high affinity [33, 34] and suppresses the DNA damage response at telomeres [35], while TIN2 bridges TRF1 and TRF2 [36, 37]. TIN2 also interacts with TPP1-POT1 bound to the G-rich 3’ single-stranded DNA overhang, providing a physical link with the core complex formed on the double-stranded DNA telomeric region and thus stabilizing a T-loop structure proposed to regulate telomere accessibility [38–40].
Figure 1. TRF2 and Rap1.
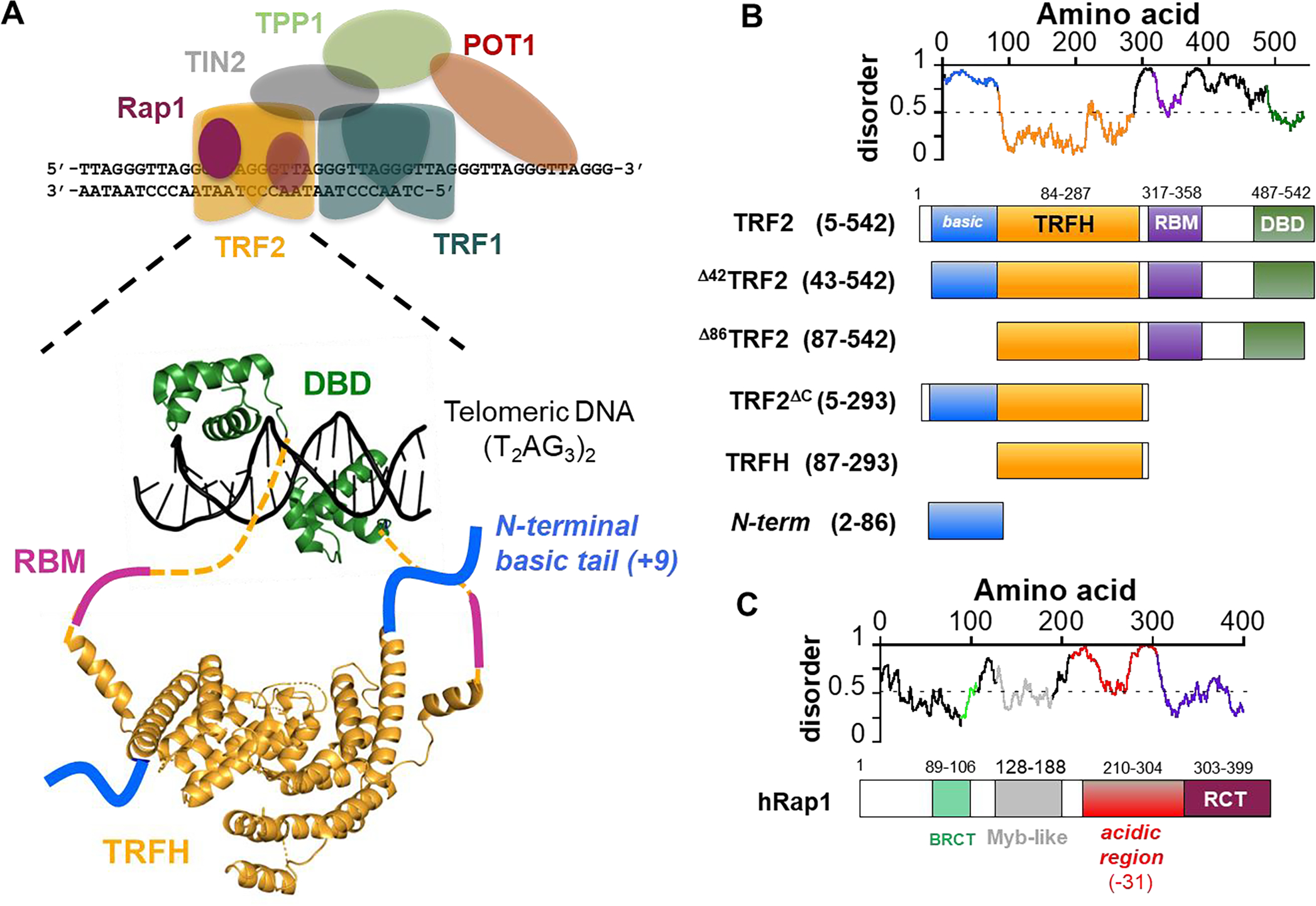
A. Schematic representation of the 6 protein shelterin complex (TRF1, TRF2, Rap1, TIN2, TPP1, POT1). Enlarged is a representation of the architecture of TRF2 dimer bound to telomeric DNA: DBD, DNA-binding domain; TRFH, homodimerization domain; RBM, Rap1 binding motif. The dashed lines represent unstructured linkers connecting the TRFH to the DBD. Structures are based on PDB codes 1h6p [106] and 1w0u [107]. B. Disorder/order predictions of TRF2 (IUPred) [108] and schematics of the TRF2 constructs studied in this work. C. Schematic of Rap1: BRCT, BRCA1 C-terminal domain; Myb domain; acidic region with a −31 net charge spanning 100 aa; RCT, Rap1 C-terminus domain that interacts with the RBM of TRF2.
While telomeres are usually thought of as the nucleoprotein complexes formed at the ends of individual chromosomes, they are not always observed as distinct and separate nuclear entities. Though usually restricted in their motion, telomeres can transiently cluster together [41, 42] and dysfunctional telomeres assemble into telomere dysfunction-induced foci [43]. Also, 10–15 % of cancer cells maintain their telomere length via Alternative Lengthening of Telomeres (ALT) [44, 45], a telomerase-independent pathway. In these cells, multiple telomeres co-localize with a subset of promyelocytic leukemia (PML) nuclear bodies to form ALT-associated promyelocytic leukemia bodies (APBs), within which telomeres are elongated [46–49]. Intriguingly, PML bodies are a type of biomolecular condensates [14, 50], and a recent study has proposed that within APBs telomeres behave as phase-separated components [51]. Whether the apparent phase-separated nature of APBs originates from an intrinsic ability of PML bodies to form biomolecular condensates or from one or more of the shelterin components to induce phase separation with telomeric DNA remains to be determined.
Of the two telomeric proteins that directly interact with DNA, TRF2 has long been implicated in DNA condensation in vitro, as monitored by gel during electrophoresis [52] or Dual Resonance Frequency Enhanced Electrostatic Microscopy (DREEM) imaging [53]. The molecular nature of these TRF2-DNA condensed states remains elusive. Single-molecule DNA curtains experiments [54] showed that pre-formed shelterin complexes do not lead to compaction of individual DNA chains or interaction between different DNA chains, suggesting that any ability of TRF2 to collapse/condense DNA may be modulated by interaction with other shelterin components. Also, individual telomeres typically appear as distinct globules of finite size, albeit T-loops where the 3’ ssDNA end is tucked into the dsDNA telomeric region can be visualized in vivo with high resolution imaging [55]. These globules seem to be more compact than expected for equivalent lengths of bare dsDNA [56, 57], suggesting that interaction of one or more shelterin component with DNA may favor condensation of telomeres. However, studies testing the idea that decompaction of telomeres may be a means to regulate telomere accessibility in vivo reached divergent conclusions [58–60].
These observations raise key and unanswered questions – do telomeric proteins induce compaction of telomeric DNA? – if so, what are the physical and molecular bases for regulation of telomere compaction? – does phase separation play a role in establishing and regulating telomeres? To begin addressing these questions, we employed a collection of biophysical methods to directly test whether specific domains of TRF2 (Figure 1B) induce the compaction of individual DNA chains and to investigate if and how binding of TRF2 to DNA leads to liquid-liquid mixing of the solutions. Our results reveal a symmetry between single-chain compaction and multi-chain condensation (i.e., phase separation), where perturbations that influence single-chain compaction are mirrored by perturbation that alters phase separation (such as salt concentration, binding of hRap1 to TRF2). Taken together, our results support the hypothesis that these emergent phenomena can be interpreted as two distinct outcomes controlled by the same set of intra- and inter-molecular interactions as well as by co-solutes.
Results.
TRF2 promotes single-chain dsDNA condensation.
We first sought to establish if TRF2 can drive single-chain DNA compaction. TRF2 has a modular architecture with an N-terminal basic disordered region, a central TRFH homo-dimerization domain, a disordered linker, and a C-terminal DNA-binding domain (DBD) that specifically recognizes T2AG3 telomeric repeats (Figure 1A). When purified homo-dimeric TRF2 (Supplementary Figure S1A–C) is combined with telomeric repeat DNA, electrophoretic mobility into the gel is impeded in a DNA-dependent manner (Supplementary Figure S1D) [52], consistent with protein-mediated DNA condensation or aggregation. To directly test whether TRF2 compacts individual DNA chains, we employed two different single-molecule force spectroscopy approaches.
First, we used two independently steerable optical traps and a laminar flow chamber to trap a single λDNA molecule in a dumb-bell configuration between two polystyrene beads (Figure 2A, and top panel in 2B). By turning off the downstream trap relative to the flow, we varied the drag force on the free bead (i.e., the bead on the right in Figure 2B) by varying the flow and measured DNA extension in the presence and absence of TRF2. In buffer alone, the DNA extension responds reversibly to changes in drag force (Figure 2C, black trace). In contrast, upon movement of the DNA into a channel containing TRF2, a dramatic reduction in DNA extension was observed when the drag force was lowered. This DNA compaction was rapid and complete, as the free bead was eventually brought all the way into the active trap, thus ending the experiment (Figure 2B and 2C). As expected for a protein-dependent phenomenon, the rate of DNA compaction increases with increasing TRF2 concentration (Figure 2C).
Figure 2. TRF2 condensation of single nucleic acids chains.
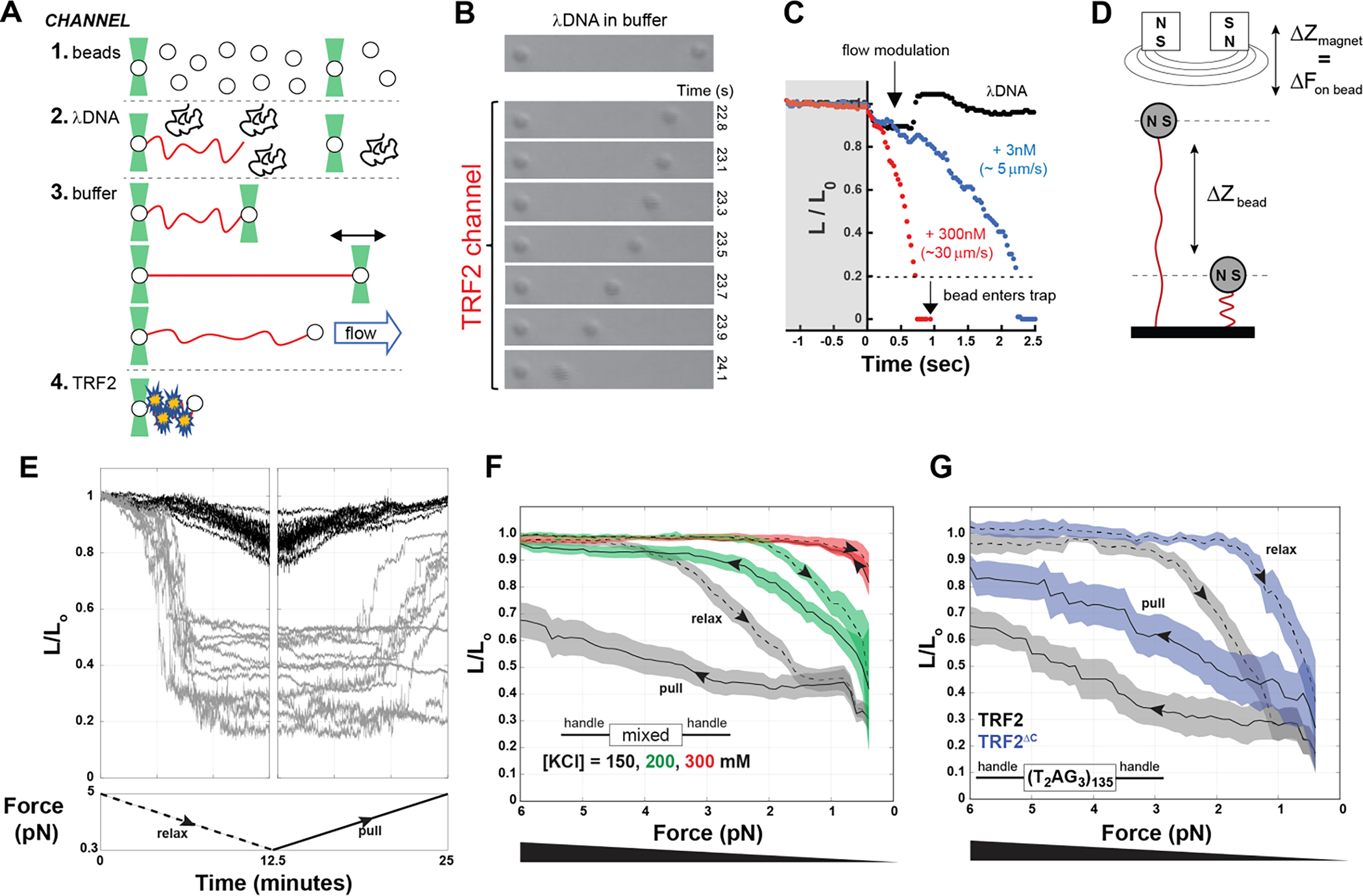
A. Schematic of optical tweezers experiments. 1 to 4 are schematics of the laminar flows used to assemble λDNA in a dumbbell configuration between two beads, before incubating it with protein. B and C. Compaction of DNA (expressed as the change in relative length L/L0) in presence of 300 nM (monomer) TRF2 as visually monitored by the decreasing distance between the beads over time (B) or quantified by tracking the position of the right bead (C), in the presence of TRF2 at 3 nM (blue) or at 300 nM (red) compared to the absence of TRF2 (black). D. Schematic of the magnetic tweezer experiment setup. E. Example of magnetic tweezer trajectories after relaxing (gray, first panel) and pulling (gray, second panel) the nucleic acid in the presence of TRF2. The DNA alone is shown as reference in black. F. Quantification of DNA compaction as function of force for dsDNA of mixed sequence in the presence of 300 nM TRF2 (dashed and solid lines are the average and color bands the standard deviation of multiple traces in either the relax or pull direction, respectively). Increasing KCl concentration decreases the hysteresis between relax and pull experiments as well as the degree of force required to expand the conformations of the DNA. G. DNA compaction measured for the specific telomeric DNA sequence in presence of 300 nM TRF2 or its variant TRF2ΔC, which lacks the DBD and the linker region.
To further examine the force-dependence of TRF2-dependent DNA compaction, we turned to a magnetic tweezers assay which offered the benefits of higher throughput and the ability to precisely and stably establish constant forces between 0.5 – 5 pN. In these experiments, dsDNA molecules of ~2 kbp length were attached at one end to a glass coverslip and at the other to a paramagnetic bead (Figure 2D). DNA tethers were initially held at > 5 pN of force, the force was ramped down to < 0.5 pN, and then back to high force, with ramping at a rate of ~ 0.6 pN / min (Figure 2E). The force dependence of the end-to-end distance of bare DNA behaves as expected [61, 62] (Figure 2E, black traces). Consistent with the optical tweezer experiments, the addition of TRF2 leads to DNA compaction (Figure 2E, gray traces, first panel). We note that, at difference with bare DNA, in the presence of TRF2 the individual DNA tethers show a wide range of behaviors, including cases in which they remained irreversibly compacted even at the highest forces we applied (~ 5 pN) (Figure 2E, gray traces, second panel). This is suggestive of a highly cooperative mode of DNA condensation. Indeed, the presence of TRF2 leads to a significant hysteresis between relaxation (i.e., lowering the force) and pulling (i.e., increasing the force) (Figure 2F), consistent with a large degree of cooperativity in the compacted state. TRF2-dependent DNA compaction and hysteresis were observed even at salt concentrations typically considered to be in the physiological range (Figure 2F) and also with DNA tethers containing 0.8 kbp of human telomeric repeats ((T2AG3)135) (Figure 2G in black).
Even though the biological function of TRF2 lies in its ability to specifically interact with telomeric DNA, our results clearly revealed that DNA compaction can occur independently of telomeric repeats. Since the DNA binding domain is known to interact specifically with telomeric repeats and these repeats appeared dispensable for compaction, we asked if the DNA binding domain (DBD) of TRF2 was also dispensable for DNA compaction. To our surprise, a TRF2 construct comprised of only the basic N-terminus and the dimerization domain (TRF2ΔC, Figure 1B) can also condense single DNA chains (Figure 2G in blue), albeit at lower forces and with less hysteresis, indicative of a more weakly-interacting system. These results suggest that the intrinsically disordered N-terminal domain directly contributes to DNA compaction, albeit perhaps in a non-specific manner.
To complement the single-molecule force spectroscopy experiments, we performed Monte Carlo simulations using a hyper coarse-grained model in which a force constant is used to extend a single “DNA” chain while “TRF2” molecules interact with the chain in a multivalent manner (Figure 3A). Simulations was performed using two different TRF2:DNA interaction strengths (Figure 3B, top). Importantly, in a first set of simulations TRF2:TRF2 interactions were completely absent, mimicking a scenario in which TRF2 does not engage in homotypic interactions. Under this regime, we observed a continuous transition in DNA extension as a function of force (Figure 3B top, Figure 3C). Such a result is in poor agreement with our force experiments (Figure 2E), which do not show a continuous dependence on extension with force. Instead, force experiments appear to reveal two regimes; a largely extended DNA molecule (observed under high force) and a largely compact DNA molecule (observed under low/no force).
Figure 3. Simulation of TRF2-dependent DNA condensation.
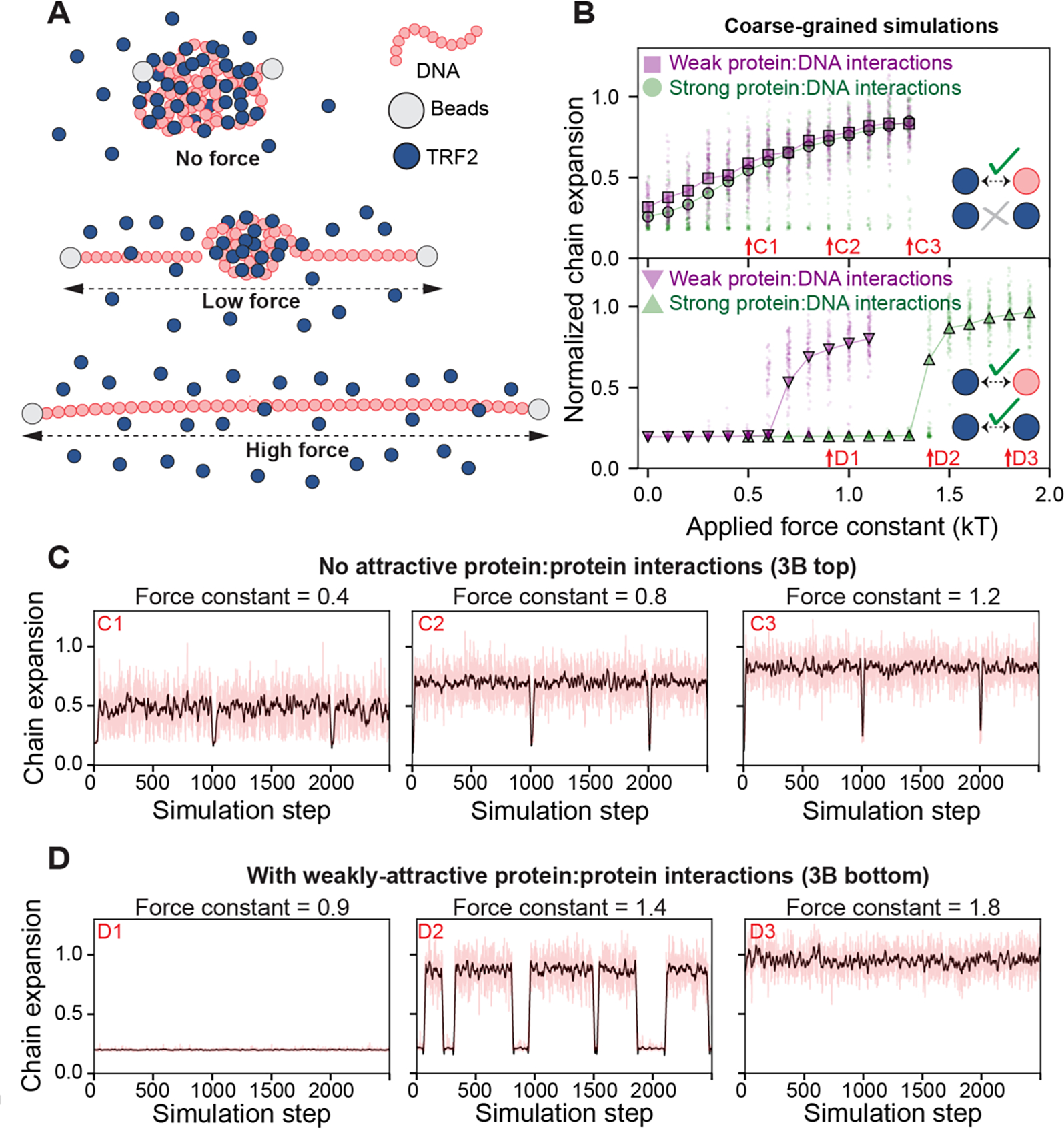
A. Schematic of setup for coarse-grained Monte Carlo simulations. A single DNA molecule of 80 beads was simulated with 4000 single-bead TRF2 molecules. The DNA end-to-end distance was constrained via a harmonic potential applied to the ends of the DNA (beads) with a variable force constant, mimicking force experiments. Under no force, TRF2 molecules lead to DNA condensation, forming a spherical globule. Under low force, parts of the DNA are extended with a single spherical globule of DNA and TRF2 in the center. Under high force, the DNA molecule is fully extended with no TRF2 bound. B. Simulation results depend on the presence or absence of weak homotypic TRF2 interactions. If TRF2:TRF2 interactions are set to zero (i.e., implying TRF2 is infinitely soluble) we observed a continuous increase in chain dimensions as a function of force constant, both under weak (purple squares) and strong (green circles) protein-DNA interactions (top). If weak TRF2:TRF2 interactions are included (leading to a solubility limit of ~0.5 mM for TRF2), we observe apparent two-state behavior (bottom). Here the DNA is either compact or expanded, as characterized by the sharp transition as a function of force and the co-existing populations giving rise to a bimodal distribution. The force constant at which expansion occurs depends on the strength of the protein:DNA interaction. Arrows reference simulation traces in panels 3C and 3D. C. Individual simulation traces for simulations performed absent TRF2:TRF2 interactions. Red lines are true data, black line is a smoothed trace. Occasional troughs are transitions into short-lived metastable compact globular states. D. Individual simulation traces for simulations performed with weak TRF2:TRF2 interactions, revealing two-state behavior. During a single simulation, the system oscillates between two fixed points (compact and expanded). This oscillation is analogous to an infinitely cooperative transition, as seen when sitting at a phase boundary. Red lines are true data, black line is a smoothed trace.
Why does our model not recapitulate the gross behavior of the force experiments? One possibility is the limiting assumption that TRF2 cannot engage in any kind of attractive interactions with itself. While TRF2 is monodisperse in isolation (Supplementary Figure S1B), it contains multiple disordered regions which may engage in both specific and non-specific protein-protein interactions [63]. More generally, proteins are typically not infinitely soluble under aqueous solutions, implying the presence of weak, non-specific intermolecular interactions [64]. This becomes especially relevant in the context of proteins that can interact at multiple degenerate sites along longer scaffolding molecules such as DNA or RNA, which can function as multivalent binding platforms to drive up the effective concentration. With this in mind, we considered an alternative model in which weakly attractive TRF2:TRF2 interactions were included. These interactions are extremely weak; in the absence of DNA, the protein remains soluble up to around 0.5 mM (~30 mg/ml, 3x more soluble than lysozyme). Despite this, the inclusion of weak TRF2:TRF2 interactions fundamentally shifts the dependence of DNA compaction on force (Fig 3B, bottom). In simulations performed with weak TRF2:TRF2 interactions, we observe a highly cooperative transition where the chain is either fully compact or fully extended, in line with the observations in force experiments (Figure 2E, Supplementary Figure S2, Figure 3B and 3D). These simulations sample from the underlying Boltzmann-weighted conformational ensemble and report on instantaneous chain dimensions, with representative traces from individual simulations shown in Figure 3C and 3D. Importantly, even within a single simulation apparent two-state behavior emerges, with the system transitioning between a compact globule and an expanded chain (Figure 3D).
Taken together, both experimental and simulated data strongly suggest that while the DBD of TRF2 contributes to an increase of interaction strength due to the specific binding to the T2AG3 repeats, weaker protein-DNA interactions in the context of the multivalent nature of TRF2 may be key drivers of the cooperative compaction of single DNA chains.
Interaction of TRF2 with multiple dsDNA molecules leads to phase separation.
Polymer theory predicts that the same molecular interactions that lead to collapse of a single DNA chain can also lead to condensation when multiple chains are present in solution at high enough concentration [18]. Indeed, TRF2 forms complexes with DNA that during electrophoresis barely migrate into the gel (Supplementary Figure S1D) [52], which is consistent with formation of either large supramolecular assemblies, amorphous aggregates or, possibly, phase separation. With these possibilities in mind, we found that while TRF2 is a well-behaved dimer in solution (Supplementary Figure S1), at a sufficiently high concentration mixing of TRF2 with non-specific dsDNA (supercoiled or linear fragments) results in an opalescent solution that contains micron-sized droplets, as revealed by Differential Interference Contrast (DIC) microscopy (Figure 4A and S3A). In addition, these droplets fuse over time (Supplementary Figure S3C), consistent with a system that has undergone phase separation to form a dynamic and liquid-like dense-phase.
Figure 4. TRF2 phase separation with specific and non-specific dsDNA.
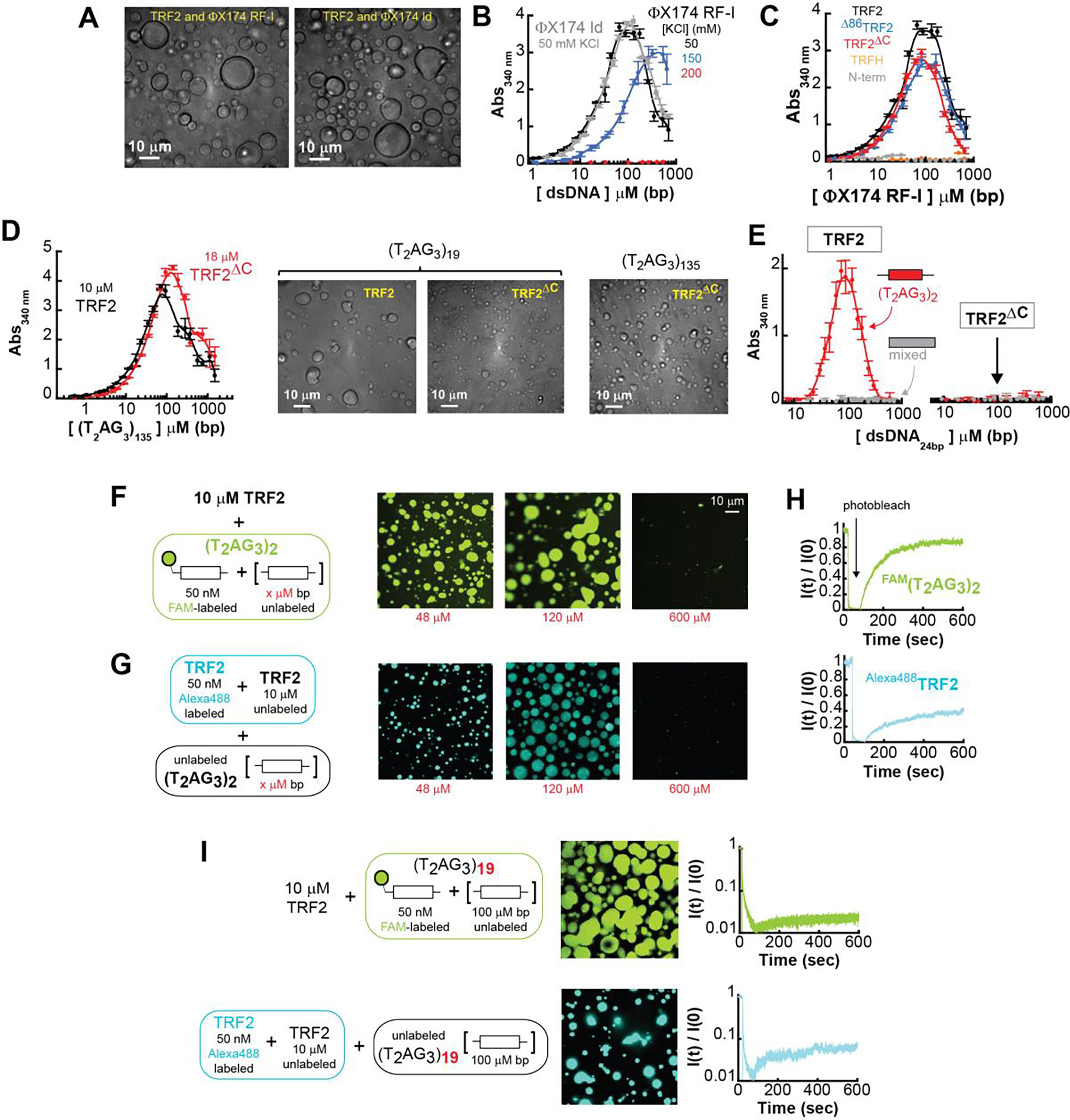
A. DIC microscopy images of phase-separated droplets in presence of ϕX174 RF-I at the indicated KCl concentration, and ϕX174 HaeIII digest (gray). B. Turbidity measurement of solutions containing a fixed concentration of TRF2 (10 μM monomer) and variable concentrations of ϕX174 RF-I (black) or ϕX174 ld (gray). Increasing salt concentration from 50 mM KCl to 150 mM KCl (blue) shifts phase boundaries to higher concentrations. A further increase to 200 mM KCl (red) suppresses phase separation at this concentration of TRF2. Error bars represent the standard deviation from three repeats, and the solid lines are interpolations of the data to facilitate visualization. C. Turbidity experiments of solution containing non-specific ϕX174 RF-I in presence of truncation variants of the protein at fixed concentration of 10 μM monomer. Turbidity is suppressed for the TRFH construct and strongly reduced for the N-terminal tail fragment. D. Turbidity measurements of TRF2 and TRF2ΔC variant reveals phase separation in presence of specific ds-DNA sequences. E. TRF2 (10 μM mon) undergoes phase separation only with a 24 bp DNA that contains a (T2AG3)2 repeat in its center, and not with a DNA of mixed sequence composition of the same length. Under identical conditions truncation of the C-terminus (TRF2ΔC) suppresses phase separation also for specific DNA. F-G. Re-entrant behavior of the phase separation with increasing concentration of specific dsDNA. H-I. FRAP of labeled TRF2 and specific DNA mixtures shows order of magnitude decrease in mobility with increasing length (from 2 to 19) of the number of specific telomeric binding sites on the dsDNA substrate.
The formation of scattering objects (i.e., droplets) was further assessed by absorbance scattering at 340 nm, where neither protein nor nucleic acid absorb. Light scattering of a multicomponent solution can reflect changes in the free-energy of mixing of the solution [65, 66] and, therefore, it may allow identification of the boundaries of the phase-diagram. The solution absorbance displayed classical re-entrant behavior as a function of DNA concentration [67] (Figure 4B). Absorbance initially increases, reflecting the formation of droplets; however, as the DNA concentration is increased further, absorbance then decreases, in line with the ‘re-entrance’ into the one-phase regime. This re-entrant behavior is expected for a two-component system governed by a closed-loop phase-diagram [67]. Whereas the re-entrant phase may be interpreted in the context of charge inversion of oppositely charged polyelectrolyte [67], the phenomenon can occur also when the interaction is realized between uncharged domains [68]. More in general, in a mixture of two different components, the re-entrant phase reflects the saturation of the binding sites of one component by the second component, such that binding across two different molecules of the same component is disfavored.
Phase separation occurs over the same concentration regime with either supercoiled DNA or its linear fragments, and at a physiological range of salt concentrations (Figure 4B). Importantly, the salt concentrations over which phase separation is observed mirror those under which TRF2-dependent DNA compaction occurs. Taken together, our results demonstrate that TRF2 can induce both DNA compaction and phase separation.
Having established that TRF2 can drive phase separation with DNA, we designed a series of TRF2 constructs to assess the contribution of specific protein regions to phase separation (Figure 1B and Supplementary Figure S1A–C). Neither deletion of the basic domain (Δ86TRF2) nor removal of the DBD and the unstructured linker (TRF2ΔC) suppressed DNA-dependent phase separation (Figure 4C). These observations indicate that TRF2-dependent phase separation is neither dictated exclusively by the presence of the disordered N-terminal basic region nor by the linker and DBD. Moreover, neither the TRFH domain nor the N-terminal region alone promote phase separation at the same protein concentrations as observed for full length protein (Fig 4C) but can be driven to phase separate with DNA at higher concentrations (Supplementary Figure S3D). These data strongly suggest that multivalent interactions with the nucleic acids are required for efficient phase separation and that multi-valency is realized via both unstructured and structured domains of TRF2 that act in a synergistic fashion.
Specificity and multi-valency of telomeric repeats.
Given the specificity and high affinity with which the DBD of TRF2 binds telomeric repeat sequences [69], we would anticipate that at cellular concentrations, telomeric repeat DNA will be preferentially bound by TRF2. With this in mind and considering TRF2 ability to undergo phase separation with non-specific dsDNA, we anticipated that TRF2 would also phase separate when mixed with telomeric repeat sequence DNA. However, in contrast to non-specific dsDNA, we expected that TRF2 phase separation with telomeric repeat DNA would be strongly dependent on the presence of the DNA binding domain. As expected, solution demixing was observed in the presence of telomeric repeat-containing DNA and TRF2 (Figure 4D). Yet, while slightly higher protein concentrations are required to achieve equivalent turbidity, the TRF2ΔC construct, which lacks the DBD, is still able to drive DNA-dependent phase separation with dsDNA that contains multiple telomeric repeats (Figure 4D). Taken at face value, these data could be interpreted to suggest that DNA-dependent phase separation is driven by non-specific interactions of TRF2 with DNA, with the specific recognition of telomeric repeats by the DBD playing a limited role in the process. Alternatively, the multivalent interactions mediated by different TRF2 domains and the DNA may mask the impact of DNA specificity encoded by the DBD in the context of telomeric repeat sequences.
To delineate between these two interpretations, we sought to test for the presence of DNA-specific interactions with shorter DNA repeat sequences. For this, we turned to a short dsDNA fragment of 24 bps that contains a single (T2AG3)2 repeat in its center and a length-matched control that lacks the (T2AG3)2 repeat (mix sequence dsDNA). Strikingly, at the protein concentration used in the assay, phase separation occurs only in the presence of telomeric repeats (Figure 4E). Furthermore, independent of the presence of repeats, a significant suppression of phase separation was observed with the TRF2ΔC construct that lacks the DBD (Figure 4E), in stark contrast to what was observed with longer DNA substrates. Thus, once the valence of the nucleic acid is reduced by decreasing its length, the importance of DBD-encoded specificity for telomeric repeats is clearly unmasked.
The importance of DBD-mediated specific interaction is further supported by fluorescence confocal imaging experiments. Droplets of TRF2 and a fluorescein-labeled 24 bp dsDNA containing the (T2AG3)2 repeat, formed in the presence of different concentrations of unlabeled DNA (Figure 4F), recapitulate the absorbance scattering experiments. As the concentration of DNA increases larger and more numerous droplets form, followed by their disappearance at high DNA concentration as expected for a re-entrant phase. Similar results are obtained when Alexa Fluor 488 labeled TRF2 is used in place of labeled DNA (Figure 4G). In addition, Fluorescence Recovery After Photobleaching (FRAP) of the fluorescent droplets indicates that a large fraction of the DNA within the droplet remains mobile (Figure 4H), consistent with a liquid-like character of the dense phase. Furthermore, TRF2 inside the droplets is mobile as well, albeit to a lesser extent than DNA. Droplet liquidity is maintained for at least 24 hours (Supplementary Figure S4A), suggesting that if further molecular re-arrangements lead to solidification (e.g., fibril formation), this occurs at significantly longer timescales [70, 71]. In stark contrast, TRF2 form significantly fewer droplets with the 24 bp dsDNA of mixed sequence composition (Supplementary Figure S4B) and TRF2ΔC shows a further reduction in droplets independently of the nature of the nucleic acid sequence (with or without telomeric repeat, Supplementary Figure S4C and S4D), consistent with the results of the turbidity experiments.
Therefore, our data support a model in which the multivalency of both the protein and the DNA dictate the phase separation propensity of the system. Once the valence of the DNA is reduced by shortening its length, non-specific TRF2-DNA interactions are no longer sufficient to drive phase separation and the specific recognition of a telomeric repeat by the DBD becomes essential. On the other hand, this model also predicts that as the valence of the DNA increases TRF2 should phase separate in a seemingly non-specific manner even in the absence of its DBD. Indeed, consistent with the absorbance scattering and DIC experiments in Figure 4D, droplets are also observed with a dsDNA that contains 19 T2AG3 repeats (Figure 4I), independent of the presence of the DNA binding domain (Supplementary Figure S4E). Interestingly, the droplets formed by TRF2 and the (T2AG3)19 telomeric substrate show limited recovery after photo-bleaching, independent of whether the DNA or TRF2 are being monitored (Figure 4I). This behavior is different to that observed from the 24 bp dsDNA and two T2AG3 repeats and it suggests that the increase in valence with multiple telomeric repeats leads to a dramatic change in the physical properties of the phase separated state. While these droplets show slow internal dynamics, they are still able to fuse over long timescales (Supplementary Figure S5).
Phase separation with single-stranded nucleic acids.
Transcription of telomeric repeats leads to telomeric repeat-containing RNA (TERRA), a long non-coding RNA proposed to play a role in telomeric protection [72, 73]. The interaction between TERRA and TRF2 has been proposed to facilitate heterochromatin formation, while TERRA depletion leads to an increase in TIFs [74]. Intriguingly, a splice-variant of TRF2 lacking the linker and DBD (similar to TRF2ΔC) co-localizes with neuronal granules, ribonuclear biomolecular condensates that facilitate the transport of mRNAs in axons [75, 76]. Based on our work here, one interpretation of this observation is that while the shorter TRF2 splice variant loses its ability to interact with telomeres it retains non-specific binding affinity for RNA, possibly via interactions within its basic N-terminus domain. With this in mind, we wondered if the DNA-dependent phase separation of TRF2 was truly dependent on dsDNA, or if phase separation could also be driven and modulated by other types of single-stranded nucleic acids (ssNAs), as has been shown for other non-specific positively charged polypeptides [77].
In line with this expectation, absorbance scattering experiments and DIC imaging indicate that TRF2 phase separation occurs with both ssDNA (Figure 5A) and ssRNA (Figure 5C) over a similar range of salt concentrations as it did with dsDNA (Figure 4B). As with dsDNA, either the N-terminal basic domain or the DBD is dispensable for phase separation (Figure 5B and 5D). However, in contrast to the lack of phase separation observed with the short 24 bp DNA of mixed sequence composition, at the same protein concentration non-specific interaction of TRF2 with an oligo-dT ssDNA as short as 20 nt is sufficient to induce phase separation (Supplementary Figure S6A). An interpretation of this result is that, because of chain flexibility, non-specific ssDNA interactions occur via a larger number of contacts than non-specific dsDNA interactions, increasing the protein-nucleic acid affinity and lowering the critical concentration for phase separation. In summary, our results demonstrate that TRF2 can phase separate with a variety of nucleic acids, where the driving force for phase separation depends on the strength and valence of interactions encoded by both proteins and nucleic acids.
Figure 5. TRF2 undergoes phase separation with ssDNA and ssRNA.
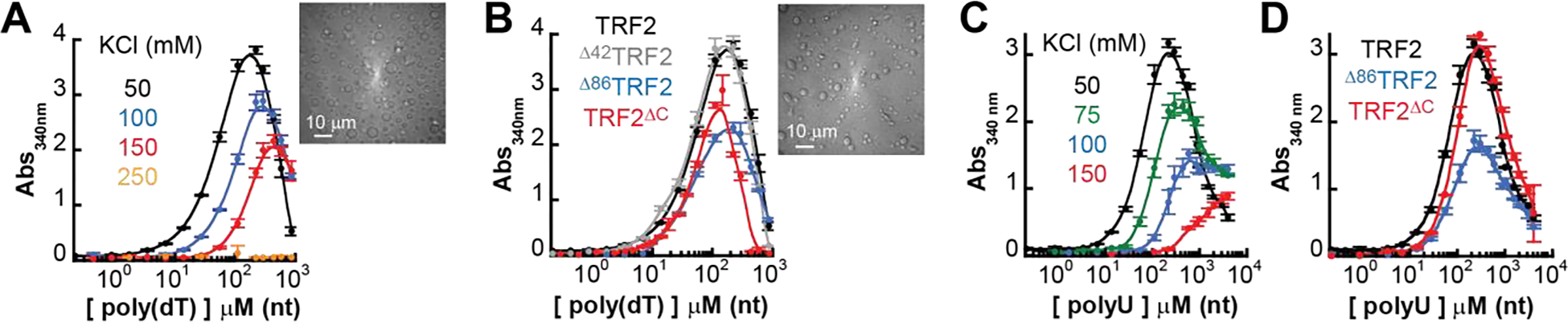
A. Turbidity experiments with mixtures of full length TRF2 (10 μM mon) and poly(dT) at increasing concentration of KCl concentration reveals a shift of the saturation concentration toward higher values and suppression of demixing at 250 mM KCl. B. Phase separation occurs also in truncated variants of the protein that lack the first 42 residues (Δ42), the first 86 residues (Δ86) or the last 294–542 amino acids (ΔC). C. Turbidity experiments with mixtures of full length TRF2 (10 μM mon) and poly-rU reveals a shift of the saturation concentration toward higher values with increasing salt as for ssDNA. D. Truncation variants of the protein maintain the phase separation propensity of the protein with poly-rU.
hRap1 modulates single DNA chain condensation and the phase-separation propensity of TRF2.
We have described the high propensity of TRF2-dependent DNA condensation and phase separation. However, given that TRF2 is only one component of the shelterin complex, we next asked how these processes might be modulated by the presence of a binding partner. More specifically, within the core-complex (TRF2-hRap1-Tin2-TRF1), hRap1 is unique in that it interacts directly and exclusively with TRF2 [29, 33, 34], making hRap1 a clear partner to test. In vitro, TRF2 and hRap1 form a tight complex [33] and, at the protein concentrations used in our experiments (> 300 nM), the TRF2-hRap1 complex can be considered as a single component. Binding of hRap1 to TRF2 lowers the DNA binding affinity of TRF2 [33] and in vivo its presence at telomeres suppresses the DNA damage response (Sfier de lange 2010). Moreover, the TRF2 interacting domain of hRap1 (RCT in Figure 1C) is preceded by a highly acidic region that may interfere with the function of the basic domain of TRF2. Thus, it is reasonable to postulate that interaction of hRap1 with TRF2 may modulate both the TRF2-dependent compaction of a single DNA chain and the DNA-dependent phase-separation propensity of TRF2. Indeed, magnetic tweezers experiments indicate that, compared to TRF2, the TRF2-hRap1 complex induces a smaller extent of compaction of DNA tethers containing the telomeric repeats and a lower degree of hysteresis (Figure 6A). These observations are consistent with hRap1 reducing the affinity of TRF2 for DNA [33], effectively altering the valence of TRF2, and modulating the properties of the collapsed state.
Figure 6. hRap1 modulates the phase-separation propensity of TRF2 with ds and ss nucleic acids.
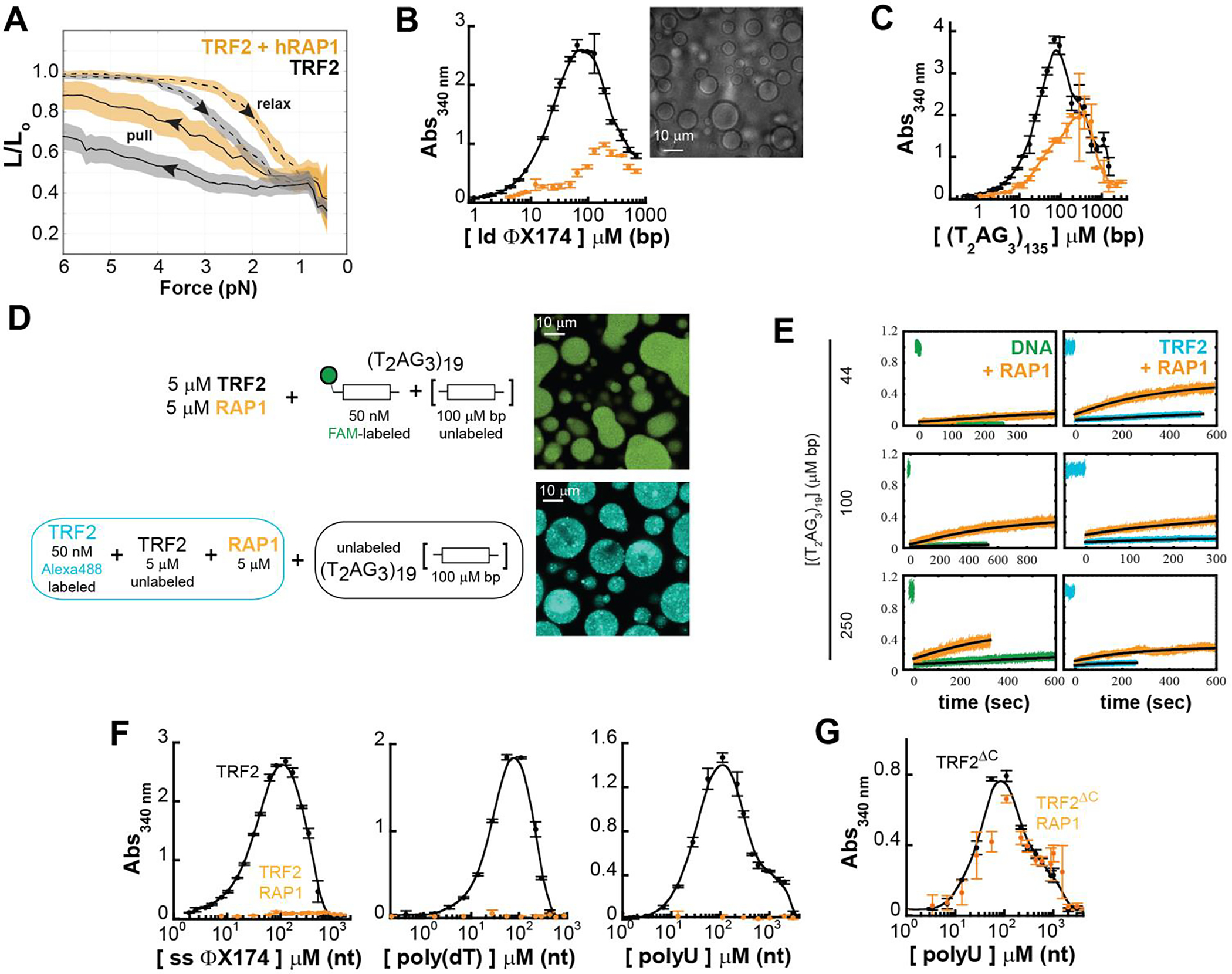
A. Force-extension experiments reveal a smaller TRF2-dependent DNA compaction in presence of stoichiometric concentration of hRap1 (dashed and solid lines are the average and color bands the standard deviation of multiple traces in either the relax or pull direction, respectively). B-C. Turbidity measurements of TRF2 and a TRF2- hRap1 stoichiometric complex in presence of non-specific (ϕX174 HaeIII digest) and specific (T2AG3)135 dsDNA. Addition of hRap1 does not ablate phase separation but modulates the phase-boundaries. D-E Phase separation of TRF2 (5 μM mon) and hRap1 (5 μM) with (T2AG3)19. Alexa Fluor 488-labeled TRF2 and FAM-labeled DNA demonstrate partitioning of the protein and DNA in the dense phase. FRAP measurements reveal that addition of hRap1 increases under all conditions the recovery of both DNA and TRF2. F. Turbidity measurements of TRF2 in presence of ss ϕX174, poly(dT), and poly(U) reveal suppression of phase separation upon addition of stoichiometric concentrations of hRap1. G. Turbidity measurements of the truncation variant TRF2ΔC, which lacks the binding site of hRap1. No change in turbidity is observed upon addition of hRap1.
Although TRF2-hRap1 interaction with non-specific double-stranded DNA still leads to formation of large micron-size droplets (Figure 6B and Supplementary Figure S3A), the change in absorbance scattering in the presence of hRap1 suggest that the phase boundaries have shifted. While binding of hRap1 to TRF2 does not suppress phase separation with dsDNA that contains telomeric repeats, there is a diminution in the driving force for assembly, mirroring changes observed in the force experiments (Figure 6C, 6D, and Supplementary Figure S3B). Interestingly, the presence of hRap1 leads to an increase in the recovery after photobleaching of the labeled DNA in the droplets (Figure 6E), consistent with its ability to reduce the strength of TRF2-DNA binding.
Finally, at the concentrations used in our experiments, interaction of hRap1 with TRF2 abolishes (or shifts to significantly higher concentrations) phase separation with single-stranded nucleic acid, either DNA or RNA (Figure 6F). These results suggests that hRap1 may function as a specificity switch for different types of nucleic acids, reducing non-specific DNA binding more severely than specific (telomeric repeat) DNA binding. It is important to note that the effect of hRap1 is mediated by its direct interaction with TRF2, as hRap1 has no effect on the phase separation of the truncated variant TRF2ΔC that lacks the interaction region with hRap1 (Figure 6G).
If TRF2 undergoes bona fide phase separation with dsDNA, then it should be possible to construct a complete two-dimensional phase diagrams with a closed-loop binodal, as predicted by analytical theories of polymer mixtures [78–81]. Moreover, if hRap1 weakens DNA compaction and phase separation by modulating protein-DNA interaction then that phase diagram should be systematically tuned by the presence of hRap1, shifting towards higher saturation concentrations [82]. At the same time, binding of hRap1 to TRF2 will introduce further excluded volume in the dense phase and therefore cause a shift toward lower concentrations of TRF2 in the dense phase.
In support of this hypothesis, we were able to infer the close-loop nature of the two-dimensional phase diagram for TRF2 with dsDNA by measuring the concentrations of TRF2 and nucleic acids in the light and dense phase (Figure 7A). Measurements are carried out using 50 nM of labeled protein or DNA mixed with excess unlabeled protein and nucleic acid (at a ratio of at least 1:200, see Supplementary Information). The use of single-photon detectors enables the determination of the concentration in the light phase with sensitivity down to the pM concentration and limits artifacts in the determination of protein and nucleic acid concentrations in the dense phase, which could arise at high concentration of fluorescent species due to quenching of the fluorophore or saturation of the detectors. The linearity range of the approach has been previously validated for a single component phase-separation system, by comparing the concentration obtained by fluorescence intensity with those measured with fluorescence correlation spectroscopy as well as independent turbidity measurements [28]. Our measurements provide an estimate for the directionality of the tie-lines, which support the notion that protein and nucleic acid favorably co-phase separate, as indicated by the positive slope in the protein-nucleic acid space. When comparing the concentration at which phase separation occurs, variation of the length of the DNA segment systematically alters the phase boundaries, as predicted by theory and in strong agreement with a three-component (solvent, DNA, protein) phase diagram (Figure 7B). Also, upon addition of hRap1, the phase boundary relative to the saturation concentrations recesses toward higher values, reporting on a systematic diminution in protein-DNA interaction driven by hRap1, as anticipated based on the force experiments (Figure 6B).
Figure 7. TRF2-DNA phase diagram and the implications for multi- and single-DNA condensation.
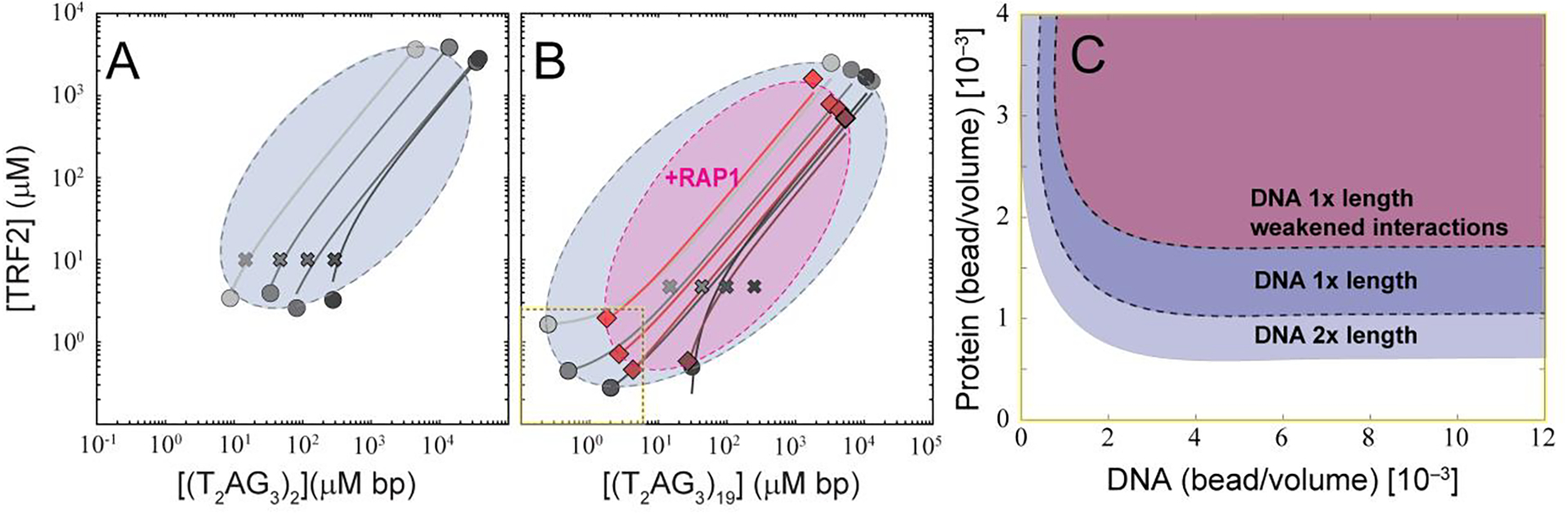
A-B. Reconstruction of the phase diagram for mixtures of TRF2 and (T2AG3)2 (A) and TRF2 and (T2AG3)19 (B) in gray as obtained by measuring the intensity of labeled protein and DNA in the dilute and dense phases (full circles) after mixing of the indicated concentrations (crosses). Solid lines connecting the dilute and dense phase concentrations represent expected tie lines across the solution, whereas the dashed lines are guide-to-the-eyes phase boundaries based on the concentration limits determined via fluorescence intensities and turbidity measurements. Addition of Rap1 (magenta area) restricts the phase boundaries. C. Lattice-based multi-component simulations reproduce the observed change in boundaries when increasing length of the DNA or decreasing multivalence of the TRF2 through a mean-field diminution in attractive interactions, mimicking the interpreted effect of hRap1. Only the lower-left portion of the phase diagram in panel B is simulated in panel C (dashed line with yellow highlight).
The phase behavior observed in experiment is recapitulated in simple coarse-grained simulations. Longer DNA molecules lead to an increased propensity to undergo phase separation as reported on by the reduction in saturation concentration (Fig 7C). Moreover, when we weakened intermolecular interactions in the same manner as in Figure 2H and Supplementary Figure S7 (which leads to a decrease in compaction) we systematically shift the saturation concentration to higher values (Figure 7C), analogous to the change observed upon addition of hRap1 (Figure 6B).
Finally, the concentration of TRF2 in the dense phase is also affected by binding of hRap1, in agreement with our excluded volume argument (Figure 7B). It is interesting to note that across the phase-diagram, addition of hRap1 alters the amplitude of FRAP (which is consistent with a weakening of interactions between TRF2 and nucleic acids), but it does not significantly affect the diffusion time of either DNA or TRF2 in the dense phase (Supplementary Figure S8). This observation is consistent with an increase in the fraction of the mobile species, where the mobility of the diffusive species is not largely dissimilar in the presence or absence of hRap1. Overall, these observations support the idea that the interaction of specific ligands (such as hRap1) can control (and even suppress) both phase separation (phase boundaries, selectivity for specific nucleic acids, transport properties) and nucleic acid compaction by modulating the set of interactions encoded by the binding partner (e.g., TRF2).
Discussion
Here we showed that two of the shelterin components, TRF2 and hRap1, modulate both protein-protein and protein-DNA interactions and lead to either compaction of individual DNA chains or phase separation of proteins and DNA at higher DNA concentrations. Similar results can be recapitulated using coarse-grained simulations that capture the overall trends of DNA compaction and phase separation propensity in the presence of a ligand. Taken all together, our observations support the original interpretation proposed by Post and Zimm [18] and are consistent with the idea that protein-dependent collapse of a single nucleic acid chain and the phase separation of mixtures of protein and nucleic acids are two distinct outcomes driven by the same set of molecular interactions [83–85].
The multi-valence encoded in both proteins and nucleic acids is clearly key for creating a network of molecular interactions that can engage both intra-molecularly (single-chain condensation) and inter-molecularly (phase separation). More specifically, our experiments reveal that the modular architecture of TRF2 encodes for a certain redundancy, such that phase separation with nucleic acids is robust even in absence of either the N-terminal basic domain or the DBD itself. Droplets also form when DNA is assembled in poly-nucleosomes (Supplementary Figure S6B), suggesting that TRF2-dependent phase separation may occur with more complex protein-DNA assembly that are formed in cells. Indeed, several recent studies have proposed or demonstrated that telomeres function as dynamic liquid-like condensates in living cells [51, 86, 87]. Given the repetitive nature of telomeric DNA, our observations suggest a model in which telomeric repeats provide a multivalent platform for the formation of dynamic, shelterin-mediated condensates at telomeres, driving compaction of telomeric DNA. This model mirrors similar conclusions arrived at independently by Jack et al [86] through a mixture of in vitro and in cell experiments.
Despite of the presence of a domain with high specificity, in our assays TRF2 is generally promiscuous in its interaction with nucleic acids, including specific and non-specific dsDNA as well as ssDNA and ssRNA, raising the question of how these types of interactions are regulated in cells. One possibility is that specificity is modulated by the presence or absence of additional binding partners. Along these lines, we find that hRap1 significantly alters TRF2’s propensity to both condense and phase-separate nucleic acids. Specifically, binding of hRap1 to TRF2 selectively abolishes phase separation with ssNAs, thereby imparting a dsDNA substrate specificity for the full-length protein. Deletion of the TRF2 region where hRap1 binds, which mimics the natural splicing form interacting with RNA, restores phase separation with single-stranded nucleic acids and suppresses specificity for dsDNA. In other words, the binding of hRap1 acts as a switch that controls the balance between promiscuous and specific interactions of TRF2 with nucleic acids. We propose that this mechanism may be important in the cell to discriminate TRF2 functions at telomeres versus the functions of its spliced variant in RNA transport in neurons.
Broadening this model, these observations suggest that perhaps different shelterin components may play a role in dictating the degree of collapse of single telomeres and favoring or disfavoring the phase separation with multiple nucleic acids. It is important to stress that though TRF2 undergoes phase separation with both non-specific and specific dsDNA, the specificity of telomeric DNA sequences and the number of specific telomeric sites have an impact on the transport properties of the phase-separated state, as monitored by FRAP. Therefore, specific sequences may have different mobilities and miscibilities, de facto contributing to the separation of different nucleic acids components.
To avoid ambiguity, we want to clarify that our results do not imply that individual telomeres are a bona fide phase-separated state. Rather, we propose that the mode of interaction between shelterin components and telomeric DNA determines whether single telomeres remain distinct entities or merge into larger bodies. Moreover, we propose that the interactions that drive bona fide phase separation in our in vitro reconstitution mirror the interactions that drive compaction of telomeric DNA, albeit perhaps with different affinities and specificities, as determined by the complement of binding partners and the local environment. This does not exclude the possibility that specific local regions of nucleic acid may favor a local partitioning of proteins, analogous to the pre-wetting demixing that occurs in the vicinity of surfaces [88] and as recently shown for the interaction of FUS in proximity of stretched DNA [89]. Furthermore, coalescence and assembly of the multiple telomeres or fragment of telomeres may explain the recent observation and proposal that telomeres in APBs have properties consistent with being a phase-separated state [51].
The phase-separation propensity of TRF2 and its modulation by interaction with hRap1 may be part of a more general mechanism for the regulation of nucleic acids in cells. Indeed, recent experimental evidence indicates that HP1a, a major component of heterochromatin, can also form liquid droplets and such a phenomenon is proposed to be the basis of heterochromatin organization [7, 8, 21]. One interpretation of these results reflects a model in which HP1a drives heterochromatin territories through bona fide liquid-liquid phase separation [7, 8, 21]. Another is that the intrinsically multivalent nature HP1a and many other DNA binding proteins (such as TRF2) drives DNA compaction through distributed multivalent interactions which will inevitably also drive phase separation with sufficiently short nucleic acid molecules in vitro [83, 85, 90]. Altering the degree of DNA compaction by modulating the same interactions that would also drive phase separation could allow for the rheostatic control of access to genomic loci. These interactions may be modulated through subtle changes in the complement of available binding partners (e.g., with hRap1, as we show here), but also by chemical modifications to proteins or nucleic acids (e.g., histone acetylation or DNA methylation). Similarly, super-enhancers have been proposed to assemble through phase separation of transcription factors at specific locations in the genome [91–94]. Another example is the recent finding that the nucleocapsid protein of the SARS-CoV-2, which plays a role in the viral RNA condensation and packaging, can also undergo phase separation and localize in biomolecular condensates within the cell [95–97]. This observation has led to a proposed model in which viral genome compaction is driven by the same interactions that can also form biomolecular condensates [85]. In this respect, the finding that telomeric components can lead to solution demixing supports the idea that the underlying physics of phase separation may be another example of a general mechanism through which the access and state of genetic material is controlled.
Our results highlight how phase behavior can be modulated by specific protein-ligand interactions, stressing the importance of investigating the contribution of linkage effects to phase separation [82]. Indeed, our results suggest that at telomeres specific interactions within the shelterin complex may be a means to regulate and suppress the phase-separation propensity of single components, such as TRF2. Similar mechanisms may have evolved in nature to modulate the multivalence of a specific protein, regulating its phase-separation propensity, specificity, or selectivity. Finally, the overall picture emerging from the parallelism between single chain condensation and multiple chain phase separation, as originally proposed by Post and Zimm [18] (see SI Appendix and Supplementary Figure S9 for additional discussion on this aspect), suggests that phase separation can be used as a readout for the condensation of single chains and, as such, can provide a convenient macroscopic tool to investigate and test therapeutics [98, 99].
Materials and Methods.
Cloning and Site-Specific Mutagenesis
All the primers sequenced used in this work are reported in Table S1. All the DNA and gene sequence used as templates were purchased from Addgene. Gene amplification was carried out using a two steps PCR protocol with Phusion DNA polymerase (NEB cat. M0530), while PfuTurbo polymerase (Agilent cat. 600250) was used for standard site-specific mutagenesis. Correct sequences for all the generated plasmids were confirmed by DNA sequencing.
hTERF2 constructs –
All the gene amplification PCR reactions were carried out in GC buffer (NEB B0519) and in the presence of 1 M betaine. All the hTERF2 constructs were cloned at EcoRI/XhoI restriction site of the pGEX-6p-1 vector. Plasmid p16-1 hTRF2 (Addgene cat. 12299) [30] was the template for the cloning of the full-length protein using primers hTERF2-F and hTERF2-R. In the resulting plasmid, the gene was not in frame with the GST and missing the Ala477. Primers h-TERF2-frame-F, h-TERF2-frame-R, hTERF2-Ala477-F and hTERF2-Ala477-R were designed to take care of these issues. The final plasmid (pGEX-hTERF2) coded for TERF2 sequence spanning from residue 5 to residue 542 (isoform 1 id:Q15554-3) and it was used as templated for the generation of all the hTERF2 constructs. Plasmids pGEX-hTERF2Δ42 and pGEX-hTERF2Δ86 were generated by regular cloning using hTERF2Δ42-F and hTERF2Δ86-F respectively as forward primer, and hTERF2-R as reverse primer. The C-terminus deletion constructs and pGEX-hTERF2ΔC were generated by site-specific mutagenesis using primers hTERF2-S294Stop-F and hTERF2-S294Stop-R. Primers hTERF2-S294Stop-F and hTERF2-S294Stop-R were also utilized for the generation of plasmid pGEX-hTERF2Δ86−ΔC, yielding the only the TRFH domain of TRF2. Plasmid pGEX-hTERF2(2–86)CC, encoding for sequence encompassing 2 and 86 residues and with the additional mutations S12C and A85C, was generated by PCR amplification of this sequence from a gBlocks-hTERF2-N-term (IDT, Corvallis) using primers hTERF2-A2-F and hTERF2-G86-R and cloning it at BamHI/XhoI site of pGEX-6p-1. Plasmid pGEX-hTERF2-Cys160 expressing hTERF2 mono-cysteine mutant at position 160 was obtained by iterative site-specific mutagenesis using primers hTERF2-C148S-F, hTERF2-C148S-R, hTERF2-C207S-F and hTERF2-C207S-R.
hRap1 construct –
Two steps were necessary in order to generate plasmid pGEX-hRap1 encoding for the wild-type hRap1 (id: Q9NYB0-1). First, the hRap1 in plasmid pLPC hRap1-FL (Addgene cat. 12542) [100] was amplified and cloned at BamHI/XhoI restriction site of the pGEX-vector using primers hRap1-F and hRap1-R. The resulting construct was missing one entire codon for a glutamate residue within the polyglutamate stretch at positions 297 to 304. The additional codon for glutamate was inserted in this region by standard PCR using primers hRap1-Glu-F and hRap1-Glu-R.
Protein Expression and Purification.
All the proteins were expressed in Rosetta2(DE3)pLysS cells by growing them at 37 °C until OD600 ≈ 0.7 in LB-Miller media; then cooled down at 4 °C for 30’ before the addition of 0.7 mM of isopropyl 1-thio-β-d-galactopyranoside, followed by overnight growing at 16 °C. Cells were harvested and stored at −80 °C.
The first steps of purification were the same for all the constructs. Briefly, cells were resuspended in lysis buffer (50 mM sodium phosphate pH 7.3, 0.4 M NaCl, 10% glycerol, 1 mM EDTA and 5 mM DTT) supplemented with 0.1 mM PMSF. Cells were lysed by sonication, debris removed by centrifugation at 14000 rpm and the supernatant incubated overnight at 4 °C with Glutahione Sepharose™ 4 Fast Flow (GE Healthcare). The resins were then washed with 3 volumes of lysis buffer followed by an additional 3 volumes wash with lysis buffer containing 1 M NaCl. Finally, the resins were equilibrated with lysis buffer and the GST-conjugated protein eluted with 25 mM reduced L-glutathione in lysis buffer. Finally, 3C-protease was added to the eluate, followed by overnight dialysis at 4 °C against the buffer used in the next purification step.
For the TRF2 constructs, the proteins were dialyzed against buffer T (20 mM Tris-HCl pH 8, 10% glycerol, 1 mM EDTA and 1 mM DTT) + 250 mM NaCl, applied onto a Macro-Prep High Q support (BIO-RAD) and the flow-through loaded on POROS 50 HE resin (Applied Biosystems) and eluted batch-wise with buffer T containing 400 mM, 600 mM and 1 M NaCl. Fractions containing the purified protein were pooled, concentrated on Amicon Ultra centrifugal filters (Millipore) and dialyzed against buffer ST (20 mM Hepes pH 7.4, 400 mM NaCl, 40% glycerol and 1 mM EDTA).
For hRap1, the protein was dialyzed against buffer T + 150 mM NaCl, loaded on Macro-Prep High Q support and eluted with buffer T + 300 mM NaCl. Pure hRap1 was concentrated with on Amicon Ultra centrifugal filters (Millipore), and dialyzed against buffer ST and stored at −80 °C. Before use the proteins were dialyzed in buffer H (20 mM Hepes-KOH pH7.4, 4% v/v glycerol, 1 mM DTT) + 100 mM KCl (unless otherwise indicated).
All the proteins were quantified by UV absorption spectra using extinction coefficients at 280 nm calculated with Protparam tool (https://web.expasy.org/protparam/). For selected protein constructs the dispersity of the sample and the oligomeric state of the protein were examined by analytical sedimentation velocity using an Optima XLA (Beckman) (see Figure S1B and 1C).
Fluorescence labeling of TRF2-Cys160.
The protein was extensively dialyzed in buffer L (20 mM sodium phosphate pH 7.4, 400 mM NaCl, 1 mM EDTA, 10% v/v glycerol, 50 μM TCEP), concentrated with Amicon Ultra centrifugal filters to ~ 300 μL ~100 μM, followed by addition of a 10-fold molar excess of Alexa Fluor 488 maleimide and incubated at 4 °C overnight under gentle rocking. The sample was then loaded on a BioGel P6 resin equilibrated in buffer L, to separate the protein from unincorporated dye. The labeled protein was quantified spectrophotometrically to ensure stoichiometric labeling, concentrated with Amicon Ultra centrifugal filters, dialyzed in buffer ST and quantified one last time before flash-freeze and storage at −80 °C.
DNA Substrates.
DNA oligonucleotides –
The unlabeled and fluorescently labeled oligonucleotides used for the preparation of the 24 bp dsDNA substrates and oligo-dTn sequences with 21 < n < 60 were purchased from IDT (Coralville, IA). For the unlabeled dsDNA substrates the two strands were mixed at equimolar ratio, while the FAM-labeled dsDNA substrates were annealed in the presence of 5% of the unlabeled strand. Annealing was carried out in 20 mM Hepes pH 7.4, 100 mM NaCl, 10% (v/v) glycerol, 2 mM MgCl2, by incubation in a preheated 95 °C water bath, followed by slow cooling to room temperature. Poly-dT longer than 250 nt (with a majority over 500 nt) and poly-U shorter than 250 nt were purchased from Midland Certified Reagent (Midland, TX).
T2AG3 Cassettes –
Plasmid pSXneo 135-T2AG3 (Addgene cat. 12402) [101] in its original Stbl3 strain was used to generate the 135-T2AG3 cassette. A sequence containing 20 T2AG3 repeats in pUC57 was purchased from GenScript (Piscataway, NJ) and cloned into pUC19 at the EcoRI/HindIII restriction sites. During cloning a full T2AG3 repeat was lost, resulting pUC19-(T2AG3)19 plasmid. The plasmid was transformed in DH5α, purified by miniprep and sequenced several times to test the stability of the (T2AG3)19 cassette in E. Coli and test for recombination events which may change the sequence.
All plasmids were purified following a modified Maniatis’s protocol [102]. Briefly, transformed cells were grown overnight at 37 °C, then harvested and resuspended in 50 mM glucose, 25 mM Tris-HCl (pH 8) and 10 mM EDTA. Lysis was achieved by adding 2 volumes of 1% SDS solution in 0.2 M NaOH and proteins were then precipitated with 0.35 volumes of 3 M potassium acetate in 12% acetic acid. After centrifugation, 0.6 volumes of isopropanol were added to the supernatant and the solution incubated in ice for 10’ before spinning. The pellet was solubilized in TE buffer (10 mM Tris-HCl pH 8 and 1 mM EDTA) and treated with 0.4 volumes of 10 M ammonium acetate; the precipitate was spun down, and the supernatant treated with 2 volumes of ethanol and incubated for 30’ at −20 °C before centrifugation. The pellet was resuspended in TE buffer and the RNA contaminants were digested with RNase A for 30’ at 37 °C. NaCl (to 1.5 M final concentration) and 0.25 volumes of 30% PEG8000 were added to the solution which was centrifuged after 30’ incubation in ice. The resulting pellet was solubilized in 100 mM Tris-HCl pH 7.5, 150 mM NaCl, 12 mM EDTA and 1% SDS, then Proteinase K was added, and the solution incubated for 30’ at 37 °C. Finally, the plasmid was phenol extracted, ethanol precipitated and resuspended in TE buffer.
pSXneo 135-T2AG3 was digested NotI-HF and SmaI while pU19-(T2AG3)19 with EcoRI and HindIII to release the cassettes from the vectors. The cassettes were separated from the vector by iterative PEG8000 fractionation in TE buffer with 600 mM NaCl. Finally, pure cassettes were ethanol precipitated, resuspended in HE and quantified. Carboxy-fluorescein labeled FAM(T2AG3)19 was generated by two step PCR using Phusion polymerase in GC buffer in the presence of 1 M betaine using pU19-19-T2AG3 as template. The annealing/amplification steps were carried out at 72°C for 30 seconds with primers FAM-pUC19-842 and pUC19-929.
DNA substrates for the magnetic tweezer –
Non-telomeric DNA was generated by Pfu PCR amplification of a 2 kbp region from (pGOHis4TATA) using primers with unique restriction sites (GO-His4-MluI-F, GO-StyI-R). Two DNA handles (1 kbp) were TAQ PCR amplified from (pSH726). One DNA handle was amplified in the presence of dUTP-biotin with primers (F-T1-DH-1kb, R-T1-DH-NheI) while the other was amplified in the presence of dUTP-digoxigenin with primers (F-T1-UH-AscI, R-T1-UH-1kb), resulting in multiple, random incorporations of label along the length of the handle DNA. PCR products were restriction digested with the appropriate restriction endonuclease and then ligated together with T4 DNA ligase following standard protocol. After each enzymatic step the DNA was purified using Qiagen PCR cleanup kit to remove contaminants. A 2:1 molar excess of DNA handles was used in the ligation reaction to drive formation of the full-length DNA substrate (~4 kbp). The full-length DNA substrate was purified by agarose gel electrophoresis (0.8% agarose in 0.5x TBE at 125 V). The gel isolated DNA was electro-eluted from the gel, purified, and concentrated using a single Qiagen spin column following manufacturer’s protocol. The DNA was eluted in 10 mM Tris-HCl pH 8.0 and supplemented with glycerol and EDTA to final concentrations of 50% (v/v) and 1 mM, respectively.
DNA containing human telomeric sequence was generated by incorporating the (T2AG3)135 cassette as follows. The (T2AG3)135 cassette was isolated by restriction digest of pSXneo 135-T2AG3 with EcoRI and NotI. Two symmetric DNA extensions of 625 bp were PCR amplified from pUC19 using primers (pUC19-927-EcoRI / pUC19-1553r-NheI) for one extension and primers (pUC19-927-NotI / pUC19-1553r-MIuI) for the other extension, following the two step PCR as described above for FAM(T2AG3)19. Two 1 kbp DNA handles were PCR amplified as described above. The full-length DNA was assembled in two ligation steps. First, the DNA handles were digested and ligated to their compatible DNA extensions (AscI:MluI and NheI:NheI). Then, the resulting ligation products were digested with EcoRI and NotI and ligated to the isolated (T2AG3)135 cassette. The full-length DNA product was isolated by agarose gel electrophoresis, purified, and concentrated as described above for the non-telomeric DNA.
Optical and Magnetic tweezers.
Optical tweezers experiments were performed using a custom-built dual-optical-trap based on a 1064 nm solid state laser split by polarization[103]. λDNA was biotinylated by T7 DNA polymerase in T7 polymerase buffer (NEB M0274) for 40’ at 12 °C in the presence of 0.6 mM dNTPs (dATP, dTTP, and dCTP) and 1 mM biotinylated-dGTP, and purified on spin column S-400 HR. A laminar flow-cell (μ-Flux, Lumicks) was used to establish DNA tethers in the absence of TRF2 by flowing biotinylated λDNA past trapped streptavidin-coated 2.1 μm beads (Spherotech, SVP-20-5) under constant flow. The binding of DNA to the bead was judged by the presence of an increase in the force exerted on that bead due to the increased drag of the DNA. Subsequently, the beads and DNA were moved to a DNA-free lamina and a tether was formed between the two beads by fishing for the other end of the biotinylated DNA using the second bead trapped in the second trap. Once the presence of a single λDNA in the dumbbell configuration was confirmed by force-extension measurements, the second trap was turned off and the DNA was held extended only by the flow force. Finally, the tether was moved into a lamina were TRF2 was present. At a constant high flow force (≥ 4 pN as judged by the end-to-end distance of the DNA) the DNA remained extended. The flow force was then gradually reduced by raising the height of exit tube. At a critical flow velocity (i.e., force), the end-to-end distance of the DNA as monitored by the bead-bead distance would decrease until the second bead made contact with the first bead and was trapped in the first optical trap. Movies were recorded at 32 fps and the position of the free bead over time was determined by a tracking routine in ImageJ. All experiments were performed at room temperature.
Magnetic tweezers experiments were performed with a custom-built instrument [104]. DNA tethers were constructed to have 1 kb biotin handles on one end and 1 kb digoxygenin handles on the other. The DNA was first bound to 1 μm streptavidin-coated paramagnetic beads (MyOne, Invitrogen) in a test tube. The DNA-bead pairs were then immobilized on an anti-digoxygenin:polyethylene glycol coated surface by flowing them into the flow cell, incubating for 5 minutes and then flowing out non-bound beads. Force-extension control experiments confirmed the presence of a single DNA tethers on individual beads in buffer HK150 (20 mM Hepes-KOH pH7.4, 150 mM KCl, 2% v/v glycerol, 1 mM DTT) supplemented with 2 mg/mL BSA and 0.1% v/v Pluronic F-127 (ThermoFisher). After addition of 300 nM TRF2 on fully extended DNA held at maximum force (~ 5 pN, magnet position 0 mm), force release experiments were performed by changing the magnet position at a constant velocity (0.004 mm/sec) over 12.5 minutes until a force of ~ 0.3 pN was reached (magnet position 3 mm). Subsequently, the force was increased by gradually increasing moving the magnets back towards the sample over another 12.5 minutes. Movies of the force-extension experiments were collected, and the xyz-position of the DNA tethered beads and surface attached reference beads were tracked off-line using the NanoBLOC software[105]. Instrumental drift in the DNA tethered beads’ position was accounted for by subtracting the xyz-displacement of surface attached reference beads[104]. Prior to the force-extension experiments, a force calibration measurement was collected by tracking DNA tethered beads at different magnet heights and calculating the force using the inverse pendulum model [104]. A mixture of constrained and unconstrained DNA tethers was used for the analysis and no major differences were observed between them. All experiments were performed at room temperature.
Turbidity experiments.
Equal volumes of 2X solutions of DNA and protein were mixed to yield the appropriate DNA to protein ratio, in a final buffer H2 (20 mM Hepes-KOH pH7.4, 2% v/v glycerol, 1 mM DTT) + 100 mM KCl (unless otherwise indicated). Absorbance of the solutions at 340 nm was measured using a NanoDrop 2000 (Fisher Scientific) after 1 minute of mixing. The reported error bars are from three independent readings. All the experiments were performed by maintaining constant the protein concentration and varying the DNA concentration. Unless otherwise indicated, the TRF2 concentration used in the experiments was 10 μM monomers. Because of the high affinity and the high protein concentrations used, the complexes of hRap1 with the TRF2 variants were formed by mixing the individual proteins at a 1:1 ratio and incubating for at least 10 minutes before starting the experiments. All experiments were performed at room temperature.
Fluorescence and Differential Interference Contrast (DIC) microscopy.
In order to obtain images and determine concentrations and changes in diffusion rates in protein-DNA condensates we employed confocal fluorescence microscopy on samples doped with either fluorescein (FAM) 3’-labeled DNA or Alexa Fluor 488 labeled full length TRF2. Confocal fluorescence measurements were performed on a Picoquant MT200 instrument (Picoquant, Germany). The microscope (Olympus IX-73, Japan) is equipped with a high numerical aperture water immersion objective (60×1.2 UPlanSApo Superapochromat, Olympus, Japan). Fluorophores were excited using a 485 nm pulsed laser (LDH P-C-485, Picoquant, Germany) with a repetition rate of 20 MHz. Excitation power was monitored before the objective with a laser photodiode and optimized to minimize photobleaching under normal measurement conditions, saturation of detectors at the maximum labeling concentration, and to get the higher signal to noise ratio, in a way that allowed us to keep a constant power for each set of measurements. Emitted photons were collected with a 60×1.2 UPlanSApo Superachromat water immersion objective (Olympus, Japan), passed through a dichroic mirror (ZT568rpc, Chroma, USA), and filtered by a 100 μm pinhole (Thorlabs, USA). Photons were separated according to polarization using a polarizer beam splitter cube (Ealing, California, USA) and further refined by a bandpass filter, either 525 nm ± 25 nm (ET525/50m, Chroma, USA) for FAM and Alexa Fluor 488 fluorophores, or 642 nm ± 40 nm (ET642/80m, Chroma, USA) for Cy3, in front of the SPAD detectors (Excelitas, USA). Photons were counted and accumulated by a HydraHarp 400 TCSPC module (Picoquant, Germany) with 16 picosecond resolution.
All measurements were performed in either uncoated polymer coverslip cuvettes (Ibidi, Wisconsin, USA) or glass coverslips with 4 mm-diameter glass cylinder attached on top. Between measurements cuvette wells were soaked with hot 10% DECONEX11 Universal cleaning solution, rubbed with CleanWIPE swaps, rinsed thoroughly with distilled water, then double distilled water and finally dried by flowing dried air. All measurements were performed at 23.0 ± 0.5 °C in a temperature-controlled room, as detected in the microscope stage.
DIC imaging of the phase-separated solutions was performed with an inverted Olympus IX81 microscope equipped with Nomarski prisms and a Hamamatsu C9100 EM-CCD camera. A drop of solution was deposited on a glass coverslip and imaged with a 10X objective as the droplets deposit by gravity onto the surface. Movies were recorded at 10 fps. Static DIC images of selected fields of view were generated in Image J by Z-average of 50 frames, and used to measure the diameter of the formed droplets. All experiments were performed at room temperature.
Detailed information on Fluorescence Recovery After Photobleaching (FRAP) analysis and phase diagram tie-lines construction is presented in Supporting Information.
Coarse-grained simulations.
Variable force constant simulations –
Coarse-grained simulations were performed using the PIMMS simulation engine[28]. Briefly, all force-dependent simulations were performed with an 80-mer “DNA” strand in an 80×80×80 box with 4,000 copies of the protein bead. Extension force was applied with a distance-dependent harmonic potential with an ideal distance of 80 lattice units and a varying force constant. Practically speaking, individual simulations were run at under equilibrium conditions with different fixed force constants (as opposed to a bona fide pulling simulation), with the resulting extension vs. force constant data shown in Figure 2 and Figure S10. Protein molecules were represented as single beads, with an excluded volume of one lattice site and short- and long- range interactions that extend one or two lattice sites away from the bead in every direction. As such, the radius of gyration for the bead is 2.5 lattice units (1/2 the diameter of the volume in which interactions are ‘felt), while the excluded volume is a single lattice voxel.
At least five independent replicas were run for each possible condition, with 10 – 20 replicas for many systems. Individual replicas were run for between 2 × 109 and 9 × 1010 Monte Carlo steps. Specifically, for force constant values in which bimodal behavior was observed, all independent replicas were run for a sufficient number of steps to ensure that multiple exchanges between the two states are observed in a single simulation. Supplementary Table S8 outlines the interaction parameters used in units of kBT−1. Simulations were performed at a temperature of T=140 with an 80:15:5 moveset ratio for crankshaft, chain translate, and chain rotate moves. Interaction parameters reported in Supplementary Table S8 reflect strengths in kBT, i.e. parameter file value divided by temperature (k=1). When applied, smoothing was performed using a Savitzky-Golay filter with a window size of 31 simulation steps and a 3rd order polynomial approximation (e.g., Figures 3C and 3D).
We estimated the TRF2 solubility threshold in simulations by performing simulations in a fixed box volume at variable TRF2 concentrations and determining the concentration at which self-assembly occurs. We converted lattice units to nanometers with a conversion factor of 4.5 nm per lattice unit. This conversion places the excluded volume of TRF2 molecules at 4.5 nm3, with the radius of gyration for the full-length protein of 5 nm. This conversion allows us to estimate molar concentrations by calculating number of molecules per unit volume. The solubility threshold was set at the concentration at which a monodisperse solution is no longer observed, which emerges a density of 800 molecules per 90 nm3. Varying the conversion factor used here changes the value between 0.5 mM and 3 mM, such that a ~1 mM order of magnitude reflects an approximate regime. The mass concentration (~30 mg/ml) was estimated by convert 0.5 mM to mass concentration using a molecular mass of 59593.50 Da for full-length TRF2. The solubility of lysozyme was taken from the lysozyme Sigma Aldritch product information (CAS RN 12650-88-3, EC 3.2.1.17, Lysozyme from chicken egg white for Molecular Biology).
Phase separation simulations –
Phase separation simulations were run with identical parameters, movesets, and setup as variable force constant simulations. A 10-mer (short) or 20-mer (long) DNA polymer was used and weaker vs. stronger interaction strengths used as in the force experiments. DNA concentration varied between 400 and 2,600 DNA beads (40 and 260 molecules) and between 200 and 1,000 protein beads (200 and 1,000 molecules). Where phase separation occurred a single protein:DNA assembly always formed and was stable for the duration of the simulation.
Supplementary Material
Highlights.
TRF2 condenses single DNA chains, while in the presence of multiple DNA chains the system undergoes phase separation.
TRF2-dependent condensation of single nucleic acid molecules and phase separation of multiple ones are two physical processes driven by the same set of molecular interactions.
Interaction of hRap1 with TRF2 modulates both DNA collapse and alters nucleic acid specificity in phase-separation.
We speculate that the physical interactions that mediate phase separation and condensation represent a possible means for controlling access to and the state of telomeres.
Acknowledgements –
We would like to thank John Cooper for access to the microscope for DIC imaging. This work was supported by the National Institutes of Health 1R35GM139508 to R.G. and 1R01AI163142 to A.S.
Footnotes
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
A.S.H. is a scientific consultant with Dewpoint Therapeutics.
REFERENCES
- [1].Riback JA, Katanski CD, Kear-Scott JL, Pilipenko EV, Rojek AE, Sosnick TR, et al. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell. 2017;168:1028–40 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lafontaine DLJ. Birth of Nucleolar Compartments: Phase Separation-Driven Ribosomal RNA Sorting and Processing. Mol Cell. 2019;76:694–6. [DOI] [PubMed] [Google Scholar]
- [3].Correll CC, Bartek J, Dundr M. The Nucleolus: A Multiphase Condensate Balancing Ribosome Synthesis and Translational Capacity in Health, Aging and Ribosomopathies. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lafontaine DLJ, Riback JA, Bascetin R, Brangwynne CP. The nucleolus as a multiphase liquid condensate. Nat Rev Mol Cell Biol. 2021;22:165–82. [DOI] [PubMed] [Google Scholar]
- [5].Smith J, Calidas D, Schmidt H, Lu T, Rasoloson D, Seydoux G. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brangwynne CP, Eckmann CR, Courson DS, Rybarska A, Hoege C, Gharakhani J, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–32. [DOI] [PubMed] [Google Scholar]
- [7].Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, et al. Liquid droplet formation by HP1alpha suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, Karpen GH. Phase separation drives heterochromatin domain formation. Nature. 2017;547:241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guillen-Boixet J, Kopach A, Holehouse AS, Wittmann S, Jahnel M, Schlussler R, et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell. 2020;181:346–61 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mitrea DM, Cika JA, Guy CS, Ban D, Banerjee PR, Stanley CB, et al. Nucleophosmin integrates within the nucleolus via multi-modal interactions with proteins displaying R-rich linear motifs and rRNA. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhu L, Brangwynne CP. Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol. 2015;34:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Banani SF, Rice AM, Peeples WB, Lin Y, Jain S, Parker R, et al. Compositional Control of Phase-Separated Cellular Bodies. Cell. 2016;166:651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fay MM, Anderson PJ. The Role of RNA in Biological Phase Separations. J Mol Biol. 2018;430:4685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hyman AA, Weber CA, Julicher F. Liquid-liquid phase separation in biology. Annu Rev Cell Dev Biol. 2014;30:39–58. [DOI] [PubMed] [Google Scholar]
- [17].Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357. [DOI] [PubMed] [Google Scholar]
- [18].Post CB, Zimm BH. Theory of DNA condensation: collapse versus aggregation. Biopolymers. 1982;21:2123–37. [DOI] [PubMed] [Google Scholar]
- [19].Quail T, Golfier S, Elsner M, Ishihara K, Murugesan V, Renger R, et al. Force generation by protein–DNA co-condensation. Nature Physics. 2021;17:1007–12. [Google Scholar]
- [20].Strom AR, Biggs RJ, Banigan EJ, Wang X, Chiu K, Herman C, et al. HP1α is a chromatin crosslinker that controls nuclear and mitotic chromosome mechanics. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Keenen MM, Brown D, Brennan LD, Renger R, Khoo H, Carlson CR, et al. HP1 proteins compact DNA into mechanically and positionally stable phase separated domains. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Morin JA, Wittmann S, Choubey S, Klosin A, Golfier S, Hyman AA, et al. Surface condensation of a pioneer transcription factor on DNA. bioRxiv. 2020:2020.09.24.311712. [Google Scholar]
- [23].Ryu J-K, Hwang D-E, Choi J-M. Current Understanding of Molecular Phase Separation in Chromosomes. International Journal of Molecular Sciences. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Renger R, Morin JA, Lemaitre R, Ruer-Gruss M, Jülicher F, Hermann A, et al. Co-condensation of proteins with single- and double-stranded DNA. bioRxiv. 2021:2021.03.17.435834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rubinstein M, & Colby RH. Polymer physics: Oxford: Oxford University Press; 2003. [Google Scholar]
- [26].Dong X, Bera S, Qiao Q, Tang Y, Lao Z, Luo Y, et al. Liquid–Liquid Phase Separation of Tau Protein Is Encoded at the Monomeric Level. The Journal of Physical Chemistry Letters. 2021;12:2576–86. [DOI] [PubMed] [Google Scholar]
- [27].Zeng X, Holehouse AS, Chilkoti A, Mittag T, Pappu RV. Connecting Coil-to-Globule Transitions to Full Phase Diagrams for Intrinsically Disordered Proteins. Biophysical journal. 2020;119:402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Martin EW, Holehouse AS, Peran I, Farag M, Incicco JJ, Bremer A, et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. 2020;367:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].de Lange T Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. [DOI] [PubMed] [Google Scholar]
- [30].Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17:231–5. [DOI] [PubMed] [Google Scholar]
- [31].Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999;18:5735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bilaud T, Brun C, Ancelin K, Koering CE, Laroche T, Gilson E. Telomeric localization of TRF2, a novel human telobox protein. Nat Genet. 1997;17:236–9. [DOI] [PubMed] [Google Scholar]
- [33].Janouskova E, Necasova I, Pavlouskova J, Zimmermann M, Hluchy M, Marini V, et al. Human Rap1 modulates TRF2 attraction to telomeric DNA. Nucleic Acids Res. 2015;43:2691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–83. [DOI] [PubMed] [Google Scholar]
- [35].Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science. 2010;327:1657–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim SH, Beausejour C, Davalos AR, Kaminker P, Heo SJ, Campisi J. TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem. 2004;279:43799–804. [DOI] [PubMed] [Google Scholar]
- [37].Ye JZ, Donigian JR, van Overbeek M, Loayza D, Luo Y, Krutchinsky AN, et al. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. 2004;279:47264–71. [DOI] [PubMed] [Google Scholar]
- [38].Frescas D, de Lange T. TRF2-tethered TIN2 can mediate telomere protection by TPP1/POT1. Mol Cell Biol. 2014;34:1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu C, Rai R, Huang C, Broton C, Long J, Xu Y, et al. Structural and functional analyses of the mammalian TIN2-TPP1-TRF2 telomeric complex. Cell Res. 2017;27:1485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Takai KK, Kibe T, Donigian JR, Frescas D, de Lange T. Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell. 2011;44:647–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Molenaar C, Wiesmeijer K, Verwoerd NP, Khazen S, Eils R, Tanke HJ, et al. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003;22:6631–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nagele RG, Velasco AQ, Anderson WJ, McMahon DJ, Thomson Z, Fazekas J, et al. Telomere associations in interphase nuclei: possible role in maintenance of interphase chromosome topology. J Cell Sci. 2001;114:377–88. [DOI] [PubMed] [Google Scholar]
- [43].Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–56. [DOI] [PubMed] [Google Scholar]
- [44].Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–30. [DOI] [PubMed] [Google Scholar]
- [45].Henson JD, Neumann AA, Yeager TR, Reddel RR. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002;21:598–610. [DOI] [PubMed] [Google Scholar]
- [46].Chung I, Osterwald S, Deeg KI, Rippe K. PML body meets telomere: the beginning of an ALTernate ending? Nucleus. 2012;3:263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Draskovic I, Arnoult N, Steiner V, Bacchetti S, Lomonte P, Londono-Vallejo A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A. 2009;106:15726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chung I, Leonhardt H, Rippe K. De novo assembly of a PML nuclear subcompartment occurs through multiple pathways and induces telomere elongation. J Cell Sci. 2011;124:3603–18. [DOI] [PubMed] [Google Scholar]
- [49].Zhang JM, Yadav T, Ouyang J, Lan L, Zou L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019;26:955–68 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Mediani L, Guillen-Boixet J, Alberti S, Carra S. Nucleoli and Promyelocytic Leukemia Protein (PML) bodies are phase separated nuclear protein quality control compartments for misfolded proteins. Mol Cell Oncol. 2019;6:e1415624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang H, Zhao R, Tones J, Liu M, Dilley RL, Chenoweth DM, et al. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol Biol Cell. 2020;31:2048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Poulet A, Pisano S, Faivre-Moskalenko C, Pei B, Tauran Y, Haftek-Terreau Z, et al. The N-terminal domains of TRF1 and TRF2 regulate their ability to condense telomeric DNA. Nucleic Acids Res. 2012;40:2566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kaur P, Wu D, Lin J, Countryman P, Bradford KC, Erie DA, et al. Enhanced electrostatic force microscopy reveals higher-order DNA looping mediated by the telomeric protein TRF2. Sci Rep. 2016;6:20513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Erdel F, Kratz K, Willcox S, Griffith JD, Greene EC, de Lange T. Telomere Recognition and Assembly Mechanism of Mammalian Shelterin. Cell Rep. 2017;18:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Doksani Y, Wu JY, de Lange T, Zhuang X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013;155:345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tree DR, Muralidhar A, Doyle PS, Dorfman KD. Is DNA a Good Model Polymer? Macromolecules. 2013;46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Robertson RM, Laib S, Smith DE. Diffusion of isolated DNA molecules: Dependence on length and topology. Proceedings of the National Academy of Sciences. 2006;103:7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Shelterin Protects Chromosome Ends by Compacting Telomeric Chromatin. Cell. 2016;164:735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Timashev LA, Babcock H, Zhuang X, de Lange T. The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes Dev. 2017;31:578–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Vancevska A, Douglass KM, Pfeiffer V, Manley S, Lingner J. The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes Dev. 2017;31:567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith SB, Cui Y, Bustamante C. Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science. 1996;271:795–9. [DOI] [PubMed] [Google Scholar]
- [62].Marko JF, Siggia ED. Stretching DNA. Macromolecules. 1995;28:8759–70. [Google Scholar]
- [63].Martin EW, Holehouse AS. Intrinsically disordered protein regions and phase separation: sequence determinants of assembly or lack thereof. Emerging topics in life sciences. 2020;4:307–29. [DOI] [PubMed] [Google Scholar]
- [64].Kramer RM, Shende VR, Motl N, Pace CN, Scholtz JM. Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophysical journal. 2012;102:1907–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Stockmayer WH. Light Scattering in Multi-Component Systems. The Journal of Chemical Physics. 1950;18:58–61. [Google Scholar]
- [66].Wahle CW, Ross DS, Thurston GM. On inferring liquid-liquid phase boundaries and tie lines from ternary mixture light scattering. J Chem Phys. 2012;137:034203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Banerjee PR, Milin AN, Moosa MM, Onuchic PL, Deniz AA. Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew Chem Int Ed Engl. 2017;56:11354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Choi JM, Dar F, Pappu RV. LASSI: A lattice model for simulating phase transitions of multivalent proteins. PLoS Comput Biol. 2019;15:e1007028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Choi KH, Farrell AS, Lakamp AS, Ouellette MM. Characterization of the DNA binding specificity of Shelterin complexes. Nucleic acids research. 2011;39:9206–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lin Y, Protter DS, Rosen MK, Parker R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol Cell. 2015;60:208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–77. [DOI] [PubMed] [Google Scholar]
- [72].Bettin N, Oss Pegorar C, Cusanelli E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Luke B, Lingner J. TERRA: telomeric repeat-containing RNA. EMBO J. 2009;28:2503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Deng Z, Norseen J, Wiedmer A, Riethman H, Lieberman PM. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell. 2009;35:403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Grammatikakis I, Zhang P, Mattson MP, Gorospe M. The long and the short of TRF2 in neurogenesis. Cell Cycle. 2016;15:3026–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Grammatikakis I, Zhang P, Panda AC, Kim J, Maudsley S, Abdelmohsen K, et al. Alternative Splicing of Neuronal Differentiation Factor TRF2 Regulated by HNRNPH1/H2. Cell Rep. 2016;15:926–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Boeynaems S, Holehouse AS, Weinhardt V, Kovacs D, Van Lindt J, Larabell C, et al. Spontaneous driving forces give rise to protein–RNA condensates with coexisting phases and complex material properties. Proceedings of the National Academy of Sciences. 2019;116:7889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Altena FW, Smolders CA. Calculation of liquid-liquid phase separation in a ternary system of a polymer in a mixture of a solvent and a nonsolvent. Macromolecules. 1982;15:1491–7. [Google Scholar]
- [79].Bergfeldt K, Piculell L, Linse P. Segregation and Association in Mixed Polymer Solutions from Flory–Huggins Model Calculations. The Journal of Physical Chemistry. 1996;100:3680–7. [Google Scholar]
- [80].Hsu CC, Prausnitz JM. Thermodynamics of Polymer Compatibility in Ternary Systems. Macromolecules. 1974;7:320–4. [Google Scholar]
- [81].Nandi SK, Heidenreich M, Levy E, Safran S. Interacting multivalent molecules: affinity and valence impact the extent and symmetry of phase separation. arXiv: Soft Condensed Matter. 2019. [DOI] [PubMed] [Google Scholar]
- [82].Ruff KM, Dar F, Pappu RV. Ligand effects on phase separation of multivalent macromolecules. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Muzzopappa F, Hertzog M, Erdel F. DNA length tunes the fluidity of DNA-based condensates. Biophysical journal. 2021;120:1288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kang H, Yoo J, Sohn B-K, Lee S-W, Lee HS, Ma W, et al. Sequence-dependent DNA condensation as a driving force of DNA phase separation. Nucleic Acids Research. 2018;46:9401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Cubuk J, Alston JJ, Incicco JJ, Singh S, Stuchell-Brereton MD, Ward MD, et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat Commun. 2021;12:1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Jack A, Kim Y, Strom AR, Lee DSW, Williams B, Schaub JM, et al. Compartmentalization of telomeres through DNA-scaffolded phase separation. Developmental Cell. 2022;57:277–90 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Min J, Wright WE, Shay JW. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019;33:814–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Morin JA, Wittmann S, Choubey S, Klosin A, Golfier S, Hyman AA, et al. Surface condensation of a pioneer transcription factor on DNA. bioRxiv. 2020. [Google Scholar]
- [89].Renger R, Morin JA, Lemaitre R, Ruer-Gruss M, Jülicher F, Hermann A, et al. Co-condensation of proteins with single- and double-stranded DNA. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Erdel F, Rademacher A, Vlijm R, Tünnermann J, Frank L, Weinmann R, et al. Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol Cell. 2020;78:236–49.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Boija A, Klein IA, Sabari BR, Dall’Agnese A, Coffey EL, Zamudio AV, et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 2018;175:1842–55 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45–54. [DOI] [PubMed] [Google Scholar]
- [93].Franklin JM, Guan KL. YAP/TAZ phase separation for transcription. Nat Cell Biol. 2020;22:357–8. [DOI] [PubMed] [Google Scholar]
- [94].Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Iserman C, Roden CA, Boerneke MA, Sealfon RSG, McLaughlin GA, Jungreis I, et al. Genomic RNA Elements Drive Phase Separation of the SARS-CoV-2 Nucleocapsid. Mol Cell. 2020;80:1078–91 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Savastano A, Ibanez de Opakua A, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS-CoV-2 phase separates into RNA-rich polymerase-containing condensates. Nat Commun. 2020;11:6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang J, Shi C, Xu Q, Yin H. SARS-CoV-2 nucleocapsid protein undergoes liquid-liquid phase separation into stress granules through its N-terminal intrinsically disordered region. Cell Discov. 2021;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].de Oliveira GAP, Cordeiro Y, Silva JL, Vieira T. Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv Protein Chem Struct Biol. 2019;118:289–331. [DOI] [PubMed] [Google Scholar]
- [99].Jack A, Ferro LS, Trnka MJ, Wehri E, Nadgir A, Costa K, et al. SARS CoV-2 nucleocapsid protein forms condensates with viral genomic RNA. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Li B, de Lange T. Rap1 affects the length and heterogeneity of human telomeres. Mol Biol Cell. 2003;14:5060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hanish JP, Yanowitz JL, de Lange T. Stringent sequence requirements for the formation of human telomeres. Proc Natl Acad Sci U S A. 1994;91:8861–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- [103].Moffitt JR, Chemla YR, Smith SB, Bustamante C. Recent advances in optical tweezers. Annu Rev Biochem. 2008;77:205–28. [DOI] [PubMed] [Google Scholar]
- [104].Galburt EA. Magnetic tweezers force spectroscopy. Handbook of Imaging in Biological Mechanics: CRC Press; 2014. p. 481–49. [Google Scholar]
- [105].Cnossen JP, Dulin D, Dekker NH. An optimized software framework for real-time, high-throughput tracking of spherical beads. Rev Sci Instrum. 2014;85:103712. [DOI] [PubMed] [Google Scholar]
- [106].Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. Structure of the TRFH Dimerization Domain of the Human Telomeric Proteins TRF1 and TRF2. Molecular Cell. 2001;8:351–61. [DOI] [PubMed] [Google Scholar]
- [107].Court R, Chapman L, Fairall L, Rhodes D. How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep. 2005;6:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.