

Abstract
We analyzed the cross-reactivity of anti-drug antibodies (ADAs) against agalsidase-alfa and -beta from 49 patients with Fabry disease (FD) against the novel PEGylated enzyme pegunigalsidase-alfa (PRX-102). The affinity of purified anti-AGAL antibodies from pooled patient sera was significantly lower for PRX-102 compared to agalsidase-alfa and -beta (both p < 0.05). Pull-down experiments revealed the presence of masked epitopes on PRX-102, possibly due to PEGylation. ADA titers in serum (μg/mL) and corresponding inhibitory capacities against agalsidase-alfa and -beta were measured in male patients with FD, showing strong correlations (r2 = 0.9978 and 0.4930, both p < 0.001). Affinities of ADAs of individual patients against PRX-102 (Kd: 3.55 ± 2.72 μmol) were significantly lower compared to agalsidase alfa (Kd: 1.99 ± 1.26 μmol) and -beta (Kd: 2.18 ± 1.51 μmol) (both p < 0.0001). Cross-ELISAs supported the presence of masked epitopes on PRX-102. Importantly, inhibition measurements also revealed a 30% reduction in inhibitory capacity of pre-existing ADAs towards PRX-102. Enzyme-uptake experiments in AGAL-deficient EA.hy926 cells demonstrated less effects of ADAs on cellular PRX-102 uptake compared with agalsidase beta. We conclude that due to the reduced affinity of pre-existing ADAs against agalsidase-alfa or -beta, ADA-affected patients might benefit from a therapy switch to PRX-102, which is currently evaluated in clinical trials.
Key words: anti-drug antibodies, affinity, enzyme replacement therapy, Fabry disease, pegunigalsidase alfa, enzyme uptake
Graphical abstract
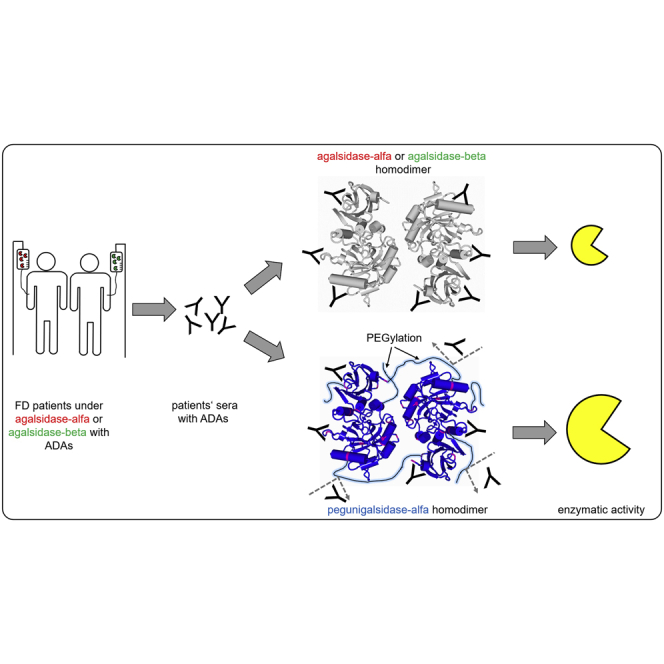
Neutralizing anti-drug antibodies lower the efficacy of enzyme replacement therapy in patients with Fabry disease. Here, Lenders et al. measured the cross-reactivity of individual ADAs from patients under agalsidase alfa or -beta against the novel component pegunigalsidase alfa, demonstrating a reduced affinity of existing ADAs against the new compound.
Introduction
Fabry disease (FD) is a rare X-linked lysosomal storage disease caused by a deficiency of the enzyme α-galactosidase A (AGAL; EC 3.2.1.22). The resulting enzyme deficiency leads to a progressive accumulation of the AGAL substrate globotriaosylceramide (Gb3), resulting in a multisystem disease with heart failure, cardiac arrhythmia, cerebrovascular events, and end-stage renal disease.1 Since 2001, FD is treatable by enzyme replacement therapy (ERT) using either agalsidase-alfa (0.2 mg/kg body weight every other week; Shire/Takeda) or -beta (1.0 mg/kg body weight every other week; Sanofi-Genzyme).2,3 Treatment with both compounds demonstrated beneficial effects on disease manifestation and progression in affected patients.4 However, classical male patients without cross-reactive immunologic material (i.e., lack of any endogenous AGAL protein, generally due to nonsense mutations) are under a high risk to form neutralizing anti-drug antibodies (ADAs) against both compounds, which significantly impairs the therapeutic efficacy of ERT.5, 6, 7, 8
Pegunigalsidase-alfa (PRX-102, Protalix BioTherapeutics, Chiesi Farmaceutici) is a PEGylated (polyethylene glycol [PEG]) and covalently cross-linked form of AGAL produced in tobacco cells and developed as potential novel ERT for FD.9, 10, 11 Currently, 10 clinical studies are being conducted to explore the safety and therapeutic efficacy of PRX-102 (https://clinicaltrials.gov/; data of last access: February 3, 2022). Importantly, preliminary studies on PRX-102 suggested less immunogenicity compared to agalsidase-alfa and -beta.11 This effect may be due to the prolonged stability and increased half-life of the enzyme in plasma, which is due to the PEGylation and stable cross-linked homodimerization. A reduced immunogenicity could lead to a better therapeutic effect in PRX-102-treated classical male ERT-naive patients and potentially in already treated patients with pre-existing ADAs, too, due to an absent immune response (anergy) in ERT-naive patients or by a tolerization, respectively. However, currently, it is unknown if pre-existing ADAs against agalsidase alfa and -beta will also recognize PRX-102 with comparable affinities, leading to similar enzyme inhibition and reduced cellular enzyme uptake, as well.
To our knowledge, we are the first to address this clinically relevant question with regard to future treatment of affected patients with FD with PRX-102. To this end, we measured and compared the individual affinities of existing ADAs against agalsidase alfa, agalsidase beta, and the new PRX-102 in a large cohort (n = 49) of classical male patients with FD with ADAs who were naive to PRX-102. In addition, we measured inhibitory capacities toward both approved agents and PRX-102 and analyzed the potential effects of pre-existing ADAs on cellular uptake of PRX-102 in endothelial cells.
Results
General ADA affinities against agalsidase-alfa, agalsidase-beta, and PRX-102
Recently, we successfully established a polyclonal human reference anti-AGAL antibody by appropriate immunoabsorption from sera of 22 male patients with FD with ADAs against infused AGAL.12 This antibody demonstrated comparable affinities for agalsidase-alfa and -beta. To analyze whether pre-existing anti-AGAL also recognizes the new potential second-generation ERT PRX-102 (pegunigalsidase-alfa, Chiesi Farmaceutici), we performed regular ELISA techniques against all three enzymes to determine ADA affinities. Interestingly, the reference antibody showed a significantly higher Kd value for PRX-102 (Kd: 1.86 ± 0.26 μM) compared with agalsidase-alfa (Kd: 0.99 ± 0.12 μM) and -beta (Kd: 1.21 ± 0.34 μM) and thus a higher affinity for both approved compounds (Figure 1A and 1B).
Figure 1.

Molecular characterization of a polyclonal human anti-AGAL antibody against 3 different recombinant α-galactosidase A compounds
(A and B) ELISA-based affinity measures versus agalsidase-alfa, agalsidase-beta, and pegunigalsidase-alfa (PRX-102). (C) Schematic overview of putative epitopes on the three AGALs (shown as monomers) and the potential impact of PEGylation on ADA binding. The PEGylation might form a shell around epitopes, resulting in a physical barrier. ADAs recognizing all three AGALs are highlighted in green. ADAs blocked by PEGylation are highlighted in black. (D) Workflow of the pull-down experiment and cross-over ELISA to detect the presence of agalsidase-beta-specific ADAs. (E) Pull-down experiment versus agalsidase-beta (Beta) and PRX-102. ADA, anti-drug antibodies. ∗p < 0.05, ∗∗p < 0.01.
Since PRX-102 is PEGylated, unlike agalsidase-alfa and -beta, some epitopes may be masked (by PEGylation), which could explain a lower affinity of pre-existing ADAs against PRX-102 (Figure 1C). To address this issue, we performed a pull-down experiment with the reference antibody in combination with PRX-102 and used the remaining unbound (and thus free) ADAs for subsequent ELISAs against agalsidase-beta and PRX-102 (Figure 1D). After pull-down, the remaining free ADAs did not recognize PRX-102 but did recognize agalsidase-beta (Figure 1E), demonstrating either the presence of agalsidase-beta-specific antibodies in pre-existing ADAs or the presence of masked epitopes by PEGylation of PRX-102.
Individual affinities against agalsidase-alfa, agalsidase-beta, and PRX-102
Reduced affinity of pre-existing ADAs against PRX-102 could lead to reduced antibody-mediated ERT inhibition on PRX-102 and eventually therapeutic benefit in affected patients. Thus, we aimed to analyze individual ADA affinities against agalsidase-alfa, agalsidase-beta, and PRX-102 in a cohort of 49 male patients with FD with pre-existing ADAs against agalsidase-alfa and -beta. To calculate appropriate Kd values, we first measured individual ADA concentrations expressed as μg anti-AGAL antibody/mL serum against agalsidase-alfa and -beta, based on our previously published method.12 As expected, patients showed comparable titers against both recombinant enzymes (r2 = 0.9978, p < 0.0001; Figure 2A). Furthermore, a higher ADA titer (μg/mL serum) was associated with a higher inhibitory capacity (mg enzyme) (r2 = 0.4930, p < 0.0001; Figure 2B). Of note, the inhibitory capacities against agalsidase-alfa and -beta were comparable, too (r2 = 0.9724; p < 0.0001; Figure S1). With known ADA concentrations, we next performed individual ELISA-based affinity assays against all three recombinant enzymes (agalsidase-alfa, agalsidase-beta, and PRX-102). The individual measurements for each patient are shown in the supplemental information (Figure S2). Overall, ADAs from the 49 patients showed lower affinities for PRX-102 (Kd: 3.55 ± 2.72 μmol) compared with agalsidase-alfa (Kd: 1.99 ± 1.26 μmol) and -beta (Kd: 2.18 ± 1.51 μmol) (both p < 0.0001; Figure 2E). However, we also identified some patients (n = 8; 16.3%) presenting with comparable or even higher affinities for PRX-102 (patient nos. 5, 8, 14, 30, 33, 39, 40, and 47).
Figure 2.

Antibody and affinity measurements in individual serum samples against agalsidase-alfa, agalsidase-beta, and pegunigalsidase-alfa
(A) Correlation of individual antibody titers against agalsidase-alfa and -beta. (B) Correlation of individual antibody titers (μg/mL serum) against agalsidase-beta versus the individual total inhibitory capacity (mg). (C and D) Representative ELISA-based affinity measures against 3 different enzymes. The horizontal lines mark respective Bmax values. (E) Comparison between individual Kd versus agalsidase-alfa, agalsidase-beta, and pegunigalsidase-alfa (PRX-102). ∗∗∗p < 0.001.
Masking of ADA epitopes by PEGylation
Next, we analyzed in sera from all patients whether PEGylation could reduce ADA binding to PRX-102 due to epitope masking, as demonstrated for the reference antibody. To analyze this adequately, we performed cross-ELISAs using supernatants from the individual affinity measurements against agalsidase-beta and PRX-102 (n = 35) in a second ELISA against agalsidase-beta and PRX-102 (Figure 3). All performed cross-ELISAs are provided in Figure 3B and within Figure S3. Overall, the cross-ELISAs showed comparable results to those of the reference antibody. In detail, samples initially incubated with PRX-102 showed lower Kd and thus higher affinity for agalsidase-beta than for PRX-102 (Kd: 0.63 [95% confidence interval (CI) 0.43–0.94] μL and Kd 1.20 [95% CI 0.68–2.13] mL, respectively; Figure 3C). Conversely, samples first incubated with agalsidase-beta showed the highest Kd and thus the lowest affinity for agalsidase-beta and PRX-102 (Kd: 1.81 [95% CI 0.99–3.35] μL and Kd: 2.36 [95% CI 1.00–6.12] μL, respectively; Figure 3C).
Figure 3.

ELISA-based detection of agalsidase-beta-specific antibodies
(A) Schematic overview of the workflow for cross-ELISAs to detect agalsidase-beta-specific antibodies (highlighted in green), which are blocked by PEGylation. (B) Representative cross-ELISA for one patient. (C) Summary of all performed cross-ELISAs (n = 35 patient samples) against agalsidase beta and PRX-102 with 95% confidence intervals. Beta, agalsidase beta; PRX, pegunigalsidase alfa.
Potential impact of PEGylation on inhibitory capacities
Previously, we demonstrated that ADA-mediated ERT inhibition results in a reduced therapeutic efficacy.6 However, patients with saturated ADAs (either due to less inhibitory capacity or higher infused dosages) showed an improved biochemical as well as clinical response compared with non-saturated patients.7 To analyze whether a reduced affinity for PRX-102 could also lead to a lower inhibitory capacity of pre-existing ADAs, we determined the individual inhibitory capacities of the patients’ ADAs against PRX-102 by titration and compared these values with the inhibitory capacity against agalsidase-beta. A selection of individual titration curves from 16 patients is provided in Figure S4.
Inhibitory capacities against agalsidase-beta positively correlated with inhibitory capacities against PRX-102 (r2 = 0.6221, p < 0.0001; Figure 4A). However, since we identified 8 (16.3%) patients with similar or increased binding affinities for PRX-102, we compared the inhibitory capacities of ADAs against agalsidase-beta and PRX-102 as a function of their respective affinities (Figures 4B and 4C). In detail, serum from patients with lower ADA binding affinities for PRX-102 showed a significantly decreased inhibitory capacity (−33.1%) against PRX-102 compared with agalsidase-beta (PRX-102: 43.9 [0.0–664.0] mg; agalsidase-beta: 66.6 [0.0–2741.0] mg; p < 0.0001; Figure 4B). In contrast, pre-existing ADAs with comparable or higher binding affinity for PRX-102 had overall similar inhibitory capacities against PRX-102 and agalsidase-beta (PRX-102: 79.7 [0.0–177.0] mg; agalsidase-beta: 86.1 [10.9–184.8] mg; p = 0.6908; Figure 4C). In fact, only 3 (37.5%) of these 8 patients had ADAs with higher inhibitory capacities against PRX-102; the remaining 5 patients showed comparable or less inhibitory capacities against PRX-102 (Figure 4C).
Figure 4.

Impact of ADAs on PRX-102 inhibition and cellular uptake
(A) Correlation between individual inhibitory capacities of pre-existing ADAs against agalsidase-beta and pegunigalsidase-alfa (PRX-102). Left: all values. Right: without the two patients with extreme high ADA titers. (B) Pre-existing ADAs with less affinity for PRX-102 have also less inhibitory capacities against PRX-102 compared to agalsidase-beta. (C) Pre-existing ADAs with comparable or higher affinity for PRX-102 have similar inhibitory capacities against PRX-102 and agalsidase-beta. (D) Impact of pre-existing ADAs on cellular AGAL uptake. (E) Impact of pre-existing ADAs on intracellular AGAL activities. Asterisks (∗) highlight differences between agalsidase-beta and PRX-102 with ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Hashtags (#) highlight differences between agalsidase-beta versus control and PRX-102 versus control with #p < 0.05, ##p < 0.01, ###p < 0.001.
Impact of pre-existing ADAs on cellular enzyme uptake
Depending on their epitopes, ADAs not only inhibit extracellular and intracellular enzyme activity but can also impair enzyme uptake.13 In this respect, we previously identified two patients with ADAs that decreased cellular agalsidase-beta uptake ex vivo.13 To investigate whether these ADAs have similar effects on PRX-102, we performed appropriate enzyme-uptake experiments (Figures 4D and 4E). In total, sera from 30 patients were tested. We identified 11 (36.7%) patients with ADAs significantly impairing PRX-102 uptake (Figures 4D) and 20 (66.6%) patients with ADAs significantly impairing agalsidase-beta uptake (Figure 4D), resulting in a significant difference (p = 0.0379; relative risk [RR]: 1.84 [95% CI 1.10–3.26]). Independent of these calculations, we identified 9 patients whose ADAs resulted in a significant difference between PRX-102 and agalsidase-beta uptake. In detail, ADAs of 8 (88.9%) patients had significantly less effect on PRX-102 uptake compared with agalsidase-beta (Figure 4D).
Furthermore, we identified 16 (53.3%) patients with ADAs significantly decreasing intracellular PRX-102 activity (Figures 4E) and 22 (73.3%) patients with ADAs significantly decreasing intracellular agalsidase-beta activity (Figure 4E). Although there seemed to be a trend, these differences were not significant (p = 0.1199; RR: 1.51 [95% CI 0.91–2.47]). Furthermore, we identified 2 (6.7%) patients with ADAs, which slightly increased intracellular agalsidase-beta activity, an effect not observed for PRX-102. Independent of these calculations, we identified 13 (43.3%) patients whose ADAs resulted in a significant difference between PRX-102 and agalsidase-beta intracellular AGAL activity. ADAs from 10 (76.9%) of these patients had less effects on intracellular PRX-102 activity (Figure 4E). Of note, individual ADA affinities or inhibitory capacities against agalsidase-beta or PRX-102 did not correlate with the effects on enzyme uptake or intracellular AGAL activities for both enzymes (data not shown).
Discussion
The formation of neutralizing ADAs against approved recombinant agalsidases in male patients with FD limits the therapeutic efficacy of ERT significantly. Thus, it is most important to establish novel treatment options with a reduced immunogenicity. Here, we characterized pre-existing ADAs against agalsidase-alfa and -beta in a cohort of classical male patients with FD compared with the novel potential next-generation ERT pegunigalsidase-alfa (PRX-102), which has a prolonged plasma half-life (∼80 h) due to PEGylation.9 In short, our main findings were that (1) pre-existing ADAs against agalsidase-alfa and -beta showed a lower affinity for PRX-102, (2) the lower affinity also resulted in a reduced inhibitory capacity and less impact on cellular uptake by ADAs, and (3) a reduced affinity could be explained by masked epitopes due to PEGylation rather than agalsidase-alfa- or -beta-specific epitopes of pre-existing ADAs.
Over the past two decades, the deleterious impact of neutralizing ADAs on therapy efficacy in FD became increasingly apparent.4,14 To overcome this therapeutic dilemma, either specific immunomodulatory protocols or novel treatment options including next-generation ERTs need to be implemented.
PRX-102 is a novel recombinant human AGAL produced in Tobacco protoplast and is currently in phase III clinical trials. One unique feature of PRX-102 is its increased half-life and stability due to PEGylation and cross-linking of the homodimer,9 potentially leading to a reduced immune response in classical male patients with FD, who are at highest risk for ADA formation. In view of a possible future treatment with PRX-102, it is important to characterize the immunogenicity of PRX-102 against pre-existing ADAs directed toward agalsidase-alfa and -beta. This will be particularly important if affected patients will be switched from one of the two currently approved enzymes to PRX-102.
Our affinity assays using a representative polyclonal human anti-AGAL antibody pool12 and individual serum samples from 49 patients with pre-existing ADAs against current ERTs demonstrated an on average 1.8-fold lower affinity for PRX-102. In line with these observations, our pull-down experiment with the reference antibody and the cross-ELISAs demonstrated that not all pre-existing ADAs recognize PRX-102. Since all three enzymes share the same amino acid sequence of human AGAL, the presence of agalsidase-alfa- and -beta-specific antibodies seems unlikely. Rather, the presence of masked epitopes by PEGylation could be assumed. This observation is in accordance with previous studies demonstrating that PEGylation reduces protein immunogenicity by forming a “shell” around the enzyme that masks antigenic determinants by presenting a flexible, unbranched hydrophilic surface.15 Our results were further supported by the fact that pre-existing ADAs had less enzyme inhibitory effects against PRX-102 than against agalsidase-beta, which might be explained by the reduced affinity and thus less ADA binding against PRX-102. Interestingly, 8 of 49 patients showed comparable or even higher ADA affinities for PRX-102 compared with agalsidase-beta. Nevertheless, only three of these eight patients (37.5%) showed a higher inhibitory capacity of PRX-102. Since PEG is generally not immunogenic16 and antibodies to PEG can only be formed when PEG residues are combined with highly immunologic proteins,17 pre-existing anti-PEG antibodies in our patients, which were naive to PRX-102 (and other PEGylated drugs), are unlikely. Rather, it could be assumed that these patients have formed (more) antibodies, which better recognize exposed epitopes on PRX-102 not masked by PEGylation. Future studies are now warranted to analyze this in more detail using appropriate epitope mapping. Therefore, treatment with PRX-102 could be a promising therapeutic option for the majority of patients with pre-existing ADAs, but patients should be tested (for ADAs and their affinities against recombinant AGALs) before the switch in order to better predict the therapeutic success.
Both agalsidase-alfa and -beta uptake is mannose-6-phosphate (M6P)-receptor dependent. In a previous study, we demonstrated that some patients may have ADAs that interfere with cellular AGAL uptake, possibly by blocking M6P-mediated mechanisms (i.e., by masking M6P residues on AGAL). These ADAs had slightly less effect on the cellular PRX-102 uptake, which is consistent with a previous study suggesting that PRX-102 uptake is independent of M6P and mannose receptors.9 More importantly, the intracellular AGAL activity of PRX-102 was markedly increased, further highlighting the reduced inhibitory effect of pre-existing anti-AGAL antibodies on PRX-102 and the protective effect of PEGylation.16 Of note, we identified 2 patients with ADAs slightly increasing intracellular agalsidase-beta activity compared with the control situation. It could theoretically be that these 2 patients have (specific) antibodies that result in larger ADA/AGAL complexes and thus more agalsidase-beta being internalized. However, it is more likely that the antibodies stabilize the enzyme through their binding, for example, by cross-linking the AGAL dimer, which could lead to increased activity and stability (at least intracellularly). Further studies are now needed to analyze whether there are more patients with similar antibodies showing comparable effects and to elucidate the underlying mechanism.
Overall, we demonstrated that antibody titers against PRX-102 in PRX-102-naive patients can be measured using the same established methods as for agalsidase-alfa or -beta. However, as demonstrated, ADAs against current ERTs had some, but not full, cross-reactivity to PRX-102. Furthermore, due to the long half-life of PRX-102, ADA measurements in patients treated by PRX-102 could need additional modifications due to interfering long-lasting PRX-102/ADA complexes.4 In addition to classical ELISAs (or inhibition assays), which measure the free and thus unbound antibodies within the serum, modified ELISAs also need to be implemented, which are either capable of detecting PRX-102/ADA complexes within the serum or require special sample preparation to dissociate existing ADA/AGAL complexes. Scientists and laboratories should be aware that due to a lower affinity, antibody titers and inhibitory capacities in patients with pre-existing ADAs in PRX-102-naive patients will be measured lower against PRX-102 than against the two other enzymes, which could lead to false negative results. This will be of particular interest for the upcoming three phase III trials for PRX-102, especially when analyzing patients with pre-existing ADAs. Therefore, it might be advisable to measure ADA titers against PRX-102 and at least agalsidase-alfa or -beta in parallel in these patients.
Another ADA-mediated mechanism that could efficiently lower the therapeutic efficacy of ERT is the clearance of ADA/AGAL complexes from circulation. In detail, complexes can be recognized by macrophages via the Fcγ-receptor and thus eliminated during infusions. In this respect, preliminary data demonstrated that AGAL activity (and therefore the AGAL amount) was increased in leucocytes in one single patient with ADAs compared with three patients negative for ADAs.5 However, additional studies with more patients are needed to confirm this and to compare the corresponding effect to the direct enzyme inhibition during infusion and ADA-mediated uptake inhibition of ERT via the M6P receptor.
In conclusion, we observed lower affinity and enzyme inhibition (including activity and cellular uptake) of pre-existing ADAs against PRX-102, resulting in a decreased ADA-mediated enzyme inhibition and reduced ADA interference on cellular enzyme uptake. Therefore, treatment with PRX-102 could be a promising therapeutic option for the majority of patients with pre-existing ADAs, but patients should be tested before the switch in order to better predict the therapeutic success. However, consistent with general recommendations for early treatment in FD, a therapy switch is likely to be most effective when irreversible organ damage, including fibrosis, has not yet occurred.
Limitations of the study
Cross-ELISAs were performed with supernatants from previous affinity ELISAs and not individually titrated, which implies that in some cases of high ADA titers, the coating amount of 100 ng agalsidase-beta or PRX-102 was not sufficient to bind all free antibodies. However, the combined data of all cross-ELISAs (n = 35) support the observations made for the reference antibody, suggesting the presence of masked epitopes by PEGylation. Future studies are now needed to analyze the effects of ADAs on enzyme uptake and intracellular AGAL activity in more detail and to establish a cut-off, based on relevant biochemical differences in terms of intracellular Gb3 depletion.
Material and methods
Patients
All investigations were performed after approval by the Medical Association of Westphalian-Lippe and the Ethics Committee of the Medical Faculty of the University of Muenster (project no. 2011-347-f, date of report: July 7, 2011) and in accordance with the Declaration of Helsinki. Written informed consent was obtained from all included patients for analysis and publication. Serum from a healthy individual without ADAs against AGAL was used as control.
ELISA-based measurements of pre-existing ADA titers
ADA titers were measured as recently described.12 In short, wells were pre-coated with either 100 ng agalsidase-alfa or -beta, blocked with 2% BSA, and incubated overnight at 4°C with serial dilutions of raw sera. A serial dilution of the anti-AGAL reference antibody was used as reference. Wells were washed five times with 0.1% Tween 20/phosphate-buffered saline (PBS), and anti-human immunoglobulin G (hIgG) antibodies conjugated with HRP (ab98624, Abcam; working concentration: 20 ng/mL) were used for 1 h at room temperature followed by five washing steps with 0.1% Tween-20/PBS. For IgG detection, 50 μL 1-Step TMB-ELISA substrate solution (Thermo Fisher Scientific) was added to the wells, followed by 50 μL 2 M sulfuric acid to stop the reaction after 15 to 30 min. Absorption was measured at 450 nm. To calculate ADA concentrations, linear regressions within serial dilutions of patients’ sera were applied. Finally, the concentrations were calculated using the equation obtained from the anti-AGAL antibody reference curve.
ELISA-based affinity assays
Wells were pre-coated with 100 ng agalsidase-alfa, agalsidase-beta, or PRX-102 overnight at 4°C and subsequently blocked with 2% BSA in PBS. Serial dilutions of raw sera were applied on wells and incubated overnight at 4°C. After five washing steps with 0.1% Tween 20/PBS, anti-hIgG antibodies conjugated with HRP (ab98624, Abcam; working concentration: 20 ng/mL) were applied and incubated for 1 h at room temperature. Wells were washed again five times with 0.1% Tween 20/PBS. For IgG detection, 50 μL 1-Step TMB-ELISA substrate solution (Thermo Fisher Scientific) was added to the wells, followed by 50 μL 2 M sulfuric acid to stop the reaction after 15 to 30 min. Absorption was measured at 450 nm.
PRX-102-mediated reference antibody pull-down and subsequent ELISAs
Two μg PRX-102 (provided by Chiesi Farmaceutici) was pre-incubated with 100 ng reference anti-AGAL antibodies12 for 4 h at room temperature. Subsequently, remaining free ADAs, which did not recognize PRX-102, were purified using MelonGel (Invitrogen, Darmstadt, Germany) as previously described.7,18 Finally, serial dilutions of purified ADAs were used in ELISAs against agalsidase-beta and PRX-102 as described above.
Cross-ELISAs to detect non-PRX-102 recognizing ADAs
Supernatants from ADA affinity measurements against agalsidase-beta and PRX-102 were transferred on 96 wells pre-coated with 100 ng agalsidase-beta or PRX-102 and additionally incubated overnight at 4°C. Again, ADA detection was performed as described above using an HRP-linked anti-hIgG antibody (ab98624, Abcam).
Titration of neutralizing ADAs to assess inhibitory capacities
The amount of enzymes (agalsidase-alfa, agalsidase-beta, or PRX-102) required to saturate ADAs in patients’ sera was determined as described previously.7,18 In short, 5 μg patients’ purified total IgGs were pre-incubated with a serial dilution of AGAL enzymes (agalsidase-alfa, agalsidase-beta, or PRX-102) for 10 min at room temperature. To express enzyme inhibition in a percentage, residual AGAL activities were normalized against inhibition-negative controls. Enzyme inhibition was plotted against the amount of agalsidase, and saturation was defined as the amount of enzyme required to reduce the neutralizing capacity of 5 μg patients’ total IgG below the ERT neutralizing threshold of 10% (background threshold).7,18
Endothelial uptake assays
AGAL-deficient EA.hy926 cells were seeded on 96-well plates (plate reader analyses) with a density of 2 × 105 cells/mL and grown until confluence. To determine the effect of neutralizing ADAs on cellular AGAL uptake, 10 μL patient and control sera were pre-incubated with 5 μg/mL AGAL (agalsidase-beta or PRX-102, respectively) for 10 min at room temperature. Subsequently, mixtures were added to cells and incubated for 4 h at 37°C. Cells were washed with PBS. For further subsequent enzyme activity assays, cells were lysed with 30 μL 1x Passive Lyse Buffer (Promega, Wisconsin, USA, E194A), and AGAL activities were determined as described above, normalized for protein concentrations. Uptake quantification was performed by ELISAs, which were performed as previously described.13 96-well plates were coated with a capture antibody (α-AGAL, R&D Systems, Minneapolis, MN, USA; no. AF6146, 2 μg/mL) overnight at 4°C. After washing and blocking, 10 μL cell lysates within 100 μL PBS were applied. For AGAL detection, anti-AGAL antibodies in PBS/BSA 2% (Abcam; no. ab168341, 0.33 μg/mL) were added for 2 h at room temperature, and subsequently secondary antibodies (anti-rabbit IgG; Merck, Darmstadt, Germany; no. 12-348, 33 ng/mL) were applied for 1 h at room temperature. After additional washes, 50 μL TMB-ELISA Substrate Solution (Thermo Fisher Scientific) was added to each well, incubated for 30 min, and stopped with 50 μL 2 M sulphuric acid. Absorption was measured at 450 nm and normalized for protein concentration.
Statistical analyses
Unless otherwise indicated, experiments were performed in triplets. Experiments with the reference antibody and cell culture experiments were replicated three times. Categorical data were expressed as numbers and relative frequencies as percentages. Two-tailed Student’s t tests or one-way analyses of variance (ANOVAs) with Dunnett’s multiple comparison tests were used to compare ADA-mediated effects on agalsidase beta and PRX-102 uptake and intracellular activity, separately. To further assess differences between PRX-102 and agalsidase beta uptake and intracellular activity, we used one-way ANOVA with post-hoc multiple comparisons. Statistical significance was considered at a two-sided p <0.05. One-site non-linear fitting models were used to calculate Bmax and Kds. GraphPad Prism v.5.0 software (GraphPad Software, La Jolla, CA, USA) was used for appropriate statistical analyses and visualization.
Data availability section
This study includes no data deposited in external repositories.
Acknowledgments
We thank the patients for their participation and for providing serum samples that made this work possible. The technical assistance of Samira Schiwek, Birgit Orlowski, and Anne Huster is gratefully acknowledged.
Author contributions
M.L. designed the concept and methodology. M.L., M.T., and S.P. conducted the experiments. M.L. wrote the paper. E.B. reviewed and edited the manuscript. M.L. and E.B. provided resources. All authors analyzed and interpreted the data and revised the manuscript critically for important intellectual content. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
Parts of this work were supported by the fund “Innovative Medical Research” of the University of Muenster Medical School (LE221801) and by Chiesi GmbH, Germany. M.L. received speaker honoraria, travel funding, and research grants from Sanofi Genzyme, Shire Corporation/Takeda, Amicus Therapeutics, and Chiesi GmbH. E.B. received research grants and speaker honoraria from Sanofi Genzyme, Shire Corporation/Takeda, Amicus Therapeutics, Chiesi GmbH, and Eleva. S.P. and M.T. have nothing to declare.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.07.009.
Supplemental information
References
- 1.Zarate Y.A., Hopkin R.J. Fabry's disease. Lancet. 2008;372:1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]
- 2.Eng C.M., Guffon N., Wilcox W.R., Germain D.P., Lee P., Waldek S., Caplan L., Linthorst G.E., Desnick R.J., International Collaborative Fabry Disease Study Group Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease. N. Engl. J. Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- 3.Schiffmann R., Kopp J.B., Austin H.A., 3rd, Sabnis S., Moore D.F., Weibel T., Balow J.E., Brady R.O. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- 4.Lenders M., Brand E. Mechanisms of neutralizing anti-drug antibody formation and clinical relevance on therapeutic efficacy of enzyme replacement therapies in Fabry disease. Drugs. 2021;81:1969–1981. doi: 10.1007/s40265-021-01621-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linthorst G.E., Hollak C.E.M., Donker-Koopman W.E., Strijland A., Aerts J.M.F.G. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- 6.Lenders M., Stypmann J., Duning T., Schmitz B., Brand S.M., Brand E. Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J. Am. Soc. Nephrol. 2016;27:256–264. doi: 10.1681/ASN.2014121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenders M., Neußer L.P., Rudnicki M., Nordbeck P., Canaan-Kühl S., Nowak A., Cybulla M., Schmitz B., Lukas J., Wanner C., et al. Dose-dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with Fabry disease. J. Am. Soc. Nephrol. 2018;29:2879–2889. doi: 10.1681/ASN.2018070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Veen S.J., van Kuilenburg A.B.P., Hollak C.E.M., Kaijen P.H.P., Voorberg J., Langeveld M. Antibodies against recombinant alpha-galactosidase A in Fabry disease: subclass analysis and impact on response to treatment. Mol. Genet. Metabol. 2019;126:162–168. doi: 10.1016/j.ymgme.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Kizhner T., Azulay Y., Hainrichson M., Tekoah Y., Arvatz G., Shulman A., Ruderfer I., Aviezer D., Shaaltiel Y. Characterization of a chemically modified plant cell culture expressed human α-Galactosidase-A enzyme for treatment of Fabry disease. Mol. Genet. Metabol. 2015;114:259–267. doi: 10.1016/j.ymgme.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Ruderfer I., Shulman A., Kizhner T., Azulay Y., Nataf Y., Tekoah Y., Shaaltiel Y. Development and analytical characterization of pegunigalsidase alfa, a chemically cross-linked plant recombinant human α-galactosidase-A for treatment of Fabry disease. Bioconjug. Chem. 2018;29:1630–1639. doi: 10.1021/acs.bioconjchem.8b00133. [DOI] [PubMed] [Google Scholar]
- 11.Schiffmann R., Goker-Alpan O., Holida M., Giraldo P., Barisoni L., Colvin R.B., Jennette C.J., Maegawa G., Boyadjiev S.A., Gonzalez D., et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1-year Phase 1/2 clinical trial. J. Inherit. Metab. Dis. 2019;42:534–544. doi: 10.1002/jimd.12080. [DOI] [PubMed] [Google Scholar]
- 12.Lenders M., Scharnetzki D., Heidari A., Di Iorio D., Wegner S.V., Brand E. Generation and characterization of a polyclonal human reference antibody to measure anti-drug antibody titers in patients with Fabry disease. Int. J. Mol. Sci. 2021;22:2680. doi: 10.3390/ijms22052680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stappers F., Scharnetzki D., Schmitz B., Manikowski D., Brand S.M., Grobe K., Lenders M., Brand E. Neutralising anti-drug antibodies in Fabry disease can inhibit endothelial enzyme uptake and activity. J. Inherit. Metab. Dis. 2020;43:334–347. doi: 10.1002/jimd.12176. [DOI] [PubMed] [Google Scholar]
- 14.Lenders M., Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. J. Am. Soc. Nephrol. 2018;29:2265–2278. doi: 10.1681/ASN.2018030329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abuchowski A., McCoy J.R., Palczuk N.C., van Es T., Davis F.F. Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J. Biol. Chem. 1977;252:3582–3586. [PubMed] [Google Scholar]
- 16.Harris J.M., Chess R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 17.Richter A.W., Akerblom E. Antibodies against polyethylene glycol produced in animals by immunization with monomethoxy polyethylene glycol modified proteins. Int. Arch. Allergy Appl. Immunol. 1983;70:124–131. doi: 10.1159/000233309. [DOI] [PubMed] [Google Scholar]
- 18.Lenders M., Schmitz B., Brand S.M., Foell D., Brand E. Characterization of drug-neutralizing antibodies in patients with Fabry disease during infusion. J. Allergy Clin. Immunol. 2018;141:2289–2292.e7. doi: 10.1016/j.jaci.2017.12.1001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study includes no data deposited in external repositories.