

Abstract
Functional studies of the RNA N6-methyladenosine (m6A) modification have been limited by an inability to map individual m6A-modified sites in whole transcriptomes. To enable such studies, here, we introduce m6A-selective allyl chemical labeling and sequencing (m6A-SAC-seq), a method for quantitative, whole-transcriptome mapping of m6A at single-nucleotide resolution. The method requires only ~30 ng of poly(A) or rRNA-depleted RNA. We mapped m6A modification stoichiometries in RNA from cell lines and during in vitro monocytopoiesis from human hematopoietic stem and progenitor cells (HSPCs). We identified numerous cell-state-specific m6A sites whose methylation status was highly dynamic during cell differentiation. We observed changes of m6A stoichiometry as well as expression levels of transcripts encoding or regulated by key transcriptional factors (TFs) critical for HSPC differentiation. m6A-SAC-seq is a quantitative method to dissect the dynamics and functional roles of m6A sites in diverse biological processes using limited input RNA.
RNA modifications have emerged as critical regulators of gene expression programs1,2. The most abundant internal mRNA modification in mammals is m6A. m6A modification exhibits physiologically important effects on a variety of biological processes1,2. Despite the importance of RNA m6A methylation, a quantitative method that maps whole-transcriptome m6A sites with stoichiometric information is lacking. Such a method would allow the evaluation of contributions of individual m6A sites to biological functions and enable analysis and comparison of potential global dynamic changes during cell differentiation, cell signaling or stress responses.
The first two transcriptome-wide mappings of m6A modifications, conducted in 2012 (m6A-seq or MeRIP-seq), used anti-m6A to enrich RNA fragments containing m6A, producing maps with a resolution of 100–200 nucleotides (nt) (refs.3,4). This antibody-based profiling approach and later improved versions, such as PA-m6A-seq5, miCLIP6 and m6A-LAIC-seq7, have enabled extensive research on m6A and its biological functions. However, the antibody-based approach has several notable limitations, including low resolution, a lack of stoichiometric information, a requirement of a large amount of input materials and a limited ability to compare m6A methylation across conditions8. A DART-seq method fuses an m6A-reader protein to an RNA-editing enzyme to induce mutations at sites adjacent to m6A residues9. This method can map m6A using limited input material, but it still lacks quantitative information regarding the modification site and stoichiometry.
MAZTER-seq10 and m6A-REF-seq11 use the MazF RNase known to selectively cleave RNA at unmethylated ACA motifs but not methylated m6ACA motifs12, allowing detection of m6A at m6ACA motifs after subtraction. However, these methods can only cover ~16% of m6A sites in this specific motif. Additionally, they lack detection sensitivity. Because the m6A-to-A ratio is typically 0.5% in most RNA samples, methods that detect A residues instead of m6A and subtract from the over 200-fold excess of unmodified A to obtain information for the much less abundant m6A demand extremely high sensitivity and selectivity for these approaches. A method that selectively detects m6A instead of A is therefore preferred.
The recently developed m6A-SEAL13 and m6A-label-seq14 offer new options for m6A-specific mapping, but they still lack stoichiometric information. A variety of antibody-independent methods15–19 have been developed to measure the abundance of m6A at a particular site, but these could not be applied across the whole transcriptome. A high-throughput sequencing method using limited input material to quantitatively map RNA m6A, similar to bisulfite sequencing for 5-methylcytosine in DNA, is critical for mechanistic investigations of m6A biology and for revealing new biological insights in eukaryotic systems.
Results
Strategy and development of m6A-SAC-seq.
To detect and quantify m6A levels across the transcriptome at single-nucleotide resolution, we developed m6A-SAC-seq. This method uses the Dim1/KsgA family of dimethyltransferases, which transfer the methyl group from S-adenosyl-l-methionine (SAM) to adenosines, forming m6A and then N6,N6-dimethyladenosine (m62A) in consecutive methylation reactions20 (Fig. 1a).
Fig. 1 |. m6A-SAC-seq strategy and development.

a, MjDim1 uses allylic-SAM as a cofactor to label m6A to a6m6A, which undergoes cyclization following I2 treatment. b, An m6A-modified 12-mer RNA probe was treated with MjDim1 and allylic-SAM, followed by matrix-assisted laser desorption ionization (MALDI) characterization. The added molecular weight is that of the allyl group. c, An m6A-free 12-mer RNA probe was treated with MjDim1 and allylic-SAM, followed by MALDI characterization. No detectable new product appeared. d, Michaelis–Menten steady-state kinetics of the MjDim1-catalyzed allyl transfer to an m6A-containing probe (MALDI_Probe_m6A in Supplementary Table 1). Data are represented as mean ± s.e.m. for two biological replicates × two technical replicates. e, Michaelis–Menten steady-state kinetics of the MjDim1-catalyzed allyl transfer to an unmodified control probe (MALDI_Probe_A in Supplementary Table 1). Data are represented as mean ± s.e.m. for two biological replicates × two technical replicates. f, Cyclized a6m6A induces higher mutation rates than cyclized a6A in various RNA sequence contexts when using HIV RT. RNA oligonucleotides containing a6A or a6m6A were synthesized by incorporating O6-phenyl-adenosine phosphoramidite into the designed sequence containing an NNXNN motif (X = a6A or a6m6A).
For m6A-SAC-seq, we selected the Methanocaldococcus jannaschii homolog MjDim1, which shows highly processive kinetics of converting m6A into m62A21, and used a chemically modified allylic-SAM as the cofactor22 (Fig. 1a and Supplementary Fig. 1a,b). We synthesized a 12-mer RNA probe with a GGA/m6ACU consensus motif containing ‘A’ or ‘m6A’ (underlined) in the middle (Fig. 1b,c and Supplementary Fig. 1c,d). In the presence of allylic-SAM, MjDim1 exhibited an approximately tenfold preference for m6A over A in a model allyl group transfer reaction (Fig. 1d,e), converting m6A into allyl-modified m6A (N6-allyl,N6-methyladenosine or a6m6A) and A into allyl-modified A (N6-allyl-adenosine or a6A). We further optimized the enzyme-labeling protocol to achieve the optimum efficiency (Supplementary Fig. 1e,f).
Subsequent I2 treatment converts a6m6 A and a6A into homologs of N1,N6-ethanoadenine and N1,N6-propanoadenine, respectively, as we have previously shown22 (Fig. 1a and Supplementary Fig. 1g). Human immunodeficiency virus 1 (HIV-1) reverse transcriptase (RT) reads through synthetic oligonucleotides containing the allyl-labeled and cyclized adducts with negligible RT stops (Supplementary Fig. 1h). To our delight, the HIV-1 RT generated ~tenfold higher mutation rates at the cyclized a6m6A sites (true-positive m6A sites) than cyclized a6A sites (unmodified A sites) in almost all sequence contexts tested using model oligonucleotides containing NNXNN (X is either cyclized a6m6A or a6A; Supplementary Table 1 and Fig. 1f). Therefore, m6A-SAC-seq directly detects m6A and exhibits high selectivity toward m6A over A at two steps of the procedure: (1) the allyl transfer from allylic-SAM catalyzed by MjDim1 is ~tenfold more selective for m6A than A, and (2) the labeled and cyclized m6A adducts generate higher mutation rates than the corresponding adducts formed from unmodified A. Even with the high selectivity of m6A-SAC-seq (~100-fold), there is a ~200-fold excess of unmodified A that could produce non-specific background noise. To confirm the specificity of m6A-SAC-seq, we included a control sample in which RNA is treated with the m6A demethylase FTO before MjDim1 labeling. FTO treatment removed a large portion of the m6A in isolated mRNAs from HeLa cells, justifying its use to erase m6A in the transcriptome as a background control (Supplementary Fig. 1i).
Next, we assessed the ability of m6A-SAC-seq (Supplementary Fig. 1j) to detect m6A in a variety of sequence contexts by using different RNA probes that contain either NNa6m6ANN or NNm6ANN in the middle (Supplementary Table 1). Using probes that contain NNa6m6ANN, we confirmed that HIV RT does not display a notable intrinsic sequence context preference, and the misincorporation occurs at the cyclized a6m6A site, with an expected mutation pattern of A to U/C > G (data not shown). We then analyzed the sequence preference of MjDim1 using RNA probes that contain NNm6ANN in the middle. We performed allyl transfer catalyzed by MjDim1 followed by chemically induced cyclization and RT. Because the RT step does not show notable sequence preference, mutation rates from this experiment mostly reflect the sequence preference of MjDim1 toward the GA motif (Supplementary Fig. 1k). We found that m6A in the most common consensus motif of Gm6AC tends to yield high mutation rates (>30%), whereas m6A in the less common Am6AC afforded lower but still clearly above background mutation rates (5–10%), indicating that our method can determine the modification stoichiometry of m6A in almost all sequence contexts (Supplementary Fig. 1l). To ensure modification stoichiometry quantification at most sequence contexts when sequencing biological samples, we added in spike-in calibration probes that contain varying fractions of m6A in NNm6ANN with different barcodes (Supplementary Table 1). These calibration probes undergo the same m6A-SAC-seq procedures as the real biological samples. Therefore, mutation rates of m6A in the calibration probes in different sequence contexts provide normalization standards to determine modification fractions of individual m6A sites in sample RNA.
Quantitative m6A maps of poly(A)-tailed RNAs from cell lines.
We comprehensively mapped m6A sites in poly(A)-tailed RNAs isolated from HeLa, HEK293 and HepG2 cells using m6A-SAC-seq with spike-in calibration RNA probes. We observed efficient labeling and mismatch conversion of m6A; the spike-in probes with 100% DRm6ACH displayed a mutation rate of ~75%, whereas those with 100% unmodified DRACH displayed a mutation rate below 5% (Supplementary Fig. 2a). FTO treatment of the spike-in probes further revealed that the non-specific mutation rate in the DRACH motif is below 5% (Supplementary Fig. 2a). The m6A sites in spike-in probes displayed characteristic mutational patterns, and the observed mutation rates are proportional to the m6A stoichiometry (an example probe is shown in Supplementary Fig. 2b). We relied on spike-in standards with different fractions to assign modification stoichiometry information. Significant linear correlations (R2 > 0.92, P < 0.05) between mutation frequency and m6A fraction of spike-ins ensured reliability of quantification for the m6A fraction on various DRACH motifs (Supplementary Fig. 2c). To ensure reliability of our identified m6A sites, we kept only the m6A sites identified in both biological replicates after comparison with demethylation controls (Supplementary Fig. 2d–f). About 60% of m6A sites identified in each technical replicate overlap with sites identified in the other technical replicate (Supplementary Fig. 2g). According to the sampling test on mapped reads, ~80% of mapped reads could recover approximately 90% of m6A sites (Supplementary Fig. 2h). As a base-resolution method, m6A-SAC-seq requires more sequencing depth than the antibody-dependent profiling approaches.
We identified more than 10,000 high-confidence m6A sites with stoichiometrical information in each of these three cell lines with two biological replicates (Supplementary Data 1 and Supplementary Fig. 3a,b). m6A stoichiometry was reproducible within biological replicates (Supplementary Fig. 3c). Consistent with previous observations3,4, most m6A sites are enriched around the stop codon and located in the 3′-untranslated region (3′-UTR) and coding DNA sequence (CDS) regions (Supplementary Fig. 3d,e), with identified m6A sites in the frequent m6A-methylated GGACU and AGACU motifs displaying the highest frequency (Supplementary Fig. 3f). We noticed that the number of identified sites in the less frequent m6A-methylated DAACH motifs was low. This is most likely caused by the fact that m6A-SAC-seq yields low mutation rates for certain Am6AC motifs (Supplementary Fig. 1l). To estimate how many DAACH sites m6A-SAC-seq might have missed, we analyzed DRACH motifs under MeRIP-seq peaks and compared them with m6A-SAC-seq sites. We could observe seven to nine DRACH motifs per peak, on average, from MeRIP-seq data3,23,24 in three cell lines that we studied (Supplementary Fig. 3g). We decided to focus on Gm6AC- or Am6AC-enriched peaks to avoid overestimating DRACH sites under MeRIP-seq peaks that are not methylated and not detected by m6A-SAC-seq. We identified ~72% of these peaks, on average, that were enriched with the Gm6AC motif (Supplementary Fig. 3h), which is consistent with previous reports6,25,26. Based on this analysis, m6A-SAC-seq likely missed ~85% of Am6AC motif occurrences (Supplementary Fig. 3i). However, the Am6AC sites detected by m6A-SAC-seq were significantly enriched in high-signal peaks (Supplementary Fig. 3j), indicating that m6A-SAC-seq can still uncover highly modified Am6AC sites.
Next, to further validate the m6A-SAC-seq results, we enriched m6A-modified poly(A)-tailed RNAs in HeLa cells and then performed m6A-SAC-seq (MeRIP-SAC-seq) (Methods). Almost 70% of the confident m6A-SAC-seq sites overlap with MeRIP-SAC-seq sites (Supplementary Fig. 3k). Moreover, around 60% of the m6A-SAC-seq sites overlap with MeRIP-seq peaks (Supplementary Fig. 3l). Notably, m6A-SAC-seq sites are significantly enriched in high-signal MeRIP-seq peaks (Supplementary Fig. 3m). We further compared m6A sites identified by m6A-SAC-seq with public data, such as data from MeRIP-seq in HeLa24 and HEK2934 cells and data from m6A-SEAL13 and miCLIP6 in HEK293 cells. The distribution of m6A sites revealed from m6A-SAC-seq resembled that of m6A peaks obtained by MeRIP-seq (Supplementary Fig. 3n). MeRIP-seq and m6A-SEAL sites overlapped better with m6A-SAC-seq sites than miCLIP sites (Supplementary Fig. 3o,p). However, we observed that miCLIP sites are enriched near the identified sites of m6A-SAC-seq (Supplementary Fig. 3q). These results, therefore, confirm that m6A-SAC-seq could specifically detect m6A-modified sites in the whole transcriptome. We applied SELECT19 and MAZF digestion10,11 to validate several randomly selected sites, including one m6A negative and four m6A positives using SELECT (Supplementary Fig. 4a) and one m6A negative and five m6A positives using MAZF digestion (Supplementary Fig. 4b), confirming the accuracy of m6A-SAC-seq. FTO− and FTO+ signal tracks for representative transcripts clearly showed the specificity of m6A-SAC-seq. (Supplementary Fig. 4c). Of note, even without FTO treatment control, we could assign more than 90% confident m6A sites (Supplementary Fig. 4d), justifying the specificity of the m6A-SAC-seq strategy even without the demethylation control. In conclusion, although m6A-SAC-seq has a limitation in the detection of the less frequently m6A-methylated Am6AC motif, it could still reveal high m6A stoichiometry sites in these motifs; it reveals more than 70% DRACH sequences with stoichiometric information.
In addition to m6A sites in the 3′-UTRs and CDSs, we also observed a considerable number of m6A sites in the 5′-UTRs and intronic regions, including sites with over 40% modification stoichiometry in all three cell lines (Fig. 2a). The average stoichiometry for m6A is notably higher in intronic regions than in the 5′-UTR, 3′-UTR and CDS (Fig. 2a). We performed unsupervised k-means clustering for transcripts with 5′-UTR-, 3′-UTR- and CDS-specific methylation, respectively. We found that the majority of m6A sites display considerably different stoichiometries across different cell lines (Fig. 2b). Gene ontology (GO) enrichment analysis showed that the cell-type-specific m6A sites (clusters C13–C15 and C24–C26) are enriched in transcripts associated with various biological processes, such as histone or chromatin modification, RNA splicing, RNA catabolism, RNA metabolism and cell cycle (Fig. 2c). Results from these three cell lines suggest that quantitative m6A fractions of a large portion of m6A sites are different among cell types. To seek further evidence of cell-type-specific m6A methylation, we collected and processed the published MeRIP-seq data from different cell types using a unified pipeline. We found only 35–50% (5,073) common peaks among the three cell lines at 50- to 500-nt (peaks) resolution (Supplementary Fig. 4e).
Fig. 2 |. Characteristics of quantitative m6A maps in poly(A)-tailed RNAs from HeLa, HeK293 and HepG2 cells.

a, Number (bar plots) and modification fractions (violin box plots) distribution of m6A sites in different RNA regions that include 3′-UTR, CDS, intronic, 5′-UTR, intergenic and promoter. In box plots, lower and upper hinges represent first and third quartiles, the center line represents the median, the red dot represents the mean, and whiskers represent ±1.5× the interquartile range (HEK293: n = 6,201 3′-UTR, n = 4,967 CDS, n = 514 5′-UTR, n = 379 intronic, n = 148 intergenic, n = 25 promoter; HeLa: n = 6,004 3′-UTR, n = 4,069 CDS, n = 377 5′-UTR, n = 324 intronic, n = 103 intergenic, n = 15 promoter; HepG2: n = 6,030 3′-UTR, n = 4,585 CDS, n = 460 5′-UTR, n = 126 intronic, n = 57 intergenic, n = 12 promoter). b, k-means clustering was performed for RNA 100-nt bins that contain m6A sites in at least one of the three cell lines. The 5′-UTR, 3′-UTR and CDS 100-nt bins were classified into seven clusters by k-means clustering on the m6A fractions sum of 100-nt bin. Each row represents a 100-nt bin, and the number of 100-nt bins is shown in the bracket. c, GO Biological Process (GOBP) enrichment analysis for the m6A clusters defined in b. Cell type-specific clusters (C5, C13–C15 and C24–C26) are highlighted by bold italic.
Region-specific m6A on mRNA decay and translation efficiency.
To reveal the relationship between region-specific m6A and mRNA metabolism, we categorized 5′-UTR-m6A-only, 3′-UTR-m6A-only and CDS-m6A-only transcripts into three groups (high, medium and low) with equal numbers of transcripts based on the sum of their m6A fractions at specific mRNA regions (3′-UTR, 5′-UTR or CDS) of the transcript and determined how their lifetimes and translation efficiencies were affected by the knockdown of the m6A writer (METTL3) or readers (YTHDF1/YTHDF2), respectively. We observed shorter lifetimes for transcripts with high m6A stoichiometries than for transcripts with low m6A stoichiometries for all transcripts and 3′-UTR-m6A-only and CDS-m6A-only transcripts (Fig. 3a and Supplementary Fig. 5a,b). The knockdown of either METTL3 or YTHDF2 led to extended RNA lifetime, especially for the more heavily modified transcripts (Supplementary Fig. 5a,b), confirming the main role of m6A in mRNA turnover27. Notably, our analysis revealed that the 5′-UTR-m6A-only transcripts tend to display a longer lifetime than unmodified transcripts and 3′-UTR-m6A-only and CDS-m6A-only transcripts (Fig. 3b,c). Furthermore, depletion of YTHDF2 caused reduced stability of these transcripts, suggesting a stabilization role of YTHDF2 on the 5′-UTR-m6A-only transcripts, either directly or indirectly (Fig. 3c).
Fig. 3 |. effects of m6A on the modified RNAs in cell lines.

a, Cumulative curves and box violin plots showing the distribution of transcript lifetime with different m6A fractions in HeLa cells. Transcripts were classified into three groups (high, medium and low) with equal numbers of transcripts based on the sum of their m6A fractions (left: n = 1,576 high, n = 1,575 medium, n = 1,575 low, n = 5,277 no m6A; right: n = 1,672 high, n = 1,671 medium, n = 1,671 low, n = 6,739 no m6A). Two lifetime data sets, GSE98856 (left) and GSE49339 (right), were used to confirm each other. The P value between two groups (the no m6A group as reference) was determined by one-tailed Wilcoxon rank-sum test in the violin box plots. b, Cumulative curves and box violin plots showing the lifetime distribution of 5′-UTR-m6A-only (n = 77), 3′-UTR-m6A-only (n = 1,579), CDS-m6A-only (n = 889) and non-m6A (n = 5,277) transcripts; lifetime data set: GSE98856. c, Cumulative curves and box violin plots showing the distribution of lifetime for 5′-UTR-m6A-only (n = 110), 3′-UTR-m6A-only (n = 2,200), CDS-m6A-only (n = 1,182) and non-m6A (n = 6,739) transcripts in siControl (control short interfering RNA) and siYTHDF2 (YTHDF2 short interfering RNA) data sets and their ratios in HeLa cells. The y axis label of the box violin plot is the same as the x axis label of the cumulative curve plot; lifetime data set: GSE49339. d, Distribution of MFE of RNA folding for a 31-nt sliding window with or without m6A on the 16th adenosines. The upstream and downstream 15 nt of adenines with or without m6A were used to generate the 31-mer window. All identified m6A sites (n = 12,234 HEK293, n = 10,892 HeLa, n = 11,270 HepG2) were used to calculate MFE, and the number of non-m6A sites was 10,000 for each cell type. e, Distribution of PhastCons scores of m6A and non-m6A sites in cell lines. In different cell lines, m6A sites were classified into two categories (low and high) based on their PhastCons scores (low ≤ 0.5, high > 0.5) (HEK293: n = 2,711 low, n = 7,074 high; HeLa: n = 2,482 low, n = 6,139 high; HepG2: n = 2,230 low, n = 6,998 high). For b–e, the P value was determined by one-tailed Wilcoxon rank-sum test. In box plots (a–e), lower and upper hinges represent first and third quartiles, the center line represents the median, the red dot represents the mean, and whiskers represent ±1.5× the interquartile range.
The m6A fractions of transcripts showed an effect on translation efficiency, with knockdown of either METTL3 or YTHDF1 further decreasing translation efficiency (Supplementary Fig. 6a,b). Translation efficiency can be notably perturbed by m6A sites located in the CDS but less so in the 3′-UTR and 5′-UTR28 (Supplementary Fig. 6c). These observations confirm the translation upregulation role discovered previously28,29 and in a recent m6A-QTL analysis30. The m6A-QTL study also revealed a heterogeneous effect of m6A on translation, with certain m6A sites recognized by RNA-binding proteins (RBPs) that suppress translation. When we analyzed a 31-mer window around the modified m6A sites, we observed that they tend to adopt predicted structures with lower minimum free energies (MFE) than non-methylated sites, suggesting that m6A tends to mark regions that form secondary structures (Fig. 3d). This result confirms previous observations that m6A marks more structured transcripts and elevates translation efficiency, potentially by relaxing the secondary structure28.
We previously reported that mRNA m6A is evolutionarily conserved and tends to cluster among species11. It is tempting to speculate that specific adenine sites subjected to m6A modification could be more conserved than those without methylation, because m6A plays important roles in mRNA stability, translation and secondary structure tuning. Using the stoichiometry information from our m6A-SAC-seq data, we explored the correlation of m6A stoichiometry and natural selection during evolution. Like previous studies3,11, we also observed that m6A sites are more conserved than unmethylated A sites (Supplementary Fig. 7a). However, we found that m6A sites with low PhastCons scores tend to be highly m6A modified, while m6A sites with high PhastCons scores tend to be lowly m6A modified (Fig. 3e), suggesting that high-stoichiometry m6A sites are more likely to evolve quickly than low-stoichiometry sites.
We also compared HepG2 m6A sites and ENCODE eCLIP data31,32 from the same cell line to explore potential connections between m6A and RBPs. We identified 11 RBPs that show site overlap of at least 5% between m6A and eCLIP binding sites (Supplementary Fig. 7b), including known components of methyltransferases (e.g., RBM15 (ref.33)), readers (e.g., IGF2BPs34) (note that the eCLIP data are not available for the YTH family readers of m6A in HepG2) and potential new reader protein candidates whose relationship with m6A would need future investigation.
m6A deposition is linked to RNA splicing.
Recent studies have uncovered the effects of m6A on pre-mRNA splicing through hnRNPG35,36, hnRNPA2B1 (ref.37) and YTHDC1 (ref.38). To explore a potential link between m6A and RNA splicing, we investigated m6A sites identified from HEK293 rRNA-depleted RNA samples. We observed a depletion of m6A close to the splice site (SS) in both introns and exons; however, we found that m6A is enriched toward the 5′-end of introns but not at 5′- or 3′-ends of exons (Supplementary Fig. 8a). We observed approximately 29.6% of m6A sites close to the 5′- or 3′-SSs (i.e., within 300 nt of intron and 200 nt of exon flanking the 5′- or 3′-SS; Supplementary Fig. 8b) and subsequently asked whether these sites could be associated with alternative splicing (AS). We selected previously identified hnRNPG-bound m6A sites36 and examined their co-occurrence with AS events after METTL3 or METTL14 knockdown. We found approximately 11.6% of hnRNPG-bound m6A sites near SSs showing potential relationships with AS events (| ΔΨ | > 5%, false-discovery rate (FDR) < 0.1) following depletion of either METTL3 or METTL14 (Supplementary Fig. 8c). The deposition of m6A near SSs is specific in distinct AS events (Supplementary Fig. 8d,e), suggesting that m6A might be responsible, at least in part, for some of these AS events.
m6A dynamics along monocytopoiesis.
Despite the crucial effects of RNA m6A methylation on cell differentiation as well as cell signaling and stimulation responses, the dynamics of m6A modification along most biological processes remain unclear10. RNA m6A modification is known to play important roles during normal and malignant hematopoiesis39. We applied m6A-SAC-seq to interrogate whole-transcriptome m6A changes during hematopoiesis, a highly controlled process during which epigenetic events and gene expression are strictly orchestrated to ensure correct fate decisions. We specifically studied monocytopoiesis starting from human umbilical cord blood-derived CD34+ HSPCs. Human monocytopoiesis is completed in vitro by day 9 (Supplementary Fig. 9a–e). We therefore performed m6A-SAC-seq with samples collected at days 0 (d0), 3 (d3), 6 (d6) and 9 (d9) during monocytopoiesis. To gain comprehensive information about m6A dynamics on pre-mRNAs and other unprocessed RNAs, we sequenced isolated RNAs after rRNA depletion. As a result, we identified approximately 20,000–32,000 high-confidence m6A sites in each of the above four time points during monocytopoiesis (Supplementary Data 1).
We identified a total of 9,654 gene transcripts containing m6A modifications at least at one time point (Supplementary Data 2). As expected, we observed enrichment of m6A sites in the CDS and 3′-UTR (Supplementary Fig. 10a). The m6A stoichiometry mainly ranges from 25 to 75%; m6A sites in introns (1,416 to 3,611 sites at different stages of monocytopoiesis) showed surprisingly the highest average modification stoichiometry (Fig. 4a). All regions of mRNAs can be m6A methylated, although the 3′-UTR and CDS tend to enrich more highly modified m6A sites, and certain non-coding RNAs (ncRNAs) also contain m6A sites (Supplementary Fig. 10b and Supplementary Data 2). Many of these m6A-modified transcripts (e.g., RUNX1 (ref.40), FOS41, ZEB2 (ref.42), ETV6 (ref.43), XIST44 and MALAT1 (ref.45)) have been reported to regulate hematopoiesis. Furthermore, transcripts of m6A writers, readers and erasers (e.g., METTL3/METTL14, WTAP, YTHDF1/YTHDF2/YTHDF3 and ALKBH5) are also m6A modified, suggesting a mechanism of m6A autoregulation (Supplementary Data 2). By exploiting expression patterns of genes with m6A modification, we found that transcripts with dynamic m6A stoichiometric changes tend to show more significant expression variations than transcripts possessing stable m6A (Fig. 4b). In addition, we observed that the stoichiometry of m6A at different RNA regions displayed weak negative or no correlations with gene expression level (Supplementary Fig. 11). A similar result could also be found in cell lines (Supplementary Fig. 12), indicating that transcript abundance does not affect m6A detection using m6A-SAC-seq.
Fig. 4 |. m6A dynamics across hematopoietic stem cell differentiation into monocytes.
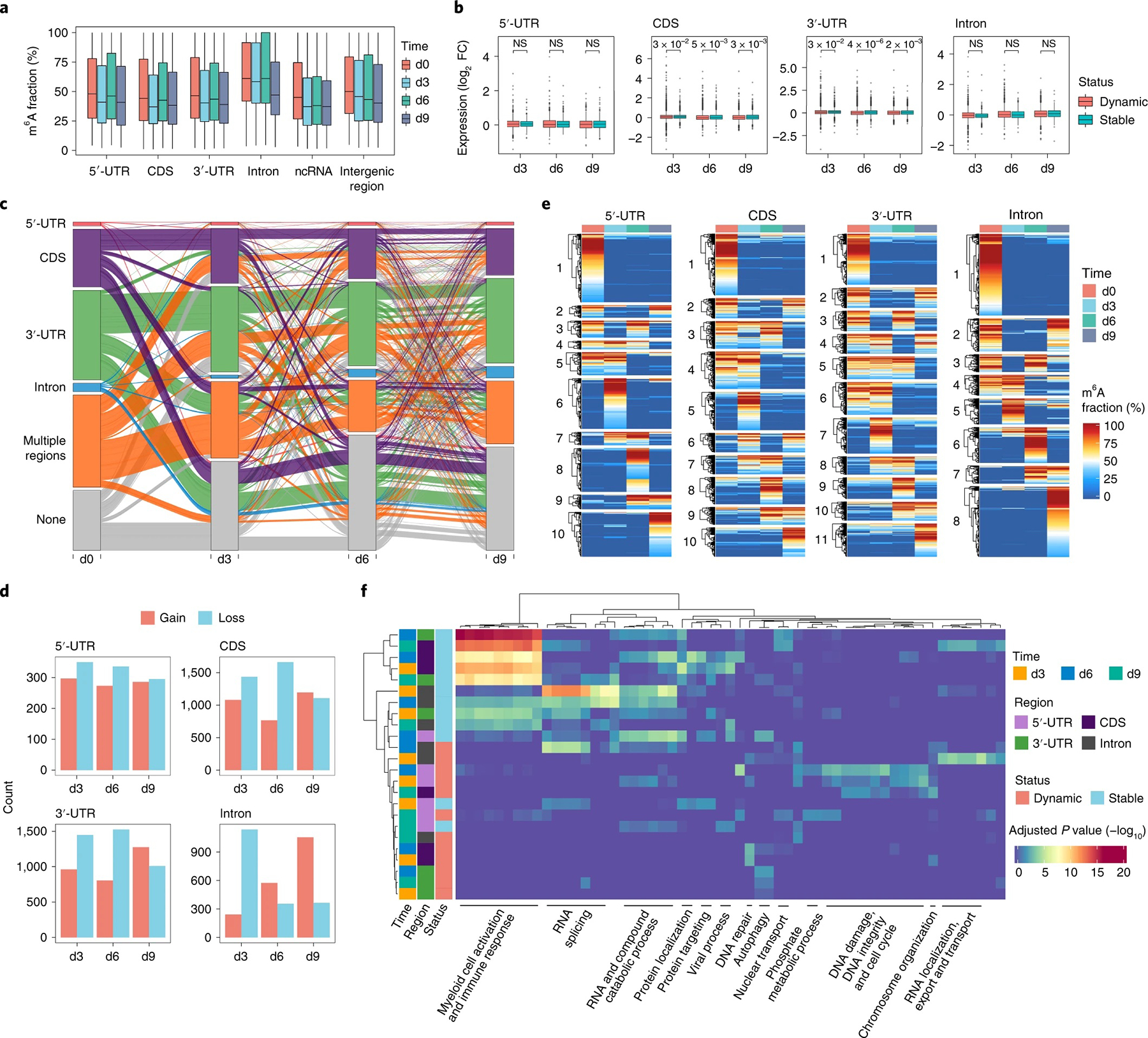
a, Distribution of m6A fractions in different genomic features. Colors indicate time points. In box plots, lower and upper hinges represent first and third quartiles, the center line represents the median, and whiskers represent ±1.5× the interquartile range (5′-UTR: n = 867 d0, n = 773 d3, n = 628 d6, n = 647 d9; CDS: n = 11,630 d0, n = 12,277 d3, n = 7,215 d6, n = 7,603 d9; 3′-UTR: n = 15,243 d0, n = 15,393 d3, n = 10,378 d6, n = 11,310 d9; intron: n = 3,611 d0, n = 1,416 d3, n = 1,481 d6, n = 3,560 d9; ncRNA: n = 1,473 d0, n = 1,232 d3, n = 948 d6, n = 1,073 d9; intergenic region: n = 590 d0, n = 424 d3, n = 414 d6, n = 581 d9). b, The log2-transformed fold changes (log2 FC) of expression levels of transcripts with dynamic or stable m6A sites in different feature regions of mRNAs between adjacent time points are shown. The dynamic or stable m6A sites were defined as those that were detected between adjacent time points, and their changes of m6A fractions were more or less than 20%, respectively. Colors indicate the dynamic or stable status. P values were determined using a two-tailed Mann–Whitney U-test; NS, not significant. In box plots, lower and upper hinges represent first and third quartiles, the center line represents the median, and whiskers represent ±1.5× the interquartile range (5′-UTR: n = 613, n = 299, n = 576, n = 259, n = 531, n = 254; CDS: n = 3,341, n = 2,220, n = 3,124, n = 1,767, n = 2,814, n = 1,611; 3′-UTR: n = 3,586, n = 2,740, n = 3,380, n = 2,304, n = 3,263, n = 2,171; intron: n = 1,558, n = 253, n = 1,050, n = 198, n = 1,657, n = 290). The order of the n number in each region is consistent with that of box plots shown in each region. c, Alluvial plots showing global m6A dynamics on feature regions of mRNAs during monocytopoiesis. Each line represents one transcript bearing m6A at different transcript regions across four time points. Colors indicate feature regions where m6A was initially installed at d0. d, Number of genes that gained or lost m6A in different regions of mRNAs were counted by comparing to the previous time point. Only genes gaining or losing m6A on the specific genomic region were considered. Colors indicate the status of gain or loss. e, Heat maps depicting clusters of m6A sites by m6A stoichiometries in different transcript regions of mRNAs. The dendrogram in each cluster was constructed using complete linkage based on Euclidean distance. Numbers near the dendrograms represent cluster identifiers. f, Heat map illustrating GO analysis on genes with dynamic or stable m6A in different transcript regions during differentiation. The dendrograms in both rows and columns were constructed using complete linkage based on Euclidean distance. Between adjacent time points, m6A sites with a stoichiometry difference more than 20% were defined as dynamic, and those less than 20% were defined as stable.
Contrary to some of previous speculations, we found that m6A displayed substantial redistribution and dynamics among different regions of mRNA during differentiation (Fig. 4c). We identified many m6A sites in the CDS, 3′-UTR and intronic regions that were gained or lost compared to the previous time point (Fig. 4d). Moreover, we observed that m6A stoichiometry near the stop codons decreased during HSPC differentiation but was restored after differentiation (Supplementary Fig. 10c), suggesting that m6A stoichiometric changes might play a role in the regulation of differentiation. To further investigate m6A dynamics across differentiation, we grouped genes based on their mRNA m6A stoichiometric changes in the 5′-UTR, CDS, 3′-UTR and intron regions and identified a total of 39 clusters (Fig. 4e and Supplementary Fig. 10d). We next examined functional differences between genes with dynamic or stable m6A deposition. GO analysis showed that genes maintaining basic cell survival functions tend to undergo dynamic m6A stoichiometric changes (pathways related to DNA repair, damage, integrity and cell cycle), whereas genes involved in pathways related to myeloid cell activation, immune response, RNA splicing and catabolic processes are more stable in m6A stoichiometry (Fig. 4f).
Dynamic m6A levels correlate with gene expression changes.
m6A modification could mark and prime transcripts for coordinated decay and translation and thus facilitate transcriptome turnover and protein production during cell differentiation2. We identified 4,406 m6A-modified transcripts that were differentially expressed between any two consecutive time points (FC > 1.5 or FC < 0.667, FDR < 0.05; Fig. 5a and Supplementary Data 3). Among them, transcripts encoding TFs critical for monocyte developmental regulation41,46–48 exhibited m6A stoichiometry changes along with expression level changes during HSPC differentiation (Fig. 5b). For example, the stoichiometry of m6A methylation in the CDS of KLF4 increased along with abundance of the transcript level from d6 to d9; a similar observation was made for EGR1 CDS m6A methylation at the d0-to-d3 transition. Additional examples include m6A stoichiometry changes of 3′-UTR m6A methylation in ERG1, SPI1 and CEBPA, which decreased in m6A stoichiometry and increased in transcript abundance at the d0-to-d3 transition.
Fig. 5 |. m6A modification impacting gene expression during monocytopoiesis.
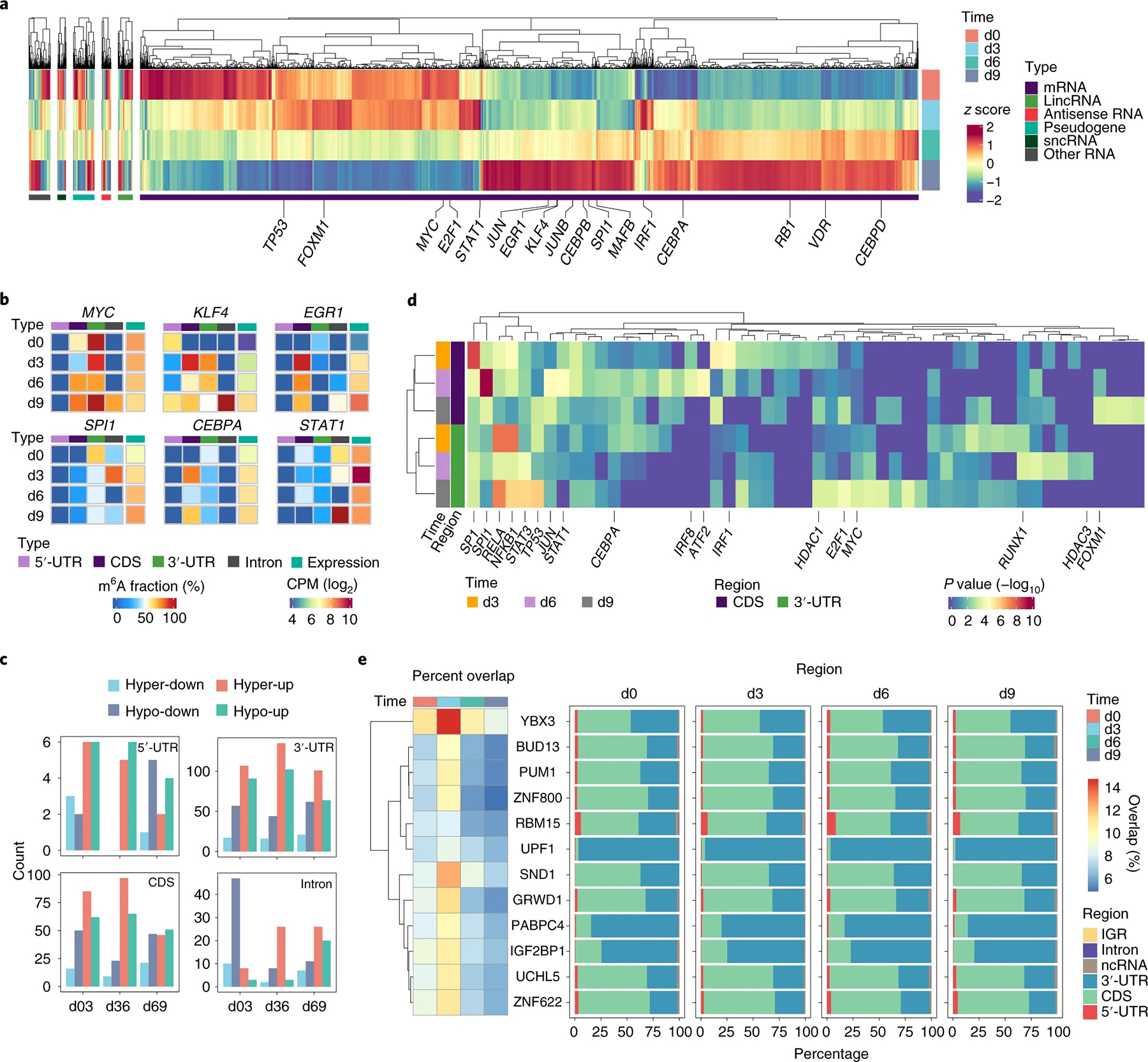
a, Heat map showing expression profiles of m6A-modified and differentially expressed genes (FC > 1.5 or FC < 0.667, FDR < 0.05) by comparing any two time points during HSPC differentiation. Gene expression levels were scaled by z score across time points. The dendrogram was constructed using complete linkage based on Euclidean distance. Master TFs involved in regulating HSPC differentiation into monocytes are labeled in the heat map; lincRNA, long intergenic non-coding RNA; sncRNA, small non-coding RNA. b, Examples displaying changes of both m6A stoichiometries in different transcript regions and expression levels of key TF transcripts; CPM, counts per million. c, Distribution of counts of genes with changes in both m6A stoichiometries in different transcript regions and expression levels between adjacent time points. Colors indicate changes, with hyper- or hypomethylation defined as having methylation difference (MD) > 10% or MD < −10%, respectively; up- or downregulated gene expression differences are defined as having FDR < 0.05 and FC > 1.5 or FC < 0.667, respectively. d, Heat map showing TF-regulated gene enrichment analysis on the gene sets with m6A in the CDS and 3′-UTR in c. The dendrograms in both rows and columns were constructed using complete linkage based on Euclidean distance. Master TFs during differentiation were labeled in the heat map. e, Heat map (left) depicting the overlap between RBP eCLIP peaks and m6A sites and bar plots (right) showing the distribution of counts of their shared binding regions, respectively. The dendrogram in rows was constructed using complete linkage based on Euclidean distance. The percent overlap was represented by the average of the fractions of overlap that was reciprocal for RNA m6A sites and RBP eCLIP peaks. The co-occurrence criteria between RBPs and m6A was defined as both of RBP and m6A reciprocal fractions of site overlap > 5% and one of them > 9% at least at one time point.
To further investigate the relationship between m6A modification and gene expression, we identified 1,240 genes with changes in both m6A stoichiometry (| MD | > 10%) and transcript level (FC > 1.5 or FC < 0.667, FDR < 0.05) between two adjacent time points (Fig. 5c and Supplementary Data 4). Notably, these genes with m6A installed in the CDS or 3′-UTR are significantly enriched in transcripts regulated by master TFs (Fig. 5d), which orchestrate the development of blood cells from HSPCs. For instance, transcripts regulated by SP1 and SPI1 are enriched with m6A in the CDS, whereas transcripts regulated by NF-κB1, RELA, STAT3 and TP53 tend to accumulate m6A in the 3′-UTR, respectively. The current quantitative maps of m6A during this process, particularly the correlation between m6A stoichiometry changes and transcript level differences for key transcripts, provide rich resources for further mechanistic understanding and modulation of HSPC differentiation in the future.
To provide additional insights, we used ENCODE eCLIP data31,32 from K562 cells, an immortalized myelogenous leukemia cell line, to investigate a potential association between individual m6A sites and RBP binding. We observed significant overlap between binding sites of a preferred set of 12 RBPs (Fig. 5e and Supplementary Data 5) or an exploratory set of 23 RBPs (Supplementary Fig. 13a and Supplementary Data 5) and m6A sites. Among them, YBX3, which we recently reported as a potential m6A effector protein30, presents the highest overlapping binding sites with m6A sites. Moreover, we observed that the YBX3-bound and m6A-modified transcripts are enriched in pathways related to cell cycle, especially at the early stages of differentiation (Supplementary Fig. 13b). Although these results suggest that YBX3 might play a translation regulation role through m6A during HSPC differentiation, more experiments to support this role need to be conducted in the future.
We then investigated the correlation between m6A and RNA splicing during monocytopoiesis. We found a similar distribution of m6A in the introns and exons and near SSs at different stages of monocytopoiesis to that of HEK293 cells, with a characteristic enrichment of m6A toward the 5′-end of introns (Supplementary Fig. 14a,b). The conserved deposition of m6A in introns suggests its potential functional role in pre-mRNA processing. We further identified 7,480 AS events in total (| ΔΨ | > 5%, FDR < 0.1) during monocytopoiesis (Supplementary Data 6). Among these events, around 14.2% correlate with changes of stoichiometry (| MD | > 10%) of SS-adjacent m6A sites (that is, m6A within 300 nt of intron and 200 nt of exon flanking the 5′- or 3′-SS; Supplementary Fig. 14c and Supplementary Data 7). For instance, TRPV2 (ref.49) and DOK2 (ref.50), involved in HSPC cell cycle progression, growth and differentiation, both showed notable AS level differences along with changes of m6A stoichiometry at one single site during d0-to-d6 and d6-to-d9 transitions, respectively (Supplementary Fig. 14d). To further analyze changes in m6A stoichiometry during monocytopoiesis that may accompany AS changes, we performed RNA-sequencing analysis of HSPC samples after knockdown of METTL3 and METTL14. After METTL3/METTL14 knockdown, we identified 9,248 AS events during HSPC differentiation (Supplementary Data 8), including the event that was shown in Supplementary Fig. 14d (left). The splicing inclusion level of the skipped exon from TRPV2 transcripts increased after METTL3/METTL14 knockdown (Supplementary Fig. 14e, left). Therefore, our results may suggest effects of m6A deposition on mRNA splicing during cell differentiation.
Discussion
While the RNA m6A modification plays critical roles during cell differentiation, tissue development and transcriptional regulation, studies of these biological processes have been hampered by the limited materials for sequencing and lack of robust methods for comparison of m6A levels across conditions.
We present a quantitative m6A-SAC-seq method that maps m6A sites in the entire transcriptome at single-base resolution. The method requires only ~30 ng of input RNA (from 300 ng total RNA), making it suitable for investigating a variety of biological systems. We detailed comprehensive maps of m6A at single-base precision and with stoichiometry information. m6A-SAC-seq does show a motif preference of GAC over AAC. Previous studies using chromatography51,52, miCLIP6 and miCLIP2 (ref.25) have reported that ~70–75% of m6A sites occur in the GAC motif. This suggests that m6A-SAC-seq could uncover up to about 80% of m6A sites even with a limitation at detecting AAC sites, although m6A-SAC-seq can still reveal highly modified m6A sites in the AAC motif. Engineering of the current methyltransferase to exhibit less sequence bias is a future direction we hope to pursue. In addition, our quantitative sequencing data revealed numerous cell-type-specific m6A sites among three different cell lines (HeLa, HEK293 and HepG2), consistent with our observation that MeRIP-seq data showing 35–50% common peaks among these cell lines. Application of m6A-SAC-seq to monocytopoiesis led to a quantitative delineation of m6A deposition and stoichiometric dynamics across cell differentiation, revealing a broad scope of m6A dynamics on key transcripts important for cell differentiation and uncovering new m6A-interacting protein candidates. This study showcases the potential for broadly applying m6A-SAC-seq to obtain whole-transcriptome m6A stoichiometry maps in various cell differentiation, early development, neuronal signaling and clinical samples. Therefore, we think m6A-SAC-seq will serve as a gold standard that overcomes the current technological bottleneck for quantitative m6A sequencing and enables new biological discoveries.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-022-01243-z.
Methods
Expression and purification of recombinant MjDim1.
The MjDim1 gene was codon optimized and synthesized by Thermo Fisher Scientific and cloned into a pET-His-SUMO vector. T7 Express Competent Escherichia coli (NEB) was transformed with the plasmid and cultured at 37 °C. When the optical density at 600 nm (OD600) reached 1, cells were cooled to 16 °C, IPTG was added to a final concentration of 0.1 mM for inducible expression, and cells were cultured at 16 °C for an additional 18 h. Cells were collected and lysed by EmulsiFlex-C3 (Avestin) in lysis buffer (50 mM Tris-HCl (pH 7.5), 300 mM NaCl). The soluble recombinant protein was purified using a nickel resin column (GE Healthcare) washed with washing buffer (50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5 mM imidazole). Ulp1 was added in the resin to cleave the SUMO tag on the column at 4 °C for 16 h. The enzyme was eluted in elution buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 20 mM imidazole) and subjected to anion-exchange chromatography (Source15-Q 10/10, GE Healthcare) on an AKTA Purifier 10 system (GE Healthcare) to get rid of the RNA and DNA bound with the enzyme. Flow-through was collected for the second round of cation-exchange chromatography (Source15-S 10/10, GE Healthcare), and the fractions coming out between conductivity 20 and 35 were collected and concentrated to ~1.6 mM. Glycerol was added in the enzyme to a final concentration of 30% and stored at −80 °C for future use.
Synthesis of allylic-SAM analog.
S-Adenosyl-l-homocysteine (20 mg, 0.05 mmol, 1 equiv.) was dissolved in formic and acetic acids (1:1, 2 ml). Allyl bromide (425 μl, 5 mmol, 100 equiv.) and AgClO4 (10.4 mg, 0.05 mmol, 1 equiv.) were added and stirred at ambient temperature (22 °C) for 8 h. The reaction was quenched with 20 ml of 0.01% trifluoroacetic acid (TFA; vol/vol) in water. The aqueous phase was washed three times with diethyl ether (3 × 10 ml) and then passed through a 0.2-μm syringe filter. The crude mixture of allyl-SAM was purified using a preparative reversed-phase high-performance liquid chromatography (RP-HPLC) column (XBridge Prep C18 5 μm OBD 19 × 150 mm). A diastereomeric mixture of the allylic-SAM analog was collected, concentrated and lyophilized. The resultant compounds were redissolved in water containing 0.01% TFA (vol/vol), aliquoted and stored at −80 °C before use.
Biochemical assay for a6m6A methyltransferase activity in vitro.
The in vitro methyltransferase activity assay was performed in a standard 20-μl reaction mixture containing the following components: 50 ng to 1 μg of RNA probe or 30 ng of mRNA, 10 nmol of fresh recombinant MjDim1 enzymes, 3 mM allylic-SAM, 40 mM HEPES (pH 8.0), 40 mM NH4 Cl, 4 mM MgCl2 and 1 U μl−1 SUPERase In RNase Inhibitor (Thermo Fisher Scientific). For the RNA probe, the reaction was incubated at 50 °C for 1 h. For mRNA or other biological samples, RNA fragments were ligated with biotin-modified 3′-adapter and bound with Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific). The reaction was performed on the solid phase using the following procedure: 50 °C for 1 h and change new reaction system for three rounds to efficiently label m6A sites with an allyl group.
MALDI-time-of-flight (MALDI-TOF) and HPLC.
The RNA reaction products were purified using Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific) and eluted by heating at 98 °C for 10 min. One microliter of the supernatant was then mixed with an equal amount of MALDI matrix, which was composed of a 9:1 (vol/vol) ratio of 2′,4′,6′-trihydroxy acetophenone (THAP; 10 mg ml−1 in 50% acetonitrile and water):diammonium citrate (50 mg ml−1 in water). The mixture was then spotted on a MALDI sample plate, dried under vacuum and analyzed by a Bruker Ultraflextreme MALDI-TOF-TOF mass spectrometer in a reflector, positive mode. The HPLC profiles were acquired using Waters e2695 equipment.
Quantification of am6A in RNA by LC–MS/MS.
RNA oligonucleotides or mRNAs were digested into nucleosides, and the amount of am6A was measured by using Agilent 6460 Triple Quad MS–MS with a 1290 UHPLC supplied with a ZORBAX Eclipse XDB-C18 column (UHPLC–QQQ–MS/MS) and calculated based on the standard curve generated by pure standards. For each sample, RNA was digested by using 1 U of nuclease P1 (Wako) in a 25-μl reaction containing 10 mM ammonium acetate at 37 °C for 16 h. Then, 1 μl of FastAP thermosensitive alkaline phosphatase and 3 μl of 10× FastAP buffer (Thermo Scientific) was added, and the reaction was incubated at 37 °C for 2 h. Samples were then filtered using a 0.22-μm filter (Millipore) and injected into LC–MS/MS. The nucleosides were quantified by using the nucleoside-to-base ion mass transitions of 282 to 150 (m6A), 323 to 191 (am6A), 268 to 136 (A) and 284 to 152 (G). Quantification was performed in comparison to the standard curve obtained from pure nucleoside standards run on the same batch of samples. The ratio of m6A to G was calculated based on the calibrated concentrations.
RNA probe synthesis and validation.
RNA probes (Supplementary Table 1) with modifications were designed and synthesized using an EXPEDIT DNA Synthesizer 8909. Unmodified probes were purchased from Integrated DNA Technologies. Probes for the calibration spike-in mix were splint ligation products of barcoded 12-mer purchased oligonucleotides with the 29-mer m6A-containing synthesized probe. All the probe sequences are listed in Supplementary Table 1. The a6m6A-containing RNA oligonucleotide was synthesized by incorporating O6-phenyl-adenosine phosphoramidite into the designed sequence containing a GGACU motif. The beads were treated with N-methyl-N-allylamine to convert O6-phenyl to the N6-methyl N6-allyl group. After the regular procedure to remove the 2′-silyl protecting group, the crude RNA oligonucleotide was ethanol precipitated and further purified by RP-HPLC. The structure of the probe was confirmed by MALDI-TOF MS with THAP as the matrix. The HPLC-purified probe was dissolved in deionized water; 1 nmol of RNA probes in 200 μL water was treated with 8 μl of 0.2 M iodine dissolved in 0.2 M KI. After incubation at room temperature for 1 h, 8 μl of 0.2 M Na2S2O3 was added to quench the reaction. The mixture was filtered and injected into HPLC with the same gradient. It can be observed that the original peak disappeared at retention time (32.3 min), while two identical peaks eluted at 29.3 and 29.6 min, which are the two isomers of the cyclized products. Both peaks show the same mass-to-charge ratio (m/z) at 3,018 in the MALDI-TOF mass spectrum, consistent with the proposed structure.
Steady-state kinetics of MjDim1-catalyzed am6A and a6A modifications.
MjDim1 (168 μM) was used for the kinetic measurements using both MALDI_Probe_m6A and MALDI_Probe_A (Supplementary Table 1) with allylic-SAM as a cofactor. Considering that allylic-SAM is not an optimal cofactor for the enzyme, we chose relatively long reaction time points as linear intervals: 0, 1, 2.5, 5, 7.5 and 10 min. The reaction products were analyzed by LC–MS/MS. Relative amounts of A, m6A, a6A and ma6A and G were calculated for each measurement according to standard curves. The amounts of the adenosine derivatives are normalized to the amount of G nucleotide. Error bars indicate s.d. for duplicate experiments from two independent assays.
m6A-SAC-seq scheme.
m6A-SAC-seq experiments require parallel construction of three libraries: (1) the input library, where the RNA is subjected to standard library construction without any treatment by MjDim1 or I2 and is used as a reference to call mutations; (2) the experimental library (FTO−), where RNA is treated with the MjDim1 enzyme with cofactor allylic-SAM to convert m6A sites into allylic-m6A by MjDim1 and (3) the background noise group (FTO+), where m6A sites were erased by FTO first, followed by allylic labeling. Both experimental groups and the background noise group were treated with I2 for the cyclization reaction, followed by reverse transcription with HIV RT (Worthington Biochemical).
RNA poly(A) tail elimination, fragmentation and 3′-adapter ligation.
The library construction strategy was modified from the m1A-MAP approach53 with some changes. Specifically, 30 to 100 ng of poly(A)+ RNA or ribo– RNA (300 ng to 1 μg of total RNA) were annealed with oligo(dT), digested with RNase H (NEB) and DNase I (NEB) to remove oligo(dT) and purified by RNA Clean & Concentrator kits (Zymo Research). The purified RNA was fragmented by sonication using Bioruptor (Diagenode; 30 cycles of 30 s on/30 s off to obtain ~150-nt fragments), followed by PNK enzyme (NEB) treatment at 37 °C for 30 min to expose the 3′-hydroxyl group. Calibration spike-in mix (0.6%) was added in the reaction and subjected to 3′-adapter (Supplementary Table 1) ligation with T4 RNA ligase 2, truncated KQ (NEB). The excessive RNA adaptor was digested by adding 1 μl of 5′ Deadenylase (NEB) into the ligation mix followed by incubation at 30 °C for 1 h. Then, 1 μl of RecJf (NEB) was added and incubated at 37 °C for 1 h. One microliter of RT primer (50 μM) was added with the following parameters: anneal 75 °C for 5 min, 37 °C for 15 min and 25 °C for 15 min.
m6A site labeling and reverse transcription.
Fifteen microliters of dynabeads C1 (Thermo Fisher Scientific) was added to the reaction to purify the 3′-adapter-ligated RNA. The beads were washed, resuspended in 6 μl of water and denatured at 70 °C for 30 s and cooled on ice. m6A enzymatic labeling was performed on beads. Two microliters of 10× buffer (400 mM HEPES (pH 8.0), 400 mM NH4Cl, 40 mM MgCl2,), 2 μl of SUPERase In RNase Inhibitor (Thermo Fisher Scientific), 6 μl of allylic-SAM and 4 μl of MjDim1 enzyme (1.6 mM) were added in the reaction and incubated at 50 °C for 1 h. The supernatant was removed, and 4 μl of water, 1 μl of 10× buffer, 1 μl of RNase inhibitor, 2 μl of allylic-SAM and 2 μl of enzyme were added in the reaction and incubated at 50 °C for 20 min (this labeling step was repeated six times). Beads were washed and resuspended in 25 μl of water. One microliter of 125 mM I2 was added and mixed thoroughly and kept in the dark at room temperature for 1 h, then 1 μl of 40 mM Na2S2SO3 was added to quench I2. Beads were washed and resuspended in 9 μl of water. Then, 2 μl of 10× RT buffer (SuperScript III First-Strand Synthesis SuperMix, Thermo Fisher Scientific), 2 μl of 10 mM dNTP, 2 μl of 25 mM MgCl2, 1.25 μl of 0.1 M DTT, 2 μl of RNaseOUT and 2 μl of HIV RT enzyme (Worthington Biochemical) were added in the tube to perform reverse transcription at 37 °C for 3 h. For input RT, 1 h with 1 μl enzyme was sufficient. Beads were washed and resuspended in 8 μl of water.
cDNA 3′-adapter ligation, library construction, purification and sequencing.
One microliter of RNase H buffer and 1 μl of RNase H were added into the resuspended reverse transcription product and placed in a thermocycler (Bio-Rad) at 37 °C for 30 min. Beads were washed and resuspended in 50 μl of water. cDNA was eluted by boiling the beads at 95 °C for 10 min, purified using a DNA Clean & Concentrator kit (Zymo Research) to remove short adapters and eluted into 10 μl of water. Two microliters of 10× T4 RNA ligase buffer, 2 μl of 10 mM ATP, 10 μl of 50% PEG8000, 1 μl of cDNA_3′adapter (50 μM; Supplementary Table 1) and 1 μl of T4 RNA ligase 1 were added into the eluted cDNA, and the ligation was performed at 25 °C overnight. The reaction was purified using a DNA Clean & Concentrator kit (Zymo Research) and eluted with 21 μl of water. One microliter of supernatant was used for quantitative real-time PCR (qPCR) testing, and the remaining 15 μl was used for library construction. NEBNext Ultra II Q5 Master Mix and NEBNext adaptors were used for library amplification. Amplified libraries were purified using 0.8× Ampure beads. The purified libraries were sent for next-generation deep sequencing. The libraries were sequenced on an Illumina HiSeq X Ten with paired-end 2 × 150 bp read length.
FTO demethylation.
The fragmented RNA materials were ligated to the biotin-modified 3′-adapter (Supplementary Table 1) and bound with Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific). RNA on beads was denatured at 70 °C for 30 s and quickly put on ice to quench secondary structure formation. Demethylation was performed on the solid phase in a 50-μl reaction system of 50 mM HEPES buffer (pH 7.0), 75 μM (NH4)2Fe (SO4)2, 2 mM l-ascorbic acid, 0.3 mM α-ketoglutarate, 2 U μl−1 RNase inhibitor and 0.2 nmol of FTO. The demethylated RNA was washed and subjected to m6A-SAC-seq.
m6A-immunoprecipitation (m6A-IP).
HeLa poly(A)+ RNA was purified with a Dynabeads mRNA DIRECT Purification kit (Thermo Fisher Scientific) and sonicated to ~150-nt fragments using Bioruptor (Diagenode) with 30 cycles of 30 s on/30 s off. An EpiMark N6-Methyladenosine Enrichment kit (NEB) was used to enrich m6A-containing RNA fragments. m6A-IP RNA fragments (120 ng) were prepared, and a 0.6% calibration spike-in mix was added. Half of the materials were subjected to m6A-SAC-seq (demethylase− group), and the other half was treated with FTO followed by m6A-SAC-seq (demethylase+ group).
Quantitation of m6A stoichiometry by standard curve construction using model oligonucleotides.
Four pairs of synthetic RNA oligonucleotides containing either m6A or A were used to examine the efficiency of m6A-SAC-seq in causing misincorporation during reverse transcription in a quantitative manner.
MeRIP-SAC-seq flow chart.
To validate our strategy, we enriched the m6A-containing poly(A)+ RNA using an m6A antibody and subjected the m6A-enriched mRNA to m6A-SAC-seq as a positive control. In addition to the enzyme-treated sample, we also perform two additional control reactions in parallel: (1) an input library of untreated RNA that will be used as a reference to exclude single-nucleotide polymorphisms and (2) a demethylase-treated control in which RNA is treated with recombinant FTO to erase m6A sites before enzyme labeling to distinguish true m6A sites from the off-target activity of the MjDim1 enzyme at a subset of adenosine residues.
Procedures of SELECT and MazF for m6A-SAC-seq validation.
The SELCET procedures were described previously19. Total RNA was mixed with 40 nM Up Primer, 40 nM Down Primer and 5 μM dNTP in 17 μl of 1× CutSmart buffer (50 mM potassium acetate, 20 mM Tris-acetic acid, 10 mM magnesium acetate, 100 μg ml−1 BSA, pH 7.9, at 25 °C). The RNA and primers were annealed by incubating the mixture at a temperature gradient: 90 °C for 1 min, 80 °C for 1 min, 70 °C for 1 min, 60 °C for 1 min, 50 °C for 1 min and 40 °C for 6 min. Subsequently, 3 μl of a mixture containing 0.01 U Bst 2.0 DNA polymerase, 0.5 U SplintR ligase and 10 nmol of ATP was added in the former mixture to a final volume of 20 μl. The final reaction mixture was incubated at 40 °C for 20 min, denatured at 80 °C for 20 min and kept at 4 °C. Afterward, qPCR was performed in high-performance real-time PCR with LightCycler Systems (Roche). The 20-μl qPCR reaction was composed of 2× qPCR SYBR Green Master Mix (Roche), 200 nM qPCRF primer, 200 nM qPCRR primer, 2 μl of the final reaction mixture and double-distilled water. qPCR was run using the following conditions: 95 °C for 5 min, 95 °C for 10 s and 60 °C for 35 s for 40 cycles, 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s and hold at 4 °C. PCR products were analyzed by PAGE.
The MazF procedures were reported previously11. Total RNA was incubated with 2.5 U of MazF (mRNA interferase MazF; Takara Bio, 2415A) in the 20-μl reaction mixture of MazF buffer (40 mM sodium phosphate (pH 7.5) and 0.01% Tween 20) at 37 °C for 30 min. Total RNA with or without MazF treatment was subjected to gene-specific quantitative PCR with reverse transcription (RT–qPCR).
Monocytic differentiation of CD34+ HSPCs.
CD34+ HSPCs were isolated from umbilical cord blood samples, and the differentiation of CD34+ cells was conducted as described previously46,54. Briefly, mononuclear cells (MNCs) were enriched from umbilical cord blood by density gradient centrifugation via Ficoll (17-1440-02, GE Healthcare), and red cells were depleted from MNCs by ammonium chloride solution (07850, STEMCELL Technologies). CD34+ cells were enriched from MNCs with a CD34 MicroBead kit (130-046-702, Miltenyi Biotec) following the manufacturer’s instructions. The CD34+ cells were maintained in SFEM medium (09650, STEMCELL Technologies) with 10 U ml−1 penicillin/streptomycin (15-140-122, Thermo Fisher Scientific), 25 μg ml−1 plasmocin prophylactic (ant-MPP, InvivoGen), 100 ng ml−1 stem cell factor (300–07, PeproTech), 10 ng ml−1 interleukin-3 (IL-3; 200–03, PeproTech) and 10 ng ml−1 IL-6 (200–06, PeproTech) for 48 h. Cells were induced into monocytic differentiation with StemSpan Myeloid Expansion Supplement II (02694, STEMCELL Technologies), collected at the indicated points and stained with CD34 (11-0349-42, eBioscience; 5 μl (0.5 μg) per test), CD11b (12-0118-42, eBioscience; 5 μl (0.5 μg) per test) and CD14 (17-0149-42, eBioscience; 5 μl (0.25 μg) per test). The flow cytometry samples were analyzed with a BD LSRFortessa cell analyzer (BD FACSDiva v8.0.1), and the related data were analyzed using FlowJo v10.
Primary and differentiated HSPC RNA purification.
RNA samples were extracted with an miRNeasy Mini kit (217004, Qiagen). Ribosomal RNA was removed with a RiboMinus Eukaryote kit (Thermo Fisher Scientific). Purified ribo− RNA was subjected to m6A-SAC-seq.
m6A-SAC-seq data preprocessing.
Adapters of all raw m6A-SAC-seq data sets from two FTO+/− replicates of HeLa, HEK293, HepG2 cells and four time points of HSPC differentiation into monocytes were clipped away by Cutadapt v1.15 (ref.55) or Trimmomatic v0.39 (ref.56). Adapter-free reads plus 5′ and 3′ barcodes were collapsed to remove PCR duplicates by using fastx_collapser (http://hannonlab.cshl.edu/fastx_toolkit/) or BBMap v38.73 (https://sourceforge.net/projects/bbmap/). After PCR deduplication, the leftmost 5 and rightmost 11 bases of reads were trimmed to discard 5′ and 3′ barcodes. Finally, R1 reads longer than 15 nt were retained for further analysis.
Read mapping.
All reads from preprocessed m6A-SAC-seq data sets were mapped to the human genome (hg38) and the GENCODE v27 gene annotation using STAR v2.5.3a57 with parameters according to the ENCODE long RNA-seq processing pipeline, except that the criteria to allow mismatches depending on read length were slightly less stringent to capture more potential base mutations (--outFilterMismatchNoverReadLmax 0.06).
Mutation calling and identification of m6A sites.
First, to maximize the read coverage, bam files from input and FTO+ data sets were merged into a single file by cell lines and time points, respectively. Merged bam files and those bam files from FTO− data sets were split by strands and piled up using the samtools subcommand mpileup. Second, mutation calling was performed using VarScan v2.3 (ref.58) subcommand somatic in two pair-wise comparisons: (1) FTO− versus input and (2) FTO− versus FTO+. For each comparison (X versus Y), mutation sites were kept if (1) the reference position was adenine (A), (2) the P value was <0.1, (3) the mutation frequency was X – Y > 5% (FTO− versus input) or X – Y > 2% (FTO− versus FTO+), (4) coverage was more than five reads, (5) the 5-mer context was DRACH and (6) only the common sites from two or three biological replicates of either FTO− or FTO+ were kept. Finally, sites in both types of comparisons (FTO− versus input and FTO− versus FTO+) were identified as m6A sites.
Spike-in analysis and calibration curve for m6A stoichiometry estimation.
Spike-in sequences were directly extracted from preprocessed m6A-SAC-seq data sets, and observed mutation rates of the target A were calculated by motifs. To correlate observed mutation rates and m6A fractions in each motif to fit the values measured from the spike-in samples, linear regression models were used: y = ax + b, where y is the observed mutation rate, and x is the m6A fraction. We fit spike-in mutation rates and m6A fractions using the above model in R 3.5.1. Thus, the best-fit calibration curve in each DRACH motif was used for the estimation of m6A stoichiometry in m6A-SAC-seq data sets.
mRNA secondary structure and m6A conservation analysis.
For each A or m6A site, a sliding window of 31 nt was used to calculate RNA MFE by ViennaRNA with default parameters. The upstream and downstream 15 nt of adenines with or without m6A were used to generate the 31-nt window. For m6A conservation analysis, the bigWig file with PhastCons scores (hg38.phastCons100way.bw) was downloaded from the University of California Santa Cruz Genome Browser. The PhastCons score of each m6A site was calculated using deepTools v3.4.3 (ref.59).
MeRIP-SAC-seq data analysis.
Raw data were preprocessed, and subsequent read mapping and mutation calling were performed as described above. For peak calling, the sequence alignment data were divided by strands, and then MACS2 v2.1.1 (ref.60) was used for the detection of m6A peaks with the following parameters: -f BAM -B --SPMR --nomodel --tsize 50 --extsize 150 --keep-dup all.
Ribosome profiling data analysis.
Reads in raw sequencing data from GSE63591 (ref.29) and GSE49339 (ref.27) were first subjected to adapter trimming and then mapped to the human genome (hg38) by HISAT2, as described above. Raw reads on each gene were counted by feature. Counts from Subread v1.6.4 (ref.61) were normalized for sequencing depth and gene length using the transcripts per million (TPM) method. Translation efficiency (TE) of each gene was calculated as a ratio of TPM values between ribosome protected fragments and input RNA samples. To reveal the correlation between TE and m6A fraction of different RNA regions in Hela cells, 5′-UTR, 3′-UTR and CDS regions with TE values were classified into three groups (low, medium and high) based on the sum of the m6A fraction on the region, respectively.
RNA lifetime profiling data analysis.
Adapter trimming, read mapping and TPM calculation in raw sequencing data from GSE98856 (ref.62) and GSE49339 (ref.27) were performed as described above. For RNA lifetime measurements, the External RNA Controls Consortium (ERCC) spike-in TPM values were first transformed to attomoles using a linear regression model, y = ax + b, where y is the attomole (in log2 transformation), and x is the TPM value (in log2 transformation). The variable y is calculated using the following equation: y = d·v·c/t, where d is the dilution factor of spike-in added to each RNA sample, v is the volume (in microliters) of diluted spike-in added to total RNA mass in each sample, c is the concentration (in attomoles per microliter) of each spike-in, and t is the mass (in micrograms) of total RNA in each sample. We fit spike-in TPM values and attomoles using the above model in R 3.5.1. Thus, the best-fit dose–response curve in each sample was used for the estimation of RNA attomoles of each gene. Finally, the half-life of each gene was estimated as describe in ref.63. To reveal the correlation between lifetime and m6A fraction of different RNA regions in Hela cells, 5′-UTR, 3′-UTR and CDS regions with lifetime values were classified into three groups (low, medium and high) based on the sum of the m6A fractions on the region, respectively.
m6A site clustering analysis.
k-means clustering was used to identify genomic regions (5′-UTR, 3′-UTR and CDS) with different m6A fractions in various cell lines by using Cluster 3.0 (v1.59)64 with parameter ‘runs=1000, similarity=centered correlation’, and the results were shown as a heat map by Java TreeView (v1.1.6r4)65. To investigate m6A-specific functions on distinct genomic regions across HSPC differentiation into monocytes, only genes with m6A installed on certain genomic regions were considered for fuzzy c-means clustering analysis by the Mfuzz v2.42.0 package66. Briefly, we used genes with changes of mean m6A fractions at any two time points not less than 20% by comparing to the other two time points (that is, variance of >133.33), and the data were scaled by z score before clustering. Clusters of time-specific m6A were decided in the function ‘mfuzz’ with 10,000 iterations. Euclidean distance was used as the clustering method.
Functional enrichment analysis.
GOBP enrichment analysis in HeLa, HEK293 and HepG2 cells was performed using clusterProfiler v3.15.367, and the top 18 GOBP terms with the lowest P values of <0.01 were clustered. For functional enrichment analysis on dynamic and stable m6A across HSPC differentiation into monocytes, 24 gene sets that represented the top 500 genes ranked by the largest changes of fractions on dynamic m6A or the smallest changes of fractions on stable m6A in each set were uploaded to the g:Profiler web server68. The size of GOBP terms, ranging from 15 to 700, was used, and the top five terms were ranked by the smallest adjusted P values (top terms were considered only if their adjusted P values were <1) in each set and were selected for visualization.
Differentially expressed gene analysis.
The differentially expressed genes were found in input data sets from HSPC samples by pair-wise comparisons between any two time points using the edgeR v3.24.3 package69 with the trimmed mean of M-values (TMM) normalization method for RNA abundance. The CPM for normalized gene expression and FC of gene expression between any two time points were calculated in edgeR and used for downstream analysis.
TF target enrichment analysis.
Genes with changes in m6A stoichiometries (as m6A MD, or | MD | > 10%) in CDSs or 3′-UTRs and expression levels (FC > 1.5 or FC < 0.667 and FDR < 0.05) between adjacent time points were uploaded to the TRRUST v2 web server70. TFs that met with their target hits of ≥3 and corresponding P value of <0.01 in at least one queried gene set were selected for visualization.
Differential AS event (DASE) analysis.
rMATS v4.0.2 (ref.71) was used to identify DASEs from GSE56010 (ref.72), HSPC METTL3/METTL14 knockdown and control RNA-sequencing samples and input data sets of HSPC m6A-SAC-seq samples by using the following pair-wise comparisons: (1) d3 versus d0, (2) d6 versus d3 and (3) d9 versus d6. The AS events whose sum of inclusion and skipping read counts was less than eight in any two samples were filtered out, and DASEs were defined as having an FDR of <0.1 and changes in inclusion levels | ΔΨ | > 0.05. The mean fractions of all m6A sites in SS regions (within 300 nt of intron and 200 nt of exon flanking the SS) of current DASEs were used to assess the links between m6A deposition and RNA splicing.
Co-occurrence of RBP binding and m6A sites.
eCLIP data of 103 and 120 RBPs in HepG2 and K562 cells were obtained from the ENCODE project31,32, respectively. All irreproducible discovery rate peaks or biological replicate peaks were considered if their fold enrichment was >2 and their P value was <0.01. Only data sets with at least 1,000 peaks after filtering were used for further analysis. Overlapping between RBP binding sites in each data set and m6A sites in cell lines and blood samples was determined similar to as described in ref.31. In brief, the percentage of m6A sites in a certain sample that overlapped with eCLIP peaks in a certain data set was calculated and vice versa, that is, calculating the percentage of eCLIP peaks in a certain data set that overlapped with m6A sites in a certain sample. The overall pair-wise percent overlap was represented by the average of those percentages in the analysis of HSPC samples.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
We thank P. Faber of the University of Chicago Genomics Facility for sequencing support and Q. Jin for helping with UHPLC–QQQ–MS/MS. We also thank T. Wu, H.L. Shi and Z.J. Zhang for discussions. L.H. was supported by a Chicago Fellows Program, Chicago Biomedical Consortium (CBC) postdoctoral award and a Leukemia & Lymphoma Society Special Fellow Award. B.T.H. was supported by an NIH fellowship (F32 CA221007). We thank support from National Institutes of Health (NIH) grants RM1 HG008935 (C.H.), R01 GM126553 (M.C.), R01 CA243386 (J.C.), R01 CA214965 (J.C.), R01 CA236399 (J.C.), R01 CA211614 (J.C.) and R01 DK124116 (J.C.), The Simms/Mann Family Foundation (J.C.) and The Margaret Early Medical Research Trust (R.S.). M.C. is supported by a Sloan Foundation Research Fellowship and a Human Cell Atlas Seed Network grant from the Chan Zuckerberg Initiative. C.H. is an investigator of the Howard Hughes Medical Institute. J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Footnotes
Reprints and permissions information is available at www.nature.com/reprints.
Code availability
For m6A-SAC-seq data processing, the code is available in the following GitHub repositories: https://github.com/shunliubio/m6A-SAC-seq and https://github.com/CTLife/m6A-SAC-seq.
Competing interests
A patent application for m6A-SAC-seq has been filed by the University of Chicago. C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc., and Inferna Green, Inc. The remaining authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-022-01243-z.
Data availability
Data have been deposited in the NCBI Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE162357.
References
- 1.Frye M, Harada BT, Behm M & He C RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roundtree IA, Evans ME, Pan T & He C Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominissini D et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Meyer KD et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen K et al. High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl 54, 1587–1590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linder B et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molinie B et al. m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat. Methods 13, 692–698 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McIntyre ABR et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep 10, 6590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer KD DART-seq: an antibody-free method for global m6A detection. Nat. Methods 16, 1275–1280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Campos MA et al. Deciphering the “m6A Code” via antibody-independent quantitative profiling. Cell 178, 731–747 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z et al. Single-base mapping of m6A by an antibody-independent method. Sci. Adv 5, eaax0250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y et al. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell 12, 913–923 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Xiao Y, Dong S, Yu Q & Jia G Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine. Nat. Chem. Biol 16, 896–903 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Shu X et al. A metabolic labeling method detects m6A transcriptome-wide at single base resolution. Nat. Chem. Biol 16, 887–895 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Liu N et al. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aschenbrenner J et al. Engineering of a DNA polymerase for direct m6A sequencing. Angew. Chem. Int. Ed. Engl 57, 417–421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong T et al. Precise antibody-independent m6A identification via 4SedTTP-involved and FTO-assisted strategy at single-nucleotide resolution. J. Am. Chem. Soc 140, 5886–5889 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Liu W et al. Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution. Chem. Sci 9, 3354–3359 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao Y et al. An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N6-methyladenosine modification. Angew. Chem. Int. Ed. Engl 57, 15995–16000 (2018). [DOI] [PubMed] [Google Scholar]
- 20.O’Farrell HC, Musayev FN, Scarsdale JN & Rife JP Binding of adenosine-based ligands to the MjDim1 rRNA methyltransferase: implications for reaction mechanism and drug design. Biochemistry 49, 2697–2704 (2010). [DOI] [PubMed] [Google Scholar]
- 21.O’Farrell HC, Pulicherla N, Desai PM & Rife JP Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA 12, 725–733 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shu X et al. N6-allyladenosine: a new small molecule for RNA labeling identified by mutation assay. J. Am. Chem. Soc 139, 17213–17216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwartz S et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8, 284–296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol 10, 93–95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kortel N et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 49, e92 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X & He C Dynamic RNA modifications in posttranscriptional regulation. Mol. Cell 56, 5–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X et al. N6-Methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao Y et al. m6A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun 10, 5332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X et al. N6-Methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z et al. Genetic analyses support the contribution of mRNA N6-methyladenosine (m6A) modification to human disease heritability. Nat. Genet 52, 939–949 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Nostrand EL et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol 21, 90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Nostrand EL et al. A large-scale binding and functional map of human RNA-binding proteins. Nature 583, 711–719 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patil DP et al. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol 20, 285–295 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N et al. N6-Methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res 45, 6051–6063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou KI et al. Regulation of co-transcriptional pre-mRNA splicing by m6A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alarcon CR et al. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao W et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Kuppers DA et al. N6-Methyladenosine mRNA marking promotes selective translation of regulons required for human erythropoiesis. Nat. Commun 10, 4596 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu YP, Thomas GD & Hedrick CC 2014 Jeffrey M. Hoeg Award Lecture: Transcriptional control of monocyte development. Arterioscler. Thromb. Vasc. Biol 36, 1722–1733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman AD Transcriptional control of granulocyte and monocyte development. Oncogene 26, 6816–6828 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Scott CL & Omilusik KD ZEBs: novel players in immune cell development and function. Trends Immunol 40, 431–446 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Hock H et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev 18, 2336–2341 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yildirim E et al. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152, 727–742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cui H et al. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 4, e124522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su R et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 172, 90–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramirez RN et al. Dynamic gene regulatory networks of human myeloid differentiation. Cell Syst 4, 416–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raghav PK & Gangenahalli G Hematopoietic stem cell molecular targets and factors essential for hematopoiesis. J. Stem Cell Res. Ther 8, 441 (2018). [Google Scholar]
- 49.Santoni G et al. The role of transient receptor potential vanilloid type-2 ion channels in innate and adaptive immune responses. Front. Immunol 4, 34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coppin E et al. Dok1 and Dok2 proteins regulate cell cycle in hematopoietic stem and progenitor cells. J. Immunol 196, 4110–4121 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Wei CM & Moss B Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16, 1672–1676 (1977). [DOI] [PubMed] [Google Scholar]
- 52.Schibler U, Kelley DE & Perry RP Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol 115, 695–714 (1977). [DOI] [PubMed] [Google Scholar]
- 53.Li X et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Su R et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene 34, 3226–3239 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 3 (2011). [Google Scholar]
- 56.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y et al. Model-based analysis of ChIP–seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao Y, Smyth GK & Shi W The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41, e108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edupuganti RR et al. N6-Methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol 24, 870–878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen CY, Ezzeddine N & Shyu AB Messenger RNA half-life measurements in mammalian cells. Methods Enzymol 448, 335–357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Hoon MJ, Imoto S, Nolan J & Miyano S Open source clustering software. Bioinformatics 20, 1453–1454 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Saldanha AJ Java Treeview—extensible visualization of microarray data. Bioinformatics 20, 3246–3248 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Futschik ME & Carlisle B Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol 3, 965–988 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Yu G et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han H et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 46, D380–D386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shen S et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl Acad. Sci. USA 111, E5593–5601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu N et al. N6-Methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature 518, 560–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data have been deposited in the NCBI Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE162357.