

Abstract
The application of whole-exome sequencing (WES) has led to the identification of high and moderate-risk variants that contribute to cutaneous melanoma susceptibility. However, confirming disease-causing variants remains challenging. We applied a gene co-expression network analysis to prioritize candidate genes identified from WES of 34 melanoma-prone families with at least three affected members sequenced per family (n=119 cases). A co-expression network was constructed from genotype-tissue expression (GTEx) project, skin melanoma from the cancer genome atlas (TCGA), and primary melanocyte cultures. We performed module-specific enrichment and focused on modules associated with pigmentation processes since they are the best-studied and most well-known risk factors for melanoma susceptibility. We found that pigmentation-associated modules across the four expression datasets examined were enriched for well-known melanoma susceptibility genes plus genes associated with pigmentation. We also used network properties to prioritize genes within pigmentation modules as candidate susceptibility genes. Integrating information from co-expression network analysis and variant prioritization, we identified 36 genes (such DCT, TPCN2, TRPM1, ATP10A and EPHA5) as potential melanoma risk genes in our families. Our approach also allowed us to link families with “private” gene mutations based on gene co-expression patterns and thereby may provide an innovative perspective in gene identification in high-risk families.
Keywords: Pigmentation and Pigment Cell Biology, Methods/Tools/Techniques, Melanoma, Melanocytes
INTRODUCTION
The identification of rare and highly penetrant pathogenic variants in CDKN2A and CDK4 was the start of a continued effort to uncover genetic susceptibility to cutaneous malignant melanoma (CMM). In the last several years, BAP1, POT1, ACD, TERF2IP, and TERT were also identified as high-risk melanoma susceptibility genes. Together, however, the pathogenic variants in these genes account for melanoma risk in only ~40 percent of familial cases (Read J et al., 2016). In addition, genome-wide association studies (GWAS) have identified a number of common and low-risk variants, including 54 significant loci in the latest largest GWAS (Landi MT et al., 2020); however, additional genetic susceptibility to CMM remains to be discovered.
Whole-exome sequencing (WES) is a valuable approach for identifying rare variants in high-risk pedigrees. However, the lack of recurrent variants or genes in multiple families, incomplete penetrance, and overwhelmingly large numbers of potential candidate variants have resulted in complexities of gene identification in many family studies. Alternative prioritization strategies followed by WES and novel analytical approaches to address the complexity of genetic susceptibility are critically needed.
The prioritization by coexpression is based on the principle that genes influencing and causing diseases are often functionally related; they participate in similar processes and pathways and are often coexpressed (Goh et al., 2007). This approach can be used for candidate disease gene prioritization, functional gene annotation, and transcriptional regulatory program identification (van Dam S et al., 2018). We constructed weighted correlation networks utilizing candidate genes from WES analysis of germline DNA from 34 CMM families and information from disease-relevant expression profiles measured in melanocytes, skin cells, and cutaneous melanoma samples (Figure 1). Our primary goal was to prioritize genes from WES that share similar functions with known melanoma risk pathways and processes and provide mechanistic insight into how they influence disease susceptibility.
Figure 1.
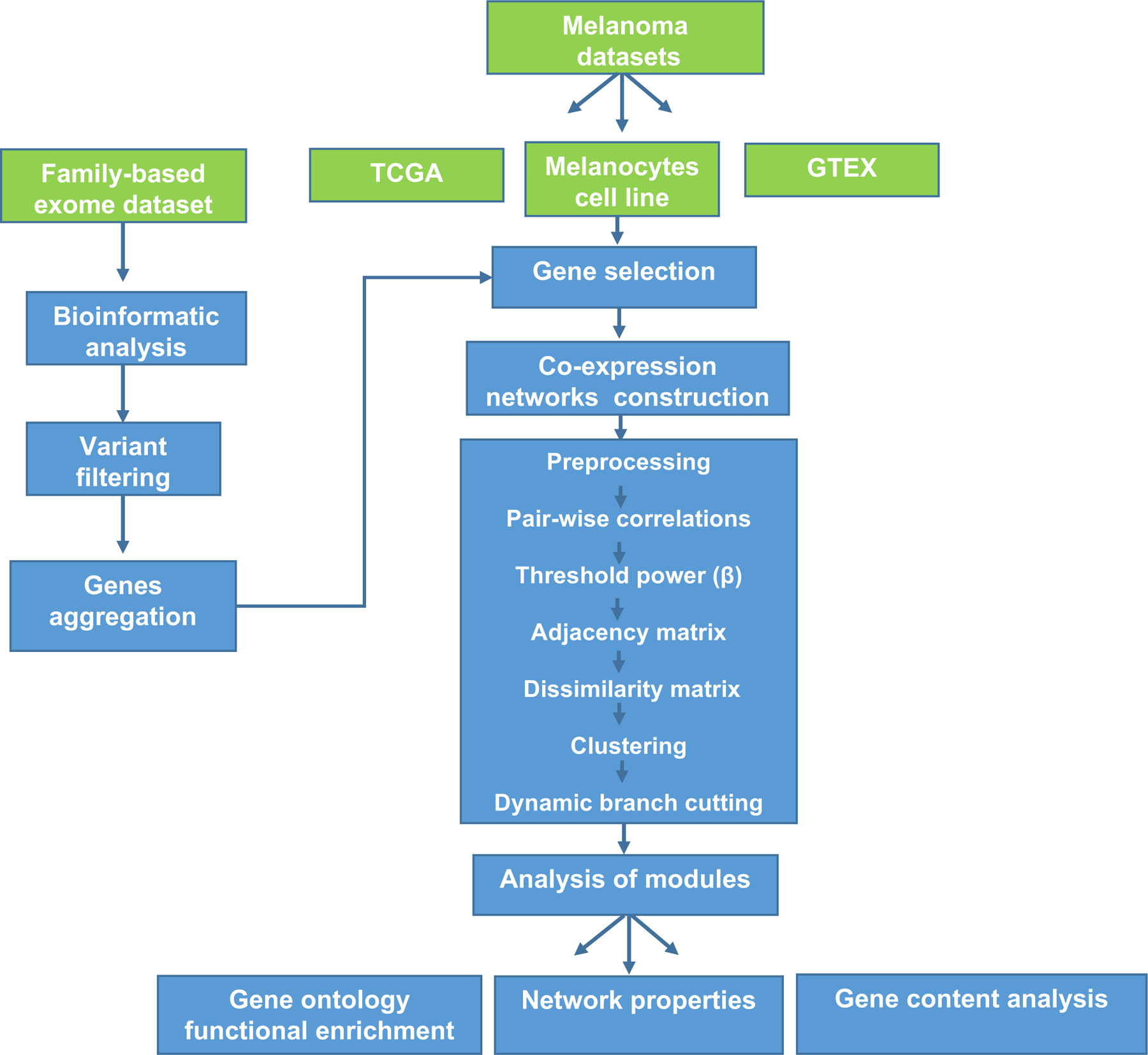
Study workflow of whole-exome analysis and co-expression network analysis. Gene co-expression networks and multi-type data integration (green boxes) were used to prioritize candidate risk genes from whole-exome analysis of melanoma-prone families. Various disease-related expression data (TCGA melanoma, primary melanocyte cultures, and skin in GTEx) were used for network construction. After the pre-processing of expression data, pairwise association was calculated for each gene pair in each dataset. The matrixes of correlations were then converted to adjacency matrixes of connection strengths. The soft threshold operation was used to identify strong correlations and remove weak or negative correlations, to make the correlation value in agreement with scale-free networks, and to identify biological significance. Modules within these networks were identified using clustering analysis. Functional enrichment, gene content analysis, and network properties were then performed on these modules.
RESULTS
Construction of gene co-expression networks
After the WES bioinformatic processing of 34 families with at least three affected members sequenced per family, nonsynonymous variants were subjected to quality control check and filtered by minor allele frequency (MAF) as described in Methods. Eight thousand one hundred fifty-three variants resulting from WES analysis were then aggregated into 5978 genes. The coexpression networks constructed from these genes in the four expression datasets, genotype-tissue expression (GTEx) human skin sun-exposed, GTEx human skin not sun-exposed, TCGA skin cutaneous melanomas, and 106 primary melanocyte cell lines, consisted of 26, 25, 37, and 20 modules, respectively. Figure 2 shows the dendrograms of genes clustered, represented by different colors, based on a dissimilarity measure. Figure S1 depicts the determination of soft-thresholding power in each dataset.
Figure 2.
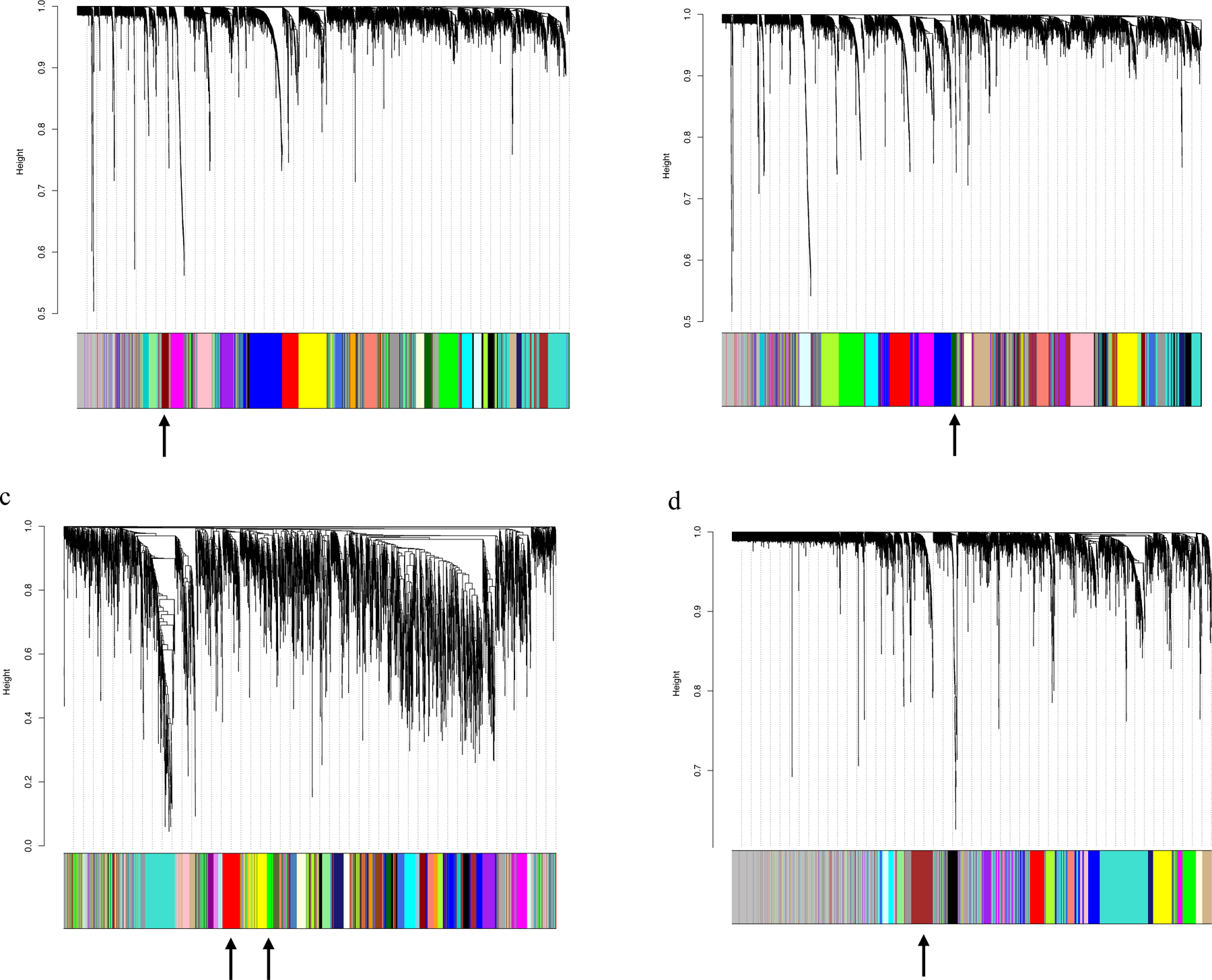
Module identification by Weighted Gene Correlation Network Analysis (WGCNA). The network analysis identified distinct modules of coexpressed genes. Dendrograms of genes were clustered based on a dissimilarity measure: (a) Sun-exposed skin, 26 modules (43 to 521 genes); (b) Sun non-exposed skin, 25 modules (44 to 378 genes); (c) Skin cutaneous melanomas, 37 modules (37 to 365 genes); (d) Primary melanocyte cultures, 20 modules (67 to 639 genes). Each leaf (vertical line) in the dendrogram corresponds to a gene (top). The branches are expression modules of highly interconnected groups of genes with the color indicating the module assignment (bottom panel). Modules enriched with pigmentation terms are indicated by arrows: dark red (GTEx sun-exposed skin), dark green (GTEx sun non-exposed skin), green and red (TCGA SKCM), and brown (Melanocyte cell line) modules.
Identification and characterization of modules associated with pigmentation
Module-specific enrichment
We performed a module-specific enrichment in search of modules significantly enriched with melanin/pigmentation terms by gene ontology (GO) analysis. We focused on studying pigmentation modules because pigmentation processes are among the best-known risk factors for CMM susceptibility. This strategy may help us identify potentially novel genes coexpressed with previously well-known pigmentation-related melanoma risk genes. Enrichment categories such as biological process, molecular function, and cellular component were analyzed in each module independently across the expression datasets, which comprised 108 modules in total. Enrichment for pigmentation was identified in the dark red (GTEx sun-exposed skin, 61 genes), dark green (GTEx sun non-exposed skin, 44 genes), green and red (TCGA SKCM, 183 genes), and brown (Melanocyte cell line, 244 genes) modules. The top 10 significantly enriched terms for the pigmentation modules in the two GTEx datasets and significant terms associated with pigmentation in selected modules in the TCGA and the melanocyte cell line are listed in Figure S2.
Module gene content
When comparing genes in these pigmentation-associated modules against a curated list of melanoma-related genes corresponding to well-known CMM susceptibility genes, GWAS loci, expression quantitative trait loci (eQTLs), genes with pleiotropic associations, and pigmentation genes from GO terms (Table S1), we found the enrichment of melanoma-related genes in pigmentation modules across the expression datasets (Figure 3). Genes mapped to pigmentation modules in multiple expression datasets included several well-known melanoma risk genes such as OCA2, MC1R, TYR, TYRP1, IRF4, MITF, and SLC45A2; newly identified GWAS loci such as MFSD12, and MSC; genes demonstrating pleiotropic associations with CMM and other risk factors such as SLC24A5, SLC24A4, and MXI1; and genes with a suggestive role in pigmentation processes from GO terms analysis but an unclear role in melanoma such as TRPM1, TPCN2, CDH3, LYST, and MYO5A. The pigmentation modules shared similar expression patterns and were enriched for functionally similar genes across datasets, with 75 genes mapped in more than two datasets in the pigmentation modules (Figure 3, Table S2, highlighted in bold). TYR, TYRP1, MFSD12, SLC24A5 and TPCN2 were identified in all four datasets. This pattern of shared genes among the expression datasets demonstrated the consistency of results and highlighted the value of using multiple disease-relevant expression datasets. These genes, especially those with disease co-segregating variants within families (Figure 3 highlighted in blue) and unknown role in melanoma susceptibility (Figure 3 highlighted in pink), are considered as strong candidates for new CMM risk genes in our families. For example, EPHA5 and ATP10A were identified in pigmentation modules of three different expression datasets and variants in these genes were shared by at least three affected relatives within a family.
Figure 3.
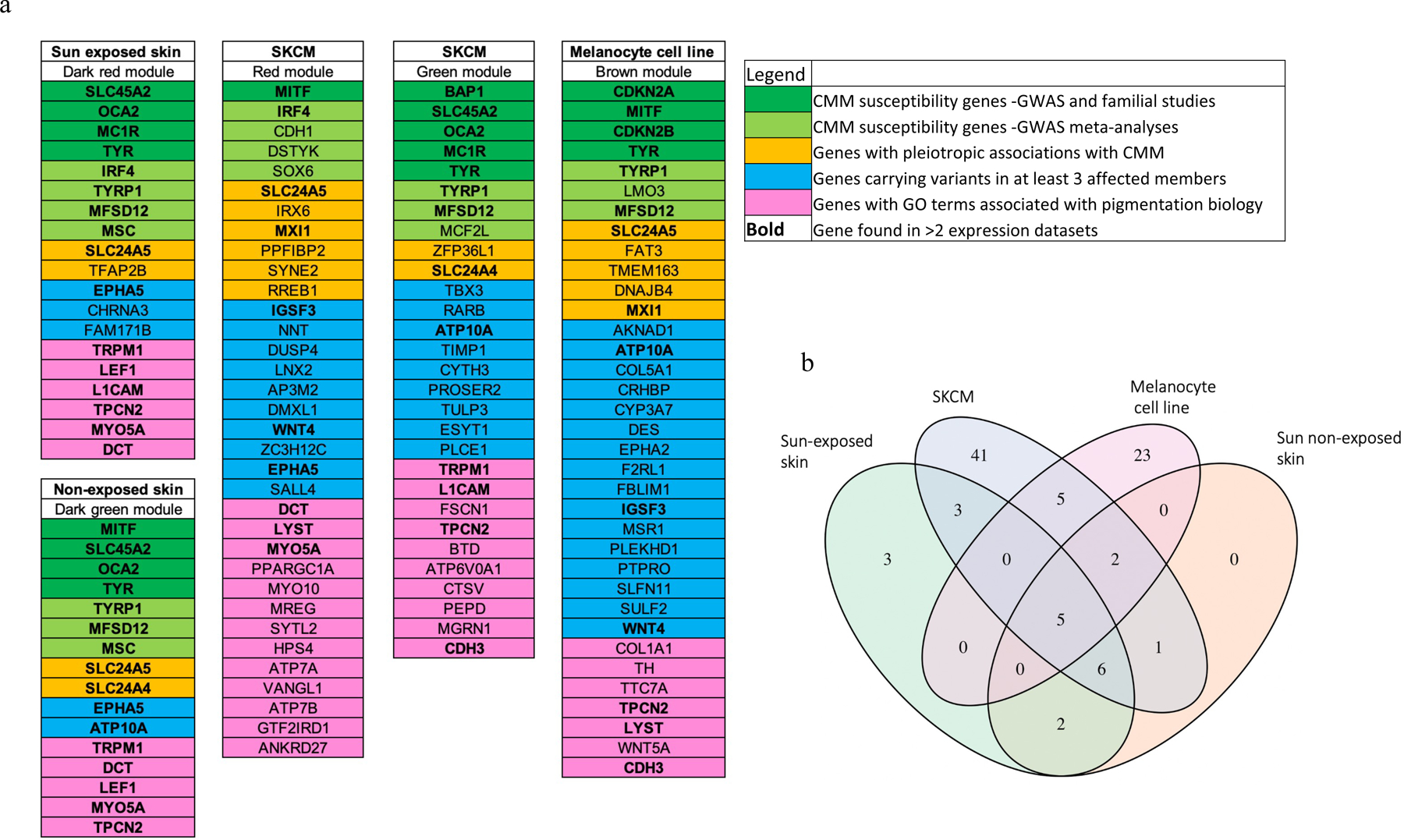
Genes included in pigmentation-enriched modules across the four expression datasets. (a) Gene categories are depicted in colors. (b) Veen diagram showing numbers of genes in common. TYR, TYRP1, MFSD12, SLC24A5 and TPCN2 were identified in all four datasets.
Network properties
We also used network properties to prioritize genes within pigmentation modules as putative susceptibility genes. We first determined if the selected modules’ network topology was relevant for the function of pigmentation. We plotted the correlation between eigengene-based connectivity (kME) and gene significance (GS), among the selected pigmentation modules (Figure S3). We identified a strong correlation between kME and GS particularly for the dark red and dark green modules from GTEx sun-exposed (r=0.96, P=2.6e−34) and GTEx sun non-exposed (r=0.95, P=7.6e−23) datasets (Figure S3). This finding is important since it suggests that not only the pigmentation genes are informative, but the topology of the modules is also important for the functions related to pigmentation. The networks corresponding to these modules consisted of 44 nodes and 364 edges for the dark green module and 61 nodes and 709 edges for the dark red module (Figure 4). Some known melanoma/pigmentation genes were grouped into a core of highly correlated genes radiating strong interconnections with candidate genes in both networks based on high topological overlap measure (TOM) values (darker edges in Figure 4).
Figure 4.
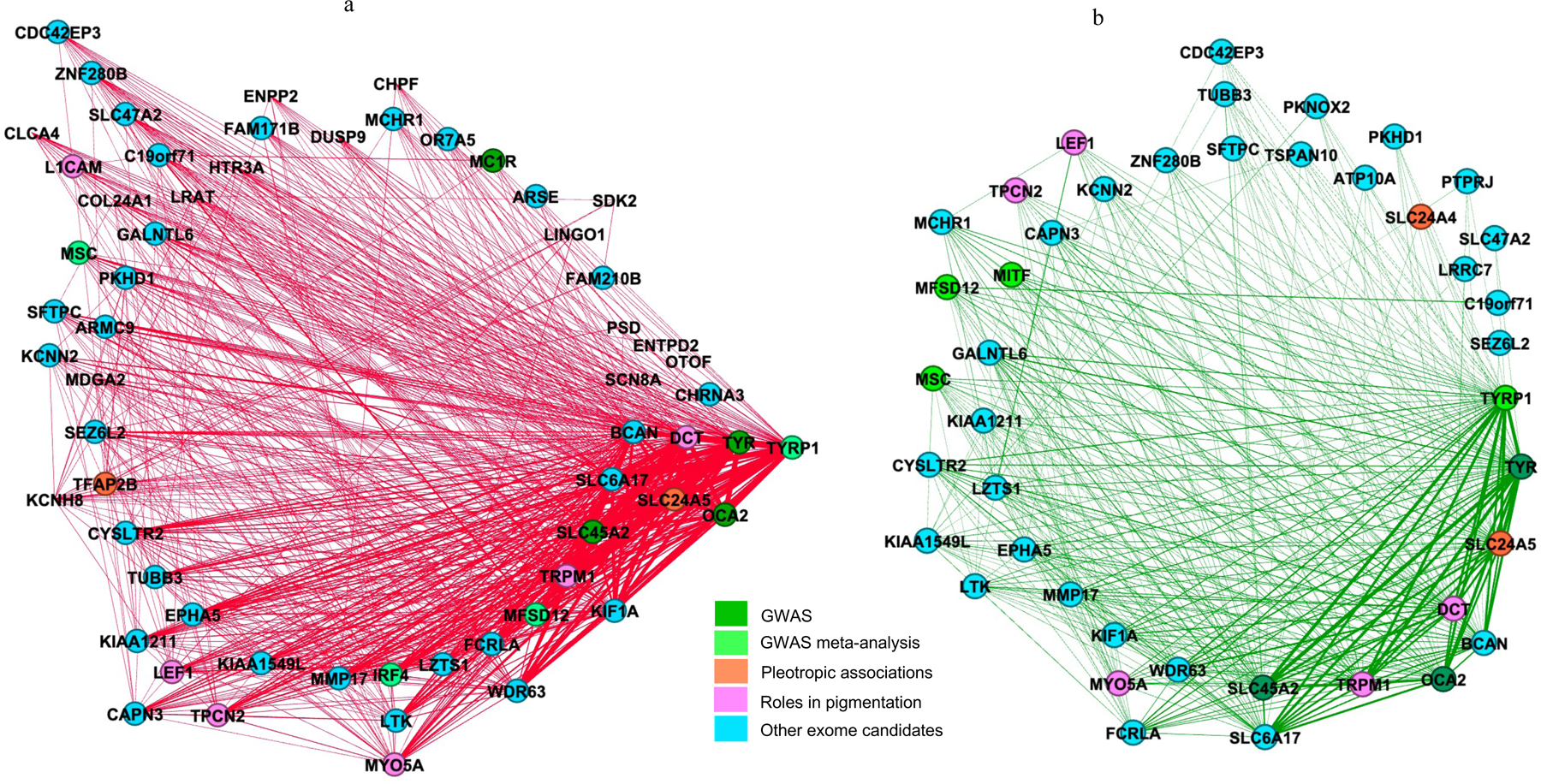
Network topology view of modules associated with pigmentation. Panel (a) dark red and panel (b) dark green modules from GTEx dataset. Edges with high topological overlapping matrices (TOM) and their corresponding nodes are displayed. Two genes have a high TOM if they are highly interconnected with the same set of genes, revealing strong co-expression relationships. Nodes are colored based on the type of genes used for gene content analysis. Some genes belong to more than one category.
We used network metrics to prioritize genes in pigmentation-related modules by selecting genes with the top 10 highest scores in kME, GS, and TOM metrics in each module. A complete list of network metrics and module assignments for all genes is included in Tables S3-S6. Notably, TYR, TYRP1, TRPM1, DCT, and TPCN2 were identified by network topology and network metrics. These and other genes that showed high scores in network metrics may relate to both pigmentation and melanoma and therefore were prioritized for further analyses.
Although the brown module in the primary melanocyte cell line showed genes associated with pigmentation and shared multiple genes related to this process with the other expression datasets, it did not show strong correlations with network metrics (Figure S3). To further explore and prioritize genes in the brown module, we analyzed the module substructure. A modularity analysis identified five distinct submodules, indicating gene subgroups with distinct expression patterns, with classic pigmentation genes represented by the blue color in Figure 5a. We therefore focused on genes in this subgroup, such as genes that showed direct coexpression with well-known melanin synthesis genes present in the module (TYR, TYRP1) and MITF, a master regulator of the melanocyte lineage. Using this strategy, we identified several genes with co-segregating variants in at least three affected members of the same family (e.g., ATP10A, EPHA2, MSR1, and WNT4) that we consider as potential CMM susceptibility genes (Figure 5b).
Figure 5.
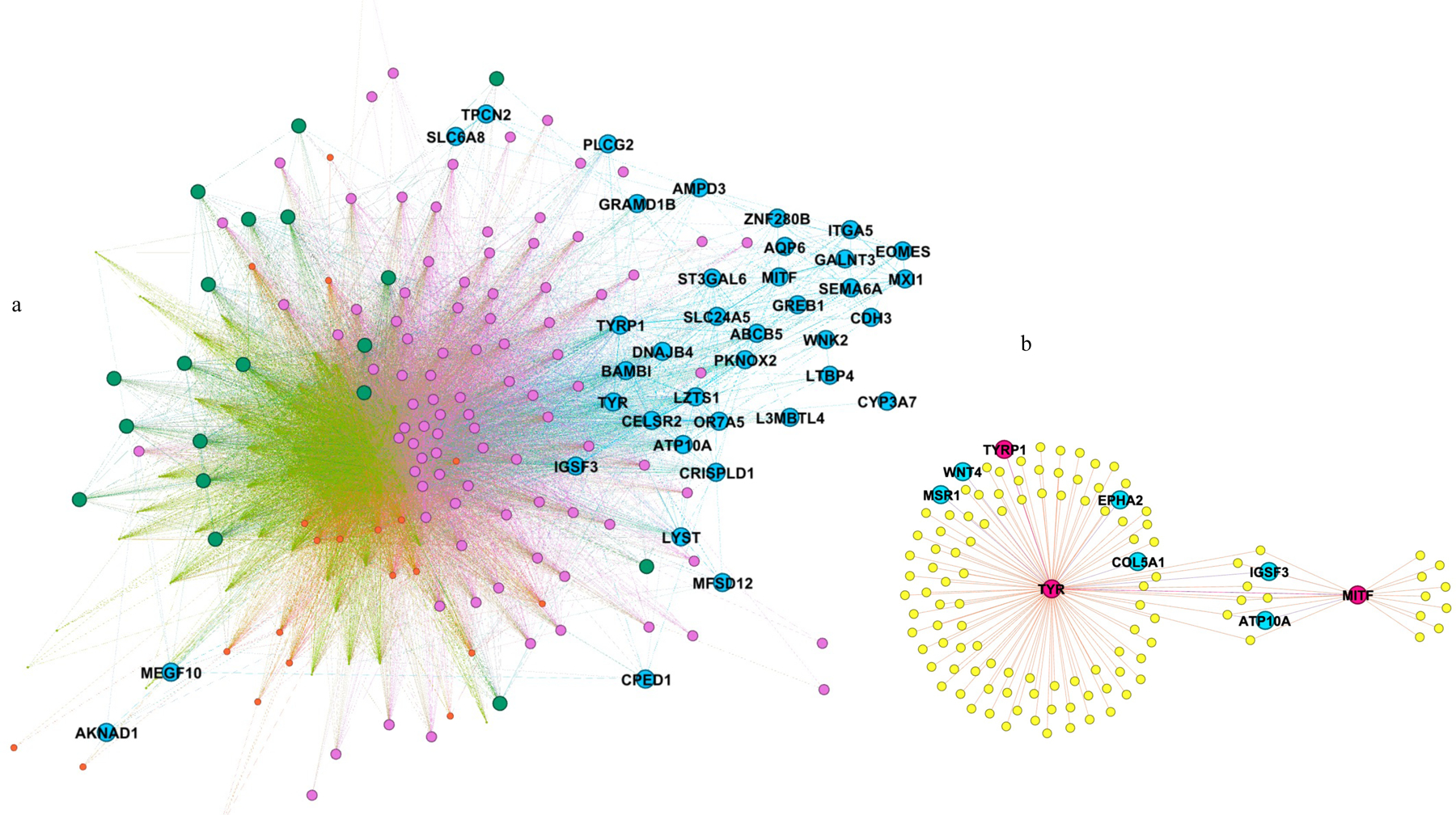
Network topology view of the pigmentation-associated brown module from the primary melanocyte cell line. (a) Modularity analysis of the brown module revealed five submodules of genes. The submodule containing classic pigmentation genes is shown on the right side of the figure (blue colored). (b) Genes with co-segregating variants showing co-expression with MITF, TYR and TYRP1.
Gene and variant prioritization in WES families
Since rare, loss-of-function and predicted deleterious missense variants (see Methods) are more likely to influence predisposition and result in disease than other types of variants, we further restricted variants based on these criteria. Rare loss-of-function and predicted deleterious missense variants were prioritized from 1) known melanoma and pigmentation-related genes; 2) genes with the highest scores in network property analyses; and 3) all genes with cosegregating variants in families.
Using this strategy, we identified 38 candidate variants in 36 genes (13 loss of function and 25 missense variants) as potential candidate genes in our families, which include some well-known CMM risk genes such as TYR and TYRP1, and new genes with previously unknown roles in melanoma risk. Table 1 shows the type, location, and frequencies of these variants.
Table 1.
Details of rare and pathogenic variants in CMM candidate susceptibility genes prioritized by gene content, network metrics, and co-segregation in families.
| Gene | Chr | Location | SNP IDS | REF | VAR | Variant type | Protein change | Variant impact | Pathogenecity prediction a | Populations MAF | Count in controls | Prioritization | Module | Family ID | Genotypes | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| METASVM | METALR | CADD | EXAC_NFE | ESP_EA | KG_AMR | PLCO | ACS | |||||||||||||
| AKNAD1 | 1 | 109358861 | rs149826017 | G | A | stop_gained | p.Gln815* | High | 2.33E-04 | 0 | 0 | co-segregation | brown | FF2 | 0\0, 0\1, 0\0, 0\1, 0\1 | |||||
| WNT4 | 1 | 22456169 | G | A | missense | p.Arg85Trp | Moderate | D | D | 35 | 0 | 0 | co-segregation | red, brown | E2, B2 | 0\1, 0\1, 0\1 | ||||
| EPHA2 | 1 | 16458599 | C | T | missense | p.Arg762His | Moderate | D | D | 21.4 | 0 | 0 | co-segregation | brown | F10 | 0\0, 0\1, 0\1, 0\1 | ||||
| DES | 2 | 220285649 | T | A | missense | p.Cys333Ser | Moderate | D | D | 22.5 | 0 | 0 | co-segregation | brown | A5 | 0\1, 0\1, 0\1 | ||||
| TTC7A | 2 | 47287965 | rs150405237 | C | T | missense | p.Ala761Val | Moderate | D | D | 20.6 | 1.05E-04 | 1.16E-04 | 0 | 0 | mapping | brown | E3 | 0\0, 0\1, 0\1 | |
| EPHA5 | 4 | 66361129 | rs200932017 | T | C | missense | p.Asp348Gly | Moderate | D | D | 28.4 | 1.50E-05 | 1.16E-04 | 0 | 0 | 0 | network properties/co-segregation | dark red, dark green, red | E1, F10 | 0\0, 0\1, 0\1, 0\1 |
| F2RL1 | 5 | 76115090 | G | A | splice_donor& intron | High | 0 | 0 | co-segregation | brown | FF2 | 0\1, 0\0, 0\0, 0\1, 0\1 | ||||||||
| CRHBP | 5 | 76251662 | C | frameshift | p.Thr173fs | High | 0 | 0 | co-segregation | brown | F10 | 0\1, 0\1, 0\0, 0\1 | ||||||||
| CRHBP | 5 | 76251666 | A | frameshift | p.Lys175fs | High | 0 | 0 | co-segregation | brown | F10 | 0\1, 0\1, 0\0, 0\1 | ||||||||
| NNT | 5 | 43651894 | C | A | missense | p.Pro591Thr | Moderate | D | D | 32 | 0 | 0 | co-segregation | red | T | 0\1, 0\1, 0\1, 0\1 | ||||
| MYO10 | 5 | 16763618 | rs372706691 | T | C | missense | p.Asn489Ser | Moderate | D | D | 26.1 | 3.00E-05 | 2.44E-04 | 0 | 0 | network properties/mapping | red | Z | 0\0, 0\0, 0\1, 0\1, 0\0 | |
| CYP3A7 | 7 | 99311149 | C | A | missense | p.Asp270Tyr | Moderate | D | D | 17.21 | 0 | 0 | co-segregation | brown | F10 | 0\1, 0\1, 0\0, 0\1 | ||||
| MSR1 | 8 | 16012621 | rs762856785 | G | A | stop_gained | p.Arg302* | High | 4.57E-05 | 0 | 0 | co-segregation | brown | E4 | 0\1, 0\1, 0\1 | |||||
| DUSP4 | 8 | 29194800 | rs770960298 | C | T | missense | p.Val310Ile | Moderate | D | D | 20.5 | 3.04E-05 | 0 | 0 | co-segregation | red | D5 | 0\1, 0\1, 0\1 | ||
| CDKN2A | 9 | 21971208 | rs730881677 | C | G | splice_acceptor&intron | High | 0 | 0 | mapping | brown | BC | 0\1, 0\0 (*), 0\1, 0\1, 0\1 | |||||||
| TYRP1 | 9 | 12698595 | rs371562555 | C | T | stop_gained | p.Gln285* | High | 1.50E-05 | 1.16E-04 | 0 | 0 | network properties/mapping | dark red, dark green, green, brown | FF2 | 0\0, 0\1, 0\1, 0\0, 0\0 | ||||
| CDKN2B | 9 | 22008767 | rs188670390 | C | G | missense | p.Gln62His | Moderate | D | D | 22.8 | 3.60E-05 | 0 | 0 | mapping | brown | M | 0\1, 0\1, 0\1, 0\1 | ||
| CDKN2B | 9 | 22008763 | C | T | missense | p.Ala64Thr | Moderate | D | D | 22.6 | 0 | 0 | mapping | brown | X | 0\1, 0\1, 0\1 | ||||
| PROSER2 | 10 | 11908784 | rs779142603 | T | C | splice_donor& intron | High | 3.17E-05 | 0 | 0 | co-segregation | green | D2 | 0\1, 0\1, 0\1 | ||||||
| PLAU | 10 | 75673056 | rs755896059 | A | T | missense | p.Asp126Val | Moderate | D | D | 17.79 | 4.50E-05 | 0 | 0 | network properties | brown | E4 | 0\1, 0\0, 0\1 | ||
| TYR | 11 | 89017960 | rs62645917 | C | T | stop_gained | p.Arg402* | High | 4.52E-05 | 0 | 0 | network properties/mapping | dark red, dark green, green, brown | A2 | 0\1, 0\0, 0\1, 0\1, 0\1 | |||||
| ZC3H12C | 11 | 110036201 | A | frameshift | p.Asn799fs | High | 0 | 0 | co-segregation | red | F10 | 0\1, 0\1, 0\1, 0\1 | ||||||||
| PPFIBP2 | 11 | 7674289 | AT | frameshift | p.Ile824fs | High | 0 | 0 | mapping | red | A1 | 0\1, 0\1, 0\0 | ||||||||
| TH | 11 | 2189810 | rs376881948 | C | T | missense | p.Arg164His | Moderate | D | D | 12.89 | 8.55E-05 | 0.00E+00 | 0 | 0 | mapping | brown | X, E4 | 0\1, 0\1, 0\0 | |
| SOX6 | 11 | 16133370 | rs76218238 | G | A | missense | p.Pro293Ser | Moderate | D | D | 16.86 | 1.65E-04 | 4.66E-04 | 0 | 0 | mapping | red | E3 | 0\1, 0\1, 0\1 | |
| TPCN2 | 11 | 68848914 | A | G | missense | p.Met546Val | Moderate | D | D | 15.94 | 0 | 0 | network properties/mapping | dark red, dark green, green, brown | F10 | 0\0, 0\1, 0\0, 0\1 | ||||
| TBX3 | 12 | 115118740 | rs745862855 | C | T | missense | p.Val201Ile | Moderate | D | D | 21.5 | 1.50E-05 | 0 | 0 | co-segregation | green | E4 | 0\1, 0\1, 0\1 | ||
| TULP3 | 12 | 3043650 | rs767887151 | C | T | missense | p.Arg283Cys | Moderate | D | T | 27.1 | 1.50E-05 | 0 | 0 | co-segregation | green | B4 | 0\1, 0\0, 0\1, 0\1 | ||
| DCT | 13 | 95112416 | T | C | missense | p.Asn370Asp | Moderate | D | D | 26.5 | 0 | 0 | network properties/mapping | dark green, dark red, red | A1 | 0\1, 0\1, 0\0 | ||||
| ATP10A | 15 | 25971215 | rs140982945 | C | G | missense | p.Ala288Pro | Moderate | D | D | 32 | 4.36E-04 | 1.16E-04 | 0 | 0 | co-segregation | dark green, green, brown | B4 | 0\1, 0\1, 0\1, 0\0 | |
| CHRNA3 | 15 | 78893642 | rs199515569 | C | A | missense | p.Val448Phe | Moderate | D | D | 27.1 | 4.50E-05 | 0 | 0 | co-segregation | dark red | A8 | 0\1, 0\1, 0\1 | ||
| TRPM1 | 15 | 31355373 | rs369237180 | C | T | missense | p.Gly322Arg | Moderate | D | D | 22.3 | 6.00E-05 | 1.19E-04 | 0 | 0 | network properties/mapping | dark red, dark green, green | A1 | 0\1, 0\0, 0\1 | |
| SLFN11 | 17 | 33689850 | rs141439855 | G | C | stop_gained | p.Ser326* | High | 3.00E-05 | 3.49E-04 | 0 | 0 | co-segregation | brown | E2 | 0\1, 0\1, 0\1 | ||||
| ATP6V0A1 | 17 | 40629682 | rs140591345 | C | T | missense | p.Ala150Val | Moderate | D | D | 23.8 | 1.80E-04 | 0.00E+00 | 0 | 0 | 0 | mapping | green | D5 | 0\1, 0\1, 0\0 |
| OSBPL1A | 18 | 21883628 | T | C | start_lost | p.Met1 | High | 0 | 0 | network properties | red | B0 | 0\0, 0\0, 0\1, 0\1 | |||||||
| SULF2 | 20 | 46300956 | T | C | missense | p.Lys521Arg | Moderate | D | D | 10.48 | 0 | 0 | co-segregation | brown | B2 | 0\1, 0\1, 0\1 | ||||
| ATP7A | X | 77296166 | A | G | missense | p.Met1246Val | Moderate | D | D | 23 | 0 | 0 | mapping | red | A6 | 0\1, 0\1, 0\0 | ||||
| TIMP1 | X | 47444391 | rs769724738 | C | T | missense | p.Arg60Cys | Moderate | D | D | 18.69 | 8.97E-05 | 0 | 0 | co-segregation | green | A8 | 0\1, 0\1, 0\1 | ||
Chr, chromosome; REF, reference allele; VAR, variant allele; MAF, minor allele frequency; T, tolerant; D, deleterious; EXAC, Exome Aggregation Consortium -frequencies in NFE; NFE, non-Finish European population; ESP, NHLBI GO Exome Sequencing Project -frequencies in European American subjects; KG, The 1,000 Genomes Project -frequencies in European sub-population; PLCO, Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial; CPS, Cancer Prevention Study
Pathogenicity prediction for missense variants based on in silico algorithms, variants with evidence of pathogenicity based on at least 2 in silico predictions (Meta Likelihood ratio: D, METASVM: D, and CADD: ≥ 20) were prioritized.
In our WES analysis, most families have a distinct set of candidate genes. Using network analysis, our data showed that although families may not harbor mutations in the same genes, they can be connected by genes in a co-expression network. Figure S4 shows an example of co-expressed genes with rare, potential pathogenic variants in families connected via pigmentation-related modules.
DISCUSSION
Using gene co-expression networks and multi-type data integration, we prioritized candidate risk genes identified from WES analysis of multiplex melanoma-prone families. We identified pigmentation-associated modules in multiple large disease-related expression datasets by weighted gene correlation network analysis (WGCNA) and functional enrichment analysis. All pigmentation-associated modules identified shared similar gene expression patterns and contained melanoma-related genes such as well-known susceptibility genes, GWAS loci, eQTLs, genes with pleiotropic associations, and pigmentation genes across four different expression datasets. Integrating information from gene content, network property, and variant analyses allowed us to identify potential new candidates with close co-expression with known melanoma risk genes but previously unknown evidence for involvement in melanoma risk (e.g., DCT, TPCN2, TRPM1, ATP10A, EPHA5). In addition, our approach allowed the connection of families that do not share variants in the same affected genes but rather involve genes that interact with each other in co-expression networks, which may provide an innovative analysis perspective in addressing the common challenge of private mutations observed in WES analysis of families.
Our approach uncovered multiple genes with previous evidence for involvement in melanoma, such as genes identified from melanoma GWAS meta-analysis (TYRP1, MFSD12, MSC, CDH1, DSTYK, SOX6, MCF2L, and LMO3), genes showing pleiotropic associations with cutaneous melanoma and nevus count or hair color (SLC24A5, TFAP2B, SLC24A4, IRX6, MXI1, PPFIBP2, SYNE2, RREB1, ZFP36L1, FAT3, TMEM163, and DNAJB4), and well-known genes previously associated with familial melanoma such as CDKN2A, OCA2, TYR, and MC1R, which strengthens the functional implication of these candidate genes in the studied modules.
We also provided a framework that helps reconstruct molecular processes and implicates genes in biological processes to prioritize genes for further studies. We were able to identify coordinated co-expression patterns of several candidate susceptibility genes that participate in pigmentation signaling processes, particularly genes identified by network metrics in pigmentation modules of GTEx datasets and a submodule in melanocytes, that demonstrated a clear role in melanin production. For example, DCT (dopachrome tautomerase) is involved in the formation of eumelanin, and with TYR and TYRP1, it is under the regulation of MITF, a master gene of melanocyte lineage (Goding CR, 2000). In the melanosome, tyrosine’s active uptake is required, which initiates a process of oxidation by TYR and other enzymes such as TYRP1 and DCT. The coupling of ion transport with SLC45A2, SLC24A5, and TPCN2 is critical in regulating the process since ion transport is necessary for melanosome function, with TYR activity being pH-dependent (Sturm RA and Duffy DL, 2012). MYO5A encodes a protein that is an actin-based motor involved in short-range movement of melanosomes, which is involved in moving melanosomes to the dendrites (Van Gele M 2009; Barral DC et al., 2004). The expression of TRPM1, which is a calcium permeable cation channel gene that is involved in malignant melanoma pathophysiology, has been shown to be inversely correlated with melanoma aggressiveness (Guo H et al., 2012).
Further, our analysis revealed molecular functions of some of the most recently identified GWAS genes. For example, MFSD12, was mapped to the pigmentation module, which is consistent with its role as a key promoter of cell proliferation in melanoma cells (Wei CY et al., 2019). This gene is also known for suppressing eumelanin biogenesis in melanocytes (Grawford NG et al., 2017).
Some genes that show co-segregation in families (blue in Figure 3) have recently been found to have putative functional roles in melanoma, as summarized in Table S7. For example, the embryonic stem cell factor SALL4 was shown to be upregulated in hyperplastic murine melanoma-prone melanocytes and that its expression was essential for melanoma primary tumor growth (Diener J et al., 2021). NNT, which encodes the mitochondrial redox-regulating enzyme nicotinamide nucleotide transhydrogenase, mediates redox-dependent tyrosinase degradation and pigmentation via a UVB- and MITF-independent mechanism (Allouche J et al., 2021). Eph receptors are the largest family of receptor tyrosine kinases that play roles in multiple cellular processes such as cell proliferation, adhesion, and various developmental processes. In particular, EPHA2 is located on 1p36, which is a region frequently altered in tumors of neuroblastoma and melanoma (Sulman EP et al., 1997). A previous study demonstrated that EPHA2 was upregulated by ultraviolet radiation and is a critical oncogene in melanoma (Udayakumar D et al., 2012). These findings further support the functional relevance of the candidate genes prioritized by our co-expression network analysis approach.
To examine whether these potential CMM susceptibility genes are enriched for somatic alterations in melanoma tumors, we investigated somatic single nucleotide variations (SNVs) and copy number alterations (SCNAs) in top candidate genes in tumor samples using data from the NCI’s Genomic Data Commons (GDC), including 13,035 tumors from The Cancer Genome Atlas (TCGA) and The Therapeutically Applicable Research to Generate Effective Treatments (TARGET) projects. Given the high tumor mutational burden in skin cutaneous melanoma (SKCM), it is not surprising that mutation rates for most of these genes are higher in SKCM as compared to other cancer types. However, some of our candidate CMM susceptibility genes, such as CDKN2A, MSR1, SLFN11, and TRPM1 for SNVs and CDKN2A, CDKN2B, TRPM1, and ZC3H12C for SCNAs, showed higher frequencies of alterations among SKCM than other tumors (Figure S5), supporting the role of these genes in melanoma involvement.
The use of network analysis provided several distinctive advantages over single gene-based approaches. By capturing the broad organization of interactions, the network analysis provides molecular insights into potential risk genes and how they interact with each other to influence disease. Our multidimensional exploration allowed integrating expression profiles in specific tissues, exome data from families, and previous knowledge on susceptibility genes with co-expression modules. Combining information from different layers of genomic data may reveal new biologically interpretable associations and improve the accuracy of variant/gene predictions. We studied modules related to pigmentation, a critical process for melanoma susceptibility; however, our approach may also be applied to other pathways and biological processes such as telomere maintenance, immune response, melanocyte differentiation, and cell adhesion that have been associated with melanoma risk (Landi MT et al., 2020). One caveat of the approach is the lack of rigorous statistical tests in gene/variant prioritization. Other limitations of our study include the inherent small number of patients in pedigree-based WES analyses and the lack of an independent dataset to replicate our results. In addition, the multiple genomic layers of information such as gene expression and WES data integrated onto the networks were not derived from the same set of samples. Further, functional characterization of the proposed candidate genes and understanding of their mechanisms of action are required to confirm whether they are indeed disease-causing.
In summary, using co-expression networks constructed from tissue-specific expression datasets and germline WES data as well as various gene/variant prioritization strategies, we identified potential risk genes for melanoma in our melanoma-prone families. Our study not only provides additional insight into melanoma susceptibility but also provides an alternative analytical perspective on gene prioritization in exome analyses of families with genetic heterogeneity. Further evaluation of candidate genes and validation in larger datasets are needed to confirm the relevance of these genes/variants in melanoma susceptibility.
MATERIALS AND METHODS
Exome analysis and variant filtering
All family members who were willing to participate in the study provided written informed consent under a National Cancer Institute (NCI) Institutional Review Board (IRB) approved protocol (NCT00040352; 02-C-0211). All methods were performed in accordance with the relevant guidelines and regulations.
WES of 34 families with at least three members affected with CMM per family (n=119 CMM patients) was performed at the Cancer Genomics Research Laboratory, National Cancer Institute (CGR, NCI). Details of the exome capture, WES, and bioinformatics pipeline used have been previously described (Goldstein AM, 2017; Pathak A et al., 2015; Yang XR et al., 2016). Briefly, exome sequencing was performed to a sufficient depth to achieve a minimum coverage of 15 reads in at least 80% of the coding sequence from the UCSC hg19 transcripts database. Variant discovery and genotype calling were performed globally using three variant callers (UnifiedGenotyper and HaplotypeCaller modules from GATK and FreeBayes). We included all targeted regions, as well as a 250-bp flanking region on each side. An Ensemble variant calling pipeline (v0.2.2) was then implemented to integrate the analysis results.
Genes were included for network reconstruction if they carry variants that met the following criteria: 1) had a minor allele frequency (MAF) of <0.001 in the 1000 Genomes Project, Exome Sequencing Project (ESP6500), and Exome Aggregation Consortium (ExAC); 2) were observed in ≤2 families from an in-house database (CGR, NCI) of ∼2000 exomes in ∼1000 cancer-prone families (excluding melanoma-prone or pancreatic cancer families); 3) were classified as non-synonymous including frameshift, stopgain, inframe deletion or insertion, missense, and splicing site variants. Variants flagged with our pipeline quality control metric (CScorefilter), with read depth < 10, ABHet < 0.2 or > 0.8, or called by only one of the three callers used were excluded. Resulting variants were then aggregated into genes for the subsequent network analysis.
Expression datasets and pre-processing
Since skin is a heterogeneous tissue and CMM is a complex disease influenced by its cellular microenvironment, we attempted to capture the actual cellular target of melanoma by using expression datasets of disease-relevant cell types in a complementary manner. The co-expression networks used for this analysis were constructed from the aggregated genes resulting from the WES analysis of our CMM families and gene expression information from an extensive collection of samples: normal skin, melanocyte cells culture, and CMM tissues (Figure 1). We downloaded the expression data for normal human skin from the GTEx project (http://www.gtexportal.org, sun-exposed skin n=605, and sun non-exposed skin n=516), primary melanocyte cultures from 106 newborns as previously reported (Zhang T et al., 2018), and skin cutaneous melanomas (SKCM) from TCGA (n=329) at the Genomic Data Commons (https://gdc.cancer.gov/access-data). Normalized gene read counts, Fragment/Reads Per Kilobase Million (FPKM) or Transcripts Per Kilobase Million (TPM) were used, and only protein-coding genes were kept. A set of covariates identified using the Probabilistic Estimation of Expression Residuals (PEER) method were calculated (Stegle et al., 2010) to control for confounding effects and hidden batch effects in expression datasets. The median absolute deviation (MAD) was used as a measure of variability, only the top 4000 most variable genes based on MAD from the expression datasets were used for network construction.
Weighted gene co-expression networks to identify gene co-expression modules
Co-expression analyses were performed using the WGCNA approach (Langfelder P, Horvath S, 2008). We first calculated Pearson correlation coefficients for all gene-gene pairs across all samples in the dataset and obtained the correlation matrix for all genes. The matrix of correlations was then converted to an adjacent matrix of connection strengths, the process uses the scale-free topology criterion (Zhang B, Horvath S. 2005) to select the soft threshold power (β), which removes the weakly correlated genes while retaining the stronger ones. Adjacent matrices were then converted to topological overlapping matrices. Then hierarchical clustering was used to make a cluster dendrogram with branches corresponding to gene co-expression modules. Modules were precisely defined using the Dynamic Hybrid branch cutting algorithm following these parameters: deep split = 4, maximum cut height = 0.95, minimal module size = 30 genes, and genes with similar expression profiles were classified into the same gene modules (Langfelder P et al., 2008). Figure 1 shows the study workflow.
To evaluate the functional relevance of the identified modules in relation to melanoma susceptibility and pigmentation processes and to rank genes within modules, we created a comprehensive list of genes that include known CMM susceptibility genes and genes that are related to pigmentation, which is a key process underlying melanoma biology. Specifically, we started by generating a list of genes located within GWAS loci, selected based on p < 2.3e−8 in a recent GWAS meta-analysis, which reported results from ~ 36,760 cases of melanoma and 375,188 controls (Landi MT et al., 2020). Loci previously identified in CMM GWAS (Macgregor S et al., 2011; Bishop, D. T et al., 2009; Barrett J. H et al., 2011; Law M. H et al., 2015; Duffy D. L et al., 2018), eQTLs, and a curated list of genes related to pigmentation and melanoma biology from GO term analysis, as well as known CMM high-, intermediate- and low-risk genes that had been identified by family studies and linkage analyses (Read J et al., 2016; Goldstein AM et al., 2017) were also included. Some genes are observed in more than one category. The list of above-mentioned genes, summarized as melanoma-related genes that were used for gene selection, is shown in Table S1.
GO Enrichment
To select modules with biological mechanisms associated with melanoma risk, such as melanin/pigmentation processes, module-specific enrichment was performed using GO enrichment analysis and the GOstats R package (Falcon S, Gentleman R 2007). Selection criteria for significance were set using a false discovery rate (FDR) and P-value less than 0.05.
Network properties
Gene significance
To study whether candidate genes are related to pigmentation, the GS for each gene was calculated. GS is defined as the absolute value of the correlation between the expression of each candidate gene and the eigengene summarizing the expression of five key pigmentation genes (TYR, TYRP1, MC1R, ASIP, and OCA2). These genes were selected based on previous evidence of their role in pigmentation and CMM risk.
Module membership also known as eigengene-based connectivity
Based on the eigenvectors of each module (the first principal component of each module’s gene expression matrix), we calculated the correlation of the expression of each gene with the corresponding module eigengene in each module (Langfelder P, Horvath S. 2008). kME is a property inherent to each gene of how tightly a particular gene fit into its module.
Network representations
We studied module topology by depicting edges and their corresponding nodes using the TOM (Zhang B, Horvath S.A 2005). Network depictions were performed in Gephi (Bastian M HS, Jacomy M, 2009).
Gene and variant prioritization in WES families
Variants were prioritized from 1) known melanoma and pigmentation-related genes included in Table S1 and found in the modules studied; 2) genes with the highest scores in network property analyses; and 3) all genes with cosegregating variants in families (variants shared by at least three affected members within a family). Variant filtering was based on frequency and pathogenicity criteria: (MAF of <0.001), high impact (frameshift indels, stop gain/loss, or known splice sites), and evidence of pathogenicity based on at least 2 in silico predictions algorithms (Meta Likelihood ratio: D, METASVM: D, and CADD: ≥ 20). The first two algorithms are Ensembl prediction scores that incorporate results from nine algorithms (SIFT, PolyPhen-2, GERP ++, Mutation Taster, Mutation Assessor, FATHMM, LRT, SiPhy, and PhyloP) and allele frequency (Dong C et al., 2015).
Supplementary Material
Figure S1. Soft-thresholding power screening. (a) sun-exposed skin β=5, (b) sun non-exposed skin β=5, (c) skin cutaneous melanomas β=3, and (d) primary melanocyte cultures β=7. R2 > 0.8 reached after the power calculations, indicating that the networks have the characteristics of scale-free topology.
Figure S2. Gene ontology (GO) enrichment analysis in pigmentation-related modules. (a) Top 10 significantly enriched terms for biological process, molecular function, and cellular component categories in GTEx sun-exposed and non-exposed skin. (b) Significant terms associated with pigmentation in selected modules from TCGA and the melanocyte cell line. P-values after adjustment for multiple comparisons.
Figure S3. Scatterplots showing the correlation between Connectivity (kme) and Gene Significance (GS)
Figure S4. Families connected by co-expressed genes in the pigmentation-related modules. Panel (a) Co-expressed genes identified in the pigmentation modules carried variants in the studied families. Vertices in the figure indicate genes identified with potential pathogenic and rare variants resulting from the gene prioritization process; some vertices are zoom in to shown gene names, squares indicate family IDs. Panel (b) shows some examples of gene variants co-segregated in families.
Figure S5. Frequencies of somatic single nucleotide variations (SNVs) and copy number alterations (SCNAs) in prioritized candidate genes. (a) SNVs and (b) SCNAs. Data are from the NCI’s Genomic Data Commons (GDC), including all tumors from The Cancer Genome Atlas (TCGA) and The Therapeutically Applicable Research to Generate Effective Treatments (TARGET) projects, 13,035 cases total, compared to TCGA-melanoma cases (SKCM, n=434 cases).
Table S1. List of melanoma-related genes
Table S2. List of all genes included in pigmentation-enriched modules
Table S3. Network metrics for GTEx sun exposed dataset
Table S4. Network metrics for GTEx sun non exposed dataset
Table S5. Network metrics for TCGA dataset
Table S6. Network metrics for primary melanocyte culture dataset
Table S7. Most relevant reference from literature on functional relevance of prioritized candidate genes
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the NIH, NCI, DCEG.
Abbreviations:
- WES
whole-exome sequencing
- TCGA
cancer genome atlas
- GTEx
genotype-tissue expression
- GWAS
genome-wide association studies
- CMM
cutaneous malignant melanoma
- MAF
minor allele frequency
- GO
gene ontology
- eQTLs
expression quantitative trait loci
- kME
eigengene-based connectivity
- TOM
topological overlap measure
- GS
gene significance
- WGCNA
weighted gene correlation network analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state no conflict of interest.
Data availability statement
Whole-exome sequencing data can be found at dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001177.v2.p1). RNA-seq expression data from 106 primary human melanocytes performed by Zhang T and colleagues can be found at dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001500.v1.p1). (Mailman M.D et al., 2007). All other Gene expression data are available from the GTEx portal (http://www.gtexportal.org) and Genomic Data Commons (https://portal.gdc.cancer.gov/).
REFERENCES
- Allouche J, Rachmin I, Adhikari K, Pardo LM, Lee JH, McConnell AM, et al. NNT mediates redox-dependent pigmentation via a UVB- and MITF-independent mechanism. Cell 2021;184(16):4268–83.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral DC, Seabra MC. The melanosome as a model to study organelle motility in mammals. Pigment Cell Res 2004;17(2):111–8. [DOI] [PubMed] [Google Scholar]
- Bastian M, Heymann S, Jacomy M. Gephi: an open source software for exploring and manipulating networks AAAI Publications, Third International AAAI Conference on Weblogs and Social Media. 2009. [Google Scholar]
- Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet 2009;41(8):920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese G, Bennett BJ, Orozco L, Kang HM, Eskin E, Dombret C, et al. Systems genetic analysis of osteoblast-lineage cells. PLoS Genet 2012;8(12):e1003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener J, Baggiolini A, Pernebrink M, Dalcher D, Lerra L, Cheng PF, et al. Epigenetic control of melanoma cell invasiveness by the stem cell factor SALL4. Nat Commun 2021;12(1):5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DL, Zhu G, Li X, Sanna M, Iles MM, Jacobs LC, et al. Novel pleiotropic risk loci for melanoma and nevus density implicate multiple biological pathways. Nat Commun 2018;9(1):4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007;23(2):257–8. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Xiao Y, Sampson J, Zhu B, Rotunno M, Bennett H, et al. Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum Mol Genet 2017;26(24):4886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Carlson JA, Slominski A. Role of TRPM in melanocytes and melanoma. Exp Dermatol 2012;21(9):650–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi MT, Bishop DT, MacGregor S, Machiela MJ, Stratigos AJ, Ghiorzo P, et al. Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet 2020;52(5):494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 2008;24(5):719–20. [DOI] [PubMed] [Google Scholar]
- Law MH, Bishop DT, Lee JE, Brossard M, Martin NG, Moses EK, et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet 2015;47(9):987–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor S, Montgomery GW, Liu JZ, Zhao ZZ, Henders AK, Stark M, et al. Genome-wide association study identifies a new melanoma susceptibility locus at 1q21.3. Nat Genet 2011;43(11):1114–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet 2007; 39(10):1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak A, Pemov A, McMaster ML, Dewan R, Ravichandran S, Pak E, et al. Juvenile myelomonocytic leukemia due to a germline CBL Y371C mutation: 35-year follow-up of a large family. Hum Genet 2015;134(7):775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read J, Wadt KA, Hayward NK. Melanoma genetics. J Med Genet 2016;53(1):1–14. [DOI] [PubMed] [Google Scholar]
- Stegle O, Parts L, Durbin R, Winn J. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 2010;6(5):e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulman EP, Tang XX, Allen C, Biegel JA, Pleasure DE, Brodeur GM, et al. ECK, a human EPH-related gene, maps to 1p36.1, a common region of alteration in human cancers. Genomics, 1997;40(2):371–74. [DOI] [PubMed] [Google Scholar]
- Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, Tsao H. EphA2 is a critical oncogene in melanoma. Oncogene 2011;30(50):4921–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam S, Võsa U, van der Graaf A, Franke L, de Magalhães JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform 2018;19(4):575–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gele M, Dynoodt P, Lambert J. Griscelli syndrome: a model system to study vesicular trafficking. Pigment Cell Melanoma Res 2009;22(3):268–82. [DOI] [PubMed] [Google Scholar]
- Yang D, Li Q, Shang R, Yao L, Wu L, Zhang M, et al. WNT4 secreted by tumor tissues promotes tumor progression in colorectal cancer by activation of the Wnt/β-catenin signalling pathway. J Exp Clin Cancer Res 2020;39(1):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XR, Rotunno M, Xiao Y, Ingvar C, Helgadottir H, Pastorino L, et al. Multiple rare variants in high-risk pancreatic cancer-related genes may increase risk for pancreatic cancer in a subset of patients with and without germline CDKN2A mutations. Hum Genet 2016;135(11):1241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 2005;4:Article 17. [DOI] [PubMed] [Google Scholar]
- Zhang T, Choi J, Kovacs MA, Shi J, Xu M, Goldstein AM, et al. Cell-type-specific eQTL of primary melanocytes facilitates identification of melanoma susceptibility genes. Genome Res 2018;28(11):1621–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Soft-thresholding power screening. (a) sun-exposed skin β=5, (b) sun non-exposed skin β=5, (c) skin cutaneous melanomas β=3, and (d) primary melanocyte cultures β=7. R2 > 0.8 reached after the power calculations, indicating that the networks have the characteristics of scale-free topology.
Figure S2. Gene ontology (GO) enrichment analysis in pigmentation-related modules. (a) Top 10 significantly enriched terms for biological process, molecular function, and cellular component categories in GTEx sun-exposed and non-exposed skin. (b) Significant terms associated with pigmentation in selected modules from TCGA and the melanocyte cell line. P-values after adjustment for multiple comparisons.
Figure S3. Scatterplots showing the correlation between Connectivity (kme) and Gene Significance (GS)
Figure S4. Families connected by co-expressed genes in the pigmentation-related modules. Panel (a) Co-expressed genes identified in the pigmentation modules carried variants in the studied families. Vertices in the figure indicate genes identified with potential pathogenic and rare variants resulting from the gene prioritization process; some vertices are zoom in to shown gene names, squares indicate family IDs. Panel (b) shows some examples of gene variants co-segregated in families.
Figure S5. Frequencies of somatic single nucleotide variations (SNVs) and copy number alterations (SCNAs) in prioritized candidate genes. (a) SNVs and (b) SCNAs. Data are from the NCI’s Genomic Data Commons (GDC), including all tumors from The Cancer Genome Atlas (TCGA) and The Therapeutically Applicable Research to Generate Effective Treatments (TARGET) projects, 13,035 cases total, compared to TCGA-melanoma cases (SKCM, n=434 cases).
Table S1. List of melanoma-related genes
Table S2. List of all genes included in pigmentation-enriched modules
Table S3. Network metrics for GTEx sun exposed dataset
Table S4. Network metrics for GTEx sun non exposed dataset
Table S5. Network metrics for TCGA dataset
Table S6. Network metrics for primary melanocyte culture dataset
Table S7. Most relevant reference from literature on functional relevance of prioritized candidate genes
Data Availability Statement
Whole-exome sequencing data can be found at dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001177.v2.p1). RNA-seq expression data from 106 primary human melanocytes performed by Zhang T and colleagues can be found at dbGaP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001500.v1.p1). (Mailman M.D et al., 2007). All other Gene expression data are available from the GTEx portal (http://www.gtexportal.org) and Genomic Data Commons (https://portal.gdc.cancer.gov/).