

Abstract
Extraintestinal manifestations are common in inflammatory bowel disease (IBD) and involve several organs, including the kidney. However, the mechanisms responsible for renal manifestation in IBD are not known. Here, we show that the Wnt-LRP5/6-signaling pathway in macrophages plays a critical role in regulating colitis-associated systemic inflammation and renal injury in a murine dextran sodium sulfate (DSS)-induced colitis model. Conditional deletion of the Wnt coreceptors low-density lipoprotein receptor-related protein 5 and 6 (LRP5/6) in macrophages in mice results in enhanced susceptibility to DSS-colitis-induced systemic inflammation and acute kidney injury (AKI). Furthermore, our studies show that aggravated colitis-associated systemic inflammation and AKI observed in LRP5/6LyzM mice are due to increased bacterial translocation to extraintestinal sites and microbiota-dependent increased proinflammatory cytokine levels in the kidney. Conversely, depletion of the gut microbiota mitigated colitis-associated systemic inflammation and AKI in LRP5/6LysM mice. Mechanistically, LRP5/6-deficient macrophages were hyperresponsive to TLR ligands and produced higher levels of proinflammatory cytokines which are associated with increased activation of MAPKs. These results reveal how the Wnt-LRP5/6 signaling in macrophages controls colitis-induced systemic inflammation and AKI.
Introduction
Extraintestinal manifestations are common in patients with inflammatory bowel disease (IBD) and involve several organs such as the kidney, liver, and central nervous system (CNS) (1–3). Increased intestinal permeability and mucosal barrier damage predispose IBD patients to enterogenous infections and the development of sepsis (4–7). Renal complications occur in 4–23% of patients with IBD, including kidney tubular damage, amyloidosis, tubulointerstitial nephritis, and glomerulonephritis(4–7). Defects in the mucosal barrier and increased intestinal permeability potentially allow the translocation of commensal bacteria, endotoxin, bacterial DNA, and uremic toxins from the gut lumen to systemic sites resulting in systemic inflammation-associated kidney injury (4–10). However, the mechanisms underlying the links between intestinal injury and changes in the kidney are mainly unknown. Using a sodium dextran sulfate (DSS)-induced colitis model, previous studies showed that acute intestinal injury leads to systemic inflammation-associated tissue injury (11–20). Moreover, inflammation-associated intestinal injury and acute kidney injury (AKI) during colitis is accompanied by increased immune cell infiltration, particularly inflammatory myeloid cells (21, 22). Additionally, macrophages have emerged among myeloid cells as principal inflammatory effectors in the pathogenesis of colitis-associated AKI (21, 22). These antigen-presenting cells (APCs) also play a pivotal role in suppressing inflammation, wound healing, and tissue repair (21, 22). However, the molecular pathways that program these cells to a regulatory state rather than an inflammatory state are unknown.
The Wnt signaling cascade plays an essential role in immune cell development and differentiation (23–25). In addition, aberrant Wnt signaling occurs in several inflammatory diseases, including IBD (24, 25). Past studies have shown that the intestinal APCs are significant producers of Wnt ligands under steady-state and inflammatory conditions (26–31). Wnt ligands in the gut environment act on immune cells and shape mucosal immune responses to commensal microbiota (26–31). Wnt signaling can be broadly classified into the canonical Wnt-β-catenin pathway and the non-canonical Wnt pathway independent of β-catenin (24, 25). The low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) are critical mediators of the canonical Wnt-β-catenin signaling pathway (32, 33). In addition, previous studies have shown that the canonical Wnt pathway in intestinal APCs plays a pivotal role in mediating mucosal tolerance and suppressing inflammatory response against commensal microbiota (31, 34). However, very little is known about the role of this pathway in regulating colitis-associated systemic inflammation and kidney injury. Therefore, we hypothesized that canonical Wnt signaling in macrophages is critical for suppressing colitis-associated systemic inflammation and kidney injury.
Here, we demonstrate that Wnt-LRP5/6-signaling in macrophages suppresses colitis-associated systemic inflammation and renal injury in the murine model of colitis. In mice, conditional deletion of the Wnt coreceptors LRP5 and LRP6 in myeloid cells enhances susceptibility to colitis-associated systemic inflammation and AKI. This is due to increased bacterial translocation to extraintestinal sites, and microbiota-dependent increased proinflammatory cytokine levels in the kidney. This condition could be improved in LRP5/6LysM mice by depleting the gut microbiota, indicating the importance of LRP5/6-signaling in regulating colitis-associated systemic inflammation and AKI. Mechanistically, LRP5/6-deficient macrophages were hyperresponsive to TLR ligands and produced higher levels of proinflammatory cytokines which are associated with increased activation of MAPKs. These results reveal how the Wnt-LRP5/6 signaling in macrophages controls systemic inflammation-associated kidney injury during colitis.
Materials and Methods
Mice
C57BL/6 male mice of 6 to 12 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME. LRP5 floxed (LRP5FL) mice and LRP6 floxed (LRP6FL) mice were crossbred to generate homozygous LRP5/6 floxed (LRP5/6FL) mice as previously described (35–37). In addition, LRP5/6FL mice were crossed to transgenic mice expressing Cre recombinase under the control of the LysM promoter (Jackson Laboratories) to generate mice in which LRP5/6 (LRP5/6LysM) were deficient in myeloid cells. Successful Cre-mediated deletion was confirmed by polymerase chain reaction (PCR) and protein expression analyses as in our previous studies (36, 37). All experiments were carried out with age-matched littermates unless specified otherwise. All mice were housed under specific pathogen-free conditions at Augusta University, with animal care protocols approved by the Institutional Animal Care and Use Committee.
Reagents and Antibodies.
Antibodies against mouse CD45 (30-F11), CD11c (N418), CD11b (M1/70), GR1 (RB6-8C5), Ly6G (1A8), Ly6C (HK1.4), CD64 (X54-5/7.1), F4/80 (BM8), and I-Ab (AF6-120.1) were purchased from BioLegend. p-p38, p38 MAPK, p-JNK, JNK, p-ERK, ERK, LRP5, and LRP6 antibodies were obtained from Cell Signaling Technology, and p-LRP5 and p-LRP6 antibodies were obtained from Thermo Fisher Scientific.
Induction of colonic inflammation
Colonic inflammation was induced as previously described (38). Briefly, mice were subjected to one cycle of DSS treatment, whereby mice were given 3.0% DSS (36–50 kDa) in their drinking water (at a dose as indicated in Results) for 8 days followed by normal drinking water. Some mice were subjected to antibiotics treatment before initiation of DSS administration. Mice were monitored for weight change, diarrhea, and rectal bleeding (38, 39).
Antibiotic-treatment of mice
Antibiotic treatment of mice was performed as described in our previous studies (30, 39). In brief, LRP5/6FL and LRP5/6LysM mice were fed with an antibiotic cocktail (1g/L ampicillin, 1g/Lmetronidazole, 1g/L neomycin sulfate, and 0.5g/L vancomycin) in drinking water for two weeks before DSS treatment. All antibiotics were purchased from Sigma-Aldrich.
Ex-vivo colon culture and ELISAs
Whole colons were excised and flushed with PBS containing penicillin and streptomycin. Approximately 1 cm-long section of the ascending colon was excised, opened longitudinally, and washed three times with sterile HBSS containing penicillin and streptomycin. Colon sections were then placed into culture in complete RPMI media (2% FBS, L-glutamine, penicillin, streptomycin, and tetracycline) and cultured for two days at 37°C with 5% CO2. Supernatants were then collected, and cytokine concentrations were determined by ELISA. IL-6, IL-10, TNF-α, IL-1α and IL- 1β were quantitated using ELISA kits procured from BioLegend.
Isolation of macrophages
CD11b+ APCs were purified from the spleen, kidney, or colon as previously described (31, 37). In brief, the spleen or kidney was cut into small fragments and then digested with collagenase type 4 (1 mg/ml) in complete DMEM plus 2% FBS for 30 min at 37 °C. Cells were washed twice and enriched for CD11b+ APCs with the CD11b-specific microbeads by elution via MACS LS columns (Miltenyi Biotec), and then FACS sorted for macrophages (CD45+I Ab+CD11b+F4/80+CD64+). After sorting, macrophages (105) were cultured in 0.2 ml RPMI 1640 complete medium in 96-well round-bottom plates for 48 h. In some experiments isolated splenic macrophages (106 cells/ml) were cultured for 24 h with Escherichia coli LPS (5 μg/ml), Pam-3-cys (0.5 μg/ml) or CpG dinucleotides (5 μg/ml). Cell culture supernatants were analyzed for indicated cytokine production by ELISA.
Measurement of intestinal permeability
Intestinal permeability assay was performed as described in our previous studies (31). In brief, mice were given fluorescein isothiocyanate (FITC)-dextran (4 kDa) by oral gavage at a dose of 0.5 mg/g of body weight. Mice were bled four hours later, and FITC-dextran was quantified in the serum via a fluorescence spectrophotometer.
Myeloperoxidase (MPO) activity measurement
Myeloperoxidase (MPO) activity was performed as described in our previous studies (31). Pieces of colon or kidney (100 mg weight) were homogenized in phosphate buffer (20 mM [pH 7.4]) and centrifuged. The pellet was re-suspended in phosphate buffer (50 mM [pH 6.0]) containing 0.5% hexadecyltrimethylammonium bromide (Sigma). The sample was freeze-thawed, sonicated, then warmed to 60°C for 2 hours before centrifuging. Redox reaction of 3,3′,5,5′-tetramethylbenzidine (Sigma) by the supernatant was used to determine myeloperoxidase (MPO) activity. The reaction was terminated with 2N HCl, and the absorbance was read at 450 nm.
Bacterial Translocation Evaluation
The animals were sacrificed on day 8 after DSS treatment and the collected mesenteric lymph nodes (MLNs), liver, spleen, and kidney were weighed and homogenized in 1 ml of sterile saline. Each tissue’s aliquots of the homogenate were plated onto TSB (Tryptic Soy Broth) agar plates (DIFCO, Detroit, Michigan, USA) and grown under aerobic conditions at 37°C for 24 hours. Representative colonies were expressed as colony-forming units per gram of organ tissue (CFU/g tissue).
Histopathology and immunohistochemistry
Sections (5 μm thick) from formalin-fixed and paraffin-embedded colons were placed onto glass slides. H&E-stained sections were blindly scored for severity of colonic inflammation as described previously (38, 39). The degree of inflammation was scored as follows: (0) no inflammation, (1) mild inflammation or prominent lymphoid aggregates, (2) moderate inflammation, (3) moderate inflammation associated with crypt loss, and (4) severe inflammation with crypt loss and ulceration. Crypt destruction was graded as follows: (0) no destruction, (1) 1%–33% of crypts destroyed, (2) 34%–66% of crypts destroyed, and (3) 67%–100% of crypts destroyed. The individual scores from inflammation and crypt damage were summed to derive histological scores for colonic inflammation (maximum score 7).
Kidney tissue was fixed in buffered 10% formalin for overnight and then embedded in paraffin wax. For assessment of renal injury, 5-μm sections were stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) followed by hematoxylin. H&E-and PASH stained sections. Renal tubular injury was assessed using a semi-quantitative scale and scoring them percentage of tubules showing epithelial cell necrosis, brush-border loss and apoptotic bodies as described previously (13, 14). Quantification of renal tubular injury was performed in a blinded manner and assigned a score of tubular injury using following scale: 0 = normal; 1 = 10–25%; 2 = 25–50%; 3= 51–75%; 4 = >75%. 10 fields at 20× magnification was examined, and the data averaged for each animal. Stained sections were photographed using an Olympus BX40 microscope (Olympus America, Melville, NY) on a bright-field setting fitted with a digital camera (Olympus DP12; Olympus America).
Assessment of kidney function and kidney injury
Renal function was assessed by serum creatinine measurements (cat no: DZ072B; Diazyme Laboratories) as described in our previous studies (13, 14). Serum neutrophil gelatinase-associated lipocalin (NGAL) was measured using ELISA (R&D Systems) following the manufacturer’s instructions.
Real-time PCR
Using the manufacturer’s protocol, total mRNA was isolated from colon, kidney, or the indicated cell type using the Omega Total RNA Kit. According to the manufacturer’s protocol, cDNA was generated using the RNA to cDNA Ecodry Premix Kit (Clontech). cDNA was used as a template for quantitative real-time PCR using SYBR Green Master Mix (Roche), and gene-specific primers (37). PCR analysis was performed using a MyiQ5 ICycler (BioRad). Gene expression was normalized relative to Gapdh.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software. An unpaired one-tailed Student’s t-test was used to determine statistical significance for mRNA expression levels, Treg percentages, and cytokines released by various cell types between different groups. A P value less than 0.05 (*) was significant, a P value less than 0.01 (**) very significant, and a P value less than 0.001 (***) extremely significant.
Results
Deletion of Wnt co-receptors LRP5 and LRP6 in myeloid cells enhances susceptibility to lethal colitis.
To delete LRP5 and LRP6 in macrophages (LRP5/6LysM), we crossed LRP5/6 floxed (LRP5/6Flox ) mice (35) with transgenic mice expressing LysM-cre (40). To confirm the efficiency of macrophage LRP5/6 deletion, we isolated macrophages from the colon and spleen of LRP5/6LysM and LRP5/6Flox mice and performed immunoblotting. As indicated in Supplemental Figure 1A, LRP5 and LRP6 protein levels were markedly reduced in LRP5/6LysM macrophages compared to the WT macrophages. We then challenged LRP5/6LysM and LRP5/6Flox mice with DSS and monitored body weight loss and recovery. Upon DSS administration, LRP5/6LysM mice showed more significant weight loss than LRP5/6Flox mice (Fig. 1A). Moreover, DSS treatment of LRP5/6LysM mice resulted in a significant reduction in the colon length than LRP5/6Flox mice (Fig. 1B). In addition, myeloperoxidase (MPO) activity, a hallmark of tissue inflammation, was markedly increased in the colons of LRP5/6LysM mice after DSS treatment (Fig. 1C). In line with these observations, histopathological analysis of colons of DSS-treated LRP5/6LysM mice showed extensive damage to the mucosa with epithelial erosion, loss of crypts, and increased infiltration of immune cells compared to the colons of DSS-treated LRP5/6Flox mice (Fig. 1D, E). However, colons from untreated LRP5/6Flox and LRP5/6LysM mice showed no morphological sign of damage or inflammation (data not shown). These results show that LRP5/6 deficiency in macrophages markedly delays functional recovery of mice in acute colitis.
FIGURE 1.
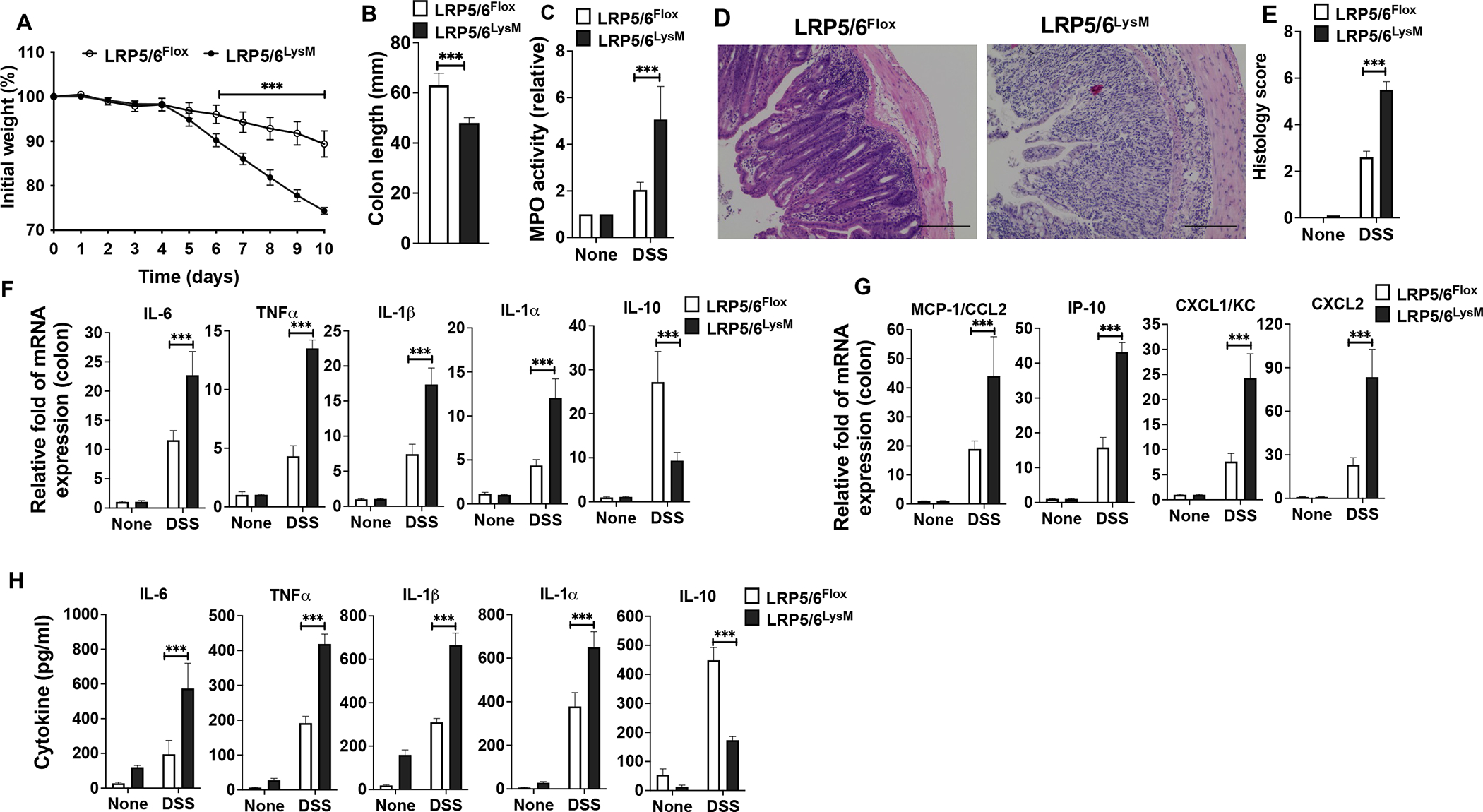
Myeloid cell-specific LRP5/6 ablation promotes colitis. LRP5/6Flox and LRP5/6LysM mice were treated with 3.0% DSS (36–50 kDa) in their drinking water for 8 days before returning to normal water, and, at day 10, the colons of mice were analyzed for inflammation. (A, B) Change in body weight and colon length of LRP5/6Flox and LRP5/6LysM mice. (C) Myeloperoxidase (MPO) activity in the colon. (D) Representative images of H&E-stained colonic sections from DSS-treated LRP5/6Flox and LRP5/6LysM mice (Scale bars, 100 μm). (E) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated LRP5/6Flox and LRP5/6LysM mice. (F, G) RNA was extracted from colons of untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice. The expression of indicated genes was quantified by qPCR. (H) Excised colon samples of untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice were cultured for 2 days ex vivo, and the indicated cytokine levels in the culture supernatants were quantified by ELISA. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. ***p<0.001
In the DSS model of intestinal inflammation, inflammatory cytokines produced by innate immune cells present in the gut microenvironment promote colitis and augment tissue injury (41). Thus, we analyzed the expression of immune regulatory and inflammatory factors that suppress or promote colonic inflammation. Colons of DSS-treated LRP5/6LysM mice expressed higher levels of inflammatory cytokines IL-6, TNF-α, IL-1β, and IL-1α, and inflammatory chemokines MCP1, IP-10, KC, and CXCL2 compared to colons of DSS-treated LRP5/6Flox mice (Fig. 1F, G). In contrast, the colons of DSS-treated LRP5/6LysM mice expressed lower levels of immune-regulatory cytokine IL-10 compared to those of colons of LRP5/6Flox mice (Fig. 1F). Consistent with these observations, colon explant cultures showed that colons of DSS-treated LRP5/6LysM mice produced higher inflammatory cytokines and lower levels of IL-10 than colons of LRP5/6Flox mice (Fig. 1H). Collectively, these findings demonstrate an imbalance in the production of anti-inflammatory molecules versus inflammatory molecules that favors increased susceptibility of LRP5/6LysM mice to DSS-induced colitis and inflammation-associated intestinal injury.
LRP5/6LysM mice show exacerbated systemic inflammation and increased bacterial translocation to systemic sites during colitis.
Intestinal epithelial barrier dysfunction and increased permeability have been linked to systemic inflammation in many diseases, including IBD (4–10). Thus, we measured epithelial barrier function by orally gavaging control and DSS-treated LRP5/6Flox and LRP5/6LysM mice with the commonly used permeability marker (FITC-dextran) and subsequently measuring their levels in the serum. Consistent with enhanced gut inflammation and delayed recovery, we also observed increased FITC-dextran in the serum of DSS-treated LRP5/6LysM mice, indicating severe intestinal injury and increased permeability (Fig. 2A). Next, we asked whether severe intestinal injury and increased permeability observed in LRP5/6LysM mice accelerate systemic inflammation. Systemic inflammation is often assessed by measuring the splenic weight and quantitating the levels of inflammatory cytokines in the serum. On day 8 after DSS treatment, LRP5/6LysM mice showed a marked increase in spleen weight compared to the LRP5/6Flox mice (Fig. 2B). Furthermore, DSS treatment of LRP5/6LysM mice resulted in a marked increase in serum proinflammatory cytokines such as IL-6, TNF-α, IL-1β, and IL-1α and decreased levels of IL-10 (Fig. 2C). Next, we asked whether exacerbated systemic inflammation observed in LRP5/6LysM mice was due to increased bacterial translocation to extraintestinal sites. Therefore, we quantified the serum endotoxin levels and bacterial load in systemic sites. As a result, we observed a marked increase in the serum endotoxin levels of LRP5/6LysM mice compared with LRP5/6Flox mice after DSS treatment (Fig. 2D). In line with this observation, we found that LRP5/6LysM mice exhibited increased bacterial burden in the MLNs, spleen, liver, and kidney after DSS treatment (Fig. 2E). These results indicate that LRP5/6 deficiency in myeloid cells exacerbates the systemic inflammation with increased bacterial translocation to systemic sites during colitis.
FIGURE 2.
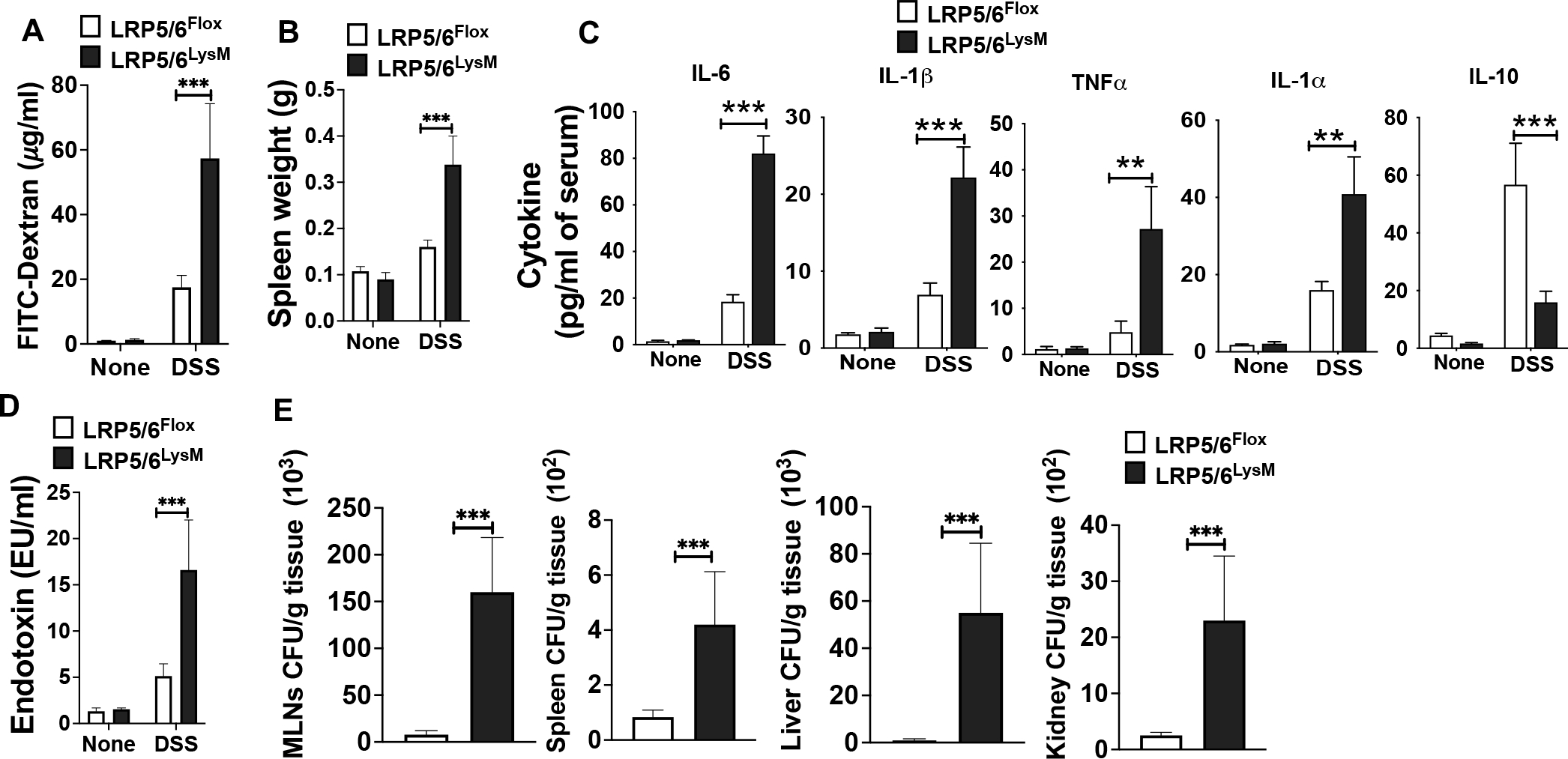
Increased intestinal permeability, systemic inflammation and tissue bacterial burden in LRP5/6LysM mice during DSS-induced colitis. (A) Serum FITC-dextran levels, (B) splenic weight and (C, D) serum cytokine and endotoxin levels of untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice on day 8. (E) Cultured bacterial colony-forming units (CFU) from the indicated tissue of DSS-treated LRP5/6Flox and LRP5/6LysM mice on day 8. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. **p<0.01; ***p<0.001.
LRP5/6LysM mice are highly susceptible to systemic inflammation-induced AKI during colitis.
Excessive systemic inflammatory responses to bacterial endotoxin and infections are major triggers of AKI (42, 43). Thus, we asked whether exacerbated systemic inflammation seen in LRP5/6LysM mice leads to renal injury and affects kidney function. We measured serum NGAL and kidney KIM-1 expression, two widely used biomarkers of AKI, and its severity (44, 45). The NGAL levels in the serum and KIM-1 expression in the kidney were markedly increased in LRP5/6LysM mice compared with the LRP5/6Flox mice, indicating inflammation-associated AKI (Fig. 3A, B). Furthermore, serum creatinine levels were used to assess renal function (45), and its levels were enhanced in DSS-treated LRP5/6LysM mice compared with DSS-treated LRP5/6Flox mice (Fig. 3C). Additionally, MPO activity, a marker for tissue inflammation, was markedly increased in the kidney of LRP5/6LysM mice compared with LRP5/6Flox mice after DSS treatment (Fig. 3D). In line with these observations, kidney histology revealed increased structural changes in the kidney, such as increased tubular injury, tubular epithelial vacuolation, loss of epithelial cells, and dilation of the tubular lumen in LRP5/6LysM mice compared to LRP5/6Flox mice (Fig. 3E). Quantification data further confirm that tubular injury score was significantly increased in LRP5/6LysM mice compared to LRP5/6Flox mice (Fig. 3F).
FIGURE 3.
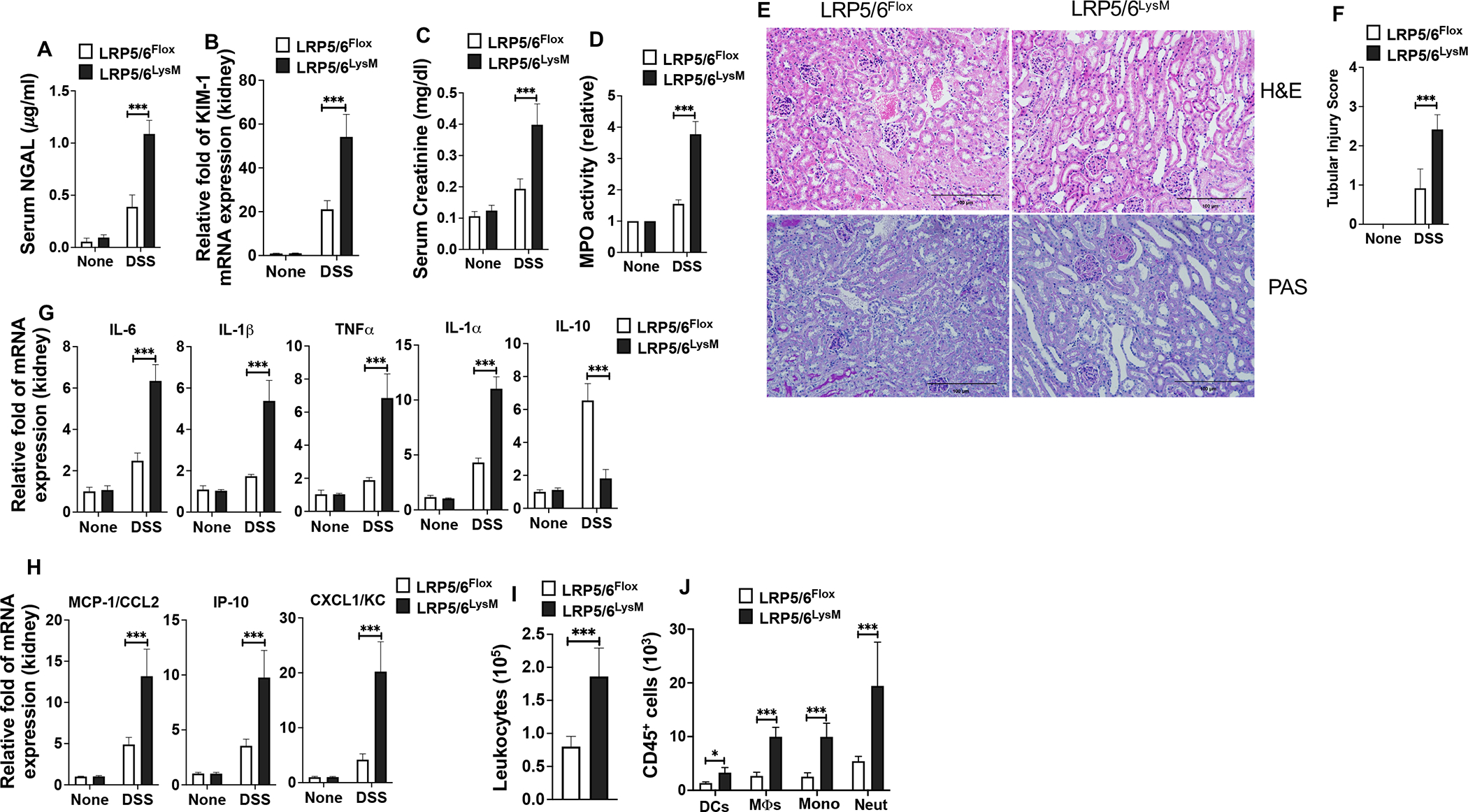
LRP5/6 deficiency in myeloid cells exacerbates DSS-colitis-associated acute kidney injury. (A) Serum NGAL levels, (B) KIM1 mRNA expression in the kidney, (C) serum creatinine levels, and (D) myeloperoxidase (MPO) activity in the kidney of untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice on day 8. (E) Representative images of H&E- or PAS-stained kidney sections from DSS-treated LRP5/6Flox and LRP5/6LysM mice (Scale bars, 100 μm). H&E- and PAS-stained sections show increased severity of the tubular injury, tubular epithelial vacuolation, dilation of the tubular lumen, and immune cell infiltration in DSS-treated LRP5/6LysM mice. (F) Tubular injury score of kidneys was graded following analysis of H&E- and PAS-stained cross-sections of kidneys of DSS-treated LRP5/6Flox and LRP5/6LysM mice. (G, H) RNA was extracted from kidneys of untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice. The expression of indicated genes was quantified by qPCR. (I, J) Total number of leukocytes (CD45+), DCs (CD45hi MHC IIhi CD11c+ CD64−), MΦs (CD45hi MHC IIhi CD11c−CD11b+ CD64+), monocytes (CD45hi MHC II− CD11c−CD11b+ Ly6Chi Ly6GLow) and neutrophils (CD45hi MHC II− CD11c−CD11b+ Ly6Clow Ly6Ghi) from the kidney of DSS-treated LRP5/6Flox and LRP5/6LysM mice on day 8 analyzed by flow cytometry. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. *p<0.05; ***p<0.001.
Increased levels of inflammatory factors cause stress and injury to kidney endothelial, tubular, and glomerular cells. Based on the above results, we asked whether increased renal injury seen in LRP5/6LysM mice is due to increased levels of inflammatory factors and enhanced infiltration of inflammatory cells into the kidney. We observed a marked increase in the mRNA levels of proinflammatory cytokines such as IL-6, TNF-α, IL-1β, and IL-1α and proinflammatory chemokines such as IP-10, KC, and MCP-1 in the kidney of LRP5/6LysM mice during colitis (Fig. 3G, H). In contrast, we observed a marked decrease in the expression levels of decreased expression levels of IL-10 in the kidney of LRP5/6LysM mice during colitis (Fig. 3G). Furthermore, increased levels of inflammatory factors were associated with enhanced infiltration of leukocytes such as DCs, monocytes, macrophages, and neutrophils in the kidney of DSS-treated LRP5/6LysM mice (Fig. 3I, J). Therefore, LRP5/6 deletion in macrophages led to increased renal inflammation and injury following colitis. These results indicate that the Wnt-LRP5/6 pathway plays an essential role in DSS-induced kidney injury.
LRP5/6 deficiency in macrophages leads to increased expression of inflammatory cytokines and activation of MAPKs during colitis.
Next, we investigated the mechanisms of systemic and renal inflammation regulation by LRP5/6 following colitis. Tissue infiltrating macrophages play a critical role in regulating inflammation-associated AKI (21, 22). Our previous studies have shown that splenic DCs and macrophages express low levels of Wnt ligands under homeostatic conditions, and the canonical Wnt/LRP5/6 pathway is not active in these APCs (30, 37). Since LRP5/6LysM mice are highly susceptible to colitis-associated systemic and renal inflammation, we asked whether colitis leads to the expression of Wnt ligands and subsequent activation of the Wnt-LRP5/6 pathway in macrophages in the spleen and kidney. In line with previous studies, macrophages isolated from the spleen and kidney of LRP5/6Flox mice expressed markedly lower levels of Wnt ligands without DSS treatment (Fig. 4A, B). However, we noted a marked increase in the expression of several Wnt ligands in splenic macrophages in response to DSS treatment (Fig. 4A). Like in the spleen, DSS treatment also resulted in a marked increase in the expression of Wnt ligands in the kidney macrophages (Fig. 4B). Next, we assessed if an increased level of Wnt ligands leads to the activation of LRP5/6 in macrophages using antibodies that recognizes the phosphorylated (active) form of LRP5 or LRP6. Splenic macrophages isolated from LRP5/6Flox (WT) mice without DSS treatment showed low/undetectable levels of active (phosphorylated) LRP5 or LRP6 (Supplementary Fig. 1B). However, splenic macrophages from LRP5/6Flox mice treated with DSS showed a marked increase in the phosphorylated form of LRP5 and LRP6 (Supplementary Fig. 1B). Collectively, these observations show intestinal inflammation leads to the expression of Wnt ligands and activation of the LRP5/6 pathway in macrophages in systemic sites.
FIGURE 4.
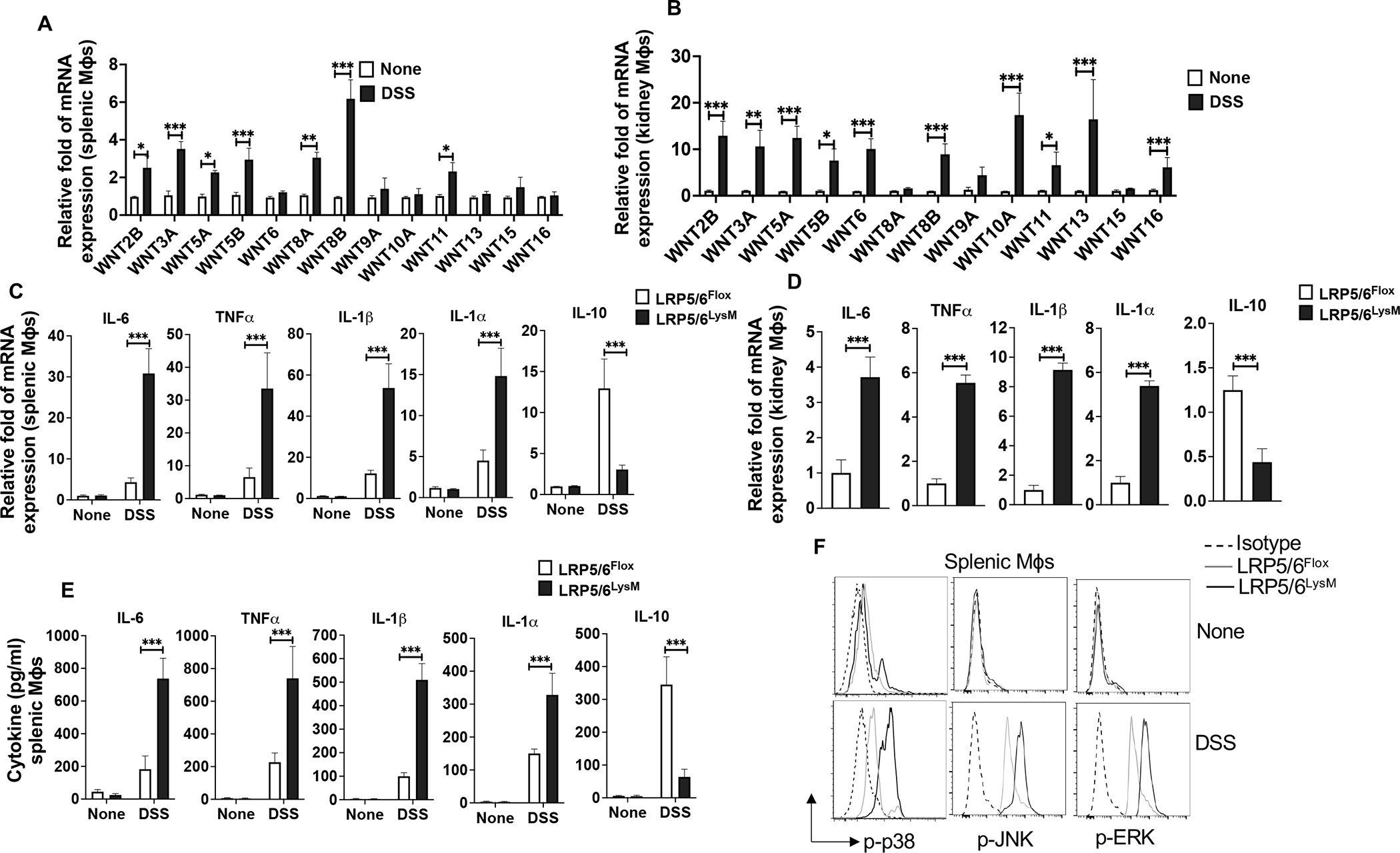
LRP5/6 signaling in macrophages regulates the expression of proinflammatory cytokines and activation of MAPKs. (A, B) Quantitative real-time PCR analysis of the expression of Wnt ligands in splenic and kidney macrophages (Mϕs) isolated from LRP5/6Flox mice treated with or without DSS on day 8. (C, D) Quantitative real-time PCR analysis of the mRNA of indicated genes in splenic and kidney macrophages from DSS-treated LRP5/6Flox and LRP5/6LysM mice (day 8). (E) Sorted splenic macrophages from untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice were cultured for 2 days ex vivo and cytokine levels in the culture supernatants were quantified by ELISA. (F) Representative histogram of phosphorylated p38, JNK, and ERK MAPKs in splenic macrophages isolated from untreated and DSS-treated LRP5/6Flox and LRP5/6LysM mice (day 8). Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. *p<0.05; **p<0.01; ***p<0.001.
Based on the above results, we hypothesized activating canonical Wnt-mediated signaling in macrophages in systemic sites following colitis suppresses systemic inflammation-associated AKI. Since macrophage-mediated inflammation has been implicated in AKI (21, 22), we analyzed the expression levels of inflammatory and anti-inflammatory factors in splenic and renal macrophages of LRP5/6Flox and LRP5/6LysM mice during colitis. Splenic macrophages isolated from LRP5/6LysM mice expressed markedly higher mRNA levels of inflammatory factors IL-6, TNF-α, IL-1β, and IL-1α and decreased levels of immune regulatory factors IL-10 compared with splenic macrophages from LRP5/6Flox mice following DSS treatment (Fig 4C). We also observed similar increases in the expression of proinflammatory factors and decreased expression of IL-10 in renal macrophages isolated from the LRP5/6LysM mice following DSS treatment (Fig. 4D). Consistent with these observations, splenic macrophages from LRP5/6LysM mice cultured ex vivo produced higher levels of IL-6, TNF-α, IL-1β, and IL-1α and lower levels of IL-10 compared to macrophages from LRP5/6Flox mice (Fig. 4E). Thus, the increased proinflammatory cytokines and decreased IL-10 expression suggest that LPR5/6-deficient macrophages exhibit a proinflammatory phenotype.
Mitogen-activated protein kinase (MAPK) pathways are critical downstream mediators of toll-like receptor (TLR) signaling, and their activation is critical for the expression of inflammatory factors (46, 47). Thus, we assessed the activation status of p38α, ERK, and JNK MAPKs in splenic and intestinal macrophages in LRP5/6LysM and LRP5/6Flox mice during colitis by intracellular flow cytometry. Macrophages isolated from the spleens of DSS-treated LRP5/6LysM mice showed a marked increase in the phosphorylated (active) form of p38α, ERK, and JNK MAPKs compared with macrophages isolated from DSS-treated LRP5/6Flox mice during colitis (Fig. 4F). Likewise, the activity of p38α, ERK, and JNK MAPKs was markedly elevated in colonic macrophages isolated from DSS-treated LRP5/6LysM mice compared to colonic macrophages isolated from DSS-treated LRP5/6Flox mice (Supplementary Fig. 1C). Thus, our data demonstrate that LRP5/6-mediated signaling in macrophages limits the expression of proinflammatory factors and induces anti-inflammatory factors by regulating the activation of MAPKs during colitis.
LRP5/6-deficient macrophages are hyperresponsive to TLR ligand.
Macrophages express several TLRs, which are critical in sensing pathogen-associated molecular patterns (PAMPs) associated with invading pathogens (48). Engagement of such receptors activates macrophages and triggers the production of proinflammatory cytokines and chemokines that are critical mediators of tissue damage. Thus, we asked whether the increased expression of inflammatory factors by LRP5/6-deficient splenic macrophages is due to hyper-responsiveness to microbial ligands and microbiota. We also asked whether Wnt ligand expression in macrophages is dependent on TLRs. Thus, we treated WT splenic macrophages with TLR2 (Pam(3)Cys), TLR4 (LPS), or TLR9 (CpG) ligands and evaluated the expression levels of Wnt ligands after 18 hours. Splenic macrophages expressed markedly lower levels of Wnt ligands without treatment with TLR ligands (Fig. 5A). However, we noted a marked increase in the expression of several Wnt ligands in response to Pam(3)Cys, LPS, and CpG treatment (Fig. 5A). Next, we asked whether LRP5/6 deficiency in macrophages alters TLR activation response and subsequent inflammatory cytokines production. LPR5/6-deficient macrophages produced markedly higher levels of IL-6, TNF-α, IL-1β, and IL-1α than WT controls in response to TLR2, TLR4, and TLR9 stimulation (Fig. 5B).
FIGURE 5. Wnt-LRP5/6 signaling in macrophages regulates responsiveness to TLR ligands.
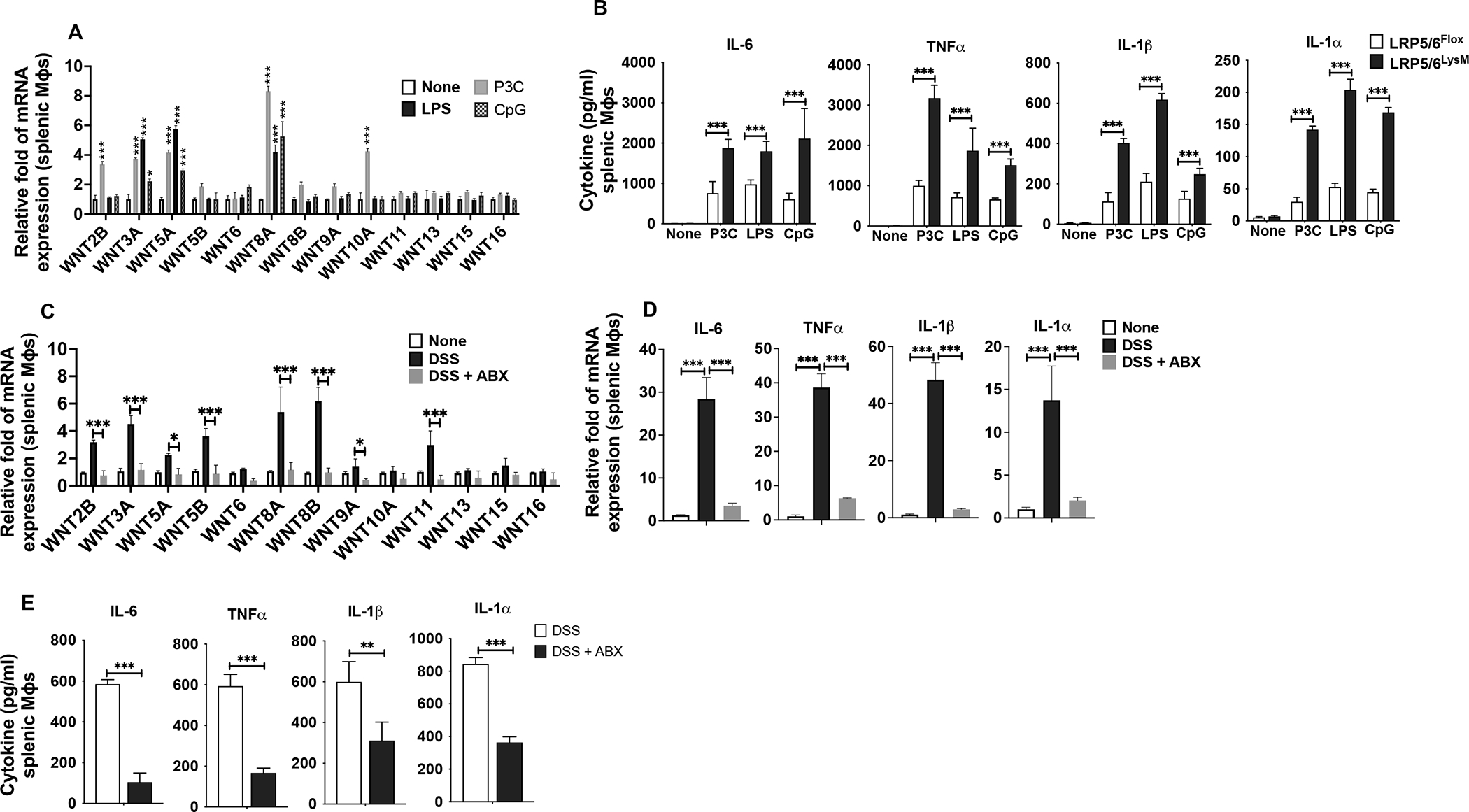
(A) Quantitative real-time PCR analysis of Wnt ligand expression in splenic macrophages (Mϕs) isolated from LRP5/6Flox mice treated with TLR2, TLR4, or TLR9 ligands for 24h and (B) Splenic macrophages (Mϕs) isolated from LRP5/6Flox and LRP5/6LysM mice treated with TLR2, TLR4 or TLR9 ligands for 24h. Cytokine levels in the culture supernatants were quantified by ELISA. (C, D) Quantitative real-time PCR analysis of the mRNA of indicated genes in splenic macrophages from LRP5/6LysM mice without any treatment (none) or treated with or without antibiotics (ABX) and then with DSS. (E) Splenic macrophages from LRP5/6LysM mice described above were cultured for 2 days ex vivo. Cytokine levels in the culture supernatants were quantified by ELISA. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. *p<0.05; **p<0.01; ***p<0.001.
To expand upon these studies, we performed microbiota depletion studies in mice to assess the expression of Wnt ligands and proinflammatory factors in splenic macrophages in response to DSS treatment. Depletion of commensal microbiota in WT mice markedly reduced the expression of Wnt ligands in splenic macrophages during colitis, suggesting that Wnt ligand expression in response to commensal microbiota translocation to systemic sites (Fig. 5C). Next, we assessed the expression of inflammatory factors in splenic macrophages isolated from antibiotic-treated LRP5/6LysM mice.
Splenic macrophages from the antibiotic-treated LRP5/6LysM mice expressed markedly lower levels of inflammatory factors than splenic macrophages from LRP5/6LysM mice without treatment during colitis (Fig. 5D). Besides, LRP5/6-deficient splenic macrophages also produced lower levels of proinflammatory cytokines IL-6, TNF-α, IL-1β, and IL-1α upon commensal depletion (Fig. 5E). These observations suggest that TLR activation induces the expression of Wnt ligands, and LRP5/6 deficiency in macrophages alters the responsiveness to TLR ligands.
The depletion of gut microbiota in LRP5/6LysM mice attenuates colitis-associated systemic inflammation.
The gut microbiota plays an essential role in regulating intestinal and systemic immunity (49–51). Thus, we asked if increased colitis-associated systemic inflammation and renal injury observed in LRP5/6LysM mice are due to increased bacterial translocation to the systemic sites during colitis. Depletion of commensal microbiota markedly decreased the DSS-induced weight loss, shortening of colon and intestinal permeability in LRP5/6LysM mice (Fig. 6A–C). Antibiotics treatment also significantly decreased colonic MPO activity in LRP5/6LysM mice, suggesting reduced infiltration of inflammatory cells (Fig. 6D). Furthermore, histological analysis of the colon showed that antibiotic treatment significantly decreased injury in LRP5/6LysM mice in response to DSS compared to mice that received no antibiotics (Fig. 6E, F). In line with these observations, colons of antibiotic-treated LRP5/6LysM mice expressed markedly lower levels of inflammatory cytokines such as IL-6, TNF-α, IL-1β, and IL-1α in response to DSS compared to mice that were not treated with antibiotics (Fig. 6G). Next, we assessed the effect of commensal depletion on systemic inflammation upon DSS treatment. Depletion of commensal microbiota resulted in a marked decrease in splenic weight in DSS-treated LRP5/6LysM mice compared to control mice without antibiotic treatment (Fig. 6H). Antibiotics treatment also significantly decreased serum endotoxin levels and proinflammatory cytokine levels in LRP5/6LysM mice (Fig. 6I, J). These data clearly show that exacerbated colitis-associated systemic inflammation in LRP5/6LysM mice is due to intestinal commensal microbiota.
FIGURE 6. Exacerbated DSS-induced colitis and systemic inflammation in LRP5/6LysM mice are driven by the gut microbiota.
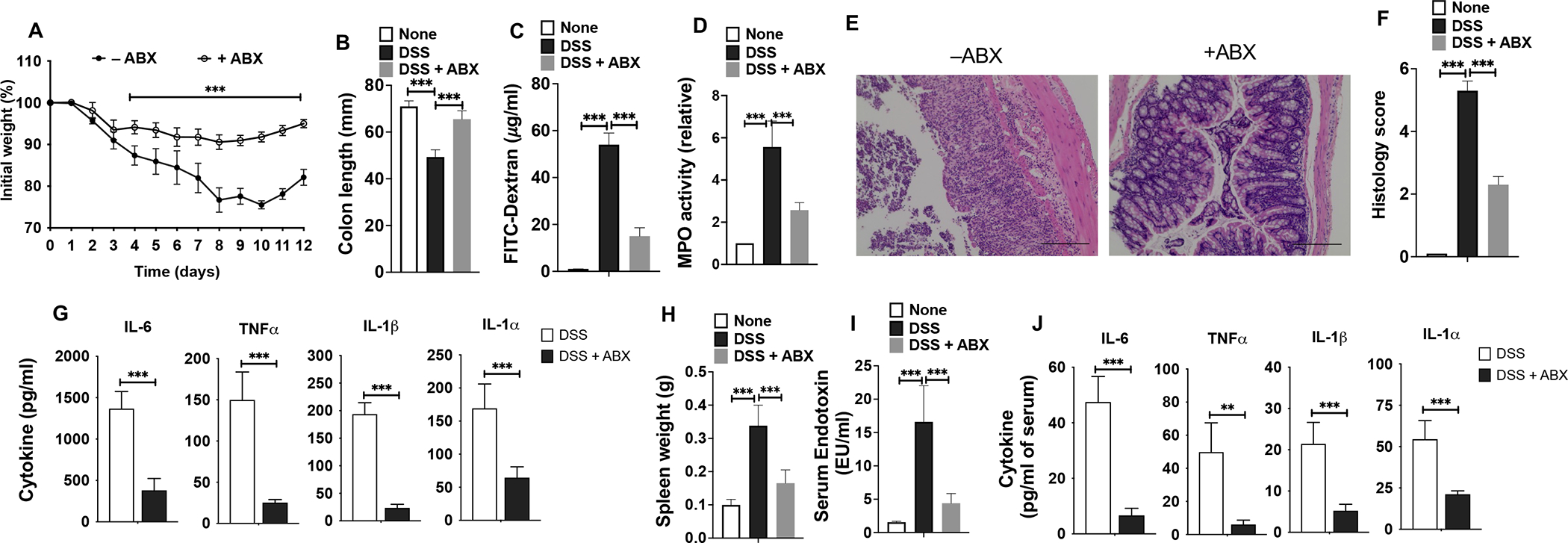
LRP5/6LysM mice were treated with (+ABX) or without antibiotic (−ABX) in addition to receiving DSS as described in the Methods. (A, B) Change in body weight and colon length of LRP5/6LysM mice. (C) Commensal bacteria-depleted LRP5/6LysM mice were fed with FITC-dextran on day 10 after DSS treatment, and 4 hr later, FITC-dextran was quantified in serum. (D) Myeloperoxidase (MPO) activity in the colon. (E) Representative images of H&E-stained colonic sections from DSS-treated LRP5/6LysM mice (Scale bars, 100 μm). (F) Histopathological score (inflammation + epithelial damage) of colons was graded following analysis of H&E-stained cross-sections of colons of DSS-treated LRP5/6LysM mice. (G) Excised colon samples of untreated and DSS-treated LRP5/6LysM mice were cultured for 2 days ex vivo, and the indicated cytokine levels in the culture supernatants were quantified by ELISA. (H) Splenic weight, (I) Endotoxin, and (J) Cytokine levels in the serum of commensal bacteria-depleted LRP5/6LysM mice treated with DSS. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. **p<0.01; ***p<0.001.
Depleting intestinal commensal microbiota mitigates colitis-associated AKI in LRP5/6LysM mice.
Based on the above results, we asked whether commensal depletion could ameliorate inflammation-associated AKI in LRP5/6LysM mice during colitis. Depletion of commensal bacteria significantly preserved renal function in LRP5/6LysM mice, as indicated by lower serum creatinine and NGAL levels and markedly decreased KIM-1 mRNA levels (Fig. 7A–C). Also, the improved renal function in commensal bacteria-depleted LRP5/6LysM mice was also accompanied by significantly reduced MPO activity suggesting reduced inflammation (Fig. 7D). In line with these observations, histological examination of LRP5/6LysM kidneys show that the renal structural integrity was better maintained in antibiotic-treated mice, as shown by reduced tubular damage and tubular dilation, brash border loss and tubular damage (Fig. 7E). Besides, the tubular injury score was significantly decreased in commensal depleted LRP5/6LysM mice (Fig. 7F). Importantly, commensal depletion resulted in a marked reduction in the renal inflammatory factors in LRP5/6LysM mice as reflected by decreased mRNA levels of IL-6, TNF-α, IL-1β, IL-1α, MCP-1, IP-10, and KC (Fig. 7G, H) and decreased infiltration of inflammatory leukocytes such as DCs, monocytes, macrophages, and neutrophils in the kidney (Fig. 7I, J). Taken together, these data clearly show that depletion of the microbiota protects LRP5/6LysM mice against systemic inflammation-associated renal injury during colitis.
FIGURE 7. Gut microbiota depletion protects LRP5/6LysM mice from colitis-associated kidney inflammation and injury.
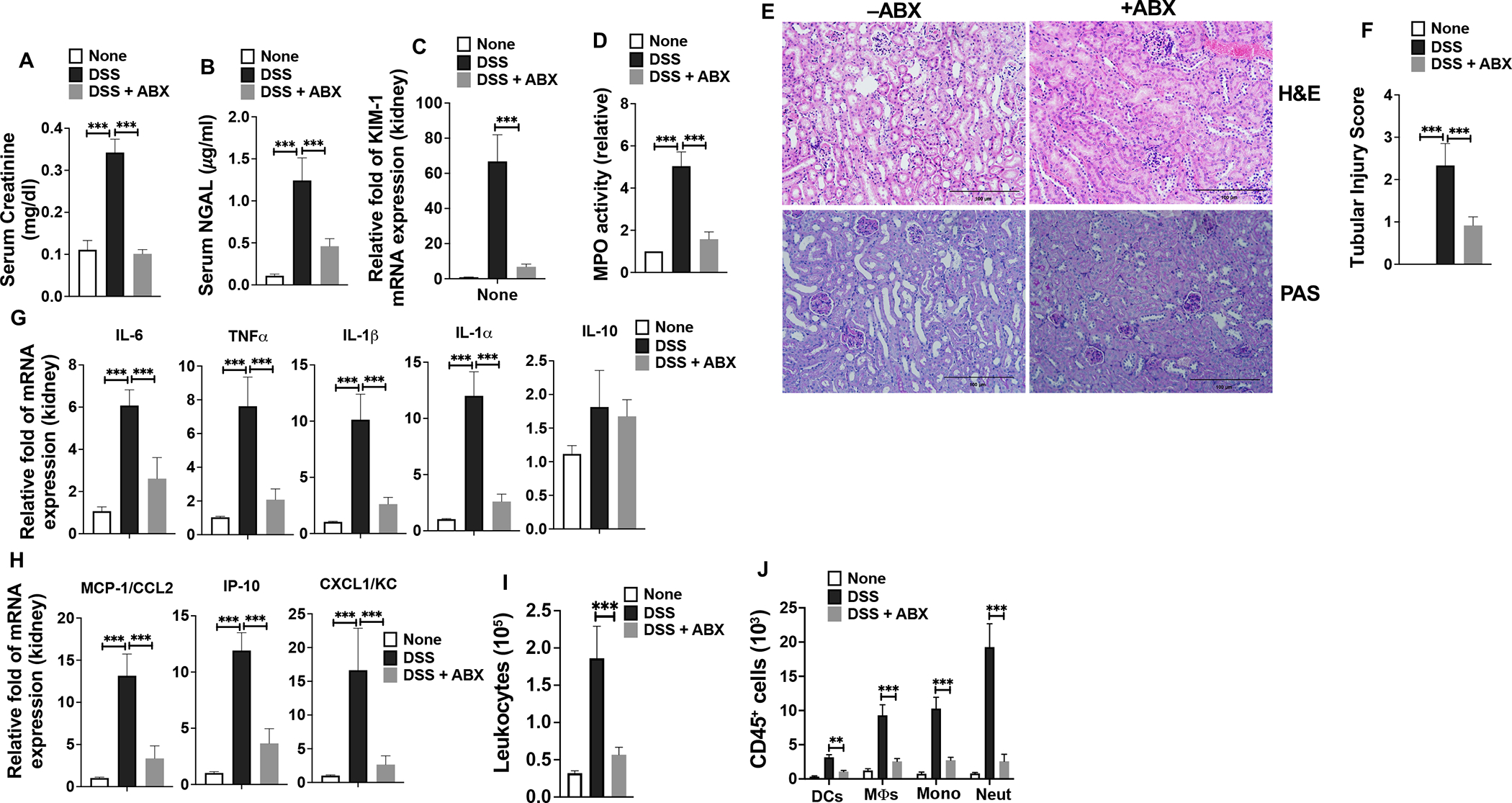
(A-C) Creatinine and NGAL levels in the serum and KIM1 mRNA expression in the kidney of control and commensal bacteria-depleted LRP5/6LysM mice on day 8 after DSS treatment. (D) Myeloperoxidase (MPO) activity in the kidney. (E) H&E-and PAS-stained sections show decreased severity of the tubular injury, tubular epithelial vacuolation, dilation of the tubular lumen, and immune cell infiltration in commensal bacteria-depleted LRP5/6LysM mice upon DSS treatment (Scale bars, 100 μm). (F) Tubular injury score of kidneys was graded following analysis of H&E- and PAS-stained cross-sections of kidney of control and commensal bacteria-depleted LRP5/6LysM mice on day 8 after DSS treatment. (G, H) RNA was extracted from the kidneys of control (without antibiotics, −ABX) or commensal bacteria-depleted (antibiotics treated, +ABX) LRP5/6LysM mice treated with DSS. The expression of indicated genes was quantified by qPCR. (I, J) The total number of leukocytes (CD45+), DCs (CD45hi MHC IIhi CD11c+ CD64−), MΦs (CD45hi MHC IIhi CD11c−CD11b+ CD64+), monocytes (CD45hi MHC II− CD11c−CD11b+ Ly6Chi Ly6GLow), and neutrophils (CD45hi MHC II− CD11c−CD11b+ Ly6Clow Ly6Ghi) from the kidney of control or commensal bacteria-depleted LRP5/6LysM mice treated with DSS and analyzed by flow cytometry on day 8. Data is representative of two experiments (n = 4–5 mice per experiment). Error bars show mean values ± SEM. **p<0.01; ***p<0.001.
Discussion
The current study defines an essential role for the Wnt-LRP5/6 signaling in macrophages in controlling systemic inflammation-associated kidney injury during colitis. Accordingly, myeloid cell-specific deletion of LRP5/6 in mice (LRP5/6LysM) led to increased proinflammatory factors with diminished production of anti-inflammatory factor IL-10 during colitis-associated systemic and renal inflammation. Consequently, mice lacking LRP5/6 in macrophages exhibited severe colitis-associated renal pathology with increased infiltration of inflammatory leukocytes in the kidney. The exacerbated colitis-associated systemic and renal inflammation in LRP5/6LysM mice was due to increased intestinal permeability and decreased barrier function resulting in increased translocation of intestinal microbial products and bacteria into systemic sites. This condition could be improved in LRP5/6LysM mice by depleting the gut microbiota, indicating the importance of LRP5/6 in controlling inflammation in the intestine and systemic sites during colitis. Mechanistically, LRP5/6-deficient macrophages were hyperresponsive to TLR ligands and produced higher levels of proinflammatory cytokines which are associated with increased activation of MAPKs. These data indicate an essential role for Wnt-LRP5/6 signaling in myeloid cells in controlling inflammation and colon-kidney crosstalk in DSS-induced acute colitis. Hence, this pathway constitutes a new therapeutic target for treating colitis-induced AKI.
Aberrant Wnt signaling is associated with many immune cell-mediated inflammatory diseases, including IBD. Our prior studies have shown that the canonical signaling in intestinal antigen-presenting cells is critical for suppressing pathological inflammatory responses and associated injury in the colon (31, 34). However, an unanswered question was whether canonical Wnt signaling in APCs is critical for suppressing systemic inflammation-associated AKI during colitis. The current study shows that the canonical Wnt signaling in macrophages is critical for controlling systemic inflammation-associated AKI during colitis. Inflammation-induced tissue injury is closely associated with macrophage activation and polarization (21, 22, 52). Macrophages displaying a proinflammatory phenotype produce higher levels of proinflammatory factors, which worsen tissue injury during inflammation (21, 22, 52). In contrast, macrophages displaying immune regulatory or anti-inflammatory phenotypes resolve inflammation and express factors that facilitate tissue repair (21, 22, 52). However, the critical receptors and signaling networks in programming macrophages in inducing inflammatory versus regulatory responses were not elucidated. Our studies show that the disruption of Wnt-LRP5/6 signaling skews the polarization of macrophages towards a proinflammatory phenotype upon activation in response to microbes. Accordingly, LRP5/6-deficient macrophages expressed markedly higher levels of proinflammatory markers such as TNFα, IL-6, IL-1β, and IL-1α, leading to more severe damage to the colon and kidney during colitis. Our study also shows that LRP5/6 deficiency results in a marked decrease in the expression of the anti-inflammatory factor IL-10 which is associated with healing and inhibition of inflammation. These findings are consistent with the studies on murine models of atherosclerosis, chronic kidney diseases (CKD), and tumors, showing that Wnt signaling imparts an anti-inflammatory phenotype on macrophages and skews activated macrophages towards M2 polarization while suppressing M1 plarization (53–55). Macrophages also express several toll-like receptors (TLR), which are critical in sensing the pathogen-associated molecular patterns (PAMPs) in invading pathogens (48). Engagement of such receptors activates macrophages and triggers the production of proinflammatory cytokines and chemokines that are critical mediators of tissue damage. Our study shows that LRP5/6-deficient macrophages are hyper-responsive to TLR ligands and produce high levels of proinflammatory factors. In line with these observations, previous studies have shown that LRP5/6 signaling programs DCs to a regulatory state, and these Wnt-conditioned DCs express higher levels of immune regulatory factors and lower levels of proinflammatory cytokines in response to TLR ligands (31, 34, 37, 56). However, further studies are warranted to understand how the canonical Wnt pathway skews activated macrophages towards regulatory phenotype during colitis-induced systemic inflammation-associated AKI.
Engagement of TLRs by specific ligands or microbes leads to the activation of three major families of MAP kinases (MAPKs), ERK, JNK, and p38, which are critical for the production of proinflammatory factors (48). Moreover, heightened activation of MAPKs in APCs is observed in IBD, AKI, and other inflammatory diseases (46, 47). Our data indicate that loss of LRP5/6 in macrophages leads to heightened activation of ERK, JNK, and p38 MAPKs and increased levels of proinflammatory factors. These observations are consistent with previous studies on DCs showing an essential role for Wnt-LRP5/6 signaling in modulating MAPK activity during inflammation (31, 34, 56). However, further studies are warranted to understand whether Wnt-LRP5/6 signaling regulates the activation of the MAPK pathway directly or indirectly. Nevertheless, these observations collectively support the hypothesis that activating the canonical Wnt signaling pathway in myeloid cells is a feedback mechanism to control systemic and renal inflammation.
Besides its well-established role in regulating intestinal immune homeostasis, the canonical Wnt pathway plays a key role in wound healing and intestinal repair during inflammation (26–28). Intestinal epithelial barrier dysfunction and increased permeability have been linked to several human diseases, including IBD, sepsis, irritable bowel syndrome (IBS), liver cirrhosis, severe acute pancreatitis (SAP), and more (4–10, 24). However, the mechanisms linking intestinal injury to systemic inflammation-associated AKI are unknown. Our study shows that DSS-induced aggravated the intestinal injury and severely impaired gut barrier function in LRP5/6LysM mice permits increased translocation of bacteria and endotoxin to the systemic circulation leading to systemic inflammation-associated AKI. In line with our findings, it has been reported that a dysfunctional intestinal barrier with increased permeability leads to sepsis (57, 58). Furthermore, other studies using murine models of intestinal inflammation have shown that increased intestinal permeability exacerbates systemic inflammation-associated AKI (12–14, 59, 60). What are the cellular sources for Wnt ligands in inflamed tissues, and how Wnt ligand expression is regulated under homeostasis and during inflammation are critical unanswered questions. In line with other studies (31, 34, 37, 56), we also observe a marked increase in the expression of Wnt ligands upon systemic inflammation in the spleen and kidney during colitis. Also, our studies show that macrophages are the major producers of Wnt ligands in inflamed tissues, dependent on TLRs. Likewise, our studies show that gut microbiota plays an essential role in expressing Wnt ligands in macrophages, as the depletion of commensal bacteria markedly reduced the expression of Wnt ligands in systemic sites during colitis. Loss of immune tolerance or hyperresponsiveness to commensal microbiota or microbial ligands results in host susceptibility to colonic inflammation (49–51) and systemic inflammation-associated AKI (61). Furthermore, the depletion of gut microbiota ameliorates tissue inflammation and injury in murine models of colitis, AKI, and CKD (7, 49, 61, 62). Likewise, antibiotic treatment mitigates systemic inflammation-associated AKI (59, 63, 64). However, the mechanisms linking gut microbiota to systemic inflammation-associated kidney injury were unknown. The present study shows that macrophages are programmed towards inflammatory phenotype in the absence of the LRP5/6 signaling as they express higher levels of proinflammatory factors and lower levels of IL-10 in response to commensal microbiota. Also, the present study shows that depletion of commensal microbiota in LRP5/6 mice markedly reduces the expression of inflammatory factors in macrophages suggesting that LRP5/6-deficient macrophages are hyper-responsive to microbial ligands. In line with these observations, the antibiotic treatment also attenuated colonic inflammation and systemic inflammation-associated AKI in LRP5/6LysM mice. These observations suggest that LRP5/6-dependent signaling in macrophages plays a vital role in restraining colitis-associated systemic inflammation and AKI in response to commensal microbiota during colitis.
Besides macrophages, neutrophils have emerged as principal inflammatory effectors in the pathogenesis of IBD and AKI (65, 66). Our studies also show a marked increase in the infiltration of neutrophils in the kidney of LRP5/6LysM mice during colitis. In addition to macrophages, neutrophils might have also contributed to the exacerbated colitis-associated systemic inflammation and AKI during colitis in LRP5/6LysM mice. Further studies may be needed to clarify the role of neutrophil-intrinsic Wnt-LRP5/6 signaling in regulating systemic inflammation associated AKI in this model.
In summary, our study reveals an important role for Wnt-LRP5/6 signaling in macrophages in exerting a protective effect on systemic inflammation-associated AKI during colitis by regulating the expression of crucial proinflammatory and anti-inflammatory mediators. Thus, manipulating the Wnt-LRP5/6 signaling pathway in macrophages could provide novel opportunities for regulating extraintestinal manifestations in response to intestinal injury and represent a potential therapeutic approach to control colitis-associated tissue injury.
Supplementary Material
Key points.
LRP5/6 signaling in myeloid cells protects against colitis-associated AKI.
LRP5/6 signaling in macrophages regulates the responsiveness to commensal microbiota.
LRP5/6 signaling limits microbiota-induced systemic and renal inflammation.
Acknowledgments
We thank Dr. Brat O Williams (Van Andel Research Institute, MI) for kindly providing LRP5 and LRP6 floxed mice. We also thank Jeanene Pihkala for technical help with FACS sorting and analysis and Janice Randall for expert technical assistance with mice used in this study.
Footnotes
This work was supported by the National Institutes of Health awards (DK123360, AI156106) to SM.
Footnotes
Conflict of Interest: Authors disclose no conflict of interest.
References:
- 1.Levine JS, and Burakoff R. 2011. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol Hepatol (N Y) 7: 235–241. [PMC free article] [PubMed] [Google Scholar]
- 2.Vavricka SR, Schoepfer A, Scharl M, Lakatos PL, Navarini A, and Rogler G. 2015. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm Bowel Dis 21: 1982–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ott C, and Scholmerich J. 2013. Extraintestinal manifestations and complications in IBD. Nat Rev Gastroenterol Hepatol 10: 585–595. [DOI] [PubMed] [Google Scholar]
- 4.Pardi DS, Tremaine WJ, Sandborn WJ, and McCarthy JT. 1998. Renal and urologic complications of inflammatory bowel disease. Am J Gastroenterol 93: 504–514. [DOI] [PubMed] [Google Scholar]
- 5.Fraser JS, Muller AF, Smith DJ, Newman DJ, and Lamb EJ. 2001. Renal tubular injury is present in acute inflammatory bowel disease prior to the introduction of drug therapy. Aliment Pharmacol Ther 15: 1131–1137. [DOI] [PubMed] [Google Scholar]
- 6.Lehto M, and Groop PH. 2018. The Gut-Kidney Axis: Putative Interconnections Between Gastrointestinal and Renal Disorders. Front Endocrinol (Lausanne) 9: 553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Ankawi G, Sun J, Digvijay K, Yin Y, Rosner MH, and Ronco C. 2018. Gut-kidney crosstalk in septic acute kidney injury. Crit Care 22: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cojocaru M, Cojocaru IM, Silosi I, and Vrabie CD. 2011. Gastrointestinal manifestations in systemic autoimmune diseases. Maedica (Buchar) 6: 45–51. [PMC free article] [PubMed] [Google Scholar]
- 9.Bergemalm D, Andersson E, Hultdin J, Eriksson C, Rush ST, Kalla R, Adams AT, Keita AV, D’Amato M, Gomollon F, Jahnsen J, Consortium IBDC, Ricanek P, Satsangi J, Repsilber D, Karling P, and Halfvarson J. 2021. Systemic Inflammation in Preclinical Ulcerative Colitis. Gastroenterology 161: 1526–1539 e1529. [DOI] [PubMed] [Google Scholar]
- 10.Magro DO, Kotze PG, Martinez CAR, Camargo MG, Guadagnini D, Calixto AR, Vasques ACJ, Ayrizono MLS, Geloneze B, Pareja JC, Saad MJ, and Coy CSR. 2017. Changes in serum levels of lipopolysaccharides and CD26 in patients with Crohn’s disease. Intest Res 15: 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han Y, Zhao T, Cheng X, Zhao M, Gong SH, Zhao YQ, Wu HT, Fan M, and Zhu LL. 2018. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci Bull 34: 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye M, Joosse ME, Liu L, Sun Y, Dong Y, Cai C, Song Z, Zhang J, Brant SR, Lazarev M, and Li X. 2020. Deletion of IL-6 Exacerbates Colitis and Induces Systemic Inflammation in IL-10-Deficient Mice. J Crohns Colitis 14: 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranganathan P, Jayakumar C, Manicassamy S, and Ramesh G. 2013. CXCR2 knockout mice are protected against DSS-colitis-induced acute kidney injury and inflammation. Am J Physiol Renal Physiol 305: F1422–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ranganathan P, Jayakumar C, Santhakumar M, and Ramesh G. 2013. Netrin-1 regulates colon-kidney cross talk through suppression of IL-6 function in a mouse model of DSS-colitis. Am J Physiol Renal Physiol 304: F1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mateer SW, Mathe A, Bruce J, Liu G, Maltby S, Fricker M, Goggins BJ, Tay HL, Marks E, Burns G, Kim RY, Minahan K, Walker MM, Callister RC, Foster PS, Horvat JC, Hansbro PM, and Keely S. 2018. IL-6 Drives Neutrophil-Mediated Pulmonary Inflammation Associated with Bacteremia in Murine Models of Colitis. Am J Pathol 188: 1625–1639. [DOI] [PubMed] [Google Scholar]
- 16.Chang CJ, Wang PC, Huang TC, and Taniguchi A. 2019. Change in Renal Glomerular Collagens and Glomerular Filtration Barrier-Related Proteins in a Dextran Sulfate Sodium-Induced Colitis Mouse Model. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Li N, Liu J, Wang T, Dong R, Ge D, and Peng G. 2022. Gegen Qinlian Decoction Alleviates Experimental Colitis and Concurrent Lung Inflammation by Inhibiting the Recruitment of Inflammatory Myeloid Cells and Restoring Microbial Balance. J Inflamm Res 15: 1273–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon J, Lee C, Heo S, Kim B, and Hyun CK. 2021. DSS-induced colitis is associated with adipose tissue dysfunction and disrupted hepatic lipid metabolism leading to hepatosteatosis and dyslipidemia in mice. Sci Rep 11: 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan S, Du X, Chen S, Liang J, Huang S, Hou S, Gao J, and Ding P. 2020. Effect of vitexin on alleviating liver inflammation in a dextran sulfate sodium (DSS)-induced colitis model. Biomed Pharmacother 121: 109683. [DOI] [PubMed] [Google Scholar]
- 20.Shen B, Wang J, Guo Y, Gu T, Shen Z, Zhou C, Li B, Xu X, Li F, Zhang Q, Cai X, Dong H, and Lu L. 2021. Dextran Sulfate Sodium Salt-Induced Colitis Aggravates Gut Microbiota Dysbiosis and Liver Injury in Mice With Non-alcoholic Steatohepatitis. Front Microbiol 12: 756299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Na YR, Stakenborg M, Seok SH, and Matteoli G. 2019. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol 16: 531–543. [DOI] [PubMed] [Google Scholar]
- 22.Huen SC, and Cantley LG. 2017. Macrophages in Renal Injury and Repair. Annu Rev Physiol 79: 449–469. [DOI] [PubMed] [Google Scholar]
- 23.Staal FJ, Luis TC, and Tiemessen MM. 2008. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol 8: 581–593. [DOI] [PubMed] [Google Scholar]
- 24.Suryawanshi A, Tadagavadi RK, Swafford D, and Manicassamy S. 2016. Modulation of Inflammatory Responses by Wnt/beta-Catenin Signaling in Dendritic Cells: A Novel Immunotherapy Target for Autoimmunity and Cancer. Front Immunol 7: 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swafford D, and Manicassamy S. 2015. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med 19: 303–310. [PMC free article] [PubMed] [Google Scholar]
- 26.Cosin-Roger J, Ortiz-Masia D, Calatayud S, Hernandez C, Esplugues JV, and Barrachina MD. 2016. The activation of Wnt signaling by a STAT6-dependent macrophage phenotype promotes mucosal repair in murine IBD. Mucosal Immunol 9: 986–998. [DOI] [PubMed] [Google Scholar]
- 27.Saha S, Aranda E, Hayakawa Y, Bhanja P, Atay S, Brodin NP, Li J, Asfaha S, Liu L, Tailor Y, Zhang J, Godwin AK, Tome WA, Wang TC, Guha C, and Pollard JW. 2016. Macrophage-derived extracellular vesicle-packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun 7: 13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cosin-Roger J, Ortiz-Masia D, Calatayud S, Hernandez C, Alvarez A, Hinojosa J, Esplugues JV, and Barrachina MD. 2013. M2 macrophages activate WNT signaling pathway in epithelial cells: relevance in ulcerative colitis. PLoS One 8: e78128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch S, Nava P, Addis C, Kim W, Denning TL, Li L, Parkos CA, and Nusrat A. 2011. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology 141: 259–268, 268 e251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, and Pulendran B. 2010. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 329: 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swafford D, Shanmugam A, Ranganathan P, Hussein MS, Koni PA, Prasad PD, Thangaraju M, and Manicassamy S. 2018. Canonical Wnt Signaling in CD11c(+) APCs Regulates Microbiota-Induced Inflammation and Immune Cell Homeostasis in the Colon. J Immunol 200: 3259–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clevers H 2006. Wnt/beta-catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- 33.Suryawanshi A, Hussein MS, Prasad PD, and Manicassamy S. 2020. Wnt Signaling Cascade in Dendritic Cells and Regulation of Anti-tumor Immunity. Front Immunol 11: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swafford D, Shanmugam A, Ranganathan P, Manoharan I, Hussein MS, Patel N, Sifuentes H, Koni PA, Prasad PD, Thangaraju M, and Manicassamy S. 2020. The Wnt-beta-Catenin-IL-10 Signaling Axis in Intestinal APCs Protects Mice from Colitis-Associated Colon Cancer in Response to Gut Microbiota. J Immunol 205: 2265–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhong Z, Baker JJ, Zylstra-Diegel CR, and Williams BO. 2012. Lrp5 and Lrp6 play compensatory roles in mouse intestinal development. Journal of cellular biochemistry 113: 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong Y, Manoharan I, Suryawanshi A, Shanmugam A, Swafford D, Ahmad S, Chinnadurai R, Manicassamy B, He Y, Mellor AL, Thangaraju M, Munn DH, and Manicassamy S. 2016. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology 5: e1115941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suryawanshi A, Manoharan I, Hong Y, Swafford D, Majumdar T, Taketo MM, Manicassamy B, Koni PA, Thangaraju M, Sun Z, Mellor AL, Munn DH, and Manicassamy S. 2015. Canonical wnt signaling in dendritic cells regulates Th1/Th17 responses and suppresses autoimmune neuroinflammation. J Immunol 194: 3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, and Ganapathy V. 2014. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manoharan I, Suryawanshi A, Hong Y, Ranganathan P, Shanmugam A, Ahmad S, Swafford D, Manicassamy B, Ramesh G, Koni PA, Thangaraju M, and Manicassamy S. 2016. Homeostatic PPARalpha Signaling Limits Inflammatory Responses to Commensal Microbiota in the Intestine. J Immunol 196: 4739–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clausen BE, Burkhardt C, Reith W, Renkawitz R, and Forster I. 1999. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8: 265–277. [DOI] [PubMed] [Google Scholar]
- 41.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, and Elson CO. 1994. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107: 1643–1652. [DOI] [PubMed] [Google Scholar]
- 42.Danoff TM 1998. Chemokines in interstitial injury. Kidney Int 53: 1807–1808. [DOI] [PubMed] [Google Scholar]
- 43.Cantaluppi V, Quercia AD, Dellepiane S, Ferrario S, Camussi G, and Biancone L. 2014. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol Dial Transplant 29: 2004–2011. [DOI] [PubMed] [Google Scholar]
- 44.Devarajan P 2010. Neutrophil gelatinase-associated lipocalin: a promising biomarker for human acute kidney injury. Biomark Med 4: 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaidya VS, Ferguson MA, and Bonventre JV. 2008. Biomarkers of acute kidney injury. Annu Rev Pharmacol Toxicol 48: 463–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krementsov DN, Thornton TM, Teuscher C, and Rincon M. 2013. The emerging role of p38 mitogen-activated protein kinase in multiple sclerosis and its models. Mol Cell Biol 33: 3728–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar S, Boehm J, and Lee JC. 2003. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2: 717–726. [DOI] [PubMed] [Google Scholar]
- 48.Manicassamy S, and Pulendran B. 2009. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol 21: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belkaid Y, and Harrison OJ. 2017. Homeostatic Immunity and the Microbiota. Immunity 46: 562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ivanov II, and Honda K. 2012. Intestinal commensal microbes as immune modulators. Cell Host Microbe 12: 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Littman DR, and Pamer EG. 2011. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cantero-Navarro E, Rayego-Mateos S, Orejudo M, Tejedor-Santamaria L, Tejera-Munoz A, Sanz AB, Marquez-Exposito L, Marchant V, Santos-Sanchez L, Egido J, Ortiz A, Bellon T, Rodrigues-Diez RR, and Ruiz-Ortega M. 2021. Role of Macrophages and Related Cytokines in Kidney Disease. Front Med (Lausanne) 8: 688060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinstock A, Rahman K, Yaacov O, Nishi H, Menon P, Nikain CA, Garabedian ML, Pena S, Akbar N, Sansbury BE, Heffron SP, Liu J, Marecki G, Fernandez D, Brown EJ, Ruggles KV, Ramsey SA, Giannarelli C, Spite M, Choudhury RP, Loke P, and Fisher EA. 2021. Wnt signaling enhances macrophage responses to IL-4 and promotes resolution of atherosclerosis. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng Y, Liang Y, Zhu X, Wang M, Gui Y, Lu Q, Gu M, Xue X, Sun X, He W, Yang J, Johnson RL, and Dai C. 2018. The signaling protein Wnt5a promotes TGFbeta1-mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J Biol Chem 293: 19290–19302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, Liu WC, and Qin HY. 2018. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis 9: 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manoharan I, Swafford D, Shanmugam A, Patel N, Prasad PD, Thangaraju M, and Manicassamy S. 2021. Activation of Transcription Factor 4 in Dendritic Cells Controls Th1/Th17 Responses and Autoimmune Neuroinflammation. J Immunol 207: 1428–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colbert JF, Schmidt EP, Faubel S, and Ginde AA. 2017. Severe Sepsis Outcomes Among Hospitalizations With Inflammatory Bowel Disease. Shock 47: 128–131. [DOI] [PubMed] [Google Scholar]
- 58.Limsrivilai J, Rao K, Stidham RW, Govani SM, Waljee AK, Reinink A, Johnson L, Briggs E, and Higgins PDR. 2018. Systemic Inflammatory Responses in Ulcerative Colitis Patients and Clostridium difficile Infection. Dig Dis Sci 63: 1801–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumar M, Leon Coria A, Cornick S, Petri B, Mayengbam S, Jijon HB, Moreau F, Shearer J, and Chadee K. 2020. Increased intestinal permeability exacerbates sepsis through reduced hepatic SCD-1 activity and dysregulated iron recycling. Nat Commun 11: 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Visitchanakun P, Saisorn W, Wongphoom J, Chatthanathon P, Somboonna N, Svasti S, Fucharoen S, and Leelahavanichkul A. 2020. Gut leakage enhances sepsis susceptibility in iron-overloaded beta-thalassemia mice through macrophage hyperinflammatory responses. Am J Physiol Gastrointest Liver Physiol 318: G966–G979. [DOI] [PubMed] [Google Scholar]
- 61.Rabb H, Pluznick J, and Noel S. 2018. The Microbiome and Acute Kidney Injury. Nephron 140: 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li F, Wang M, Wang J, Li R, and Zhang Y. 2019. Alterations to the Gut Microbiota and Their Correlation With Inflammatory Factors in Chronic Kidney Disease. Front Cell Infect Microbiol 9: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Moturi KR, Wang L, Zhang K, and Yu C. 2019. Gut derived-endotoxin contributes to inflammation in severe ischemic acute kidney injury. BMC Nephrol 20: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Emal D, Rampanelli E, Stroo I, Butter LM, Teske GJ, Claessen N, Stokman G, Florquin S, Leemans JC, and Dessing MC. 2017. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol 28: 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fournier BM, and Parkos CA. 2012. The role of neutrophils during intestinal inflammation. Mucosal Immunol 5: 354–366. [DOI] [PubMed] [Google Scholar]
- 66.Bolisetty S, and Agarwal A. 2009. Neutrophils in acute kidney injury: not neutral any more. Kidney Int 75: 674–676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.