

Summary
Many insects depend on ancient associations with intracellular bacteria for essential nutrition. The genomes of these bacteria are often highly reduced. Although drift is a major driver of symbiont evolution, other evolutionary forces continue to influence them. To understand how ongoing molecular evolution and gene loss shape symbiont genomes, we sequenced two of the most ancient symbionts known, Sulcia and Nasuia, from 20 Hawaiian Nesophrosyne leafhoppers. We leveraged the parallel divergence of Nesophrosyne lineages throughout Hawaii as a natural experimental framework. Sulcia and Nasuia experience ongoing—but divergent—gene loss, often in a convergent fashion. Although some genes are under relaxed selection, purifying and positive selection are also important drivers of genome evolution, particularly in maintaining certain nutritional and cellular functions. Our results further demonstrate that symbionts experience dramatically different evolutionary environments, as evidenced by the finding that Sulcia and Nasuia have one of the slowest and fastest rates of molecular evolution known.
Subject areas: Bacteriology, Entomology, evolutionary biology
Graphical abstract
Highlights
-
•
Ancient, reduced symbiont genomes are evolutionarily dynamic
-
•
Leafhopper symbionts have two of the slowest and fastest molecular evolution rates
-
•
Shared symbionts exhibit divergent and convergent gene losses among related hosts
-
•
Multiple symbionts in the same host experience different evolutionary pressures
Bacteriology; Entomology; Evolutionary biology.
Introduction
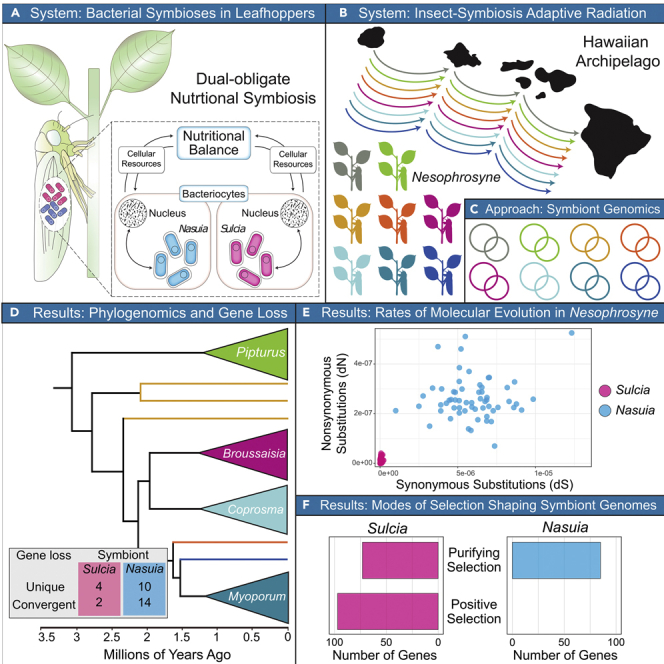
Bacterial symbionts have enabled many animal groups to take advantage of unsuitable ecological niches leading to their biological diversification (Takiya et al., 2006; Moran, 2007; Sudakaran et al., 2015; Hendry et al., 2016; Sogin et al., 2020; Myers et al., 2021). In insects, bacterial endosymbionts are a key source of essential nutrition for many species, and even entire orders, that specialize in diets limited in essential nutritional resources (Moran, 1996; Baumann, 2005; Douglas, 2009). These symbionts are generally restricted to within specialized insect organs (bacteriomes) and cells (bacteriocytes) that enable host-bacterial interaction and strict vertical transmission from mother to offspring (Buchner, 1965; Koga et al., 2012). However, owing to their intracellular lifestyle, bacteria often lose over 90% of their genes (McCutcheon and Moran, 2012; McCutcheon et al., 2019). As symbioses age, bacteria continue to experience ongoing gene losses from their most basic—and essential—cellular processes and metabolisms (e.g., DNA replication and repair). Thus, endosymbionts require extensive resources from their hosts and other bacterial partners to function (Nakabachi et al., 2005; Hansen and Moran, 2011; McCutcheon and von Dohlen, 2011; Sloan et al., 2014; Luan et al., 2015; Mao et al., 2018; Weglarz et al., 2018; Kobiałka et al., 2018). Although we have a good picture of how symbiont genomes shrink on the scale of 10-100s millions years, we understand comparatively little about how this process continues to shape the tiny genomes of ancient symbionts among closely related host species (Wernegreen, 2002; McCutcheon et al., 2009a, 2009b; Moran et al., 2009; Mao et al., 2017; Chong et al., 2019).
In many cases, our understanding and inference of the evolutionary processes that influence the structure and function of symbiont genomes are derived from a single, or just a few, representative genomes (McCutcheon et al., 2009a, 2009b; Bennett and Moran, 2013; Koga and Moran, 2014; Bennett and Mao, 2018; Michalik et al., 2021). These processes include selection to retain essential functions, selection to adapt to changing host and environmental conditions, and strong genetic drift owing to small population sizes and strong intergenerational bottlenecks (Moran, 1996; Wernegreen, 2002; Woolfit and Bromham, 2003; Campbell et al., 2015; Perreau et al., 2021). These processes are ongoing among related symbiont lineages that are separated into distinct host species and their populations. The intensity of these forces may vary depending on the bacterial symbiotic roles and cellular environments, as well as the biology and ecology of their different host insect species (Wernegreen, 2002; Sabater-Muñoz et al., 2017; Chong et al., 2019). As a result, there is likely to be tremendous variation among the symbiont genomes of closely related host species. For example, the process of drift is known to cause independent symbiont lineages to differ widely in their genetic capabilities even between closely related host species (McCutcheon and Moran, 2010; Patiño-Navarrete et al., 2013; Husnik et al., 2013; Campbell et al., 2015, 2017; Bennett et al., 2016b; Husnik and McCutcheon, 2016; Boscaro et al., 2017; Łukasik et al., 2018; Chong et al., 2019; Monnin et al., 2020; Santos-Garcia et al., 2020). It is less clear how drift and selection work together to shape and maintain the genes and functions of symbionts as they diversify along with their hosts. Thus, an investigation into the patterns of gene loss and molecular evolution (e.g., the relative roles of selection vs. drift in gene evolution) among closely related host sister species has the potential to illuminate the fine-scale evolutionary processes that underlie ongoing symbiont genome evolution and diversification.
To better understand the evolutionary processes that shape ancient symbiont genome evolution, we analyzed the genomes of “Candidatus Sulcia muelleri” (Bacteroidetes; hereafter referred to as Sulcia) and “Ca. Nasuia deltocephalinicola” (Betaproteobacteria; hereafter Nasuia) lineages from insect sister species belonging to the endemic Hawaiian leafhopper genus, Nesophrosyne (Hemiptera: Auchenorrhyncha: Cicadellidae). Nesophrosyne is one of the largest insect adaptive radiations in Hawaii (Hembry et al., 2021). It originated ∼3.2 million years ago, rapidly diversifying into over 200 species that specialize in endemic plant species in a one-to-one fashion (Zimmerman, 1948; Bennett and O’Grady, 2012, 2013). Nesophrosyne lineages established these host-plant relationships early in their diversification on the archipelago. These lineages then maintained their host-plant associations as they diversified across newly formed islands in a parallel and replicated fashion (Bennett and O’Grady, 2013). Thus, Nesophrosyne can provide a natural evolutionary experiment to test questions of how evolutionary processes shape symbiont genome evolution across diverging insect lineages and to further understand to what extent evolutionary processes are predictable.
Sulcia and Nasuia, like most other symbionts in the Auchenorrhyncha suborder, complement each other to provide their hosts with the 10 essential amino acids (EAAs) that are lacking in their xylem and phloem plant sap diets (Bennett and Moran, 2013). Genomic evidence suggests that both bacteria are ancient, having partnered with insects ∼300 million years ago (Moran et al., 2005; Bennett and Mao, 2018). As a result, Sulcia and Nasuia have highly reduced genomes of 190 kilobases (kb) and 112 kb, respectively (Bennett and Moran, 2013). Both genomes maintain a core set of essential nutritional genes, but are lacking genes in most other essential functions that include translation and transcription, energy synthesis, and DNA replication and repair (Bennett and Moran, 2013; Bennett et al., 2014; Mao et al., 2017).
Here, we leverage the Nesophrosyne leafhopper radiation and their endosymbionts to understand (i.) whether and how ongoing gene losses continue to shape ancient symbiont genomes, (ii.) how selection and drift lead to symbiont genome diversification, and (iii.) whether these evolutionary forces are shared or distinct between multiple symbionts in a shared host. We further test our questions in absolute time provided by the Nesophrosyne radiation to gain general insights into the tempo and mode of bacterial symbiont genome evolution.
Results and discussion
Host-symbiont taxon sampling and nomenclature for genomic analysis
To compare the genomes of symbionts associated with Nesophrosyne, we strategically sub-sampled 20 species that span the ecological and phylogenetic diversity of the leafhopper genus. Our selected species comprise monophyletic groups that specialize in eight distinct endemic Hawaiian plants, encompassing the diversity of host-plant associations known in the Nesophrosyne (Bennett and O’Grady, 2012; see Figure 1, Figure 2 and 2). The sister species of each host lineage diversified across the Hawaiian Archipelago, with distinct species occurring on each island and even on a volcanic mountain (Bennett and O’Grady, 2013).
Figure 1.

Convergent and unique gene loss among Sulcia genomes from endemic Hawaiian leafhoppers (Nesophrosyne)
Patterns of gene loss were estimated with maximum likelihood ancestral state reconstruction on an absolute time-calibrated phylogenetic tree in millions of years, using complete host mitochondrial genes (see Supplemental information). Convergent gene losses (i.e., multiple repeated losses) are indicated by blue boxes. Unique gene losses (i.e., losses that occurred once) are indicated by yellow boxes. Shorthand gene names are provided in each box. Posterior support for each node is shown as colored circles as follows: black >95, gray = 90-95, and white <90. See section methods for details on tree search parameters. See Table 1 for species shorthand nomenclature. MYA = million years ago.
Figure 2.

Convergent and unique gene loss among Nasuia genomes from endemic Hawaiian leafhoppers (Nesophrosyne)
Patterns of gene loss were estimated with maximum likelihood ancestral state reconstruction on an absolute time-calibrated phylogenetic tree in millions of years, using complete host mitochondrial genes (see Supplemental information). Convergent gene losses (i.e., multiple repeated losses) are indicated by blue boxes. Unique gene losses (i.e., losses that occurred once) are indicated by yellow boxes. Shorthand gene names are provided in each box. Posterior probability support for each node is shown as colored circles as follows: black >95, gray = 90-95, and white <90. See section methods for details on tree search parameters. See Table 1 for species shorthand nomenclature. MYA = million years ago. hypo = hypothetical protein.
Sulcia and Nasuia strains are hereafter identified by the island and host plant genera associated with the leafhopper host as follows: the initial for the island location (e.g., Hawaiʻi Island = HI) and the first two letters of the plant genus (e.g., Pipturus = PI). For example, the Sulcia strain associated with the Nesophrosyne species restricted to Hawaiʻi Island and the plant genus Pipturus is referred to as Sulcia-HIPI (Table 1).
Table 1.
Shorthand nomenclature used for bacterial symbiont strains
| Host species | Island location | Habitat type | Host-plant group | Shorthand naming |
|---|---|---|---|---|
| N. sp. 295 | East Hawaiʻi Island | Rain Forest | Broussaisia | EHBR |
| N. ogradyi | East Maui Island | EMBR | ||
| N. kanawao | North Hawaiʻi Island | NHBR | ||
| N. makaihe | West Oʻahu Island | WOBR | ||
| N. sp. 23 | Kauaʻi Island | Rain Forest | Clermontia | KICL |
| N. haleakala | Haleakalā Mtn, Maui | Spanning all habitats | Coprosma | HMCO |
| N. sp. 302 | Hawaiʻi Island | HICO | ||
| N. sp. 58 | Maui Island | MICO | ||
| N. sp. 29 | Kauaʻi Island | Dry-Mesic | Dodonea | KIDO |
| N. maratima | Oʻahu Island | OIDO | ||
| N. sp. 281 | Hawaiʻi Island | Spanning all habitats | Myoporum | HIMY |
| N. sp. 126 | Kauaʻi Island | KIMY | ||
| N. sp. 242 | Molokaʻi Island | MIMY | ||
| N. sp. 246 | Oʻahu Island | OIMY | ||
| N. montium | Hawaiʻi Island | Rain Forest | Pipturus | HIPI |
| N. sp. 17 | Kauaʻi Island | KIPI | ||
| N. sp. 48 | Maui Island | MIPI | ||
| N. ponapona | Oʻahu Island | OIPI | ||
| N. sp. 15 | Molokaʻi Island | Dry-Mesic | Psychotria | MIPS |
| N. sp. 21 | Kauaʻi Island | Spanning all habitats | Scaevola | KISC |
Shorthand naming are the first letters of the island location and the first two letters of the host-plant group. Habitat type of each host-plant group is added as well. (N. = Nesophrosyne, sp. = species).
Phylogenetic relationships among Nesophrosyne and their symbionts are congruent
To test our questions in absolute time, we generated a time-calibrated phylogeny from complete Nesophrosyne mitochondrial genomes (15 genes, 14,304 sites) using absolute time calibration points determined previously by Bennett and O’Grady (2013). To compare phylogenetic topologies of the host and symbionts, we also reconstructed phylogenies for Nasuia (99 genes, 86,095 sites) and Sulcia (184 genes, 181,781 sites) independently using their complete genomes. The relationships among our sub-sampled host species agree with previous phylogenetic work and there is strong support for the monophyly of leafhoppers associated with their host-plant groups (Bennett and O’Grady, 2012, 2013; see Figure S1). However, two Nesophrosyne species are weakly supported in their placements and vary between trees derived from the symbiont and mitochondrial datasets (KIDO and MIPS; see Figures S1–S3). Previous work also observed a similar mid-depth polytomy, suggesting a rapid early diversification in the Nesophrosyne that is unable to be split by available genetic and genomic data (i.e., a hard polytomy; see Bennett and O’Grady 2012), or the possible introgression of symbiont lineages between hybridizing hosts. Hybridization events are known to occur among rapidly diversifying, host-plant restricted auchenorrhynchan lineages in Hawaiʻi (Roesch Goodman et al., 2012). However, we do not currently have a suitable sampling of host species and their populations to thoroughly test this hypothesis in the Nesophrosyne.
Gene content of Nesophrosyne’s symbiont genomes varies despite their highly reduced size
From the Illumina sequenced Nesophrosyne host species, we recovered 20 Sulcia and 18 Nasuia genomes (see Table S1). All symbiont genomes are complete and circular. Even coverage across all circularized genomes was verified with read mapping that verified complete, high-quality assemblies (see Methods). Two Nasuia genomes (Nasuia-HIPI and Nasuia-OIDO) were omitted because sequencing coverage was too low to assemble reliable contigs and complete genomes required for our downstream molecular assays.
The average genome size of Sulcia is 190 kilobases (kb; range = 190.3-190.9 kb) with an average of 190 protein-coding genes (range = 188-192 genes). These genomes further retain a single conserved 16S/23S/5S rRNA operon and 30 tRNAs. In contrast, Nasuia exhibits more variation between host species. Its average genome size is 112 kb (range = 107.7-116.1 kb) with an average of 132 protein-coding genes (range = 125-139 genes), a single 16S/23S/5S rRNA operon, and 18-21 tRNAs (see Table S1). Nesophrosyne’s Sulcia genomes are highly conserved, varying by up to six genes in the most extreme cases (∼3% of its genome). In contrast, Nasuia’s genome is highly variable among the Nesophrosyne, differing by up to 24 genes (>20% of its genome).
Globally, patterns of genome evolution and gene retention among Sulcia and Nasuia are similar to patterns observed in other Auchenorrhyncha lineages. Both retain complementary essential amino acid (EAA) pathways in an 8 + 2 arrangement for Sulcia and Nasuia, respectively, as observed in other leafhoppers and related insects (Chang et al., 2015; Bennett et al., 2016a; Mao et al., 2017; see also Bennett and Moran, 2013 for a list and pathways in Nasuia and Sulcia). The highly conserved nature of Sulcia’s genome has been widely observed across the other major auchenorrhynchan lineages that retain it (e.g., sharpshooter leafhoppers, cicadas, spittlebugs; McCutcheon and Moran, 2007, 2010; Koga and Moran, 2014; Campbell et al., 2015; Łukasik et al., 2018; Matsuura et al., 2018). The most dramatic differences observed among Sulcia genomes occur between the major infraorders Fulgomorpha (planthoppers) and Cicadomorpha (leafhoppers and kin). Among the planthoppers, Sulcia genomes are much smaller than in the cicadomorphan lineages (<149 kb in Fulgomorpha vs. an avg. of 251 kb in Cicadomorpha [range = 179-288 kb]; Bennett and Mao, 2018; Michalik et al., 2021). Among other gene losses, planthopper Sulcia lineages provide only three of the seven to eight EAAs typically retained in strains found in cicadomorphan hosts (McCutcheon and Moran, 2010; Michalik et al., 2021).
Nasuia’s genomes, in contrast to Sulcia, exhibit more variation in the genes they retain. The number of gene losses involves >20% of Nasuia’s genome (n = 24 genes) among the Nesophrosyne. This variation is significant considering that Nasuia’s genome is among the smallest known of any bacterium. The loss of any single gene that Nasuia lineages still retain likely requires direct adaptation by the host, or its partner symbionts, to support lost genetic and functional capabilities (Moran and Bennett, 2014; McCutcheon et al., 2019). The diversity of co-primary symbionts associated with Sulcia also showsimilarly higher genomic variation. For example, the cicada co-primary symbiont, “Ca. Hodgkinia” (hereafter Hodgkinia), has the most dramatic genomic variation among a symbiont yet observed. Although some cicada species harbor Hodgkinia with typical circular chromosomes (avg. size = 142 kb), in other hosts its genome is broken into mini circles of varying size and complexity (e.g., fragments range from 71 to 150 kb in 13-year Magicicadas; Van Leuven et al., 2014; Campbell et al., 2015; Łukasik et al., 2018). Similarly, the lineages of the co-primary symbiont “Ca. Baumannia,” which replaced Nasuia in sharpshooter leafhoppers >60 million years ago, vary by the loss of large chunks of its genome spanning >100 kb and >100 protein-coding genes (Wu et al., 2006; Bennett et al., 2014, 2016b).
Ancestral gene losses are ongoing and to some extent evolutionarily convergent
To determine patterns of gene loss (i.e., unique vs. convergent losses) among Sulcia and Nasuia genomes, we reconstructed ancestral patterns of gene loss and retention with maximum likelihood approaches (see Figures S4 and S5 and STAR Methods). We also used Sulcia and Nasuia lineages from two previously sequenced species from the Membracoidea superfamily, the aster leafhopper (Macrosteles quadrilineatus) and the keeled treehopper (Entylia carinata), to determine ancestral patterns of gene loss leading to the Nesophrosyne lineage (Bennett and Moran, 2013; Mao et al., 2017). In general, the membracoidean Sulcia and Nasuia lineages are structurally conserved and perfectly syntenic, aside from differences in their patterns of individual gene losses (reviewed by Mao et al., 2017). Later in the discussion, we summarize gene losses in both symbionts. We caution that gene loss counts presented here are minimum counts, as we did not sequence the symbiont genomes from all of Nesophrosyne’s 200 + species.
Sulcia in the common ancestor to Membracoidea likely retained at least 210 protein-coding genes and 30 tRNAs (reviewed by Mao et al., 2017). Prior to the diversification of the Deltocephalinae leafhoppers (Nesophrosyne’s leafhopper subfamily), Sulcia’s genome was reduced to 192 genes and 30 tRNAs (Shcherbakov, 2002; Mao et al., 2017). Among the Nesophrosyne, it has further undergone at least six instances of gene loss (infC, bamAD, rbfA, rpsO, and pheT; see Figure 1). The loss of these genes impacts a range of key cellular functions in Sulcia, which have significant implications for how the symbiosis functions and is maintained (Wilson and Duncan, 2015; Bublitz et al., 2019; Mao and Bennett, 2020). Some of these gene losses appear to be unique independent events among individual species, while others have been convergently lost multiple times during Nesophrosyne diversification.
Two genes removed from Sulcia’s genomes were lost multiple times in convergent evolutionary events: infC and bamA (Figure 1). The outer membrane protein assembly factor gene (bamA) was lost at least three independent times among our sampled Nesophrosyne species (Figure 1). It is part of a multi-gene complex essential for bacterial outer membrane assembly and metabolite exchange (Malinverni et al., 2006; Charles et al., 2011). Interestingly, the loss of bamA in Sulcia-KISC co-occurs with the loss of its interacting partner protein (bamD) and may be linked (Wu et al., 2005). Thus, some gene losses may instigate the loss of others in a domino-like fashion, as has been proposed to occur in the Blattabacterium-cockroach system (Kinjo et al., 2021). Perhaps more remarkable is the loss of the translation initiation factor IF-3 gene (infC) at least six times during the diversification of Nesophrosyne (see Figure S4). The infC gene is part of a three protein complex involved in translation initiation (Sabol et al., 1970). All Nesophrosyne Sulcia lineages still retain the other two infAB genes, suggesting support from partner symbionts or that they have moonlighting functions. The infABC gene set is generally retained in most other bacterial symbiont genomes across the Hemiptera, indicating that the gene has an essential functional role (Nakabachi et al., 2006; McCutcheon et al., 2009a, 2009b; McCutcheon and Moran, 2010; McCutcheon and von Dohlen, 2011).
Nasuia in the common ancestor to Membracoidea retained at least 163 genes and 29 tRNAs (Mao et al., 2017). Early on in the divergence of the Deltocephalinae leafhoppers, Nasuia’s genome was further reduced to a mere ∼142 genes and just over 112 kb in size (Bennett and Moran, 2013). Across the Nesophrosyne, Nasuia has undergone at least 24 instances of gene loss (>20% of its genome). Ten of these genes have been lost once among our sampled Nesophrosyne host species, including genes involved in ribosome function (rpsKRS and rpmB), tRNA synthesis (tilS in Nasuia-MICO), and histidine synthesis (hisD from Nasuia-EMBR). The latter two genes are essential for EAA and general protein synthesis (Soma et al., 2003; Van Leuven et al., 2019). The concentrated losses of ribosome-associated genes (8 out of 24) suggest that the host either easily replaces them, or the ribosomal holoenzyme can adapt to their absence (Akanuma et al., 2012; Galperin et al., 2021; Nikolaeva et al., 2021).
More than half of gene losses in Nasuia (n = 14) have been convergently lost in at least two or more host insect species (Figure 2). The most extreme case of convergent loss is that of a gene cassette that includes four complete genes: 30S ribosomal subunit protein S21 (rpsU), tyrosine-tRNA ligase (tyrS), tRNA-specific 2-thiouridylase (mnmA), and flavodoxin/ferredoxin-NADP + reductase (fpr) (see ancestral state reconstructions in Figure 3B). Additionally, an essential component of the DNA replication holoenzyme, dnaN, has been lost at least five times and is missing from more than half of our Nasuia genomes (Johanson and McHenry, 1980; Figure 3C). Finally, several ribosomal proteins (rsmD, rpmJG, and rpsQ) have been convergently lost across our sampled Nasuia species.
Figure 3.

Nasuia gene losses of rpsU, tyrS, mnmA, fpr and dnaN cassette
(A) Nasuia gene losses of rpsU, tyrS, mnmA, fpr, and dnaN cassette for three representative genomes, as well as the genes maintained in Nasuia-HMCO (N-HMCO) sequence. The genes rpsU, tyrS, mnmA, and fpr have been lost in all genomes except N-HMCO (top). The dnaN gene has been convergently lost in nine sequences. Ancestral state reconstruction of rpsU, tyrS, mnmA and fpr (B) suggests that these genes have been lost at least five times. Ancestral state reconstruction of dnaN (C) suggests that this gene has been lost at least five times. Colors correspond to the same gene in each genome segment (hypo = hypothetical protein). See Table 1 for species shorthand nomenclature. (B and C) Ancestral state reconstruction of rpsU, tyrS, mnmA, and fpr, as well as dnaN from phytools v.1.0-1 package on genes convergently lost in Nasuia (Revell, 2012). The loss of rpsU, tyrS, mnmA, and fpr may have instigated the loss of dnaN. See section methods for details on ancestral state reconstruction.
The ongoing loss of more than 20% of Nasuia’s genes among these lineages similarly presents major challenges to its hosts and partner symbionts, particularly because several of these genes are essential to its nutritional role in the symbiosis and its cellular functions (e.g., hisD and tilS, respectively). Although the number of genes Nasuia is capable of losing stands in stark contrast to Sulcia, both symbionts require the host or companion symbionts to adapt stabilizing support mechanisms (Mao et al., 2018). However, Nasuia is apparently a more demanding partner requiring independent host lineages to innovate novel support strategies. The reason for Nasuia’s more exaggerated rates of gene losses over Sulcia is likely owing to its rapid rates of molecular evolution discussed below (Bourguignon et al., 2020).
Sulcia and Nasuia have among the slowest and fastest rates of symbiont molecular evolution
To determine the underlying drivers of symbiont genome evolution among Nesophrosyne, we estimated genome-wide substitution rates in absolute time for Sulcia and Nasuia. We further compared these against host mitochondrial rates (Figure 4). The average substitution rates for Sulcia genomes are 3.24 × 10−9 substitutions/site/year. In contrast, Nasuia genomes exhibit a 34.8-fold higher rate of molecular evolution (avg. = 1.13 × 10−7 substitutions/site/year). The evolutionary rates of both symbiont genomes do not exceed that of the mitochondria (avg. = 2.15 × 10−7 substitutions/site/year: 1.9-fold from Nasuia and 66-fold from Sulcia). Nesophrosyne mitochondrial rates of evolution are in-line with observations from other hemipteran insects, which are generally elevated relative to other insects (Dowton et al., 2009; Song et al., 2012; Cui et al., 2013; Cameron, 2014; Li et al., 2017).
Figure 4.

Summary of uncorrelated rates of evolution between symbiont and host genes
(A) Linear regression between pairwise distances and age of divergence among all Nasuia, Sulcia, and mitochondrial genes for Hawaiian leafhoppers (Nesophrosyne). Colors indicate pairwise distance of protein-coding genes in each genome. Genomes are separated by the host-plant group that the host species has specialized in. A linear regression line is mapped between pairwise distance values from the closest related species to the most divergent species in each host-plant group. The regression equation and the coefficient of determination (R2) are also reported and colorized by the genome. Outliers with a pairwise distance >0.5 were removed (4 from Nasuia, 3 from Mitochondria).
(B) Associated p values of statistical tests between evolutionary rates of genomes across all plant groups and within plant groups. Significant p value between groups indicates no correlation between means of evolutionary rate.
To compare the rate of molecular evolution in Sulcia and Nasuia to available symbionts from other insect hosts, we estimated nonsynonymous (dN) and synonymous (dS) substitutions over their divergence times. We used the general M0 model, which averages substitution rates across whole genes and phylogeny. We further converted rates to dN/time (dN/t) and dS/time (dS/t) (Yang 2007; Silva and Santos-Garcia, 2015). The rates for Sulcia among the Nesophrosyne are 9.46 × 10−9 dN/t and 4.21 × 10−8 dS/t (time = 3.2 MYA). In contrast, Nasuia’s rates are highly elevated, averaging 1.65 × 10−7 for dN/t and 3.76 × 10−6 for dS/t. The average dN/t in Nasuia is 17.5-fold higher than in Sulcia, while the differences in average dS/t are even higher (89.3-fold).
Compared to other insect symbionts, for which data are available, Nasuia has among the highest evolutionary rates yet identified. The well-known symbiont of aphids, Buchnera, has an average rate of 2.58 × 10−9 dN/t and 1.43 × 10−8 dS/t, which is 64.2 and 262-fold less than Nasuia’s rates, respectively (using 20 Buchnera protein coding genes; Clark et al., 1999). Several of the highest rates of molecular evolution previously documented for insect symbionts belong to Baumannia, which replaced Nasuia in sharpshooter leafhoppers >60 million years ago, and Blochmannia found in carpenter ants (Silva and Santos-Garcia, 2015). Nasuia exceeds these, with 42 and 341-fold higher for dS/t and 20 and 165-fold higher for dN/t than Blochmannia and Baumannia, respectively.
One factor that may influence the rates of evolution in Nasuia and Sulcia, as well as other symbionts more broadly, is differences in host and symbiont generation times (e.g., bacteria with shorter, more frequent generations can incur more mutations and substitutions per some unit of time; Degnan et al., 2005; Silva and Santos-Garcia, 2015). However, we do not have a clear understanding of generation times in our insects for several reasons. First, they are difficult to rear owing to their highly restricted habitat ranges and species further experience differences in rain-fall-associated seasonality (Bennett and O’Grady, 2012; see also Degnan et al., 2005). In addition, it is not known whether symbiont replication rates are even coupled with host generations in our system, nor among most other insect symbioses. It is, however, worth noting that Sulcia and Nasuia exhibit dramatically different rates of evolution despite sharing the same host lineages. Thus, host generation time alone cannot explain observed differences.
In contrast to Nasuia’s highly elevated rates of molecular evolution, Sulcia has one of the most depressed rates of any biological system (McCutcheon et al., 2009a, 2009b; Bennett et al., 2014; see Figure 4). It has been widely observed among the Auchenorrhyncha (e.g., in cicadas and spittlebugs), that Sulcia has a nearly inert rate of molecular evolution even across divergences spanning 100s of millions of years (Takiya et al., 2006; Koga et al., 2013; Bennett et al., 2014; Bennett and Mao, 2018; Waneka et al., 2021; Arab and Lo, 2021; Michalik et al., 2021). Sulcia’s depressed evolutionary rates are an enigmatic biological phenomenon. One possible explanation for Sulcia’s reduced rates of molecular evolution may be its retention of mutation repair systems. In a Macrosteles leafhopper, Sulcia was found to have an overall low rate of mutagenesis compared to Nasuia, possibly owing to its retention of the DNA mismatch repair protein, mutS (Waneka et al., 2021). The mutS gene recognizes and initiates the repair of mismatched bases and small indels, which can lower substitution rates (Dettman et al., 2016; Long et al., 2018; Waneka et al., 2021). Most Sulcia lineages in the Auchenorrhyncha still retain mutS. This gene retention pattern may explain Sulcia’s universally conserved rates of molecular evolution (McCutcheon and Moran, 2007, 2010; Woyke et al., 2010; Bennett and Moran, 2013; Koga and Moran, 2014; Bennett and Mao, 2018). In contrast, the mutS gene is widely lost from most of Sulcia’s co-primary symbiont partners, which have characteristically elevated rates of molecular evolution—with the exception of “Candidatus Zinderia insecticola” in spittlebugs, which still maintains the gene (Takiya et al., 2006; McCutcheon and Moran, 2012; Bennett et al., 2014, 2016b; Campbell et al., 2015; Arab and Lo, 2021). These higher rates may drive genomic volatility and variation among Sulcia's partner symbionts, which are more frequently lost or replaced among the Auchenorrhyncha (Bennett and Moran, 2015; Sudakaran et al., 2017).
Rates of molecular evolution are uncorrelated between each symbiont
To further test whether Sulcia and Nasuia show correlated rates of evolution with each other and the host mitochondria, suggesting a shared evolutionary environment (Arab and Lo, 2021), we estimated substitution rates for both symbionts and mitochondria globally across and within the Nesophrosyne’s monophyletic host-plant associated clades (Broussaisia, Coprosma, Pipturus, and Myoporum; see Figure 4A). Correlations between substitutions rates among host and symbiont genomes could be explained if similar forces of selection are acting on the genomes, such as shared population bottlenecks and dependence on shared genes (e.g., host mutation repair genes taking over for those lost in symbiont genes; McCutcheon and Moran, 2012; Mao et al., 2018; McCutcheon et al., 2019) Overall, substitution rates between Nesophrosyne’s Sulcia, Nasuia, and the mitochondria are not correlated across host-plant affiliated clades (Figure 4B). Rates between symbionts and mitochondrial genes within host-plant clades are also uncorrelated (Figure 4B). Our results support a recent analysis of Sulcia from more widely divergent auchenorrhynchan clades that found similar decoupling of rates (see Arab and Lo, 2021). However, both of these findings are in contrast to mono-symbiont systems (Blochmannia-Carpenter Ants, Blattabacterium-Cockroaches, and Buchnera-Aphids) that tend to show a significant correlation between the rate of evolution in mitochondrial and symbiont genes (Degnan et al., 2004; Arab et al., 2020; Arab and Lo, 2021).
The disparity in rates of molecular evolution between Sulcia and its partners strongly suggests that different molecular and cellular processes, and evolutionary pressures, are likely shaping symbiont genomes (see Figure 4; Takiya et al., 2006; Bennett et al., 2014; Campbell et al., 2015; Arab and Lo, 2021). Although there is evidence that more ancient mitochondrial and plastid symbiont genomes do tend to show correlated rates of molecular evolution, these genomes are highly integrated into the general biology of most eukaryotic cellular and metabolic processes (Smith and Lee, 2010; Sloan et al., 2012; Hua et al., 2012). In contrast, the biological roles of insect symbionts are arguably less integrated into system-wide biological functions of their host insects. Nutritional symbionts are sequestered to distinct organs in the host, retaining relatively enriched genetic autonomy, distinct population sizes, and distinct cellular replication and repair capabilities (Buchner, 1965; Mira and Moran, 2002; Koga et al., 2012; Bennett et al., 2014; Chong and Moran, 2016; Mao and Bennett, 2020; Stever et al., 2021). As a result, they likely do not experience the same patterns and processes of molecular evolution as their partner symbionts, mitochondria, or host nuclear genes. Nevertheless, more fine-scale analyses that include a broadscale sampling of host nuclear and symbiont genes, as well as focus on protein domains, may further find rate correlations on interacting genes.
Sulcia and Nasuia experience differential selection patterns across their genomes
To understand how different modes of selection are shaping the evolution and function of Sulcia and Nasuia genomes, we tested for selection both across genes and across sites using the ratio of nonsynonymous to synonymous substitutions (denoted by ω; see STAR Methods). To determine whether symbiont genes are generally under strong purifying selection (ω < 0.1), relaxed purifying selection (0.95 < ω > 0.1), or positive selection (ω > 1), we initially used the M0 model in codeml (ω estimated across the whole gene; Yang et al., 2000; Z. Yang 2007; Sloan and Moran, 2012; Sabater-Muñoz et al., 2017; Perreau et al., 2021). In Sulcia, a small subset of genes are undergoing strong purifying selection (avg. ω = 0.0615 [range = 0.0001-0.0970, n = 34]), while most genes are undergoing relaxed purifying selection (avg. ω = 0.331 [range = 0.112-0.931, n = 133]). Additionally, three genes in Sulcia show signatures of positive selection (atpH, putA, and trpC; ω > 1). In Nasuia, most genes are undergoing strong purifying selection (avg. ω = 0.0449 [range = 0.0162-0.0988, n = 73]), while comparatively few genes are experiencing relaxed purifying selection (avg. ω = 0.243 [range = 0.105-0.555, n = 12]). This approach did not detect any Nasuia genes under positive selection.
Although it is useful to obtain an average ω value for a gene, it is not a sufficiently realistic model to detect signatures of positive selection that operate at finer scales (Anisimova et al., 2001). Specific codons related to intrinsic protein function may be under positive selection, while the majority of the gene can experience relaxed purifying selection (Yang et al., 2000; Anisimova et al., 2001). Thus, to test for positive selection on different codon sites within symbiont genes, we used two nested models: M1a-M2a and M7-M8 in codeml (Yang 2007). We interpret consistent results between the two models (and also the M0 model from above) as strong global support for positive selection operating on sites within a gene (Anisimova et al., 2001; Padhi et al., 2009; Price et al., 2011; Alves et al., 2013). We applied this approach to Sulcia and Nasuia across our sampled Nesophrosyne.
Sulcia site-based selection analyses recovered the same genes undergoing positive selection from the M0 model, as well as an additional 94 genes (97 in total, p < 0.05 with BH correction). Genes under positive selection are functionally enriched for the Clusters of Orthologous Groups (COG) Translation (J; 31 genes; (Fisher exact test, p = 0.0008; Figure 5) and Energy Production and Conversion (C; 14 genes; p = 0.0486; Figure 5). It is notable that, although not significant, 27 genes under positive selection are involved in essential amino acid synthesis (E; p = 0.1030; Figure 5).
Figure 5.

Bar chart showing genes undergoing positive selection or purifying/neutral selection across Sulcia and Nasuia genomes from Hawaiian leafhoppers (Nesophrosyne)
Genes are binned into their Clusters of Orthologous Groups (COGs) functional categories (Tatusov et al., 2000). Bars are color coded according to their genome (i.e., Sulcia or Nasuia) and selection (i.e., Positive or Neutral/Purifying). We used two nested models, M1a-M2a and M7-M8, to determine overall support for selection among genes (See section methods for further explanation; Anisimova et al., 2001). The likelihood scores were compared within paired models (chi-squared test; p ≤ 0.05) to indicate significant positive selection for genes.
Sulcia’s genes under positive selection are primarily involved in essential amino acid synthesis, buffering degraded protein function, and other cellular processes. Some examples of proteins with positive sites include protein chaperonins (groEL and dnaK), EAA metabolite synthesis (e.g., arginine [argBDEG], lysine [asd, dapBD, and lysAC], valine [ilvBCEN], and phenylalanine and tryptophan [aroABCEGK and trpABCE]), transcription and translation (rplA and rpsDFMNPT), transcription release factors (prfAB), aminoacyl tRNA synthetases (glnS, leuS, serS, tyrS, and valS), and energy synthesis (gapA and atpBCFGH). In addition to genes involved in the incomplete TCA cycle of Sulcia (aceF, acoA, lpdA, korB, and sucA) and the pathway for the conversion of glutamine to carbamoyl phosphate (carAB). Although some of these genes are clearly important to the host (e.g., amino acid-related genes), others are key components of Sulcia’s independent cellular stability. For example, Sulcia’s groEL gene, which assists in the folding of damaged proteins (reviewed in Kupper et al., 2014), exhibits an overall pattern of strong purifying selection, but some sites within it are under positive selection. As suggested previously in pea-aphids, the sites within groEL under positive selection are involved in gene interaction domains that likely improve its ability to bind with other rapidly evolving proteins (Fares et al., 2002, 2004).
In Nasuia, only four genes are predicted to be under positive selection with site-based models. This result is congruent with results from the M0 model that show most Nasuia genes are under strong purifying selection (Figure 5). None of these genes are associated with amino acid synthesis and are too few for COG enrichment analyses. Given Nasuia’s elevated rates of molecular evolution, selection seems to be operating to maintain function, even though this symbiont has a higher rate of gene loss compared with its partner. The ongoing widespread gene losses from Nasuia’s genome may generally result from its high rates of molecular evolution. In this scenario, the probability of a random mutation disabling a gene that can become fixed through genetic drift is much higher (Wernegreen, 2015). Similar results have also been demonstrated in the genomes of Blattabacterium from cockroaches and Buchnera from pea-aphids, where the rate of genome reduction is also associated with an increased mutation rate rather than selection acting on genes (Bourguignon et al., 2020; Kinjo et al., 2021).
Finally, to further understand whether patterns of selection can predict convergent loss of particular genes, we performed a test of selection with the M0 model (described above) on genes that are not universally retained in Nesophrosyne’s Sulcia and Nasuia genomes. Some symbiotic genes that are only retained in one host species (e.g., the gene cassette containing rpsU, tyrS, mnmA, and fpr) could not be tested owing to the lack of information to make pairwise comparisons possible. In Nasuia, genes that have been lost in some taxa, but not in others, are generally undergoing strong purifying selection (n = 4; rpmG, rpmJ, rpsQ, and rsmD; avg. dN/dS = 0.033), while one gene is undergoing relaxed purifying selection (dnaN; dN/dS = 0.183). In Sulcia, we see a similar pattern (n = 2). One gene is undergoing strong purifying selection (infC; dN/dS = 0.0001), while the other is undergoing relaxed purifying selection (bamA; dN/dS = 0.222). These results suggest that types of selection alone are not strong predictors of whether genes will eventually be lost.
Conclusion: Divergent evolutionary forces shape co-primary symbiont genomes
Our sampling of Nesophrosyne leafhoppers and their symbionts provides a fine-scale look into the processes that underlie symbiont genome evolution. Gene losses among the Nesophrosyne’s Sulcia and Nasuia lineages demonstrate that there is still ongoing volatility, even among symbionts with two of the smallest known genomes. However, these patterns vary widely between these symbiont species. Sulcia has one of the slowest evolving genomes known, while Nasuia has an exceptionally fast evolving one. Sulcia also has far more genes under positive or relaxed purifying selection (80% of genes tested) than Nasuia’s genome, which is largely under strong purifying selection (86% of genes tested). Taken together, our results indicate that the two symbionts experience independent cellular, metabolic, and evolutionary pressures. These differences may further lead to a high level of retention of Sulcia among the Auchenorrhyncha and the relatively high turnover of its partner symbionts (reviewed in Koga et al., 2013; Bennett and Moran, 2015; Sudakaran et al., 2017; Bourguignon et al., 2020).
Regardless of the differences between Sulcia and Nasuia, our results show that there is repeatability in gene losses that may be more easily accommodated if mutations render them non-functional. Both symbionts exhibit convergent gene loss events, particularly regarding genes involved in transcription and translation. These findings suggest a pre-adapted genetic or cellular host environment that permits these genes to be repeatedly lost (see Figure 1, Figure 2 and Figure 2). The host, or partner symbionts, may be capable of filling gaps in these bacterial cellular processes, as is predicted to occur in a wide range of hemipteran insect systems (Hansen and Moran, 2011; Sloan et al., 2014; Luan et al., 2015; Mao et al., 2018; Van Leuven et al., 2019; Mao and Bennett, 2020).
Finally, even though ancient symbiont genomes of insects show some level of conservation among host lineages, a closer look among host sister species reveals complex patterns of gene loss and modes of selection. The evolutionary processes acting on even the tiniest symbiont genomes are clearly dynamic and highly variable. This understanding can be overlooked when comparing lineages among disparately related taxonomic groups.
Limitations of the study
While we sequenced leafhopper species that range the diversity of the genus Nesophrosyne, we do not have symbiont lineages sequenced from all 200 + species. Therefore, the range of gene loss exhibited with our species may be greater in other lineages. Additionally, we assembled the mitochondrial sequences to compare rates of evolution; however, we do not have insight into the role of the host nuclear genome in supporting symbiont genome loss or correlated rates of evolution. Finally, a more thorough investigation within and between populations of leafhopper species would provide more information on how host-symbiont co-evolve on an ecological scale.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| N. sp. 295 | USA: East Hawaii Island | EHBR |
| N. ogradyi | USA: East Maui Island | EMBR |
| N. kanawao | USA: North Hawaii Island | NHBR |
| N. makaihe | USA: West Oahu Island | WOBR |
| N. sp. 23 | USA: Kauai Island | KICL |
| N. haleakala | USA: Haleakala Mtn, Maui | HMCO |
| N. sp. 302 | USA: Hawaii Island | HICO |
| N. sp. 58 | USA: Maui Island | MICO |
| N. sp. 29 | USA: Kauai Island | KIDO |
| N. maratima | USA: Oahu Island | OIDO |
| N. sp. 281 | USA: Hawaii Island | HIMY |
| N. sp. 126 | USA: Kauai Island | KIMY |
| N. sp. 242 | USA: Molokai Island | MIMY |
| N. sp. 246 | USA: Oahu Island | OIMY |
| N. montium | USA: Hawaii Island | HIPI |
| N. sp. 17 | USA: Kauai Island | KIPI |
| N. sp. 48 | USA: Maui Island | MIPI |
| N. ponapona | USA: Oahu Island | OIPI |
| N. sp. 15 | USA: Molokai Island | MIPS |
| N. sp. 21 | USA: Kauai Island | KISC |
| Orosius sp. | ||
| Deposited Data | ||
| BioProject | NCBI – GenBank | PRJNA816609 |
| Sulcia sequence data | NCBI – GenBank | CP093890 to CP093909 |
| Nasuia sequence data | NCBI – GenBank | CP094180 to CP094197 |
| Insect mitochondrial sequence data | NCBI – GenBank | ON135504 to ON135524 |
| Software and algorithms | ||
| Trimmomatic | Bolger et al., 2014 | v0.39 |
| FastQC | Andrews, 2010 | v0.11.9 |
| SPAdes | Bankevich et al., 2012 | v3.14 |
| GLIMMER | Delcher et al., 2007 | v3.02 |
| Geneious Pro | Drummond et al., 2011 | |
| BLAST | Altschul et al., 1990 | |
| Bowtie2 | Langmead and Salzberg, 2012 | v2.3.5.1 |
| RAST | Overbeek et al., 2014 | v2 |
| RNAmmer | Lagesen et al., 2007 | v1.2 |
| tRNAscan-SE | Lowe and Chan, 2016 | v2.0 |
| MITOS | Bernt et al., 2013 | |
| MAFFT | Katoh and Standley, 2013 | v7.455 |
| PartitionFinder 2 | Lanfear et al., 2017 | |
| BEAST | Drummond and Rambaut, 2007 | v.1.10.4 |
| Tracer | Rambaut et al., 2018 | v1.7.1 |
| RWTY | Warren et al., 2017 | v1.0.2 |
| Phytools | Revell, 2012 | v.1.0–1 |
| RStudio | RStudio Team, 2018 | |
| MEGAX | Kumar et al., 2018 | v.10.2.4 |
| JModelTest2 | Darriba et al., 2012 | v2.1.10 |
| PAML (codeml) | Yang 2007 | v4.8 |
| Other | ||
| Sulcia genome assembly from Macrosteles quadrilineatus | Bennett and Moran (2013) | NCBI: CP006060 |
| Nasuia genome assembly from Macrosteles quadrilineatus | Bennett and Moran (2013) | NCBI: CP006059 |
| Sulcia genome assembly from Entylia carinata | Mao et al., 2017 | NCBI: CP021172 |
| Nasuia genome assembly from Entylia carinata | Mao et al., 2017 | NCBI: CP021173 |
Resource availability
Lead contact
Further information and requests for resources, data, and codes should be directed to and will be fulfilled by the lead contact, Gordon Bennett (gbennett2@ucmerced.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Sample collection
We sampled a targeted set of 20 species that span the ecological and phylogenetic diversity of the genus. Adult female and male leafhoppers were field-collected and stored in ethanol to be used for downstream analysis. The selected species are also specific to eight different host-plant species that encompass the diversity of host-plant families and genera that the genus is associated with (see Table 1).
Method details
Genome sequencing
For each target species, ten field-collected individuals were pooled to obtain enough DNA for genomic sequencing. DNA was extracted with a DNAeasy kit (Qiagen) and concentration quantified with a Quibit 3.0 fluorometer (ThermoFisher). Libraries and Illumina MiSeq sequencing were conducted at UC Berkeley qB3 Functional Genomics Lab for 4 million 2 × 300 base pair (bp), paired-end reads.
Genome assembly and annotation
Raw reads were quality filtered and cleaned of adapters using Trimmomatic v0.39 and checked with FastQC v0.11.9 (Andrews, 2010; Bolger et al., 2014). Assembly of symbiont genomes was done using SPAdes v3.14 (program settings: -k 127, --only-assembler, --meta; Bankevich et al., 2012). Since extracts contain both host and bacterial DNA, symbiont and mitochondrial genomes were manually extracted by using features unique to each symbiont (i.e., high relative coverage and high AT content). Extracted contigs were verified and confirmed with BLAST searches of open reading frames predicted with GLIMMER v3.02 in Geneious Pro (Altschul et al., 1990; Drummond et al., 2011). To confirm consistent assembly coverage and circularization of bacterial genomes, quality filtered reads were aligned to the completed symbiont genome using Bowtie2 v2.3.5.1 (program settings: --local; Langmead and Salzberg, 2012). Linear chromosomes were circularized by breaking contigs and attaching ends. High, consistent coverage across these ends were verified to confirmed closure of the circular bacterial chromosomes. No plasmids were identified, as is to be expected for tiny symbiont genomes (Bennett and Moran 2013).
Initial genome annotations were done with RAST v2 (Overbeek et al., 2014). Annotations were then verified with GLIMMER v3.02 gene predictions that were checked with BLASTP searches against the nr database (Altschul et al., 1990; Delcher et al., 2007). Bacterial RNA genes were further identified with RNAmmer v1.2 and tRNAscan-SE v2.0 (Lagesen et al., 2007; Lowe and Chan, 2016). Mitochondrial genes were identified with MITOS (Bernt et al., 2013).
Quantification and statistical analysis
Phylogenetic tree construction
To construct a phylogenetic tree in absolute time for downstream analysis, and to verify relationships with the Nesophrosyne genus, we extracted and aligned complete mitochondrial genomes of our sampled insect species. We included the mitochondrial DNA from Orosius sp. as the known outgroup for Nesophrosyne (Bennett and O’Grady, 2012; Fletcher et al., 2017). We also tested co-cladogenesis between host and symbiont by extracting and aligning all Nasuia and Sulcia genes. For the mitochondria and symbiont genomes, each gene was individually aligned with MAFFT v7.455 using the L-INS-i model (Katoh and Standley, 2013). Genes that did not occur in all genomes, or that were difficult to align with confident site homology, were omitted.
The resulting phylogenetic datasets included concatenated protein coding and ribosomal genes for a total of 184 genes (181,781 sites) from Sulcia, 99 genes (86,095 sites) from Nasuia, and 15 genes (14,304 sites) from leafhopper mitochondria. Best-fit models of nucleotide substitutions and partitioning schemes were determined using PartitionFinder 2 (program settings: branchlengths = unlinked, models = all, model_selection = bic; Lanfear et al., 2017). Bayesian time calibrated phylogenies were then inferred using BEAST v.1.10.4, using the generated partition scheme and corresponding molecular substitution models (Drummond and Rambaut, 2007). The tree prior included the yule process speciation with a random starting tree. Five internal node calibrations were selected following our previous phylogenetic study of the Nesophrosyne (Bennett and O’Grady, 2013). Briefly, internal node calibrations were determined from Nesophrosyne species divergences that match the sequential geological formation of the Hawaiian Islands (i.e., progression rule). Calibrations were applied with a normal prior distribution since absolute species divergence could have occurred earlier or after island formation (see Bennett and O’Grady, 2012 for additional information; Bennett and O’Grady, 2013). Multiple BEAST chains were run per genome alignment and sampled every 1000 generations following Bayesian recommendations (two chains with four million generations; Huelsenbeck et al., 2002). Runs were performed with an uncorrelated relaxed clock with a lognormal distribution. Convergence and stationarity of chains were verified with ESS values were >200 using Tracer v1.7.1 and RWTY v1.0.2 (Rambaut et al., 2018; Warren et al., 2017).
Ancestral genome reconstruction and ancestral state reconstruction
To determine patterns of gene loss (i.e., unique vs convergent gene losses) among symbionts between host lineages, we reconstructed the ancestral gene retention across Nesophrosyne’s symbionts. We further included symbiont genomes from previously sequenced lineages including the aster leafhopper (Macrosteles quadrilineatus) and the keeled treehopper (Entylia carinata) (Bennett and Moran, 2013; Mao et al., 2017). Maximum likelihood ancestral state reconstructions were estimated with phytools v.1.0–1 package (Revell, 2012; RStudio Team, 2018). We used a custom model that allows for the loss of genes to occur (no gene gain) to account for the inability of symbionts to recombine with other environmental or symbiotic bacteria. Posterior density of stochastic character maps was generated by simulating 100 trees.
Patterns of molecular evolution
To test for genome wide substitution rates across Sulcia and Nasuia genes, as well as host mitochondria, we used MEGAX v.10.2.4 (Kumar et al., 2018). Model selection for pairwise evolutionary distances were selected with JModelTest2 v2.1.10 with constricted model selection to those available for MEGAX analyses (e.g., Jukes-Cantor, Tamura-Nei, etc.; Darriba et al., 2012). To test the rate of substitutions between islands within the same plant family, two pairwise analyses were done: (i.) the oldest diverging species (e.g., Kauaʻi and Hawaiʻi species) and (ii.) the closest diverging species (e.g., Maui and Hawaiʻi species). These ages ranged from the most recent divergence (0.351 MYA in Broussaisia) to the most ancient divergence (2.4239 MYA in Pipturus; see Figure 4A). Rates of substitutions were graphed across absolute time of divergence. To test whether Sulcia and Nasuia show correlation in their evolutionary rates with Nesophrosyne mitochondrial genes, we performed a Kruskal-Wallis test and a pairwise comparison with a Wilcoxon rank sum test and a Benjamini-Hochberg correction for multiple tests (Wilcoxon, 1945; Benjamini and Hochberg, 1995; McKight and Najab, 2010; RStudio Team, 2018). Additionally, in our analysis, we consider the rate of evolution in absolute time by testing across host-plant groups (specifically Broussaisia, Coprosma, Pipturus and Myoporum) and within host-plant clades (Figure 4).
Selection among symbiont genomes
In order to test for selection among symbiont genes, we measured rates of synonymous (dS) and nonsynonymous (dN) substitutions with codeml v4.8 using the mitochondrial time-calibrated phylogeny (Yang 2007 ). The ratio of nonsynonymous to synonymous substitutions (ω = dN/dS) were calculated for each aligned Sulcia and Nasuia gene. Selection is calculated by measuring the ratio of nonsynonymous to synonymous substitutions (denoted by ω), where ω > 1, ω = 0 and ω < 1 indicate positive, neutral and purifying selection, respectively (Yang and Bielawski, 2000).
We estimated rates of synonymous and nonsynonymous substitutions using three models. The M0 model was used to test for selection (ω) across all codon sites. This generated a single ω value that was evaluated further. Additionally, we used two nested models, M1a-M2a and M7-M8, to determine strong support for selection in codon sites (Anisimova et al., 2001). Models M1a and M7 are constrained and disallow positive selection while the M2a and M8 models are unconstrained, allowing for positive selection. The M7-M8 models offer a more stringent test of positive selection (Anisimova et al., 2001). However, by using multiple nested models, we verify positive selection in genes that are highly supported in both models (Anisimova et al., 2001). The likelihood scores were compared within paired models (chi-squared test; p ≤ 0.05) to indicate significant positive selection for genes. To confirm the specificity of our results, we only consider genes that were identified as being under significant positive selection by both nested models for downstream analysis. Genes were further separated into Clusters of Orthologous Groups (COG) to test for functional group enrichment within functional categories, using a Fisher exact test with Benjamini-Hochberg Procedure multiple-testing correction (Benjamini and Hochberg, 1995; Fisher, 1992).
Acknowledgments
We would like to thank Kirsten Poff for the initial dissection and DNA extraction of bacteriomes, Dr. Allen Yang for the initial assembly of symbionts, and the UC Merced Cluster for providing support for bioinformatic approaches. We also thank the anonymous reviewers for their valuable comments and suggestions that helped to improve this work. This project was supported by the National Science Foundation under grant NSF-1347116.
Author contributions
This study was conceived by G.M.B. and Y.M.V., field species collection was conducted by G.M.B., dissections were conducted by G.M.B. and Y.M.V., genome assembly and bioinformatic analyses were performed by Y.M.V., and the article was written by Y.M.V. and G.M.B.
Decalration of interests
The authors declare no competing interests.
Published: August 19, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104786.
Contributor Information
Yumary M. Vasquez, Email: yvasquez8@ucmerced.edu.
Gordon M. Bennett, Email: gbennett2@ucmerced.edu.
Supplemental information
Data and code availability
-
•
Sequence data have been deposited with NCBI under the BioProject number: PRJNA816609 and are publicly available as of the date of publication. Sulcia genomes can be found under the accession numbers GenBank: CP093890 to GenBank: CP093909. Nasuia genomes can be found under the accession numbers GenBank: CP094180 to GenBank: CP094197. Insect mitochondrial genomes can be found under the accession numbers GenBank: ON135504 to GenBank: ON135524. These accession numbers are also listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Akanuma G., Nanamiya H., Natori Y., Yano K., Suzuki S., Omata S., Ishizuka M., Sekine Y., Kawamura F. Inactivation of ribosomal protein genes in Bacillus subtilis reveals importance of each ribosomal protein for cell proliferation and cell differentiation. J. Bacteriol. 2012;194:6282–6291. doi: 10.1128/JB.01544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Alves J.M.P., Serrano M.G., Maia da Silva F., Voegtly L.J., Matveyev A.V., Teixeira M.M.G., Camargo E.P., Buck G.A. Genome evolution and phylogenomic analysis of Candidatus Kinetoplastibacterium, the betaproteobacterial endosymbionts of Strigomonas and Angomonas. Genome Biol. Evol. 2013;5:338–350. doi: 10.1093/gbe/evt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: A Quality Control Tool for High Throughput Sequence Data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- Anisimova M., Bielawski J.P., Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol. Biol. Evol. 2001;18:1585–1592. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- Arab D.A., Bourguignon T., Wang Z., Ho S.Y.W., Lo N. Evolutionary rates are correlated between cockroach symbionts and mitochondrial genomes. Biol. Lett. 2020;16:20190702. doi: 10.1098/rsbl.2019.0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arab D.A., Lo N. Evolutionary rates are correlated between Buchnera endosymbionts and the mitochondrial genomes of their aphid hosts. J. Mol. Evol. 2021;89:238–248. doi: 10.1007/s00239-021-10001-9. [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 2005;59:155–189. doi: 10.1146/annurev.micro.59.030804.121041. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- Bennett G.M., Abbà S., Kube M., Marzachì C. Complete genome sequences of the obligate symbionts “Candidatus Sulcia muelleri” and “Ca. Nasuia deltocephalinicola” from the pestiferous leafhopper Macrosteles quadripunctulatus (Hemiptera: cicadellidae) Genome Announc. 2016;4:e01604–e01615. doi: 10.1128/genomeA.01604-15. https://10.1128/genomeA.01604-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.M., Mao M. Comparative genomics of a quadripartite symbiosis in a planthopper host reveals the origins and rearranged nutritional responsibilities of anciently diverged bacterial lineages. Environ. Microbiol. 2018;20:4461–4472. doi: 10.1111/1462-2920.14367. [DOI] [PubMed] [Google Scholar]
- Bennett G.M., McCutcheon J.P., MacDonald B.R., Romanovicz D., Moran N.A. Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. mBio. 2014;5:e01697-14. doi: 10.1128/mBio.01697-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.M., McCutcheon J.P., McDonald B.R., Moran N.A. Lineage-specific patterns of genome deterioration in obligate symbionts of sharpshooter leafhoppers. Genome Biol. Evol. 2016;8:296–301. doi: 10.1093/gbe/evv159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.M., Moran N.A. Heritable symbiosis: the advantages and perils of an evolutionary rabbit hole. Proc. Natl. Acad. Sci. USA. 2015;112:10169–10176. doi: 10.1073/pnas.1421388112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.M., Moran N.A. Small, smaller, smallest: the origins and evolution of ancient dual symbioses in a phloem-feeding insect. Genome Biol. Evol. 2013;5:1675–1688. doi: 10.1093/gbe/evt118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett G.M., O’Grady P.M. Historical biogeography and ecological opportunity in the adaptive radiation of native Hawaiian leafhoppers (Cicadellidae: Nesophrosyne) J. Biogeogr. 2013;40:1512–1523. doi: 10.1111/jbi.12099. [DOI] [Google Scholar]
- Bennett G.M., O’Grady P.M. Host–plants shape insect diversity: phylogeny, origin, and species diversity of native Hawaiian leafhoppers (Cicadellidae: Nesophrosyne) Mol. Phylogenet. Evol. 2012;65:705–717. doi: 10.1016/j.ympev.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Bernt M., Donath A., Jühling F., Externbrink F., Florentz C., Fritzsch G., Pütz J., Middendorf M., Stadler P.F. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscaro V., Kolisko M., Felletti M., Vannini C., Lynn D.H., Keeling P.J. Parallel genome reduction in symbionts descended from closely related free-living bacteria. Nat. Ecol. Evol. 2017;1:1160–1167. doi: 10.1038/s41559-017-0237-0. [DOI] [PubMed] [Google Scholar]
- Bourguignon T., Kinjo Y., Villa-Martín P., Coleman N.V., Tang Q., Arab D.A., Wang Z., Tokuda G., Hongoh Y., Ohkuma M., et al. Increased mutation rate is linked to genome reduction in prokaryotes. Curr. Biol. 2020;30:3848–3855.e4. doi: 10.1016/j.cub.2020.07.034. [DOI] [PubMed] [Google Scholar]
- Bublitz D.C., Chadwick G.L., Magyar J.S., Sandoz K.M., Brooks D.M., Mesnage S., Ladinsky M.S., Garber A.I., Bjorkman P.J., Orphan V.J., McCutcheon J.P. Peptidoglycan production by an insect-bacterial mosaic. Cell. 2019;179:703–712.e7. doi: 10.1016/j.cell.2019.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner P. John Wiley & Sons; New York: 1965. Endosymbiosis of Animals with Plant Microorganisms. [Google Scholar]
- Cameron S.L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 2014;59:95–117. doi: 10.1146/annurev-ento-011613-162007. [DOI] [PubMed] [Google Scholar]
- Campbell M.A., Łukasik P., Simon C., McCutcheon J.P. Idiosyncratic genome degradation in a bacterial endosymbiont of periodical cicadas. Curr. Biol. 2017;27:3568–3575.e3. doi: 10.1016/j.cub.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M.A., Van Leuven J.T., Meister R.C., Carey K.M., Simon C., McCutcheon J.P. Genome expansion via lineage splitting and genome reduction in the cicada endosymbiont Hodgkinia. Proc. Natl. Acad. Sci. USA. 2015;112:10192–10199. doi: 10.1073/pnas.1421386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.-H., Cho S.-T., Canale M.C., Mugford S.T., Lopes J.R.S., Hogenhout S.A., Kuo C.-H. Complete genome sequence of “Candidatus Sulcia muelleri” ML, an obligate nutritional symbiont of maize leafhopper ( Dalbulus maidis ) Genome Announc. 2015;3 doi: 10.1128/genomeA.01483-14. e01483–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles H., Balmand S., Lamelas A., Cottret L., Pérez-Brocal V., Burdin B., Latorre A., Febvay G., Colella S., Calevro F., Rahbé Y. A genomic reappraisal of symbiotic function in the aphid/buchnera symbiosis: reduced transporter sets and variable membrane organisations. PLoS One. 2011;6:e29096. doi: 10.1371/journal.pone.0029096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong R.A., Moran N.A. Intraspecific genetic variation in hosts affects regulation of obligate heritable symbionts. Proc. Natl. Acad. Sci. USA. 2016;113:13114–13119. doi: 10.1073/pnas.1610749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong R.A., Park H., Moran N.A. Genome evolution of the obligate endosymbiont Buchnera aphidicola. Mol. Biol. Evol. 2019;36:1481–1489. doi: 10.1093/molbev/msz082. [DOI] [PubMed] [Google Scholar]
- Clark M.A., Moran N.A., Baumann P. Sequence evolution in bacterial endosymbionts having extreme base compositions. Mol. Biol. Evol. 1999;16:1586–1598. doi: 10.1093/oxfordjournals.molbev.a026071. [DOI] [PubMed] [Google Scholar]
- Cui Y., Xie Q., Hua J., Dang K., Zhou J., Liu X., Wang G., Yu X., Bu W. Phylogenomics of Hemiptera (Insecta: paraneoptera) based on mitochondrial genomes. Syst. Entomol. 2013;38:233–245. doi: 10.1111/j.1365-3113.2012.00660.x. [DOI] [Google Scholar]
- Darriba D., Taboada G.L., Doallo R., Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan P.H., Lazarus A.B., Brock C.D., Wernegreen J.J. Host–symbiont stability and fast evolutionary rates in an ant–bacterium association: cospeciation of Camponotus species and their endosymbionts, Candidatus Blochmannia. Syst. Biol. 2004;53:95–110. doi: 10.1080/10635150490264842. [DOI] [PubMed] [Google Scholar]
- Degnan P.H., Lazarus A.B., Wernegreen J.J. Genome sequence of Blochmannia pennsylvanicus indicates parallel evolutionary trends among bacterial mutualists of insects. Genome Res. 2005;15:1023–1033. doi: 10.1101/gr.3771305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A.L., Bratke K.A., Powers E.C., Salzberg S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettman J.R., Sztepanacz J.L., Kassen R. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics. 2016;17:27. doi: 10.1186/s12864-015-2244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009;23:38–47. doi: 10.1111/j.1365-2435.2008.01442.x. [DOI] [Google Scholar]
- Dowton M., Cameron S.L., Austin A.D., Whiting M.F. Phylogenetic approaches for the analysis of mitochondrial genome sequence data in the Hymenoptera–a lineage with both rapidly and slowly evolving mitochondrial genomes. Mol. Phylogenet. Evol. 2009;52:512–519. doi: 10.1016/j.ympev.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Drummond A., Ashton B., Buxton S., Cheung M., Cooper A., Duran C., Field M., Heled J., Kearse M., Markowitz S. Vol. 5.6. 2011. Geneious Pro V5. [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares M.A., Barrio E., Sabater-Muñoz B., Moya A. The Evolution of the Heat-Shock Protein GroEL from Buchnera, the Primary Endosymbiont of Aphids, Is Governed by Positive Selection. Mol. Biol. Evol. 2002;19:1162–1170. doi: 10.1093/oxfordjournals.molbev.a004174. In this issue. [DOI] [PubMed] [Google Scholar]
- Fares M.A., Moya A., Barrio E. GroEL and the maintenance of bacterial endosymbiosis. Trends Genet. 2004;20:413–416. doi: 10.1016/j.tig.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Fisher R.A. Breakthroughs in Statistics. Springer; 1992. Statistical methods for research workers; pp. 66–70. [Google Scholar]
- Fletcher M., Löcker H., Mitchell A., Gopurenko D. A revision of the genus Orosius Distant (Hemiptera: cicadellidae) based on male genitalia and DNA barcoding: revision of Orosius. Austral Entomology. 2017;56:198–217. doi: 10.1111/aen.12224. [DOI] [Google Scholar]
- Galperin M.Y., Wolf Y.I., Garushyants S.K., Vera Alvarez R., Koonin E.V. Nonessential ribosomal proteins in bacteria and archaea identified using clusters of orthologous genes. J. Bacteriol. 2021;203 doi: 10.1128/JB.00058-21. JB.00058-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A.K., Moran N.A. Aphid genome expression reveals host–symbiont cooperation in the production of amino acids. Proc. Natl. Acad. Sci. USA. 2011;108:2849–2854. doi: 10.1073/pnas.1013465108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hembry D.H., Bennett G., Bess E., Cooper I., Jordan S., Liebherr J., Magnacca K.N., Percy D.M., Polhemus D.A., Rubinoff D., et al. Insect radiations on islands: biogeographic pattern and evolutionary process in Hawaiian insects. Q. Rev. Biol. 2021;96:247–296. doi: 10.1086/717787. [DOI] [Google Scholar]
- Hendry T.A., de Wet J.R., Dougan K.E., Dunlap P.V. Genome evolution in the obligate but environmentally active luminous symbionts of flashlight fish. Genome Biol. Evol. 2016;8:2203–2213. doi: 10.1093/gbe/evw161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua J., Smith D.R., Borza T., Lee R.W. Similar relative mutation rates in the three genetic compartments of mesostigma and chlamydomonas. Protist. 2012;163:105–115. doi: 10.1016/j.protis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P., Larget B., Miller R.E., Ronquist F. Potential applications and pitfalls of Bayesian inference of phylogeny. Syst. Biol. 2002;51:673–688. doi: 10.1080/10635150290102366. [DOI] [PubMed] [Google Scholar]
- Husnik F., McCutcheon J.P. Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proc. Natl. Acad. Sci. USA. 2016;113:E5416–E5424. doi: 10.1073/pnas.1603910113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnik F., Nikoh N., Koga R., Ross L., Duncan R.P., Fujie M., Tanaka M., Satoh N., Bachtrog D., Wilson A.C.C., et al. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell. 2013;153:1567–1578. doi: 10.1016/j.cell.2013.05.040. [DOI] [PubMed] [Google Scholar]
- Johanson K.O., McHenry C.S. Purification and characterization of the beta subunit of the DNA polymerase III holoenzyme of Escherichia coli. J. Biol. Chem. 1980;255:10984–10990. doi: 10.1016/S0021-9258(19)70404-9. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinjo Y., Lo N., Martín P.V., Tokuda G., Pigolotti S., Bourguignon T. Enhanced mutation rate, relaxed selection, and the “domino effect” are associated with gene loss in Blattabacterium , A cockroach endosymbiont. Mol. Biol. Evol. 2021;38:3820–3831. doi: 10.1093/molbev/msab159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobiałka M., Michalik A., Walczak M., Szklarzewicz T. Dual “bacterial-fungal” symbiosis in Deltocephalinae leafhoppers (insecta, Hemiptera, Cicadomorpha: cicadellidae) Microb. Ecol. 2018;75:771–782. doi: 10.1007/s00248-017-1075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga R., Bennett G.M., Cryan J.R., Moran N.A. Evolutionary replacement of obligate symbionts in an ancient and diverse insect lineage. Environ. Microbiol. 2013;15:2073–2081. doi: 10.1111/1462-2920.12121. [DOI] [PubMed] [Google Scholar]
- Koga R., Meng X.-Y., Tsuchida T., Fukatsu T. Cellular mechanism for selective vertical transmission of an obligate insect symbiont at the bacteriocyte-embryo interface. Proc. Natl. Acad. Sci. USA. 2012;109:E1230–E1237. doi: 10.1073/pnas.1119212109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga R., Moran N.A. Swapping symbionts in spittlebugs: evolutionary replacement of a reduced genome symbiont. ISME J. 2014;8:1237–1246. doi: 10.1038/ismej.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Li M., Knyaz C., Tamura K. Mega X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupper M., Gupta S.K., Feldhaar H., Gross R. Versatile roles of the chaperonin GroEL in microorganism-insect interactions. FEMS Microbiol. Lett. 2014;353:1–10. doi: 10.1111/1574-6968.12390. [DOI] [PubMed] [Google Scholar]
- Lagesen K., Hallin P., Rødland E.A., Stærfeldt H.-H., Rognes T., Ussery D.W. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017;34:772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Leavengood J.M., Chapman E.G., Burkhardt D., Song F., Jiang P., Liu J., Zhou X., Cai W. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. Biol. Sci. 2017;284:20171223. doi: 10.1098/rspb.2017.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H., Miller S.F., Williams E., Lynch M. Specificity of the DNA mismatch repair system (MMR) and mutagenesis bias in bacteria. Mol. Biol. Evol. 2018;35:2414–2421. doi: 10.1093/molbev/msy134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Chan P.P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–W57. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan J.-B., Chen W., Hasegawa D.K., Simmons A.M., Wintermantel W.M., Ling K.-S., Fei Z., Liu S.-S., Douglas A.E. Metabolic coevolution in the bacterial symbiosis of whiteflies and related plant sap-feeding insects. Genome Biol. Evol. 2015;7:2635–2647. doi: 10.1093/gbe/evv170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łukasik P., Nazario K., Van Leuven J.T., Campbell M.A., Meyer M., Michalik A., Pessacq P., Simon C., Veloso C., McCutcheon J.P. Multiple origins of interdependent endosymbiotic complexes in a genus of cicadas. Proc. Natl. Acad. Sci. USA. 2018;115:E226–E235. doi: 10.1073/pnas.1712321115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinverni J.C., Werner J., Kim S., Sklar J.G., Kahne D., Misra R., Silhavy T.J. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol. Microbiol. 2006;61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- Mao M., Bennett G.M. Symbiont replacements reset the co-evolutionary relationship between insects and their heritable bacteria. ISME J. 2020;14:1384–1395. doi: 10.1038/s41396-020-0616-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao M., Yang X., Bennett G.M. Evolution of host support for two ancient bacterial symbionts with differentially degraded genomes in a leafhopper host. Proc. Natl. Acad. Sci. USA. 2018;115:E11691–E11700. doi: 10.1073/pnas.1811932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao M., Yang X., Poff K., Bennett G. Comparative genomics of the dual-obligate symbionts from the treehopper, entylia carinata (Hemiptera: membracidae), provide insight into the origins and evolution of an ancient symbiosis. Genome Biol. Evol. 2017;9:1803–1815. doi: 10.1093/gbe/evx134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura Y., Moriyama M., Łukasik P., Vanderpool D., Tanahashi M., Meng X.-Y., McCutcheon J.P., Fukatsu T. Recurrent symbiont recruitment from fungal parasites in cicadas. Proc. Natl. Acad. Sci. USA. 2018;115:E5970–E5979. doi: 10.1073/pnas.1803245115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J.P., Boyd B.M., Dale C. The life of an insect endosymbiont from the cradle to the grave. Curr. Biol. 2019;29:R485–R495. doi: 10.1016/j.cub.2019.03.032. [DOI] [PubMed] [Google Scholar]
- McCutcheon J.P., McDonald B.R., Moran N.A. Convergent evolution of metabolic roles in bacterial co-symbionts of insects. Proc. Natl. Acad. Sci. USA. 2009;106:15394–15399. doi: 10.1073/pnas.0906424106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J.P., McDonald B.R., Moran N.A. Origin of an alternative genetic code in the extremely small and GC–rich genome of a bacterial symbiont. PLoS Genet. 2009;5:e1000565. doi: 10.1371/journal.pgen.1000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J.P., Moran N.A. Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- McCutcheon J.P., Moran N.A. Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol. Evol. 2010;2:708–718. doi: 10.1093/gbe/evq055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J.P., Moran N.A. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc. Natl. Acad. Sci. USA. 2007;104:19392–19397. doi: 10.1073/pnas.0708855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J.P., von Dohlen C.D. An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Curr. Biol. 2011;21:1366–1372. doi: 10.1016/j.cub.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKight P.E., Najab J. Kruskal-Wallis Test. The Corsini Encyclopedia of Psychology. 2010 doi: 10.1002/9780470479216.corpsy0491. [DOI] [Google Scholar]
- Michalik A., Castillo Franco D., Kobiałka M., Szklarzewicz T., Stroiński A., Łukasik P. Alternative transmission patterns in independently acquired nutritional cosymbionts of dictyopharidae planthoppers. mBio. 2021;12:e0122821. doi: 10.1128/mBio.01228-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A., Moran N.A. Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria. Microb. Ecol. 2002;44:137–143. doi: 10.1007/s00248-002-0012-9. [DOI] [PubMed] [Google Scholar]
- Monnin D., Jackson R., Kiers E.T., Bunker M., Ellers J., Henry L.M. Parallel evolution in the integration of a co-obligate aphid symbiosis. Curr. Biol. 2020;30:1949–1957.e6. doi: 10.1016/j.cub.2020.03.011. [DOI] [PubMed] [Google Scholar]
- Moran N.A. Symbiosis as an adaptive process and source of phenotypic complexity. Proc. Natl. Acad. Sci. USA. 2007;104:8627–8633. doi: 10.1073/pnas.0611659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N.A. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran N.A., Bennett G.M. The tiniest tiny genomes. Annu. Rev. Microbiol. 2014;68:195–215. doi: 10.1146/annurev-micro-091213-112901. [DOI] [PubMed] [Google Scholar]
- Moran N.A., McLaughlin H.J., Sorek R. The dynamics and time scale of ongoing genomic erosion in symbiotic bacteria. Science. 2009;323:379–382. doi: 10.1126/science.1167140. [DOI] [PubMed] [Google Scholar]
- Moran N.A., Tran P., Gerardo N.M. Symbiosis and insect diversification: an ancient symbiont of sap-feeding insects from the bacterial phylum bacteroidetes. Appl. Environ. Microbiol. 2005;71:8802–8810. doi: 10.1128/AEM.71.12.8802-8810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers K.N., Conn D., Brown A.M.V. Essential amino acid enrichment and positive selection highlight endosymbiont’s role in a global virus-vectoring pest. mSystems. 2021;6 doi: 10.1128/mSystems.01048-20. e01048-20–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabachi A., Shigenobu S., Sakazume N., Shiraki T., Hayashizaki Y., Carninci P., Ishikawa H., Kudo T., Fukatsu T. Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc. Natl. Acad. Sci. USA. 2005;102:5477–5482. doi: 10.1073/pnas.0409034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabachi A., Yamashita A., Toh H., Ishikawa H., Dunbar H.E., Moran N.A., Hattori M. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. doi: 10.1126/science.1134196. [DOI] [PubMed] [Google Scholar]
- Nikolaeva D.D., Gelfand M.S., Garushyants S.K. Simplification of ribosomes in bacteria with tiny genomes. Mol. Biol. Evol. 2021;38:58–66. doi: 10.1093/molbev/msaa184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R., Olson R., Pusch G.D., Olsen G.J., Davis J.J., Disz T., Edwards R.A., Gerdes S., Parrello B., Shukla M., et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42:D206–D214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A., Verghese B., Otta S.K. Detecting the form of selection in the outer membrane protein C of Enterobacter aerogenes strains and Salmonella species. Microbiol. Res. 2009;164:282–289. doi: 10.1016/j.micres.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Patiño-Navarrete R., Moya A., Latorre A., Peretó J. Comparative genomics of Blattabacterium cuenoti: the frozen legacy of an ancient endosymbiont genome. Genome Biol. Evol. 2013;5:351–361. doi: 10.1093/gbe/evt011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreau J., Zhang B., Maeda G.P., Kirkpatrick M., Moran N.A. Strong within-host selection in a maternally inherited obligate symbiont: Buchnera and aphids. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2102467118. e2102467118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price D.R.G., Duncan R.P., Shigenobu S., Wilson A.C.C. Genome expansion and differential expression of amino acid transporters at the aphid/Buchnera symbiotic interface. Mol. Biol. Evol. 2011;28:3113–3126. doi: 10.1093/molbev/msr140. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell L.J. phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol. Evol. 2012;3:217–223. doi: 10.1111/j.2041-210X.2011.00169.x. [DOI] [Google Scholar]
- Roesch Goodman K., Welter S.C., Roderick G.K. Genetic divergence is decoupled from ecological diversification in the Hawaiian nesosydne planthoppers: divergence decoupled from diversification. Evolution. 2012;66:2798–2814. doi: 10.1111/j.1558-5646.2012.01643.x. [DOI] [PubMed] [Google Scholar]
- RStudio Team . 2018. RStudio: Integrated Development Environment for R. RStudio, Inc. [Google Scholar]
- Sabater-Muñoz B., Toft C., Alvarez-Ponce D., Fares M.A. Chance and necessity in the genome evolution of endosymbiotic bacteria of insects. ISME J. 2017;11:1291–1304. doi: 10.1038/ismej.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabol S., Sillero M.A., Iwasaki K., Ochoa S. Purification and properties of initiation factor F3. Nature. 1970;228:1269–1273. doi: 10.1038/2281269a0. [DOI] [PubMed] [Google Scholar]
- Santos-Garcia D., Mestre-Rincon N., Ouvrard D., Zchori-Fein E., Morin S. Portiera gets wild: genome instability provides insights into the evolution of both whiteflies and their endosymbionts. Genome Biol. Evol. 2020;12:2107–2124. doi: 10.1093/gbe/evaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakov D. 2002. The 270 Million Year History of Auchenorrhyncha (Homoptera) [Google Scholar]
- Silva F.J., Santos-Garcia D. Slow and fast evolving endosymbiont lineages: positive correlation between the rates of synonymous and non-synonymous substitution. Front. Microbiol. 2015;6:1279. doi: 10.3389/fmicb.2015.01279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan D.B., Alverson A.J., Wu M., Palmer J.D., Taylor D.R. Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus silene. Genome Biol. Evol. 2012;4:294–306. doi: 10.1093/gbe/evs006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan D.B., Moran N.A. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol. Biol. Evol. 2012;29:3781–3792. doi: 10.1093/molbev/mss180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan D.B., Nakabachi A., Richards S., Qu J., Murali S.C., Gibbs R.A., Moran N.A. Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol. Biol. Evol. 2014;31:857–871. doi: 10.1093/molbev/msu004. https://10.1093/molbev/msu004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.R., Lee R.W. Low nucleotide diversity for the expanded organelle and nuclear genomes of volvox carteri supports the mutational-hazard hypothesis. Mol. Biol. Evol. 2010;27:2244–2256. doi: 10.1093/molbev/msq110. [DOI] [PubMed] [Google Scholar]
- Sogin E.M., Leisch N., Dubilier N. Chemosynthetic symbioses. Curr. Biol. 2020;30:R1137–R1142. doi: 10.1016/j.cub.2020.07.050. [DOI] [PubMed] [Google Scholar]
- Soma A., Ikeuchi Y., Kanemasa S., Kobayashi K., Ogasawara N., Ote T., Kato J.i., Watanabe K., Sekine Y., Suzuki T. An RNA-modifying enzyme that governs both the codon and amino acid specificities of isoleucine tRNA. Mol. Cell. 2003;12:689–698. doi: 10.1016/S1097-2765(03)00346-0. [DOI] [PubMed] [Google Scholar]
- Song N., Liang A.-P., Bu C.-P. A molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS One. 2012;7:e48778. doi: 10.1371/journal.pone.0048778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stever H., Eiben J., Bennett G.M. Hawaiian nysius insects rely on an obligate symbiont with a reduced genome that retains a discrete nutritional profile to match their plant seed diet. Genome Biol. Evol. 2021;13:evab189. doi: 10.1093/gbe/evab189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakaran S., Kost C., Kaltenpoth M. Symbiont acquisition and replacement as a source of ecological innovation. Trends Microbiol. 2017;25:375–390. doi: 10.1016/j.tim.2017.02.014. [DOI] [PubMed] [Google Scholar]
- Sudakaran S., Retz F., Kikuchi Y., Kost C., Kaltenpoth M. Evolutionary transition in symbiotic syndromes enabled diversification of phytophagous insects on an imbalanced diet. ISME J. 2015;9:2587–2604. doi: 10.1038/ismej.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiya D.M., Tran P.L., Dietrich C.H., Moran N.A. Co-cladogenesis spanning three phyla: leafhoppers (Insecta: Hemiptera: cicadellidae) and their dual bacterial symbionts. Mol. Ecol. 2006;15:4175–4191. doi: 10.1111/j.1365-294X.2006.03071.x. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leuven J.T., Mao M., Xing D.D., Bennett G.M., McCutcheon J.P. Cicada endosymbionts have tRNAs that are correctly processed despite having genomes that do not encode all of the tRNA processing machinery. mBio. 2019;10:e01950-18. doi: 10.1128/mBio.01950-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leuven J.T., Meister R.C., Simon C., McCutcheon J.P. Sympatric speciation in a bacterial endosymbiont results in two genomes with the functionality of one. Cell. 2014;158:1270–1280. doi: 10.1016/j.cell.2014.07.047. [DOI] [PubMed] [Google Scholar]
- Waneka G., Vasquez Y.M., Bennett G.M., Sloan D.B. Mutational pressure drives differential genome conservation in two bacterial endosymbionts of sap-feeding insects. Genome Biol. Evol. 2021;13:evaa254. doi: 10.1093/gbe/evaa254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren D.L., Geneva A.J., Lanfear R. RWTY (R We There Yet): an R package for examining convergence of Bayesian phylogenetic analyses. Mol. Biol. Evol. 2017;34:1016–1020. doi: 10.1093/molbev/msw279. [DOI] [PubMed] [Google Scholar]
- Weglarz K.M., Havill N.P., Burke G.R., von Dohlen C.D. Partnering with a pest: genomes of hemlock woolly adelgid symbionts reveal atypical nutritional provisioning patterns in dual-obligate bacteria. Genome Biol. Evol. 2018;10:1607–1621. doi: 10.1093/gbe/evy114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen J.J. Endosymbiont evolution: predictions from theory and surprises from genomes: endosymbiont genome evolution. Ann. N. Y. Acad. Sci. 2015;1360:16–35. doi: 10.1111/nyas.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen J.J. Genome evolution in bacterial endosymbionts of insects. Nat. Rev. Genet. 2002;3:850–861. doi: 10.1038/nrg931. [DOI] [PubMed] [Google Scholar]
- Wilcoxon F. Individual comparisons by ranking methods. Biometrics Bulletin. 1945;1:80–83. [Google Scholar]
- Wilson A.C.C., Duncan R.P. Signatures of host/symbiont genome coevolution in insect nutritional endosymbioses. Proc. Natl. Acad. Sci. USA. 2015;112:10255–10261. doi: 10.1073/pnas.1423305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfit M., Bromham L. Increased rates of sequence evolution in endosymbiotic bacteria and fungi with small effective population sizes. Mol. Biol. Evol. 2003;20:1545–1555. doi: 10.1093/molbev/msg167. [DOI] [PubMed] [Google Scholar]
- Woyke T., Tighe D., Mavromatis K., Clum A., Copeland A., Schackwitz W., Lapidus A., Wu D., McCutcheon J.P., McDonald B.R., et al. One bacterial cell, one complete genome. PLoS One. 2010;5:e10314. doi: 10.1371/journal.pone.0010314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D., Daugherty S.C., Van Aken S.E., Pai G.H., Watkins K.L., Khouri H., Tallon L.J., Zaborsky J.M., Dunbar H.E., Tran P.L., et al. Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol. 2006;4:e188. doi: 10.1371/journal.pbio.0040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T.J., Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Yang Z. Paml 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. https://10.1093/molbev/msm088 [DOI] [PubMed] [Google Scholar]
- Yang Z., Bielawski J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Nielsen R., Goldman N., Pedersen A.M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman E.C. Insects of Hawaii. Vol. 4. University of Hawaii Press,; Honolulu, Hawaii: 1948. Homoptera: Auchenorrhyncha. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sequence data have been deposited with NCBI under the BioProject number: PRJNA816609 and are publicly available as of the date of publication. Sulcia genomes can be found under the accession numbers GenBank: CP093890 to GenBank: CP093909. Nasuia genomes can be found under the accession numbers GenBank: CP094180 to GenBank: CP094197. Insect mitochondrial genomes can be found under the accession numbers GenBank: ON135504 to GenBank: ON135524. These accession numbers are also listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.