

Abstract
INTRODUCTION:
Understanding and treating disease requires deep, systematic characterization of different cells and their interactions across human tissues and organs, along with characterization of the genetic variants that causally contribute to disease risk. Recent studies have combined single-cell atlases of specific human tissues and organs with genes associated with human disease to relate risk variants to likely cells of action. However, it has been challenging to extend these studies to profile multiple tissues and organs across the body, conduct studies at population scale, and integrate cell atlases from multiple organs to yield unified insights.
RATIONALE:
Because of the pleiotropy and specificity of disease-associated variants, systematically relating variants to cells and molecular processes requires analysis across multiple tissues and individuals. Prior cell atlases primarily relied on fresh tissue samples from a single organ or tissue. Single-nucleus RNA sequencing (snRNA-seq) can be applied to frozen, archived tissue and captures cell types that do not survive dissociation across many tissues. Deep learning methods can integrate data across individuals and tissues by controlling for batch effects while preserving biological variation.
RESULTS:
We established a framework for multi-tissue human cell atlases and generated an atlas of 209,126 snRNA-seq profiles from eight tissue types across 16 individuals, archived as frozen tissue as part of the Genotype-Tissue Expression (GTEx) project. We benchmarked four protocols and show how to apply them in a pooled setting to enable larger studies. We integrated the cross-tissue atlas using a conditional variational autoencoder, annotated it with 43 broad and 74 fine categories, and demonstrated its use to decipher tissue residency, such as a macrophage dichotomy and lipid associations that are preserved across tissues, and tissue-specific fibroblast features, including lung alveolar fibroblasts with likely roles in mechanosensation. We relate cells to human disease biology and disease-risk genes for both rare and common diseases, including rare muscle disease gene groups enriched in distinct subsets of myonuclei and nonmyocytes, and cell type–specific enrichment of expression and splicing quantitative trait locus (QTL) target genes mapped to genome-wide association study loci.
CONCLUSION:
Our framework will empower large, cross-tissue population and/or disease studies at single-cell resolution. These frameworks and the cross-tissue perspective provided here will form a basis for larger-scale future studies to improve our understanding of cross-tissue and cross-individual variation of cellular phenotypes in relation to disease-associated genetic variation.
Graphical Abstract
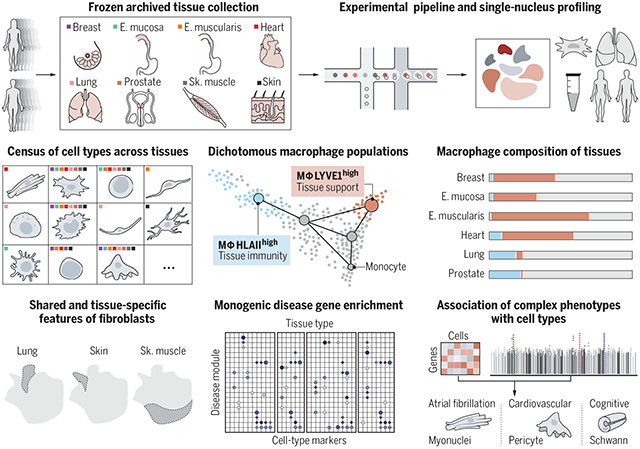
Cross-tissue snRNA-seq atlas in eight frozen, archived adult human tissues. Tissue sites and experimental pipeline (top row). The resulting atlas enables a cross-tissue census of tissue-specific and shared cell types (middle left). Differentiation trajectories and compositional analysis of dichotomous macrophage populations improve our understanding of tissue residency (middle center and right). Analyses of fibroblasts across tissues reveal tissue-specific and shared fibroblast features and their functional interpretation (bottom left). Robust and scalable computational methods enable comprehensive associations of monogenic and complex diseases to tissue-specific and shared cell populations (bottom center and right). E. mucosa, esophagus mucosa; E. muscularis, esophagus muscularis; Sk. muscle, skeletal muscle.
Understanding gene function and regulation in homeostasis and disease requires knowledge of the cellular and tissue contexts in which genes are expressed. Here, we applied four single-nucleus RNA sequencing methods to eight diverse, archived, frozen tissue types from 16 donors and 25 samples, generating a cross-tissue atlas of 209,126 nuclei profiles, which we integrated across tissues, donors, and laboratory methods with a conditional variational autoencoder. Using the resulting cross-tissue atlas, we highlight shared and tissue-specific features of tissue-resident cell populations; identify cell types that might contribute to neuromuscular, metabolic, and immune components of monogenic diseases and the biological processes involved in their pathology; and determine cell types and gene modules that might underlie disease mechanisms for complex traits analyzed by genome-wide association studies.
Tissue homeostasis and pathology arise from an intricate interplay between different cell types, such that disease risk is influenced by variation in genes that affect the cells’ functions and interactions. Human genetics studies, to date, have mapped tens of thousands of loci that either underlie rare monogenic disease or are associated with complex polygenic disease risk (1-3), including many in regulatory regions, whereas single-cell genomics has become instrumental in constructing cell atlases of both healthy organs and diseased tissues (4-6).
Coupling these advances in human genetics and single-cell genomics should enhance our understanding of cell type–specific changes in the function and regulation of disease genes. In particular, tissue (7), cell type (8-11), time point, and stimulation (12-14) all affect gene expression in disease-associated genetic loci. Recently, studies combining single-cell expression atlases and genetic signals have been able to associate risk genes with specific cell types and states in relevant tissues (15-18).
Because complex diseases often manifest in and implicate cells across multiple tissues, fully understanding the way in which genetic variation affects disease requires generating atlases from diverse tissues across the body and from many individuals, spanning different populations. This poses several challenges. First, collecting fresh tissue samples at scale is logistically challenging, and some tissues, such as brain, muscle, and adipose, are difficult to process into single-cell suspensions (19-23). As a result, large-scale single-cell profiling studies in human populations (24, 25) have focused on peripheral blood mononuclear cells, which can be frozen and thawed for multiplexed single-cell analysis. Single-nucleus RNA sequencing (snRNA-seq) offers a compelling alternative because it can be applied to archived, frozen tissues (26, 27) from multiple organs and captures diverse cell types. Second, annotation and classification of cell types and states require defining biological relationships between parenchymal, immune, and stromal cells across tissue types. Finally, data integration and interpretation require cross-tissue analytical frameworks to remove unwanted variation while preserving biological differences; identify cell types and states; and relate cell types and states to monogenic and complex trait genetics.
Results
A multitissue, multi-individual single-nucleus reference atlas from archived, frozen human tissues
We constructed a cross-tissue snRNA-seq atlas from 25 archived, frozen tissue samples, previously collected and banked by the Genotype-Tissue Expression (GTEx) project (7), that span three or four samples from each of eight tissue sites—breast, esophagus mucosa, esophagus muscularis, heart, lung, prostate, skeletal muscle, and skin—from 16 individuals (seven males and nine females) (Fig. 1A). We selected the samples by RNA quality, tissue autolysis score, and the availability of existing bulk RNA-seq and genome sequencing data [(28); table S1]. Histology slides corresponding to each tissue were reviewed by a pathologist to provide detailed annotations (table S1). Because different nucleus extraction protocols can be optimal for different tissues (26, 29), we isolated nuclei from each sample using four protocols that vary in detergents, salt, buffer, and mechanical preparation methodology [CST (0.49% CHAPS detergent, salts, and Tris buffer), NST (NP-40, salts, and Tris buffer), TST (0.03% Tween 20 detergent, salts, and Tris buffer), and the EZ nuclei isolation kit (proprietary composition; NUC101, Sigma-Aldrich); (26, 28, 29); table S1], followed by droplet-based single-cell RNA-seq (scRNA-seq) (28).
Fig. 1. Cross-tissue snRNA-seq atlas in eight archived, frozen adult human tissues.

(A) Study design. (B to F) Cross-tissue single-nucleus atlas. Uniform manifold approximation and projection (UMAP) representation of single-nucleus profiles (dots) colored by main compartments (B), broad cell types (C), tissues (D), isolation protocol (E), and individual donors (F). (G) Cell-type composition across tissues. The overall proportion of cells (%) of each type and number of nuclei profiled in each tissue (rows) are shown. Numbers in circles indicate the corresponding broad cell type. Black vertical lines indicate the relative proportion of nuclei from each individual.
We processed the initial snRNA-seq profiles to retain high-quality nuclei profiles and remove the effects of contaminant transcripts from ambient RNA (28). In breast and skin, the majority of nuclei profiles were recovered from only one individual sample for each tissue (breast: 61.3%, skin: 93.1%; table S1). Some tissues and protocols had higher ambient RNA contamination, reflected as spurious expression of highly expressed transcripts from one cell type in nuclei profiles of other cell types. Such effects were more prominent in skeletal muscle and heart [false discovery rate (FDR) < 0.05], irrespective of protocol, but were also present in other tissues [(28); fig. S1]. We corrected for ambient RNA contamination with CellBender v2.1 (30) (fig. S1) and further applied standard quality control metrics (28), retaining 209,126 nuclei profiles across the eight tissues, with a mean of 918 genes and 1519 transcripts (unique molecular identifiers) detected per profile.
Cross-tissue atlas annotation recovers diverse cell types, including difficult-to-profile and rare cell subsets
We integrated data from all samples and methods using a conditional variational autoencoder (cVAE), which is designed to correct for multiple sources of variation in expression, such as individual-, sex-, and protocol-specific effects, while preserving tissue- and cell typey–specific variation [(28); Fig. 1, B to F, and figs. S2A and S3]. We benchmarked the cVAE against several other data integration methods, obtaining comparable or improved results and providing guidelines for future integration efforts [fig. S4 and supplementary text note S1; (28)]. Cells grouped first by cell type and then by tissue-specific subclusters (Fig. 1, B to D), suggesting that the variation between cell types is larger than the variation within a cell type across tissues.
We annotated cell types within each tissue alter dimensionality reduction and graph-based clustering (28) by identifying genes that are differentially expressed between clusters and comparing them with literature-based marker genes [(28); tables S2 and S3]. We curated comprehensive lists of cell-type markers from the literature for each tissue (figs. S5 and S6 and table S3), including markers for relatively poorly characterized cells, such as interstitial cells of Cajal (ICCs). We defined cellular compartments shared across tissues (e.g., adipose, endothelial, epithelial, fibroblast, immune, muscle) (Fig. 1B), broad cell types (e.g., luminal epithelial cells, vascular endothelial cells) (Fig. 1C and fig. S5), and granular cell subsets (e.g., luminal epithelial cell 1 and 2) (fig. S6). The annotations were consistent across extraction protocols, tissues, and donors (figs. S2, B and D, and S7).
The atlas features 43 broad cell classes (Fig. 1C and tables S2 and S3), with both tissue-shared cell types and tissue-specific subsets (e.g., Fig. 1G and figs. S2, B and D, and S5). For example, tissue-specific cell types such as pneumocytes (alveolar type I and II) and keratinocytes were the predominant cell types in the lung and skin, respectively. Many shared broad cell types such as immune and stromal cells were detected across all tissues (fig. S2, D and E), but with tissue-specific specializations (discussed later in the text). For example, macrophages made up the largest immune population, with diverse subsets of tissue-resident cells.
The atlas captured profiles from cell classes that are difficult to profile by dissociation-based scRNA-seq (23, 31, 32), including 2350 adipocytes, 21,607 skeletal muscle myonuclei, and 9619 cardiac myonuclei. We detected adipocytes in five of the eight tissue types (breast, muscle, heart, esophagus muscularis, and skin), with 86% of adipocytes from breast tissue (fig. S2D), making up 18% of all breast nuclei profiles (Fig. 1G) (28). Skeletal and cardiac myonuclei included key subsets (33, 34). Cardiac myonuclei primarily included the previously distinguished classical myonuclei as well as the recently reported “cytoplasmic myonuclei” (33) (fig. S8 and supplementary text note S2). Other myonuclei subsets included neuromuscular junction (NMJ)–localized skeletal muscle myonuclei, which have also been observed in scRNA-seq and snRNA-seq studies in mice (21, 31, 35), and “fast-twitch” and “slow-twitch” subtypes (Fig. 1, B and C, and fig. S6G), which are characterized by differentially expressed genes (fig. S9, A and B) that are concordant with previously reported markers (36) (fig. S9, C and D).
Cross-tissue and cross-sample integration enhanced our ability to resolve multiple rare cell subsets (Fig. 1C and figs. S2, B and D, S5, and S6). For example, we detected Schwann cells that support peripheral nerves (37) in multiple tissues (esophagus mucosa and muscularis, heart, prostate, and skeletal muscle), rare neuroendocrine cells in the prostate (38), and rare (26) ICCs and enteric neurons in the esophagus. Because these rare cells can contribute to various pathologies, their profiling in human tissues will enable disease studies (26, 37).
snRNA-seq protocols perform well across tissues and correspond to scRNA-seq
We benchmarked the performance of our nucleus extraction and profiling protocols (26, 29) relative to each other across all eight profiled tissues and to other snRNA-seq, scRNA-seq, and bulk RNA-seq datasets in relevant tissues. For each dataset, we compared standard quality control metrics per profiled cell or nucleus, as well as the diversity and proportions of captured cell types.
Of the four tested nucleus isolation protocols (CST, NST, TST, and EZ; table S1), the EZ protocol displayed lower performance in each of the eight profiled tissues by multiple quality metrics (28, 39) (Fig. 2A and fig. S10). These included the lowest total number of nuclei captured (fig. S10, A and B), higher levels of ambient RNA (FDR < 0.05; fig. S1, A and B), and separate grouping of EZ-profiled samples [fig. S2C; (28)].
Fig. 2. Concordance of cell-type diversity and cell-intrinsic profiles between snRNA-seq and scRNA-seq.

(A) Cell-type diversity (Shannon entropy, y axis) of each protocol (color) in each sample (dot) and tissue (x axis). Dashed lines indicate the average across samples. (B) Differences in cell proportions. The proportions (y axis) of cells from major categories (color) in each individual by tissue and protocol (x axis) are shown. (C to E) Concordance of cell-intrinsic programs. Proportions of cells (dot color and size) of a manually annotated group (rows) predicted to belong to a given nucleus profile annotation label (columns) by a random forest classifier trained on nuclei and applied to cells of the same tissue for skin (C), lung (D) or prostate (E) are shown. (F) Tissue dissociation expression signatures in scRNA-seq. Scores [y axis, average background corrected log(TP10K+1)] of a dissociation-related stress signature (41) in scRNA-seq (pink) and snRNA-seq (blue) profiles in each major lung cell type (x axis) are shown (***Benjamini-Hochberg FDR < 10−16, Wilcoxon rank sum test). Box plots show median, quartiles, and whiskers at 1.5 times the interquartile range (IQR). (G) Divergent genes between cell and nucleus profiles. Averaged pseudobulk expression (28) of protein-coding genes (dots) in skin basal keratinocyte nuclei (x axis) and cells (y axis) is shown. Divergent genes are represented by a black dot outline. The color scale shows the total length of polyA stretches with at least 20 adenine bases in log2 scale. Epi., epithelial; sm., smooth; SMC, smooth muscle cell.
The extraction protocols also varied in the proportion of nuclei recovered from each cell type (figs. S2, B and D, and S11A; supplementary text note S3), consistent with our previous observations in tumors (29). The TST, CST, and NST protocols had comparable cell-type diversity as measured by Shannon entropy [Fig. 2A; (28)], whereas the EZ protocol resulted in significantly lower diversity (Fig. 2A; linear mixed-effects model effect size of −1.08, P = 5 × 10−11). Overall, TST yielded the highest cell-type diversity, on average, across tissues (Fig. 2A) and significantly higher proportions of T cells, fibroblasts, and vascular endothelial cells (FDR < 10%; fig. S11A). Because the protocols varied by their performance (most diverse, high capture of the desired cell types), users should choose protocols by matching protocol features to scientific goal, tissue type, and complexity; and further protocol optimization may still be required (29) (for further details and guidance, see supplementary text note S3).
We compared cell-type compositions between our four protocols and other snRNA-seq studies, focusing on our frozen heart left-ventricle samples, for which two recently published snRNA-seq studies evaluated similar samples (33, 34). We found agreement in broad cell types such as mast cells, adipocytes, “cytoplasmic” cardiac myonuclei, and Schwann cells (fig. S11, B and C, and table S4)—but differences in some of their proportions (fig. S11, D and E). Protocols used in the published studies and the EZ protocol in our study captured a higher proportion of muscle nuclei, whereas CST, NST, and TST yielded a higher proportion of endothelial cells (fig. S11E). There was also high concordance between the expression profiles of bulk RNA-seq [from GTEx; (7)] and pseudobulk profiles derived from our snRNA-seq (accuracy 92.3%; fig. S12). A few samples showed lower agreement (heart-EZ, breast-EZ, and two breast-TST samples), suggesting that these particular tissue-protocol combinations may not reflect cellular composition as accurately.
We next compared snRNA-seq data to fresh-tissue scRNA-seq data from lung (40), skin [current study; (28)], and prostate (38). For cell composition (Fig. 2B), we recovered the same main cell groups across compartments. We confirmed the accuracy of our annotations by training a multiclass random forest classifier on snRNA-seq data and predicting cell types on scRNA-seq data [(28); Fig. 2, C to E], and vice versa (fig. S11, F to H). In addition, cell-type intrinsic (pseudobulk) profiles of protein-coding genes were overall similar between snRNA-seq and scRNA-seq [average Spearman ρ across cell types of 0.58 (skin), 0.69 (prostate), and 0.47 (lung); table S4]. Moreover, integrating cell and nuclei profiles from prostate, skin, and lung and annotating the cells with a random forest classifier that is trained on our nuclei profiles with our granular annotations yielded well-mixed groupings, similar marker genes, and high concordance between protocols (fig. S13).
Divergences observed include the greater expression in cells versus nuclei of a dissociation-induced stress signature (41, 42) (Wilcoxon rank sum test, Benjamini-Hochberg FDR < 10−16; Fig. 2F and fig. S14, A and B), as reported (29), and of ribosomal and nuclear-encoded mito-chondrial protein genes [Fig. 2G; (28); linear model], consistent with their longer half-lives and higher cytoplasmic levels (43, 44). Conversely, nuclei profiles had higher levels of longer transcripts (fig. S14, H and I) and of transcripts with a larger number of adenine stretches (Fig. 2G and fig. S14, C to G), consistent with previous reports (45).
Notably, our snRNA-seq generally captured relatively lower proportions of lymphocytes. For example, for lung and skin, respectively, T cells represented 1.7 and 1.4% of all cells (aggregated) compared with 8.73 and 6.83% by scRNA-seq. We observed similar patterns for B cells in skin. Furthermore, these immune cell proportions varied across samples and protocols. A study comparing snRNA-seq and in situ measurements (46) suggested that scRNA-seq may oversample immune cells.
Myeloid populations across tissues
Our cross-tissue atlas allowed us to characterize tissue-specific and shared features of tissue-resident immune cells, which play key roles in immune surveillance and tissue support (47, 48). Integration and annotation of 14,156 myeloid nuclei profiles (28) (60% of immune nuclei) revealed 14 distinct monocyte, macrophage, and dendritic cell (DC) subsets (Fig. 3A; fig. S15, A and B; and table S5). These included CD16+ monocytes, CD14+ monocytes, two transitional Mo/MΦ FCGR3Alow and Mo/MΦ FCGR3Ahigh populations with coexpression of both monocyte and macrophage markers (see next section), DC1s, DC2s (49), mature DCs, and Langerhans cells (Fig. 3B). Tissue macrophage states further included lung macrophages expressing an alveolar macrophage signature (50) (fig. S15C), proliferating macrophages, cytokine- and chemokine-expressing inflammatory macrophages, and two additional macrophage subsets: MΦ LYVE1high and MΦ HLAIIhigh (Fig. 3B), where HLAII is HLA class II. Finally, lipid-associated macrophage (LAM)–like nuclei highly expressed LAM signature (Fig. 3B and fig. S15C) (51) as well as lipid metabolism-related, myeloid cell immune activation, and macrophage migration genes (fig. S15D). Although most myeloid subsets were present in multiple tissues, notable exceptions included PPARGhigh lung macrophages, which were present only in lung, and CD207/Langerin+ Langerhans cells, which were present only (97%) in skin and esophagus mucosa, consistent with their role in antigen sampling within stratified epithelia (52, 53) (Fig. 3C and fig. S15, F and I).
Fig. 3. A dichotomy between LYVE1- and HLAII–expressing macrophages and LAM-like populations across tissues.

(A) Myeloid profiles (dots), colored by cell type and state and overlaid with a PAGA graph of myeloid states (large nodes). (B) Expression of marker genes (columns) associated with each subset (rows). (C) Myeloid cell distribution across tissues. The overall proportion of myeloid cell subsets (colors) in each tissue (bars) is shown at the top, and the overall proportion of cells from each tissue in each subset (bars) is shown at the bottom. (D) LYVE1high and HLAIIhigh macrophages are end points of two differentiation trajectories. A diffusion map of monocytes, macrophages, and transitional subsets (colors) is shown. Large circles represent centroids (sizes are proportional to population size). (E) Cross-tissue and tissue-specific markers. Expression of marker genes (columns) associated with two myeloid subsets (left) in each tissue (rows) is shown. The right bar plot shows the number of nuclei. (F to H) LAM-like cells across tissues. Myeloid cells (dots) colored by their classification [legend; (28)] are shown in (F). Classification scores (y axis) of LAM-like and other macrophages across tissues (x axis) are shown in (G). Expression of LAM marker genes (columns) in LAM-like profiles from other studies (rows) is shown in (H). (I and J) Inferred TFs regulating the LAM-like program. TF differential activity scores between LAMs and other macrophages (y axis) for each TF (dot) ranked by score (x axis) are shown in (I). TF differential activity scores (x axis) for three TFs with significantly high scores (two tailed t test; Benjamini-Hochberg *FDR < 0.05, **FDR < 0.01, and ***FDR < 0.001) in LAMs or other macrophages are shown in (J). Box plots show median, quartiles, and whiskers at 1.5 times the IQR. E., esophagus; ENS, enteric nervous system; Sk., skeletal.
Myeloid cell proportions were more highly correlated between samples within a tissue type (fig. S15, E and F) than between different tissues (fig. S15, E to H), confirming the reproducibility of tissue-specific myeloid state proportions. Moreover, related tissues—such as muscle (heart, esophagus muscularis, skeletal muscle) or epithelial barriers (esophagus mucosa, skin)—grouped by their myeloid composition profiles (fig. S15H). Macrophage types and proportions varied by tissue, with breast, esophagus mucosa, esophagus muscularis, heart, and skeletal muscle having significantly higher proportions of MΦ LYVE1high macrophages [P < 10−6, Dirichlet regression likelihood ratio test (LRT); (28)] and lung and prostate having significantly higher proportions of MΦ HLAIIhigh [P < 10−8, Dirichlet regression LRT; (28)] (Fig. 3C and fig. S15, F and I).
A dichotomy between LYVE1- and HLAII–expressing macrophages is preserved across tissues
Two expression states of LYVE1high and HLAIIhigh macrophage populations were dichotomous—either LYVE1highHLAIIlow or LYVE1lowHLAIIhigh—and represented the end points of two alternative branches. Specifically, low-dimensional representation of monocytes, macrophages, and Mo/MΦ populations as a continuum with diffusion maps captures the HLAIIhigh and LYVE1high cells as “terminal” points in two branches that emanate from CD14+ monocytes at the root. Each of the two terminals is preceded by distinct earlier putative transitional states: a Mo/MΦ FCGR3Alow state between CD14+ monocytes and the LYVE1highHLAIIlow population and a Mo/MΦ FCGR3Ahigh state between the monocytes and the LYVE1low HLAIIhigh cells (Fig. 3D and fig. S16, A and B). (There is also a putative secondary path between FCGR3Alow and LYVE1lowHLAIIhigh cells through a FCGR3Ahigh intermediate.) The position of the FCGR3Ahigh transitional state is consistent with lung data from a humanized mouse model (54, 55). These key features are consistent overall in the map that is constructed only from macrophage nuclei from a single tissue (fig. S16A). Thus, FCGR3Alow and FCGR3Ahigh states might be Mo/MΦ populations that are less-differentiated or less-activated states of LYVE1high and HLAIIhigh MΦs, respectively.
Each of the LYVE1high and HLAIIhigh subsets expressed a combination of a common signature and tissue-specific markers (Fig. 3E, fig. S16C, and table S5) and was enriched for distinct functions, mirroring those of Lyve1high MHCIIlow and Lyve1lowMHCIIhigh resident macrophage populations in mouse tissues (47) (fig. S16, C and E). HLAIIhigh cells were enriched for immune-related processes and expressed higher levels of complement components APOE and C1QA, C1QB, and C1QC (Fig. 3B and fig. S16, C and D). Genes differentially expressed between human HLAIIhigh and LYVE1high subsets corresponded to those in murine counterparts (fig. S16E), but with higher expression in human HLAIIhigh macrophages of C1QB and C1QC complement genes in lung and phagocytic receptors MARCO and CD36 in heart (fig. S16E). HLAIIhigh macrophages were also enriched for immune interactions with B cells, DCs, mast cells, natural killer (NK) cells, and T cells (fig. S16G). LYVE1high profiles were enriched for tissue-supporting modules and had putative receptor-ligand interactions (28) with lymphatic endothelial cells, fibroblasts, adipocytes, and myocytes (fig. S16F). In mice, MΦ Lyve1high cells were located near blood vessels (47) and regulated vascular tone (56), and cardiac LYVE1+ macrophages have been implicated in regulating the lymphatic network (57). Thus, LYVE1high macrophages may have a homeostatic role in the human heart, lung, and esophagus.
LAM-like macrophages are prevalent across human tissues and share a regulatory program
In our atlas, we identified LAM-like cells as widely distributed across healthy human tissues, with the vast majority of LAMs (97%, 268 of 283) from the breast, heart, lung, and prostate (Fig. 3C). LAMs and LAM-like cells have been previously reported in disease contexts in adipose tissue from obese humans and mice (51), injured and fibrotic liver (58-60), obese liver (61), fibrotic lung (50, 62), atherosclerotic aortic tissue (63, 64), leprosy (65), and the brains of individuals with Alzheimer’s disease (66-68). However, an understanding of their distribution and heterogeneity across human tissues is still lacking.
To characterize LAMs across the body, we analyzed LAM-like cells in the expanded context of our study and 17 other published atlases spanning 14 tissues. We trained a linear classifier with published omental adipose scRNA-seq containing LAMs (28, 51) and classified each myeloid profile in our dataset and the published compendium as LAM-like macrophages, non-LAM macrophages, and non-macrophages. From this, we recovered 283 LAM-like cells in our study and 4285 LAM-like cells in the 17 published studies (Fig. 3, F to H; fig. S17, A to D; and table S6).
LAM-like cells were present among tissue-resident macrophages across a broad range of tissues and pathologies. These included adipose (51, 69) and atherosclerotic (70, 71) tissue, as reported, as well as healthy tissues [placenta (72), testis (73), kidney (74), pancreas (75), prostate (38), decidua (72), liver (76), ovary (76, 77), skeletal muscle (78), and intestine (26)] and other disease contexts [acne (79), leprosy (79) and atopic dermatitis (80, 81) (skin), and Crohn’s disease (ileum) (82)]. Microglia from the central nervous system of epileptic patients (83) were also classified as LAM-like cells, indicating that microglia that express LAM signature genes extend beyond Alzheimer’s disease (66-68). LAM signature genes were enriched for genome-wide association study (GWAS) genes associated with levels of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol, triglycerides, type 2 diabetes, and the tau to Aβ1-42 ratio in cerebrospinal fluid (fig. S17H and table S7), further supporting their role in lipid homeostasis.
Although a core set of signature LAM genes was expressed across most tissues and studies, many other genes varied across tissues. For example, both CHIT1 and CTSK were highly expressed in LAMs from leprosy skin samples (but had very low expression in other skin LAMs) (Fig. 3H and fig. S17F), in line with higher serum chitotriosidase activity in leprosy patients (84). Select lipid pathway genes, including fatty-acid binding protein FABP4, lipoprotein lipase LPL, and phagocytic lipid receptor CD36, were highly expressed in LAMs from tissues with high adipose content, including adipose, atherosclerotic lesions, and intestine creeping fat samples, possibly reflecting increased lipid-induced transcriptional stimulation of these target genes under these conditions (85). LAMs in creeping fat from Crohn’s intestine and atherosclerotic lesions were additionally characterized by higher expression of interleukin IL1B and multiple chemokines (CXCL8, CXCL3, CCL4), possibly reflecting the inflammatory environment under these conditions (fig. S17F).
We predicted transcription factors (TFs) that could mediate LAM-like gene expression by inferring TF activity from target expression (28) and ranking TFs by the mean difference between their activities in LAMs versus non-LAM macrophages [(28); Fig. 3I]. LAM-associated TFs inferred across all classified LAM-like cells included PPARG, USF1, and NR1H3 (LXRA) (Fig. 3J and fig. S17G), suggesting a shared core regulatory mechanism. These are major regulators of lipid metabolism–related expression (86) and have been proposed to regulate Trem2 expression in mice (87).
Shared and tissue-specific features of fibroblasts
To characterize fibroblast heterogeneity (88), we analyzed 32,421 fibroblast nuclei profiles across the eight profiled tissues (fig. S18A), identifying shared and tissue-specific signatures. The cross-tissue, shared fibroblast expression program consisted of markers that were significantly more highly expressed in fibroblasts than in nonfibroblast cell types within each tissue (FDR < 0.05, Welch’s t test) and included multiple extracellular matrix (ECM) constituents (Fig. 4A and table S8). Conversely, the tissue-enriched fibroblast signatures were defined based on genes exclusive to or highly enriched in fibroblasts from a given tissue versus fibroblasts from all other tissues (Fig. 4, B to D, and fig. S18B).
Fig. 4. Shared and tissue-specific fibroblast features.

(A and B) Expression in each tissue subset (rows) of marker genes (columns) distinguishing fibroblasts from nonfibroblasts across all tissues (A) or enriched in fibroblasts in one versus other tissues (B). (C and D) Fibroblast profiles (dots) colored by tissue (C) or expression of the most exclusive marker (D). (E) Significance [−log10(FDR), x axis] of gene sets (y axis) enriched (FDR < 5%) in genes covarying with the lung-specific fibroblast signature. (F) Expression of ECM and cation transport genes (columns) in the covarying gene module in each granular fibroblast subtype in each tissue (rows). (G) ITGA8 and PIEZO2 (columns) expression in granular cell types (rows) in lung. (H) Significance (x axis) and Open Targets Genetics locus-to-gene score (color) of the most significant variants mapped to NPNT with a high (>0.5) locus-to-gene score in GWASs (y axis). FEV, forced expiratory volume.
Tissue-enriched fibroblast features were consistent with the specific functions and interactions required in the respective tissues. For example, the esophagus mucosa fibroblast signature (table S8) included genes involved in neuron and axon development (e.g., NTN1, PLXNB1, FGF13), suggesting interactions with the enteric nervous system, possibly through NTN1-DCC and NTN1-UNC5C interactions (fig. S18E). The cardiac fibroblast signature genes included TFs involved in cardiac development (e.g., GATA4 and GATA6), revealing that expression of these developmental TFs is retained in the adult cardiac fibroblast compartment (89-92). The skeletal muscle signature showed increased expression of the CXCL14 and CXCL12 chemokines and of components of the renin-angiotensin-aldosterone system (AGTR1 and MME), which regulate skeletal muscle mass (93, 94) [reminiscent of the expression of the same components by WNT2B+ fibroblasts in a local renin-angiotensin system in the colon (16)].
Lung fibroblast signatures were enriched for ECM, cation transport, and contractile functions (Fig. 4E), including multiple components of the basement membrane (BM) that are required for epithelial-mesenchymal adhesion [nephronectin (NPNT), FRAS1, hemicentin-1 (HMCN1), and integrin ITGA8 (Fig. 4, F and G), which form a protein complex (95-98) that anchors NPNT to the BM (99-103)]. Mutations in ITGA8, FRAS1, and HMCN1 have been linked to Fraser syndrome, a congenital disorder that affects cell adhesion and results in skin, kidney, lung, and craniofacial abnormalities (104-107), and common variants in NPNT are significantly associated with chronic obstructive pulmonary disease (COPD) and forced expiratory volume (FEV) in GWASs (Fig. 4H). Granular annotation of the lung fibroblast profiles (40, 108) (fig. S18C) suggests that the adhesion complex is expressed by alveolar fibroblasts (Fig. 4F and fig. S18D). Alveolar fibroblasts also specifically expressed FGFR4 (Fig. 4B), which is essential for alveologenesis in mice (109) and is genetically linked with bronchopulmonary dysplasia that affects alveoli in humans (110).
Of all tissue fibroblasts, lung alveolar fibroblasts distinctively expressed a calcium signaling and actomyosin contractility program (Fig. 4, E and F, and fig. S18D), including the mechanosensitive calcium ion channel PIEZO2 (Fig. 4G), which has been proposed to sense pulmonary stretch (111, 112); multiple calcium channels and adrenergic and purinergic receptors, which have been implicated in stimulating calcium release from intracellular stores (CACNA1D, TRPC6, MCOLN2/TRPML2); and myosin light chain kinase (MYLK), which is involved in mediating calcium-induced actomyosin contraction (113). This suggests that alveolar fibroblasts could integrate mechanical stretch and forces (through PIEZO2) and neuronal excitatory signals (40), which could affect their migratory or mechanical properties. Notably, although alveolar fibroblasts express myofibroblast markers (e.g., ACTA2; fig. S18D), they are distinct from previously reported myofibroblasts (40), and our data suggest that they constitute a distinctive contractile and excitable fibroblast state.
Intra- and cross-tissue cell-type associations with monogenic disorders
Human genetics has identified numerous rare monogenic disease genes, and many have been experimentally mapped to cell type(s) of action (114, 115). To characterize the expression of monogenic disease genes across cell types, we related disease genes associated with different phenotypes or disease categories in the Online Mendelian Inheritance in Man (OMIM) database (116) to the cell populations in which they are expressed in our cross-tissue atlas. Because OMIM entries are not organized by disease categories, we leveraged topic modeling to aggregate 5812 genotype-phenotype associations based on similarities in text descriptions of clinical features, resulting in 229 distinct disease topics [(28); figs. S19 and S20). We then related topics to a cell type based on the enriched expression of the topic genes in the cell type (Fig. 5A, figs. S21 and S22, and table S9).
Fig. 5. Monogenic muscle disease genes related to cell types and interactions across cardiac, skeletal, and smooth muscle tissues.

(A) Enrichment of monogenic disease groups to broad cell types. Effect size (log odds ratio, dot color) and significance [−log10(FDR), dot size] of enrichment of genes from disease topics [rows; (28)] in broad cell-type markers in each tissue (columns) are shown. A red outline indicates an FDR less than 0.1. Topic names consist of the topic identifier and five words with the highest loadings. Red stars indicate highlighted topics. (B) Relation of broad cell types to monogenic muscle disease groups. Effect size and significance of enrichment of genes from monogenic muscle disease groups (rows) for broad cell type markers in each tissue (columns) are shown. A red outline indicates an FDR less than 0.1. Color shading indicates disease groups associated with only nonmyocytes (green), only myonuclei (yellow), or both (light purple). (C and D) DMD expression in human (C) and mouse (D) muscle. Cell types (x axis) are ordered, left to right, such that the cell types that are shared between human and mouse within a tissue are presented first and species-specific cell types follow. (E and F) Putative cell-cell interactions in muscle implicating muscle disease genes. Shown are cell types (inner color) from muscle tissues (outer color) connected by putative interactions (dotted edges) between a receptor (left square) expressed in one cell type and a ligand (right square) expressed in the other in interactions involving myocytes (E) or only nonmyocytes (F). Black and gray connecting lines between cell types and genes indicate high and low expression, respectively. Bold formatting indicates a muscle disease gene. (G) Diseases highlighted in (E) and (F). ALS, amyotrophic lateral sclerosis; AD, autosomal dominant; AR, autosomal recessive; CMT, Charcot-Marie-Tooth disease; XR, X-linked recessive.
Many topics mapped to their expected cell populations. For example, cardiac disease topics (topics 65 and 66) mapped to cardiac myonuclei and/or endothelial cells, immune and infection topics (topics 132, 205, and 217) mapped to immune cells across tissues, and a diabetes and lipodystrophy topic (topic 222) mapped to adipocytes in skeletal muscle and skin (Fig. 5A and fig. S23A). Male infertility and sperm motility topic (topic 129) was associated with ciliated lung cells and stromal cells (fig. S23B). The link between sperm dysmotility and bronchitis is well established, with genes common to flagella and cilia perturbed across both sperm and lung (117); we further detect this specific link in topic 155, which associates ciliary dyskinesias and the lung ciliated epithelium (fig. S23C).
Genes from monogenic muscle disease groups are enriched in distinct subsets of myocyte and nonmyocyte nuclei in three muscle types
Among the monogenic disorders, muscle disease phenotypes are a well characterized subset and are known to arise from mutations in genes that are expressed in myocytes (e.g., structural genes involved in contraction) and/or other cells in the surrounding tissue (e.g., NMJ, ECM, and adipose tissue) (118-121). We leveraged the three muscle types represented in our atlas—cardiac, skeletal, and smooth muscle—to map 605 well-curated monogenic muscle disease genes (118) (table S10), recovering known biology and extending hypotheses beyond those obtained from bulk-tissue RNA-seq (122, 123). We tested disease groups (e.g., hereditary cardiomyopathies, motor neuron diseases) for their enrichment with cell type–specific markers across our muscle tissues (FDR < 0.1) [(28); Fig. 5B, fig. S24, and table S11].
As expected, different disease gene sets were associated with different myonuclei subsets (113 of 605 genes; table S11) in patterns that recapitulated known disease mechanisms (Fig. 5B). For example, skeletal muscle myonuclei were associated with congenital myopathy genes (FDR = 7.07 × 10−5), and cardiac myonuclei were associated with hereditary cardiomyopathy genes (FDR = 4.24 × 10−12). Some associations highlighted finer myonuclei subsets. For example, genes linked to congenital myasthenic syndrome, a disorder affecting neuromuscular transmission (124), were specifically expressed in NMJ-localized myonuclei but not in other skeletal myonuclei (tables S2 and S12). These included acetylcholine receptor subunits (CHRNE, CHRNA1, CHRND), the NMJ-organizing receptor tyrosine kinase MUSK, and NMJ-enriched ECM components (COL13A1, LAMA2).
Other disease gene sets were associated with nonmyocyte accessory cells, including neurons, Schwann cells, fibroblasts, and adipocytes (127 genes; table S12), often mirroring clinical features, such as nervous system cells in neuropathies or adipocytes in metabolic myopathies. In particular, Schwann cells were associated with hereditary motor and sensory neuropathies in all three tissues (FDR = 0.015 to 0.06), but their association with Dejerine-Sottas hypertrophic neuropathy (a subtype of Charcot-Marie-Tooth disease) was specific to skeletal muscle (FDR = 2.28 × 10−5), consistent with the need to maintain innervation to prevent muscle atrophy (125). Thus, distinctive cell expression patterns can provide insight into onset (Dejerine-Sottas is an early-onset disease), severity, and the predominant peripheral nerves that are affected (i.e., motor, sensory, or autonomic), where heart can possibly act as a proxy for autonomic nerves. In other examples, adipocytes from esophagus muscularis were associated with metabolic myopathies related to lipid metabolism (FDR = 0.0003); the congenital muscular dystrophies genes COL6A1, COL6A2, and COL6A3 are expressed in fibroblasts, consistent with joint laxity and progressive contractures along with muscle weakness (126); and the recessive limb girdle muscular dystrophies gene DYSF is expressed in immune cells, consistent with the role of more aggressive monocytes in disease progression (127). Some of the enrichments in nonmyocytes are also present in the same cell types in other nonmuscle tissues (e.g., breast adipocytes for metabolic myopathies or breast and skin pericytes for cardiomyopathies; fig. S25I), highlighting that tissue-specific pathology may arise from the relation between an accessory cell’s broader function and specialized tissue demand.
There were differences in the expression of monogenic muscle disease genes between slow and fast myocyte subsets for both classical and cytoplasmic myonuclei (fig. S9). As expected, slow myocytes preferentially expressed type 1 fiber markers (36, 128) and disease genes MYH7 (FDR < 10−15 in cytoplasmic myonuclei) and PPARGC1A (FDR < 10−15 in regular myonuclei). Fast myocytes specifically expressed the type 2 fiber marker MYH2 (FDR < 10−15 in cytoplasmic myonuclei), in line with respective clinical myosinopathy phenotypes (129). Congenital myopathy disease genes, which include genes known to be active in slow and fast myocytes, were enriched in both groups. The myotonic dystrophy (MD) type 2 gene CNBP, was enriched in regular myonuclei from fast myocytes [FDR < 10−12; (28); fig. S9, E and F], which are preferentially affected in MD type 2 (130, 131). By contrast, MD type 1 disease gene DMPK was preferentially expressed in cytoplasmic myonuclei from slow myocytes (P < 0.036, Welch’s t test), which are perturbed in MD type 1 (132). Regular myonuclei subsets from slow myocytes were also enriched in TPM3 [FDR < 10−15; (28); fig. S9, E and F], which has been linked to congenital fiber type disproportion, a condition characterized by smaller slow myocyte fibers (133). Metabolic myopathy disease genes (GBE1, ETFDH, and SLC25A20) were also enriched in slow myocyte markers (P < 0.05, Fisher’s exact test; fig. S9, E and F).
Many of the cell-type associations for monogenic muscle disease genes were conserved between our human muscle atlas and corresponding mouse snRNA-seq data (26, 28) (fig. S25), but there were also notable differences. In both mouse and human, there were significant associations between skeletal muscle myonuclei and various dystrophies and myopathies (FDR < 0.1, Fisher’s exact test), between cardiac myonuclei and cardiomyopathies (FDR < 0.1), between adipocytes (in skeletal muscle, esophagus, and heart) and metabolic myopathies (FDR < 0.1), and between Schwann cells in skeletal muscle and hereditary motor and sensory neuropathies (FDR < 0.1) (fig. S25H). However, dystrophin (DMD) expression across accessory cell types varied between human and mouse. In humans, high levels of DMD expression (comparable to that in myonuclei) were observed in adipocytes in all muscle types; pericytes and Schwann cells in skeletal muscle and esophagus muscularis; and enteric neurons in esophagus muscularis [mean expression log(TP10K+1) > 2.0; Fig. 5C]. Lower, intermediate levels of DMD expression were observed in skeletal muscle fibroblasts and satellite cells, as well as esophagus muscularis ICCs [mean expression log(TP10K+1) < 2.0; Fig. 5C]. DMD expression in adipocytes supports the possibility of local metabolic perturbation (134) (Fig. 5C), whereas its expression in the enteric nervous system and ICCs raises the possibility that perturbation of these cells contributes to the gastrointestinal dysfunction phenotype in Duchenne muscular dystrophy (135). In mouse, whereas Dmd expression is high in myonuclei, pericytes, satellite cells, and Schwann cells, it is low in adipocytes and fibroblasts across all three muscle tissue types [mean expression log(TP10K+1) < 1.0; Fig. 5D]. These differences suggest that the effects of DMD mutation on accessory cells and their contribution to disease (135, 136) may differ between human patients and mouse disease models (137).
Cell-composition and cell-intrinsic secondary effects in muscle disease tissue
Muscle dystrophies commonly display a secondary shift in cellular composition, with fibrotic or adipogenic replacement of muscle tissue (138), which can introduce secondary pathologic processes that exacerbate muscle loss (139). To characterize the cellular expression patterns of disease genes whose bulk-tissue expression levels are altered in muscle diseases, we analyzed the cell-type specificity of genes up-regulated in bulk RNA-seq data from muscle tissues from 43 patients with rare muscle disorders (122) compared with healthy muscle tissues (140) (fig. S26). Some of the cell type–specific expression patterns reflected known biology, including up-regulation of hallmark fibroblast, adipocyte, and immune genes, likely because of changes in cell proportions (e.g., COL6A2, ADIPOQ, and HLA-A, respectively). Other patterns suggest additional, cell-intrinsic regulatory events (beyond cell composition changes) that may modify disease progression. For example, dermatopontin (DPT), an adipokine that promotes ECM remodeling and inflammation (141), is expressed in adipocytes but is not correlated with ADIPOQ expression across adipocytes from different tissues. This suggests that its expression is regulated and is not merely reflecting changes in cell composition. Similarly, ELK3, which is expressed in endothelial cells, suppresses angiogenesis (142) and may contribute to functional muscle ischemia in muscle disease (143), which is otherwise typically attributed to nitric oxide signaling (136). Thus, the increased resolution of a single-cell atlas can help disentangle secondary effects related to cell-composition and cell-regulatory events in accessory cells during disease.
Disease genes may affect receptor-ligand interactions
Some disease genes encode receptors or ligands that participate in cell-cell interactions, such that loss-of-function mutations can affect tissue function through non–cell autonomous effects through these cell-cell interactions. We related cell types in muscle tissue to one another through receptor-ligand interactions (16, 26, 144) that included at least one monogenic disease gene (28) in every tissue in our atlas (table S13).
Our analysis suggests that mutations in some disease-causing genes may disrupt interactions between myocytes and other cell types. For example, mutations in ERBB3 (the disease gene for lethal congenital contracture syndrome) may disrupt interactions between myocytes and Schwann cells (Fig. 5E and table S13) and contribute to joint contractures as an associated, but not primary, disease phenotype (145). Mutations in DAG1 (the disease gene for congenital muscular dystrophy), although known to interact primarily with laminin produced by fibroblasts (146, 147), may additionally disrupt interactions with immune cells through LGALS9 and alter their function (148). Putative cell-cell interactions involving only nonmyocytes included the disease genes L1CAM (MASA syndrome), MET (arthrogryposis and muscular dysplasia), and NGF (hereditary sensory and autonomic neuropathy), each potentially affecting multiple cell pairs, including neurons and Schwann, satellite, immune, and stromal cells (Fig. 5, F and G, and table S13).
Cell type–specific enrichment of QTL genes mapped to GWAS loci
Single-cell atlases can also provide insights into cell-type specificity and mechanisms of action of the genes in disease-associated loci identified by GWASs. Studies associating genetic variants to changes in gene expression or splicing quantitative trait loci (eQTL or sQTL, respectively) showed tissue-specific colocalization with multiple loci from GWASs of human traits, including disease risk (7, 149-151), but lacked cellular resolution (11). To prioritize cell types of action and causal genes for complex diseases and traits in specific cells and tissues, we used ECLIPSER (28, 152) to test whether GWAS loci from 21 complex traits (table S14), with likely effects in at least one of the eight tissues analyzed, are enriched for genes with high cell type–specific expression in each tissue. We defined putative causal genes for each GWAS locus as the set of genes whose eQTLs and sQTLs (7) were in linkage disequilibrium [squared genotype correlation (r2) > 0.8] with the lead GWAS variant(s) [Fig. 6A; (28)]. We further included genes prioritized by additional genomic data [e.g., Hi-C and protein QTLs (pQTLs)] and linkage to predicted deleterious protein-coding variants (153, 154). Because more than one gene typically maps to a GWAS locus using this approach (mean = 2, and maximum = 37 for selected traits and 170 for null traits), we scored loci by the fraction of cell type–specific genes in the locus [Fig. 6A; (28)]. We assessed enrichment for each GWAS locus set against a null distribution of GWAS loci associated with tissue-unrelated traits using a Fisher’s exact test [Fig. 6A; (28)].
Fig. 6. Cell type–specific enrichment of eQTL and sQTL target genes mapped to GWAS loci.

(A) Schematic of the method (ECLIPSER). (B) Cell-type enrichment of genes mapped to GWAS loci for 17 of the 21 complex traits tested with at least one tissue-wide significant result (FDR < 0.05, correcting for all cell types tested per tissue per trait) across eight GTEx tissues. Gray, orange, and red borders indicate nominal, tissue-wide, and experiment-wide significance (FDR < 0.05, correcting for all cell types tested across eight tissues and 21 traits), respectively. Only cell types with at least one tissue-wide enrichment are shown. (C and D) Myonuclei and pericyte genes enriched in atrial fibrillation GWAS loci (tissue-wide FDR < 0.05, Bayesian Fisher’s exact test). Fold-enrichment (x axis) of cell types (y axis) for atrial fibrillation GWAS in heart (top) and skeletal muscle (bottom) is shown in (C). Error bars represent 95% credible intervals. Red indicates tissue-wide significance, orange indicates nominal significance, and blue indicates nonsignificance (P ≥ 0.05, Bayesian Fisher’s exact test). Differential expression in myonuclei versus other cell types from heart (red), skeletal muscle (blue), esophagus muscularis (orange), and prostate (brown) of the genes (x axis) driving enrichment of atrial fibrillation GWAS loci in heart cardiac myonuclei is shown in (D). Gray and pink vertical lines indicate log2(fold change) > 0.5 and FDR < 0.1 in myonuclei in all four tissues or only in heart, respectively. FC, fold change.
Seventeen of the traits were enriched in both expected and previously undescribed cell types at a tissue-wide FDR less than 0.05 (Benjamini-Hochberg), 16 of which were significant (FDR < 0.05) across tissues (Fig. 6B, figs. S27 and S28, and tables S15 and S16). Among the expected associations are skin pigmentation traits in melanocytes, autoimmune and inflammatory diseases in T and NK cells, COPD in lung fibroblasts, prostate cancer in luminal epithelial cells, atrial fibrillation and heart rate in myonuclei, and heart rate in lymphatic endothelial cells (155) (Fig. 6B). Type 2 diabetes loci were enriched in skeletal muscle adipocytes and in lymphatic endothelial cells in multiple tissues, which might contribute to the predisposition of type 2 diabetes to vascular disease (156, 157) (Fig. 6B). Less well-characterized cell type–trait associations included DCs (in almost all tissues) with non-melanoma skin cancer (158) and adipocytes (breast) with atrial fibrillation (table S15). GWAS loci enriched in a specific cell type from a known tissue of action frequently showed similar enrichment in the same cell type from other uninvolved tissues. For example, atrial fibrillation GWAS loci were enriched in myonuclei in heart, skeletal muscle, esophagus muscularis, and prostate (Fig. 6, B and C); coronary artery disease and heart rate loci were enriched in pericytes in five or six tissues in addition to heart; and prostate cancer loci were enriched in luminal epithelial cells in both prostate and breast (Fig. 6B and figs. S29 and S30).
Cell-type enrichment helped identify putative causal genes in GWAS loci with multiple QTL-mapped genes (tables S15 and S16). On average, about two-thirds of genes driving the cell type–specific enrichment for a given trait in a relevant tissue [mean = 66%, 60 to 71.4% (95% confidence interval)] were also driving the enrichment in the same cell type in other tissues. For example, in the case of atrial fibrillation and cardiac myonuclei, 26 out of 31 myonuclei-specific genes that drove the enrichment signal in myonuclei in heart and were shared with at least one other tissue (Fig. 6D) were enriched in muscle system–related processes, such as muscle contraction (FDR < 0.05; table S17). The five myonuclei-specific genes that were specific to heart only (pink vertical lines in Fig. 6D) were instead enriched in heart development processes, such as cardiac muscle tissue development (FDR < 0.05; table S18). Cardiac myonuclei were also found to be the most relevant cell type for atrial fibrillation in two separate snRNA-seq studies of the human heart (33, 34) and based on ECLIPSER analysis of these two studies [fig. S31 and table S19; (28)]. CASQ2, which encodes a cardiac muscle member of the calsequestrin family, and the myosin heavy chain 6 and 7 genes (MYH6 and MYH7) were the top myonuclei-specific genes driving the enrichment signal for atrial fibrillation in all three studies (table S19).
Associating GWAS genes with gene programs across cell types reveals six main trait groups
To chart cellular programs and processes that may be affected by genetic variants, we associated a larger set of >2000 complex phenotypes with cell types and the covarying gene modules that these cell types express (28). We defined gene modules in the cells in our atlas by hierarchically clustering genes based on correlation across all cells, as well as within cell types. We then scored modules for their overlap with GWAS genes [defined by variant to gene mapping in Open Targets Genetics (153, 154)] that are also highly expressed by cell type [(28); fig. S32 and table S20]. Next, we grouped GWAS phenotypes into major groups by the similarity of their module enrichment across cell types (Fig. 7, A and B, and fig. S33). Finally, for each major group of traits, we identified the relevant cell types associated with the underlying modules (Fig. 7, A and C) and tested the GWAS genes that overlapped with gene modules for functional enrichments (Fig. 7, A, D, and E).
Fig. 7. Cell types and gene modules relevant for trait and disease groups by GWAS module enrichment.

(A) Schematic of the module-based enrichment method. Shaded edges indicate associations between cell types and phenotypes through modules (middle). (B to E) Trait and disease groups identified by GWAS-cell type relationships. Similarity (Spearman correlation coefficient) between GWAS traits and diseases (rows and columns) by enriched cell types is shown in (B). Dashed lines demarcate trait and disease groups. Shown in (C) is the cell-type enrichment for each of the GWAS-enriched modules. Significance [−log10(FDR), circle size] and F score (circle color) of enrichment of gene sets (columns) with the genes in the intersection of a gene module and GWAS genes for each trait or disease in (B) (rows) are shown in (D). A red outline indicates an FDR less than 0.1. Shown in (E) is the number of traits (x axis) in each module in (B) where a gene (y axis) is detected as the driver of the association in the intersection of the gene modules, a trait or disease enriched in the module, and a functional gene set for the top 10 most frequently identified genes in the enrichment analysis of each module. EA, East Asian; SCZ, Schizophrenia; UKBB, UK Biobank.
Traits and diseases partitioned into six major groups, spanning immune hypersensitivity, cardiovascular, calcium channel–related, cognitive and psychiatric, pigmentation, and HDL cholesterol-related based on their associations with cell types (Fig. 7B). The immune hypersensitivity disorders were associated with T cells, including the expected relation between lung T cells and hay fever, allergic rhinitis, and respiratory disease [(159, 160); Fig. 7C]. GWAS genes in the modules associated with these traits were enriched for lymphocyte activation and differentiation and T cell receptor signaling and cell-cell adhesion (Fig. 7D), with interleukin-35 (IL-35) signaling genes enriched in hypothyroidism and inflammatory bowel disease (IBD), consistent with IL-35 up-regulation in Hashimoto’s thyroiditis (161) and IBD (162) and down-regulation in Graves’ disease (163). The cardiovascular traits group was associated with pericytes and smooth muscle cells and enriched with blood circulation, smooth muscle contraction, muscle structure development, and cardiocyte differentiation genes. The cardiovascular group overlapped with the calcium channel–related group—which included blood pressure medication, pulse rate, medication use of calcium-channel blockers, and vascular system traits, as well as schizophrenia and autism spectrum disorder, which are psychiatric disorders with known calcium channel associations (164)—and was enriched with membrane depolarization and calcium ion channel genes (Fig. 7D). Other cognitive and psychiatric phenotypes grouped separately and were enriched with neuronal synapse organization, structure, and activity genes (Fig. 7D) Finally, a group of HDL cholesterol traits was associated with adipocytes in all three muscle tissues and enriched for fatty acid, triglyceride, and lipid homeostasis and related metabolic processes (monocarboxylic and glycerol metabolism), including ANGPTL8 and PNPLA3 (Fig. 7E and table S20), which are genes involved in lipolysis regulation in adipocytes that might affect extrahepatic cholesterol transport via HDLs (165, 166).
Toward large-scale snRNA-seq of human tissues with pooling
To enable future studies at the population scale, we tested whether frozen samples from different individuals can be pooled for snRNA-seq, followed by computational demultiplexing, as a cost-effective and scalable approach (167), as previously applied to large-scale studies of human peripheral blood mononuclear cells (24). We processed lung or prostate samples jointly from three individuals using the CST and TST protocols. We pooled tissue samples from three individuals and processed the pool for single-nucleus extraction, thus minimizing technical batch effects and wet-lab time. After sequencing, we removed ambient RNA (30) and performed de novo genotype-based demultiplexing to assign nuclei to donors [using souporcell (168); fig. S34; (28)]. We validated the demultiplexing by comparing the genotype-based assignments to those from an expression-based multinomial logistic classifier that assigned donor identity to each nucleus profile after training with unpooled samples of the same donors, which showed high concordance between the two approaches (accuracy 88 to 96%; fig. S34). Moreover, genotype-based doublet calls were concordant with expression-based doublet calls in both lung and prostate (mean balanced accuracy of 63%) (fig. S34).
Discussion
Cross-tissue atlases allow us to characterize tissue-specific and tissue-agnostic features of cells of a common type that serve accessory roles in tissues, such as immune and stroma cells (fig. S35A). For example, for LYVE1- and HLAII–expressing macrophages, our results reinforce the notion of functional specification of these two macrophage states into tissue support and tissue immunity, respectively (47), and propose a model for their differentiation (fig. S35, B and C). In mice, Lyve1high macrophages are localized perivascularly, whereas MHCIIhigh macrophages are found in proximity to neurons (47). Future studies can address this localization in humans, and signals that govern the tissue-specific ratios of LYVE1- versus HLAII–expressing cells.
Our data demonstrate the prevalence of LAM-like cells across tissue contexts and pathologies, including breast and heart, where we recovered both adipocytes and LAM-like cells (Figs. 1 and 3C and fig. S2). In line with a model of lipid-induced differentiation of macrophages toward the LAM state (85), our classifier recovered LAMs in pathologies characterized by lipid accumulation: atherosclerosis, Crohn’s disease, and acne. The inferred role for PPARG and NR1H3 in driving the LAM expression program suggests a model in which signaling through lipid-bound receptors on macrophages, such as TREM2 or CD36, up-regulates the expression of more lipid receptors, as well as of lipid-modifying enzymes through PPARG and NR1H3. Because we observed LAMs in healthy organs and in conditions linked to lipid accumulation, future studies may identify other tissue-specific signals or conditions that can trigger LAM-like states.
In lung alveolar fibroblasts, we identified an interconnected functional module that may enable an adequate response to alveolar distortion, through transduction of mechanical cues from the ECM to cytosolic calcium-induced cytoskeletal contraction (fig. S35, D and E). These include BM adhesion genes, including ITGA8 and its interacting partner NPNT (95-100); a calcium transport module featuring neurotransmitter receptors and multiple calcium ion channels, including the mechanosensitive channel PIEZO2; and an actomyosin contractility program that features MYLK and mediates cytosolic calcium-induced cytoskeletal contraction, which is required for the transduction of mechanical cues from the ECM (169). PIEZO2 and ITGA8 are also coexpressed in intraglomerular mesangial cells (170, 171), where PIEZO2 may sense mechanical forces resulting from changes in blood flow and ITGA8 confers contractility and adhesion (172). Because the alveolus and glomerulus are stretch sensors of air pressure and blood pressure, respectively, PIEZO2+/ITGA8+ alveolar fibroblasts may contribute to mechanosensing of alveolar tension, which so far has been mainly attributed to Piezo2+ sensory neurons in mice (111), suggesting conserved features in mechanosensing across organs. Both lung alveolar fibroblasts and myofibroblasts have been localized to alveoli (40), suggesting a possible relationship, possibly through FGFR4 in alveolar fibroblasts and its ligand FGF18 in myofibroblasts (fig. S18D) (109, 173).
We further demonstrated the utility of our tissue atlas for monogenic and polygenic disease biology. Many monogenic disease gene modules that are defined by comorbidity [e.g., diabetes and lipodystrophy (174)] or similarity of clinical phenotypes (e.g., muscle diseases) were enriched in expected cell types (fig. S35F). Focusing on the pathobiology of monogenic muscle diseases (fig. S35G), we highlighted nonmyocyte cell populations with a potential role in muscle diseases, including nervous system, immune, and stromal cells (118), as well as specific myonuclei subsets that express muscle disease genes. Whether the multiple myonuclei subsets we observed are related to multinucleation and specialization of different nuclei in one syncytium (31) remains unclear. Some disease-risk genes may also disrupt cell-cell interactions in the muscle. Notably, we observed varying levels of DMD expression across cell types, as well as between human and mouse. Variation in DMD isoform expression, which has critical implications in Duchenne muscular dystrophy (135), can be investigated in future studies. For common complex diseases, we found significant enrichment in specific cell groups for multiple traits (fig. S35H). For more than half of the traits, there was enrichment for the same cell type in different tissues driven by both common and tissue-specific genes. Future work will be needed to extend these analyses across a broad set of tissues and cell types and examine the role of disease-associated genes and cell types in a disease context in patient samples.
Advances in single-cell epigenomics (175) and multi-omics (176-178) should further enable linking GWAS variants to their target genes and the cell types and programs in which they act. Recent findings indicating that a large fraction of genetic regulatory effects linked to GWAS variants can only be detected at the cellular level (11, 25) suggest that cell-level eQTL maps will be essential. The experimental and computational methods we developed for a cross-tissue atlas, and the biological queries we defined, will provide a basis for scaling such efforts to hundreds of individuals and diverse populations.
Methods summary
Tissue samples were selected from among a subset of GTEx project samples that were flash frozen and banked. Nuclei from each sample were extracted using the EZ, CST, NST, and TST protocols described in (26). Libraries for snRNA-seq were generated using the Chromium Single Cell 3′ v2 Reagent Kit (10x Genomics), and sequencing was performed with Illumina HiSeq X (96 samples) or NextSeq (three samples), according to the manufacturer’s protocols. The resulting snRNA-seq data were aligned and quantified using CellRanger v2.1.0 (10x Genomics), ambient RNA correction was performed using CellBender (30), and low-quality nuclei were filtered out using standard criteria (28). The resulting snRNA-seq expression profiles were integrated across samples using a total correlation variational autoencoder (28). Detailed descriptions of all computational analyses are provided in (28).
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Gaffney and A. Hupalowska for help with figure preparation. We thank T. Harvey, B. Cummings, B. Weisburd, A. O. Luria, and D. G. MacArthur for sharing expertise on muscle diseases. We thank A. R. Hamel for assistance with running GeneEnrich. This publication is part of the Human Cell Atlas (www.humancellatlas.org/publications).
Funding:
This project has been funded in part with funds from the Manton Foundation, Klarman Family Foundation, and Howard Hughes Medical Institute (A.R.). This work was also supported in part by the Common Fund of the Office of the Director, US National Institutes of Health (NIH), and by the National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH), National Institute on Aging (NIA), National Institute of Allergy and Infectious Diseases (NIAID), and National Institute of Neurological Disorders and Stroke (NINDS) through NIH contract HHSN268201000029C (S.A., G.G., F.A., and K.G.A.); 5U41HG009494 (S.A., G.G., F.A., and K.G.A.); NEI R01 EY031424-01 (A.V.S. and J.M.R.), and Chan Zuckerberg Initiative (CZI) Seed Network for the Human Cell Atlas award CZF2019-002459 (A.V.S. and J.M.R.). A.M.T. is funded by NHLBI grants R56HL157632 and R21HL156124.
Footnotes
Competing interests: A.R. is a co-founder and equity holder of Celsius Therapeutics, is an equity holder in Immunitas, and was a scientific advisory board member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics, and Asimov until 31 July 2020. Since 1 August 2020, A.R. is an employee of Genentech with equity in Roche. G.G. was partially funded by the Paul C. Zamecnik Chair in Oncology at the Massachusetts General Hospital Cancer Center. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, POLYSOLVER, and TensorQTL. G.G. is a founder of and consultant to, and holds privately held equity in, Scorpion Therapeutics. F.A. is an inventor on a patent application related to TensorQTL. F.A. has been an employee of Illumina, Inc., since 8 November 2021. E.D. is an employee of Bristol Myers Squibb. G.E. has been an employee of Genentech since 4 April 2022. O.R.-R. has been an employee of Genentech since 19 October 2020. She has given numerous lectures on the subject of single-cell genomics to a wide variety of audiences and, in some cases, has received remuneration to cover time and costs. O.R.-R. and A.R. are co-inventors on patent applications filed at the Broad Institute of MIT and Harvard related to single-cell genomics. G.E., E.D., O.R.-R., and A.R. are co-applicants on patent WO 2020/232271 relating to the work in this manuscript.
Data and materials availability: Raw sequence data are available at AnVIL (https://anvil.terra.bio/#workspaces/anvil-datastorage/AnVIL_GTEx_V9_hg38; dbGaP accession phs000424). Gene expression matrices are available from the GTEx Portal (www.gtexportal.org) and the Single Cell Portal (https://singlecell.broadinstitute.org/single_cell/study/SCP1479). The code used in the analysis is available at Zenodo (179).
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Tam V et al. , Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 (2019). doi: 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- 2.Mills MC, Rahal C, A scientometric review of genome-wide association studies. Commun. Biol 2, 9 (2019). doi: 10.1038/s42003-018-0261-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cano-Gamez E, Trynka G, From GWAS to function: Using functional genomics to identify the mechanisms underlying complex diseases. Front. Genet 11, 424 (2020). doi: 10.3389/fgene.2020.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camp JG, Platt R, Treutlein B, Mapping human cell phenotypes to genotypes with single-cell genomics. Science 365, 1401–1405 (2019). doi: 10.1126/science.aax6648 [DOI] [PubMed] [Google Scholar]
- 5.Sun G et al. , Single-cell RNA sequencing in cancer: Applications, advances, and emerging challenges. Mol. Ther. Oncolytics 21, 183–206 (2021). doi: 10.1016/j.omto.2021.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potter SS, Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol 14, 479–492 (2018). doi: 10.1038/s41581-018-0021-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). doi: 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kasela S et al. , Pathogenic implications for autoimmune mechanisms derived by comparative eQTL analysis of CD4+ versus CD8+ T cells. PLOS Genet. 13, e1006643 (2017). doi: 10.1371/journal.pgen.1006643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Wijst MGP et al. , Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet 50, 493–497 (2018). doi: 10.1038/s41588-018-0089-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhernakova DV et al. , Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet 49, 139–145 (2017). doi: 10.1038/ng.3737 [DOI] [PubMed] [Google Scholar]
- 11.Kim-Hellmuth S et al. , Cell type-specific genetic regulation of gene expression across human tissues. Science 369, eaaz8528 (2020). doi: 10.1126/science.aaz8528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye CJ et al. , Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014). doi: 10.1126/science.1254665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuomo ASE et al. , Single-cell RNA-sequencing of differentiating iPS cells reveals dynamic genetic effects on gene expression. Nat. Commun 11, 810 (2020). doi: 10.1038/s41467-020-14457-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strober BJ et al. , Dynamic genetic regulation of gene expression during cellular differentiation. Science 364, 1287–1290 (2019). doi: 10.1126/science.aaw0040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skene NG et al. , Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833 (2018). doi: 10.1038/s41588-018-0129-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smillie CS et al. , Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22 (2019). doi: 10.1016/j.cell.2019.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weeks EM et al. , Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. medRxiv 2020.09.08.20190561 [Preprint] (2020); 10.1101/2020.09.08.20190561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagadeesh KA et al. , Identifying disease-critical cell types and cellular processes across the human body by integration of single-cell profiles and human genetics. bioRxiv 2021.03.19.436212 [Preprint] (2021); 10.1101/2021.03.19.436212. [DOI] [Google Scholar]
- 19.Habib N et al. , Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). doi: 10.1038/nmeth.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habib N et al. , Div-Seq: Single-nucleus RNA-seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016). doi: 10.1126/science.aad7038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petrany MJ et al. , Single-nucleus RNA-seq identifies transcriptional heterogeneity in multinucleated skeletal myofibers. Nat. Commun 11, 6374 (2020). doi: 10.1038/s41467-020-20063-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H, Kirita Y, Donnelly EL, Humphreys BD, Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: Rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol 30, 23–32 (2019). doi: 10.1681/ASN.2018090912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun W et al. , snRNA-seq reveals a subpopulation of adipocytes that regulates thermogenesis. Nature 587, 98–102 (2020). doi: 10.1038/s41586-020-2856-x [DOI] [PubMed] [Google Scholar]
- 24.Kang HM et al. , Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol 36, 89–94 (2018). doi: 10.1038/nbt.4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Wijst M et al. , The single-cell eQTLGen consortium. eLife 9, e52155 (2020). doi: 10.7554/eLife.52155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drokhlyansky E et al. , The human and mouse enteric nervous system at single-cell resolution. Cell 182, 1606–1622.e23 (2020). doi: 10.1016/j.cell.2020.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delorey TM.et al. , COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021). doi: 10.1038/s41586-021-03570-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Materials and methods are available as supplementary materials.
- 29.Slyper M et al. , A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802 (2020). doi: 10.1038/s41591-020-0844-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming SJ, Marioni JC, Babadi M, CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. bioRxiv 791699 [Preprint] (2019); 10.1101/791699. [DOI] [Google Scholar]
- 31.Kim M et al. , Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nat. Commun 11, 6375 (2020). doi: 10.1038/s41467-020-20064-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfien M et al. , Single-nucleus sequencing of an entire mammalian heart: Cell type composition and velocity. Cells 9, 318 (2020). doi: 10.3390/cells9020318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tucker NR et al. , Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482 (2020). doi: 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Litviňuková M et al. , Cells of the adult human heart. Nature 588, 466–472 (2020). doi: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verma RX et al. , Single cell RNA-seq analysis of the flexor digitorum brevis mouse myofibers. Skelet. Muscle 11, 13 (2021). doi: 10.1186/s13395-021-00269-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rubenstein AB et al. , Single-cell transcriptional profiles in human skeletal muscle. Sci. Rep 10, 229 (2020). doi: 10.1038/s41598-019-57110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jessen KR, Mirsky R, The success and failure of the Schwann cell response to nerve injury. Front. Cell. Neurosci 13, 33 (2019). doi: 10.3389/fncel.2019.00033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henry GH et al. , A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep. 25, 3530–3542.e5 (2018). doi: 10.1016/j.celrep.2018.11.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luecken MD, Theis FJ, Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol 15, e8746 (2019). doi: 10.15252/msb.20188746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Travaglini KJ et al. , A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020). doi: 10.1038/s41586-020-2922-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Brink SC et al. , Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). doi: 10.1038/nmeth.4437 [DOI] [PubMed] [Google Scholar]
- 42.Denisenko E et al. , Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21, 130 (2020). doi: 10.1186/s13059-020-02048-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaghlool A et al. , Characterization of the nuclear and cytosolic transcriptomes in human brain tissue reveals new insights into the subcellular distribution of RNA transcripts. Sci. Rep 11, 4076 (2021). doi: 10.1038/s41598-021-83541-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rabani M et al. , High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159, 1698–1710 (2014). doi: 10.1016/j.cell.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solnestam BW et al. , Comparison of total and cytoplasmic mRNA reveals global regulation by nuclear retention and miRNAs. BMC Genomics 13, 574 (2012). doi: 10.1186/1471-2164-13-574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hwang WL et al. , Single-nucleus and spatial transcriptomics of archival pancreatic cancer reveals multi-compartment reprogramming after neoadjuvant treatment. bioRxiv 2020.08.25.267336 [Preprint] (2020); 10.1101/2020.08.25.267336. [DOI] [Google Scholar]
- 47.Chakarov S et al. , Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964 (2019). doi: 10.1126/science.aau0964 [DOI] [PubMed] [Google Scholar]
- 48.Varol C, Mildner A, Jung S, Macrophages: Development and tissue specialization. Annu. Rev. Immunol 33, 643–675 (2015). doi: 10.1146/annurev-immunol-032414-112220 [DOI] [PubMed] [Google Scholar]
- 49.Villani A-C et al. , Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). doi: 10.1126/science.aah4573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reyfman PA et al. , Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med 199, 1517–1536 (2019). doi: 10.1164/rccm.201712-2410OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaitin DA et al. , Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell 178, 686–698.e14 (2019). doi: 10.1016/j.cell.2019.05.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Capucha T et al. , Distinct murine mucosal langerhans cell subsets develop from pre-dendritic cells and monocytes. Immunity 43, 369–381 (2015). doi: 10.1016/j.immuni.2015.06.017 [DOI] [PubMed] [Google Scholar]
- 53.Deckers J, Hammad H, Hoste E, Langerhans cells: Sensing the environment in health and disease. Front. Immunol 9, 93 (2018). doi: 10.3389/fimmu.2018.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evren E et al. , Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity 54, 259–275.e7 (2021). doi: 10.1016/j.immuni.2020.12.003 [DOI] [PubMed] [Google Scholar]
- 55.Patel AA et al. , The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med 214, 1913–1923 (2017). doi: 10.1084/jem.20170355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lim HY et al. , Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018). doi: 10.1016/j.immuni.2018.06.008 [DOI] [PubMed] [Google Scholar]
- 57.Bizou M et al. , Cardiac macrophage subsets differentially regulate lymphatic network remodeling during pressure overload. Sci. Rep 11, 16801 (2021). doi: 10.1038/s41598-021-95723-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran P et al. , Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518 (2019). doi: 10.1038/s41586-019-1631-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perugorria MJ et al. , Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut 68, 533–546 (2019). doi: 10.1136/gutjnl-2017-314107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiong X et al. , Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e5 (2019). doi: 10.1016/j.molcel.2019.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guilliams M et al. , Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 185, 379–396.e38 (2022). doi: 10.1016/j.cell.2021.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayaub EA et al. , Single cell RNA-seq and mass cytometry reveals a novel and a targetable population of macrophages in idiopathic pulmonary fibrosis. bioRxiv 2021.01.04.425268 [Preprint] (2021); 10.1101/2021.01.04.425268. [DOI] [Google Scholar]
- 63.Cochain C et al. , Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res 122, 1661–1674 (2018). doi: 10.1161/CIRCRESAHA.117.312509 [DOI] [PubMed] [Google Scholar]
- 64.Willemsen L, de Winther MPJ, Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol 250, 705–714 (2020). doi: 10.1002/path.5392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma F et al. , The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol 22, 839–850 (2021). doi: 10.1038/s41590-021-00956-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Keren-Shaul H et al. , A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 67.Grubman A et al. , A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097 (2019). doi: 10.1038/s41593-019-0539-4 [DOI] [PubMed] [Google Scholar]
- 68.Mathys H et al. , Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). doi: 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muus C et al. , Single-cell meta-analysis of SARS-CoV-2 entry genes across tissues and demographics. Nat. Med 27, 546–559 (2021). doi: 10.1038/s41591-020-01227-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wirka RC et al. , Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med 25, 1280–1289 (2019). doi: 10.1038/s41591-019-0512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alsaigh T, Evans D, Frankel D, Torkamani A, Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. bioRxiv 2020.03.03.968123 [Preprint] (2020); 10.1101/2020.03.03.968123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vento-Tormo R et al. , Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353 (2018). doi: 10.1038/s41586-018-0698-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tan K et al. , Transcriptome profiling reveals signaling conditions dictating human spermatogonia fate in vitro. Proc. Natl. Acad. Sci. U.S.A 117, 17832–17841 (2020). doi: 10.1073/pnas.2000362117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liao J et al. , Single-cell RNA sequencing of human kidney. Sci. Data 7, 4 (2020). doi: 10.1038/s41597-019-0351-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qadir MMF et al. , Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. U.S.A 117, 10876–10887 (2020). doi: 10.1073/pnas.1918314117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacParland SA et al. , Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun 9, 4383 (2018). doi: 10.1038/s41467-018-06318-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chitiashvili T et al. , Female human primordial germ cells display X-chromosome dosage compensation despite the absence of X-inactivation. Nat. Cell Biol 22, 1436–1446 (2020). doi: 10.1038/s41556-020-00607-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Micheli AJ, Spector JA, Elemento O, Cosgrove BD, A reference single-cell transcriptomic atlas of human skeletal muscle tissue reveals bifurcated muscle stem cell populations. Skelet. Muscle 10, 19 (2020). doi: 10.1186/s13395-020-00236-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hughes TK et al. , Second-strand synthesis-based massively parallel scRNA-Seq reveals cellular states and molecular features of human inflammatory skin pathologies. Immunity 53, 878–894.e7 (2020). doi: 10.1016/j.immuni.2020.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rojahn TB et al. , Single-cell transcriptomics combined with interstitial fluid proteomics defines cell type-specific immune regulation in atopic dermatitis. J. Allergy Clin. Immunol 146, 1056–1069 (2020). doi: 10.1016/j.jaci.2020.03.041 [DOI] [PubMed] [Google Scholar]
- 81.Rindler K et al. , Spontaneously resolved atopic dermatitis shows melanocyte and immune cell activation distinct from healthy control skin. Front. Immunol 12, 630892 (2021). doi: 10.3389/fimmu.2021.630892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ha CWY et al. , Translocation of viable gut microbiota to mesenteric adipose drives formation of creeping fat in humans. Cell 183, 666–683.e17 (2020). doi: 10.1016/j.cell.2020.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tome-Garcia J et al. , Cell type-specific isolation and transcriptomic profiling informs glial pathology in human temporal lobe epilepsy. bioRxiv 2020.12.11.421370 [Preprint] (2020); 10.1101/2020.12.11.421370. [DOI] [Google Scholar]
- 84.Iyer A et al. , Increased chitotriosidase activity in serum of leprosy patients: Association with bacillary leprosy. Clin. Immunol 131, 501–509 (2009). doi: 10.1016/j.clim.2009.02.003 [DOI] [PubMed] [Google Scholar]
- 85.Deczkowska A, Weiner A, Amit I, The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181, 1207–1217 (2020). doi: 10.1016/j.cell.2020.05.003 [DOI] [PubMed] [Google Scholar]
- 86.Kidani Y, Bensinger SJ, Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol. Rev 249, 72–83 (2012). doi: 10.1111/j.1600-065X.2012.01153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Savage JC et al. , Nuclear receptors license phagocytosis by Trem2+ myeloid cells in mouse models of Alzheimer’s disease. J. Neurosci 35, 6532–6543 (2015). doi: 10.1523/JNEUROSCI.4586-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Plikus MV et al. , Fibroblasts: Origins, definitions, and functions in health and disease. Cell 184, 3852–3872 (2021). doi: 10.1016/j.cell.2021.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H, GATA transcription factors in the developing and adult heart. Cardiovasc. Res 63, 196–207 (2004). doi: 10.1016/j.cardiores.2004.03.025 [DOI] [PubMed] [Google Scholar]
- 90.Dittrich GM et al. , Fibroblast GATA-4 and GATA-6 promote myocardial adaptation to pressure overload by enhancing cardiac angiogenesis. Basic Res. Cardiol 116, 26 (2021). doi: 10.1007/s00395-021-00862-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Forte E et al. , Adult fibroblasts retain organ-specific transcriptomic identity. bioRxiv 2021.06.03.446915 [Preprint] (2021); 10.1101/2021.06.03.446915. [DOI] [Google Scholar]
- 92.Furtado MB et al. , Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res 114, 1422–1434 (2014). doi: 10.1161/CIRCRESAHA.114.302530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Forrester SJ et al. , Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev 98, 1627–1738 (2018). doi: 10.1152/physrev.00038.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Powers SK, Morton AB, Hyatt H, Hinkley MJ, The renin-angiotensin system and skeletal muscle. Exerc. Sport Sci. Rev 46, 205–214 (2018). doi: 10.1249/JES.0000000000000158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kiyozumi D et al. , Basement membrane assembly of the integrin α8β1 ligand nephronectin requires Fraser syndrome-associated proteins. J. Cell Biol 197, 677–689 (2012). doi: 10.1083/jcb.201203065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kiyozumi D, Sugimoto N, Sekiguchi K, Breakdown of the reciprocal stabilization of QBRICK/Frem1, Fras1, and Frem2 at the basement membrane provokes Fraser syndrome-like defects. Proc. Natl. Acad. Sci. U.S.A 103, 11981–11986 (2006). doi: 10.1073/pnas.0601011103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brandenberger R et al. , Identification and characterization of a novel extracellular matrix protein nephronectin that is associated with integrin α8β1 in the embryonic kidney. J. Cell Biol 154, 447–458 (2001). doi: 10.1083/jcb.200103069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sato Y et al. , Molecular basis of the recognition of nephronectin by integrin α8β1. J. Biol. Chem 284, 14524–14536 (2009). doi: 10.1074/jbc.M900200200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Müller U et al. , Integrin α8β1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell 88, 603–613 (1997). doi: 10.1016/S0092-8674(00)81903-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benjamin JT et al. , The role of integrin α8β1 in fetal lung morphogenesis and injury. Dev. Biol 335, 407–417 (2009). doi: 10.1016/j.ydbio.2009.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vrontou S et al. , Fras1 deficiency results in cryptophthalmos, renal agenesis and blebbed phenotype in mice. Nat. Genet 34, 209–214 (2003). doi: 10.1038/ng1168 [DOI] [PubMed] [Google Scholar]
- 102.Petrou P, Pavlakis E, Dalezios Y, Galanopoulos VK, Chalepakis G, Basement membrane distortions impair lung lobation and capillary organization in the mouse model for Fraser syndrome. J. Biol. Chem 280, 10350–10356 (2005). doi: 10.1074/jbc.M412368200 [DOI] [PubMed] [Google Scholar]
- 103.Linton JM, Martin GR, Reichardt LF, The ECM protein nephronectin promotes kidney development via integrin α8β1-mediated stimulation of Gdnf expression. Development 134, 2501–2509 (2007). doi: 10.1242/dev.00503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McGregor L et al. , Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat. Genet 34, 203–208 (2003). doi: 10.1038/ng1142 [DOI] [PubMed] [Google Scholar]
- 105.Shafeghati Y, Kniepert A, Vakili G, Zenker M, Fraser syndrome due to homozygosity for a splice site mutation of FREM2. Am. J. Med. Genet. A 146A, 529–531 (2008). doi: 10.1002/ajmg.a.32091 [DOI] [PubMed] [Google Scholar]
- 106.Alazami AM et al. , FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am. J. Hum. Genet 85, 414–418 (2009). doi: 10.1016/j.ajhg.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carney TJ et al. , Genetic analysis of fin development in zebrafish identifies Furin and Hemicentin1 as potential novel Fraser syndrome disease genes. PLOS Genet. 6, e1000907 (2010). doi: 10.1371/journal.pgen.1000907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buechler MB et al. , Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021). doi: 10.1038/s41586-021-03549-5 [DOI] [PubMed] [Google Scholar]
- 109.Weinstein M, Xu X, Ohyama K, Deng CX, FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125, 3615–3623 (1998). doi: 10.1242/dev.125.18.3615 [DOI] [PubMed] [Google Scholar]
- 110.Rezvani M et al. , Association of a FGFR-4 gene polymorphism with bronchopulmonary dysplasia and neonatal respiratory distress. Dis. Markers 35, 633–640 (2013). doi: 10.1155/2013/932356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nonomura K et al. , Piezo2 senses airway stretch and mediates lung inflation-induced apnoea. Nature 541, 176–181 (2017). doi: 10.1038/nature20793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Delle Vedove A et al. , Biallelic loss of proprioception-related PIEZO2 causes muscular atrophy with perinatal respiratory distress, arthrogryposis, and scoliosis. Am. J. Hum. Genet 99, 1206–1216 (2016). doi: 10.1016/j.ajhg.2016.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kuo IY, Ehrlich BE, Signaling in muscle contraction. Cold Spring Harb. Perspect. Biol 7, a006023 (2015). doi: 10.1101/cshperspect.a006023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Montoro DT et al. , A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018). doi: 10.1038/s41586-018-0393-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Plasschaert LW et al. , A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). doi: 10.1038/s41586-018-0394-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Amberger JS, Bocchini CA, Scott AF, Hamosh A, OMIM. org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038–D1043 (2019). doi: 10.1093/nar/gky1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Inaba K, Mizuno K, Sperm dysfunction and ciliopathy. Reprod. Med. Biol 15, 77–94 (2015). doi: 10.1007/s12522-015-0225-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Benarroch L, Bonne G, Rivier F, Hamroun D, The 2021 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord 30, 1008–1048 (2020). doi: 10.1016/j.nmd.2020.11.009 [DOI] [PubMed] [Google Scholar]
- 119.Csapo R, Gumpenberger M, Wessner B, Skeletal muscle extracellular matrix – What do we know about its composition, regulation, and physiological roles? A narrative review. Front. Physiol 11, 253 (2020). doi: 10.3389/fphys.2020.00253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ohlendieck K, Swandulla D, Complexity of skeletal muscle degeneration: Multi-systems pathophysiology and organ crosstalk in dystrophinopathy. Pflugers Arch. 473, 1813–1839 (2021). doi: 10.1007/s00424-021-02623-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Verschuuren J, Strijbos E, Vincent A, Neuromuscular junction disorders. Handb. Clin. Neurol 133, 447–466 (2016). doi: 10.1016/B978-0-444-63432-0.00024-4 [DOI] [PubMed] [Google Scholar]
- 122.Cummings BB et al. , Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med 9, eaal5209 (2017). doi: 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gonorazky HD et al. , Expanding the boundaries of RNA sequencing as a diagnostic tool for rare mendelian disease. Am. J. Hum. Genet 104, 466–483 (2019). doi: 10.1016/j.ajhg.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vanhaesebrouck AE, Beeson D, The congenital myasthenic syndromes: Expanding genetic and phenotypic spectrums and refining treatment strategies. Curr. Opin. Neurol 32, 696–703 (2019). doi: 10.1097/WCO.0000000000000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maggs AM, Huxley C, Hughes SM, Nerve-dependent changes in skeletal muscle myosin heavy chain after experimental denervation and cross-reinnervation and in a demyelinating mouse model of Charcot-Marie-Tooth disease type 1A. Muscle Nerve 38, 1572–1584 (2008). doi: 10.1002/mus.21106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bönnemann CG, The collagen VI-related myopathies: Muscle meets its matrix. Nat. Rev. Neurol 7, 379–390 (2011). doi: 10.1038/nrneurol.2011.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.de Morrée A et al. , Dysferlin regulates cell adhesion in human monocytes. J. Biol. Chem 288, 14147–14157 (2013). doi: 10.1074/jbc.M112.448589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin J et al. , Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 418, 797–801 (2002). doi: 10.1038/nature00904 [DOI] [PubMed] [Google Scholar]
- 129.Tajsharghi H et al. , Recessive myosin myopathy with external ophthalmoplegia associated with MYH2 mutations. Eur. J. Hum. Genet 22, 801–808 (2014). doi: 10.1038/ejhg.2013.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pisani V et al. , Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2. Muscle Nerve 38, 1405–1411 (2008). doi: 10.1002/mus.21122 [DOI] [PubMed] [Google Scholar]
- 131.Vihola A et al. , Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 60, 1854–1857 (2003). doi: 10.1212/01.WNL.0000065898.61358.09 [DOI] [PubMed] [Google Scholar]
- 132.Krivickas LS, Ansved T, Suh D, Frontera WR, Contractile properties of single muscle fibers in myotonic dystrophy. Muscle Nerve 23, 529–537 (2000). doi: [DOI] [PubMed] [Google Scholar]
- 133.Clarke NF, Congenital fiber-type disproportion. Semin. Pediatr. Neurol 18, 264–271 (2011). doi: 10.1016/j.spen.2011.10.008 [DOI] [PubMed] [Google Scholar]
- 134.Strakova J et al. , Integrative effects of dystrophin loss on metabolic function of the mdx mouse. Sci. Rep 8, 13624 (2018). doi: 10.1038/s41598-018-31753-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A, Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7, 13 (2021). doi: 10.1038/s41572-021-00248-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sander M et al. , Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A 97, 13818–13823 (2000). doi: 10.1073/pnas.250379497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.t Hoen PAC et al. , Generation and characterization of transgenic mice with the full-length human DMD gene. J. Biol. Chem 283, 5899–5907 (2008). doi: 10.1074/jbc.M709410200 [DOI] [PubMed] [Google Scholar]
- 138.Hogarth MW et al. , Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun 10, 2430 (2019). doi: 10.1038/s41467-019-10438-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lemos DR et al. , Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med 21, 786–794 (2015). doi: 10.1038/nm.3869 [DOI] [PubMed] [Google Scholar]
- 140.GTEx Consortium, Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Unamuno X et al. , Dermatopontin, a novel adipokine promoting adipose tissue extracellular matrix remodelling and inflammation in obesity. J. Clin. Med 9, 1069 (2020). doi: 10.3390/jcm9041069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Heo S-H, Cho J-Y, ELK3 suppresses angiogenesis by inhibiting the transcriptional activity of ETS-1 on MT1-MMP. Int. J. Biol. Sci 10, 438–447 (2014). doi: 10.7150/ijbs.8095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Podkalicka P, Mucha O, Dulak J, Loboda A, Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci 76, 1507–1528 (2019). doi: 10.1007/s00018-019-03006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R, CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506 (2020). doi: 10.1038/s41596-020-0292-x [DOI] [PubMed] [Google Scholar]
- 145.Narkis G et al. , Lethal congenital contractural syndrome type 2 (LCCS2) is caused by a mutation in ERBB3 (Her3), a modulator of the phosphatidylinositol-3-kinase/Akt pathway. Am. J. Hum. Genet 81, 589–595 (2007). doi: 10.1086/520770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ibraghimov-Beskrovnaya O et al. , Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 (1992). doi: 10.1038/355696a0 [DOI] [PubMed] [Google Scholar]
- 147.Gillies AR, Lieber RL, Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44, 318–331 (2011). doi: 10.1002/mus.22094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dai S-Y et al. , Galectin-9 induces maturation of human monocyte-derived dendritic cells. J. Immunol 175, 2974–2981 (2005). doi: 10.4049/jimmunol.175.5.2974 [DOI] [PubMed] [Google Scholar]
- 149.Finucane HK et al. , Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). doi: 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gamazon ER et al. , Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet 50, 956–967 (2018). doi: 10.1038/s41588-018-0154-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barbeira AN et al. , Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 22, 49 (2021). doi: 10.1186/s13059-020-02252-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rouhana JM et al. , ECLIPSER: identifying causal cell types and genes for complex traits through single cell enrichment of e/sQTL-mapped genes in GWAS loci. bioRxiv 2021.11.24.469720 [Preprint] (2021); 10.1101/2021.11.24.469720. [DOI] [Google Scholar]
- 153.Ghoussaini M et al. , Open Targets Genetics: Systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021). doi: 10.1093/nar/gkaa840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mountjoy E et al. , An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet 53, 1527–1533 (2021). doi: 10.1038/s41588-021-00945-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brakenhielm E, Alitalo K, Cardiac lymphatics in health and disease. Nat. Rev. Cardiol 16, 56–68 (2019). doi: 10.1038/s41569-018-0087-8 [DOI] [PubMed] [Google Scholar]
- 156.Paneni F, Beckman JA, Creager MA, Cosentino F, Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J 34, 2436–2443 (2013). doi: 10.1093/eurheartj/eht149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Einarson TR, Acs A, Ludwig C, Panton UH, Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol 17, 83 (2018). doi: 10.1186/s12933-018-0728-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kothari C, Diorio C, Durocher F, The importance of breast adipose tissue in breast cancer. Int. J. Mol. Sci 21, 5760 (2020). doi: 10.3390/ijms21165760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kay AB, Allergy and allergic diseases. First of two parts. N. Engl. J. Med 344, 30–37 (2001). doi: 10.1056/NEJM200101043440106 [DOI] [PubMed] [Google Scholar]
- 160.Galli SJ, Tsai M, Piliponsky AM, The development of allergic inflammation. Nature 454, 445–454 (2008). doi: 10.1038/nature07204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yilmaz H, Cakmak M, Ceydilek B, Demir C, Aktas A, Role of interlekin-35 as a biomarker in patients with newly diagnosed Hashimoto’s thyroiditis. Endocr. Regul 50, 55–61 (2016). doi: 10.1515/enr-2016-0009 [DOI] [PubMed] [Google Scholar]
- 162.Li Y et al. , The possible role of the novel cytokines IL-35 and IL-37 in inflammatory bowel disease. Mediators Inflamm. 2014, 136329 (2014). doi: 10.1155/2014/136329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Saeed M-H et al. , Decreased serum levels of IL-27 and IL-35 in patients with Graves’ disease. Arch. Endocrinol. Metab 64, 521–527 (2021). doi: 10.20945/2359-3997000000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pinggera A et al. , New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet 26, 2923–2932 (2017). doi: 10.1093/hmg/ddx175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mysore R, Liebisch G, Zhou Y, Olkkonen VM, Nidhina Haridas PA, Angiopoietin-like 8 (Angptl8) controls adipocyte lipolysis and phospholipid composition. Chem. Phys. Lipids 207, 246–252 (2017). doi: 10.1016/j.chemphyslip.2017.05.002 [DOI] [PubMed] [Google Scholar]
- 166.Yang A, Mottillo EP, Adipocyte lipolysis: From molecular mechanisms of regulation to disease and therapeutics. Biochem. J 477, 985–1008 (2020). doi: 10.1042/BCJ20190468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Xu J et al. , Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biol. 20, 290 (2019). doi: 10.1186/s13059-019-1852-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Heaton H et al. , Souporcell: Robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods 17, 615–620 (2020). doi: 10.1038/s41592-020-0820-1 [DOI] [PubMed] [Google Scholar]
- 169.Kechagia JZ, Ivaska J, Roca-Cusachs P, Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol 20, 457–473 (2019). doi: 10.1038/s41580-019-0134-2 [DOI] [PubMed] [Google Scholar]
- 170.Lake BB et al. , A single-nucleus RNA-sequencing pipeline to decipher the molecular anatomy and pathophysiology of human kidneys. Nat. Commun 10, 2832 (2019). doi: 10.1038/s41467-019-10861-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lake BB et al. , An atlas of healthy and injured cell states and niches in the human kidney. bioRxiv 2021.07.28.454201 [Preprint] (2021); 10.1101/2021.07.28.454201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Marek I, Hilgers KF, Rascher W, Woelfle J, Hartner A, A role for the alpha-8 integrin chain (itga8) in glomerular homeostasis of the kidney. Mol. Cell Pediatr 7, 13 (2020). doi: 10.1186/s40348-020-00105-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Usui H et al. , Fgf18 is required for embryonic lung alveolar development. Biochem. Biophys. Res. Commun 322, 887–892 (2004). doi: 10.1016/j.bbrc.2004.07.198 [DOI] [PubMed] [Google Scholar]
- 174.Zammouri J et al. , Molecular and cellular bases of lipodystrophy syndromes. Front. Endocrinol 12, 803189 (2022). doi: 10.3389/fendo.2021.803189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kelsey G, Stegle O, Reik W, Single-cell epigenomics: Recording the past and predicting the future. Science 358, 69–75 (2017). doi: 10.1126/science.aan6826 [DOI] [PubMed] [Google Scholar]
- 176.Ma S et al. , Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020). doi: 10.1016/j.cell.2020.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mimitou EP et al. , Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol 39, 1246–1258 (2021). doi: 10.1038/s41587-021-00927-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fiskin E et al. , Single-cell profiling of proteins and chromatin accessibility using PHAGE-ATAC. Nat. Biotechnol 40, 374–381 (2022). doi: 10.1038/s41587-021-01065-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Eraslan G, The code used in Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Zenodo; (2022); 10.5281/zenodo.5775379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.