

Abstract
Background:
Ca2+ signals in smooth muscle cells (SMCs) contribute to vascular resistance and control blood pressure. Increased vascular resistance in hypertension has been attributed to impaired SMC Ca2+-signaling mechanisms. In this regard, transient receptor potential vanilloid 4 (TRPV4SMC) ion channels are a crucial Ca2+-entry pathway in SMCs. However, their role in blood pressure regulation has not been identified.
Methods:
We used SMC-specific TRPV4-/- (TRPV4SMC−/−) mice to assess the role of TRPV4SMC channels in blood pressure regulation. Further, we determined the contribution of TRPV4SMC channels to the constrictor effect of α1 adrenergic receptor (α1AR) stimulation and elevated intraluminal pressure—two main physiological stimuli that constrict resistance-sized arteries. The contribution of spatially separated TRPV4SMC channel subpopulations to elevated blood pressure in hypertension was evaluated in angiotensin II-infused mice and hypertensive patients.
Results:
We provide first evidence that TRPV4SMC channel activity elevates resting blood pressure in normal mice. α1AR stimulation activated TRPV4SMC channels through protein kinase Cα (PKCα) signaling, which contributed significantly to vasoconstriction and blood pressure elevation. Surprisingly, intraluminal pressure-induced TRPV4SMC channel activity opposed vasoconstriction through activation of Ca2+-sensitive K+ (BK) channels, indicating functionally opposite pools of TRPV4SMC channels. Superresolution imaging of SMCs revealed spatially separated α1AR:TRPV4 and TRPV4:BK nanodomains in SMCs. These data suggest that spatially separated α1AR–TRPV4SMC and intraluminal pressure–TRPV4SMC–BK channel signaling have opposite effects on blood pressure, with α1AR–TRPV4SMC signaling dominating under resting conditions. Further, in hypertensive patients and a mouse model of hypertension, constrictor α1AR–PKCα–TRPV4 signaling was upregulated, whereas dilator pressure–TRPV4–BK channel signaling was disrupted, thereby increasing vasoconstriction and elevating blood pressure.
Conclusions:
Collectively, our data identify novel smooth muscle Ca2+-signaling nanodomains that regulate blood pressure and demonstrate their impairment in hypertension.
Keywords: Calcium signaling, vascular, smooth muscle cell, ion channels, blood pressure, hypertension
Introduction
Intracellular Ca2+ is a crucial contributor to vascular smooth muscle cell (SMC) contraction and vascular resistance. Indeed, abnormal Ca2+ handling in SMCs from resistance-sized arteries has been implicated in blood pressure elevation in hypertension1–3. Ca2+ signals in SMCs occur mainly through ion channels on the cell membrane (voltage-gated Ca2+ channels, transient receptor potential [TRP] channels) or the sarcoplasmic reticular membrane (ryanodine receptors [RyR], IP3 receptors [IP3Rs])4. The specific coupling of a Ca2+ signal to its Ca2+-sensitive target determines its effect on vascular resistance. For example, activation of IP3Rs by voltage-gated Ca2+ channels/TRP channels promotes vasoconstriction and elevates blood pressure, whereas localized coupling of RyR Ca2+ signals with Ca2+-activated K+ (BK) channels lowers vascular resistance and blood pressure4. Therefore, identifying the specific Ca2+ signal–target linkages that control SMC contractility is essential for understanding Ca2+ signaling abnormalities in hypertension and designing novel therapeutic strategies.
The vanilloid 4-type TRP channel (TRPV4) has recently emerged as a crucial Ca2+-influx pathway in SMCs5–7. However, the role of SMC TRPV4 (TRPV4SMC) channels in regulating resting blood pressure has remained elusive, mainly due to a paucity of studies in SMC-specific knockout mice. Functional TRPV4 channels are present in both endothelial and smooth muscle cells in the vascular wall5,8. Studies using endothelium-specific TRPV4−/− mice indicate a blood pressure-lowering effect of endothelial TRPV4 channels9. As for TRPV4SMC channels, current evidence suggests that these channels have varying effects on arterial diameter, depending on the physiological vasoconstrictor stimulus. TRPV4SMC channels oppose pressure-induced (myogenic) vasoconstriction5, but contribute to Gq protein-coupled receptor (GqPCR) agonist-induced vasoconstriction10. These divergent findings raise the interesting possibility that TRPV4SMC channels may reside in separate pools, where they are activated by distinct physiological stimuli and have opposing effects on arterial diameter.
Hypercontraction of resistance arteries is a critical contributor to the pathogenesis of hypertension11. In small arteries, nerve stimulation and intraluminal pressure are the primary endogenous stimuli that contract SMCs and determine vascular resistance12,13. Both of these mechanisms are abnormally active in hypertension11,14.
Sympathetic nerve stimulation constricts arteries when norepinephrine binds to α1 adrenergic receptors (α1ARs) expressed by SMCs to activate GqPCR signaling15. In multiple other cell types, GqPCR signaling is known to open TRPV4 channels6,16. Therefore, we hypothesized that Ca2+ influx through TRPV4SMC channels contributes to α1AR-induced constriction of small arteries. Conversely, TRPV4SMC channels also signal through BK channels to oppose myogenic constriction of cerebral arteries5. Therefore, we postulated that TRPV4SMC channels exist in two functionally distinct pools that have opposing effects on arterial diameter and blood pressure. It has been proposed that TRPV4SMC channel expression is increased in mouse models of hypertension17. Therefore, an in-depth understanding of the physiological stimuli that activate constrictor and dilator TRPV4SMC channels, and their downstream signaling targets and relative contributions to the pathogenesis of hypertension could assist in developing new therapeutic strategies targeting only the pathogenic subpopulation of TRPV4SMC channels.
Using inducible TRPV4SMC−/− mice, we provide direct evidence that TRPV4SMC channels increase resting blood pressure and contribute to blood pressure elevation in hypertension. Moreover, we identify two spatially separated pools of TRPV4SMC channels that have opposite effects on vascular diameter: 1) α1AR-activated TRPV4SMC channels cause vasoconstriction; and 2) intraluminal pressure-activated TRPV4SMC channels signal through BK channels to cause vasodilation. Constrictor α1AR–TRPV4SMC channel signaling is upregulated in hypertension, whereas dilator pressure–TRPV4SMC–BK channel signaling is reduced. The discovery of two functionally opposing TRPV4SMC channel nanodomains advances our mechanistic understanding of blood pressure regulation. Moreover, our findings identify a novel mechanism that increases blood pressure in hypertension and may be relevant in other vascular pathologies.
Methods
The data, analytical methods, and materials are available from the corresponding author on reasonable request. An expanded Materials and Methods section can be found in the Data Supplement.
Animal Models and Human Tissue
All animal protocols were approved by the University of Virginia Animal Care and Use Committee. Normal or hypertensive (angiotensin II-infused) C57BL6/J mice and TRPV4SMC−/− mice were used. Isolation of paraspinal muscle tissue from non-hypertensive and hypertensive individuals was approved by the University of Virginia Institutional Review Board (Protocol #18699), and the subjects gave informed consent.
Pressure Myography
Third-order mesenteric Arteries (MAs, ~ 100 μm) were cannulated onto 2 glass micropipettes and pressurized to 80 mm Hg. Internal diameter was recorded in response to various treatments.
Blood Pressure Measurement
A radiotelemetry catheter was implanted in the left carotid artery under isoflurane anesthesia. Continuous blood pressure measurements were performed after a 7-day recovery period.
Ca2+ Imaging
Ca2+ imaging studies were acquired in pressurized arteries (80 mm Hg) with a spinning-disk confocal imaging system and electron-multiplying charge-coupled device camera.
Patch Clamp in SMCs
Currents through TRPV4 and BK channels were recorded in SMCs freshly isolated from mouse and human arteries.
Superresolution microscopy
A dSTORM superresolution imaging system was used for acquisition of superresolution images of TRPV4 channel, BK channel, and α1AR in SMCs.
Statistics
Data were analyzed with 2-tailed paired or independent t test, 1-way ANOVA (Tukey correction for multiple comparisons), or 2-way ANOVA (Bonferroni correction for multiple comparisons). Statistical significance was determined as a value of P<0.05.
Results
Inducible TRPV4 deletion from SMCs lowers resting blood pressure.
Strong TRPV4 immunostaining was observed in SMC and EC layers in small, third-order MAs from tamoxifen-injected TRPV4fl/fl Cre-control (TRPV4fl/fl) mice (Figure 1A). TRPV4 immunostaining was absent in SMCs from TRPV4SMC−/− (tamoxifen-injected TRPV4fl/fl Cre+) mice. Consistent with this finding, quantitative polymerase chain reaction (qPCR) studies revealed that TRPV4 mRNA levels were reduced by ~80% in EC-denuded MAs from TRPV4SMC−/− mice (Figure 1A; Figure S1). TRPV4 immunofluorescence in the EC layer, mostly localized at endothelial projections to SMCs (black holes in the autofluorescence of the internal elastic lamina), was unaltered in TRPV4SMC−/− mice, confirming SMC-specific knockdown. Elementary Ca2+-influx signals through TRPV4 channels, optically detected in the form of TRPV4 sparklets, have been recorded in the intact endothelium and enzymatically isolated SMCs6,18. However, TRPV4 sparklet activity has not been demonstrated in intact SMCs of pressurized arteries. Accordingly, we performed high-speed Ca2+ imaging in Fluo-4–loaded, pressurized (80 mm Hg) MAs (Figure 1B). Experiments were performed in the presence of 1 μM nifedipine (L-type Ca2+ channel [LTCC] inhibitor) and 20 μM cyclopiazonic acid (CPA; sarco/endoplasmic reticulum ATPase [SERCA] inhibitor) to eliminate interference from Ca2+-influx events through LTCCs and intracellular Ca2+-release signals, respectively. The presence of CPA did not alter TRPV4SMC channel activity (Figure S2). MAs from control mice displayed TRPV4SMC sparklet activity at baseline (Figure 1C, Figure S3A). This activity, expressed as sparklet activity per site and number of sparklet sites per cell, was increased by the TRPV4 channel agonist GSK1016790A (30 nM; hereafter, GSK101) and inhibited by the TRPV4 channel inhibitor GSK2193874 (100 nM; hereafter, GSK219) (Figure 1D, Figure S3C). An all-points histogram of TRPV4SMC sparklet traces showed a quantal level (unitary amplitude) of 0.3 ΔF/F0 (Figure S3B), similar to that for TRPV4 sparklets in ECs18. TRPV4SMC sparklet activity also was abolished on exposure to a Ca2+-free extracellular solution (Figure S4A, S4B), confirming that TRPV4SMC sparklets represent Ca2+-influx signals. Endothelial denudation did not alter TRPV4SMC sparklet activity (Figure 1D, Figure S5A, S5B; Figure S6A, S6B), suggesting that endothelial TRPV4 channels do not alter TRPV4SMC channel activity. Finally, overall TRPV4SMC sparklet activity per cell (activity per site × sites per cell) was diminished by ~ 95% in MAs from TRPV4SMC−/− mice (Figure 1D). The remaining 5% TRPV4SMC sparklet activity in TRPV4SMC−/− mice may indicate an incomplete channel knockdown, which is commonly observed in inducible knockout mice9,19,20. These data represent the first recordings of TRPV4SMC sparklets in pressurized arteries and serve as a functional validation of TRPV4SMC−/− mice.
Fig. 1. Genetic deletion of SMC TRPV4 (TRPV4SMC) channels lowers resting blood pressure.
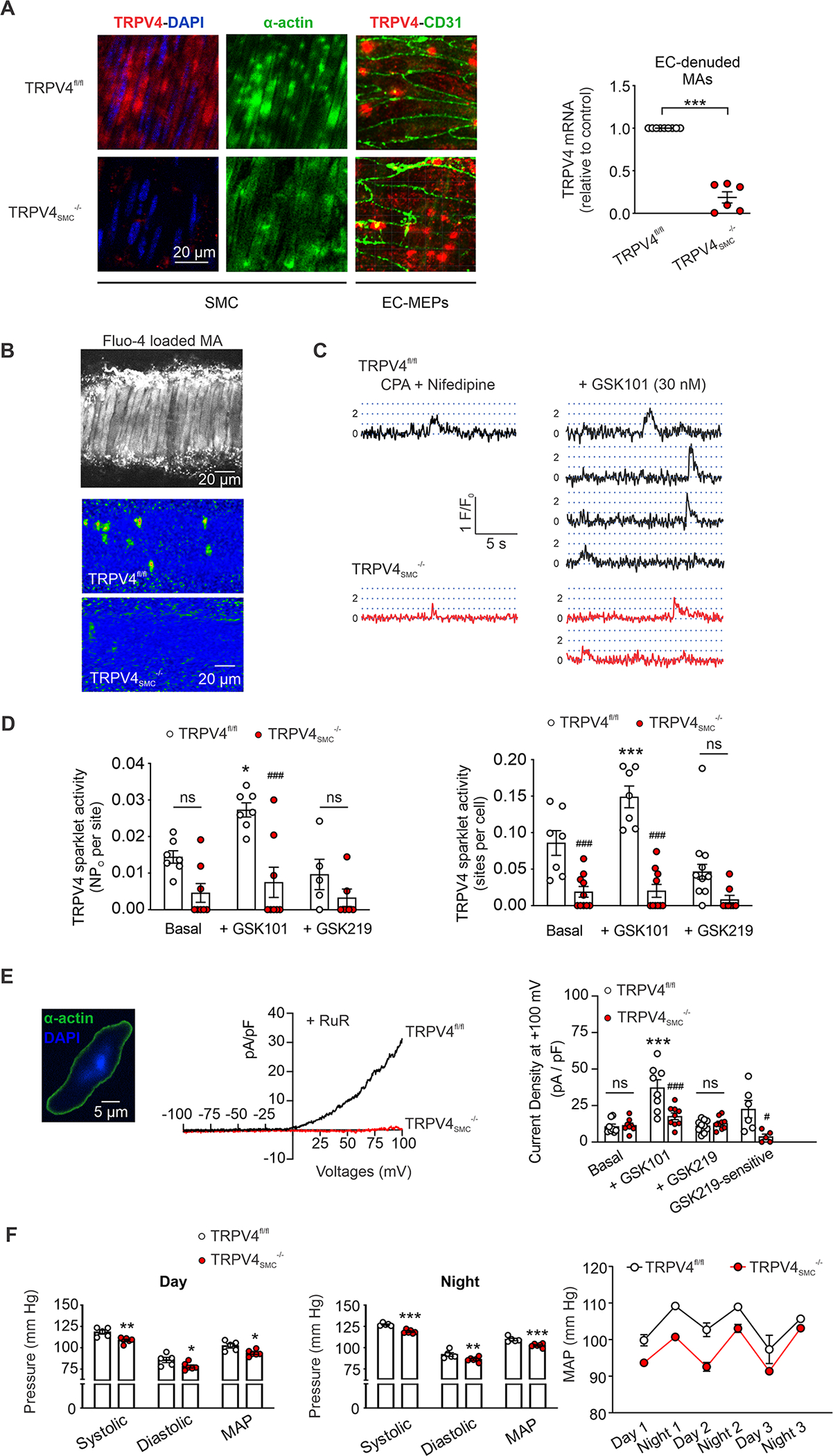
(A) Left, representative images for TRPV4 (red), α-actin (green; SMC marker), and CD31 immunostaining (green; endothelial cell marker) and nuclear staining (DAPI, blue) in en face preparations of MAs from TRPV4fl/fl and TRPV4SMC−/− mice. MEPs, myoendothelial projections. Right, TRPV4 mRNA levels in endothelium-denuded MAs of TRPV4fl/fl (n = 9) and TRPV4SMC-/- (n = 6) mice. Elementary Ca2+-influx events through TRPV4 channels (TRPV4 sparklets) were recorded in MAs loaded with Fluo-4AM (10 μM) in the presence of cyclopiazonic acid (CPA, 20 μM) to eliminate interference from Ca2+ release from the sarcoplasmic reticulum (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (B) Grayscale image of a field of view containing ~26 SMCs from a Fluo-4AM–loaded MA (top). Pseudocolor overlay images showing all GSK1016790A (GSK101, TRPV4 agonist, 30 nM)-elicited TRPV4 sparklet events detected in a field of view in MAs from TRPV4fl/fl (middle) and TRPV4SMC-/- (bottom) mice over a 30-second interval. (C) Fluo-4AM-loaded, pressurized MAs were pretreated with CPA (20 μM), nifedipine (L-type Ca2+ channel blocker, 1 μM), and GSK101 (30 nM). Representative fractional fluorescence (F/F0) traces reflecting sparklet activity in MAs from TRPV4fl/fl and TRPV4SMC−/− mice. Dotted lines represent quantal levels (single-channel amplitudes) determined from all-points histograms. (D) Left, TRPV4 sparklet activity per site (NPo, where N is the number of channels and PO is the open state probability) and (right) TRPV4 sparklet sites per cell under basal conditions (CPA + nifedipine), with GSK101 (30 nM), or with GSK101+GSK219 (100 nM) in MAs from TRPV4fl/fl (n = 7) and TRPV4SMC−/− (n = 8) mice (*P < 0.05, ***P < 0.001 vs. CPA alone; ###P < 0.001 vs. TRPV4fl/fl; ns, not significant; two-way ANOVA). (E) Left, widefield image of an isolated SMC from a mouse MA stained for α-actin (green); blue indicates nuclear staining with DAPI. The fusiform shape (positive α-actin staining) is characteristic of isolated SMCs selected for patch-clamp experiments. Middle, representative whole-cell patch-clamp traces of GSK219-sensitive TRPV4 currents (GSK101 [30 nM] minus GSK101+GSK2193874 (GSK219; TRPV4 inhibitor; 100 nM) in freshly isolated SMCs from MAs of TRPV4fl/fl and TRPV4SMC−/− mice. Experiments were performed in the presence of ruthenium red (RuR; TRPV4 inhibitor, 1 μM) to prevent Ca2+ influx through TRPV4 channels and subsequent activation of BK currents. Right, averaged outward currents in freshly isolated SMCs from MAs of TRPV4fl/fl (n = 8) and TRPV4SMC−/− (n = 8) mice at +100 mV under basal conditions in the presence of GSK101 (30 nM) or GSK101+GSK219 (100 nM), and GSK219-sensitive currents (GSK101 minus GSK101+GSK219) (***P < 0.001 vs. Basal; ###P < 0.001 vs. TRPV4fl/fl; ns, not significant; two-way ANOVA). (F) Quantification of systolic and diastolic blood pressure, and mean arterial pressure (MAP; mean ± SEM) during (left) daytime and (middle) nighttime in TRPV4SMC−/− (n = 6) and TRPV4fl/fl (n = 5) mice. (right) Resting MAP (mean ± SEM) in TRPV4fl/fl (n = 5) and TRPV4SMC−/− (n = 6) mice recorded over 3 days and 3 nights (*P < 0.05; **P < 0.01; ***P < 0.001 vs. TRPV4fl/fl; two-way ANOVA).
GSK219 (TRPV4 inhibitor)-sensitive outward currents at +100 mV were compared in freshly isolated SMCs from MAs of control and TRPV4SMC−/− mice (Figure 1E). The experiments were performed in the presence of ruthenium red (1 μM, TRPV4 inhibitor)18, which prevents Ca2+ entry through TRPV4 channels at negative potentials and subsequent activation of Ca2+-sensitive K+ (BK) channels (Figure S7). TRPV4 activation with GSK101 (30 nM) increased outward currents in SMCs from control mice, which were inhibited by GSK219 (Figure 1E). GSK101 failed to increase outward currents in SMCs from TRPV4SMC−/− mice (Figure 1E). These data further confirm that TRPV4 channel activity is absent in SMCs from TRPV4SMC−/− mice.
L-type (LTCCs) and T-type (TTCCs) Ca2+ channels are the main voltage-gated Ca2+ channels involved in SMC contraction. Nifedipine (LTCC inhibitor, 1 μm; Figure S8A)- or mibefradil (TTCC inhibitor, 1 μm; Figure S8B)-induced decrease in SMC Ca2+ was not different between control and TRPV4SMC−/− mice, supporting the idea that LTCCs and TTCCs activity is not altered in SMCs from TRPV4SMC−/− mice. However, the activity of IP3R Ca2+ signals (Figure S9A) and RyR Ca2+ signals (Figure S9B) was lower in SMCs from TRPV4SMC−/− mice than control mice, pointing to TRPV4–IP3R and TRPV4–RyR signaling in SMCs.
Resting systolic pressure, diastolic pressure, and mean arterial pressure (MAP) recorded by radiotelemetry, were lower in TRPV4SMC−/− mice compared to TRPV4fl/fl control mice (Figure 1F). The decrease in resting arterial pressure in TRPV4SMC−/− mice was observed in both daytime and nighttime recordings (Figure 1F). However, resting heart rate and cardiac function as measured with functional cardiac MRI (fMRI, Table 1), were not different between TRPV4fl/fl and TRPV4SMC−/− mice. These new findings provide the first evidence that TRPV4SMC channels elevate resting blood pressure without altering cardiac function.
Table 1. Cardiac functional MRI data for TRPV4fl/fl and TRPV4SMC−/− mice.
ESWT, end systolic wall thickness; EDWT, end diastolic wall thickness; ESV, end systolic volume; EDV, end diastolic volume; EF, ejection fraction; SV, stroke Volume; RR, R-R interval; CO, cardiac output; HR, heart rate.
| TRPV4fl/fl (n = 5) |
TRPV4SMC−/− (n = 5) |
|
|---|---|---|
| Mass (mg) | 80 ± 3 | 75 ± 4 |
| ESWT (mm) | 1.1 ± 0.04 | 1.1 ± 0.04 |
| EDWT (mm) | 0.9 ± 0.04 | 0.8 ± 0.04 |
| ESV (μL) | 22 ± 4 | 19 ± 3 |
| EDV (μL) | 51 ± 6 | 50 ± 5 |
| EF (%) | 59 ± 3 | 62 ± 3 |
| SV (μL) | 30 ± 2 | 31 ± 2 |
| RR (ms) | 124 ± 6 | 129 ± 10 |
| CO (mL/min) | 15 ± 1 | 14 ± 1 |
| HR (beat/min) | 532 ± 17 | 569 ± 11 |
α1AR signaling causes vasoconstriction and elevates blood pressure by activating TRPV4SMC channels.
Synaptic release of norepinephrine from nerve terminals and subsequent activation of SMC α1ARs is a crucial mechanism for blood pressure regulation21. Consistent with this idea, the α1AR antagonist prazosin (1 mg/kg, intraperitoneal [i.p.]) reduced resting blood pressure in control mice (Figure 2A, Figure S10), confirming a blood pressure-elevating effect of α1ARs under resting conditions. Prazosin-induced decreases in blood pressure were diminished in TRPV4SMC−/− mice (Figure 2A), supporting a critical role for TRPV4SMC channels in α1AR-induced increases in resting blood pressure. Exposure to prazosin (1 μM) also resulted in ~10% dilation of pressurized MAs from control mice but did not affect the diameter of MAs from TRPV4SMC−/− mice, confirming the constrictor effect of α1ARs under basal conditions and the crucial contribution of TRPV4SMC channels to this effect (Figure 2B, 2C). An acute injection of the α1AR agonist phenylephrine (PE; 10 mg/kg, i.p.) elevated systolic, diastolic, and mean blood pressure in both control and TRPV4SMC−/− mice. However, the PE-induced increase in blood pressure was significantly lower in TRPV4SMC−/− mice compared with TRPV4fl/fl control mice (Figure 2D). Heart rate following PE administration, however, was not different between TRPV4fl/fl and TRPV4SMC−/− mice (Figure S11). In pressure myography experiments, PE (10−8 M to 10−5 M) induced a concentration-dependent constriction of MAs that was completely inhibited by prazosin (Figure 2E). PE-induced constriction was significantly less in MAs from TRPV4SMC−/− mice (Figure 2E) and in the presence of TRPV4 channel inhibitor GSK219, further confirming a central role for TRPV4SMC channels in α1AR-induced vasoconstriction (Figure 2E).
Fig. 2. α1AR signaling causes vasoconstriction and elevates blood pressure through TRPV4SMC channel activation.
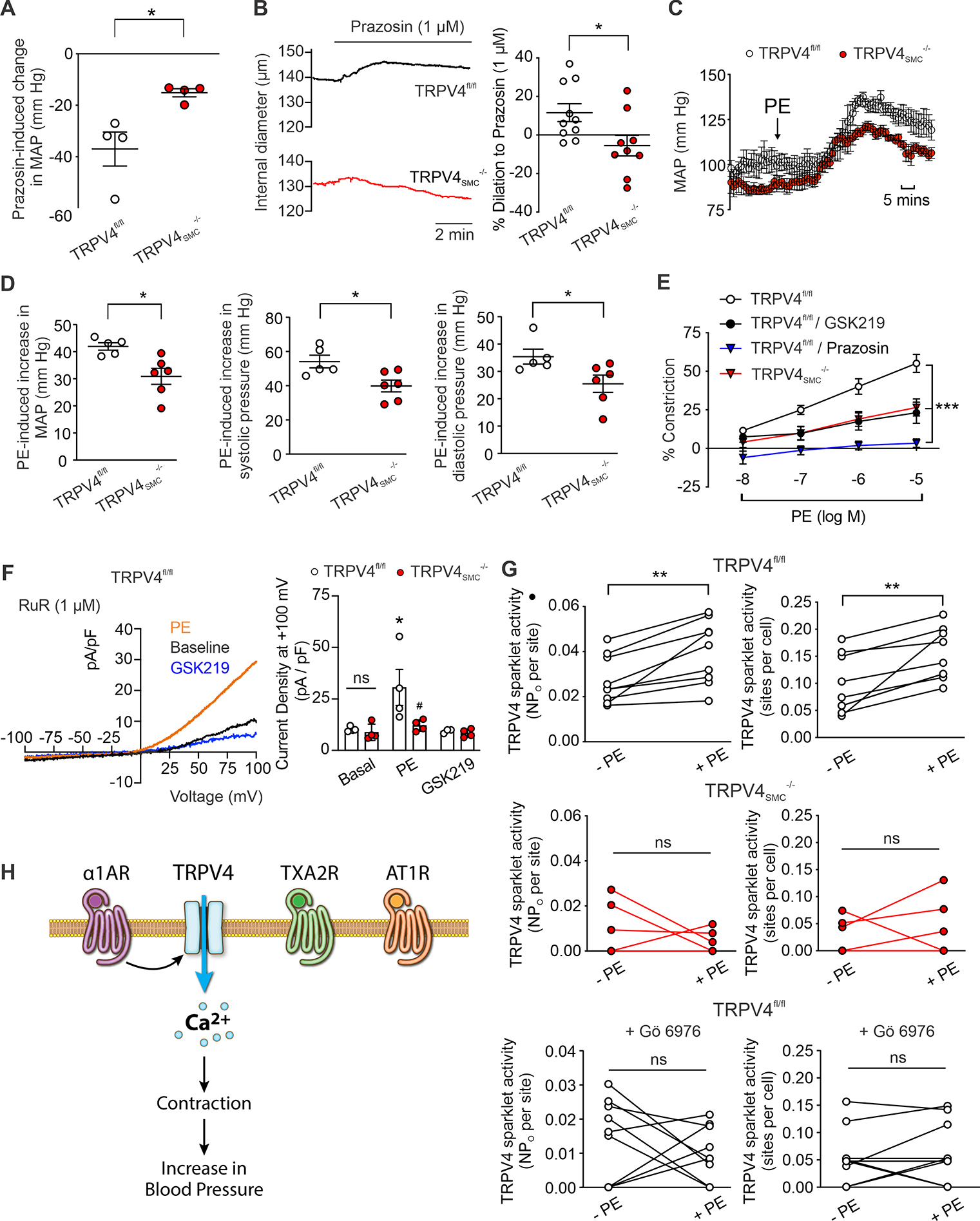
(A) Decrease in resting MAP after a single bolus injection of prazosin (α1 adrenergic receptor [α1AR] blocker; 1 mg/kg; i.p.). (TRPV4fl/fl, n = 4; TRPV4SMC−/−, n = 4; *P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (B) Left, representative diameter traces for prazosin (1 μM)-induced dilation in resistance MAs of TRPV4fl/fl and TRPV4SMC−/− mice. Right, percent dilation of MAs from TRPV4fl/fl (n = 10) and TRPV4SMC−/− (n = 9) mice in response to prazosin (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (C) Mean arterial pressure (MAP) in TRPV4fl/fl (n = 5) and TRPV4SMC−/− (n = 6) mice, averaged before and after the administration of phenylephrine (PE; 10 mg/kg; i.p.). (D) Left, increase in MAP after a single bolus injection of PE. (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test.) Middle, increase in systolic pressure after a single bolus injection of PE (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). Right, increase in diastolic pressure after a single bolus injection of PE (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (E) PE-induced constriction of resistance MAs in the absence or presence of the TRPV4 inhibitor GSK219 (100 nM) or α1AR antagonist prazosin (1 μM). (TRPV4fl/fl, n = 8; TRPV4SMC−/−, n = 7; TRPV4fl/fl+GSK219, n = 5; TRPV4fl/fl+prazosin, n = 4; ***P < 0.001 vs. TRPV4fl/fl; two-way ANOVA). (F) Left, representative traces of ionic currents through TRPV4 channels in isolated SMCs from MAs of control mice at +100 mV under baseline conditions and in the presence of PE (1 μM) or GSK219 (100 nM), recorded in the whole-cell patch-clamp configuration. Experiments were performed in the presence of RuR (1 μM) to block Ca2+ entry at negative voltages. Right, averaged outward currents of isolated SMCs from MAs of TRPV4fl/fl mice (n = 4) and TRPV4SMC−/− mice (n = 4) before and after administration of PE (1 μM) or GSK219 (100 nM) at +100 mV (*P < 0.05 vs. Basal; ns, not significant; two-way ANOVA). (G) TRPV4SMC sparklets in MAs from TRPV4fl/fl and TRPV4SMC−/− mice, expressed as activity per site (top left, middle left, bottom left) and sites per cell (top right, middle right, bottom right). Fluo-4AM-loaded, pressurized MAs from TRPV4fl/fl (n = 9) and TRPV4SMC−/− (n = 6) mice were pretreated with CPA (20 μM), nifedipine (1 μM), and GSK101 (30 nM). Experiments were performed before and after adding PE, with and without Gö 6976 (PKCα/β inhibitor, 1 μM) pretreatment (**P < 0.01 vs. –PE; ns, not significant; paired t-test). (H) Schematic diagram showing that α1AR, but not thromboxane A2 (TXA2R) or angiotensin II type 1 receptors (AT1R), activates TRPV4SMC channels, causing subsequent contraction of SMCs and increasing blood pressure.
Addition of the α1AR agonist PE (1 μM) increased the GSK219-sensitive currents through TRPV4 channels in SMCs from normal C57BL6 mice, but not TRPV4SMC−/− mice (Figure 2F). PE also increased the activity of TRPV4SMC sparklets in pressurized MAs (Figure 2G). PE-induced increase in TRPV4SMC sparklet activity was abolished by the TRPV4 channel inhibitor GSK219 and was absent in TRPV4−/− mice (Figure 2G; Figure S12), further supporting the activation of TRPV4SMC channels by α1AR signaling. To test the hypothesis that α1ARs activate TRPV4SMC channels via phospholipase C (PLC) signaling, we tested the effect of PE in the presence of the PLC inhibitor U37122 (3 μM). U73122 alone did not affect TRPV4SMC sparklet activity (Figure S13A, S13B), but completely abolished the effect of PE on TRPV4SMC sparklets (Figure S14A, S14B). Protein kinase Cα (PKCα), but not PKCβ, has been shown to activate TRPV4 channels in ECs19,20. A PKCα/β inhibitor, Gö 6976 (1 μM), abolished PE-induced activation of TRPV4SMC sparklets (Figure 2G), confirming PLC–PKCα–dependent activation of TRPV4SMC channels by α1ARs.
To test whether TRPV4SMC channel-dependent vasoconstriction is specific to Gq/11α-coupled 1ARs in SMCs or is a general property of GqPCR activation, we assessed the effect of SMC TRPV4 deletion on vasoconstriction to the SMC GqPCR activators, U46619 (thromboxane A2 receptor agonist) and angiotensin II (Ang II; Ang II receptor 1 agonist). The constriction of MAs to U46619 (1–300 nM) (Figure S15A) or Ang II (Figure S16A) was not altered by the TRPV4 inhibitor GSK219 or in MAs from TRPV4SMC−/− mice. Moreover, both the thromboxane receptor agonist U46619 (100 nM) (Figure S15B, S15C) and Ang II (Figure S16B, S16C) were unable to activate TRPV4SMC sparklets, providing further evidence for a selective α1AR–TRPV4 signaling linkage that mediates SMC contraction (Fig. 2H).
Intraluminal pressure-induced activation of TRPV4SMC–BK channel signaling opposes MA constriction.
Intraluminal pressure is another critical endogenous mechanism that increases SMC contractility and elevates blood pressure. However, the effect of intraluminal pressure on TRPV4SMC channel activity has not been investigated. Increasing intraluminal pressure from 20 mm Hg to 80 mm Hg increased sparklet activity in pressurized MAs, whereas decreasing pressure from 80 mm Hg to 20 mm Hg decreased sparklet activity (Figure 3A), supporting the efficacy of intraluminal pressure as a stimulus for sparklet activity. No pressure-induced increase in TRPV4SMC sparklet activity was observed in the presence of the TRPV4 inhibitor GSK219 (Figure 3A). Surprisingly, pressure-induced (myogenic) constriction was higher in MAs from TRPV4SMC−/− mice than in MAs from control mice (Figure 3B), indicating that intraluminal pressure-induced TRPV4SMC channel activity opposes myogenic constriction. Thus, α1AR–TRPV4 signaling and pressure–TRPV4SMC signaling exert opposing effects on arterial contraction.
Fig. 3. Intraluminal pressure-activated TRPV4SMC–BK channel signaling opposes myogenic vasoconstriction and lowers blood pressure.
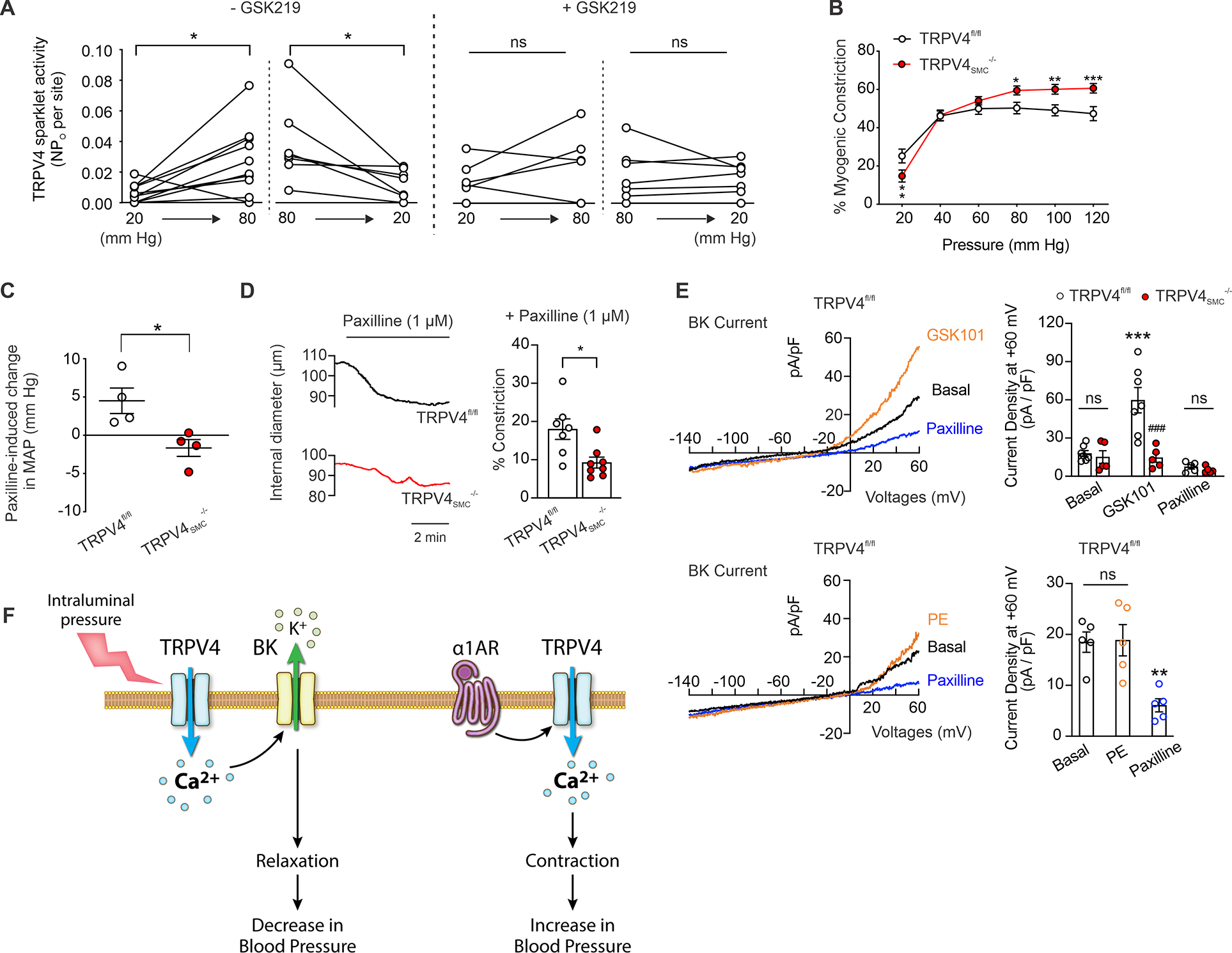
(A) TRPV4SMC sparklets (activity per site) in MAs from control mice were performed. Pressurized MAs were pretreated with CPA (20 μM), nifedipine (1 μM) and GSK101 (30 nM). Intraluminal pressure was increased from 20 mm Hg to 80 mm Hg and back to 20 mm Hg in the absence or presence of GSK219 (100 nM) in MAs from TRPV4fl/fl (n = 7) mice (*P < 0.05; ns, not significant; paired t-test). (B) Averaged data showing the development of myogenic constriction in MAs from TRPV4fl/fl (n = 8) and TRPV4SMC−/− (n = 8) mice in response to different intraluminal pressures (*P < 0.05 vs. TRPV4fl/fl, **P < 0.01 vs. TRPV4fl/fl, ***P < 0.001 vs. TRPV4fl/fl; two-way ANOVA). (C) Increase in resting MAP in TRPV4fl/fl and TRPV4SMC−/− mice (n = 4 each) after a single bolus injection of paxilline (8 mg/kg, i.p.; *P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (D) Left, representative pressure myography traces of paxilline (1 μM)-induced constriction of MAs from TRPV4fl/fl and TRPV4SMC−/− mice. Right, averaged data showing the constriction to paxilline (1 μM) in MAs from TRPV4fl/fl (n = 7) and TRPV4SMC−/− (n = 8) mice (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (E) Top left, representative traces of ionic currents through BK channels in isolated SMCs from MAs of TRPV4fl/fl mice under baseline conditions and in the presence of GSK101 (30 nM), recorded in the whole-cell patch-clamp configuration. Top right, averaged outward currents in isolated SMCs from MAs of TRPV4fl/fl mice (n = 7) and TRPV4SMC−/− mice (n = 5) at +60 mV under basal conditions and in the presence of GSK101 (30 nM) before and after addition of paxilline (BK channel inhibitor; 8 mg/kg; i.p.; *P < 0.05 vs. TRPV4fl/fl; unpaired t-test). Bottom left, representative traces of ionic currents through BK channels in isolated SMCs under baseline conditions and in the presence of PE (1 μM), recorded in the whole-cell patch-clamp configuration. Bottom right, averaged outward currents in isolated SMCs from MAs of TRPV4fl/fl mice (n = 5) at +60 mV under basal conditions and in the presence of PE (1 μM) before and after the addition of paxilline (1 μM; **P < 0.01 vs. Basal; ns, not significant; one-way ANOVA). (F) Schematic diagram showing discrete TRPV4–BK and α1AR–TRPV4SMC signaling in SMCs.
A previous study attributed the vasodilatory effect of TRPV4SMC channels to downstream activation of BK channels5, although the effect of TRPV4SMC–BK signaling on blood pressure was not reported. In this context, we found that the BK channel inhibitor paxilline (8 mg/kg, i.p.) induced an increase in resting blood pressure 15 minutes after paxilline administration, an effect that was significantly lower in TRPV4SMC−/− mice compared with TRPV4fl/fl control mice (Figure 3C, Figure S17), confirming a blood pressure-lowering effect of TRPV4SMC–BK signaling under resting conditions. Paxilline (1 μM) also caused constriction of MAs ex vivo under basal conditions, and this constriction was lower in MAs from TRPV4SMC-/- mice (Figure 3D), supporting the idea that TRPV4SMC–BK channel signaling attenuates SMC contraction and lowers resting blood pressure.
In patch-clamp studies, the TRPV4 channel activator GSK101 induced an increase in outward currents through BK channels that was inhibited by the BK channel inhibitor paxilline (1 μM) and was absent in SMCs from TRPV4SMC−/− mice (Figure 3E). While the α1AR agonist PE increased TRPV4SMC currents (Figure 2F), it did not increase BK channel currents (Figure 3E), thus ruling out α1AR–TRPV4–BK channel or α1AR–BK channel signaling. Collectively, these results support the idea of two distinct pools of TRPV4SMC channels with opposing functional effects: an α1AR-activated constrictor TRPV4SMC channel pool and an intraluminal pressure-activated dilator TRPV4SMC channel pool (Figure 3F).
Co-localization of α1AR or BK channels with TRPV4 channels in SMCs.
Our results raise the interesting possibility that distinct TRPV4SMC channel-dependent signaling microdomains have opposing functional effects. We first tested the possibility that α1ARs and BK channels exist in nanometer proximity with TRPV4SMC channels using an in situ proximity ligation assay (PLA), which identifies two proteins within ~40 nm of one another. PLA puncta were observed for both α1AR:TRPV4SMC and TRPV4SMC:BK pairs (Figure 4A), but not for α1AR:BK pair, providing evidence for distinct pools of TRPV4SMC channels co-localizing with α1ARs and BK channels. PLA assays also detected PKCα:TRPV4SMC co-localization, which was increased following α1AR stimulation (Figure S18A). Notably, PLA puncta were not detected for the PKCα:BK pair (Figure S19), pointing to α1AR–PKCα–TRPV4SMC signaling at constrictor nanodomains. α1AR stimulation did not affect α1AR:TRPV4SMC co-localization (Figure S18A), and increase in intraluminal pressure did not alter TRPV4SMC:BK co-localization (Figure S18B).
Fig. 4. Discrete α1AR:TRPV4SMC and TRPV4SMC:BK channel signaling nanodomains in SMCs.
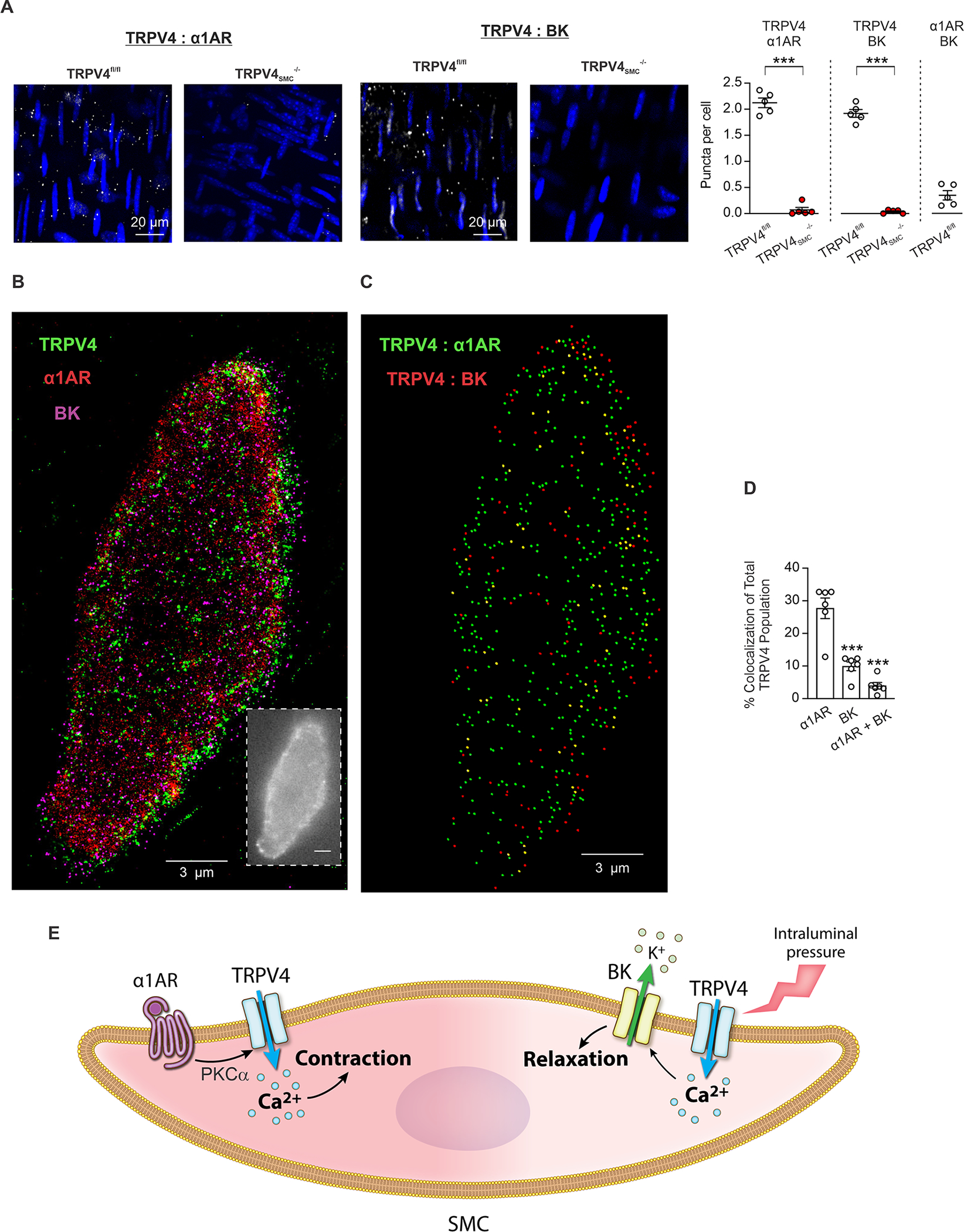
(A) Left, representative in situ proximity ligation assay (PLA) images showing SMC nuclei (blue) and α1AR:TRPV4SMC co-localization (white puncta) in en face preparations of MAs from TRPV4fl/fl (left) and TRPV4SMC−/− (right) mice. Middle, representative PLA images of SMC nuclei (blue) and TRPV4SMC:BK co-localization (white puncta) in en face preparations of MAs from TRPV4fl/fl (left) and TRPV4SMC−/− (right) mice. Right, quantification of α1AR:TRPV4SMC (left) and TRPV4SMC:BK co-localization (right) in MAs from TRPV4fl/fl and TRPV4SMC−/− mice (***P < 0.001 vs. TRPV4fl/fl; unpaired t-test). (B) Superresolution localization maps for TRPV4, α1AR and BK channels in native SMCs from control mice. The inset shows a widefield snapshot of one SMC. Scale bars, 3 μm. (C) Co-localization analysis (Imaris) results for localization of TRPV4 channels with α1ARs (green dots), BK channels (red dots), or both (yellow dots). (D) Summary of co-localization analysis showing the percentage of total TRPV4 channels coupling with α1ARs, BK channels, or both in SMCs from control mice (n = 6; ***P < 0.001 vs. α1AR-paired; one-way ANOVA). (E) Schematic diagram showing the divergent roles of TRPV4SMC channels in regulating arterial diameter. Pressure-induced TPRV4SMC:BK channel signaling causes vasodilation. In contrast, α1AR:TPRV4SMC channel signaling elicits vasoconstriction.
Next, we used dSTORM superresolution imaging (15 nm resolution) to confirm the presence of exclusive coupling of TRPV4SMC channels with α1ARs and BK channels. SMCs from MAs were co-immunolabeled with primary antibodies targeting the TRPV4 channel, α1AR and BK channel to generate superresolution localization maps (Figure 4B, Figure S20). A co-localization analysis was further performed for TRPV4:α1AR and TRPV4:BK pairs, and the overlap between the two pairs (Figure 4C, 4D). This analysis indicated that ~30% of total TRPV4SMC channels co-localize with α1ARs and ~10% of TRPV4SMC channels co-localize with BK channels. Importantly, only ~3% of TRPV4SMC channels co-localize with both α1ARs and BK channels, supporting the concept of spatially distinct constrictor (α1AR:TRPV4SMC) and dilator (TRPV4SMC:BK) TRPV4SMC channels (Figure 4E).
Elevated α1AR–TRPV4SMC signaling and impaired TRPV4SMC–BK channel signaling contributes to elevated blood pressure in hypertension.
Our data indicated that TRPV4SMC channel activity elevates resting blood pressure. Therefore, we tested the possibility that TRPV4SMC channels contribute to the increase in blood pressure in hypertension. Blood pressure was recorded in Ang II-infused mice after 7 and 14 days of infusion (1 μg/kg/min). MAP after 7 and 14 days of Ang II infusion was lower in TRPV4SMC−/− mice compared with TRPV4fl/fl control mice (Figure 5A). Moreover, the increase in blood pressure following Ang II infusion was less in TRPV4SMC-/- mice than in control mice (Figure 5A), suggesting that a lower blood pressure in Ang II-infused TRPV4SMC−/− mice was not due to lower starting blood pressure on day 0. Ang II infusion did not result in a significant change in heart rate in either group (Figure 5A).
Fig. 5. Upregulated α1AR–TRPV4SMC signaling and impaired TRPV4SMC–BK channel signaling elevates blood pressure in hypertension.
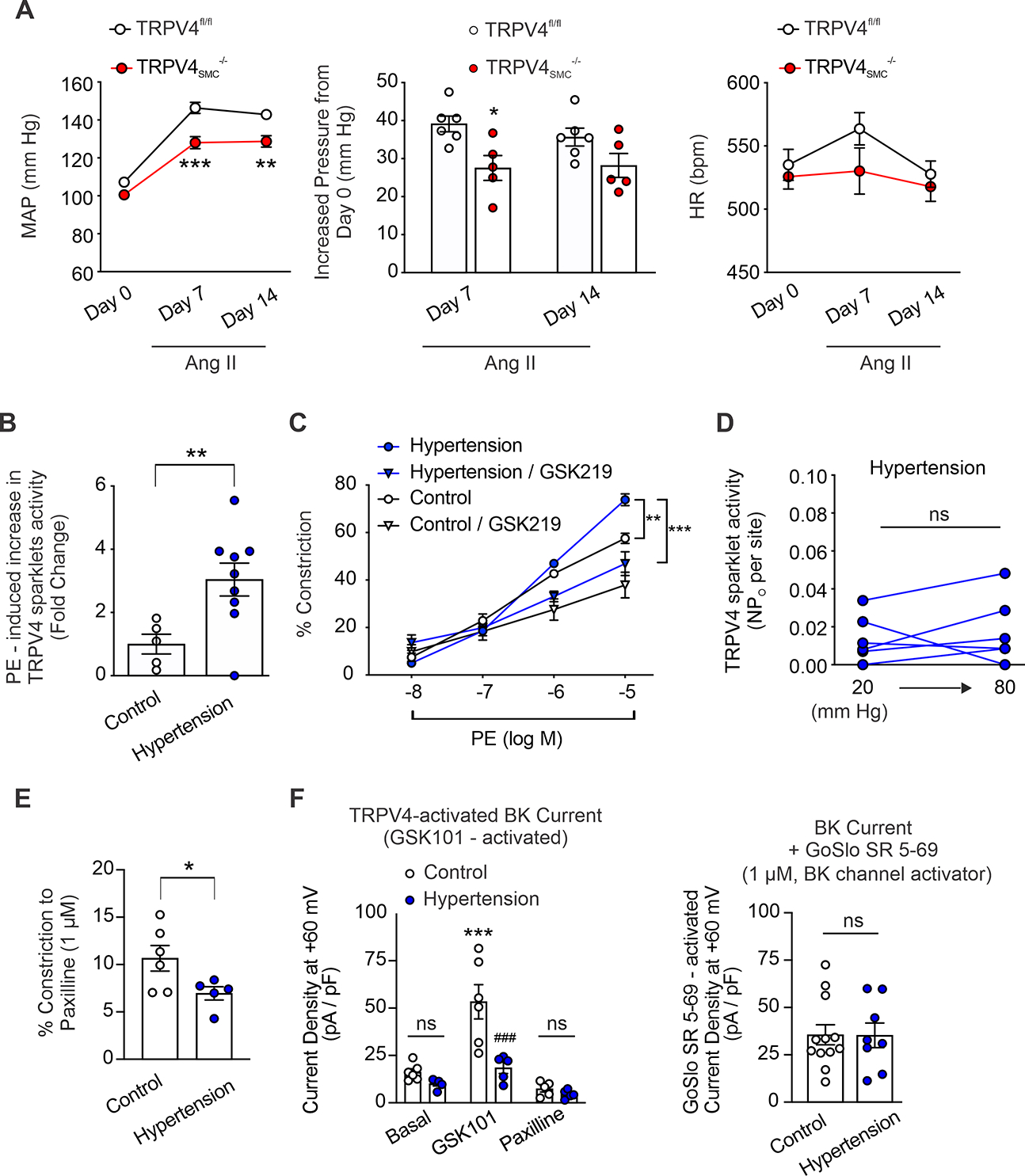
Mice were implanted with osmotic minipumps containing angiotensin II (Ang II; Ang II receptor 1 agonist; 1 μg/kg/min). (A) Left, mean arterial pressure (MAP) in TRPV4fl/fl (n = 6) and TRPV4SMC−/− (n = 5) mice, recorded before the start of Ang II infusion (day 0) and 7 and 14 days following Ang II infusion via osmotic minipumps. Middle, increased MAP in TRPV4fl/fl (n = 6) and TRPV4SMC−/− (n = 5) mice after 7 and 14 days of Ang II infusion (*P < 0.05 vs. TRPV4fl/fl, **P < 0.01 vs. TRPV4fl/fl, ***P < 0.001 vs. TRPV4fl/fl; two-way ANOVA). Right, heart rate (HR) in TRPV4fl/fl (n = 6) and TRPV4SMC−/− (n = 5) mice, analyzed before Ang II infusion (day 0) and after 7 and 14 days of infusion. (B) PE-induced TRPV4SMC sparklet activity (fold change) in Ang II-induced hypertensive mice (n = 9) and control mice (n = 5). Pressurized MAs from control (infused with 0.9% saline; n = 5) and Ang II-induced hypertensive (n = 6) mice were pretreated with CPA (20 μM), nifedipine (1 μM), and GSK101 (30 nM) (**P < 0.01 vs. Control; unpaired t-test). (C) PE-induced constriction of MAs from Ang II-induced hypertensive mice (n = 5) and control mice (n = 9), studied in the absence or presence of the TRPV4 inhibitor GSK219 (100 nM) (**P < 0.01 vs. Control; two-way ANOVA). (D) TRPV4 sparklet activity induced by increasing intraluminal pressure from 20 mm Hg to 80 mm Hg in MAs from Ang II-induced hypertensive mice (n = 6; ns, not significant; paired t-test). (E) Paxilline (1 μM)-induced vasoconstriction in control (n = 6) and Ang II-induced hypertensive mice (n = 5) (*P < 0.05 vs. TRPV4fl/fl; unpaired t-test). (F) Left, averaged outward currents in SMCs isolated from MAs of Control mice (n = 6) and Ang II-induced hypertensive mice (n = 5) at +60 mV under basal conditions and in the presence of GSK101 (30 nM) and GSK101 + paxilline (1 μM) (***P < 0.001 vs. Basal, ###P < 0.001 vs. Control; ns, not significant; two-way ANOVA). Right, averaged GoSlo SR 5–69 (BK channel activator; 1 μM)-activated outward currents (GoSlo SR 5–69 minus baseline current) in SMCs isolated from MAs of Control (n = 12) and Ang II-induced hypertensive mice (n = 8) at +60 mV (ns, not significant; unpaired t-test).
We postulated that an imbalance between constrictor (α1AR-associated) and dilator (BK channel-associated) pools of TRPV4SMC channels contributes to elevated blood pressure in hypertension. qPCR studies showed that mRNA levels for TRPV4 channels in SMCs are not different between saline-infused control and Ang II-infused hypertensive C57BL6 mice, providing evidence that expression of TRPV4 channels is not altered with hypertension (Figure S21). However, α1AR-induced increase in TRPV4SMC sparklet activity was higher in resistance arteries of C57BL6 mice infused with Ang II than in control mice (Figure 5B), suggesting an increase in constrictor α1AR–TRPV4SMC signaling in hypertension. Consistent with these data, PE-induced constriction was increased in MAs from hypertensive C57BL6 mice compared with saline-infused control mice, and the increase in PE-induced constriction was eliminated by the TRPV4 inhibitor GSK219 (Figure 5C). Moreover, acute treatment with prazosin (1 mg/kg, i.p.) lowered blood pressure in hypertensive mice, and the blood pressure-lowering effect of prazosin was reduced in hypertensive mice pre-treated with TRPV4 inhibitor GSK219 (1 mg/kg, i.p.) (Figure S22). Together, these results supported the idea that α1AR–TRPV4SMC signaling contributes to blood pressure elevation in hypertension.
Notably, the increase in intraluminal pressure was unable to activate TRPV4SMC sparklets in hypertensive mice (Figure 5D), suggesting the loss of pressure-induced activation of dilatory TRPV4SMC channels in hypertension. Moreover, constriction to the BK channel inhibitor, paxilline, was reduced in MAs from hypertensive mice compared with MAs from control mice (Figure 5E), possibly suggesting impaired dilatory TRPV4SMC–BK channel signaling in hypertension. The TRPV4SMC channel activator GSK101 increased BK channel currents in SMCs from control mice but was unable to activate BK channel currents in SMCs from hypertensive mice (Figure 5F). However, the BK channel current density elicited by the direct BK channel activator, GoSlo SR 5–69 (1 μM), was not different between SMCs from normal and hypertensive mice (Figure 5F), pointing to an impairment in TRPV4SMC–BK channel signaling rather than downregulation of BK channels in hypertension.
Hypertensive patients show increased α1AR–TRPV4SMC signaling and reduced TRPV4SMC–BK channel signaling.
To establish the clinical relevance of our findings obtained in a mouse model of hypertension, we recorded α1AR-activated TRPV4SMC channel currents and TRPV4SMC-activated BK channel currents in freshly isolated SMCs from paraspinal muscle arteries (Figure 6A) from non-hypertensive and hypertensive individuals (Table S1). Baseline TRPV4SMC currents were not different between non-hypertensive and hypertensive individuals, and PE increased GSK219-sensitive outward currents in SMCs from non-hypertensive as well as hypertensive individuals. However, PE-induced TRPV4SMC currents were higher in SMCs from hypertensive individuals compared to non-hypertensive individuals (Figure 6A). Moreover, PE-induced increase in TRPV4SMC sparklet activity was higher in the arteries from hypertensive subjects than non-hypertensive subjects (Figure 6B), suggesting increased constrictor α1AR–TRPV4SMC signaling in human hypertension. The TRPV4 agonist GSK101 also activated paxilline-sensitive BK channel currents in SMCs from both non-hypertensive and hypertensive individuals. Interestingly, GSK101-induced BK channel currents were smaller in SMCs from hypertensive individuals than non-hypertensive individuals (Figure 6C). The BK channel current density induced by a direct activation of BK channels by GoSlo (1 μM) was not different between SMCs from non-hypertensive and hypertensive individuals (Figure 6D), suggesting that direct activation of BK channels can increase channel activity to a similar extent in the two groups. Notably, intraluminal pressure-induced activation of TRPV4SMC sparklets was also attenuated in hypertensive subjects (Figure 6E), indicating impaired pressure–TRPV4SMC–BK channel signaling in human hypertension. Additionally, α1ARs, TRPV4SMC, and BK channel immunofluorescence was not different between non-hypertensive and hypertensive subjects (Figure S23). Taken together, these data confirm that α1AR–TRPV4SMC constrictor signaling is increased and TRPV4SMC–BK channel dilator signaling is reduced in hypertension, which collectively may contribute to increased vasoconstriction and elevated blood pressure (Figure 6F).
Fig. 6. SMCs from hypertensive patients show increased α1AR–TRPV4SMC and reduced TRPV4SMC–BK channel signaling.
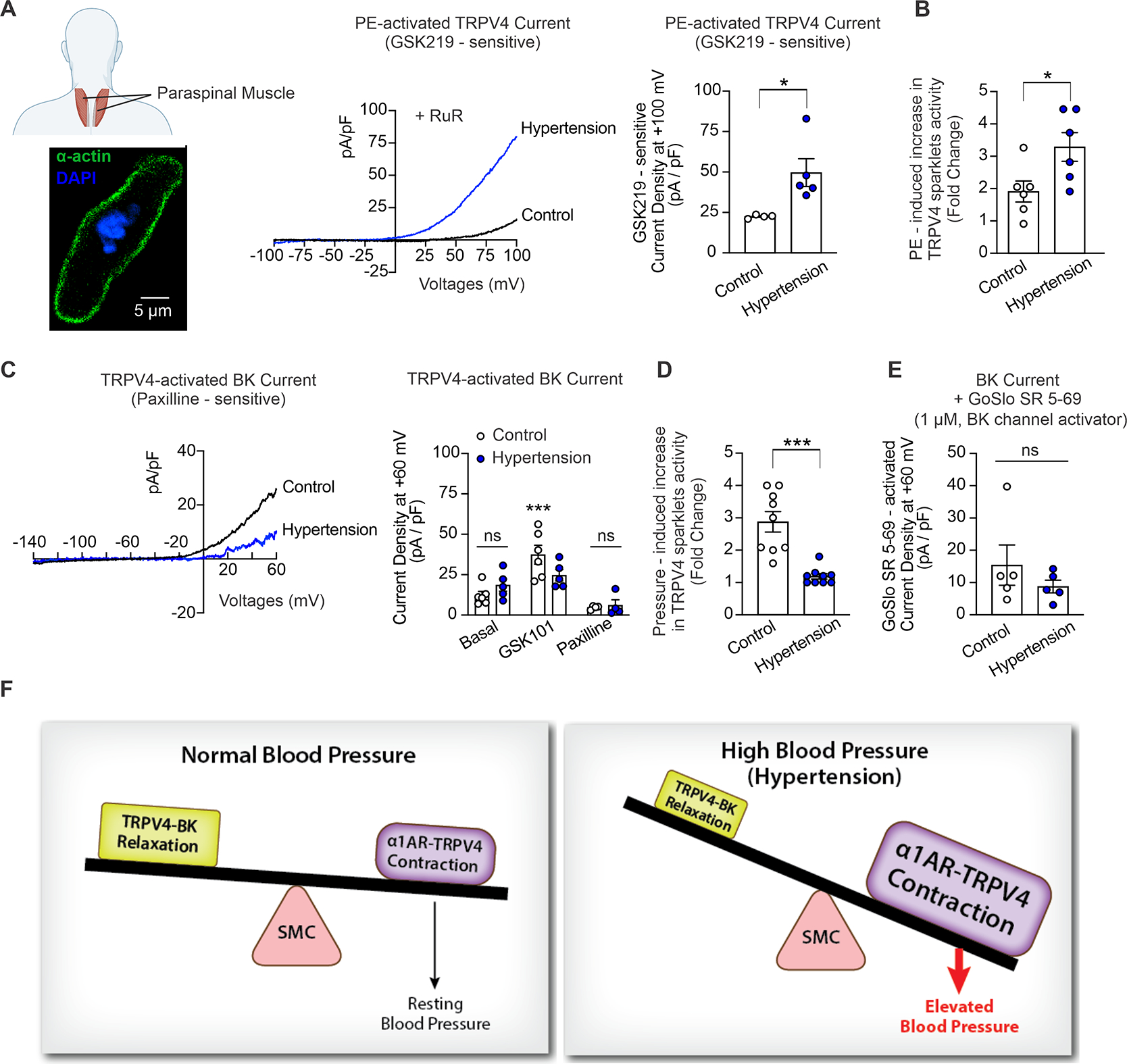
(A) Human vascular SMCs were isolated from paraspinal muscle arteries from non-hypertensive and hypertensive patients (1–2 SMCs per subject per treatment group for functional studies). The inset (Left bottom) shows widefield images of an isolated SMC from a human paraspinal muscle artery, stained for α-actin (green) and counterstained with the nuclear dye DAPI (blue). The fusiform shape (positive α-actin staining) is characteristic of isolated SMCs selected for patch-clamp experiments. Middle, representative whole-cell patch-clamp traces for PE (1 μM)-induced, GSK219 (100 nM)-sensitive ionic currents at +100 mV in SMCs freshly isolated from paraspinal muscle arteries of non-hypertensive (control) subjects and hypertensive patients at +100 mV. Right, averaged GSK219-sensitive outward currents (current at +100 mV in the presence of PE minus current in the presence of PE + GSK219 [100 nM]) in SMCs isolated from paraspinal muscle arteries from non-hypertensive (control, n = 4) subjects and hypertensive patients (n = 5) (*P < 0.05 vs. control, unpaired t-test). (B) PE-induced TRPV4SMC sparklet activity in non-hypertensive (n = 6) and hypertensive patients (n = 6). Pressurized human arteries were pretreated with CPA (20 μM), nifedipine (1 μM), and GSK101 (30 nM) (*P < 0.05 vs. Control; unpaired t-test). (C) Left, representative traces of GSK101 (30 nM)-induced, paxilline (1 μM)-sensitive BK currents in SMCs isolated from paraspinal muscle arteries of non-hypertensive (control) subjects and hypertensive patients, recorded in the whole-cell patch-clamp configuration. Right, averaged outward currents at +60 mV in SMCs isolated from paraspinal muscle arteries of non-hypertensive control subjects (n = 6) and hypertensive patients (n = 5) under basal conditions and in the presence of GSK101 (30 nM) and GSK101 + paxilline (1 μM) (***P < 0.001 vs. Basal; ns, not significant; two-way ANOVA). (D) Increased intraluminal pressure (from 20 mmHg to 80 mmHg)-induced TRPV4SMC sparklet activity in non-hypertensive (n = 9) and hypertensive patients (n = 9). Pressurized human arteries were treated with CPA (20 μM), nifedipine (1 μM), and GSK101 (30 nM) (***P < 0.001 vs. Control; unpaired t-test). (E) Averaged GoSlo SR 5–69-activated outward currents at +60 mV (current in the presence of GoSlo SR 5–69 minus currents before addition of GoSlo SR 5–69) in SMCs isolated from paraspinal muscle arteries from non-hypertensive control subjects (n = 5) and hypertensive patients (n = 5) (ns, not significant; unpaired t-test). (F) Schematic diagram showing the imbalance between α1AR:TRPV4SMC and TRPV4SMC:BK channel signaling in hypertension, which leads to increased constriction of resistance arteries and elevated blood pressure.
PKCα:TRPV4SMC co-localization is increased and TRPV4SMC:BK co-localization is reduced in hypertension.
We hypothesized that altered localization of the signaling elements impairs the balance between constrictor and dilator TRPV4SMC signaling nanodomains in hypertension. PLA studies showed that α1AR:TRPV4SMC co-localization is not altered in Ang II-infused mice and hypertensive individuals (Figure S24), but PKCα:TRPV4SMC co-localization is higher than non-hypertensive controls (Figure 7). Moreover, TRPV4SMC:BK channel co-localization was significantly reduced in Ang II-infused mice and hypertensive individuals (Figure 7), suggesting that increased PKCα:TRPV4SMC co-localization and reduced TRPV4SMC:BK channel co-localization may underlie the imbalance between the constrictor and dilator TRPV4SMC signaling nanodomains in hypertension.
Fig. 7. PKCα:TRPV4SMC co-localization is increased and TRPV4SMC:BK co-localization is reduced in hypertension.
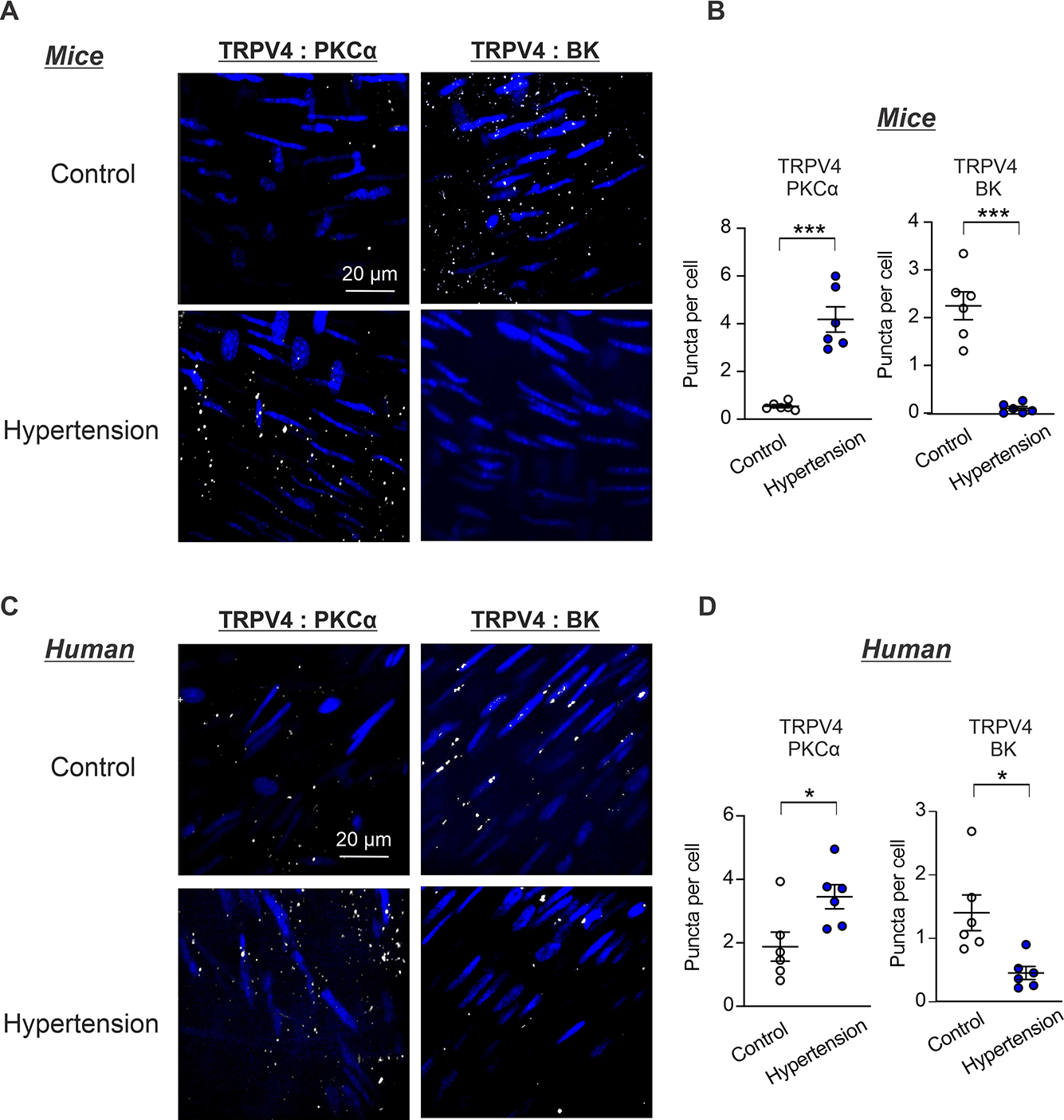
(A) Representative in situ PLA images showing SMC nuclei (blue), TRPV4SMC:PKCα and TRPV4SMC:BK co-localization (white puncta) in en face preparations of MAs from Ang II-induced hypertensive mice and non-hypertensive control mice. (B) Quantification of TRPV4SMC:PKCα (Control, n = 6; Hypertension, n = 6) and TRPV4SMC:BK co-localization (Control, n = 6; Hypertension, n = 6) co-localization in MAs from Ang II-induced hypertensive mice and control mice (***P < 0.001 vs. Control; unpaired t-test). (C) Representative in situ PLA images showing SMC nuclei (blue), TRPV4SMC:PKCα and TRPV4SMC:BK co-localization (white puncta) in en face preparations of paraspinal muscle arteries from non-hypertensive (control) and hypertensive individuals. (D) Quantification of TRPV4SMC:PKCα (Control, n = 6; Hypertension, n = 6) and TRPV4SMC:BK (Control, n = 6; Hypertension, n = 6) co-localization in paraspinal muscle arteries from non-hypertensive (control) and hypertensive individuals (*P < 0.05 vs. Control; unpaired t-test).
Discussion
Our study identifies two novel SMC Ca2+-signaling microdomains that control resting blood pressure and demonstrate an imbalance that may favor arterial contraction during hypertension. The TRPV4SMC−/− mouse has proven to be an invaluable tool, yielding unprecedented insights into the role of SMC TRPV4 channels in regulating resting blood pressure. Our studies provide evidence that TRPV4SMC channels are present in two distinct signaling microdomains that are activated by distinct physiological stimuli and have opposing effects on arterial diameter and blood pressure. The α1AR–TRPV4SMC signaling microdomains, activated by sympathetic stimulation, increase arterial contraction and elevate resting blood pressure. The TRPV4SMC–BK channel signaling microdomains, activated by intravascular pressure, reduce arterial contraction and lower blood pressure. Notably, the effect of α1AR-TRPV4SMC signaling predominates under resting conditions, increasing blood pressure. In hypertensive mice, vasoconstrictor α1AR–TRPV4SMC signaling is accentuated whereas vasodilator TRPV4SMC-BK channel signaling is reduced. This imbalance between the two TRPV4SMC channel signaling microdomains increases vasoconstriction and elevates blood pressure in hypertension. Overall, our data reveal the essential roles of these new TRPV4SMC channel-dependent signaling microdomains in blood pressure regulation and show that altered signaling in these microdomains contributes to blood pressure elevation in hypertension. The findings from human arterial SMCs provide the first evidence for α1AR–TRPV4SMC and TRPV4SMC–BK channel signaling in human vascular SMCs. Furthermore, patch-clamp data from SMCs of non-hypertensive and hypertensive individuals support the idea that an imbalance between functionally opposing subpopulations of TRPV4SMC channels is a clinically relevant finding in human hypertension.
TRPV4SMC channels have received significant research attention in the past decade, yet their physiological roles remain elusive. Earley et al. showed that Ca2+ influx through TRPV4SMC channels could activate BK channels and oppose pressure-induced constriction of cerebral arteries5. In contrast, Xia and colleagues reported that TRPV4SMC channels contribute to serotonin-induced constriction of pulmonary arteries17. Whether dilator or constrictor effects of TRPV4SMC channels alter resting blood pressure remained unknown. Global TRPV4−/− mice are not useful in this regard because they do not display a blood pressure phenotype, possibly owing to the absence of TRPV4 channels in multiple cell types or compensatory upregulation of other TRP channels22. Using endothelial TRPV4−/− mice, we recently showed that endothelial TRPV4 channels lower resting blood pressure9. Thus, endothelial and SMC TRPV4 channels have opposite effects on resting blood pressure, exemplifying the unique cellular activation/function of TRPV4 channels in ECs or SMCs, which would be difficult to decipher using global knockout mice. Our results also suggest that direct and general activation or inhibition of TRPV4 channels is likely associated with undesirable effects because of the diverse physiological roles of these channels and their presence in multiple cell types.
The α1AR–PKCα–TRPV4SMC signaling pathway is a novel mechanism for blood pressure regulation. Sympathetic nerve stimulation causes vasoconstriction and elevates blood pressure, mainly through activation of α1ARs on SMC membranes15,21. α1ARs increase resting MAP by ~20 mm Hg, an effect that is predominantly mediated by activation of TRPV4SMC channels. Earlier, Mercado et al. showed that PKCα anchoring by AKAP150 (A kinase anchoring protein 150) increases TRPV4SMC channel activity in cerebral arteries6. Global AKAP150−/− mice have reduced resting blood pressure, and show a smaller increase in blood pressure in response to chronic Ang II infusion3. Therefore, it is plausible that AKAP150 anchoring of PKCα also mediates α1AR activation of TRPV4SMC channels, although this possibility has not been verified. Sympathetic overactivation is commonly observed in hypertensive patients and mouse models of hypertension23,24. Our data raise the possibility that excessive α1AR–TRPV4SMC signaling may amplify the effect of sympathetic overactivation to contribute to the elevated blood pressure in hypertension.
SMC BK channels are well-known regulators of intravascular pressure-induced constriction and blood pressure25,26. Under normal conditions, RyR Ca2+ sparks activate BK channels, which exert a dilatory effect that opposes intravascular pressure-induced vasoconstriction27,28. Our data confirmed that TRPV4SMC channels increase the activity of RyR Ca2+ sparks. Collectively, these data are consistent with previously published findings5 on vasodilatory TRPV4–RyR–BK channel signaling in SMCs. Intravascular pressure has been shown to increase Ca2+ spark–BK channel signaling29–31 and our data show that intravascular pressure activates TRPV4SMC channels. Together, these data imply that TRPV4SMC channel–Ca2+ spark–BK channel signaling opposes intravascular pressure-induced vasoconstriction. In hypertension, the negative regulation of α1AR–TRPV4SMC channel signaling by TRPV4–BK channel signaling is minimized, resulting in accentuated vasoconstriction and elevated blood pressure.
The cellular mechanisms responsible for spatial separation of constrictor and dilator TRPV4SMC subpopulations are not clear. Scaffolding proteins play a central role in facilitating the co-localization of signaling elements at SMC membranes3,9,32. The association of TRPV4SMC channels with distinct scaffolding proteins, including AKAPs (A-kinase anchoring proteins) and caveolins, may underlie their separation into two functionally opposite subpopulations. In this regard, AKAP150 and caveolin-1 have been shown to co-localize with PKCα and BK channels3,32,33. Co-localization with AKAP150 increases TRPV4SMC channel activity6,16. Moreover, we recently showed that caveolin-1 increases the activity of endothelial TRPV4 channels19, although the caveolin-1–TRPV4 association has not been confirmed in SMCs. Several studies also described the assembly of caveolin-1 and α1ARs in close proximity to sympathetic nerves on the cell membranes34,35. Impaired expression or mis-localization of scaffolding proteins may also underlie increased PKCα:TRPV4SMC and reduced TRPV4SMC:BK co-localization in hypertension, although this possibility has not been verified.
Our Ca2+-imaging data represent the first recordings of TRPV4 sparklet activity in intact SMCs under physiological conditions of intravascular pressure, temperature, and ionic environment. Previous studies have measured TRPV4 channel currents and TRPV4 sparklet activity in isolated SMCs from cerebral arteries6. The ability to record TRPV4SMC sparklets in pressurized arteries has enabled studies of intravascular pressure-induced activation of TRPV4SMC channels. The upstream mechanisms underlying intravascular pressure-induced activation of TRPV4SMC sparklets are unknown. TRPV4SMC channels can be activated mechanically, although the consensus of recent studies is that TRPV4 channels are not direct mechanosensors36–38. Intravascular pressure stimulates GqPCR signaling in SMCs39–41, which in turn, reduces levels of the TRPV4 channel inhibitor PIP242. Whether reduced PIP2 levels or another upstream mechanosensor underlies intravascular pressure-induced activation of TRPV4SMC sparklets has not been investigated.
In summary, our data identify two novel, spatially separated, and functionally opposing Ca2+-signaling microdomains in SMCs: α1AR–TRPV4SMC microdomains that elevate blood pressure, and TRPV4SMC–BK channel microdomains that lower blood pressure. Increased α1AR–TRPV4SMC signaling and reduced TRPV4SMC–BK channel signaling contribute to excessive vasoconstriction and blood pressure elevation in hypertension. A detailed understanding of the mechanisms that regulate TRPV4SMC channels may help in the design of therapeutic strategies that target abnormalities in SMC Ca2+ signaling to normalize vascular function in hypertension.
Supplementary Material
Acknowledgement.
We thank the Molecular Imaging Core and Robert M. Berne Cardiovascular Research Center Small Animal Blood Pressure Core at the University of Virginia for fMRI studies and help with blood pressure measurements, respectively.
Funding Support.
This work was supported by funding from the American Heart Association to YLC (POST833691), an award from the American Physiological Society to ZD, the National Institutes of Health to SKS (HL146914, HL142808, and HL147555), and an award from Neurosurgery Research and Education Foundation to JDS and MSP.
Non-standard Abbreviations and Acronyms:
- SMCs
smooth muscle cells
- TRPV4SMC
smooth muscle cell transient receptor potential vanilloid 4
- α1AR
α1 adrenergic receptor
- TRPV4SMC−/−
SMC-specific TRPV4 knockout
- BK channels
large conductance calcium-activated potassium channels
- RyR
ryanodine receptors, IP3Rs, IP3 receptors
- GqPCR
Gq protein-coupled receptor
- PLA
proximity ligation assay
- dSTORM
direct stochastic optical reconstruction microscopy
- MAs
mesenteric arteries
- MAP
mean arterial pressure
- PE
phenylephrine
- CPA
cyclopiazonic acid
- PLC
phospholipase C
- Ang II
angiotensin II
- LTCC
L-type calcium channels
- TTCC
T-type calcium channels
Footnotes
Disclosures. None.
References
- 1.Bannister JP, Bulley S, Narayanan D, Thomas-Gatewood C, Luzny P, Pachuau J, Jaggar JH. Transcriptional upregulation of alpha2delta-1 elevates arterial smooth muscle cell voltage-dependent Ca2+ channel surface expression and cerebrovascular constriction in genetic hypertension. Hypertension. 2012;60:1006–1015. doi: 10.1161/HYPERTENSIONAHA.112.199661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW, Rusch NJ. The beta3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension. 2013;61:137–142. doi: 10.1161/HYPERTENSIONAHA.112.197863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 is required for stuttering persistent Ca2+ sparklets and angiotensin II-induced hypertension. Circ Res. 2008;102:e1–e11. doi: 10.1161/CIRCRESAHA.107.167809 [DOI] [PubMed] [Google Scholar]
- 4.Ottolini M, Sonkusare SK. The Calcium Signaling Mechanisms in Arterial Smooth Muscle and Endothelial Cells. Compr Physiol. 2021;11:1831–1869. doi: 10.1002/cphy.c200030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6 [DOI] [PubMed] [Google Scholar]
- 6.Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. J Gen Physiol. 2014;143:559–575. doi: 10.1085/jgp.201311050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, Liedtke W, Sham JS. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol. 2013;305:C704–715. doi: 10.1152/ajpcell.00099.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ottolini M, Hong K, Cope EL, Daneva Z, DeLalio LJ, Sokolowski JD, Marziano C, Nguyen NY, Altschmied J, Haendeler J, et al. Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation. 2020;141:1318–1333. doi: 10.1161/CIRCULATIONAHA.119.043385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, Liedtke W, Sham JS. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol. 2013;305:C704–C715. doi: 10.1152/ajpcell.00099.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cowley AW Jr. Long-term control of arterial blood pressure. Physiol Rev. 1992;72:231–300. doi: 10.1152/physrev.1992.72.1.231 [DOI] [PubMed] [Google Scholar]
- 12.Anschutz S, Schubert R. Modulation of the myogenic response by neurogenic influences in rat small arteries. Br J Pharmacol. 2005;146:226–233. doi: 10.1038/sj.bjp.0706323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan CO, Hamner JW, Taylor JA. The role of myogenic mechanisms in human cerebrovascular regulation. J Physiol. 2013;591:5095–5105. doi: 10.1113/jphysiol.2013.259747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, Montezano AC. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529–539. doi: 10.1093/cvr/cvy023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyner MJ, Charkoudian N, Wallin BG. Sympathetic nervous system and blood pressure in humans: individualized patterns of regulation and their implications. Hypertension. 2010;56:10–16. doi: 10.1161/HYPERTENSIONAHA.109.140186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, Santana LF, Nelson MT. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Science signaling. 2014;7:ra66. doi: 10.1126/scisignal.2005052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldenberg NM, Wang L, Ranke H, Liedtke W, Tabuchi A, Kuebler WM. TRPV4 Is Required for Hypoxic Pulmonary Vasoconstriction. Anesthesiology. 2015;122:1338–1348. doi: 10.1097/ALN.0000000000000647 [DOI] [PubMed] [Google Scholar]
- 18.Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daneva Z, Marziano C, Ottolini M, Chen YL, Baker TM, Kuppusamy M, Zhang A, Ta HQ, Reagan CE, Mihalek AD, et al. Caveolar peroxynitrite formation impairs endothelial TRPV4 channels and elevates pulmonary arterial pressure in pulmonary hypertension. Proc Natl Acad Sci U S A. 2021;118. doi: 10.1073/pnas.2023130118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daneva Z, Ottolini M, Chen YL, Klimentova E, Kuppusamy M, Shah SA, Minshall RD, Seye CI, Laubach VE, Isakson BE, et al. Endothelial pannexin 1-TRPV4 channel signaling lowers pulmonary arterial pressure in mice. Elife. 2021;10. doi: 10.7554/eLife.67777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanoue A, Nasa Y, Koshimizu T, Shinoura H, Oshikawa S, Kawai T, Sunada S, Takeo S, Tsujimoto G. The alpha(1D)-adrenergic receptor directly regulates arterial blood pressure via vasoconstriction. J Clin Invest. 2002;109:765–775. doi: 10.1172/JCI14001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017;13:e1006780. doi: 10.1371/journal.pgen.1006780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mancia G, Grassi G. The autonomic nervous system and hypertension. Circ Res. 2014;114:1804–1814. doi: 10.1161/CIRCRESAHA.114.302524 [DOI] [PubMed] [Google Scholar]
- 24.Fisher JP, Paton JF. The sympathetic nervous system and blood pressure in humans: implications for hypertension. J Hum Hypertens. 2012;26:463–475. doi: 10.1038/jhh.2011.66 [DOI] [PubMed] [Google Scholar]
- 25.Krishnamoorthy G, Sonkusare SK, Heppner TJ, Nelson MT. Opposing roles of smooth muscle BK channels and ryanodine receptors in the regulation of nerve-evoked constriction of mesenteric resistance arteries. Am J Physiol Heart Circ Physiol. 2014;306:H981–988. doi: 10.1152/ajpheart.00866.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brayden JE, Nelson MT. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 1992;256:532–535. doi: 10.1126/science.1373909 [DOI] [PubMed] [Google Scholar]
- 27.Knot HJ, Standen NB, Nelson MT. Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+-dependent K+ channels. J Physiol. 1998;508 (Pt 1):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. [DOI] [PubMed] [Google Scholar]
- 29.Perez GJ, Bonev AD, Patlak JB, Nelson MT. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J Gen Physiol. 1999;113:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaggar JH. Intravascular pressure regulates local and global Ca(2+) signaling in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C439–448. doi: 10.1152/ajpcell.2001.281.2.C439 [DOI] [PubMed] [Google Scholar]
- 31.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. 1998;508 (Pt 1):199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mercado J, Baylie R, Navedo MF, Yuan C, Scott JD, Nelson MT, Brayden JE, Santana LF. Local control of TRPV4 channels by AKAP150-targeted PKC in arterial smooth muscle. The Journal of general physiology. 2014;143:559–575. doi: 10.1085/jgp.201311050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashad AM, Harraz OF, Brett SE, Romero M, Kassmann M, Puglisi JL, Wilson SM, Gollasch M, Welsh DG. Caveolae Link CaV3.2 Channels to BKCa-Mediated Feedback in Vascular Smooth Muscle. Arterioscler Thromb Vasc Biol. 2018;38:2371–2381. doi: 10.1161/ATVBAHA.118.311394 [DOI] [PubMed] [Google Scholar]
- 34.DeLalio LJ, Keller AS, Chen J, Boyce AKJ, Artamonov MV, Askew-Page HR, Keller TCSt, Johnstone SR, Weaver RB, Good ME, et al. Interaction between pannexin 1 and caveolin-1 in smooth muscle can regulate blood pressure. Arterioscler Thromb Vasc Biol. 2018;38:2065–2078. doi: 10.1161/ATVBAHA.118.311290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris JB, Huynh H, Vasilevski O, Woodcock EA. Alpha1-adrenergic receptor signaling is localized to caveolae in neonatal rat cardiomyocytes. J Mol Cell Cardiol. 2006;41:17–25. doi: 10.1016/j.yjmcc.2006.03.011 [DOI] [PubMed] [Google Scholar]
- 36.Swain SM, Liddle RA. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J Biol Chem. 2021;296:100171. doi: 10.1074/jbc.RA120.015059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Elias A, Mrkonjic S, Pardo-Pastor C, Inada H, Hellmich UA, Rubio-Moscardo F, Plata C, Gaudet R, Vicente R, Valverde MA. Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proc Natl Acad Sci U S A. 2013;110:9553–9558. doi: 10.1073/pnas.1220231110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vasquez V, Laver DR, Martinac B. Mammalian TRP ion channels are insensitive to membrane stretch. J Cell Sci. 2019;132. doi: 10.1242/jcs.238360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauffenstein G, Laher I, Matrougui K, Guerineau NC, Henrion D. Emerging role of G protein-coupled receptors in microvascular myogenic tone. Cardiovasc Res. 2012;95:223–232. doi: 10.1093/cvr/cvs152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schleifenbaum J, Kassmann M, Szijarto IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N, et al. Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res. 2014;115:263–272. doi: 10.1161/CIRCRESAHA.115.302882 [DOI] [PubMed] [Google Scholar]
- 41.Gonzales AL, Yang Y, Sullivan MN, Sanders L, Dabertrand F, Hill-Eubanks DC, Nelson MT, Earley S. A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Science signaling. 2014;7:ra49. doi: 10.1126/scisignal.2004732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. eLife. 2018;7:e38689. doi: 10.7554/eLife.38689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, et al. Postnatal deletion of the type II transforming growth factor-beta receptor in smooth muscle cells causes severe aortopathy in mice. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:2647–2656. doi: 10.1161/ATVBAHA.115.306573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. American journal of physiology Heart and circulatory physiology. 2007;292:H2613–H2622. doi: 10.1152/ajpheart.01286.2006 [DOI] [PubMed] [Google Scholar]
- 45.Chennupati R, Wirth A, Favre J, Li R, Bonnavion R, Jin YJ, Wietelmann A, Schweda F, Wettschureck N, Henrion D, et al. Myogenic vasoconstriction requires G12/G13 and LARG to maintain local and systemic vascular resistance. Elife. 2019;8. doi: 10.7554/eLife.49374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong K, Cope EL, DeLalio LJ, Marziano C, Isakson BE, Sonkusare SK. TRPV4 (Transient Receptor Potential Vanilloid 4) Channel-Dependent Negative Feedback Mechanism Regulates Gq Protein-Coupled Receptor-Induced Vasoconstriction. Arterioscler Thromb Vasc Biol. 2018;38:542–554. doi: 10.1161/ATVBAHA.117.310038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ottolini M, Daneva Z, Chen YL, Cope EL, Kasetti RB, Zode GS, Sonkusare SK. Mechanisms underlying selective coupling of endothelial Ca2+ signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. Journal of Physiology. 2020;598:3577–3596. doi: 10.1113/JP279570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A. 2013;110:6157–6162. doi: 10.1073/pnas.1216514110 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.