

ABSTRACT
Mycoplasma pneumoniae and Streptococcus pneumoniae are the most common bacterial causes of pneumonia in children. The clinical characteristics of pneumonia differ significantly between the two bacteria. We aimed to elucidate the differences in pathogenesis between M. pneumoniae and S. pneumoniae by characterizing the respiratory epithelial cell immune response to both pathogens. Using primary human bronchial epithelial cells in air-liquid interface cultures, we observed lower production of the proinflammatory cytokines interleukin-6 (IL-6) and IL-8 in response to M. pneumoniae than to S. pneumoniae. In contrast to the differences in proinflammatory cytokine production, Toll-like receptor 2 (TLR2)-mediated signaling in response to M. pneumoniae was stronger than to S. pneumoniae. This difference largely depended on TLR1 and not TLR6. We found that M. pneumoniae, but not S. pneumoniae, also induced signaling of TLR10, a coreceptor of TLR2 that has inhibitory properties. M. pneumoniae-induced TLR10 signaling on airway epithelial cells was partially responsible for low IL-8 production, as blocking TLR10 by specific antibodies increased cytokine production. M. pneumoniae maintained Th2-associated cytokine production by epithelial cells, which concurs with the known association of M. pneumoniae infection with asthma. M. pneumoniae left IL-33 levels unchanged, whereas S. pneumoniae downregulated IL-33 production both under homeostatic and Th2-promoting conditions. By directly comparing M. pneumoniae and S. pneumoniae, we demonstrate that M. pneumoniae avoids induction of proinflammatory cytokine response despite its ability to induce robust TLR2 signaling. Our new findings suggest that this apparent paradox may be partially explained by M. pneumoniae-induced signaling of TLR2/TLR10.
KEYWORDS: host response, IL-33, IL-8, Mycoplasma pneumoniae, primary bronchial epithelial cells, Streptococcus pneumoniae, TLR10, TLR2, pneumonia, respiratory pathogens
INTRODUCTION
Bacterial pneumonia in children is a major cause of death in low- and middle-income countries and a common cause of morbidity and hospitalization in high-income countries (1). Currently Mycoplasma pneumoniae is the most common cause of bacterial pneumonia in hospitalized children, followed by Streptococcus pneumoniae (2). There are several clinical differences between respiratory tract infections (RTIs) induced by M. pneumoniae and those induced by S. pneumoniae: M. pneumoniae respiratory tract infections present as subacute infections with fluctuating fever, cough, and moderate dyspnea, whereas S. pneumoniae respiratory tract infections cause acute, high fever (3, 4). C-reactive protein and peripheral blood leukocyte numbers are typically elevated in infections caused by S. pneumoniae, whereas they remain low in M. pneumoniae infections (5). M. pneumoniae usually causes mild infections, whereas S. pneumoniae pneumonias are generally more severe and more often lead to mortality and intensive care unit (ICU) admission. The immunological basis for these clinical differences is not known.
The epithelium of the respiratory tract separates the airway lumen from lung tissue and plays an essential role in the defense against microorganisms. Apart from providing a physical barrier, respiratory epithelial cells have important roles in the host immune response. Epithelial cells can recognize bacteria by expressing a diverse array of innate pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), NOD-like receptors (NLR), and RIG-I-like receptors (6). PRR-induced signaling not only leads to the production of defensins and mucus by the epithelial cells but also to the production of chemokines and cytokines. These chemokines and cytokines recruit and activate immune cells, such as monocytes, neutrophils, and lymphocytes, which further contribute to barrier function. However, these responses need to be tightly regulated to balance pathogen clearance with collateral tissue damage due to overzealous inflammation. Furthermore, inappropriate activation of the immune system in the respiratory tract can lead to chronic pulmonary conditions, such as asthma.
The cell wall of S. pneumoniae contains TLR2- and TLR4-recognized lipids and lipopeptides, and although M. pneumoniae does not possess a cell wall, its cell membrane does contain TLR2- and TLR4-binding ligands (7–10). Not only can bacterial lipids and lipopeptides be bound by TLR2 and TLR4 homodimers, heterodimers of TLR2 with TLR1, TLR6, and possibly TLR10 have similar capacities, albeit with different specificities for particular lipid moieties (11–14). Furthermore, lipids can require loading on TLRs by scavenger receptors such as CD36 (15), and the assembly of TLR heterodimers can require coreceptors, such as CD14 (16). Importantly, TLR10, either as a homodimer or a heterodimer with TLR1 or TLR6, is the only TLR that relays inhibitory signals and can thereby dampen inflammatory responses (14, 17).
In this study, we directly compared M. pneumoniae- and S. pneumoniae-induced epithelial cell responses to gain better insight in the underlying immune response during respiratory tract infections in children. We describe the first direct comparison of the epithelial response to these two major causes of pneumonia in children. Understanding the mechanisms of the epithelial response will allow for the discovery of new diagnostic biomarkers that can predict severity of infection, as well as the design of immunomodulatory therapies to reduce the severity of RTIs.
RESULTS
Moderate activation of human primary bronchial epithelial cells by M. pneumoniae compared to S. pneumoniae.
The respiratory tract epithelium recruits and activates immune cells in response to microorganisms by secreting chemokines and cytokines, such as interleukin-8 (IL-8), IL-6, and CCL20 (18, 19). We set out to evaluate chemokine and cytokine production by primary bronchial epithelial cells (PBECs) upon coculture with M. pneumoniae or S. pneumoniae in air-liquid interface (ALI) cultures. We detected increased levels of IL-8 in supernatant of PBECs that were incubated with the highest dose of S. pneumoniae, i.e., 100 bacteria/cell (Fig. 1A). Although M. pneumoniae induced IL-8 production, these levels were significantly lower than those of S. pneumoniae (Fig. 1A). Similarly, CCL2 and CCL20 production were markedly increased in PBECs stimulated with S. pneumoniae, whereas PBECs stimulated with M. pneumoniae only showed a trend toward increased CCL2 and CCL20 levels (Fig. 1B and C; see also Fig. S1B and C in the supplemental material). Overall, these data show that PBECs produce significantly higher amounts of proinflammatory chemokines upon interaction with S. pneumoniae than with M. pneumoniae.
FIG 1.
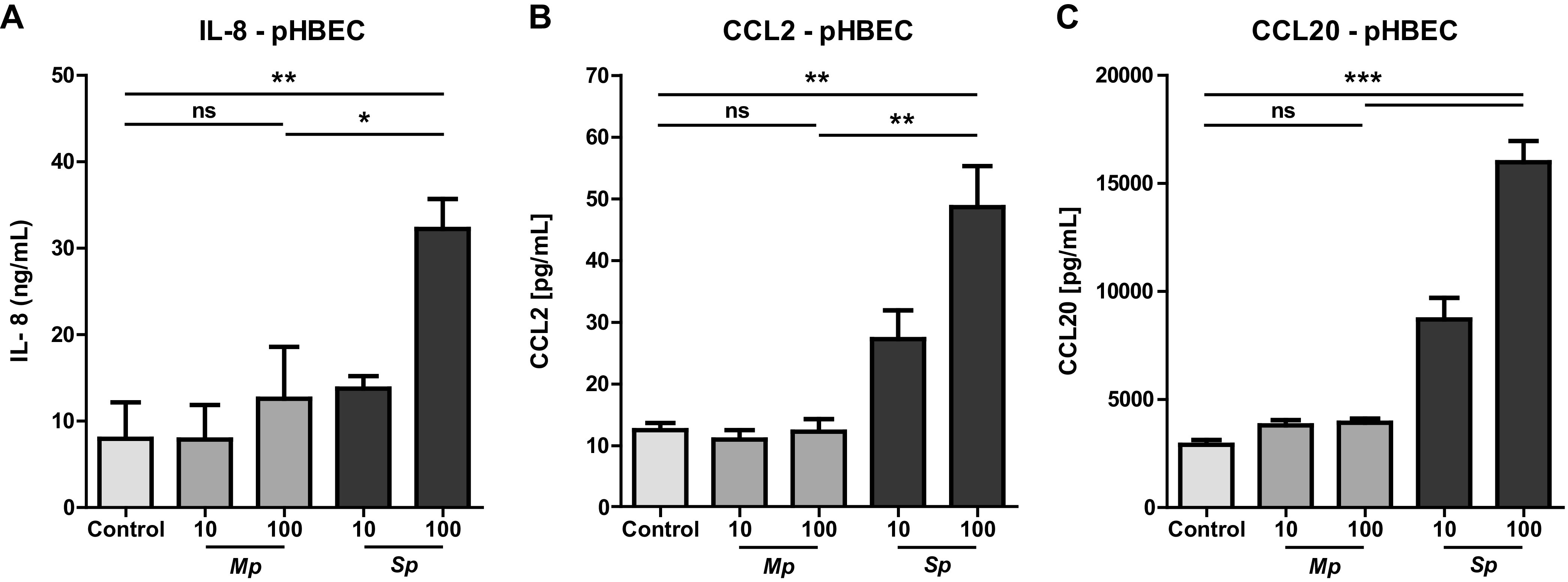
Reduced activation of primary human bronchial epithelial cells by M. pneumoniae compared to S. pneumoniae. (A to C) Primary human bronchial epithelial cells cultured in air-liquid interface were stimulated with live M. pneumoniae or S. pneumoniae at a multiplicity of infection of 10 or 100. To reduce potential influence of patient-specific characteristics, primary cells of three different donors were used for all experiments. (A) Levels of IL-8 in culture medium after 24 h of stimulation. (B) Levels of CCL-2 in culture medium after 24 h of stimulation. (C) Levels of CCL20 in culture medium after 24 h of stimulation. Combined data of three individual donors from two independent experiments. For every donor, we performed two biological replicates of which we took two technical replicates. Bars represent means, and error bars show standard error of the mean (SEM). *, P < 0.05; **, P < 0.01 (repeated measures ANOVA with post hoc Bonferroni correction).
M. pneumoniae-induced cytokine production is lower than S. pneumoniae-induced production in both upper and lower respiratory tract epithelial cells.
Having established that PBECs display a different response to M. pneumoniae and S. pneumoniae, we next determined whether epithelial cells from the upper and lower respiratory tract responded differently, as this has been shown for other respiratory pathogens (20). We therefore compared IL-8 production by Detroit 562 cells derived from the nasopharynx with IL-8 production by alveolar A549 cells. IL-8 levels in supernatants of both epithelial cell types were significantly higher after stimulation with S. pneumoniae than M. pneumoniae (Fig. 2A and B; see also Fig. S2A and B in the supplemental material). We observed a similar pattern for IL-6, with M. pneumoniae consistently inducing lower cytokine levels (Fig. 2C and D; see also Fig. S2C and D). Furthermore, production of CCL20 was strongly elevated in both Detroit 562 and A549 cells after S. pneumoniae stimulation in contrast to M. pneumoniae-induced CCL20 production. (Fig. 2E and F). Similar results were obtained for the monocyte attractant CCL2 (Fig. S2F to H). Together, these data show that proinflammatory cytokine and chemokine production induced by M. pneumoniae was significantly lower than that induced by S. pneumoniae and that this difference was present in epithelial cells derived from both the upper respiratory tract and the lower respiratory tract. This difference was not explained by differences in growth speed between M. pneumoniae and S. pneumoniae, since growth of S. pneumoniae with the experimental time frame was not substantial (Fig. S2I). Furthermore, even after stimulation with M. pneumoniae at an multiplicity of infection (MOI) of 2,500, IL-8 production by A549 cells did not exceed production in response to S. pneumoniae at and MOI of 100 (Fig. S2J).
FIG 2.

M. pneumoniae-induced cytokine production is reduced compared to S. pneumoniae-induced production in both upper and lower respiratory tract epithelial cells. Respiratory epithelial cell lines Detroit 562 and A549 were stimulated with live M. pneumoniae and S. pneumoniae at a multiplicity of infection of 10 or 100. (A to D) IL-8/IL-6 protein concentration in culture supernatant harvested after 24 h of stimulation (n = 5/condition). (E and F) CCL20 protein levels in culture supernatant harvested after 24 h of stimulation. (A to D) Data of at least two independent experiments. (E and F) Data of three experiments. All measurements consist of two technical replicates. (A to F) Bars represent means, and error bars show standard error of the mean. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (ANOVA with post hoc Bonferroni correction).
M. pneumoniae induces stronger TLR2 signaling than S. pneumoniae.
We wondered if the difference in epithelial cytokine responses to M. pneumoniae and S. pneumoniae was associated with differences in TLR activation. Both bacteria contain structures that bind to TLR1/2 and TLR2/6 heterodimers, and TLR-induced signaling can lead to cytokine production by respiratory epithelial cells (7–10, 21). TLR1, TLR2, TLR4, and TLR6 mRNA transcripts were expressed by all respiratory epithelial cell lines tested, although mRNA levels of TLR2 and TLR4 in A549 cells were lower than those in Detroit 562 and Calu-3 cells (Fig. 3A to D). Using specific TLR1/2, TLR2/6, and TLR4 ligands (i.e., PAM3CSK4, FSL-1, and lipopolysaccharide (LPS), respectively), we confirmed that TLR-induced activation alone is sufficient to trigger cytokine production by Detroit 562 and A549 cells (see Fig. S3A to E in the supplemental material). Next, we directly compared TLR2-mediated NF-κB activation induced by heat-killed M. pneumoniae and S. pneumoniae using TLR2 reporter cells. M. pneumoniae induced stronger TLR2-mediated signaling than S. pneumonia (Fig. 3E), which was in contrast to the cytokine responses (Fig. 1 and 2). Results were similar for both heat-killed and living M. pneumoniae and S. pneumoniae (Fig. S3F). Since S. pneumoniae cells are five times larger than M. pneumoniae cells, we also assessed TLR2-mediated signaling upon incubation of the reporter cells with equal weights of bacteria (22). Under these conditions, the difference between M. pneumoniae- and S. pneumoniae-induced TLR2-mediated signaling was even larger (Fig. 3F). Next, we evaluated if TLR2-mediated signaling in response to M. pneumoniae involved TLR1 or TLR6. In both TLR1- and TLR6-deficient TLR2 reporter lines, both bacteria still induced TLR2-mediated signaling, although the responses were markedly reduced (Fig. 3G and H). In TLR1-deficient TLR2 reporter lines, there was almost no difference in TLR2-mediated signaling between M. pneumoniae and S. pneumoniae (Fig. 3G), while in TLR6-deficient TLR2 reporter lines, this difference was still present (Fig. 3H). Combined, these findings show that M. pneumoniae more potently induces TLR2-mediated signaling than S. pneumoniae and suggest that TLR2/1 ligands in M. pneumoniae are largely responsible for this difference.
FIG 3.
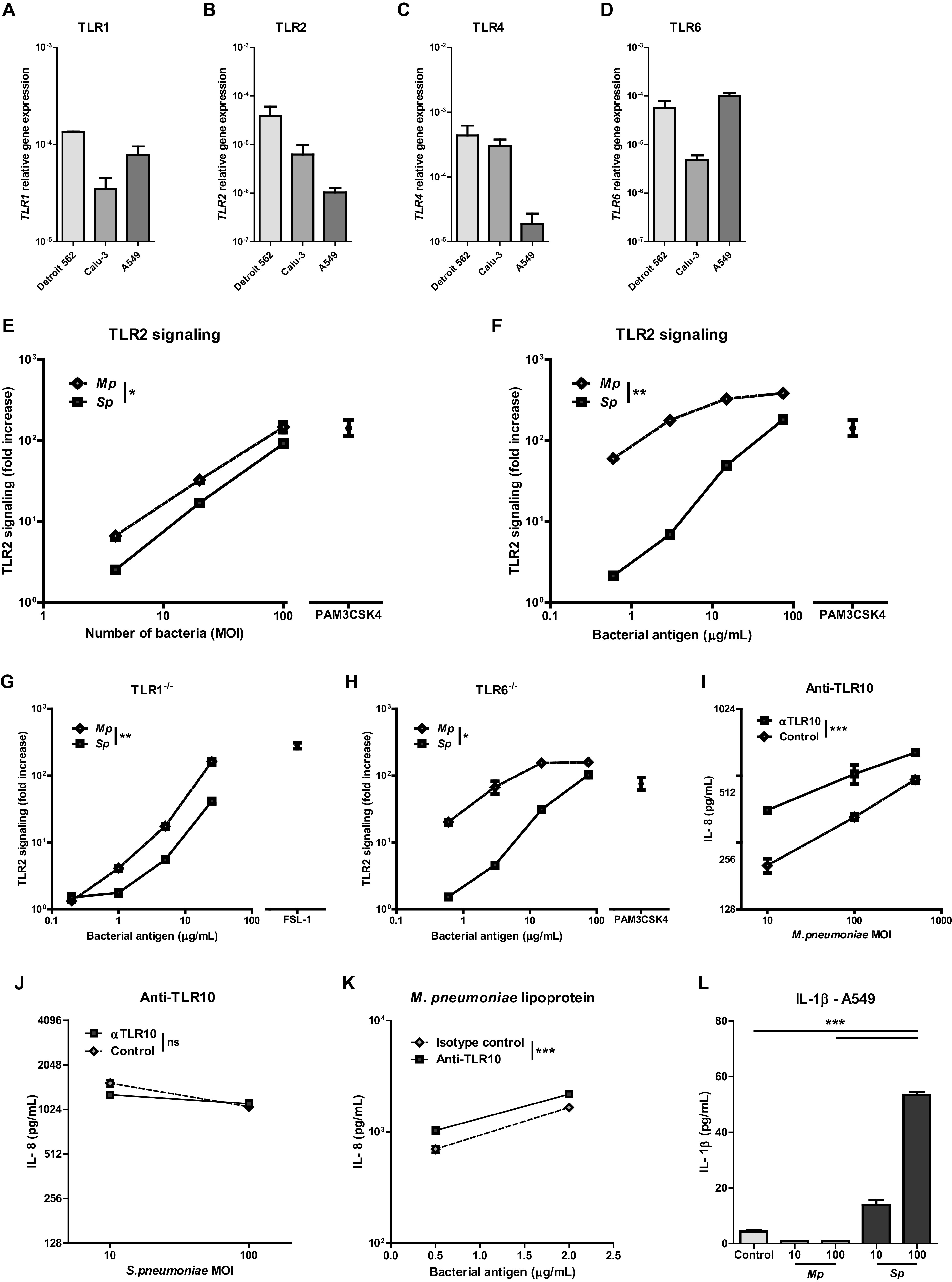
In contrast to M. pneumoniae, S. pneumoniae lowered Th2-associated cytokine production in resting and IL-1 alpha stimulated respiratory epithelial cells. (A) TLR1, TLR2, TLR4, and TLR6 relative gene expression in unstimulated respiratory epithelial cells (n = 3 to 4/group). (E and F) TLR2 signaling as assessed by fold increase in bioluminescence of TLR2-luciferase reporter cells upon stimulation with different doses of heat-killed bacteria compared to vehicle controls (n = 3/dose). (G) TLR2 signaling as assessed by fold increase in bioluminescence of TLR1−/− TLR2-luciferase reporter cells stimulated with multiple doses of bacteria (n = 3/dose). (H) TLR2 signaling as assessed by fold increase in bioluminescence of TLR6−/− TLR2-luciferase reporter cells stimulated with multiple doses of bacteria (n = 3/dose). (I and J) IL-8 levels in culture medium after 24 h of stimulation of A549 cells with M. pneumoniae or S. pneumoniae in the presence of TLR10 blocking antibody or isotype control. (K) IL-8 levels in culture medium after 24 h of stimulation of M. pneumoniae lipoproteins with either a TLR10-blocking antibody or isotype control. (L) IL-1β levels in culture medium after 24 h of stimulation with live M. pneumoniae or S. pneumoniae at an MOI of 10 or 100. (A to D) Data of two independent experiments. (E to L) Data shown of one representative experiment. Bars and dots represent group means, and error bars show SEM. **, P ≤ 0.01; ***, P ≤ 0.001 (comparing 50% effective concentration [EC50] of fitted dose-response curves or 2-way ANOVA).
Since TLR2/1 heterodimers only partly explained the differences between M. pneumoniae and S. pneumoniae, we looked at TLR4, as some reports have indicated that M. pneumoniae and S. pneumoniae express ligands that bind TLR4 (8, 10). A549 cells were responsive to TLR4-mediated signaling as shown by upregulating IL6 and CXCL8 gene expression after stimulation with LPS (Fig. S3D and E). However, neither heat-killed M. pneumoniae nor S. pneumoniae induced TLR4-mediated signaling in our TLR4 reporter cell system in contrast to the Gram-negative bacterium Haemophilus influenzae (Fig. S3G).
Since TLR10 has been shown to be an inhibitory coreceptor of TLR2, we set out to examine its potential involvement in M. pneumoniae-induced epithelial responses (17, 23). TLR10 gene expression was present in A549 respiratory epithelial cells and PBECs (data not shown). Blocking TLR10 using specific antibodies increased IL-8 production of A549 cells in response to M. pneumoniae but not S. pneumoniae (Fig. 3I and J). In contrast, IL-6 production in response to M. pneumoniae did not increase when blocking TLR10 signaling (Fig. S3H). Since TLR10 has been reported to bind similar ligands as TLR2/1, we next examined if M. pneumoniae lipoproteins could be the ligand for TLR10. Incubation of A549 respiratory epithelial cells with M. pneumoniae lipoproteins inducted high amounts of IL-8 (Fig. 3K). Coincubation with a TLR10-blocking antibody yielded a further increase in IL-8 levels.
We also evaluated the role of the intracellular bacterial sensors NOD1 and NOD2, as M. pneumoniae and S. pneumoniae have been shown to be capable of an intracellular lifestyle in epithelial cells (24, 25). However, incubation with heat-killed M. pneumoniae and S. pneumoniae did not induce NOD1- or NOD2-mediated NF-κB signaling in NOD signaling reporter cells (data not shown). We next assessed if M. pneumoniae and S. pneumoniae were capable of activating Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing receptors (NLRPs) and inducing inflammasome activation by measuring IL-1β levels in response to bacterial stimulation. M. pneumoniae did not lead to the production of IL-1β, whereas S. pneumoniae led to inflammasome activation as demonstrated by potent induction of IL-1β (Fig. 3L).
In contrast to M. pneumoniae, S. pneumoniae lowered Th2-associated cytokine production in resting and IL-1 alpha stimulated respiratory epithelial cells.
We observed that M. pneumoniae could robustly activate TLR2 signaling but did not evoke a strong proinflammatory response in respiratory epithelial cells, which was partly explained by inhibitory signaling by TLR10. Apart from this quantitative difference, we speculated that M. pneumoniae and S. pneumoniae would induce qualitatively different responses. M. pneumoniae did not activate an anti-inflammatory program in epithelial cells as gene expression of IL10 and TGFB1 were undetectable in our system (data not shown). Neither did we observe a Th17-associated response, as IL17C mRNA levels were below the detection limit, even in S. pneumoniae-stimulated cells (data not shown).
Next, we looked at the production of Th2-associated cytokines by respiratory epithelial cells, since several studies have suggested that M. pneumoniae infections are associated with the onset of asthma and/or predispose to asthma exacerbations (26, 27). Respiratory epithelial cells are known to be important players in asthma by producing Th2-associated cytokines, such as IL-25 and IL-33 that drive Th2-associated inflammation (28). We evaluated the effect of M. pneumoniae on the production of the Th2-associated cytokine IL-33 by respiratory epithelial cells. However, incubation of Calu-3 or A549 cells with M. pneumoniae did not result in increased IL33 gene expression (Fig. 4A and B). In contrast, S. pneumoniae showed a 4-fold downregulation of IL33 mRNA levels in Calu-3 cells (Fig. 4A). Although expression of IL25 mRNA was not detectable in our model (data not shown), IL-25 receptor (IL17RB) mRNA levels were also downregulated after S. pneumoniae stimulation in both Calu-3 and A549 cells. Again, incubation of epithelial cells with M. pneumoniae did not change IL17RB mRNA expression levels compared to those of control epithelial cell cultures (Fig. 4C and D). These data suggest that under homeostatic conditions, M. pneumoniae does not induce production of Th2-associated cytokines in respiratory epithelial cells. This could be different when M. pneumoniae interacts with respiratory epithelial cells concomitantly with a Th2-associated stimulus, such as IL-1α. IL-1α is known to be essential for the Th2 response to house dust mite in vivo by inducing IL-33 in respiratory epithelial cells (29). We confirmed that IL-1α led to increased amounts of IL33 mRNA in Detroit 562 cells (Fig. 4E). M. pneumoniae stimulation of respiratory epithelial cells in the presence of IL-1α changed IL33 gene expression less than 2-fold (Fig. 4E). Whereas, in Detroit 562 cells stimulated with S. pneumoniae in the presence of IL-1α, we again observed a downregulation of IL33 gene expression compared with that of IL-1α alone (Fig. 4E). Importantly, IL-1α did not lead to overstimulation of epithelial cells, rendering them insensitive for further stimulation, since IL-1α in combination with FSL-1 did increase IL33 gene expression (see Fig. S4A in the supplemental material). M. pneumoniae in the presence of IL-1α did not increase CXCL8 mRNA levels (Fig. 4F), whereas without IL-1α M. pneumoniae did increase IL-8 (Fig. 2A). Simultaneous with IL33 downregulation, upregulated CXCL8 gene expression was detected after S. pneumoniae stimulation (Fig. 4F). Consequently, S. pneumoniae strongly promoted respiratory epithelial cells to trigger a Th1 environment, as demonstrated by the high CXCL8/IL33 ratio, even under Th2-promoting conditions (Fig. 4G).
FIG 4.
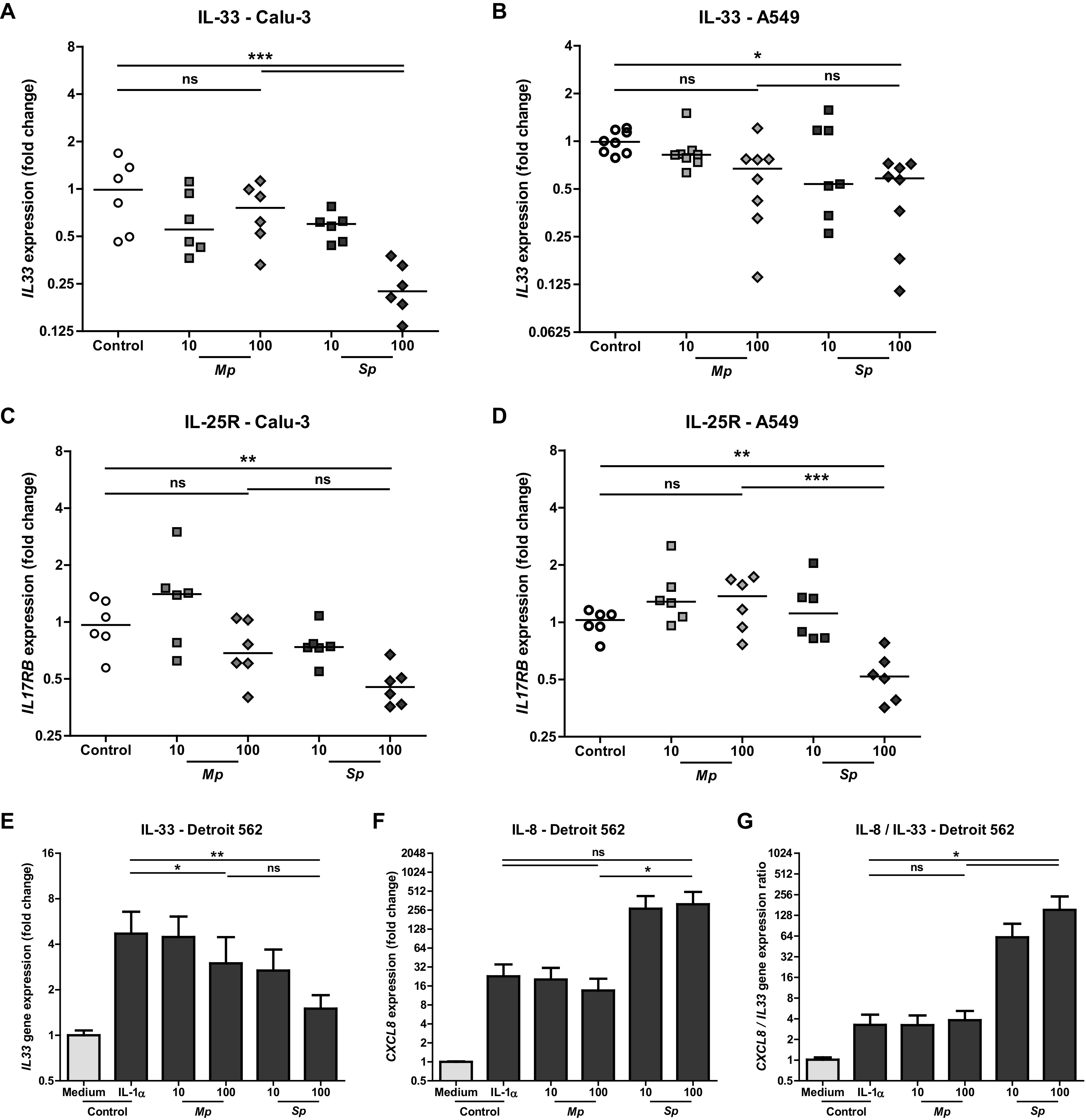
S. pneumoniae lowered Th2-associated cytokine production in resting and IL-1 alpha stimulated respiratory epithelial cells, whereas M. pneumoniae did not. Respiratory epithelial cell lines Detroit 562, Calu-3, and A549 were stimulated with live M. pneumoniae and S. pneumoniae with a multiplicity of infection of 10 or 100. (A and B) IL33 mRNA as fold increase to controls after 5 h of stimulation (n = 6 to 8/condition). (C and D) IL17RB mRNA as fold increase to controls after 5 h of stimulation (n = 6/condition). (E to G) Detroit 562 cells were incubated with IL-1 alpha and simultaneously stimulated with live M. pneumoniae or S. pneumoniae at a multiplicity of infection of 10 or 100. IL33 and CXCL8 gene expressions were assessed after 5 h. (n = 4/condition). Combined data of at least two independent experiments. All measurements consist of two technical replicates. (A to D) Data points represent biological replicates, and lines group medians. ANOVA with post hoc Bonferroni correction. (E to G) Bars represent group means, and error bars show SEM. Repeated measures ANOVA with post hoc Bonferroni correction. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
DISCUSSION
The immunological basis for the clinical differences between M. pneumoniae and S. pneumoniae pneumonia is unknown. Yet, understanding these mechanisms will give better insight in pneumonia pathogenesis and potentially unveil targets for immunomodulatory therapies that can lower pneumonia severity. We show that both M. pneumoniae and S. pneumoniae induce proinflammatory cytokine production in respiratory epithelial cells, confirming previous studies (30, 31). We extend these observations by showing that respiratory epithelial cell responses to M. pneumoniae and S. pneumoniae differ both in quantity and in quality. Proinflammatory cytokine and chemokine production by respiratory epithelial cells in response to M. pneumoniae was lower than that in response to S. pneumoniae, as demonstrated by lower IL-8, IL-6, CCL20, and CCL2 levels and the absence of IL-1β. In contrast, M. pneumoniae induced substantially stronger TLR2 signaling than S. pneumoniae. This difference depended mostly on TLR1 and not on TLR6. Furthermore, TLR10 signaling was only induced by M. pneumoniae and dampened proinflammatory IL-8 production but not IL-6. There were marked qualitative differences between M. pneumoniae- and S. pneumoniae-induced levels of Th2-associated cytokines. Only S. pneumoniae showed a downregulation of IL-33 in respiratory epithelial cells under homeostatic conditions as well as in the presence of Th2-skewing cytokines. Meanwhile, after stimulation with M. pneumoniae, respiratory epithelial cells maintained production of Th2-associated cytokines.
IL-8 levels were consistently higher in supernatants of respiratory epithelial cells that were stimulated with S. pneumoniae compared to M. pneumoniae. This quantitative difference might be explained by the differential activation of TLR10 by both pathogens. Blocking TLR10 increased IL-8 production by A549 respiratory epithelial cells after M. pneumoniae stimulation, which demonstrates that M. pneumoniae contains ligands for TLR10, which has not been shown previously. Our data on TLR10 blockade shows that M. pneumoniae lipoproteins are a ligand for TLR10. In contrast, proinflammatory cytokine production by S. pneumoniae was unchanged by blocking TLR10, suggesting S. pneumoniae does not contain TLR10 ligands. By engaging with this inhibitory TLR, M. pneumoniae thus dampens IL-8 production by epithelial cells. This fits with the clinical observation that M. pneumoniae lower respiratory tract infections generally have a milder disease course, as systemic IL-8 levels have been shown to correlate with the severity of pneumonia in children (32, 33). Pneumonia caused by M. pneumoniae is associated with lower levels of the acute phase response protein CRP than S. pneumoniae lower respiratory tract infections (34). Indeed, we found that IL-6, which mediates the acute phase response, was less elevated in cocultures of epithelial cells and M. pneumoniae compared to S. pneumoniae (35). We did not observe differences in proinflammatory cytokine production between respiratory tract cell lines derived from the upper versus the lower respiratory tract. However, our model system does not account for the influence of microbiota and the presence of cell types other than respiratory epithelial cells. These factors likely contribute to a local niche that could influence differences in cytokine responses between the upper and lower respiratory tract.
To our surprise, we found that M. pneumoniae more potently induces TLR2 signaling compared to S. pneumoniae, whereas proinflammatory cytokine production in response to M. pneumoniae was in fact lower. This suggests that the differences in cytokine production by respiratory epithelial cells may be explained by cotriggering of TLR2 and another PRR by M. pneumoniae, which could result in dampening rather than activating innate immunity in epithelial cells. Indeed, we observed that TLR10, which has been shown to recognize triacylated lipoproteins and to have inhibitory effects on TLR2 signaling, was partially responsible for inhibition of immunity to M. pneumoniae (17, 23). TLR10 signaling in response to M. pneumoniae lowered IL-8 production, which could limit recruitment of neutrophils during M. pneumoniae infection, thereby dampening inflammation.
The difference in TLR2 signaling between M. pneumoniae and S. pneumoniae was larger when correcting for bacterial size. Possibly, the small size of M. pneumoniae and subsequent lower TLR2 signaling can be helpful to evade an immune response and maintain a state of asymptomatic carriage. This effect could be even more pronounced because bacterial growth of M. pneumoniae in vivo is lower than that of S. pneumoniae. This suggests that higher M. pneumoniae bacterial loads would lead to stronger immune activation and subsequently to more severe disease. However, studies on the relationship between microbial load and disease severity are conflicting (36, 37).
Most surprisingly TLR4 signaling was not induced by M. pneumoniae or S. pneumoniae, whereas other studies suggest both bacteria contain TLR4 ligands (8, 10). In this study, we used heat-killed bacteria to assess TLR signaling, which could have affected our results. However, we obtained similar results when using ethanol-treated bacteria, suggesting that the lack of TLR4 signaling was not due to heat inactivation of lipoproteins. The main TLR4 binding component of S. pneumoniae is pneumolysin, which could be produced in a phase-dependent way, explaining different findings in literature (10). Furthermore, we directly assessed the ability of S. pneumoniae to induce TLR4 signaling using a TLR4 reporter cell line, which does not recapitulate any indirect effects of TLR4 in vivo.
TLR2/1 and TLR2/6 heterodimers recognize triacylated and diacylated lipoproteins, respectively, and both lipoproteins are present in M. pneumoniae (11, 38, 39). Observations in the TLR1-deficient TLR2 reporter system suggest that TLR2 signaling by M. pneumoniae largely depends on TLR1 and that M. pneumoniae contains triacylated lipoproteins capable of strongly activating TLR2. Although TLR2 signaling in the TLR1- and TLR6-deficient reporter systems was markedly reduced, we still observed substantial TLR2 signaling at higher concentrations of bacterial antigen. This could indicate that although TLR1 and TLR6 contribute to TLR2 signaling in response to M. pneumoniae and S. pneumoniae, neither are essential. Perhaps other coreceptors on respiratory epithelial cells, such as CD14, LBP, and CD36, or the formation of TLR2 homodimers can compensate for the loss of TLR1 or TLR6 (9, 15, 40, 41). However, the highest concentrations of bacterial antigen used in our assay could be supraphysiological and not reflect levels in vivo. Our data certainly do not rule out the possibility that M. pneumoniae signals via TLR2/6, but TLR2/1 heterodimers could be more important than previously realized.
Levels of IL17C mRNA were undetectable in respiratory epithelial cells. However, there could still be a role for respiratory epithelial cells in the Th17 response via the production of IL-6. IL-6 was produced in respiratory epithelial cells (Fig. 2) and has been shown to enhance the production of IL-17A by lymphocytes in response to stimulation with M. pneumoniae (42). The same study showed IL-10 production in response to M. pneumoniae, which coincided with increased levels of FOXP3 mRNA, suggesting that T cells could be the source of IL-10 during M. pneumoniae infection and not respiratory epithelial cells.
The correlation found in the literature between M. pneumoniae pneumonia and asthma can be explained by the following two phenomena: (i) M. pneumoniae triggers and/or contributes to the development of asthma, and (ii) having asthma makes patients more susceptible to M. pneumoniae pneumonia. We found no evidence for induction of Th2-associated cytokines in respiratory epithelial cells by M. pneumoniae even though respiratory epithelial cells are known to be key players in asthma by producing IL-25 and IL-33 that drive Th2-associated inflammation (28). Alternatively, it could be that asthma increases patient susceptibility to M. pneumoniae infection, since asthma has been shown to be a risk factor for several infections, including invasive pneumococcal disease (43). Asthma patients are known to have reduced Th1 responses after infectious challenges (44), whereas Th1 responses are thought to be important for the clearance of M. pneumoniae infections (45). We show that M. pneumoniae stimulation of resting epithelial cells hardly instigates production of proinflammatory, Th1-associated cytokines. When combined with already reduced responses in asthma patients, this could lead to increased susceptibility to M. pneumoniae infection. Additionally, established asthma in a murine model decreased TLR2 expression, which coincided with decreased clearance of M. pneumoniae (46). In our study, we showed that M. pneumoniae strongly induces TLR2 signaling in human respiratory epithelial cells and that TLR2 activation leads to proinflammatory cytokine production (Fig. 3), which could indicate that TLR2 is needed for clearance of M. pneumoniae. Through these mechanisms, asthma patients could have an increased susceptibility to M. pneumoniae infection, which would explain the correlation between asthma and M. pneumoniae found in clinical studies (26, 27, 47).
By directly comparing M. pneumoniae and S. pneumoniae, we show that there are important differences in the immune response by respiratory epithelial cells to both pathogens, which provide insight into the regulation of the immune system during respiratory tract infection. These differences in the height and the quality of the innate epithelial response matched with clinical differences between M. pneumoniae and S. pneumoniae RTIs and provide new targets for diagnostic and prognostic biomarkers and opportunities for immunotherapy of RTIs.
MATERIALS AND METHODS
Bacterial culture and lipoprotein isolation.
M. pneumoniae strain M129 (ATCC 29342) was cultured in SP4 medium at 37°C/5% CO2 until color change of the medium (48). CFU counts were determined by culture on PPLO agar plates (BD Difco) at 37°C. S. pneumoniae strain D39 (NCTC 7466) was cultured in Todd-Hewitt broth (Sigma-Aldrich, St. Louis, USA) until an optical density at 600 nm (OD600) of 0.6 was reached. CFU counts were determined using Todd-Hewitt agar plates (Sigma-Aldrich). Haemophilus influenzae strain 576 (ATCC 9334) was cultured in Müller-Hinton broth supplemented with 15 μg/mL NAD (Sigma-Aldrich) and 15 μg/mL hemin (Sigma-Aldrich) at 37°C/5% CO2 until an OD600 of 0.5 was reached. CFU counts were determined using Müller-Hinton agar plates (Sigma-Aldrich). Homogenous bacterial suspensions were aliquoted and stored at −80°C until use. Bacteria for TLR reporter assays were heat-killed by incubating at 56°C for 30 min. Protein concentrations of the bacterial suspensions were determined using Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, USA) according to manufacturer’s instructions. For isolation of M. pneumoniae lipoproteins, M. pneumoniae bacterial suspension was added to TX-114 at 0 to 4°C. After centrifugation at 15,000 × g, the supernatant was harvested. Supernatant was then incubated at 37°C and centrifuged. The lower TX phase was harvested, and lipoproteins were precipitated using methanol at −80°C.
Air-liquid interface culture of primary human bronchial epithelial cells.
Primary bronchial epithelial cells (PBECs) were isolated from surgically resected lung tissue obtained from patients who had underlying conditions such as cancer. The resected tissues were macroscopically and microscopically checked by a pathologist for the absence of (suspected) lesions (Erasmus MC, Rotterdam, The Netherlands; METC 2012-512). To reduce potential influence of patient-specific characteristics, primary cells of three different donors were used for experiments. After excision, the tissues were trimmed, washed, and incubated overnight at 4°C with 0.15% protease XIV in Hanks’ balanced salt solution (HBSS). Thereafter, cell culture was performed as described previously (49, 50). In brief, cells were resuspended in serum-free keratinocyte medium (KSFM) (Life Technologies Europe B.V.) supplemented with 0.2 ng/mL human epidermal growth factor, 25 μg/mL bovine pituitary extract, and 1 μM isoproterenol and cultured until confluent in plates coated with 10 μg/mL human fibronectin, 30 μg/mL bovine serum albumin (BSA), and 10 μg/mL PureCol. Before seeding the epithelial cells in the air-liquid interface (ALI) culture system, transepithelial resistance (TEER) was measured. When there was sufficient barrier function, as evidenced by TEER, epithelial cells were trypsinized and used in ALI cultures. Hereto, 80,000 cells/well were seeded onto coated 12-well transwell inserts (Corning Inc., Corning, USA) and cultured in bronchial epithelial growth medium (BEGM) (ScienCell Research Laboratories, Carlsbad, USA) supplemented with 1 nM retinoic acid (RA) at 37°C in a humidified incubator with 5% CO2. When cells reached full confluence, the BEGM medium was removed from the apical chamber and only supplied to the basal chamber and supplemented with 50 nM RA. The medium was changed every other day, and the apical chamber was rinsed with phosphate-buffered saline (PBS). Stimuli were applied to the apical chambers (50).
Culture and stimulation of respiratory epithelial cell lines.
Detroit 562 (CCL-138; ATCC, Manassas, USA) and Calu-3 (HTB-55; ATCC) cell lines were cultured on a collagen (Advanced BioMatrix, San Diego, CA, USA) and fibronectin (Merck Millpore, Burlington, USA) coat in modified Eagle’s medium (MEM) containing Earl’s salts and l-glutamine (Gibco, Carlsbad, USA) and supplemented with 20% heat-inactivated fetal calf serum (FCShi; Bodinco, Alkmaar, The Netherlands) at 37°C/5% CO2 until confluent. Cells were harvested using 5 mg/mL trypsin (Sigma-Aldrich) and seeded at 800,000 cells/well in collagen/fibronectin-coated 24-well plates (Greiner Bio-One). A549 cells (CCL-185; ATCC) were cultured in RPMI 1640 medium containing l-glutamine (Gibco) and supplemented with 10% FCShi until confluent. After trypsinization, A549 cells were seeded at 800,000 cells/well in a 24-well plate. After 1 day of culture, medium was replaced by medium with 1% FCShi. Two days after seeding, cells were incubated with homogenous bacterial cell suspensions at 10 to 1,000 bacteria per cell. Furthermore, cells were stimulated with 100 ng/mL FSL-1 (Sigma-Aldrich), 1 mg/mL Pam3CSK4 (InvivoGen, San Diego, USA), or 10 to 100 ng/mL LPS (InvivoGen). TLR10-blocking was performed using a monoclonal anti-TLR10 antibody and isotype antibody as a control, clones 3C10C5 and MOPC-21, respectively (Hycult Biotech, Uden, The Netherlands).
In some experiments, Detroit 562 cells were incubated with bacteria in the presence of IL-1 alpha (5 ng/mL; Immunotools, Friesoythe, Germany). Cells were harvested 4 to 6 h after stimulation to isolate RNA for gene expression analysis. Culture supernatants were harvested at 24 h after stimulation to assess cytokine levels.
RNA extraction and quantitative PCR of gene expression.
RNA was isolated using NucleoSpin RNA extraction kit (Macherey-Nagel, Düren, Germany). cDNA was synthesized using the SensiFast cDNA synthesis kit according to manufacturer’s instructions (Bioline Reagents, London, UK). Real-time PCR was performed using specific primers and SYBR green (Bioline Reagents) with the CFX96 real-time system C1000 (Bio-Rad Laboratories, Hercules, USA). The quantification cycle (Cq) was determined using Bio-Rad CFX manager algorithm (Bio-Rad Laboratories). GAPDH was used as a reference gene for normalizing gene expression. The following primer sets were used: GAPDH, Fw 5′-GTCGGAGTCAACGGATT-3′, Rv 5′-AAGCTTCCCGTTCTCAG-3′; CCL2 (MCP-1), Fw 5′-TCCAGCATGAAAGTCTCTG-3′, Rv 5′-CGAGCCTCTGCACTGA-3′; CCL20 (MIP-3α), Fw 5′-GAAGGCTGTGACATCAATG-3′, Rv 5′-CCCCAGCAAGGTTCTT-3′; IL33,Fw 5′-AACACCCCTCAAATGAATC-3′, Rv 5′-CTTGCATTCAAATGAAACAC-3′; CXCL8 (IL-8), Fw 5′-CCGGAAGGAACCATCT-3′, Rv 5′-TTGGGGTGGAAAGGTT-3′; IL17RB (IL-25 receptor), Fw 5′-GGCACGAAAGGATCAAG-3′, Rv 5′-CTGCAATGGTTTTGAAGAA-3′. More detailed information can be found in the supplemental material.
Protein quantification (ELISA and LEGENDplex).
Interleukin-6 (IL-6) and IL-8 in culture supernatants were measured by enzyme-linked immunosorbent assay (ELISA) using specific antibody pairs (Thermo Fisher Scientific) following manufacturer’s instructions. 3,3′,5,5′-Tetramethylbenzidine (Sigma-Aldrich, USA) was used as a substrate. Optical density was measured at 450 nm using a microplate reader (SpectraMax iD3; Molecular Devices, San José, CA, USA). CCL2 and CCL20 levels were measured using the LEGENDplex bead-based immunoassay (BioLegend, San Diego, CA, USA) according to manufacturer’s instructions.
Toll-like receptor and NOD-like receptor signaling reporter assays.
Human endothelial kidney cells (HEK293) were transfected using PolyFect (Qiagen Benelux, Venlo, The Netherlands) with TLR/NLR-encoding constructs, i.e., TLR2, TLR4, NOD1, and NOD2 (InvivoGen, Toulouse, France). After selection, these cells were subsequently transfected with a reporter vector expressing luciferase under the control of a NF-κB-responsive promoter (pNifty2-luc; InvivoGen). Stably transfected clones were selected and used in bioassays. Cells were plated at 105 cells/well in flatbottomed 96-well plates and incubated with indicated amounts of heat-killed bacteria or TLR-ligands for 16 h at 37°C. Subsequently, HEK293 cells were lysed in Steadylite solution (PerkinElmer, Waltham, MA), and bioluminescence was measured using a Victor3 reader (PerkinElmer). As a positive control for the presence of the pNifty2-luc vector, recombinant tumor necrosis factor alpha (TNF-α) (InvivoGen) was used. Specific synthetic TLR ligands (InvivoGen) were used as positive controls for TLR signaling. TLR1−/− and TLR6−/− lines were generated on a HEK293-TLR2/CD14/pNifty2-luc background by transfecting them with CRISPR/Cas9 all-in-one plasmids (GeneCopoeia, Rockville, MD). Clonal lines were generated, and knockouts were validated using Sanger sequencing and confirmed in bioassays using TLR1/2- and TLR2/6-specific ligands.
Statistical analysis.
GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) was used for statistical analysis. Technical duplicates were taken of every sample and averaged before data analysis. ELISA and quantitative PCR (qPCR) data were assumed to be log-normally distributed. All data were log(10)-transformed before statistical hypothesis testing. Repeated measures analysis of variance (ANOVA) with post hoc Bonferroni multiple comparison test was used to compare different stimulations on PBECs of multiple individual donors. ANOVA with post hoc Bonferroni multiple comparison test was used to determine statistical significance when comparing stimulation with various bacteria to the medium control. Nonlinear regression (dose-response curves) was used to compare differences between M. pneumoniae and S. pneumoniae in TLR triggering at different bacterial antigen concentrations. Statistical significance was defined as P < 0.05. Choice of statistical test is reported in figure legends. Exact P values are reported in the supplemental material.
ACKNOWLEDGMENTS
We thank all patients who participated in this study.
R.C.A.D.G., A.M.C.V.R., and W.W.J.U. conceived the project, interpreted data, and drafted the manuscript. R.C.A.D.G., H.Z., T.H., A.B., E.E., A.E.J.T.J., A.C.J.M.D.B., and S.C.E. conducted and analyzed experiments. J.J.B., R.J.R., and M.K. interpreted data. All authors critically reviewed the manuscript and made intellectual contributions to the study.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
R. C. A. de Groot, Email: r.degroot@erasmusmc.nl.
W. W. J. Unger, Email: w.unger@erasmusmc.nl.
Liise-anne Pirofski, Albert Einstein College of Medicine.
REFERENCES
- 1.World Health Organization. 2016. Pneumonia fact sheet. World Health Organization, Geneva, Switzerland. Accessed 16 April 2018. [Google Scholar]
- 2.Jain S, Williams DJ, Arnold SR, Ampofo K, Bramley AM, Reed C, Stockmann C, Anderson EJ, Grijalva CG, Self WH, Zhu Y, Patel A, Hymas W, Chappell JD, Kaufman RA, Kan JH, Dansie D, Lenny N, Hillyard DR, Haynes LM, Levine M, Lindstrom S, Winchell JM, Katz JM, Erdman D, Schneider E, Hicks LA, Wunderink RG, Edwards KM, Pavia AT, McCullers JA, Finelli L, Team CES. 2015. Community-acquired pneumonia requiring hospitalization among U.S. children. N Engl J Med 372:835–845. doi: 10.1056/NEJMoa1405870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Groot RCA, Meyer Sauteur PM, Unger WWJ, van Rossum AMC. 2017. Things that could be Mycoplasma pneumoniae. J Infect 74(Suppl 1):S95–S100. doi: 10.1016/S0163-4453(17)30198-6. [DOI] [PubMed] [Google Scholar]
- 4.Meyer Sauteur PM, Krautter S, Ambroggio L, Seiler M, Paioni P, Relly C, Capaul R, Kellenberger C, Haas T, Gysin C, Bachmann LM, van Rossum AMC, Berger C. 2020. Improved diagnostics help to identify clinical features and biomarkers that predict Mycoplasma pneumoniae community-acquired pneumonia in children. Clin Infect Dis 71:1645–1654. doi: 10.1093/cid/ciz1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishida T, Miyashita N, Nakahama C. 2007. Clinical differentiation of atypical pneumonia using Japanese guidelines. Respirology 12:104–110. doi: 10.1111/j.1440-1843.2006.00927.x. [DOI] [PubMed] [Google Scholar]
- 6.Leiva-Juarez MM, Kolls JK, Evans SE. 2018. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunol 11:21–34. doi: 10.1038/mi.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimizu T, Kida Y, Kuwano K. 2005. A dipalmitoylated lipoprotein from Mycoplasma pneumoniae activates NF-κB through TLR1, TLR2, and TLR6. J Immunol 175:4641–4646. doi: 10.4049/jimmunol.175.7.4641. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu T, Kimura Y, Kida Y, Kuwano K, Tachibana M, Hashino M, Watarai M. 2014. Cytadherence of Mycoplasma pneumoniae induces inflammatory responses through autophagy and Toll-like receptor 4. Infect Immun 82:3076–3086. doi: 10.1128/IAI.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. 1999. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol 163:1–5. [PubMed] [Google Scholar]
- 10.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci USA 100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. 2009. Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31:873–884. doi: 10.1016/j.immuni.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. 2002. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol 169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. 2001. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol 13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 14.Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Briere F, Vlach J, Lebecque S, Trinchieri G, Bates EE. 2005. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174:2942–2950. doi: 10.4049/jimmunol.174.5.2942. [DOI] [PubMed] [Google Scholar]
- 15.Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B. 2005. CD36 is a sensor of diacylglycerides. Nature 433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 16.Manukyan M, Triantafilou K, Triantafilou M, Mackie A, Nilsen N, Espevik T, Wiesmuller KH, Ulmer AJ, Heine H. 2005. Binding of lipopeptide to CD14 induces physical proximity of CD14, TLR2 and TLR1. Eur J Immunol 35:911–921. doi: 10.1002/eji.200425336. [DOI] [PubMed] [Google Scholar]
- 17.Oosting M, Cheng SC, Bolscher JM, Vestering-Stenger R, Plantinga TS, Verschueren IC, Arts P, Garritsen A, van Eenennaam H, Sturm P, Kullberg BJ, Hoischen A, Adema GJ, van der Meer JW, Netea MG, Joosten LA. 2014. Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci USA 111:E4478–E4484. doi: 10.1073/pnas.1410293111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schutyser E, Struyf S, Van Damme J. 2003. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev 14:409–426. doi: 10.1016/S1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto K, Ahyi AN, Pepper-Cunningham ZA, Ferrari JD, Wilson AA, Jones MR, Quinton LJ, Mizgerd JP. 2014. Roles of lung epithelium in neutrophil recruitment during pneumococcal pneumonia. Am J Respir Cell Mol Biol 50:253–262. doi: 10.1165/rcmb.2013-0114OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hillyer P, Shepard R, Uehling M, Krenz M, Sheikh F, Thayer KR, Huang L, Yan L, Panda D, Luongo C, Buchholz UJ, Collins PL, Donnelly RP, Rabin RL. 2018. Differential responses by human respiratory epithelial cell lines to respiratory syncytial virus reflect distinct patterns of infection control. J Virol 92:e02202-17. doi: 10.1128/JVI.02202-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bals R, Hiemstra PS. 2004. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J 23:327–333. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 22.Waites KB, Talkington DF. 2004. Mycoplasma pneumoniae and its role as a human pathogen. Clin Microbiol Rev 17:697–728. doi: 10.1128/CMR.17.4.697-728.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guan Y, Ranoa DR, Jiang S, Mutha SK, Li X, Baudry J, Tapping RI. 2010. Human TLRs 10 and 1 share common mechanisms of innate immune sensing but not signaling. J Immunol 184:5094–5103. doi: 10.4049/jimmunol.0901888. [DOI] [PubMed] [Google Scholar]
- 24.Meseguer MA, Alvarez A, Rejas MT, Sanchez C, Perez-Diaz JC, Baquero F. 2003. Mycoplasma pneumoniae: a reduced-genome intracellular bacterial pathogen. Infect Genet Evol 3:47–55. doi: 10.1016/s1567-1348(02)00151-x. [DOI] [PubMed] [Google Scholar]
- 25.Weight CM, Venturini C, Pojar S, Jochems SP, Reine J, Nikolaou E, Solorzano C, Noursadeghi M, Brown JS, Ferreira DM, Heyderman RS. 2019. Microinvasion by Streptococcus pneumoniae induces epithelial innate immunity during colonisation at the human mucosal surface. Nat Commun 10:3060. doi: 10.1038/s41467-019-11005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeh JJ, Wang YC, Hsu WH, Kao CH. 2016. Incident asthma and Mycoplasma pneumoniae: a nationwide cohort study. J Allergy Clin Immunol 137:1017–1023. doi: 10.1016/j.jaci.2015.09.032. [DOI] [PubMed] [Google Scholar]
- 27.Atkinson TP, Duffy LB, Pendley D, Dai Y, Cassell GH. 2009. Deficient immune response to Mycoplasma pneumoniae in childhood asthma. Allergy Asthma Proc 30:158–165. doi: 10.2500/aap.2009.30.3207. [DOI] [PubMed] [Google Scholar]
- 28.Hammad H, Lambrecht BN. 2015. Barrier epithelial cells and the control of type 2 immunity. Immunity 43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Willart MA, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, Hammad H. 2012. Interleukin-1alpha controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med 209:1505–1517. doi: 10.1084/jem.20112691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sohn MH, Lee KE, Choi SY, Kwon BC, Chang MW, Kim KE. 2005. Effect of Mycoplasma pneumoniae lysate on interleukin-8 gene expression in human respiratory epithelial cells. Chest 128:322–326. doi: 10.1016/S0012-3692(15)37964-2. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Hooper WC, Phillips DJ, Talkington DF. 2002. Regulation of proinflammatory cytokines in human lung epithelial cells infected with Mycoplasma pneumoniae. Infect Immun 70:3649–3655. doi: 10.1128/IAI.70.7.3649-3655.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kutty PK, Jain S, Taylor TH, Bramley AM, Diaz MH, Ampofo K, Arnold SR, Williams DJ, Edwards KM, McCullers JA, Pavia AT, Winchell JM, Schrag SJ, Hicks LA. 2019. Mycoplasma pneumoniae among children hospitalized with community-acquired pneumonia. Clin Infect Dis 68:5–12. doi: 10.1093/cid/ciy419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saghafian-Hedengren S, Mathew JL, Hagel E, Singhi S, Ray P, Ygberg S, Nilsson A. 2017. Assessment of cytokine and chemokine signatures as potential biomarkers of childhood community-acquired pneumonia severity: a nested cohort study in India. Pediatr Infect Dis J 36:102–108. doi: 10.1097/INF.0000000000001364. [DOI] [PubMed] [Google Scholar]
- 34.Lehtomaki K. 1988. Clinical diagnosis of pneumococcal, adenoviral, mycoplasmal and mixed pneumonias in young men. Eur Respir J 1:324–329. [PubMed] [Google Scholar]
- 35.Castell JV, Gomez-Lechon MJ, David M, Andus T, Geiger T, Trullenque R, Fabra R, Heinrich PC. 1989. Interleukin-6 is the major regulator of acute phase protein synthesis in adult human hepatocytes. FEBS Lett 242:237–239. doi: 10.1016/0014-5793(89)80476-4. [DOI] [PubMed] [Google Scholar]
- 36.Nilsson AC, Bjorkman P, Welinder-Olsson C, Widell A, Persson K. 2010. Clinical severity of Mycoplasma pneumoniae (MP) infection is associated with bacterial load in oropharyngeal secretions but not with MP genotype. BMC Infect Dis 10:39. doi: 10.1186/1471-2334-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spuesens EB, Fraaij PL, Visser EG, Hoogenboezem T, Hop WC, van Adrichem LN, Weber F, Moll HA, Broekman B, Berger MY, van Rijsoort-Vos T, van Belkum A, Schutten M, Pas SD, Osterhaus AD, Hartwig NG, Vink C, van Rossum AM. 2013. Carriage of Mycoplasma pneumoniae in the upper respiratory tract of symptomatic and asymptomatic children: an observational study. PLoS Med 10:e1001444. doi: 10.1371/journal.pmed.1001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. 2007. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Kurokawa K, Ryu KH, Ichikawa R, Masuda A, Kim MS, Lee H, Chae JH, Shimizu T, Saitoh T, Kuwano K, Akira S, Dohmae N, Nakayama H, Lee BL. 2012. Novel bacterial lipoprotein structures conserved in low-GC content Gram-positive bacteria are recognized by Toll-like receptor 2. J Biol Chem 287:13170–13181. doi: 10.1074/jbc.M111.292235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, Van Heeckeren A, Prince A. 2004. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol 30:777–783. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 41.Ranoa DR, Kelley SL, Tapping RI. 2013. Human lipopolysaccharide-binding protein (LBP) and CD14 independently deliver triacylated lipoproteins to Toll-like receptor 1 (TLR1) and TLR2 and enhance formation of the ternary signaling complex. J Biol Chem 288:9729–9741. doi: 10.1074/jbc.M113.453266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kurata S, Osaki T, Yonezawa H, Arae K, Taguchi H, Kamiya S. 2014. Role of IL-17A and IL-10 in the antigen induced inflammation model by Mycoplasma pneumoniae. BMC Microbiol 14:156. doi: 10.1186/1471-2180-14-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talbot TR, Hartert TV, Mitchel E, Halasa NB, Arbogast PG, Poehling KA, Schaffner W, Craig AS, Griffin MR. 2005. Asthma as a risk factor for invasive pneumococcal disease. N Engl J Med 352:2082–2090. doi: 10.1056/NEJMoa044113. [DOI] [PubMed] [Google Scholar]
- 44.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, Contoli M, Sanderson G, Kon OM, Papi A, Jeffery PK, Stanciu LA, Johnston SL. 2008. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc Natl Acad Sci USA 105:13562–13567. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodhankar S, Sun X, Woolard MD, Simecka JW. 2010. Interferon gamma and interleukin 4 have contrasting effects on immunopathology and the development of protective adaptive immunity against mycoplasma respiratory disease. J Infect Dis 202:39–51. doi: 10.1086/653121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Q, Martin RJ, Lafasto S, Efaw BJ, Rino JG, Harbeck RJ, Chu HW. 2008. Toll-like receptor 2 down-regulation in established mouse allergic lungs contributes to decreased mycoplasma clearance. Am J Respir Crit Care Med 177:720–729. doi: 10.1164/rccm.200709-1387OC. [DOI] [PubMed] [Google Scholar]
- 47.Biscardi S, Lorrot M, Marc E, Moulin F, Boutonnat-Faucher B, Heilbronner C, Iniguez JL, Chaussain M, Nicand E, Raymond J, Gendrel D. 2004. Mycoplasma pneumoniae and asthma in children. Clin Infect Dis 38:1341–1346. doi: 10.1086/392498. [DOI] [PubMed] [Google Scholar]
- 48.Sluijter M, Hoogenboezem T, Hartwig NG, Vink C. 2008. The Mycoplasma pneumoniae MPN229 gene encodes a protein that selectively binds single-stranded DNA and stimulates recombinase A-mediated DNA strand exchange. BMC Microbiol 8:167. doi: 10.1186/1471-2180-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amatngalim GD, Schrumpf JA, Dishchekenian F, Mertens TCJ, Ninaber DK, van der Linden AC, Pilette C, Taube C, Hiemstra PS, van der Does AM. 2018. Aberrant epithelial differentiation by cigarette smoke dysregulates respiratory host defence. Eur Respir J 51:1701009. doi: 10.1183/13993003.01009-2017. [DOI] [PubMed] [Google Scholar]
- 50.Eenjes E, van Riet S, Kroon AA, Slats AM, Khedoe P, Boerema-de Munck A, Buscop-van Kempen M, Ninaber DK, Reiss IKM, Clevers H, Rottier RJ, Hiemstra PS. 2021. Disease modeling following organoid-based expansion of airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 321:L775–L786. doi: 10.1152/ajplung.00234.2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download iai.00129-22-s0001.pdf, PDF file, 1.1 MB (1.1MB, pdf)