

Abstract
The accelerated appearance of drug-resistant bacteria poses an ever-growing threat to modern medicine’s capacity to fight infectious diseases. Gram-positive species such as methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pneumoniae continue to contribute significantly to the global burden of antimicrobial resistance. For decades, the treatment of serious Gram-positive infections relied upon the glycopeptide family of antibiotics, typified by vancomycin, as a last line of defense. With the emergence of vancomycin resistance, the semisynthetic glycopeptides telavancin, dalbavancin, and oritavancin were developed. The clinical use of these compounds is somewhat limited due to toxicity concerns and their unusual pharmacokinetics, highlighting the importance of developing next-generation semisynthetic glycopeptides with enhanced antibacterial activities and improved safety profiles. This Review provides an updated overview of recent advancements made in the development of novel semisynthetic glycopeptides, spanning the period from 2014 to today. A wide range of approaches are covered, encompassing innovative strategies that have delivered semisynthetic glycopeptides with potent activities against Gram-positive bacteria, including drug-resistant strains. We also address recent efforts aimed at developing targeted therapies and advances made in extending the activity of the glycopeptides toward Gram-negative organisms.
Keywords: vancomycin, glycopeptides, antibiotic resistance, semisynthesis, mechanism of action
Introduction: Antimicrobial Resistance and Glycopeptide Antibiotics
The rise of multi-drug-resistant (MDR) bacteria, paired with the decrease in the discovery of novel antibiotics, is a major threat to world health. A recent study reported that 1.27 million deaths were directly attributable to antimicrobial resistance (AMR) in 2019, with an additional 4.95 million deaths estimated to be associated with AMR.1 The Gram-positive pathogens methicillin-resistant Staphylococcus aureus (MRSA) and Streptococcus pneumoniae accounted for 0.5 million deaths alone in 2019.1 Among the therapeutic options available for treatment of such Gram-positive infections, the glycopeptide antibiotics, typified by vancomycin, have been a mainstay for many years.2 While the glycopeptides are among the most potent anti-Gram-positive agents available, resistance to these antibiotics is also widespread, spurring the continued search for new analogues with enhanced activities and safety profiles. From the time of its discovery, vancomycin’s structural complexity tantalized synthetic chemists, posing a monumental challenge that was ultimately met in the late 1990s, when a series of total syntheses were reported.3−5 In the years since, vancomycin has continued to inspire total synthesis efforts most notably aimed at generating new analogues to address and understand resistance.6−9 While total synthesis can provide access to glycopeptide variants otherwise unavailable in nature, semisynthesis currently presents the most practical means for accessing novel analogues in quantities suitable for clinical development. To date, a number of reviews have been published on the broad topic of the glycopeptide antibiotics.10−17 In this Review we provide an updated overview of recent advancements in the field, specifically as relates to the development of novel semisynthetic glycopeptides spanning the period from 2014 to today.
Vancomycin
Vancomycin (1, Figure 1) was discovered in 1952, when a missionary stationed in Borneo provided E. C. Kornfield of Eli Lilly with a soil sample containing Streptomyces orientalis, the microorganism that produces vancomycin.18 Early attempts at purifying vancomycin for clinical use were challenging, leading to the nickname “Mississippi mud” due to the presence of impurities and brown color.18 Success in clinical trials ultimately led to the improved isolation of vancomycin, which derived its name from the word “vanquish”, given its potent antibacterial activity against a variety of Gram-positive strains, including penicillin-resistant S. aureus.18 In 1958, this novel antimicrobial agent was approved for use in the clinic.18 Interestingly, while aspects of vancomycin’s chemical structure were partially assigned by researchers in the 1960s and 1970s,19−22 it was not until 1982—some 30 years after its discovery—that a full structural elucidation was published.23,24 Notably, vancomycin’s clinical application was initially limited due to its less convenient intravenous (IV) route of administration, side effects, and the availability of alternative treatments such as methicillin and other β-lactams antibiotics. However, the rise of drug-resistant pathogens in the 1980s and 1990s, most notably MRSA, led to the emergence of vancomycin as the standard of care for many Gram-positive infections.12 The success of vancomycin subsequently led to the discovery and development of teicoplanin (2, Figure 1) as the only other natural product glycopeptide antibiotic to be used clinically.
Figure 1.
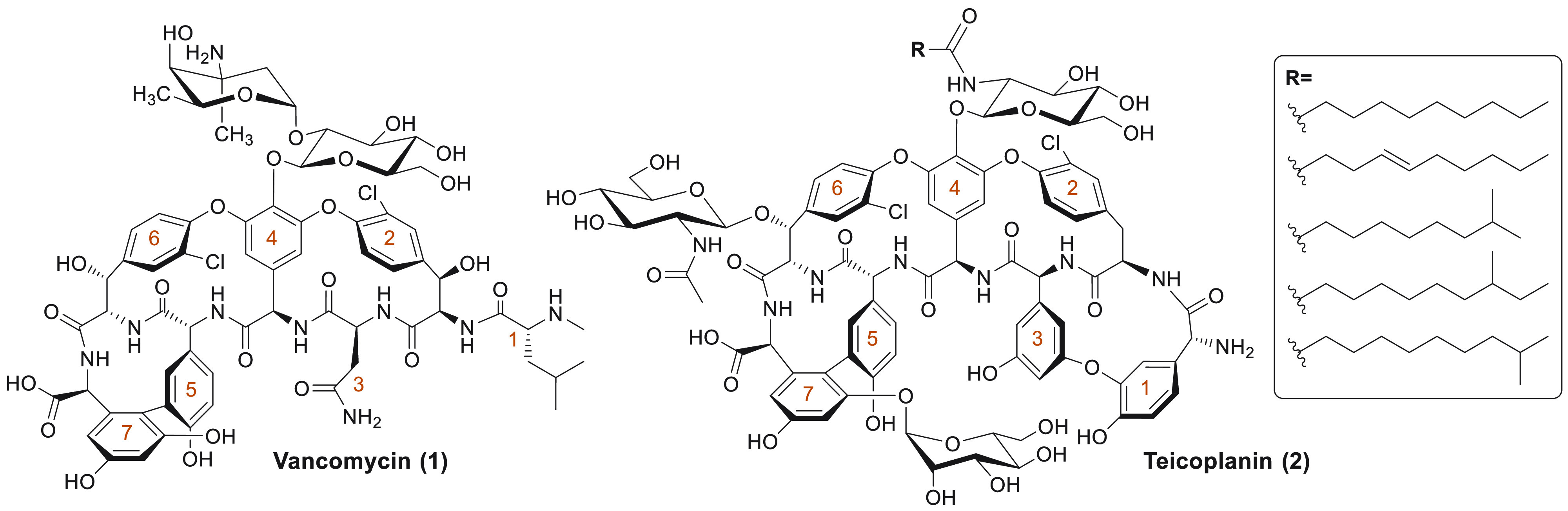
Structures of vancomycin and teicoplanin, the two clinically used natural glycopeptide antibiotics. The amino acids of the peptide are numbered in orange, starting at the N-terminus.
The antibacterial activity of vancomycin is attributable to its capacity to tightly bind the bacterial cell-wall precursor lipid II (Figure 2A) and, in turn, inhibit cell-wall biosynthesis. More specifically, vancomycin interacts with the d-Ala-d-Ala terminus of the lipid II stem pentapeptide via a well-defined network of five hydrogen bonds (Figure 2B). This interaction effectively sequesters lipid II and sterically hinders subsequent transglycosylation and transpeptidation steps, ultimately leading to the inhibition of cell-wall biosynthesis.15,19,25−27 The interaction of vancomycin with its target is further promoted by non-covalent cooperative self-dimerization, which serves to lower the energy barrier required to bind a second lipid II molecule on the bacterial cell surface due to co-localization.28−30
Figure 2.
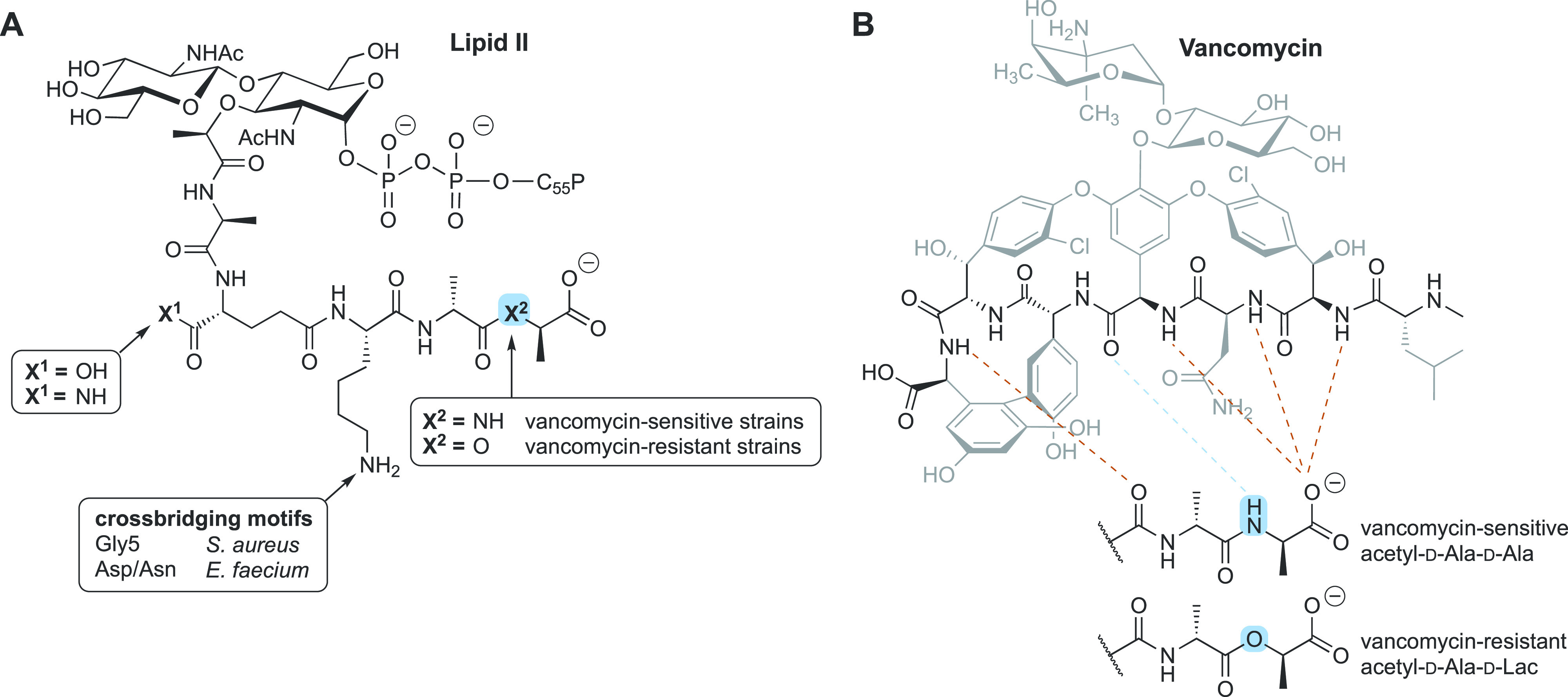
(A) Structures of lipid II found in vancomycin-sensitive and -resistant strains. Features specific to bacterial species and associated resistance are indicated. (B) Binding of vancomycin to d-Ala-d-Ala via hydrogen bonding (dotted lines). Target modification to d-Ala-d-Lac in vancomycin-resistant strains results in loss of one hydrogen bond (indicated in blue).
While the clinical use of vancomycin was accompanied by an increase in the incidence of acquired resistance to it,2 samples of vancomycin-resistant strains date back over 10 000 years ago, also suggesting the presence of an innate resistance reservoir.31 The first vancomycin-resistant enterococci (VRE) strains were reported in Europe and the U.S. in 1986 and 1987, respectively.32−34 Today, multiple vancomycin resistance patterns have been elucidated, with the plasmid-mediated vanA and vanB gene clusters being the predominant drivers. Expression of these resistance operons leads to target modification of the peptidoglycan precursor termini from d-Ala-d-Ala to d-Ala-d-Lac (for vanA, vanB, vanD, vanF, vanM) or d-Ala-d-Ser (for vanC, vanE, vanG, vanL, vanN).2,12,35−38 In the former case, the structural change leads to a >1000-fold reduction in the binding affinity of vancomycin, which can be attributed to the loss of a hydrogen bond (Figure 2B) and, more prominently, to the establishment of strong electrostatic repulsions.39,40 In the latter case, the effect of the d-Ser mutation is less pronounced, as it leads to only a 6-fold reduction in binding affinity.41,42 The vanA resistance operon has also been detected in S. aureus strains (VRSA), although it is not believed to be the main mechanism of resistance in staphylococci.12,43,44 Instead, the reduced vancomycin susceptibility in S. aureus, without the acquisition of foreign genetic material typified by vancomycin-intermediate S. aureus (VISA) and heteroresistant VISA (hVISA) strains, is characterized by thickened cell walls and decreased transpeptidation cross-linking activity. These phenomena lead to the accumulation of monomeric d-Ala-d-Ala-containing decoy targets, effectively hindering vancomycin in reaching the membrane surface.12,25,45−51
Today, vancomycin remains a first-line treatment for a variety of Gram-positive infections, including MRSA (MIC = 0.5–2 μg/mL), S. pneumoniae (MIC = 0.06–2 μg/mL), and Clostridioides difficile infections (MIC = 0.125–4 μg/mL).2,52 Vancomycin has been found effective in the treatment of many conditions, including endocarditis, skin and skin structure infections (SSSIs), bone infections, and airway infections.53 Although vancomycin can be taken orally with the purpose of reaching the colon for the treatment of C. difficile-associated diseases,54 it is preferably administered IV due to its poor oral bioavailability55 and the risk of VRE colonization linked to oral use.54 Vancomycin has a relatively low protein binding (<50%)56−58 and a half-life of 6–12 h in healthy adults,56 and is primarily eliminated unmetabolized (>80%) through renal excretion.56,59 Prolonged and slow infusion with vancomycin is recommended, given that one of the main toxicity concerns associated with its use is the so-called “red-man syndrome”, a histamine-mediated hypersensitivity reaction caused by mast-cell degranulation that predominantly occurs upon rapid infusion.60−62 Vancomycin treatment has also been linked to nephrotoxicity, particularly in patients with moderate to severe renal impairment.63
Teicoplanin
Approximately 30 years after the discovery of vancomycin, the lipoglycopeptide antibiotic teicoplanin (2, Figure 1) was isolated from Actinoplanes teichomyceticus. Subsequently, teicoplanin was approved for clinical use in Europe but never for the U.S. market.15 Its chemical structure, elucidated in 1984,64,65 differs from that of vancomycin in a number of ways, including additional glycosylation sites (at positions 6 and 7), an ether-linked 4-hydroxyphenylglycine portion (position 1), and the presence of a 3,5-dihydroxyphenylglycine residue (position 3). Teicoplanin is most significantly differentiated from vancomycin by the presence of a hydrophobic acyl tail linked to the central monosaccharide moiety (at amino acid 4), which is a non-acylated disaccharide group in vancomycin.66 Notably, the teicoplanin fatty acid motif is actually introduced as a mixture of five related lipids, giving rise to teicoplanin A2-1 through A2-5, the ratio of which can be somewhat dictated by fermentation conditions.67 Generally administered as a mixture of these five similar compounds, teicoplanin has potent antibacterial activity against a variety of Gram-positive strains, including MRSA (MIC = 0.25–2 μg/mL), S. pneumoniae (MIC = 0.06–0.25 μg/mL), and, of particular note, VanB-type VRE (MIC = 0.25–8 μg/mL).52,68
Like vancomycin, teicoplanin binds the d-Ala-d-Ala motif of lipid II through a network of five hydrogen bonds30,69,70 but, unlike vancomycin, does not show cooperative dimerization. Any potential loss of activity due to the lack of teicoplanin self-association appears to be compensated for by the hydrophobic tail, which is hypothesized to anchor the antibiotic into the bacterial membrane, enabling localization of teicoplanin’s glycopeptide core to its lipid II target.30,69 While teicoplanin is generally active against VanB-type VRE strains, in which the resistance phenotype is induced exclusively by vancomycin, for VanA-type VRE and VRSA strains the resistance phenotype is also induced by teicoplanin, rendering the antibiotic inactive.71,72 In line with what is observed for vancomycin, reduced susceptibility to teicoplanin can also occur in a non-plasmid-mediated fashion in S. aureus, either as vancomycin-susceptible but teicoplanin-resistant MRSA73 or by displaying cross-resistance to vancomycin as in VISA/hVISA,74 typified by cell-wall thickening and overproduction of decoy d-Ala-d-Ala targets.51,75
In Europe, teicoplanin is approved for intravenous and intramuscular use in conditions caused by susceptible Gram-positive infections, including SSSIs, endocarditis, complicated urinary tract infections, bone and joint infections, pneumonia, and bacteremia.76 Furthermore, oral formulations are available to treat C. difficile infections.76 As opposed to vancomycin, the hydrophobic tail makes teicoplanin highly plasma protein bound (90%),77 and this feature is responsible for the long half-life of 100–170 h.76 Like vancomycin, teicoplanin is primarily excreted renally as the unchanged drug (80%).76 However, it is considered to have a more favorable toxicity profile compared to vancomycin, given the lower overall occurrence of adverse events, including reduced nephrotoxicity, and its limited propensity to promote histamine release.61,78,79
Clinically Used Semisynthetic Lipoglycopeptide Antibiotics
The discovery of the natural lipoglycopeptide teicoplanin spiked interest in the development of semisynthetic lipoglycopeptide antibiotics. To date, three members of this class have been approved for clinical use: telavancin (3), dalbavancin (4), and oritavancin (5) (Figure 3). As noted above, a number of review articles covering the development of glycopeptide antibiotics, including telavancin, dalbavancin, and oritavancin, have been published over the years.10−13 However, given that these compounds present examples of successfully developed semisynthetic glycopeptide antibiotics, we will here also briefly touch upon their approval, structure, antibacterial activity, mechanism of action, resistance, clinical indications, pharmacokinetics (PK), and toxicity.
Figure 3.
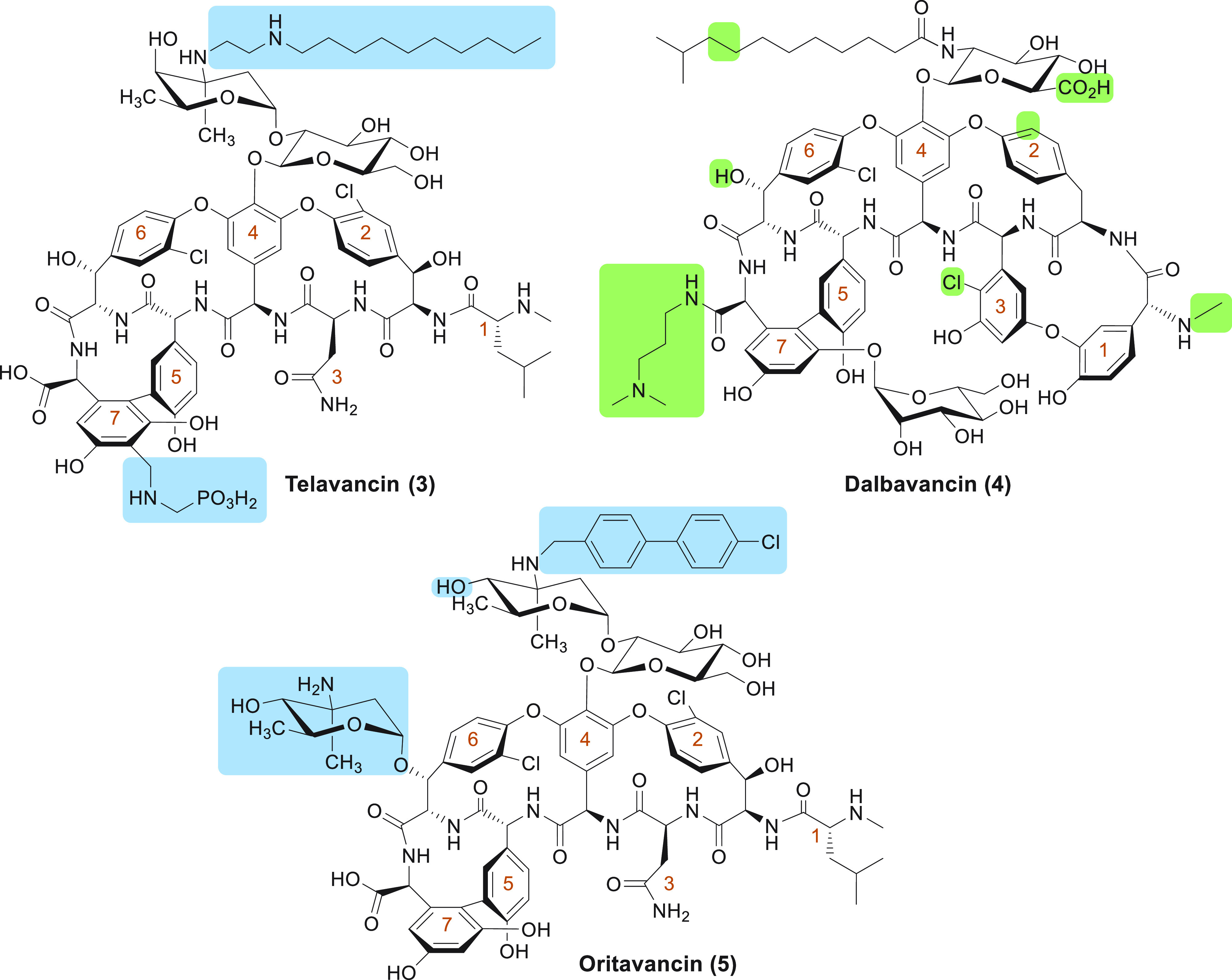
Clinically used lipoglycopeptide antibiotics. Structural differences of telavancin and oritavancin compared to vancomycin are indicated in blue. Structural differences of dalbavancin compared to teicoplanin are indicated in green. The amino acids of the peptides are numbered in orange, starting at the N-terminus.
Telavancin
Telavancin (Vibrativ, 3, Figure 3), developed by Theravance Inc., was introduced to the clinic in 2009.80 It is the only clinically approved semisynthetic glycopeptide antibiotic derived from vancomycin and differs most significantly from its parent structure by the decylaminoethyl modification on the vancosamine unit, a modification that is responsible for telavancin’s enhanced potency against Gram-positive strains.81,82 This modification alone was found to introduce unfavorable excretion and distribution properties, and so an additional (phosphonomethyl)aminomethyl moiety was appended to ring 7, leading to an improved ADME profile.81,82 Telavancin is active against a variety of Gram-positive species, including MRSA (MIC = 0.016–0.125 μg/mL), VanB-type VRE (MIC = 2 μg/mL), and S. pneumoniae (MIC = 0.008–0.03 μg/mL).52,83,84 Unlike teicoplanin, it is also potent against VISA strains.84,85
Telavancin has a dual mode of action. First, it retains the mechanism of action of vancomycin by binding lipid II and thereby inhibiting bacterial cell wall biosynthesis.86,87 This interaction is promoted by the decylaminoethyl lipid, which anchors into the cytoplasmic membrane and brings telavancin into close proximity with peptidoglycan precursors. As a consequence, telavancin displays a higher binding affinity for the bacterial cell surface and increased inhibition of transglycosylation.88 Telavancin’s lipid moiety is also responsible for a secondary mode of action, namely the concentration-dependent dissipation of bacterial cell membrane potential (at 10-fold MIC), leading to membrane permeabilization and leakage of ATP and potassium ions.13,86,88 Telavancin displays a low propensity to induce spontaneous resistance in staphylococci and enterococci.89 Similar to teicoplanin, telavancin does not induce vanB, but it does effectively induce the vanA resistance operon.13 Although this leads to reduced telavancin susceptibility in VanA-type strains, this moderate increase in MIC (from ≤2 to 4–16 μg/mL)84 is not as drastic as the complete loss of activity seen for vancomycin and teicoplanin against these strains.84,90,91
Telavancin is approved to treat complicated SSSIs caused by susceptible Gram-positive species such as S. aureus, Streptococcus agalactiae, Streptococcus pyogenes, and Enterococcus faecalis.80,85,92,93 Furthermore, telavancin has been approved to treat hospital-acquired and ventilator-associated pneumonia when alternative treatment is not suitable.93,94 Due to its poor oral bioavailability, telavancin is administered IV. It is extensively plasma protein bound (93%) and has a half-life of approximately 7–9 h in healthy adults, enabling once-a-day dosing.85,93,95,96 Telavancin is mainly excreted through the kidneys as the intact drug (∼70%),13 which results in extended half-lives for patients with renal dysfunction, potentially leading to adverse effects.97 In relation to that, telavancin was issued a black-box warning from the FDA due to its associated nephrotoxicity concerns as well as for pregnancy-related toxicity.93,98
Dalbavancin
Dalbavancin (Dalvance, 4, Figure 3) was brought to market by Durata Therapeutics/Allergan in 2014. This semisynthetic glycopeptide is synthesized from the natural product A40926, which has a teicoplanin-like structure.99 However, A40926 still has significant differences in its glycopeptide core compared to teicoplanin, including the presence of a terminal methylamino group at the N-terminus (amino acid 1), the location of a chlorine atom at ring 3 rather than ring 2, decoration of residue 4 with an N-acylaminoglucuronic acid carbohydrate rather than with an N-acylglucosamine, and finally the absence of the acetylglucosamine at position 6. Furthermore, the hydrophobic acyl tail is one carbon atom longer compared to that of teicoplanin A2-5 (Figure 3). Dalbavancin is synthesized from A40926 by a three-step sequence, resulting in amidation of the C-terminus with 3-(dimethylamino)-1-propylamine.100 Dalbavancin exhibits potent activity toward Gram-positive strains, including MRSA (MIC = 0.06–1 μg/mL), streptococci (MIC ≤0.03 μg/mL), and VanB-type VRE (MIC ≤0.03–4 μg/mL).52,101−104
As for other glycopeptide antibiotics, dalbavancin binds to the d-Ala-d-Ala termini of cell wall precursors. While dalbavancin’s hydrophobic acyl tail may play a role similar to that found for teicoplanin in membrane anchoring and localization,11 the cationic dimethylaminopropyl moiety is also believed to interact with the negatively charged phospholipid head groups of the bacterial surface.105 Interestingly, while vancomycin dimerization is cooperative and favored upon ligand binding, dalbavancin adopts a closed conformation upon interaction with lipid II, subsequently preventing dimerization.105,106In vitro selection for resistance to dalbavancin has also been successfully demonstrated employing a S. aureus strain, although resistance was slower to appear than for vancomycin and teicoplanin.107−109 Also of note, dalbavancin-induced non-susceptible VSSA and VISA strains have also been isolated from patients; however, such accounts remain relatively uncommon.110,111 In line with the features of the previously discussed lipoglycopeptide antibiotics, dalbavancin is potent against VanB-type VRE strains103 but ineffective against VanA-type strains, as it induces the vanA operon.103 Furthermore, continuous exposure to sub-lethal dalbavancin concentrations does cause resistance selection to dalbavancin in vitro in VanB-type VRE over a 20-day period (MIC from 0.12 to >16 μg/mL).112
At present, dalbavancin is only clinically approved for the treatment of acute bacterial SSSIs,113 although it is increasingly used off-label for endocarditis and osteomyelitis.114 Similarly to other lipoglycopeptides, dalbavancin is administered IV due to its poor oral bioavailability. It has high plasma protein binding (93–98%) and displays unusual PK properties, with half-lives spanning multiple days (8.5 days),113,115 resulting in once-a-week dosing. Dalbavancin has a long elimination time, eventually being excreted as unaltered drug through feces (20%, 70 days) and urine (33%) or as the hydroxyl-dalbavancin metabolite through renal clearance (12%, 42 days).113,116 Despite its unusual PK properties, dalbavancin has an acceptable safety profile and is suited for use in patients with hepatic or mild to moderate renal impairment, with dose adjustment only required for patients with severe renal impairment.10,116,117
Oritavancin
Oritavancin (Orbactiv, 5, Figure 3) was originally developed by Eli Lilly118 and eventually brought to the clinic by The Medicines Company in 2014.12 It is derived from the naturally occurring glycopeptide chloroeremomycin and is generated semisynthetically by attachment of the 4′-chlorobiphenylmethyl group to the disaccharide moiety. Compared to vancomycin, oritavancin also bears an additional 4-epi-vancosamine monosaccharide unit attached to amino acid 6.118 Oritavancin has potent antibacterial activity against MRSA (MIC ≤0.008–0.5) as well as against both vancomycin-sensitive (MIC ≤0.008–0.25 μg/mL) and -resistant enterococci (MIC VanA ≤0.008–1, VanB ≤0.008–0.03).52,119
Besides the classical glycopeptide mechanism of action resulting from its binding to the d-Ala-d-Ala terminus of lipid II, oritavancin’s enhanced activity relative to vancomycin is ascribed to its ability to engage with secondary binding sites on lipid II. Specifically, in S. aureus and Enterococcus faecium, oritavancin is reported to also bind to the pentaglycine (Gly5) and the Asp/Asn crossbridge portion of lipid II, respectively (Figure 2B). As a result, its antibacterial activity is significantly increased and maintained even in the case of VRE strains which produce modified d-Ala-d-Lac peptidoglycan building blocks.120−123 Interestingly, in the case of VRSA, while the Gly5 bridge is largely absent,124 binding of oritavancin to the amidated α-carbonyl group of the d-glutamate residue at position 2 of lipid II appears to compensate for the loss of the key hydrogen bond associated with the d-Ala-d-Lac form of lipid II.123 The enhanced affinity for amidated d-Ala-d-Ala lipid II-Gly5 compared to unmodified lipid II suggests that oritavancin’s ability to target additional binding sites is responsible for its increased potency against vancomycin-sensitive strains as well.123 Furthermore, the tendency of oritavancin to form tight homodimers increases its affinity for the target sites.122,125,126 In addition to its enhanced lipid II binding, the 4′-chlorobiphenylmethyl substituent of oritavancin is thought to be involved in anchoring to the bacterial membrane, leading to localization of the antibiotic in close proximity to the membrane as well as causing dissipation of the membrane potential.125,127−129 Owing to its multiple modes of action, oritavancin retains activity against VRSA and VanA-type VRE, as opposed to the other clinically used glycopeptide antibiotics.130−132 Its multiple mechanisms of action could also lead to a lower propensity to induce resistance: while in vitro oritavancin resistance induction has been observed,112,133in vivo oritavancin non-susceptible strains have not been reported to date.13,134
Oritavancin is used clinically to treat acute bacterial SSSIs in adults caused by a variety of Gram-positive strains, including MRSA and enterococci.135 It is typically administered IV, displays high protein binding (>85%), and has a long half-life of 245–393 h (10.3 days), which allows for single dosing.135,136 Oritavancin has high tissue accumulation and prolonged retention (mainly in the liver, ≥59%), resulting in slow excretion from tissue sites, with only <5% and 1% (unmetabolized) recovery in urine and feces, respectively, after 7 days.137 While oritavancin generally shows low incidence of serious adverse events, when compared with a vancomycin treatment group, patients treated with oritavancin did experience higher rates of osteomyelitis as a side effect.135,138,139 Oritavancin is therefore not approved for the treatment of bone or bone marrow infections, and given its long terminal half-life, patients should be monitored for signs and symptoms of osteomyelitis following treatment with oritavancin.135,138
Recent Developments in Semisynthetic Glycopeptide Antibiotics
Glycopeptide Modification Sites and Chemistry
In addition to the chemical modifications associated with the clinically used semisynthetic glycopeptide antibiotics described above, many other approaches have been explored toward the development of novel semisynthetic glycopeptides. For extensive reviews on such glycopeptide derivatives, including discoveries before 2014, we refer the reader to the previous literature.14−17 The present Review focuses on recent advancements in the discovery of new semisynthetic glycopeptide antibiotics reported in the interval between 2014 and the present. The structural modifications made in generating novel semisynthetic glycopeptides occur largely at four defined positions: the vancosamine primary amino group (Vv), the C-terminus (Vc), the N-terminus (Vn), and the resorcinol moiety (Vr) (Figure 4). While these positions are most readily modified, structural elaboration at other sites has also been reported.140 The introduction of substituents at the vancosamine (Vv) motif typically relies on the selective modification of the primary amine by means of reductive amination using aldehyde-functionalized compounds. The C-terminus (Vc) is readily altered by coupling of an amine to the carboxylic acid by means of peptide bond formation. Similarly, the N-terminus (Vn) can be conjugated to carboxylic acids using strategies for making amides. Finally, the resorcinol moiety (Vr) can be functionalized using the Mannich reaction with formaldehyde and the desired amine. These four modification sites have been used to introduce a wide diversity of structural modifications aimed at (1) improving binding to the bacterial cell surface, (2) enabling multiple modes of action by adding additional binding moieties, (3) driving glycopeptide dimerization to enhance localization to the target site, (4) delivering the drug to specific target sites in the body, and (5) expanding the antibacterial spectrum of activity toward Gram-negative strains.
Figure 4.

Main modification sites on vancomycin. Modifications on vancomycin are common on the vancosamine (Vv), the C-terminus (Vc), the N-terminus (Vn), and the resorcinol (Vr). The amino acids of the peptide are numbered in orange, starting at the N-terminus.
Cationic (Lipo)glycopeptide Antibiotics with Enhanced Bacterial Surface Binding
Design strategies aimed at conferring semisynthetic glycopeptides with activity against vancomycin-resistant strains are usually focused on enhancing their binding to the bacterial cell surface. One of the most common approaches employed to achieve this goal is the inclusion of lipophilic substituents, as seen in the clinically used lipoglycopeptides, and/or the installation of cationic moieties that are positively charged at physiological pH, as a means of generating favorable interactions with the negatively charged bacterial cell surface. To this end, in 2014 the group of Haldar, one of the key players in the lipoglycopeptide field, appended a lipid tail to the vancosamine position and a lactobionolactone moiety to the C-terminus of vancomycin to generate compound 6 (Figure 5).141 Compound 6 shows potent in vitro activity against MRSA (MIC = 0.4 μg/mL) and VRE (MIC = 1.4–2 μg/mL) (see Table 1 for a comparative overview of the activity of the semisynthetic glycopeptides covered in this Review). Shortly thereafter, the same group conjugated two different lipophilic ammonium moieties to the C-terminus of vancomycin, yielding analogues 7 and 8 (Figure 5).142 Compound 8 shows potent in vitro bactericidal activity against MRSA (MIC = 1.1 μg/mL) and VanA-type VRE (MIC = 1.2 μg/mL) (Table 1). The enhanced potency against vancomycin-resistant strains was proposed by the authors to be due to the presence of a permanent positive charge. Subsequently, the Haldar group refined their previous findings by combining the strategies used for 6 (addition of a lipid and a carbohydrate) and compounds 7 and 8 (installation of a permanent cationic lipid), culminating in the development of the lipidated pyridinium analogue 9 (Figure 5).143 While inclusion of the cationic lipid alone is enough to confer excellent activity against MRSA (MIC = 0.2 μg/mL) and VRE (MIC = 4–10 μg/mL), the added carbohydrate moiety found in 9 further enhances this analogue’s potency against VanA- and VanB-type VRE strains (MIC = 0.2 and 2.7 μg/mL, respectively) (Table 1).143 Furthermore, 9 displays anti-MRSA-biofilm activity that leads to a 3-log titer reduction compared to vancomycin.143 Mechanistically, the lipophilic substituents in 6–9 drive the enhanced potency, while the permanent positive charges found in 7–9 confer membrane-disruptive properties, and the carbohydrate moiety at the C-terminus in 6 and 9 is proposed to enhance d-Ala-d-Lac binding affinity.141−143 Furthermore, analogues 7–9 show no resistance selection against MRSA.142,143 Given that 7 and 9 have the most favorable toxicity profiles,142,143 both compounds were progressed to efficacy studies, where 7 was found to exhibit a more pronounced reduction in MRSA titer in a murine thigh infection model compared to vancomycin and linezolid.144 In addition, 9 outperformed linezolid in a murine VRE kidney infection model by further reducing the bacterial titer 2-log.143 In the case of 7, a series of further studies were aimed at evaluating its efficacy, PK, and toxicity, revealing a 50% effective dose (ED50) of 3.3 mg/kg and a 50% lethal dose (LD50) of 78 mg/kg. Moreover, compound 7 displays a prolonged half-life of 1.6 h, sustained plasma drug concentrations above MIC for at least >4 h, and no major kidney or liver damage.144 More recently, in 2021, Haldar and co-workers developed analogue 10, containing a single-site vancosamine modification consisting of an aryl-ammonium-alkyl substituent, which exhibits bactericidal activity against MRSA (MIC = 1.7 μg/mL), VRSA (MIC = 0.8–3.4 μg/mL), and VRE (MIC = 0.8–6.7 μg/mL) (Figure 5, Table 1) while displaying no hemolysis or mammalian cytotoxicity.145 In addition to binding to d-Ala-d-Ala and delocalizing cell division proteins in cells during the exponential phase, 10 also depolarizes and permeabilizes the membrane of exponential, stationary, and persister cells. Analogue 10, even when used at low concentrations, is able to more effectively reduce the MRSA titer and viability within biofilms compared to vancomycin.145 The results of these in vitro studies were also reflected in in vivo studies in mice, where 10 was found to be tolerated up to at least 55.5 mg/kg and shown to be efficacious in reducing murine MRSA thigh burden by almost 3-log compared to vehicle.145 Finally, analogue 10 was also found to show no resistance induction and a prolonged post-antibiotic effect.145
Figure 5.
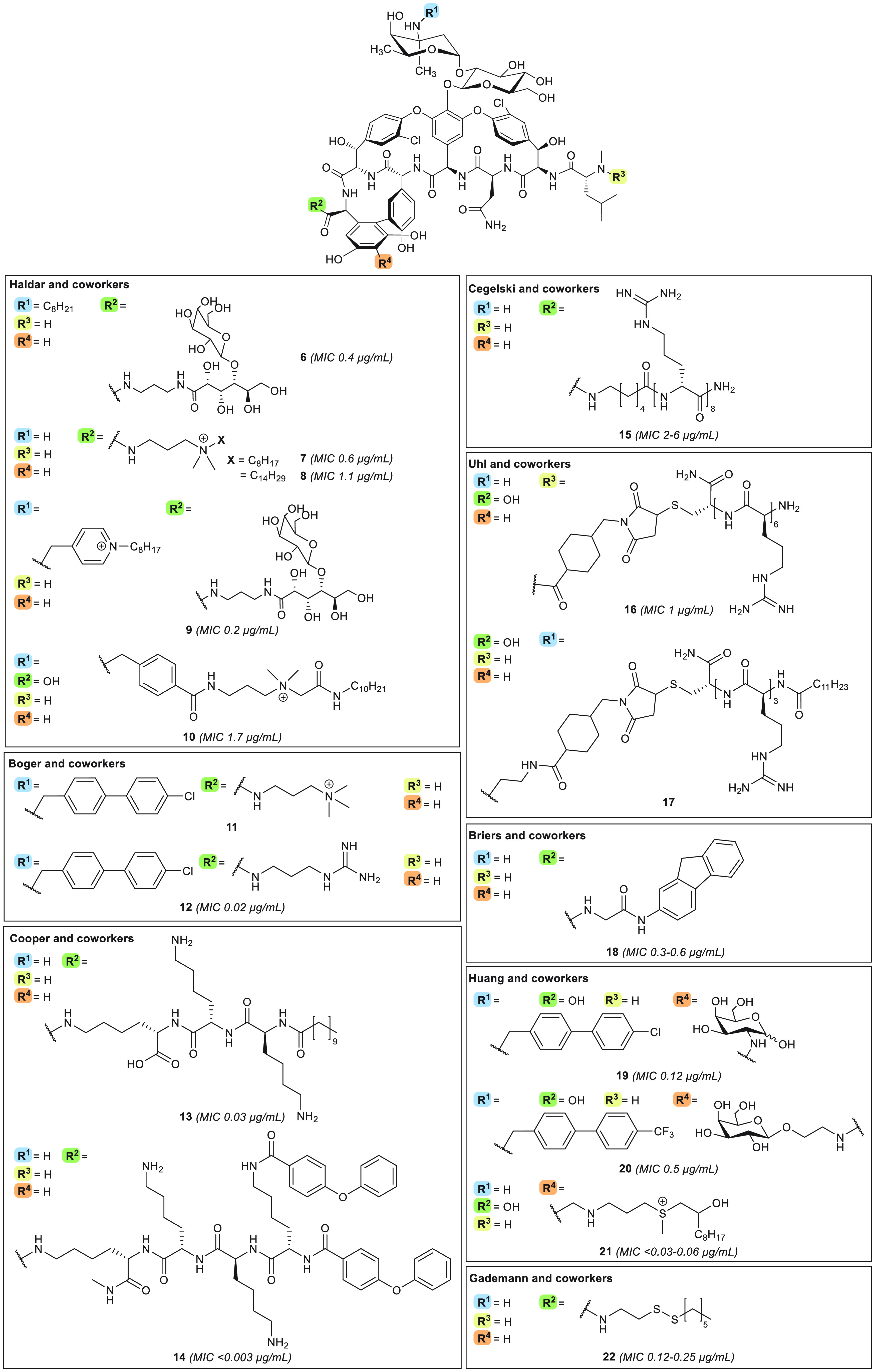
Cationic and/or lipophilic semisynthetic vancomycin analogues with enhanced cell surface binding. Compounds are organized according to research group. MIC values are indicated for MRSA strains, allowing for comparison.
Table 1. In Vitro Antibacterial Activity against Gram-Positive Strainsa.
| MIC
(μg/mL) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| category | compound | MRSA | VanA VRE | VanB VRE | refs | ||||
| Clinically used | vancomycin (1) | 0.5–2b | >32 | >32 | (52) | ||||
| teicoplanin (2) | 0.25–2b | >32 | 0.25–8 | (52, 68) | |||||
| telavancin (3) | 0.016–0.125b | 4–16 | 2 | (52, 84) | |||||
| dalbavancin (4) | 0.06–1 | >32 | ≤0.03–4 | (52, 101−104) | |||||
| oritavancin (5) | ≤0.008–0.5 | ≤0.008–1 | ≤0.008–0.03 | (52, 119) | |||||
| Cationic (lipo)glycopeptide antibiotics with enhanced bacterial surface binding | 6 | 0.4 | 1.4 | 2 | (141) | ||||
| 7 | 0.6 | 23.8 | 2.4 | (142) | |||||
| 8 | 1.1 | 1.2 | nd | (142) | |||||
| 9 | 0.2 | 0.2 | 2.7 | (143) | |||||
| 10 | 1.7 | 0.8–6.7e | (145) | ||||||
| 11 | nd | 0.25–0.5 | nd | (146) | |||||
| 12 | 0.02 | 0.15–0.6 | 0.04 | (149) | |||||
| 13, 14 | 0.03, <0.003 | 6, 0.5 | nd | (151) | |||||
| 15 | 2–6 | 11 | 90 | (152) | |||||
| 16 | 1 | ≤2.7 | <2.7 | (153) | |||||
| 17 | nd | 0.24 | 4.7 | (154) | |||||
| 18 | 0.3–0.6 | 1.3–21 | 5.2 | (155) | |||||
| 19, 20 | 0.12, 0.5 | 2, 0.5–1 | 0.25, ≤0.06 | (156) | |||||
| 21 | ≤0.03–0.06 | 8 | ≤0.0625 | (157) | |||||
| 22 | 0.12–0.25 | 16 | 0.5 | (158) | |||||
| 23 | 0.5 | 0.31 to >20 | 0.31–1.25 | (160, 161) | |||||
| 24 | 0.3 | 0.15–2.5 | 0.15 | (162) | |||||
| 25 | 0.4 | 0.1–12.5 | 0.4 | (163) | |||||
| 26 | 8 | 8 | 4 | (166) | |||||
| 27 | 0.5 | 2 | 1 | (167) | |||||
| 28 | 0.125–1 | ≤4e | (169) | ||||||
| Pyrophosphate targeting | 29 | 0.9 | 3.5 | 2.6 | (173) | ||||
| 30 | 4c | 4 | 4 | (178) | |||||
| Hybrids | 31 | 0.06–8d | 8–16e | (185) | |||||
| 32 | 1.5 | 6.2 | nd | (188) | |||||
| 33 | 0.6 | nd | 0.8 | (190) | |||||
| 34 | 6.25–12.5 | 12.5–25e | (191) | ||||||
| 35 | 4 | 4 | 8 | (192) | |||||
| 36 | 4 | 8 | 4 | (192) | |||||
| Targeted drug delivery | 37 | 0.79f | 28.9g | 28.9g | (200) | ||||
| 38 | 2 | nd | nd | (202) | |||||
| 39 | nd | nd | nd | (205) | |||||
| 40 | 0.015 | 0.03–2 | 0.03 | (210) | |||||
| Gram-negative active | 8 | 1.1 | 1.2 | nd | (142, 211) | ||||
| 41 | 0.7 | 3.8 | 6.9 | (212) | |||||
| 42 | 15–30 | nd | nd | (213) | |||||
| 43 | 8c | 32 | nd | (214) | |||||
| 44 | 0.25 | 64 to >128 | 2–64 | (215) | |||||
| 45 | 0.8d | nd | nd | (216) | |||||
| 46 | 0.5 | nd | nd | (217) | |||||
| 47 | nd | nd | nd | (218) | |||||
| 48 | 4d | nd | nd | (219) | |||||
MIC = minimum inhibitory concentration. nd = not determined.
MIC values of >10 observations are included in the reported MIC range from EUCAST.52
MRSA strain tested was also VISA.
No MRSA strain was tested; therefore, an MIC range for MSSA is indicated here.
MIC reported is from VRE in general, as literature did not specify VanA- or VanB-type resistance. Note the possibility of solely one van resistance type being present.
Low-density loading of nanoparticles (0.2 μg/mL vancomycin per 1 mg of 37).
High-density loading of nanoparticles (11.75 μg/mL vancomycin per 1 mg of 37).
In 2017, Boger and co-workers appended the 4′-chlorobiphenylmethyl (CBP) unit, also found in oritavancin, to the vancosamine site of vancomycin and added a quaternary ammonium at the C-terminus (Figure 5). These modifications resulted in compound 11, which was found to display in vitro antibacterial activity against VanA-type VRE (MIC = 0.25–0.5 μg/mL) (Table 1).146 Analogue 11 also binds the d-Ala-d-Ala motif of lipid II, inhibits cell wall biosynthesis via direct competitive inhibition of transglycosylases (owing to the CBP motif), rapidly permeabilizes and depolarizes the bacterial cell membrane (by virtue of the trimethylammonium portion), and binds to teichoic acids (due to the trimethylammonium moiety).146−148 In a follow-up publication, the same group further optimized compound 11 by retaining the CBP unit but replacing the trimethylammonium group with a guanidine moiety, hypothesized to serve as a beneficial hydrogen bond donor, to yield analogue 12.149 Analogue 12 was found to display in vitro potency against MRSA (MIC = 0.02 μg/mL), VanA-type VRE (MIC = 0.15–0.6 μg/mL), and VanB-type VRE (MIC = 0.04 μg/mL) (Figure 5, Table 1). Mechanistically, compounds 11 and 12 are comparable149 and share the key feature of a positively charged substituent (at physiological pH) situated at the vancomycin C-terminus. The importance of this structural trait is demonstrated by the fact that relocating motifs of a cationic nature elsewhere on the antibiotic core does not enhance potency and only slightly alters the initial rate of membrane permeabilization.147,149,150 While both analogues showed no mammalian cytotoxicity146,148 and exhibited good in vivo tolerability (≥50 mg/kg in mice),146,149 compound 12 appears superior to 11 by virtue of having (1) a lower propensity to induce resistance against VRE (>10-fold MIC increase for 11, marginal changes for 12)148,149 and (2) superior in vivo efficacy in a murine VRSA thigh infection model at 12.5 mg/kg (4-log versus 5-log reduction for 11 and 12, respectively, when compared to vancomycin).148,149 The half-lives of 11 and 12 in mice are 6–7 and 4.3 h, respectively.
Also with an eye to introducing cationic and lipophilic features onto the vancomycin core, Blaskovich and Cooper designed the vancaptins.151 The vancaptins feature an additional C-terminal peptide, bearing numerous positively charged functionalities, followed by a lipophilic membrane-insertive element, and are represented by compounds 13 and 14 (Figure 5). Against MRSA, the vancaptins were found to be 20- to 100-fold more active than vancomycin and daptomycin (MIC 13 and 14 < 0.003–0.03 μg/mL) (Table 1), along with having enhanced potencies against VISA (0.125–0.5 μg/mL), VRSA (0.08–1 μg/mL), S. pneumoniae (<0.003–0.06 μg/mL), and VanA-type VRE (0.5–6 μg/mL).151 These in vitro data were also found to correlate well with the in vivo activity of the vancaptins, where treatment with 13 and 14 led to 100% survival in a S. pneumoniae murine infection model. Furthermore, 13 was shown to effectively reduce murine MRSA thigh burden by 6-log compared to vehicle when employing a dose 8 times lower than that required of vancomycin to gain the same effect. Interestingly, compound 14 was found to be less effective in vivo, which was ascribed to its high protein binding, given that PK studies indicated that both 13 and 14 reach an in vivo concentration above their MIC values for more than 8 h. Additionally, the vancaptins were shown to be bactericidal, non-hemolytic, and non-toxic to mammalian cells (CC50 ≥ 100 μM) and to cause minimal resistance induction in MRSA. Mechanistic studies further revealed that the vancaptins exert their antibiotic effect through multiple modes of action by (1) inhibiting cell-wall biosynthesis by binding to d-Ala-d-Ala, (2) increasing membrane binding and cooperative dimerization similar to vancomycin, and (3) depolarizing and perturbing the cell membrane (most prominently in the case of compound 14).151
While the strategies described above mainly focused on appending cationic and lipophilic substituents to vancomycin, other groups have opted to focus solely on the introduction of additional positive charges, leading to conjugation of polyarginine motifs to vancomycin as in analogues 15(152) and 16(153) (Figure 5). To this end, the groups of Wender and Cegelski generated 15, modified at the C-terminal position with an octaarginine peptide, which was found to exhibit good potency against MRSA (MIC = 2–6 μg/mL).152 Using a similar approach, Uhl and co-workers examined the effect of introducing a hexaarginine moiety at the four different sites of vancomycin indicated in Figure 4.153 This led to identification of the N-terminally modified 16 as the most potent variant, with good activity against MRSA (MIC = 1 μg/mL) and VRE (MIC ≤2.7 μg/mL) (Table 1).153 Interestingly, the activity of 16 is not antagonized by d-Ala-d-Ala, suggesting that an alternative mode of action is responsible for the enhanced potency of this derivative.153 The mechanism of action of the hexaarginine-substituted compound is likely similar to that of analogue 15, for which enhanced binding to the membrane, driven by strong electrostatic interactions, facilitates cellular association, along with internalization to give access to intracellular peptidoglycan precursors.152 Additionally, these compounds also display rapid membrane permeabilization, although only during cell growth.152 Both 15 and 16 are active in vivo, with 16 reducing murine MRSA thigh burden similarly to vancomycin.153 Compound 15 was found to display a 6-fold potency enhancement in a murine MRSA biofilm wound model when compared with a similar dose of vancomycin.152 The in vivo anti-biofilm activity of 15 was also demonstrated with in vitro experiments, wherein treatment of pre-formed MRSA biofilms with 15 resulted in significantly reduced cell viability to 8.4% after 5 h, compared to 65% viability for vancomycin-treated biofilms. Furthermore, the unique ability of 15 to target biofilms was demonstrated by the finding that combinations of vancomycin with an octaarginine peptide failed to show any anti-biofilm activity.152 Building upon their findings with compound 16, Uhl and co-workers also examined the impact of adding lipophilic moieties by conjugating lipidated triarginine motifs at three different sites on vancomycin (Vv, Vc, Vn). From this series of analogues, vancosamine-modified 17 was found to be the most potent derivative, with MIC = 0.24–4.7 μg/mL against VRE (Figure 5, Table 1). This result is in stark contrast with the finding that, when appending a hexaarginine moiety, the best antibiotic activity was seen for compound 16, modified at the N-terminus.154 Both 16 and 17 are non-hemolytic and non-toxic toward liver and kidney cells. Moreover, in vivo mice experiments with 16 and 17 revealed that the compounds reside in the liver for several hours and do not primarily distribute to the kidneys, unlike vancomycin,153,154 a behavior which could alleviate the risk of nephrotoxicity in patients with renal impairment.63
The design of vancomycin derivatives that focus exclusively on the incorporation of lipophilic moieties has also been explored, resulting, for example, in fluorenyl-substituted compound 18 reported by Briers and co-workers in 2018 (Figure 5).155 Analogue 18 is bactericidal against MRSA (MIC = 0.3–0.6 μg/mL) and bacteriostatic against VanA-type VRE (MIC = 1.3–21 μg/mL) and VanB-type VRE (MIC = 5.2 μg/mL) (Table 1), while displaying low toxicity against mammalian cell lines (CC50 = 172 μM) and minimal resistance selection against VRE.155 In the same year, the Huang group investigated the effect of attaching additional carbohydrate moieties onto lipophilic vancomycin analogues, culminating in compounds 19 and 20, both bearing a carbohydrate substituent at the resorcinol position along with hydrophobic p-Cl- or p-CF3-biphenylmethyl moieties attached at the vancosamine site (Figure 5).156 Both 19 and 20 exhibit strong in vitro activity against MRSA (MIC = 0.12 and 0.5 μg/mL, respectively), VanA-type VRE (MIC = 2 and 0.5–1 μg/mL respectively), and VanB-type VRE (MIC = 0.25 and ≤0.06 μg/mL, respectively) (Table 1). When evaluated in an in vivo murine MRSA survival study, 19 and 20 respectively led to a 14/15 and 13/15 survival after 10 days as well as a >1-log reduction of liver colony-forming units compared to vehicle and vancomycin in a VISA abscess formation assay.156 The in vivo PK properties of compounds 19 and 20 were also assessed, revealing prolonged half-lives (∼3–4 h), with retained plasma concentrations of >1 μg/mL for 4 h. These studies also showed that incorporation of the carbohydrate moiety at the resorcinol position can be used to attenuate the compound’s half-life.156 Mechanistic studies employing NMR and molecular modeling indicate that the added carbohydrate motif might also contribute to antibacterial activity by interaction with d-Ala-d-Ala,156 a finding in line with the enhanced target binding Haldar and co-workers also reported for their carbohydrate-modified analogues 6 and 9.141,143
The Huang group also explored the addition of cationic functionalities to vancomycin, but instead of the commonly employed ammonium or guanidinium moieties, they assessed the effect of adding sulfonium groups.157 The series’ lead compound 21 (Figure 5), consisting of a resorcinol-linked alkyl-sulfonium moiety, was shown to have potent activity against MRSA (MIC ≤0.03–0.06 μg/mL) and VanB-type VRE (≤0.0625) as well as moderate MIC reductions relative to vancomycin against VanA-type VRE (to 8 μg/mL) and Escherichia coli (to 32 μg/mL) (Table 1). Murine MRSA and VRSA infection survival studies found that treatment with 21 led to 13/15 and 12/15 survival, respectively, at 14 days, a significant improvement compared to vancomycin (3/15 survival). To investigate the specific impact of the sulfonium group on PK and toxicity, compound 21 was compared to the corresponding thioether analogue. This showed that 21 has a shorter half-life (1.13 h), an unchanged MIC in the presence of human serum albumin, and less of an effect on mammalian cell viability relative to the thioether.157 The authors hypothesized that analogue 21 interacts with the negatively charged bacterial membrane via the sulfonium motif, subsequently facilitating permeabilization by means of the lipophilic tail. As the thioether-linked compound does not show membrane permeabilization, it can be concluded that the charged sulfonium portion is essential to enable this mechanism of action.157
Gademann and colleagues also designed sulfur-modified vancomycin derivatives, but these do not comprise positively charged substituents.158 Compound 22 (Figure 5), bearing a disulfide-linked lipid at the C-terminal position, was found to possess potent activity against MRSA (MIC = 0.12–0.25 μg/mL), S. pneumoniae (MIC = 0.06 μg/mL), and VanB-type VRE (0.5 μg/mL) (Table 1). Furthermore, 22 was also shown to suppress MRSA and VRE biofilm formation (MBIC = 1 and 2 μg/mL, respectively).158 Given these positive results, it would be interesting to study the influence of the disulfide on PK and toxicity relative to that of the all-carbon-based compound: the potential reductive lability of 22 might be expected to lead to decomposition in vivo to generate more hydrophilic metabolites, thereby reducing tissue accumulation and promoting excretion, as previously noted by researchers at Theravance Inc. working with similar vancomycin analogues.159
In addition to semisynthetic analogues of vancomycin, derivatives of teicoplanin and eremomycin have also been explored in recent years. Herczegh and co-workers designed a series of teicoplanin pseudo-aglycon compounds featuring N-terminal conjugation with various hydrophobic substituents which were introduced through azide–alkyne cycloaddition (Figure 6).160,161 Among the analogues thus prepared, compound 23 was found to have good activity against MRSA (MIC = 0.5 μg/mL) and VanB-type VRE (MIC = 0.31–1.25 μg/mL) (Table 1). Furthermore, some but not all VanA-type VRE isolates were found to be susceptible to this novel teicoplanin derivative (MIC = 0.31 to >20 μg/mL), as well as some strains carrying both vanA and vanB (MIC = 1.25 to >20 μg/mL).161 Optimization of 23 led to compound 24, characterized by the addition of a basic moiety at the C-terminus, which displayed improved activity against VanA-type VRE (MIC = 0.15–2.5 μg/mL) while retaining potency against MRSA (MIC = 0.3 μg/mL) and VanB-type VRE (MIC = 0.15 μg/mL) (Table 1).162 In another attempt to confer anti-VanA-type VRE activity to teicoplanin-like compounds, analogue 25, bearing an N-terminal guanidine moiety, was also synthesized.163 This led to a vast improvement in potency toward vanA VRE isolates, with most strains tested showing susceptibility (MIC = 0.1–1.6 μg/mL) and with only a few strains exhibiting higher MIC values (6.25–12.5 μg/mL). The ability of compound 25 to engage in additional hydrogen bonding via the guanidine moiety is assumed to contribute to the enhanced activity, although experimental evidence in support of this claim is yet to be reported.163 Interestingly, analogue 23 was also found to possess antiviral activity against several influenza strains,160 leading Herczegh and colleagues to design teicoplanin derivatives with structural features aimed at potentiating their antiviral action.164−168 Some of these compounds, modified at the N-terminus with lipophilic moieties linked through a triazole, still retain some antibacterial activity (see compounds 26 and 27) (Figure 6, Table 1).166,167 Of these dual antibacterial and antiviral derivatives, compound 27 displays the most favorable toxicity profile (CC50 = 97–100 μM)160,166,167 while maintaining potent antibacterial activity against MRSA (MIC = 0.5 μg/mL) and VRE (MIC = 1–2 μg/mL).167
Figure 6.
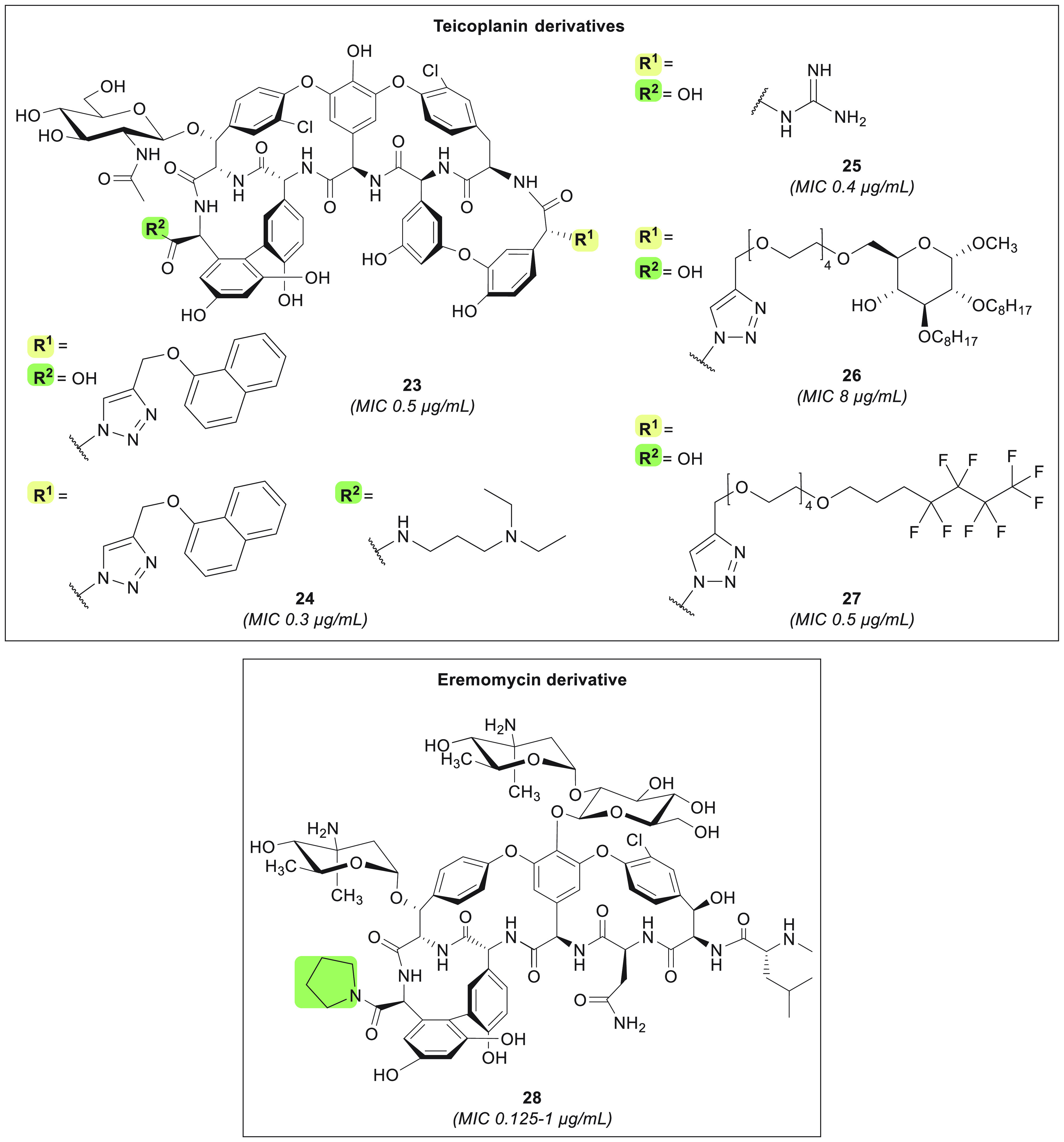
Teicoplanin and eremomycin derivatives with enhanced cell surface binding. MIC values are indicated for MRSA strains, allowing for comparison.
In a study involving the preparation of semisynthetic eremomycin analogues, Olsufyeva et al. showed that coupling small substituents to the C-terminus can be sufficient to enhance potency (Figure 6).169 Using this approach, they identified eremomycin pyrrolidide analogue 28, which was found to exhibit good in vitro activity against MRSA (MIC = 0.125–1 μg/mL) and VRE (MIC ≤4 μg/mL) (Table 1) along with in vivo activity against S. aureus (ED50 = 0.8 mg/kg, 100% survival at 2.5 mg/kg). Moreover, analogue 28 was shown to be superior to vancomycin and eremomycin in a murine sepsis model, maintaining similar in vivo acute toxicity but eliciting reduced histamine release.169
As illustrated in the preceding section, a number of the recently reported semisynthetic glycopeptides exhibit enhanced activity that is associated with an increase in net positive charge most commonly achieved by incorporation of (1) permanently positively charged substituents (e.g., tertaalkylammonium, sulfonium) and/or (2) functional groups that are positively charged at physiological pH (e.g., amine, guanidine). While many of these compounds show promising in vitro and, in some cases, in vivo potency, special attention should be paid to their toxicity and PK profiles. Another structural modification commonly associated with improved antibacterial potency is the introduction of lipophilic substituents that confer these semisynthetic glycopeptides with membrane depolarizing and permeabilizing properties. However, this can also lead to enhanced toxicity and unusual PK behavior. That said, it is possible that such issues can be addressed by structure–relationship activity studies to establish optimal lipid lengths or by the use of reductively labile disulfide-linked lipids. In addition, the introduction of hydrophilic moieties, such as carbohydrates, also provides a means for fine-tuning the PK properties of semisynthetic glycopeptides.
Pyrophosphate-Targeting Glycopeptides
As demonstrated by oritavancin, the design of glycopeptide antibiotics capable of binding to lipid II at multiple sites is a viable strategy for enhancing antibacterial activity: this approach can increase potency against vancomycin-sensitive strains as well as compensate for the loss in binding affinity to the d-Ala-d-Lac motif in vancomycin-resistant strains. One such additional binding site explored in this regard is the pyrophosphate moiety of lipid II, a target that is exploited by natural product antibiotics such as nisin, ramoplanin, and teixobactin.170−172 To this end, Haldar and co-workers reported the design of Dipi-van (29) (Figure 7). Compound 29 bears a C-terminal zinc-binding dipicolyl-1,6-hexadiamine moiety,173 a functionality known to have a high affinity for pyrophosphates.174 Compound 29 was found to exhibit potent activity against VISA as well as VanA-type and VanB-type VRE (MIC = 1.8–3.5 μg/mL) (Table 1),173 an effect that was shown to be further enhanced some 2- to 3-fold by the exogenous addition of Zn2+.173 The expected dual mode of action, based on binding to both the pyrophosphate and the d-Ala-d-Ala motifs of lipid II, was confirmed.173 Analogue 29 displays no resistance selection in MRSA (MIC remained ∼0.9 μg/mL), no hemolytic activity or mammalian cytotoxicity (at 1 mM), and no systemic in vivo toxicity (at 100 mg/kg).173,175 Furthermore, in a murine renal VanB-type VRE infection model, 29 (dosed at 12 mg/kg) reduces the bacterial titer up to 5-log compared to vehicle and 3-log compared to the same dose of vancomycin.173 Interestingly, the Zn2+-binding properties of 29 not only enhance its potency against Gram-positive species but also resensitize several NDM-1-producing Gram-negative strains to meropenem by removing the zinc ions bound to the metallo-β-lactamase, a well-documented mode of action exploited by anti-NDM antibiotic potentiators such as aspergillomarasmine A176 and dipicolinic acid derivatives.177 In this regard, co-administration of vancomycin derivative 29 with meropenem was found to cause a reduction in the MIC of meropenem from >100 to 1.5–3.1 μg/mL in Klebsiella pneumoniae and 12 μg/mL in E. coli (FIC ≤0.5).175 This in vitro synergy was also further substantiated in vivo, specifically in a sepsis model of an NDM-positive K. pneumoniae infection, where a combination treatment of meropenem and compound 29 reduces the bacterial load by 3–4 log compared to vehicle in the liver, kidneys, spleen, and lungs of mice. These results are on par with those obtained with colistin treatment but superior to those gathered using 29 or meropenem monotherapy, which resulted in a maximum 1.5-log reduction in the organs assessed.175
Figure 7.

Pyrophosphate-targeting glycopeptides 29 and 30. Derivative 30 was assessed as a Cu2+ chelation complex as well as a non-metal DPA analogue, in both cases displaying equipotent in vitro activity. MIC values are relative to experiments carried out on MRSA strains.
Huang and co-workers also explored the possibility of developing semisynthetic glycopeptides capable of targeting the pyrophosphate group of lipid II by conjugating Cu2+-dipicolylamine (DPA) complexes to either the resorcinol position or the C-terminus of vancomycin.178 Representative compound 30 (Figure 7) was shown to have enhanced activity against VRE strains (MIC = 4 μg/mL) but not against MSSA and VISA (Table 1).178 A dye displacement assay confirmed that both Cu(II)- and Zn(II)-30 complexes bind to pyrophosphoric acid, suggesting a dual mechanism of action wherein the decreased affinity for d-Ala-d-Lac is compensated for by pyrophosphate binding. Interestingly, the copper-containing 30 and the corresponding metal-free ligand are equipotent in vitro, but the presence of copper results in reduced cell viability (at >50 μM), suggesting that the latter DPA derivative shows more promise.178 Overall, pyrophosphate-targeting glycopeptide derivatives 29 and 30 display significant improvements in VanA-type VRE activity, while maintaining potency against other Gram-positive species.
Glycopeptide Hybrid Antibiotics
Another strategy often explored to achieve antibiotics with a dual mode of action is based on the design of hybrids wherein two different antibiotic molecules are covalently linked together. A suggested benefit of this approach is the reduced likelihood of resistance induction, which is minimized by the inherent difficulties in simultaneously mutating multiple targets.179 Earlier strategies in this field resorted to conjugating glycopeptides to β-lactam antibiotics180−182 or antimicrobial peptides such as nisin(1–12) and tridecaptin.183,184 More recently, the group of Batta and co-workers reported the development of glycopeptide–azithromycin hybrids (Figure 8).185 Coupling azithromycin, a macrolide antibiotic that inhibits the assembly of the 50S ribosomal subunit used to treat Gram-positive infections,186 to the C-terminus of eremomycin resulted in derivative 31, which displays in vitro activity against S. aureus and S. pneumoniae (MIC = 0.06–8 μg/mL) and moderate potency against VRE (MIC = 8–16 μg/mL).185 Compound 31 retains the mechanism of action of the azithromycin fragment and, in an in vitro setting, is 4-fold more potent than vancomycin against S. aureus. During in vivo experiments in a murine sepsis model with the same strain, hybrid 31 was shown to be equipotent to vancomycin, with both having an ED50 of 4 mg/kg.185
Figure 8.

Glycopeptide–azithromycin hybrid. The eremomycin–azithromycin hybrid 31 is the most potent representative of a panel of glycopeptide–azithromycin analogues designed by Batta and co-workers.185 MIC values are relative to experiments carried out on MSSA strains.
In addition to the hybridization of glycopeptides with other antibiotics endowed with a complementary mode of action, covalent homodimerization is another strategy for improving antibacterial potency. An exemplary example of this behavior is inspired by vancomycin, which cooperatively self-associates to form non-covalent dimers as part of its inherent mode of action. The presence of dimers leads to co-localization of the glycopeptide to its target site and reduces the energy required for a second binding event to lipid II, which results in an improved antimicrobial activity.29,30 The fact that this self-association occurs only weakly (700 M–1) in solution187 prompted the scientific community to explore the covalent dimerization of vancomycin, of which the first examples were reported in 1996 by Griffin and colleagues.187 More recently, Haldar and co-workers revisited this approach by synthesizing a number of bis(vancomycin aglycon)carboxamides, which are composed by homodimers of vancomycin aglycon linked through the C-terminus by lipophilic cationic spacers.188 One of the members of this series, compound 32, was found to retain activity against MRSA (MIC = 1–1.5 μg/mL) and displayed a 300-fold enhanced potency against VRE (MIC = 6.2 μg/mL) compared to vancomycin (Figure 9, Table 1).188 The binding affinity of 32 for N,N′-diacetyl-Lys-d-Ala-d-Ala was demonstrated to be similar to that of vancomycin, while notably a >10-fold enhancement toward N,N′-diacetyl-Lys-d-Ala-d-Lac was also measured.188 Interestingly, this result is in stark contrast to the absence of d-Ala-d-Lac binding displayed by previously studied vancomycin dimers, as reported by Ellman and co-workers.189 Further assessment of the activity of dimer 32 in an ex vivo whole blood study showed that 32 (dosed at 2 μM) causes a 1.5-log reduction of bacterial MRSA titer in comparison to vancomycin (dosed at 4 μM), suggesting that antibacterial activity is not significantly impacted by binding to plasma proteins. These results were also in line with the different in vitro killing kinetics the Haldar group observed wherein compound 32 was found to be bactericidal while vancomycin functions as bacteriostatic against higher-inoculum stationary phase MRSA.188
Figure 9.

Glycopeptide dimers. MIC values are relative to experiments carried out on MRSA strains.
Another convenient approach for generating vancomycin dimers is through the use of the copper-catalyzed azido-alkyne cycloaddition (CuAAC), as applied by the group of Sharpless, who prepared a panel of vancomycin homo- and heterodimers characterized by different alkyl and PEG spacers (Figure 9).190 The heterodimers, constructed by linking the C-terminus (Vc) of one vancomycin unit to the vancosamine (Vv) moiety of the other, showed no enhanced potency relative to vancomycin itself. However, in the case of the homodimers prepared, improved activity was observed, with the most potent C-terminal homodimer 33 exhibiting strong in vitro activity against MRSA (MIC = 0.6 μg/mL) compared to vancomycin (MIC = 2.5 μg/mL) (Table 1).190 In addition, 33 is >30-fold more active than vancomycin against a VanB-type VRE strain (MIC = 0.8 μg/mL).190 In a similar study, Sun and colleagues also utilized CuAAC chemistry to obtain covalent glycopeptide dimers typified by compound 34 (Figure 9).191 In preparing their dimers, the Sun group elected to convert the N-terminal amine of demethylvancomycin into the corresponding azide to facilitate dimerization via triazole formation with a variety of bis-alkynes. In addition, a lipophilic group was appended to the vancosamine (Vv) site. The dimers this formed were found to have no enhancement of potency against MRSA and S. pneumoniae (MIC = 6.25–25 μg/mL), whereas against VRE the activity of dimer 34 did exceed that of demethylvancomycin by ≥2–4 fold.191
In another recent report describing glycopeptide dimers, Herczegh and co-workers synthesized and characterized the first teicoplanin pseudo-aglycon N,N-terminal homodimers, 35 and 36 (Figure 9).192 As noted above, unlike vancomycin, teicoplanin does not exhibit cooperative dimerization as part of its mechanism of action. The lack of dimerizing activity for teicoplanin is hypothesized to be due to the presence of the large acyl tail appended to the amino sugar at position 4 (Figure 1), which is speculated to anchor in the bacterial membrane and make binding to nascent lipid II more favorable.29,30 Herczegh and colleagues therefore hypothesized that, by removing this hydrophobic moiety and covalently linking the corresponding pseudo-aglycon, the resulting dimers could have improved activities.192 To this end, two strategies were employed: In the first, the teicoplanin pseudo-aglycon, lacking the carbohydrate at position 4 and bearing a C-terminal diethylaminopropylamide, was dimerized via a PEG linker featuring a lipophilic substituent to yield analogue 35. In the second strategy, a histidine residue was first coupled to the N-terminus of the teicoplanin pseudo-aglycon lacking the carbohydrates at amino acid 4 and 7, followed by coordination with a simple Co3+ Schiff base complex to form the dimeric species 36.192 Disappointingly, dimers 35 and 36 both showed diminished potency against MRSA (MIC = 4 μg/mL) when compared to teicoplanin (MIC = 0.5 μg/mL).192 Only against a VanA-type VRE strain did the activities of 35 and 36 improve, with MICs of 4–8 μg/mL relative to that of teicoplanin (MIC = 256 μg/mL).192 Although derivatives 34–36 show improved activities against VRE strains compared to their respective parent compounds, these N-terminal dimers are not as potent against MRSA when compared to the C-terminally linked homodimers of Sharpless190 and Haldar188 (32 and 33), highlighting the importance of the ligation site for antibacterial activity.
Targeted Glycopeptide Delivery
Glycopeptide antibiotics are generally administered systemically, potentially leading to unwanted side effects and to the development of resistant strains. To overcome these issues, efforts directed toward delivering vancomycin and its analogues in a targeted and controlled fashion have been reported in recent years. In this context, the use of technologies such as liposomes193,194 and dendrimers195 has been investigated. In addition to these non-covalent drug delivery systems, progress has also been made in covalently loading vancomycin on dendrimers or metal nanoparticles (NPs).196-−199 Cooper and colleagues conjugated an N-hydroxysuccinimide (NHS)-activated PEG-dibenzocyclooctyne (DBCO) to a human serum albumin monolayer bound to the surface of super-paramagnetic carboxylated 170 NPs.200 Subsequently, the NPs were loaded with vancomycin-PEG-N3 at different densities, using a copper-free azide–alkyne cycloaddition reaction, yielding derivative 37 (Figure 10). Low-density 37 was found to retain potent activity against MRSA (MIC = 0.79 μg/mL), and high-density 37 exhibited an 18-fold improved activity compared to vancomycin against VanA/B-type VRE (MIC = 28.9 μg/mL).200 The improved in vitro antibacterial potency of these nanoparticle-bound vancomycin derivatives is ascribed to two factors: (1) the enhanced binding affinity of 37 to the bacteria’s cell surface (for high density particles), highlighted by the fact that antagonization of bacterial inhibition requires a 64-fold molar excess of acetyl-Lys-d-Ala-d-Ala, and (2) the membrane permeabilization properties of 37, which lead to membrane rupture for all density particles at 10-fold MIC.200
Figure 10.
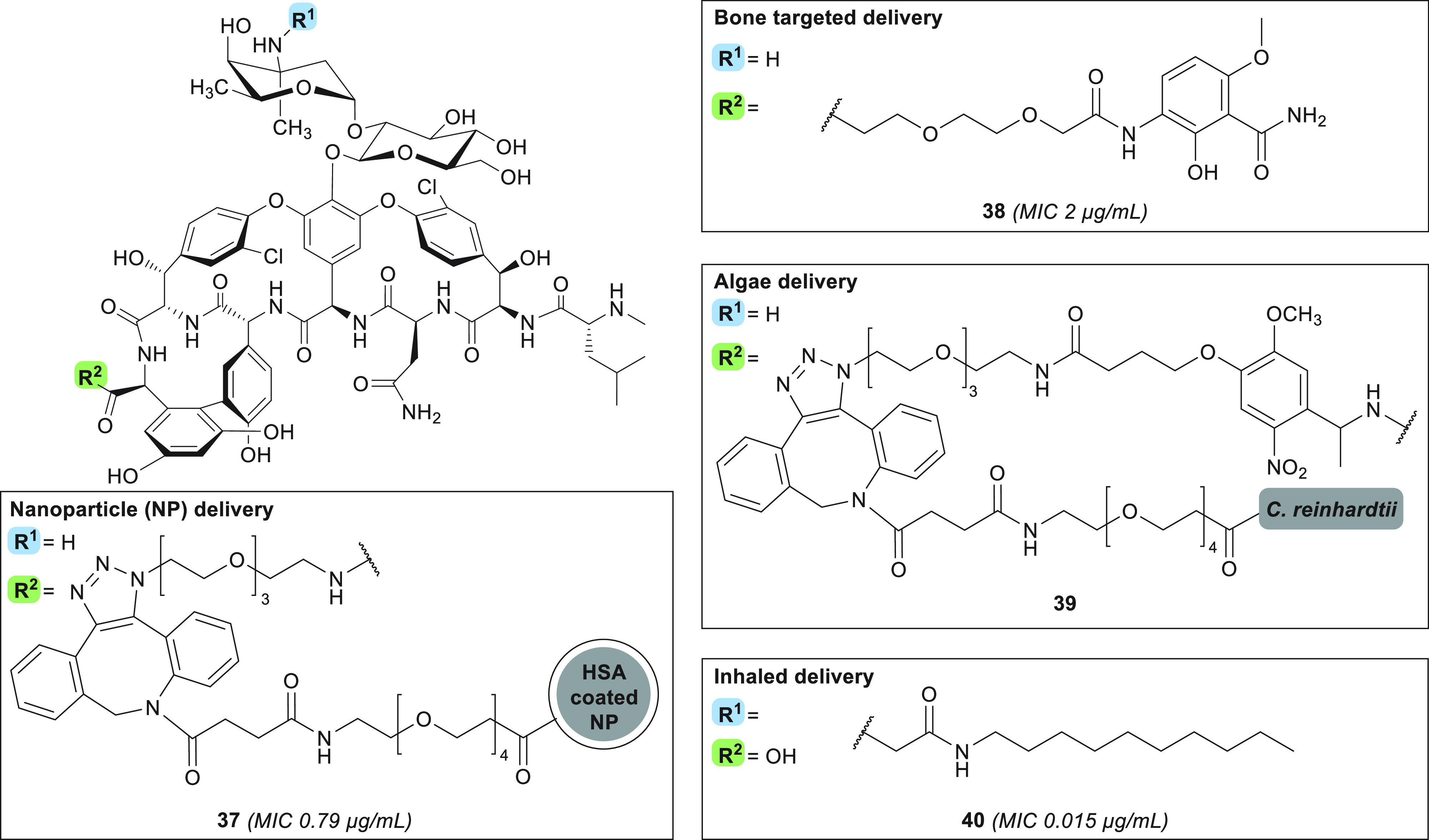
Glycopeptides designed for targeted drug delivery. MIC values are relative to experiments carried out on MRSA strains.
In addition to NP conjugation for improved drug delivery, vancomycin has also been modified with substituents designed to direct targeting to specific tissues and organs. The development of such approaches is of particular interest for those indications where vancomycin is advised as a first-line treatment, such as for targeting the bones in treating osteomyelitis, the skin for SSSIs, and the lungs in case of pulmonary infections. In one such strategy to specifically tackle osteomyelitis, for which S. aureus is a leading cause,201 researchers at the University of Louisville coupled a functional group with known hydroxyapatite affinity and enhanced bone accumulation abilities to the vancomycin C-terminus (compound 38, Figure 10).202 Given vancomycin’s poor distribution to the skeletal tissue, the local concentration of the therapeutic agent at the target site is low, and prolonged administration is required, diminishing efficacy and increasing the potential for resistance development.201,203 By comparison, compound 38 was found to maintain in vitro antibacterial activity against MRSA (2 μg/mL)202 and in rats has a 1-log-reduced MRSA titer in an osteomyelitis model compared to the same dosing of vancomycin.204 Localization of 38 to the target site was confirmed in rats, with ∼5-fold higher concentrations in the bone compared to vancomycin after 12 h and 47-fold higher after 168 h. However, this particularly long exposure time can also lead to adverse events such as renal toxicity and leukocytosis.203,204
In 2020, Gademann and co-workers developed a light-irradiation-triggered release system by functionalizing the surface of Chlamydomonas reinhardtii with vancomycin, specifically aimed at SSSI treatment, as local and light-triggered release was hypothesized to minimize resistance selection.205 This living functionalized algae carrier was chosen as it is biodegradable206 and does not trigger immune response in mice,207 and chemical engineering of the surface had been demonstrated previously.208 The algae were functionalized using the well-established DBCO handle, allowing for copper-free azide–alkyne cycloaddition. Vancomycin was modified at the C-terminus via the installation of a PEG spacer containing the photocleavable o-nitrobenzyl moiety and a terminal azide handle. The azide-modified vancomycin species was subsequently conjugated to the DBCO-decorated algae, resulting in species 39 (Figure 10).205 While the covalent linkage of vancomycin to the algae surface was demonstrated to prevent the antibiotic from exerting its antimicrobial effect, upon light irradiation and subsequent linker cleavage, 39 was shown to inhibit growth of B. subtilis at both the lag phase (at 2.5 μM loading) and the exponential phase (at 5 μM loading) (MIC = 0.06 μg/mL), with release of free vancomycin-NH2 upon UV irradiation of 39 also confirmed.205 In order to establish the clinical potential of delivery system 39 for the intended SSSI treatment, it will need to be further assessed against relevant pathogens for this disease profile, such as S. aureus and β-hemolytic streptococci.209
In addition to the treatment of osteomyelitis and SSSIs, vancomycin is also used as a front-line therapy for persistent pulmonary MRSA infections. The drawbacks associated with vancomycin therapy for this indication, which requires high-dose systemic administration, include insufficient accumulation in the lungs and risk of renal toxicity. To address this, the group of Konicek set out to design derivatives of vancomycin suitable for inhalation.210 These analogues resemble telavancin but contain a carbonyl linker at the vancosamine position and no resorcinol modification. Representative amide 40 (Figure 10) was selected for extensive investigation due to (1) its potent in vitro activity against target bacteria MRSA (MIC = 0.015 μg/mL), S. pneumoniae (MIC = 0.008 μg/mL), C. difficile (MIC = 0.015–0.06 μg/mL), VanA-type VRE (MIC = 0.03–2 μg/mL), and VanB-type VRE (MIC = 0.03 μg/mL) (Table 1) and (2) its prolonged exposure time after inhalation in rats, with a half-life of 108 h, minimal conversion to the hydrolysis product, and minimal systemic toxicity.210 Amide 40 was also found to have enhanced anti-biofilm activity compared to vancomycin. Furthermore, nebulized 40 was assessed in an in vivo acute pulmonary MRSA infection model in neutropenic rats, where it demonstrated antibacterial activity that was superior to that of inhaled vancomycin.210 Overall, targeted glycopeptide strategies do show promise; however, care and attention are required to ensure that such constructs are tailored to have optimal PK profiles that allow them to reach their designated specific target sites while displaying minimal systemic toxicity.
Glycopeptides Active against Gram-Negative Bacteria
Although most semisynthetic glycopeptide antibiotics target Gram-positive strains, the primary target of this class of antimicrobial agents—lipid II—is also present in Gram-negative bacteria. Vancomycin and other glycopeptides are inactive against Gram-negative bacteria due to their inability to cross the outer membrane (OM). However, the ability of vancomycin to bind to E. coli’s lipid II has been established previously.220 Potentiation of vancomycin by OM disruption by means of serum supplementation221 or the addition of synergists as adjuvants has also been demonstrated.222,223 While co-administration of lipopolysaccharide (LPS)-active OM disruptors potentiates vancomycin, these agents can also be covalently linked to the glycopeptide (Figure 11). In this regard, the previously discussed lipophilic cationic vancomycin analogue 8 was further investigated for activity against Gram-negative strains. The in vitro potency of 8 was assessed, where it showed moderate activity against E. coli (MIC = 2.1–7.8 μg/mL) and Acinetobacter baumannii (MIC = 5.2–9.0 μg/mL), as well as K. pneumoniae (MIC = 15.6 μg/mL) and MDR Pseudomonas aeruginosa (MIC = 10.6 μg/mL) (Figure 11, Table 2).211 The efficacy of this vancomycin derivative is reduced 2-fold in the presence of bovine serum albumin, likely due to its lipophilic nature and the consequent high protein binding.211 Notably, the anti-A. baumannii activity was also demonstrated in an in vivo murine thigh infection model, where compound 8 was found to reduce the bacterial titer by 3-log compared to vehicle. Building upon these findings, the Haldar group went on to design derivative 41, containing an amide bond between the lipid and ammonium moiety envisioned to engage in additional hydrogen bonding. This semisynthetic vancomycin derivative was found to have activity against a panel of A. baumannii clinical isolates (MIC = 6.8–13.3 μg/mL) (Figure 11, Table 2).212 Furthermore, when administered at 50 μM, compound 41 reduces A. baumannii biofilm thickness in a concentration-dependent fashion, with 4–5-fold thinner biofilm formed compared to both vancomycin-treated and untreated biofilms. The results of subsequent in vivo experiments also indicate that the inclusion of the extra amide functionality improves the toxicity profile compared to 8 when administered IV. Furthermore, no propensity for resistance selection against A. baumannii was observed for either 8 or 41.211,212 Mechanistically, both of these compounds are thought to inhibit cell-wall biosynthesis and exhibit outer and inner membrane permeabilization of both exponential and stationary phase cells, for which the permanent positive charge carried by the ammonium moiety appears essential.211 Like 8, vancomycin analogue 41 retains in vitro activity against MRSA (0.7 μg/mL) while also showing activity against VISA (0.17 μg/mL) and VRE (MIC = 3.8–6.9 μg/mL).212
Figure 11.
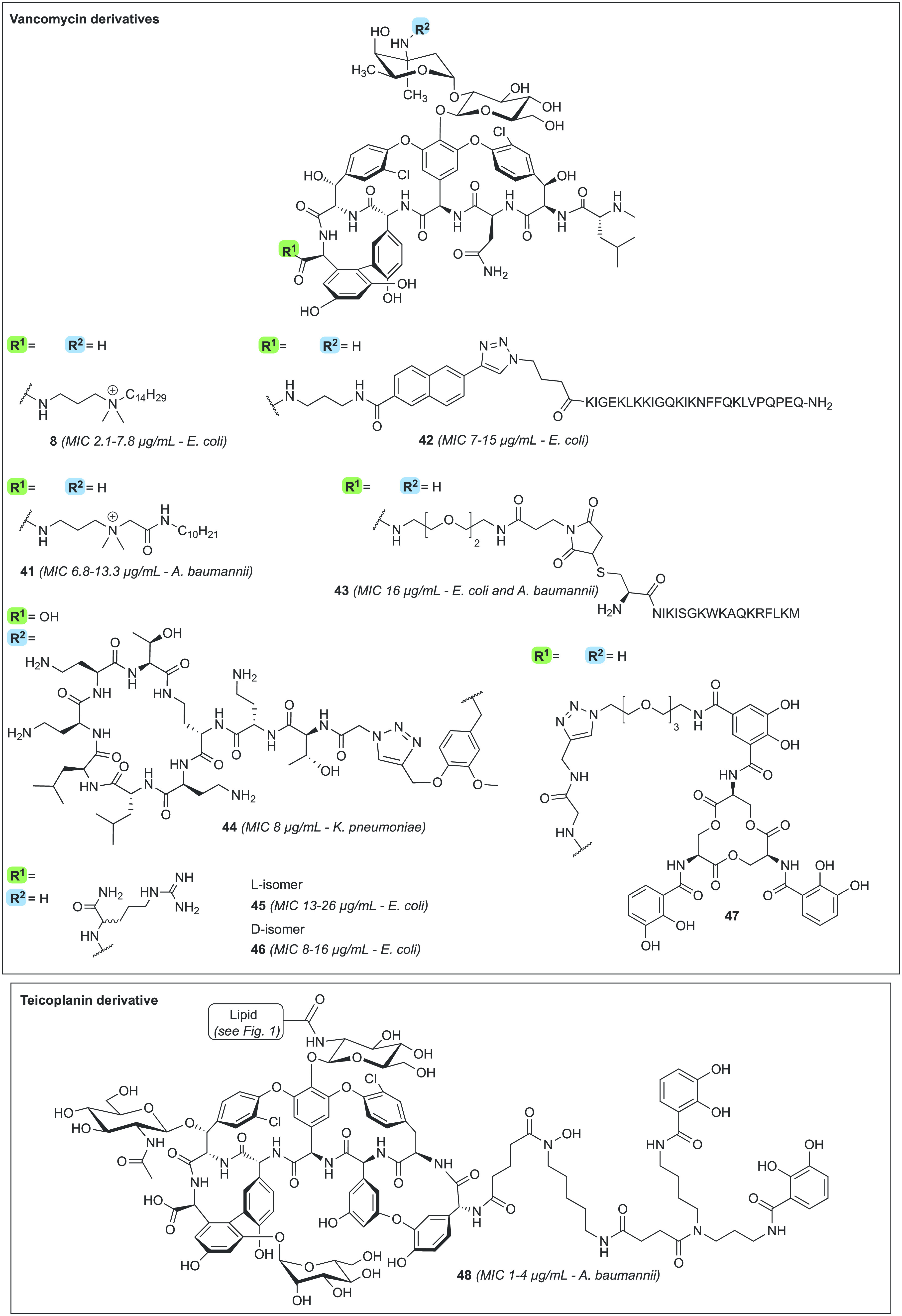
Glycopeptides with activity against Gram-negative bacteria.
Table 2. In Vitro Antibacterial Activity against Gram-Negative Strainsa.
| MIC
(μg/mL) |
||||||
|---|---|---|---|---|---|---|
| category | compound | E. coli | K. pneumoniae | A. baumannii | P. aeruginosa | refs |
| Gram-negative active | 8 | 2.1–7.8 | 15.6 | 5.2–9.0 | 10.6 | (211) |
| 41 | 22–43 | >173 | 6.8–13.3 | 22 to >173 | (212) | |
| 42 | 7–15 | nd | nd | nd | (213) | |
| 43 | 16 | 64 | 16 | 128 | (214) | |
| 44 | 16 | 8 | 32 | 16–64 | (215) | |
| 45 | 13–26 | nd | 51 | 103 | (216) | |
| 46 | 8–16 | nd | 8–32 | nd | (217) | |
| 47 | nd | nd | nd | nd | (218) | |
| 48 | >128 | nd | 1–4 | >128 | (219) | |
nd = not determined.
Following similar approaches, the van der Eycken and Huang groups independently reported the conjugation of lysine-rich antimicrobial peptides to the vancomycin C-terminus.213,214 The resulting derivatives 42 and 43 (Figure 11) were envisioned to cause OM disruption by interfering with divalent cation binding of LPS. While both compounds displayed reduced potency against the Gram-positive S. aureus (8–30 μg/mL),213,214 their ability to target Gram-negative strains is noteworthy. Analogue 42 was shown to be active against E. coli, Yerisina enterocolitica, Pseudomonas putida, and Salmonella typhimurium (MIC ≤4–30 μg/mL) (Table 2), for which anti-biofilm activity was also established (IC50 = 4–8 μg/mL).213 Compound 43 displays significant enhancement in antibacterial activity (MIC = 16 μg/mL) compared to vancomycin (MIC > 128 μg/mL) against E. coli and A. baumannii (Table 2).214 The enhanced activity of 43 toward Gram-negative species indeed appears to be the result of an OM-specific effect, given that the compound showed no reduction in cell viability in mammalian cell lines.214
In 2021, our team developed a panel of OM-disrupting vancomycin derivatives by linking the known OM disruptor and LPS-binder polymyxin E nonapeptide (PMEN) to the C-terminus or vancosamine portion of vancomycin using CuAAC conjugation.215 These derivatives, termed the vancomyxins, show improved in vitro potency compared to vancomycin alone or vancomycin supplemented with PMEN against Gram-negative bacterial strains. For example, derivative 44 (Figure 11) exhibited MIC values against K. pneumoniae and E. coli of 8 and 16 μg/mL, respectively (Table 2).215 The activity of the vancomyxins was also shown to be antagonized by LPS, suggesting that they do exert their activity via LPS binding, with OM disruption contributing to their mode of action due to the conjugation to PMEN.215 Besides showing activity against a panel of Gram-negative strains, and contrary to analogues 42 and 43, vancomyxins such as 44 retain potent activity against a variety of Gram-positive bacteria, including MRSA (MIC = 0.25 μg/mL) and VRE, for which an up to 16 000-fold improvement compared to vancomycin was measured.215 Compound 44 displays no hemolysis and has a TD50 of 0.23 mM in proximal tubule epithelial cells, a concentration several orders of magnitude higher than the corresponding MIC values.215
While the analogues described above are the result of extensive structural modifications, small adjustments to vancomycin can also enhance activity against Gram-negative bacteria. During their studies on octaarginine conjugation via the C-terminus, culminating in vancomycin analogue 15, Wender and Cegelski serendipitously discovered derivatives 45 and 46, featuring the presence of a single l/d-arginine amide at the same position (Figure 11).216,217 Compounds 45 and 46 were found to display activity against Gram-negative bacteria (Table 2), including against MDR E. coli, with MIC values of 13–26216 and 8–16 μg/mL,217 respectively. Moreover, derivative 46 was also shown to have activity against some A. baumannii species (MIC = 8–32 μg/mL).217 These conjugates retain activity against Gram-positive isolates, prove non-hemolytic, and notably cause little permeabilization of the OM.216 The authors attribute the anti-Gram-negative activity of 45 and 46 to their ability to displace the LPS-stabilizing Mg2+ cations, a feature which is usually linked to self-promoted uptake.216 Furthermore, the in vitro activity of 46 was reflected in vivo, where it reduced the E. coli thigh burden in a murine model in a dose-dependent manner (4- to 7-log greater reduction compared to vancomycin or vehicle). Also of note is the finding that the relatively small structural difference between analogue 46 and the parent antibiotic results in an increased half-life in mice (1.29 h versus 0.89 h for vancomycin).217
Another strategy to transport glycopeptide antibiotics to their target site is facilitating active transport across the OM by covalent linkage to siderophores. Siderophores are iron-chelating agents produced by microorganisms to sequester iron from the microenvironment. After binding iron, siderophores are trafficked back into the bacterial cell through dedicated transporters, after which they release the iron, which is used in key cellular processes.224 These iron uptake pathways have also been hijacked by microorganisms in generating a class of naturally occurring Trojan horse antibacterial agents known as the sideromycins. Sideromycins are siderophore-conjugated antibiotics that are actively transported past the OM through siderophore uptake receptors and into the bacterial cell whereby they can elicit their antibacterial effect.224 This strategy has inspired several research groups to design semisynthetic glycopeptide-based sideromycins with anti-Gram-negative activity. The first vancomycin-containing sideromycin was reported by Miller and co-workers in 1996.225 More recently, the group of Nolan used CuAAC to connect enterobactin, a triscatecholate siderophore with unparalleled affinity for iron,226−228 to the C-terminus of vancomycin.218 The resulting conjugate 47 (Figure 11) was shown to inhibit the growth of siderophore-deficient E. coli and P. aeruginosa. Given that the cargo size of compound 47 was deemed too large for active uptake, its antibacterial effect was ascribed to extracellular iron chelation and nutrient deprivation.218 Miller and co-workers also employed a similar strategy in developing bis-catechol/mono-hydroxymate teicoplanin analogues such as compound 48, wherein the siderophore was introduced at the N-terminus (Figure 11).219 Compound 48 exhibited in vitro antibacterial activity against A. baumannii (MIC = 1–4 μg/mL), with impressive activity against a carbapenemase-positive strain (MIC = 1 μg/mL) (Table 2).219 Also of note, while 48 was found to retain some potency against Gram-positive S. aureus (MIC = 4 μg/mL), its anti-Gram-negative activity appears specific for A. baumannii, as it had no impact on E. coli and P. aeruginosa proliferation.219 In summary, conjugating cationic groups or siderophores to glycopeptides is a viable strategy to make Gram-negative strains more susceptible to this class of antibiotics, although the resulting MIC values usually still fall in the “intermediate activity range”.
Conclusion and Perspectives
In order to address resistance to glycopeptides like vancomycin, much effort has been applied in designing semisynthetic analogues of natural occurring glycopeptides. As opposed to total synthesis, semisynthetic approaches are more time- and cost-effective and have already resulted in the introduction of three novel glycopeptide antibiotics to the clinic. While these glycopeptides display enhanced potency, telavancin (3) has a black box warning due to its associated toxicity,93,98 and dalbavancin (4) and oritavancin (5) have unusual PK properties owing to their extremely long half-lives.113,115,135,136 While this can be considered a feature in that it allows for simplification in dosing regimen,113,115,135,136 it also carries the risk that any adverse reaction may persist for weeks post treatment. Moreover, in vivo exposure to sub-therapeutic levels of these antibiotics can also confer selection for resistant sub-populations.110,111,135,138 Thus, there remains a need for novel glycopeptide antibiotics with both improved potencies and enhanced PK and safety profiles.
This Review highlights recent developments in the field of semisynthetic glycopeptides. In addition to covering new glycopeptides with enhanced activity against Gram-positive bacteria, we also summarize recent efforts at extending the activity of these antibiotics toward Gram-negative organisms. Also of note are recent reports describing glycopeptide analogues as a starting point for the design of novel antiviral agents (against, for example, influenza or COVID-19) as well as in the development of innovative diagnostics probes.16,229−232 Most research on semisynthetic glycopeptide derivatives revolves around the modification of vancomycin at one or more of the following sites: the vancosamine (Vv), C-terminus (Vc), N-terminus (Vn), and resorcinol (Vr). To date, a limited number of studies have attempted to elucidate which modification site gives the most potent analogues, revealing a subtle interplay between the nature and the positioning of the substituent(s) and their impact on antibacterial activity.
The majority of the strategies employed toward the development of novel glycopeptide antibiotics relies on enhancing the bacterial cell surface binding, which often translates into the design of glycopeptide derivatives containing additional positively charged groups. Not only has this approach proven successful in tackling Gram-positive bacteria, but it can also confer activity against Gram-negative strains. While in Gram-positive strains the presence of positively charged moieties on the antibiotic molecule is presumed to favorably impact the interaction with the negatively charged membrane, the precise mechanism by which this phenomenon occurs is yet to be explored in depth. In Gram-negative strains, the antibiotic’s cationic portions likely displace the LPS-stabilizing divalent cations, thus disrupting the OM.215,216 While the exogenous supplementation of vancomycin with positively charged small-molecule or peptide-based synergists is an established strategy to enhance its anti-Gram-negative activity,223 many of the derivatives presented in this Review provide evidence for the advantage of covalently linking the glycopeptide to a cationic OM-disrupting moiety. Covalent conjugation may facilitate co-localization to the bacterial cell surface, thus bringing the glycopeptide structure in close proximity to its target. Also of note is the fact that minor structural modifications of the cationic portion—as small as a single guanidine moiety or arginine amide—have the power of conferring enhanced potencies against Gram-positive bacteria and, in some cases, Gram-negative strains.163,216,217 Furthermore, lipidated moieties, alone or in combination with cationic substituents, have been widely demonstrated to improve antibacterial activity against resistant strains. Glycopeptides with such hydrophobic substituents are assumed to have the ability to anchor in the membrane and have been shown to depolarize or permeabilize the bacterial membrane.88,125,127−129,142,145−148,151 Also of note are recent studies elaborating the mechanism of semisynthetic glycopeptides by the introduction of groups aimed at bacterial targets other than the traditional Lipid II d-Ala-d-Ala termini. Such strategies include conjugation to pyrophosphate-targeting groups or linking to antibiotics with alternative targets, both of which have shown promise.173,175,178,185 Moreover, the covalent dimerization of glycopeptide antibiotics,187,188,190−192 inspired by vancomycin’s natural cooperative dimerization, can result in enhanced surface binding due to co-localization to the target site.28−30 Finally, while the introduction of additional carbohydrate units has also been explored primarily to address PK and toxicity issues, such modifications have also been found to result in improved target binding to d-Ala-d-Lac, likely facilitated by the introduction of favorable hydrogen-bonding interactions.141,143,156
In an effort to confer selectivity to glycopeptide antibiotics and to minimize their toxicity, targeted approaches have been investigated wherein conjugation to large systems (nanoparticles or living organisms such as algae) or specific tissue-targeting moieties allows for preferential delivery to the target site.200,202,205,210 In addition, exploitation of specific Gram-negative bacterial uptake receptors has also been investigated through the conjugation of glycopeptides antibiotics to siderophores.218,219,225 As different bacteria employ a multitude of different siderophore transporters, this approach has the potential to generate species- or even strain-selective antibiotics.
Overall, while a large number of promising new semi-synthetic glycopeptides have been described in recent years, the characterization of most remains limited to preliminary studies of in vitro potency and cell-based toxicity. In order for these new glycopeptide antibiotics to progress toward clinical trials and eventually into the clinic, further investigations and additional translational studies showing an improved therapeutic window compared to the currently clinically used glycopeptides will be necessary. Despite these challenges, the broad collection of potent semisynthetic derivatives disclosed in the literature since 2014 provides a source of optimism for the discovery of tomorrow’s antibiotics. As this overview shows, while the low-hanging fruit in antibiotic discovery may have been plucked a long time ago, judicious semi-synthetic modifications of glycopeptides still hold great promise as a means of further optimizing and expanding the clinical relevance of this important class of antibacterial agents.
Acknowledgments
Financial support provided by the European Research Council (ERC consolidator grant to N.I.M., grant agreement no. 725523).
The authors declare no competing financial interest.
References
- Murray C. J. L.; Ikuta K. S.; Sharara F.; Swetschinski L.; Robles Aguilar G.; Gray A.; Han C.; Bisignano C.; Rao P.; Wool E.; Johnson S. C.; Browne A. J.; Chipeta M. G.; Fell F.; Hackett S.; Haines-Woodhouse G.; Kashef Hamadani B. H.; Kumaran E. A. P.; McManigal B.; Agarwal R.; Akech S.; Albertson S.; Amuasi J.; Andrews J.; Aravkin A.; Ashley E.; Bailey F.; Baker S.; Basnyat B.; Bekker A.; Bender R.; Bethou A.; Bielicki J.; Boonkasidecha S.; Bukosia J.; Carvalheiro C.; Castañeda-Orjuela C.; Chansamouth V.; Chaurasia S.; Chiurchiù S.; Chowdhury F.; Cook A. J.; Cooper B.; Cressey T. R.; Criollo-Mora E.; Cunningham M.; Darboe S.; Day N. P. J.; De Luca M.; Dokova K.; Dramowski A.; Dunachie S. J.; Eckmanns T.; Eibach D.; Emami A.; Feasey N.; Fisher-Pearson N.; Forrest K.; Garrett D.; Gastmeier P.; Giref A. Z.; Greer R. C.; Gupta V.; Haller S.; Haselbeck A.; Hay S. I.; Holm M.; Hopkins S.; Iregbu K. C.; Jacobs J.; Jarovsky D.; Javanmardi F.; Khorana M.; Kissoon N.; Kobeissi E.; Kostyanev T.; Krapp F.; Krumkamp R.; Kumar A.; Kyu H. H.; Lim C.; Limmathurotsakul D.; Loftus M. J.; Lunn M.; Ma J.; Mturi N.; Munera-Huertas T.; Musicha P.; Mussi-Pinhata M. M.; Nakamura T.; Nanavati R.; Nangia S.; Newton P.; Ngoun C.; Novotney A.; Nwakanma D.; Obiero C. W.; Olivas-Martinez A.; Olliaro P.; Ooko E.; Ortiz-Brizuela E.; Peleg A. Y.; Perrone C.; Plakkal N.; Ponce-de-Leon A.; Raad M.; Ramdin T.; Riddell A.; Roberts T.; Robotham J. V.; Roca A.; Rudd K. E.; Russell N.; Schnall J.; Scott J. A. G.; Shivamallappa M.; Sifuentes-Osornio J.; Steenkeste N.; Stewardson A. J.; Stoeva T.; Tasak N.; Thaiprakong A.; Thwaites G.; Turner C.; Turner P.; van Doorn H. R.; Velaphi S.; Vongpradith A.; Vu H.; Walsh T.; Waner S.; Wangrangsimakul T.; Wozniak T.; Zheng P.; Sartorius B.; Lopez A. D.; Stergachis A.; Moore C.; Dolecek C.; Naghavi M. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399 (10325), 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. P. Vancomycin: A History. Clin. Infect. Dis. 2006, 42 (Supplement_1), S5–S12. 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- Evans D. A.; Wood M. R.; Trotter B. W.; Richardson T. I.; Barrow J. C.; Katz J. L. Total Syntheses of Vancomycin and Eremomycin Aglycons. Angew. Chemie Int. Ed. 1998, 37 (19), 2700–2704. . [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Takayanagi M.; Jain N. F.; Natarajan S.; Koumbis A. E.; Bando T.; Ramanjulu J. M. Total Synthesis of Vancomycin Aglycon—Part 3: Final Stages. Angew. Chemie Int. Ed. 1998, 37 (19), 2717–2719. . [DOI] [PubMed] [Google Scholar]
- Boger D. L.; Miyazaki S.; Kim S. H.; Wu J. H.; Castle S. L.; Loiseleur O.; Jin Q. Total Synthesis of the Vancomycin Aglycon. J. Am. Chem. Soc. 1999, 121 (43), 10004–10011. 10.1021/ja992577q. [DOI] [Google Scholar]
- Xie J.; Okano A.; Pierce J. G.; James R. C.; Stamm S.; Crane C. M.; Boger D. L. Total Synthesis of [Ψ[C(=S)NH]Tpg4]Vancomycin Aglycon, [Ψ[C(=NH)NH]Tpg4]Vancomycin Aglycon, and Related Key Compounds: Reengineering Vancomycin for Dual d-Ala-d-Ala and d-Ala-d-Lac Binding. J. Am. Chem. Soc. 2012, 134 (2), 1284–1297. 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M. J.; Qu S.; Tan C.; Cai Y.; Mogi Y.; Jamin Keith D.; Boger D. L. Next-Generation Total Synthesis of Vancomycin. J. Am. Chem. Soc. 2020, 142 (37), 16039–16050. 10.1021/jacs.0c07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Nakayama A.; Schammel A. W.; Boger D. L. Total Synthesis of [Ψ[C(=NH)NH]Tpg4]Vancomycin and Its (4-Chlorobiphenyl)Methyl Derivative: Impact of Peripheral Modifications on Vancomycin Analogues Redesigned for Dual d-Ala-d-Ala and d-Ala-d-Lac Binding. J. Am. Chem. Soc. 2014, 136 (39), 13522–13525. 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. Total Syntheses of Vancomycin-Related Glycopeptide Antibiotics and Key Analogues. Chem. Rev. 2017, 117 (18), 11952–11993. 10.1021/acs.chemrev.6b00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Trapp S.; Gin A. S.; DeCorby M.; Lagacé-Wiens P. R. S.; Rubinstein E.; Hoban D. J.; Karlowsky J. A. Dalbavancin and Telavancin: Novel Lipoglycopeptides for the Treatment of Gram-Positive Infections. Expert Rev. Anti. Infect. Ther. 2008, 6 (1), 67–81. 10.1586/14787210.6.1.67. [DOI] [PubMed] [Google Scholar]
- Zeng D.; Debabov D.; Hartsell T. L.; Cano R. J.; Adams S.; Schuyler J. A.; McMillan R.; Pace J. L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6 (12), a026989. 10.1101/cshperspect.a026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S.; Hansford K. A.; Blaskovich M. A. T.; Halai R.; Cooper M. A. Glycopeptide Antibiotics: Back to the Future. J. Antibiot. 2014, 67 (9), 631–644. 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G.; Calic D.; Schweizer F.; Zelenitsky S.; Adam H.; Lagacé-Wiens P. R. S.; Rubinstein E.; Gin A. S.; Hoban D. J.; Karlowsky J. A. New Lipoglycopeptides. Drugs 2010, 70 (7), 859–886. 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Dhanda G.; Sarkar P.; Samaddar S.; Haldar J. Battle against Vancomycin-Resistant Bacteria: Recent Developments in Chemical Strategies. J. Med. Chem. 2019, 62 (7), 3184–3205. 10.1021/acs.jmedchem.8b01093. [DOI] [PubMed] [Google Scholar]
- Blaskovich M. A. T.; Hansford K. A.; Butler M. S.; Jia Z.; Mark A. E.; Cooper M. A. Developments in Glycopeptide Antibiotics. ACS Infect. Dis. 2018, 4, 715–735. 10.1021/acsinfecdis.7b00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya Y.; Bhattacharyya S.; Dhanda G.; Haldar J. Emerging Roles of Glycopeptide Antibiotics: Moving beyond Gram-Positive Bacteria. ACS Infect. Dis. 2022, 8 (1), 1–28. 10.1021/acsinfecdis.1c00367. [DOI] [PubMed] [Google Scholar]
- Acharya Y.; Dhanda G.; Sarkar P.; Haldar J. Pursuit of Next-Generation Glycopeptides: A Journey with Vancomycin. Chem. Commun. 2022, 58 (12), 1881–1897. 10.1039/D1CC06635H. [DOI] [PubMed] [Google Scholar]
- Griffith R. S. Vancomycin Use—an Historical Review. J. Antimicrob. Chemother. 1984, 14 (suppl_D), 1–5. 10.1093/jac/14.suppl_D.1. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Kalman J. R. Structural and Mode of Action Studies on the Antibiotic Vancomycin. Evidence from 270-MHz Proton Magnetic Resonance. J. Am. Chem. Soc. 1977, 99 (8), 2768–2774. 10.1021/ja00450a058. [DOI] [PubMed] [Google Scholar]
- Marshall F. J. Structure Studies on Vancomycin. J. Med. Chem. 1965, 8 (1), 18–22. 10.1021/jm00325a004. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M.; Jones P. G.; Kennard O.; Williams D. H.; Smith G. A. Structure of Vancomycin and Its Complex with Acetyl-D-Alanyl-D-Alanine. Nature 1978, 271 (5642), 223–225. 10.1038/271223a0. [DOI] [PubMed] [Google Scholar]
- Pfeiffer R. R. Structural Features of Vancomycin. Rev. Infect. Dis. 1981, 3, S205–S209. 10.1093/clinids/3.Supplement_2.S205. [DOI] [PubMed] [Google Scholar]
- Harris C. M.; Harris T. M. Structure of the Glycopeptide Antibiotic Vancomycin. Evidence for an Asparagine Residue in the Peptide. J. Am. Chem. Soc. 1982, 104 (15), 4293–4295. 10.1021/ja00379a062. [DOI] [Google Scholar]
- Schäfer M.; Schneider T. R.; Sheldrick G. M. Crystal Structure of Vancomycin. Structure 1996, 4 (12), 1509–1515. 10.1016/S0969-2126(96)00156-6. [DOI] [PubMed] [Google Scholar]
- Pootoolal J.; Neu J.; Wright G. D. Glycopeptide Antibiotic Resistance. Annu. Rev. Pharmacol. Toxicol. 2002, 42 (1), 381–408. 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- Walsh C. T.; Fisher S. L.; Park I. S.; Prahalad M.; Wu Z. Bacterial Resistance to Vancomycin: Five Genes and One Missing Hydrogen Bond Tell the Story. Chem. Biol. 1996, 3, 21–28. 10.1016/S1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- Barna J. C.; Williams D. H. The Structure and Mode of Action of Glycopeptide Antibiotics of the Vancomycin Group. Annu. Rev. Microbiol. 1984, 38, 339–357. 10.1146/annurev.mi.38.100184.002011. [DOI] [PubMed] [Google Scholar]
- Westwell M. S.; Bardsley B.; Dancer R. J.; Try A. C.; Williams D. H. Cooperativity in Ligand Binding Expressed at a Model Cell Membrane by the Vancomycin Group Antibiotics. Chem. Commun. 1996, 5, 589–590. 10.1039/cc9960000589. [DOI] [Google Scholar]
- Mackay J. P.; Gerhard U.; Beauregard D. A.; Williams D. H.; Westwell M. S.; Searle M. S. Glycopeptide Antibiotic Activity and the Possible Role of Dimerization: A Model for Biological Signaling. J. Am. Chem. Soc. 1994, 116 (11), 4581–4590. 10.1021/ja00090a006. [DOI] [Google Scholar]
- Beauregard D. A.; Williams D. H.; Gwynn M. N.; Knowles D. J. C. Dimerization and Membrane Anchors in Extracellular Targeting of Vancomycin Group Antibiotics. Antimicrob. Agents Chemother. 1995, 39 (3), 781–785. 10.1128/AAC.39.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Costa V. M.; King C. E.; Kalan L.; Morar M.; Sung W. W. L.; Schwarz C.; Froese D.; Zazula G.; Calmels F.; Debruyne R.; Golding G. B.; Poinar H. N.; Wright G. D. Antibiotic Resistance Is Ancient. Nature 2011, 477 (7365), 457–461. 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- Leclercq R.; Derlot E.; Duval J.; Courvalin P. Plasmid-Mediated Resistance to Vancomycin and Teicoplanin in Enterococcus Faecium. N. Engl. J. Med. 1988, 319 (3), 157–161. 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- Sahm D. F.; Kissinger J.; Gilmore M. S.; Murray P. R.; Mulder R.; Solliday J.; Clarke B. In Vitro Susceptibility Studies of Vancomycin-Resistant Enterococcus Faecalis. Antimicrob. Agents Chemother. 1989, 33 (9), 1588–1591. 10.1128/AAC.33.9.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttley A. H. C.; Collins C. H.; Naidoo J.; George R. C. Vancomycin-Resistant Enterococci. The Lancet 1988, 331, 57–58. 10.1016/S0140-6736(88)91037-9. [DOI] [PubMed] [Google Scholar]
- Arthur M.; Quintiliani R. Regulation of VanA-and VanB-Type Glycopeptide Resistance in Enterococci. Antimicrob. Agents Chemother. 2001, 45 (2), 375–381. 10.1128/AAC.45.2.375-381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courvalin P. Resistance of Enterococci to Glycopeptides. Antimicrob. Agents Chemother. 1990, 34 (12), 2291–2296. 10.1128/AAC.34.12.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courvalin P. Vancomycin Resistance in Gram-Positive Cocci. Clin. Infect. Dis. 2006, 42 (Suppl 1), S25–S34. 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- Stogios P. J.; Savchenko A. Molecular Mechanisms of Vancomycin Resistance. Protein Sci. 2020, 29 (3), 654–669. 10.1002/pro.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComas C. C.; Crowley B. M.; Boger D. L. Partitioning the Loss in Vancomycin Binding Affinity for D-Ala-d-Lac into Lost H-Bond and Repulsive Lone Pair Contributions. J. Am. Chem. Soc. 2003, 125 (31), 9314–9315. 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- Bugg T. D. H.; Wright G. D.; Dutka-Malen S.; Arthur M.; Courvalin P.; Walsh C. T. Molecular Basis for Vancomycin Resistance in Enterococcus Faecium BM4147: Biosynthesis of a Depsipeptide Peptidoglycan Precursor by Vancomycin Resistance Proteins VanH and VanA. Biochemistry 1991, 30 (43), 10408–10415. 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- Billot-Klein D.; Gutmann L.; Sablé S.; Guittet E.; van Heijenoort J. Modification of Peptidoglycan Precursors Is a Common Feature of the Low-Level Vancomycin-Resistant VanB-Type Enterococcus D366 and of the Naturally Glycopeptide-Resistant Species Lactobacillus Casei, Pediococcus Pentosaceus, Leuconostoc Mesenteroides, And. J. Bacteriol. 1994, 176 (8), 2398–2405. 10.1128/jb.176.8.2398-2405.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.-S.; Lin C.-H.; Walsh C. T. Bacterial Resistance to Vancomycin: Overproduction, Purification, and Characterization of VanC2 from Enterococcus Casseliflavus as a d-Ala-d-Ser Ligase. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (19), 10040–10044. 10.1073/pnas.94.19.10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Cristino J.; Resina C.; Manuel V.; Lito L.; Ramirez M. First Case of Infection with Vancomycin-Resistant Staphylococcus Aureus in Europe. Lancet 2013, 382 (9888), 205. 10.1016/S0140-6736(13)61219-2. [DOI] [PubMed] [Google Scholar]
- Sievert D. M.; Rudrik J. T.; Patel J. B.; McDonald L. C.; Wilkins M. J.; Hageman J. C. Vancomycin-Resistant Staphylococcus Aureus in the United States, 2002–2006. Clin. Infect. Dis. 2008, 46 (5), 668–674. 10.1086/527392. [DOI] [PubMed] [Google Scholar]
- McGuinness W. A.; Malachowa N.; Deleo F. R. Vancomycin Resistance in Staphylococcus Aureus. Yale J. Biol. Med. 2017, 90, 269–281. [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Ma X.; Sato K.; Okuma K.; Tenover F. C.; Mamizuka E. M.; Gemmell C. G.; Kim M.-N.; Ploy M.-C.; El Solh N.; Ferraz V.; Hiramatsu K. Cell Wall Thickening Is a Common Feature of Vancomycin Resistance in Staphylococcus Aureus. J. Clin. Microbiol. 2003, 41 (1), 5–14. 10.1128/JCM.41.1.5-14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howden B. P.; Davies J. K.; Johnson P. D. R.; Stinear T. P.; Grayson M. L. Reduced Vancomycin Susceptibility in Staphylococcus Aureus, Including Vancomycin-Intermediate and Heterogeneous Vancomycin-Intermediate Strains: Resistance Mechanisms, Laboratory Detection, and Clinical Implications. Clin. Microbiol. Rev. 2010, 23 (1), 99–139. 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieradzki K.; Tomasz A. Alterations of Cell Wall Structure and Metabolism Accompany Reduced Susceptibility to Vancomycin in an Isogenic Series of Clinical Isolates of Staphylococcus Aureus. J. Bacteriol. 2003, 185 (24), 7103–7110. 10.1128/JB.185.24.7103-7110.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Murakami H.; Kuwahara-Arai K.; Hanaki H.; Hiramatsu K. Contribution of a Thickened Cell Wall and Its Glutamine Nonamidated Component to the Vancomycin Resistance Expressed by Staphylococcus Aureus Mu50. Antimicrob. Agents Chemother. 2000, 44 (9), 2276–2285. 10.1128/AAC.44.9.2276-2285.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaki H.; Labischinski H.; Inaba Y.; Kondo N.; Murakami H.; Hiramatsu K. Increase in Glutamine-Non-Amidated Muropeptides in the Peptidoglycan of Vancomycin-Resistant Staphylococcus Aureus Strain Mu50. J. Antimicrob. Chemother. 1998, 42 (3), 315–320. 10.1093/jac/42.3.315. [DOI] [PubMed] [Google Scholar]
- Cui L.; Ma X.; Sato K.; Okuma K.; Tenover F. C.; Mamizuka E. M.; Gemmell C. G.; Kim M. N.; Ploy M. C.; El-Solh N.; Ferraz V.; Hiramatsu K. Cell Wall Thickening Is a Common Feature of Vancomycin Resistance in Staphylococcus Aureus. J. Clin. Microbiol. 2003, 41 (1), 5–14. 10.1128/JCM.41.1.5-14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Committee on Antimicrobial Susceptibility Testing. Data from the EUCAST MIC distribution website, http://www.eucast.org.
- Food and Drug Administration. FDA labeling information - Vancomycin Injection, USP. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/050671s023lbl.pdf (accessed 2022-03-01).
- Al-Nassir W. N.; Sethi A. K.; Li Y.; Pultz M. J.; Riggs M. M.; Donskey C. J. Both Oral Metronidazole and Oral Vancomycin Promote Persistent Overgrowth of Vancomycin-Resistant Enterococci during Treatment of Clostridium Difficile-Associated Disease. Antimicrob. Agents Chemother. 2008, 52 (7), 2403–2406. 10.1128/AAC.00090-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moellering R. C. Jr. Pharmacokinetics of Vancomycin. J. Antimicrob. Chemother. 1984, 14 (suppl_D), 43–52. 10.1093/jac/14.suppl_D.43. [DOI] [PubMed] [Google Scholar]
- Rybak M. J. The Pharmacokinetic and Pharmacodynamic Properties of Vancomycin. Clin. Infect. Dis. 2006, 42 (Supplement_1), S35–S39. 10.1086/491712. [DOI] [PubMed] [Google Scholar]
- Albrecht L. M.; Rybak M. J.; Warbasse L. H.; Edwards D. J. Vancomycin Protein Binding in Patients with Infections Caused by Staphylococcus Aureus. DICP 1991, 25 (7–8), 713–715. 10.1177/106002809102500701. [DOI] [PubMed] [Google Scholar]
- Ackerman B. H.; Taylor E. H.; Olsen K. M.; Abdel-Malak W.; Pappas A. A. Vancomycin Serum Protein Binding Determination by Ultrafiltration. Drug Intell. Clin. Pharm. 1988, 22 (4), 300–303. 10.1177/106002808802200404. [DOI] [PubMed] [Google Scholar]
- Matzke G. R.; Zhanel G. G.; Guay D. R. P. Clinical Pharmacokinetics of Vancomycin. Clin. Pharmacokinet. 1986, 11 (4), 257–282. 10.2165/00003088-198611040-00001. [DOI] [PubMed] [Google Scholar]
- Wallace M. R.; Mascola J. R.; Oldfield E. C. Red Man Syndrome: Incidence, Etiology, and Prophylaxis. J. Infect. Dis. 1991, 164 (6), 1180–1185. 10.1093/infdis/164.6.1180. [DOI] [PubMed] [Google Scholar]
- Sahai J.; Healy D. P.; Shelton M. J.; Miller J. S.; Ruberg S. J.; Polk R. Comparison of Vancomycin- and Teicoplanin-Induced Histamine Release and “Red Man Syndrome”. Antimicrob. Agents Chemother. 1990, 34 (5), 765–769. 10.1128/AAC.34.5.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivagnanam S.; Deleu D. Red Man Syndrome. Crit. Care 2003, 7 (2), 119. 10.1186/cc1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippone E. J.; Kraft W. K.; Farber J. L. The Nephrotoxicity of Vancomycin. Clin. Pharmacol. Ther. 2017, 102 (3), 459–469. 10.1002/cpt.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna J. C. J.; Williams D. H.; Stone D. J. M.; Leung T. W. C.; Doddrell D. M. Structure Elucidation of the Teicoplanin Antibiotics. J. Am. Chem. Soc. 1984, 106 (17), 4895–4902. 10.1021/ja00329a044. [DOI] [Google Scholar]
- Malabarba A.; Strazzolini P.; Depaoli A.; Landi M.; Berti M.; Cavalleri B. Teicoplanin, Antibiotics from Actinoplanes Teichomyceticus Nov. Sp. VI., Chemical Degradation: Physico-Chemical and Biological Properties of Acid Hydrolysis Products. J. Antibiot. 1984, 37 (9), 988–999. 10.7164/antibiotics.37.988. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Boddy C. N. C.; Bräse S.; Winssinger N. Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew. Chem., Int. Ed. 1999, 38 (15), 2096–2152. . [DOI] [PubMed] [Google Scholar]
- Borghi A.; Edwards D.; Zerilli L. F.; Lancini G. C. Factors Affecting the Normal and Branched-Chain Acyl Moieties of Teicoplanin Components Produced by Actinoplanes Teichomyceticus. Microbiology 1991, 137 (3), 587–592. 10.1099/00221287-137-3-587. [DOI] [PubMed] [Google Scholar]
- Baltch A. L.; Smith R. P.; Ritz W. J.; Bopp L. H. Comparison of Inhibitory and Bactericidal Activities and Postantibiotic Effects of LY333328 and Ampicillin Used Singly and in Combination against Vancomycin-Resistant Enterococcus Faecium. Antimicrob. Agents Chemother. 1998, 42 (10), 2564–2568. 10.1128/AAC.42.10.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna J. C. J.; Williams D. H.; Williamson M. P. Structural Features That Affect the Binding of Teicoplanin, Ristocetin A, and Their Derivatives to the Bacterial Cell-Wall Model N-Acetyl-D-Alanyl-D-Alanine. J. Chem. Soc., Chem. Commun. 1985, 5, 254–256. 10.1039/c39850000254. [DOI] [Google Scholar]
- Economou N. J.; Zentner I. J.; Lazo E.; Jakoncic J.; Stojanoff V.; Weeks S. D.; Grasty K. C.; Cocklin S.; Loll P. J. Structure of the Complex between Teicoplanin and a Bacterial Cell-Wall Peptide: Use of a Carrier-Protein Approach. Acta Crystallogr. D: Biol. Crystallogr. 2013, 69 (Pt 4), 520–533. 10.1107/S0907444912050469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista M.; Depardieu F.; Courvalin P.; Arthur M. Specificity of Induction of Glycopeptide Resistance Genes in Enterococcus Faecalis. Antimicrob. Agents Chemother. 1996, 40 (10), 2291–2295. 10.1128/AAC.40.10.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Périchon B.; Courvalin P. VanA-Type Vancomycin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2009, 53 (11), 4580–4587. 10.1128/AAC.00346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaatz G. W.; Seo S. M.; Dorman N. J.; Lerner S. A. Emergence of Teicoplanin Resistance During Therapy of Staphylococcus Aureus Endocarditis. J. Infect. Dis. 1990, 162 (1), 103–108. 10.1093/infdis/162.1.103. [DOI] [PubMed] [Google Scholar]
- Shlaes D. M.; Shlaes J. H. Teicoplanin Selects for Staphylococcus Aureus That Is Resistant to Vancomycin. Clin. Infect. Dis. 1995, 20 (4), 1071–1072. 10.1093/clinids/20.4.1071. [DOI] [PubMed] [Google Scholar]
- Sieradzki K.; Tomasz A. Suppression of Glycopeptide Resistance in a Highly Teicoplanin-Resistant Mutant of Staphylococcus Aureus by Transposon Inactivation of Genes Involved in Cell Wall Synthesis. Microb. Drug Resist. 1998, 4 (3), 159–168. 10.1089/mdr.1998.4.159. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Annex III: Summary of product characteristics, labelling and package leaflet - Targocid, https://www.ema.europa.eu/en/medicines/human/referrals/targocid-associated-names (accessed 2022-03-02).
- Campoli-Richards D. M.; Brogden R. N.; Faulds D. Teicoplanin. Drugs 1990, 40 (3), 449–486. 10.2165/00003495-199040030-00007. [DOI] [PubMed] [Google Scholar]
- Svetitsky S.; Leibovici L.; Paul M. Comparative Efficacy and Safety of Vancomycin versus Teicoplanin: Systematic Review and Meta-Analysis. Antimicrob. Agents Chemother. 2009, 53 (10), 4069–4079. 10.1128/AAC.00341-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. P. R. Comparative Safety of Teicoplanin and Vancomycin. Int. J. Antimicrob. Agents 1998, 10 (2), 143–152. 10.1016/S0924-8579(98)00025-9. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration. FDA labeling information - VIBATIV (telavancin), http://www.accessdata.fda.gov/drugsatfda_docs/label/2009/022110s000lbl.pdf.
- Leadbetter M. R.; Adams S. M.; Bazzini B.; Fatheree P. R.; Karr D. E.; Krause K. M.; Lam B. M. T.; Linsell M. S.; Nodwell M. B.; Pace J. L.; Quast K.; Shaw J.-P.; Soriano E.; Trapp S. G.; Villena J. D.; Wu T. X.; Christensen B. G.; Judice J. K. Hydrophobic Vancomycin Derivatives with Improved ADME Properties: Discovery of Telavancin (TD-6424). J. Antibiot. 2004, 57 (5), 326–336. 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- Judice J. K.; Pace J. L. Semi-Synthetic Glycopeptide Antibacterials. Bioorg. Med. Chem. Lett. 2003, 13 (23), 4165–4168. 10.1016/j.bmcl.2003.08.067. [DOI] [PubMed] [Google Scholar]
- Jansen W. T. M.; Verel A.; Verhoef J.; Milatovic D. In Vitro Activity of Telavancin against Gram-Positive Clinical Isolates Recently Obtained in Europe. Antimicrob. Agents Chemother. 2007, 51 (9), 3420–3424. 10.1128/AAC.00100-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghi D. C.; Benton B. M.; Krause K. M.; Thornsberry C.; Pillar C.; Sahm D. F. Comparative Surveillance Study of Telavancin Activity against Recently Collected Gram-Positive Clinical Isolates from across the United States. Antimicrob. Agents Chemother. 2008, 52 (7), 2383–2388. 10.1128/AAC.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B.; Sarkar C.; Das D.; Gupta A.; Kalra A.; Sahni S. Telavancin: A Novel Semisynthetic Lipoglycopeptide Agent to Counter the Challenge of Resistant Gram-Positive Pathogens. Ther. Adv. Infect. Dis. 2017, 4 (2), 49–73. 10.1177/2049936117690501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde C. S.; Hartouni S. R.; Janc J. W.; Mammen M.; Humphrey P. P.; Benton B. M. Telavancin Disrupts the Functional Integrity of the Bacterial Membrane through Targeted Interaction with the Cell Wall Precursor Lipid II. Antimicrob. Agents Chemother. 2009, 53 (8), 3375–3383. 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D. L.; Chang R.; Debabov D. V.; Leung J.; Wu T.; Krause K. M.; Sandvik E.; Hubbard J. M.; Kaniga K.; Schmidt D. E.; Gao Q.; Cass R. T.; Karr D. E.; Benton B. M.; Humphrey P. P. Telavancin, a Multifunctional Lipoglycopeptide, Disrupts Both Cell Wall Synthesis and Cell Membrane Integrity in Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2005, 49 (3), 1127–1134. 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D. L.; Chang R.; Debabov D. V.; Leung J.; Wu T.; Krause K. M.; Sandvik E.; Hubbard J. M.; Kaniga K.; Schmidt D. E.; Gao Q.; Cass R. T.; Karr D. E.; Benton B. M.; Humphrey P. P. Telavancin, a Multifunctional Lipoglycopeptide, Disrupts Both Cell Wall Synthesis and Cell Membrane Integrity in Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 2005, 49 (3), 1127–1134. 10.1128/AAC.49.3.1127-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosowska-Shick K.; Clark C.; Pankuch G. A.; McGhee P.; Dewasse B.; Beachel L.; Appelbaum P. C. Activity of Telavancin against Staphylococci and Enterococci Determined by MIC and Resistance Selection Studies. Antimicrob. Agents Chemother. 2009, 53 (10), 4217–4224. 10.1128/AAC.00742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill C. M.; Krause K. M.; Lewis S. R.; Blais J.; Benton B. M.; Mammen M.; Humphrey P. P.; Kinana A.; Janc J. W. Specificity of Induction of the VanA and VanB Operons in Vancomycin-Resistant Enterococci by Telavancin. Antimicrob. Agents Chemother. 2010, 54 (7), 2814–2818. 10.1128/AAC.01737-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlowsky J. A.; Nichol K.; Zhanel G. G. Telavancin: Mechanisms of Action, In Vitro Activity, and Mechanisms of Resistance. Clin. Infect. Dis. 2015, 61 (suppl_2), S58–S68. 10.1093/cid/civ534. [DOI] [PubMed] [Google Scholar]
- Draghi D. C.; Benton B. M.; Krause K. M.; Thornsberry C.; Pillar C.; Sahm D. F. Comparative Surveillance Study of Telavancin Activity against Recently Collected Gram-Positive Clinical Isolates from across the United States. Antimicrob. Agents Chemother. 2008, 52 (7), 2383–2388. 10.1128/AAC.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theravance Biopharma US. VIBATIV (telavancin) for injection, for intravenous use. Full Prescribing Information, https://vibativ.com/wp-content/uploads/2022/01/Vibativ-PI-final-Dec-2020-clean.pdf (accessed 2022-03-03).
- Niederman M. S.; Lee P. C.; Barriere S. L.; Barnes C. N.; Castaneda-Ruiz B. Telavancin in Hospital-Acquired and Ventilator-Associated Pneumonia (HAP/VAP) Caused by Staphylococcus Aureus: Post Hoc Analysis of 2 Randomized, Controlled Trials. Infect. Dis. Ther. 2019, 8 (3), 445–452. 10.1007/s40121-019-0255-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. P.; Seroogy J.; Kaniga K.; Higgins D. L.; Kitt M.; Barriere S. Pharmacokinetics, Serum Inhibitory and Bactericidal Activity, and Safety of Telavancin in Healthy Subjects. Antimicrob. Agents Chemother. 2005, 49 (1), 195–201. 10.1128/AAC.49.1.195-201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. L.; Barriere S. L.; Kitt M. M.; Goldberg M. R. Multiple-Dose Pharmacokinetics of Intravenous Telavancin in Healthy Male and Female Subjects. J. Antimicrob. Chemother. 2008, 62 (4), 780–783. 10.1093/jac/dkn273. [DOI] [PubMed] [Google Scholar]
- Worboys P. D.; Wong S. L.; Barriere S. L. Pharmacokinetics of Intravenous Telavancin in Healthy Subjects with Varying Degrees of Renal Impairment. Eur. J. Clin. Pharmacol. 2015, 71 (6), 707–714. 10.1007/s00228-015-1847-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere S. L. The ATTAIN Trials: Efficacy and Safety of Telavancin Compared with Vancomycin for the Treatment of Hospital-Acquired and Ventilator-Associated Bacterial Pneumonia. Future Microbiol. 2014, 9 (3), 281–289. 10.2217/fmb.14.4. [DOI] [PubMed] [Google Scholar]
- Goldstein B. P.; Selva E.; Gastaldo L.; Berti M.; Pallanza R.; Ripamonti F.; Ferrari P.; Denaro M.; Arioli V.; Cassani G. A40926, a New Glycopeptide Antibiotic with Anti-Neisseria Activity. Antimicrob. Agents Chemother. 1987, 31 (12), 1961–1966. 10.1128/AAC.31.12.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabarba A.; Goldstein B. P. Origin, Structure, and Activity in Vitro and in Vivo of Dalbavancin. J. Antimicrob. Chemother. 2005, 55 (Suppl 2), ii15–ii20. 10.1093/jac/dki005. [DOI] [PubMed] [Google Scholar]
- Azrad M.; Baum M.; Rokney A.; Levi Y.; Peretz A. In Vitro Activity of Tedizolid and Dalbavancin against MRSA Strains Is Dependent on Infection Source. Int. J. Infect. Dis. 2019, 78, 107–112. 10.1016/j.ijid.2018.11.011. [DOI] [PubMed] [Google Scholar]
- Riccobono E.; Giani T.; Baldi G.; Arcangeli S.; Antonelli A.; Tellone V.; Del Vecchio A.; De Joannon A. C.; Rossolini G. M. Update on Activity of Dalbavancin and Comparators against Clinical Isolates of Gram-Positive Pathogens from Europe and Russia (2017–2018), and on Clonal Distribution of MRSA. Int. J. Antimicrob. Agents 2022, 59 (2), 106503. 10.1016/j.ijantimicag.2021.106503. [DOI] [PubMed] [Google Scholar]
- Candiani G.; Abbondi M.; Borgonovi M.; Romanò G.; Parenti F. In-Vitro and in-Vivo Antibacterial Activity of BI 397, a New Semi-Synthetic Glycopeptide Antibiotic. J. Antimicrob. Chemother. 1999, 44 (2), 179–192. 10.1093/jac/44.2.179. [DOI] [PubMed] [Google Scholar]
- Biedenbach D. J.; Bell J. M.; Sader H. S.; Turnidge J. D.; Jones R. N. Activities of Dalbavancin against a Worldwide Collection of 81,673 Gram-Positive Bacterial Isolates. Antimicrob. Agents Chemother. 2009, 53 (3), 1260–1263. 10.1128/AAC.01453-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economou N. J.; Nahoum V.; Weeks S. D.; Grasty K. C.; Zentner I. J.; Townsend T. M.; Bhuiya M. W.; Cocklin S.; Loll P. J. A Carrier Protein Strategy Yields the Structure of Dalbavancin. J. Am. Chem. Soc. 2012, 134 (10), 4637–4645. 10.1021/ja208755j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.; Ziora Z. M.; Hansford K. A.; Blaskovich M. A.; Butler M. S.; Cooper M. A. Anti-Cooperative Ligand Binding and Dimerisation in the Glycopeptide Antibiotic Dalbavancin. Org. Biomol. Chem. 2014, 12 (16), 2568–2575. 10.1039/C3OB42428F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez S.; Hackbarth C.; Romanò G.; Trias J.; Jabes D.; Goldstein B. P. In Vitro Antistaphylococcal Activity of Dalbavancin, a Novel Glycopeptide. J. Antimicrob. Chemother. 2005, 55 (Suppl 2), ii21–ii24. 10.1093/jac/dki007. [DOI] [PubMed] [Google Scholar]
- Goldstein B. P.; Draghi D. C.; Sheehan D. J.; Hogan P.; Sahm D. F. Bactericidal Activity and Resistance Development Profiling of Dalbavancin. Antimicrob. Agents Chemother. 2007, 51 (4), 1150–1154. 10.1128/AAC.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth B. J.; Ashford N. K.; Penewit K.; Waalkes A.; Holmes E. A.; Ross D. H.; Shen T.; Hines K. M.; Salipante S. J.; Xu L. Dalbavancin Exposure in Vitro Selects for Dalbavancin-Non-Susceptible and Vancomycin-Intermediate Strains of Methicillin-Resistant Staphylococcus Aureus. Clin. Microbiol. Infect. 2021, 27 (6), 910.e1–910.e8. 10.1016/j.cmi.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth B. J.; Jain R.; Hahn A.; Cummings L.; Weaver T.; Waalkes A.; Sengupta D.; Salipante S. J.; Rakita R. M.; Butler-Wu S. M. Emergence of Dalbavancin Non-Susceptible, Vancomycin-Intermediate Staphylococcus Aureus (VISA) after Treatment of MRSA Central Line-Associated Bloodstream Infection with a Dalbavancin- and Vancomycin-Containing Regimen. Clin. Microbiol. Infect. 2018, 24 (4), 429.e1–429.e5. 10.1016/j.cmi.2017.07.028. [DOI] [PubMed] [Google Scholar]
- Kussmann M.; Karer M.; Obermueller M.; Schmidt K.; Barousch W.; Moser D.; Nehr M.; Ramharter M.; Poeppl W.; Makristathis A.; Winkler S.; Thalhammer F.; Burgmann H.; Lagler H. Emergence of a Dalbavancin Induced Glycopeptide/Lipoglycopeptide Non-Susceptible Staphylococcus Aureus during Treatment of a Cardiac Device-Related Endocarditis. Emerg. Microbes Infect. 2018, 7 (1), 1–10. 10.1038/s41426-018-0205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arhin F. F.; Seguin D. L.; Belley A.; Moeck G. In Vitro Stepwise Selection of Reduced Susceptibility to Lipoglycopeptides in Enterococci. Diagn. Microbiol. Infect. Dis. 2017, 89 (2), 168–171. 10.1016/j.diagmicrobio.2017.06.023. [DOI] [PubMed] [Google Scholar]
- Durata Therapies Limited. Prescribing Information for DALVANCE (dalbavancin) for injection, for intravenous use, https://www.allergan.com/assets/pdf/dalvance_pi (accessed 2022-03-03).
- Morrisette T.; Miller M. A.; Montague B. T.; Barber G. R.; McQueen R. B.; Krsak M. On- and off-Label Utilization of Dalbavancin and Oritavancin for Gram-Positive Infections. J. Antimicrob. Chemother. 2019, 74 (8), 2405–2416. 10.1093/jac/dkz162. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G.; Calic D.; Schweizer F.; Zelenitsky S.; Adam H.; Lagacé-Wiens P. R. S.; Rubinstein E.; Gin A. S.; Hoban D. J.; Karlowsky J. A. New Lipoglycopeptides: A Comparative Review of Dalbavancin, Oritavancin and Telavancin. Drugs 2010, 70 (7), 859–886. 10.2165/11534440-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Leighton A.; Gottlieb A. B.; Dorr M. B.; Jabes D.; Mosconi G.; VanSaders C.; Mroszczak E. J.; Campbell K. C.; Kelly E. Tolerability, Pharmacokinetics, and Serum Bactericidal Activity of Intravenous Dalbavancin in Healthy Volunteers. Antimicrob. Agents Chemother. 2004, 48 (3), 940–945. 10.1128/AAC.48.3.940-945.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti O.; Rizzetto G.; Molinelli E.; Cirioni O.; Offidani A. Review: A Safety Profile of Dalbavancin for On- and Off-Label Utilization. Ther. Clin. Risk Manag. 2021, 17, 223–232. 10.2147/TCRM.S271445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R. D.; Snyder N. J.; Zweifel M. J.; Staszak M. A.; Wilkie S. C.; Nicas T. I.; Mullen D. L.; Butler T. F.; Rodriguez M. J.; Huff B. E.; Thompson R. C. Reductive Alkylation of Glycopeptide Antibiotics: Synthesis and Antibacterial Activity. J. Antibiot. 1996, 49 (6), 575–581. 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- Mendes R. E.; Farrell D. J.; Sader H. S.; Jones R. N. Oritavancin Microbiologic Features and Activity Results From the Surveillance Program in the United States. Clin. Infect. Dis. 2012, 54 (suppl_3), S203–S213. 10.1093/cid/cir923. [DOI] [PubMed] [Google Scholar]
- Patti G. J.; Kim S. J.; Yu T.-Y.; Dietrich E.; Tanaka K. S. E.; Parr T. R. J.; Far A. R.; Schaefer J. Vancomycin and Oritavancin Have Different Modes of Action in Enterococcus Faecium. J. Mol. Biol. 2009, 392 (5), 1178–1191. 10.1016/j.jmb.2009.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. J.; Cegelski L.; Stueber D.; Singh M.; Dietrich E.; Tanaka K. S. E.; Parr T. R.; Far A. R.; Schaefer J. Oritavancin Exhibits Dual Mode of Action to Inhibit Cell-Wall Biosynthesis in Staphylococcus Aureus. J. Mol. Biol. 2008, 377 (1), 281–293. 10.1016/j.jmb.2008.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Schweizer F.; Karlowsky J. A. Oritavancin: Mechanism of Action. Clin. Infect. Dis. 2012, 54 (suppl_3), S214–S219. 10.1093/cid/cir920. [DOI] [PubMed] [Google Scholar]
- Münch D.; Engels I.; Müller A.; Reder-Christ K.; Falkenstein-Paul H.; Bierbaum G.; Grein F.; Bendas G.; Sahl H.-G.; Schneider T. Structural Variations of the Cell Wall Precursor Lipid II and Their Influence on Binding and Activity of the Lipoglycopeptide Antibiotic Oritavancin. Antimicrob. Agents Chemother. 2015, 59 (2), 772–781. 10.1128/AAC.02663-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin A.; Tabei K.; Tenover F.; Chung M.; Clarke N.; Tomasz A. High Level Oxacillin and Vancomycin Resistance and Altered Cell Wall Composition in Staphylococcus Aureus Carrying the Staphylococcal MecA and the Enterococcal VanA Gene Complex. J. Biol. Chem. 2004, 279 (5), 3398–3407. 10.1074/jbc.M309593200. [DOI] [PubMed] [Google Scholar]
- Allen N. E.; Nicas T. I. Mechanism of Action of Oritavancin and Related Glycopeptide Antibiotics. FEMS Microbiol. Rev. 2003, 26 (5), 511–532. 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- Beauregard D. A.; Williams D. H.; Gwynn M. N.; Knowles D. J. Dimerization and Membrane Anchors in Extracellular Targeting of Vancomycin Group Antibiotics. Antimicrob. Agents Chemother. 1995, 39 (3), 781–785. 10.1128/AAC.39.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech O.; Dufrêne Y. F.; Van Bambeke F.; Tukens P. M.; Mingeot-Leclercq M.-P. Interactions of Oritavancin, a New Semi-Synthetic Lipoglycopeptide, with Lipids Extracted from Staphylococcus Aureus. Biochim. Biophys. Acta - Biomembr. 2010, 1798 (10), 1876–1885. 10.1016/j.bbamem.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Belley A.; Neesham-Grenon E.; Mckay G.; Arhin F. F.; Harris R.; Beveridge T.; Parr Jr T. R.; Moeck G. Oritavancin Kills Stationary-Phase and Biofilm Staphylococcus Aureus Cells In Vitro. Antimicrob. Agents Chemother. 2009, 53 (3), 918–925. 10.1128/AAC.00766-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech O.; Francius G.; Tulkens P. M.; Van Bambeke F.; Dufrêne Y.; Mingeot-Leclercq M.-P. Interactions of Oritavancin, a New Lipoglycopeptide Derived from Vancomycin, with Phospholipid Bilayers: Effect on Membrane Permeability and Nanoscale Lipid Membrane Organization. Biochim. Biophys. Acta - Biomembr. 2009, 1788 (9), 1832–1840. 10.1016/j.bbamem.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Brade K. D.; Rybak J. M.; Rybak M. J. Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections. Infect. Dis. Ther. 2016, 5 (1), 1–15. 10.1007/s40121-016-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes R. E.; Woosley L. N.; Farrell D. J.; Sader H. S.; Jones R. N. Oritavancin Activity against Vancomycin-Susceptible and Vancomycin-Resistant Enterococci with Molecularly Characterized Glycopeptide Resistance Genes Recovered from Bacteremic Patients, 2009–2010. Antimicrob. Agents Chemother. 2012, 56 (3), 1639–1642. 10.1128/AAC.06067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay G. A.; Beaulieu S.; Arhin F. F.; Belley A.; Sarmiento I.; Parr Jr T.; Moeck G. Time-Kill Kinetics of Oritavancin and Comparator Agents against Staphylococcus Aureus, Enterococcus Faecalis and Enterococcus Faecium. J. Antimicrob. Chemother. 2009, 63 (6), 1191–1199. 10.1093/jac/dkp126. [DOI] [PubMed] [Google Scholar]
- Arthur M.; Depardieu F.; Reynolds P.; Courvalin P. Moderate-Level Resistance to Glycopeptide LY333328 Mediated by Genes of the VanA and VanB Clusters in Enterococci. Antimicrob. Agents Chemother. 1999, 43 (8), 1875–1880. 10.1128/AAC.43.8.1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. N.; Moeck G.; Arhin F. F.; Dudley M. N.; Rhomberg P. R.; Mendes R. E. Results from Oritavancin Resistance Surveillance Programs (2011 to 2014): Clarification for Using Vancomycin as a Surrogate To Infer Oritavancin Susceptibility. Antimicrob. Agents Chemother. 2016, 60 (5), 3174–3177. 10.1128/AAC.03029-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Medicines Company. Prescribing information for ORBACTIV (oritavancin) for injection, for intravenous use. http://www.orbactiv.com/pdfs/orbactiv-prescribing-information.pdf (accessed 2022-03-04).
- Rubino C. M.; Van Wart S. A.; Bhavnani S. M.; Ambrose P. G.; McCollam J. S.; Forrest A. Oritavancin Population Pharmacokinetics in Healthy Subjects and Patients with Complicated Skin and Skin Structure Infections or Bacteremia. Antimicrob. Agents Chemother. 2009, 53 (10), 4422–4428. 10.1128/AAC.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavnani S. M.; Owen J. S.; Loutit J. S.; Porter S. B.; Ambrose P. G. Pharmacokinetics, Safety, and Tolerability of Ascending Single Intravenous Doses of Oritavancin Administered to Healthy Human Subjects. Diagn. Microbiol. Infect. Dis. 2004, 50 (2), 95–102. 10.1016/j.diagmicrobio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Saravolatz L. D.; Stein G. E. Oritavancin: A Long-Half-Life Lipoglycopeptide. Clin. Infect. Dis. 2015, 61 (4), 627–632. 10.1093/cid/civ311. [DOI] [PubMed] [Google Scholar]
- Corey G. R.; Good S.; Jiang H.; Moeck G.; Wikler M.; Green S.; Manos P.; Keech R.; Singh R.; Heller B.; Bubnova N.; O’Riordan W. Single-Dose Oritavancin Versus 7–10 Days of Vancomycin in the Treatment of Gram-Positive Acute Bacterial Skin and Skin Structure Infections: The SOLO II Noninferiority Study. Clin. Infect. Dis. 2015, 60 (2), 254–262. 10.1093/cid/ciu778. [DOI] [PubMed] [Google Scholar]
- Yoganathan S.; Miller S. J. Structure Diversification of Vancomycin through Peptide-Catalyzed, Site-Selective Lipidation: A Catalysis-Based Approach To Combat Glycopeptide-Resistant Pathogens. J. Med. Chem. 2015, 58 (5), 2367–2377. 10.1021/jm501872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V.; Konai M. M.; Manjunath G. B.; Ghosh C.; Haldar J. Tackling Vancomycin-Resistant Bacteria with “Lipophilic-Vancomycin-Carbohydrate Conjugates. J. Antibiot. 2015, 68 (5), 302–312. 10.1038/ja.2014.144. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Akkapeddi P.; Manjunath G. B.; Haldar J. Membrane Active Vancomycin Analogues: A Strategy to Combat Bacterial Resistance. J. Med. Chem. 2014, 57 (11), 4558–4568. 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Samaddar S.; Paramanandham K.; Shome B. R.; Haldar J. Membrane Disruption and Enhanced Inhibition of Cell-Wall Biosynthesis: A Synergistic Approach to Tackle Vancomycin-Resistant Bacteria. Angew. Chemie Int. Ed. 2015, 54 (46), 13644–13649. 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Konai M. M.; Manjunath G. B.; Prakash R. G.; Mani B.; Paramanandham K.; Ranjan S. B.; Ravikumar R.; Chakraborty S. P.; Roy S.; Haldar J. In Vivo Antibacterial Activity and Pharmacological Properties of the Membrane-Active Glycopeptide Antibiotic YV11455. Int. J. Antimicrob. Agents 2015, 45 (6), 627–634. 10.1016/j.ijantimicag.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Sarkar P.; Basak D.; Mukherjee R.; Bandow J. E.; Haldar J. Alkyl-Aryl-Vancomycins: Multimodal Glycopeptides with Weak Dependence on the Bacterial Metabolic State. J. Med. Chem. 2021, 64 (14), 10185–10202. 10.1021/acs.jmedchem.1c00449. [DOI] [PubMed] [Google Scholar]
- Okano A.; Isley N. A.; Boger D. L. Peripheral Modifications of [Ψ[CH2NH]Tpg4]Vancomycin with Added Synergistic Mechanisms of Action Provide Durable and Potent Antibiotics. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (26), E5052–E5061. 10.1073/pnas.1704125114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.-C.; Isley N. A.; Boger D. L. N-Terminus Alkylation of Vancomycin: Ligand Binding Affinity, Antimicrobial Activity, and Site-Specific Nature of Quaternary Trimethylammonium Salt Modification. ACS Infect. Dis. 2018, 4 (10), 1468–1474. 10.1021/acsinfecdis.8b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.-C.; Isley N. A.; Okano A.; Weiss W. J.; Boger D. L. C1-CBP-Vancomycin: Impact of a Vancomycin C-Terminus Trimethylammonium Cation on Pharmacological Properties and Insights into Its Newly Introduced Mechanism of Action. J. Org. Chem. 2020, 85 (3), 1365–1375. 10.1021/acs.joc.9b02314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.-C.; Cameron M. D.; Boger D. L. Vancomycin C-Terminus Guanidine Modifications and Further Insights into an Added Mechanism of Action Imparted by a Peripheral Structural Modification. ACS Infect. Dis. 2020, 6 (8), 2169–2180. 10.1021/acsinfecdis.0c00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.-C.; Boger D. L. Exploration of the Site-Specific Nature and Generalizability of a Trimethylammonium Salt Modification on Vancomycin: A-Ring Derivatives. Tetrahedron 2019, 75 (24), 3160–3165. 10.1016/j.tet.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich M. A. T.; Hansford K. A.; Gong Y.; Butler M. S.; Muldoon C.; Huang J. X.; Ramu S.; Silva A. B.; Cheng M.; Kavanagh A. M.; Ziora Z.; Premraj R.; Lindahl F.; Bradford T. A.; Lee J. C.; Karoli T.; Pelingon R.; Edwards D. J.; Amado M.; Elliott A. G.; Phetsang W.; Daud N. H.; Deecke J. E.; Sidjabat H. E.; Ramaologa S.; Zuegg J.; Betley J. R.; Beevers A. P. G.; Smith R. A. G.; Roberts J. A.; Paterson D. L.; Cooper M. A. Protein-Inspired Antibiotics Active against Vancomycin- and Daptomycin-Resistant Bacteria. Nat. Commun. 2018, 9 (1), 22. 10.1038/s41467-017-02123-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonoplis A.; Zang X.; Huttner M. A.; Chong K. K. L.; Lee Y. B.; Co J. Y.; Amieva M. R.; Kline K. A.; Wender P. A.; Cegelski L. A Dual-Function Antibiotic-Transporter Conjugate Exhibits Superior Activity in Sterilizing MRSA Biofilms and Killing Persister Cells. J. Am. Chem. Soc. 2018, 140 (47), 16140–16151. 10.1021/jacs.8b08711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umstätter F.; Domhan C.; Hertlein T.; Ohlsen K.; Mühlberg E.; Kleist C.; Zimmermann S.; Beijer B.; Klika K. D.; Haberkorn U.; Mier W.; Uhl P. Vancomycin Resistance Is Overcome by Conjugation of Polycationic Peptides. Angew. Chem. Int. Ed. 2020, 59 (23), 8823–8827. 10.1002/anie.202002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlberg E.; Umstätter F.; Domhan C.; Hertlein T.; Ohlsen K.; Krause A.; Kleist C.; Beijer B.; Zimmermann S.; Haberkorn U.; Mier W.; Uhl P. Vancomycin-Lipopeptide Conjugates with High Antimicrobial Activity on Vancomycin-Resistant Enterococci. Pharmaceuticals 2020, 13 (6), 110. 10.3390/ph13060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N. M.; Stolarzewicz I.; Cannaerts D.; Schuermans J.; Lavigne R.; Looz Y.; Landuyt B.; Schoofs L.; Schols D.; Paeshuyse J.; Hickenbotham P.; Clokie M.; Luyten W.; Van der Eycken E. V.; Briers Y. Iterative Chemical Engineering of Vancomycin Leads to Novel Vancomycin Analogs With a High in Vitro Therapeutic Index. Front. Microbiol. 2018, 9, 1175. 10.3389/fmicb.2018.01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D.; Chen F.; Xiong L.; Tang F.; Faridoon; Qiu Y.; Zhang N.; Gong L.; Li J.; Lan L.; Huang W. Extra Sugar on Vancomycin: New Analogues for Combating Multidrug-Resistant Staphylococcus Aureus and Vancomycin-Resistant Enterococci. J. Med. Chem. 2018, 61 (1), 286–304. 10.1021/acs.jmedchem.7b01345. [DOI] [PubMed] [Google Scholar]
- Guan D.; Chen F.; Qiu Y.; Jiang B.; Gong L.; Lan L.; Huang W. Sulfonium, an Underestimated Moiety for Structural Modification, Alters the Antibacterial Profile of Vancomycin Against Multidrug-Resistant Bacteria. Angew. Chemie Int. Ed. 2019, 58 (20), 6678–6682. 10.1002/anie.201902210. [DOI] [PubMed] [Google Scholar]
- Shchelik I. S.; Gademann K. Thiol- and Disulfide-Containing Vancomycin Derivatives Against Bacterial Resistance and Biofilm Formation. ACS Med. Chem. Lett. 2021, 12 (12), 1898–1904. 10.1021/acsmedchemlett.1c00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y. Q.; Nodwell M.; Pace J. L.; Shaw J. P.; Judice J. K. Vancomycin Disulfide Derivatives as Antibacterial Agents. Bioorg. Med. Chem. Lett. 2004, 14 (3), 735–738. 10.1016/j.bmcl.2003.11.040. [DOI] [PubMed] [Google Scholar]
- Szűcs Z.; Csávás M.; Rőth E.; Borbás A.; Batta G.; Perret F.; Ostorházi E.; Szatmári R.; Vanderlinden E.; Naesens L.; Herczegh P. Synthesis and Biological Evaluation of Lipophilic Teicoplanin Pseudoaglycon Derivatives Containing a Substituted Triazole Function. J. Antibiot. 2017, 70 (2), 152–157. 10.1038/ja.2016.80. [DOI] [PubMed] [Google Scholar]
- Szűcs Z.; Bereczki I.; Csávás M.; Rőth E.; Borbás A.; Batta G.; Ostorházi E.; Szatmári R.; Herczegh P. Lipophilic Teicoplanin Pseudoaglycon Derivatives Are Active against Vancomycin- and Teicoplanin-Resistant Enterococci. J. Antibiot. 2017, 70 (5), 664–670. 10.1038/ja.2017.2. [DOI] [PubMed] [Google Scholar]
- Szűcs Z.; Ostorházi E.; Kicsák M.; Nagy L.; Borbás A.; Herczegh P. New Semisynthetic Teicoplanin Derivatives Have Comparable in Vitro Activity to That of Oritavancin against Clinical Isolates of VRE. J. Antibiot. 2019, 72 (7), 524–534. 10.1038/s41429-019-0164-1. [DOI] [PubMed] [Google Scholar]
- Szűcs Z.; Bereczki I.; Rőth E.; Milánkovits M.; Ostorházi E.; Batta G.; Nagy L.; Dombrádi Z.; Borbás A.; Herczegh P. N-Terminal Guanidine Derivatives of Teicoplanin Antibiotics Strongly Active against Glycopeptide Resistant Enterococcus Faecium. J. Antibiot. 2020, 73 (9), 603–614. 10.1038/s41429-020-0313-6. [DOI] [PubMed] [Google Scholar]
- Szűcs Z.; Naesens L.; Stevaert A.; Ostorházi E.; Batta G.; Herczegh P.; Borbás A. Reprogramming of the Antibacterial Drug Vancomycin Results in Potent Antiviral Agents Devoid of Antibacterial Activity. Pharmaceuticals 2020, 13 (7), 139. 10.3390/ph13070139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szűcs Z.; Kelemen V.; Le Thai S.; Csávás M.; Rőth E.; Batta G.; Stevaert A.; Vanderlinden E.; Naesens L.; Herczegh P.; Borbás A. Structure-Activity Relationship Studies of Lipophilic Teicoplanin Pseudoaglycon Derivatives as New Anti-Influenza Virus Agents. Eur. J. Med. Chem. 2018, 157, 1017–1030. 10.1016/j.ejmech.2018.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereczki I.; Kicsák M.; Dobray L.; Borbás A.; Batta G.; Kéki S.; Nikodém É. N.; Ostorházi E.; Rozgonyi F.; Vanderlinden E.; Naesens L.; Herczegh P. Semisynthetic Teicoplanin Derivatives as New Influenza Virus Binding Inhibitors: Synthesis and Antiviral Studies. Bioorg. Med. Chem. Lett. 2014, 24 (15), 3251–3254. 10.1016/j.bmcl.2014.06.018. [DOI] [PubMed] [Google Scholar]
- Bereczki I.; Csávás M.; Szűcs Z.; Rőth E.; Batta G.; Ostorházi E.; Naesens L.; Borbás A.; Herczegh P. Synthesis of Antiviral Perfluoroalkyl Derivatives of Teicoplanin and Vancomycin. ChemMedChem. 2020, 15 (17), 1661–1671. 10.1002/cmdc.202000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereczki I.; Mándi A.; Rőth E.; Borbás A.; Fizil Á.; Komáromi I.; Sipos A.; Kurtán T.; Batta G.; Ostorházi E.; Rozgonyi F.; Vanderlinden E.; Naesens L.; Sztaricskai F.; Herczegh P. A Few Atoms Make the Difference: Synthetic, CD, NMR and Computational Studies on Antiviral and Antibacterial Activities of Glycopeptide Antibiotic Aglycon Derivatives. Eur. J. Med. Chem. 2015, 94, 73–86. 10.1016/j.ejmech.2015.02.028. [DOI] [PubMed] [Google Scholar]
- Olsufyeva E. N.; Shchekotikhin A. E.; Bychkova E. N.; Pereverzeva E. R.; Treshalin I. D.; Mirchink E. P.; Isakova E. B.; Chernobrovkin M. G.; Kozlov R. S.; Dekhnich A. V.; Preobrazhenskaya M. N. Eremomycin Pyrrolidide: A Novel Semisynthetic Glycopeptide with Improved Chemotherapeutic Properties. Drug Des. Devel. Ther. 2018, 12, 2875–2885. 10.2147/DDDT.S173923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu S.-T. D.; Breukink E.; Tischenko E.; Lutters M. A. G.; de Kruijff B.; Kaptein R.; Bonvin A. M. J. J.; van Nuland N. A. J. The Nisin-Lipid II Complex Reveals a Pyrophosphate Cage That Provides a Blueprint for Novel Antibiotics. Nat. Struct. Mol. Biol. 2004, 11 (10), 963–967. 10.1038/nsmb830. [DOI] [PubMed] [Google Scholar]
- Cudic P.; Kranz J. K.; Behenna D. C.; Kruger R. G.; Tadesse H.; Wand A. J.; Veklich Y. I.; Weisel J. W.; McCafferty D. G. Complexation of Peptidoglycan Intermediates by the Lipoglycodepsipeptide Antibiotic Ramoplanin: Minimal Structural Requirements for Intermolecular Complexation and Fibril Formation. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (11), 7384–7389. 10.1073/pnas.102192099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla R.; Medeiros-Silva J.; Parmar A.; Vermeulen B. J. A.; Das S.; Paioni A. L.; Jekhmane S.; Lorent J.; Bonvin A. M. J. J.; Baldus M.; Lelli M.; Veldhuizen E. J. A.; Breukink E.; Singh I.; Weingarth M. Mode of Action of Teixobactins in Cellular Membranes. Nat. Commun. 2020, 11 (1), 2848. 10.1038/s41467-020-16600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V.; Sarkar P.; Samaddar S.; Haldar J. A Vancomycin Derivative with a Pyrophosphate-Binding Group: A Strategy to Combat Vancomycin-Resistant Bacteria. Angew. Chem., Int. Ed. Engl. 2016, 55 (27), 7836–7840. 10.1002/anie.201601621. [DOI] [PubMed] [Google Scholar]
- Ngo H. T.; Liu X.; Jolliffe K. A. Anion Recognition and Sensing with Zn(Ii)-Dipicolylamine Complexes. Chem. Soc. Rev. 2012, 41 (14), 4928–4965. 10.1039/c2cs35087d. [DOI] [PubMed] [Google Scholar]
- Yarlagadda V.; Sarkar P.; Samaddar S.; Manjunath G. B.; Mitra S. D.; Paramanandham K.; Shome B. R.; Haldar J. Vancomycin Analogue Restores Meropenem Activity against NDM-1 Gram-Negative Pathogens. ACS Infect. Dis. 2018, 4 (7), 1093–1101. 10.1021/acsinfecdis.8b00011. [DOI] [PubMed] [Google Scholar]
- King A. M.; Reid-Yu S. A.; Wang W.; King D. T.; De Pascale G.; Strynadka N. C.; Walsh T. R.; Coombes B. K.; Wright G. D. Aspergillomarasmine A Overcomes Metallo-β-Lactamase Antibiotic Resistance. Nature 2014, 510 (7506), 503–506. 10.1038/nature13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. Y.; Thomas P. W.; Cheng Z.; Xu N. Y.; Tierney D. L.; Crowder M. W.; Fast W.; Cohen S. M. Investigation of Dipicolinic Acid Isosteres for the Inhibition of Metallo-β-Lactamases. ChemMedChem. 2019, 14 (13), 1271–1282. 10.1002/cmdc.201900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan D.; Chen F.; Faridoon; Liu J.; Li J.; Lan L.; Huang W. Design and Synthesis of Pyrophosphate-Targeting Vancomycin Derivatives for Combating Vancomycin-Resistant Enterococci. ChemMedChem. 2018, 13 (16), 1644–1657. 10.1002/cmdc.201800252. [DOI] [PubMed] [Google Scholar]
- Pokrovskaya V.; Baasov T. Dual-Acting Hybrid Antibiotics: A Promising Strategy to Combat Bacterial Resistance. Expert Opin. Drug Discovery 2010, 5 (9), 883–902. 10.1517/17460441.2010.508069. [DOI] [PubMed] [Google Scholar]
- Leuthner K. D.; Vidaillac C.; Cheung C. M.; Rybak M. J. In Vitro Activity of the New Multivalent Glycopeptide-Cephalosporin Antibiotic TD-1792 against Vancomycin-Nonsusceptible Staphylococcus Isolates. Antimicrob. Agents Chemother. 2010, 54 (9), 3799–3803. 10.1128/AAC.00452-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais J.; Lewis S. R.; Krause K. M.; Benton B. M. Antistaphylococcal Activity of TD-1792, a Multivalent Glycopeptide-Cephalosporin Antibiotic. Antimicrob. Agents Chemother. 2012, 56 (3), 1584–1587. 10.1128/AAC.05532-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D. D; Aggen J. B; Chinn J.; Choi S.-K.; Christensen B. G; Fatheree P. R; Green D.; Hegde S. S; Judice J K.; Kaniga K.; Krause K. M; Leadbetter M.; Linsell M. S; Marquess D. G; Moran E. J; Nodwell M. B; Pace J. L; Trapp S. G; Turne S D. Exploring the Positional Attachment of Glycopeptide/Beta-Lactam Heterodimers. J. Antibiot. 2008, 61 (10), 603–614. 10.1038/ja.2008.80. [DOI] [PubMed] [Google Scholar]
- Arnusch C. J.; Bonvin A. M. J. J.; Verel A. M.; Jansen W. T. M.; Liskamp R. M. J.; de Kruijff B.; Pieters R. J.; Breukink E. The Vancomycin-Nisin(1–12) Hybrid Restores Activity against Vancomycin Resistant Enterococci. Biochemistry 2008, 47 (48), 12661–12663. 10.1021/bi801597b. [DOI] [PubMed] [Google Scholar]
- Cochrane S. A.; Li X.; He S.; Yu M.; Wu M.; Vederas J. C. Synthesis of Tridecaptin-Antibiotic Conjugates with in Vivo Activity against Gram-Negative Bacteria. J. Med. Chem. 2015, 58 (24), 9779–9785. 10.1021/acs.jmedchem.5b01578. [DOI] [PubMed] [Google Scholar]
- Tevyashova A. N.; Bychkova E. N.; Korolev A. M.; Isakova E. B.; Mirchink E. P.; Osterman I. A.; Erdei R.; Szücs Z.; Batta G. Synthesis and Evaluation of Biological Activity for Dual-Acting Antibiotics on the Basis of Azithromycin and Glycopeptides. Bioorg. Med. Chem. Lett. 2019, 29 (2), 276–280. 10.1016/j.bmcl.2018.11.038. [DOI] [PubMed] [Google Scholar]
- Champney W. S.; Burdine R. Macrolide Antibiotics Inhibit 50S Ribosomal Subunit Assembly in Bacillus Subtilis and Staphylococcus Aureus. Antimicrob. Agents Chemother. 1995, 39 (9), 2141–2144. 10.1128/AAC.39.9.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundram U. N.; Griffin J. H.; Nicas T. I. Novel Vancomycin Dimers with Activity against Vancomycin-Resistant Enterococci. J. Am. Chem. Soc. 1996, 118 (51), 13107–13108. 10.1021/ja9621298. [DOI] [Google Scholar]
- Yarlagadda V.; Sarkar P.; Manjunath G. B.; Haldar J. Lipophilic Vancomycin Aglycon Dimer with High Activity against Vancomycin-Resistant Bacteria. Bioorg. Med. Chem. Lett. 2015, 25 (23), 5477–5480. 10.1016/j.bmcl.2015.10.083. [DOI] [PubMed] [Google Scholar]
- Jain R. K.; Trias J.; Ellman J. A. D-Ala-D-Lac Binding Is Not Required for the High Activity of Vancomycin Dimers against Vancomycin Resistant Enterococci. J. Am. Chem. Soc. 2003, 125 (29), 8740–8741. 10.1021/ja0359761. [DOI] [PubMed] [Google Scholar]
- Silverman S. M.; Moses J. E.; Sharpless K. B. Reengineering Antibiotics to Combat Bacterial Resistance: Click Chemistry [1,2,3]-Triazole Vancomycin Dimers with Potent Activity against MRSA and VRE. Chem. - A Eur. J. 2017, 23 (1), 79–83. 10.1002/chem.201604765. [DOI] [PubMed] [Google Scholar]
- Jiang Y.-W.; Xu L.; Fu W.; Lin H.; Yu J.-M.; Sun X. Design, Synthesis and Biological Activity of Novel Demethylvancomycin Dimers against Vancomycin-Resistant Enterococcus Faecalis. Tetrahedron 2018, 74 (27), 3527–3533. 10.1016/j.tet.2018.04.091. [DOI] [Google Scholar]
- Bereczki I.; Szűcs Z.; Batta G.; Nagy T. M.; Ostorházi E.; Kövér K. E.; Borbás A.; Herczegh P. The First Dimeric Derivatives of the Glycopeptide Antibiotic Teicoplanin. Pharmaceuticals 2022, 15, 77. 10.3390/ph15010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez Gomez A.; Xu C.; Hosseinidoust Z. Preserving the Efficacy of Glycopeptide Antibiotics during Nanoencapsulation in Liposomes. ACS Infect. Dis. 2019, 5 (10), 1794–1801. 10.1021/acsinfecdis.9b00232. [DOI] [PubMed] [Google Scholar]
- Sande L.; Sanchez M.; Montes J.; Wolf A. J.; Morgan M. A.; Omri A.; Liu G. Y. Liposomal Encapsulation of Vancomycin Improves Killing of Methicillin-Resistant Staphylococcus Aureus in a Murine Infection Model. J. Antimicrob. Chemother. 2012, 67 (9), 2191–2194. 10.1093/jac/dks212. [DOI] [PubMed] [Google Scholar]
- Sonawane S. J.; Kalhapure R. S.; Rambharose S.; Mocktar C.; Vepuri S. B.; Soliman M.; Govender T. Ultra-Small Lipid-Dendrimer Hybrid Nanoparticles as a Promising Strategy for Antibiotic Delivery: In Vitro and in Silico Studies. Int. J. Pharm. 2016, 504 (1), 1–10. 10.1016/j.ijpharm.2016.03.021. [DOI] [PubMed] [Google Scholar]
- Choi S. K.; Myc A.; Silpe J. E.; Sumit M.; Wong P. T.; McCarthy K.; Desai A. M.; Thomas T. P.; Kotlyar A.; Holl M. M. B.; Orr B. G.; Baker J. R. Dendrimer-Based Multivalent Vancomycin Nanoplatform for Targeting the Drug-Resistant Bacterial Surface. ACS Nano 2013, 7 (1), 214–228. 10.1021/nn3038995. [DOI] [PubMed] [Google Scholar]
- Rashid M.; Rabbi M. A.; Ara T.; Hossain M. M.; Islam M. S.; Elaissari A.; Ahmad H.; Rahman M. M. Vancomycin Conjugated Iron Oxide Nanoparticles for Magnetic Targeting and Efficient Capture of Gram-Positive and Gram-Negative Bacteria. RSC Adv. 2021, 11 (57), 36319–36328. 10.1039/D1RA04390K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N.; Otsuka K.; Sawada H.; Maejima T.; Shirotake S. Bacteriolysis by Vancomycin-Conjugated Acryl Nanoparticles and Morphological Component Analysis. Drug Dev. Ind. Pharm. 2014, 40 (6), 813–818. 10.3109/03639045.2013.788012. [DOI] [PubMed] [Google Scholar]
- Jiang G.; Liu S.; Yu T.; Wu R.; Ren Y.; van der Mei H. C.; Liu J.; Busscher H. J. PAMAM Dendrimers with Dual-Conjugated Vancomycin and Ag-Nanoparticles Do Not Induce Bacterial Resistance and Kill Vancomycin-Resistant Staphylococci. Acta Biomater. 2021, 123, 230–243. 10.1016/j.actbio.2021.01.032. [DOI] [PubMed] [Google Scholar]
- Hassan M. M.; Ranzoni A.; Phetsang W.; Blaskovich M. A. T.; Cooper M. A. Surface Ligand Density of Antibiotic-Nanoparticle Conjugates Enhances Target Avidity and Membrane Permeabilization of Vancomycin-Resistant Bacteria. Bioconjugate Chem. 2017, 28 (2), 353–361. 10.1021/acs.bioconjchem.6b00494. [DOI] [PubMed] [Google Scholar]
- Lew D. P.; Waldvogel F. A. Osteomyelitis. Lancet 2004, 364 (9431), 369–379. 10.1016/S0140-6736(04)16727-5. [DOI] [PubMed] [Google Scholar]
- Pierce W. M.; Grant Taylor K.; Waite L. C.. WO2008103951A1. Methods and Compounds for the Targeted Delivery of Agents to Bone for Interaction Therewith. US8071575B2, 2008.
- Albayati Z. A. F.; Sunkara M.; Schmidt-Malan S. M.; Karau M. J.; Morris A. J.; Steckelberg J. M.; Patel R.; Breen P. J.; Smeltzer M. S.; Taylor K. G.; Merten K. E.; Pierce W. M.; Crooks P. A. Novel Bone-Targeting Agent for Enhanced Delivery of Vancomycin to Bone. Antimicrob. Agents Chemother. 2016, 60 (3), 1865–1868. 10.1128/AAC.01609-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karau M. J.; Schmidt-Malan S. M.; Greenwood-Quaintance K. E.; Mandrekar J.; Cai J.; Pierce Jr W. M.; Merten K.; Patel R. Treatment of Methicillin-Resistant Staphylococcus Aureus Experimental Osteomyelitis with Bone-Targeted Vancomycin. Springerplus 2013, 2, 329. 10.1186/2193-1801-2-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchelik I. S.; Sieber S.; Gademann K. Green Algae as a Drug Delivery System for the Controlled Release of Antibiotics. Chem. - Eur. J. 2020, 26 (70), 16644–16648. 10.1002/chem.202003821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdy A.; Mendez L.; Ballesteros M.; González-Fernández C. Enhanced Methane Production of Chlorella Vulgaris and Chlamydomonas Reinhardtii by Hydrolytic Enzymes Addition. Energy Convers. Manag. 2014, 85, 551–557. 10.1016/j.enconman.2014.04.097. [DOI] [Google Scholar]
- Schenck T. L.; Hopfner U.; Chávez M. N.; Machens H.-G.; Somlai-Schweiger I.; Giunta R. E.; Bohne A. V.; Nickelsen J.; Allende M. L.; Egaña J. T. Photosynthetic Biomaterials: A Pathway towards Autotrophic Tissue Engineering. Acta Biomater. 2015, 15, 39–47. 10.1016/j.actbio.2014.12.012. [DOI] [PubMed] [Google Scholar]
- Kerschgens I. P.; Gademann K. Antibiotic Algae by Chemical Surface Engineering. ChemBioChem 2018, 19 (5), 439–443. 10.1002/cbic.201700553. [DOI] [PubMed] [Google Scholar]
- Sader H. S.; Streit J. M.; Carvalhaes C. G.; Huband M. D.; Pfaller M. A. Frequency and Antimicrobial Susceptibility of Bacterial Isolates from Patients Hospitalised with Community-Acquired Skin and Skin-Structure Infection in Europe, Asia and Latin America. J. Glob. Antimicrob. Resist. 2019, 17, 103–108. 10.1016/j.jgar.2018.11.013. [DOI] [PubMed] [Google Scholar]
- Plaunt A. J.; Rose S. J.; Kang J. Y.; Chen K.-J.; LaSala D.; Heckler R. P.; Dorfman A.; Smith B. T.; Chun D.; Viramontes V.; Macaluso A.; Li Z.; Zhou Y.; Mark L.; Basso J.; Leifer F. G.; Corboz M. R.; Chapman R. W.; Cipolla D.; Perkins W. R.; Malinin V. S.; Konicek D. M. Development and Preclinical Evaluation of New Inhaled Lipoglycopeptides for the Treatment of Persistent Pulmonary Methicillin-Resistant Staphylococcus Aureus Infections. Antimicrob. Agents Chemother. 2021, 65 (7), e0031621 10.1128/AAC.00316-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarlagadda V.; Manjunath G. B.; Sarkar P.; Akkapeddi P.; Paramanandham K.; Shome B. R.; Ravikumar R.; Haldar J. Glycopeptide Antibiotic To Overcome the Intrinsic Resistance of Gram-Negative Bacteria. ACS Infect. Dis. 2016, 2 (2), 132–139. 10.1021/acsinfecdis.5b00114. [DOI] [PubMed] [Google Scholar]
- Sarkar P.; Samaddar S.; Ammanathan V.; Yarlagadda V.; Ghosh C.; Shukla M.; Kaul G.; Manjithaya R.; Chopra S.; Haldar J. Vancomycin Derivative Inactivates Carbapenem-Resistant Acinetobacter Baumannii and Induces Autophagy. ACS Chem. Biol. 2020, 15 (4), 884–889. 10.1021/acschembio.0c00091. [DOI] [PubMed] [Google Scholar]
- Mishra N. M.; Briers Y.; Lamberigts C.; Steenackers H.; Robijns S.; Landuyt B.; Vanderleyden J.; Schoofs L.; Lavigne R.; Luyten W.; Van der Eycken E. V. Evaluation of the Antibacterial and Antibiofilm Activities of Novel CRAMP-Vancomycin Conjugates with Diverse Linkers. Org. Biomol. Chem. 2015, 13 (27), 7477–7486. 10.1039/C5OB00830A. [DOI] [PubMed] [Google Scholar]
- Shi W.; Chen F.; Zou X.; Jiao S.; Wang S.; Hu Y.; Lan L.; Tang F.; Huang W. Design, Synthesis, and Antibacterial Evaluation of Vancomycin-LPS Binding Peptide Conjugates. Bioorg. Med. Chem. Lett. 2021, 45, 128122. 10.1016/j.bmcl.2021.128122. [DOI] [PubMed] [Google Scholar]
- van Groesen E.; Slingerland C. J.; Innocenti P.; Mihajlovic M.; Masereeuw R.; Martin N. I. Vancomyxins: Vancomycin-Polymyxin Nonapeptide Conjugates That Retain Anti-Gram-Positive Activity with Enhanced Potency against Gram-Negative Strains. ACS Infect. Dis. 2021, 7 (9), 2746–2754. 10.1021/acsinfecdis.1c00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonoplis A.; Zang X.; Wegner T.; Wender P. A.; Cegelski L. Vancomycin-Arginine Conjugate Inhibits Growth of Carbapenem-Resistant E.Coli and Targets Cell-Wall Synthesis. ACS Chem. Biol. 2019, 14 (9), 2065–2070. 10.1021/acschembio.9b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville L. F.; Shalit I.; Warn P. A.; Scheetz M. H.; Sun J.; Chosy M. B.; Wender P. A.; Cegelski L.; Rendell J. T. In Vivo Targeting of Escherichia Coli with Vancomycin-Arginine. Antimicrob. Agents Chemother. 2021, 65 (4), e02416-20. 10.1128/AAC.02416-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T.; Bullock J. L.; Nolan E. M. Siderophore-Mediated Cargo Delivery to the Cytoplasm of Escherichia Coli and Pseudomonas Aeruginosa: Syntheses of Monofunctionalized Enterobactin Scaffolds and Evaluation of Enterobactin-Cargo Conjugate Uptake. J. Am. Chem. Soc. 2012, 134 (44), 18388–18400. 10.1021/ja3077268. [DOI] [PubMed] [Google Scholar]
- Ghosh M.; Miller P. A.; Miller M. J. Antibiotic Repurposing: Bis-Catechol- and Mixed Ligand (Bis-Catechol-Mono-Hydroxamate)-Teicoplanin Conjugates Are Active against Multidrug Resistant Acinetobacter Baumannii. J. Antibiot. 2020, 73 (3), 152–157. 10.1038/s41429-019-0268-7. [DOI] [PubMed] [Google Scholar]
- Shlaes D. M.; Shlaes J. H.; Davies J.; Williamson R. Escherichia Coli Susceptible to Glycopeptide Antibiotics. Antimicrob. Agents Chemother. 1989, 33 (2), 192–197. 10.1128/AAC.33.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesterbeek D. A. C.; Martin N. I.; Velthuizen A.; Duijst M.; Ruyken M.; Wubbolts R.; Rooijakkers S. H. M.; Bardoel B. W. Complement-Dependent Outer Membrane Perturbation Sensitizes Gram-Negative Bacteria to Gram-Positive Specific Antibiotics. Sci. Rep. 2019, 9 (1), 3074. 10.1038/s41598-019-38577-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Cebrián R.; Montalbán-López M.; Ren H.; Wu W.; Kuipers O. P. Outer-Membrane-Acting Peptides and Lipid II-Targeting Antibiotics Cooperatively Kill Gram-Negative Pathogens. Commun. Biol. 2021, 4, 31. 10.1038/s42003-020-01511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes J. M.; MacNair C. R.; Ilyas B.; French S.; Côté J.-P.; Bouwman C.; Farha M. A.; Sieron A. O.; Whitfield C.; Coombes B. K.; Brown E. D. Pentamidine Sensitizes Gram-Negative Pathogens to Antibiotics and Overcomes Acquired Colistin Resistance. Nat. Microbiol. 2017, 2, 17028. 10.1038/nmicrobiol.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negash K. H.; Norris J. K.S.; Hodgkinson J. T. Siderophore-Antibiotic Conjugate Design: New Drugs for Bad Bugs?. Molecules 2019, 24 (18), 3314. 10.3390/molecules24183314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M.; Miller M. J. Synthesis and in Vitro Antibacterial Activity of Spermidine-Based Mixed Catechol- and Hydroxamate-Containing Siderophore-Vancomycin Conjugates. Bioorg. Med. Chem. 1996, 4 (1), 43–48. 10.1016/0968-0896(95)00161-1. [DOI] [PubMed] [Google Scholar]
- Sato T.; Yamawaki K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 2019, 69 (Supplement_7), S538–S543. 10.1093/cid/ciz826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miethke M.; Marahiel M. A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol. Mol. Biol. Rev. 2007, 71 (3), 413–451. 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris W. R.; Carrano C. J.; Cooper S. R.; Sofen S. R.; Avdeef A. E.; McArdle J. V.; Raymond K. N. Coordination Chemistry of Microbial Iron Transport Compounds. 19. Stability Constants and Electrochemical Behavior of Ferric Enterobactin and Model Complexes. J. Am. Chem. Soc. 1979, 101 (20), 6097–6104. 10.1021/ja00514a037. [DOI] [Google Scholar]
- Baron S. A.; Devaux C.; Colson P.; Raoult D.; Rolain J.-M. Teicoplanin: An Alternative Drug for the Treatment of COVID-19?. Int. J. Antimicrob. Agents 2020, 55 (4), 105944. 10.1016/j.ijantimicag.2020.105944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyasu S.; Too P. C.; Mu J.; Goh C. C.; Ding Y.; Tnay Y. L.; Yeow E. K. L.; Yang L.; Ng L. G.; Chiba S.; Xing B. Glycopeptide Antibiotic Analogs for Selective Inactivation and Two-Photon Imaging of Vancomycin-Resistant Strains. Chem. Commun. 2016, 52 (25), 4667–4670. 10.1039/C5CC10230H. [DOI] [PubMed] [Google Scholar]
- Apostolos A. J.; Ferraro N. J.; Dalesandro B. E.; Pires M. M. SaccuFlow: A High-Throughput Analysis Platform to Investigate Bacterial Cell Wall Interactions. ACS Infect. Dis. 2021, 7 (8), 2483–2491. 10.1021/acsinfecdis.1c00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.-S. A.; Chen P.-L.; Chen Y.-C. S.; Hung H.-M.; Huang J.-Y. Selectively Targeting and Differentiating Vancomycin-Resistant Staphylococcus Aureus via Dual Synthetic Fluorescent Probes. ACS Infect. Dis. 2021, 7 (9), 2584–2590. 10.1021/acsinfecdis.1c00235. [DOI] [PubMed] [Google Scholar]