

SUMMARY
Disruption of circadian glucocorticoid oscillations in Cushing’s disease and chronic stress results in obesity and adipocyte hypertrophy, which is believed to be a main source of the harmful effects of obesity. Here, we recapitulate stress due to jet lag or work-life imbalances by flattening glucocorticoid oscillations in mice. Within 3 days, mice achieve a metabolic state with persistently high insulin, but surprisingly low glucose and fatty acids in the bloodstream, that precedes a more than 2-fold increase in brown and white adipose tissue mass within 3 weeks. Transcriptomic and Cd36-knockout mouse analyses show that hyperinsulinemia-mediated de novo fatty acid synthesis and Cd36-mediated fatty acid uptake drive fat mass increases. Intriguingly, this mechanism by which glucocorticoid flattening causes acute hyperinsulinemia and adipocyte hypertrophy is unexpectedly beneficial in preventing high levels of circulating fatty acids and glucose for weeks, thus serving as a protective response to preserve metabolic health during chronic stress.
Graphical Abstract
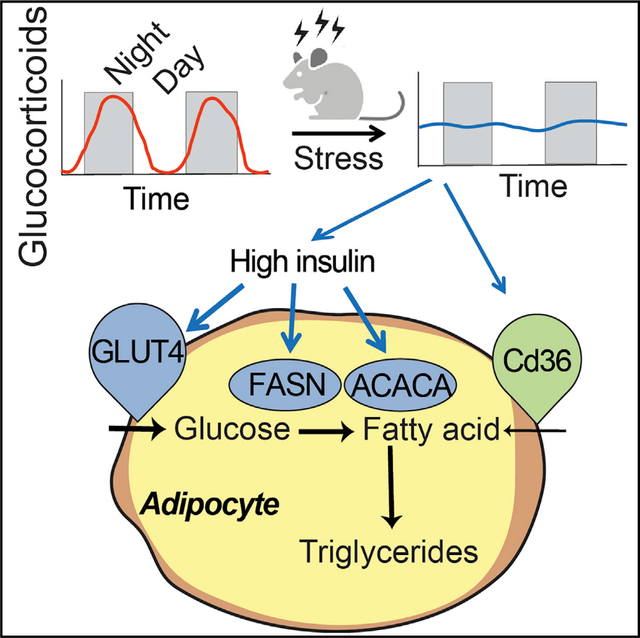
In brief
Tholen et al. show that flattening circadian glucocorticoids drives adipocyte hypertrophy via acute hyperinsulinemia, which keeps circulating fatty acids and glucose low and is unexpectedly protective to preserve metabolic health for weeks during chronic stress.
INTRODUCTION
Glucocorticoids (GCs) are steroid hormones that are capable of causing tremendous shifts in carbohydrate and fatty acid metabolism throughout the body (Macfarlane et al., 2008). GCs increase available energy substrates in the circulation by acting on multiple tissues to promote catabolism and defer anabolism. A fundamental aspect of regulation is that GCs are secreted in a circadian pattern (Weitzman et al., 1971) (Figure 1A, gray pattern). The daily increase provides a wake-up signal and turns on whole-body metabolism, and the daily trough, or off-period, allows a rest period in which catabolic GC-driven processes are not activated and the body can recover and reset (Dallman et al., 2000; Oster et al., 2017; Sapolsky et al., 1986a).
Figure 1. Flattening of circadian glucocorticoid oscillations causes rapid adipocyte hypertrophy.
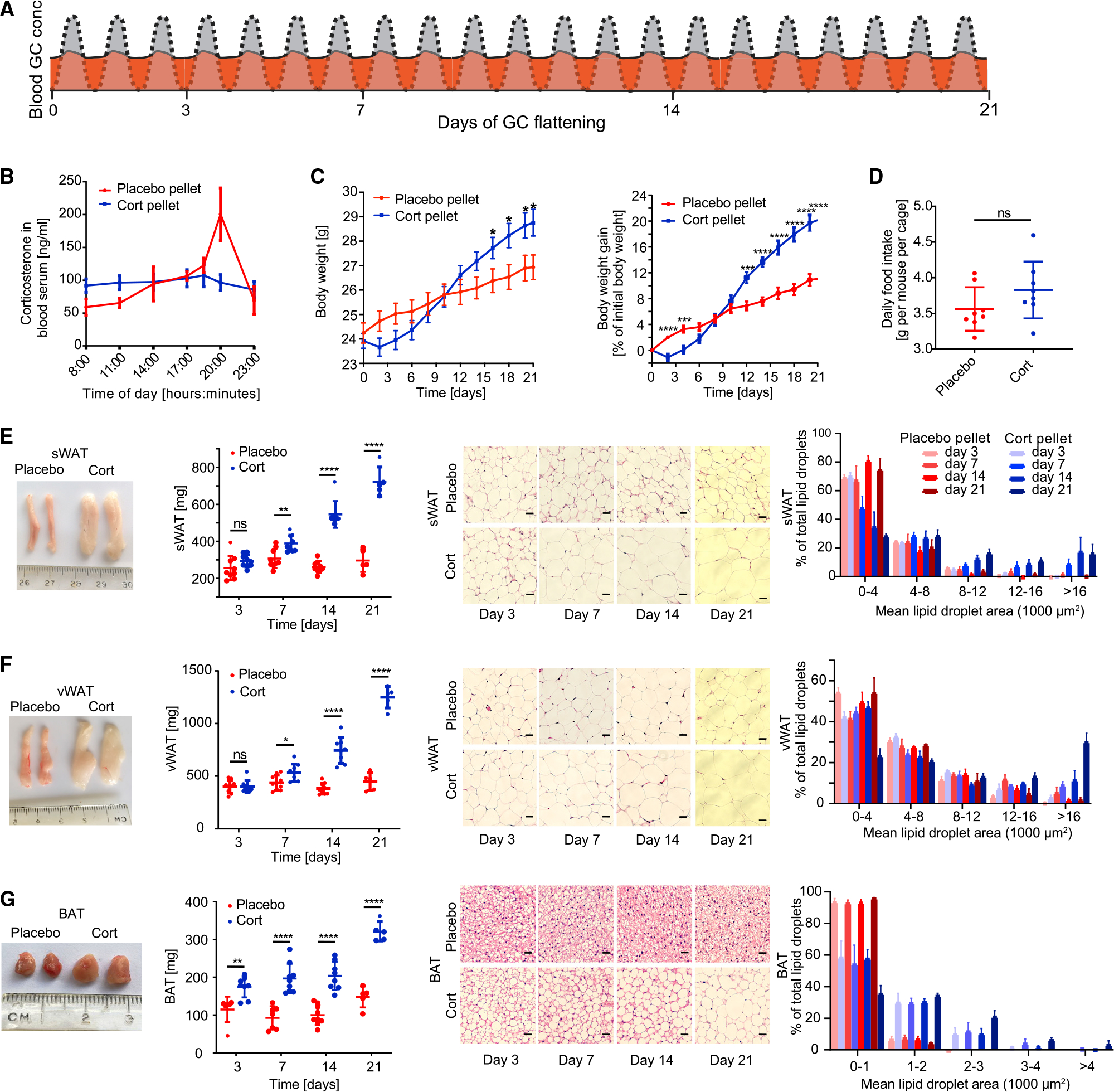
(A) Schematic of approach used to determine how flattening of GC oscillations affects adipocyte hypertrophy over time. The normal circadian GC oscillation pattern is shown in gray. The flattened GC pattern is shown in orange.
(B) Time course of average corticosterone levels in mice implanted with corticosterone pellets (cort-pellet mice) compared to mice implanted with placebo pellets (placebo-pellet mice) 7 days after pellet implantation. n = 8, means ± SEMs.
(C) Time course showing actual body weight and body weight gain of cort-pellet compared to placebo-pellet mice; n = 11, means ± SEMs. Unpaired t test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(D) Average daily food intake of each mouse per cage (n = 8). Up to 5 mice were housed in each cage. Food intake was monitored for 21 days, means ± SDs. Unpaired t test; ns, not significant.
(E–G) sWAT, vWAT, and BAT fat pad images, fat pad weight, and hematoxylin and eosin (H&E) staining analysis. Fat pad images were taken 3, 7, 14, and 21 days after pellet implantation. Fat pad weight was measured at 3, 7, 14, and 21 days after pellet implantation; n = 5–9, means ± SDs. Unpaired t test; **p < 0.01; ****p < 0.0001. One representative H&E staining is shown for each time point. Scale bar, 50 μm.
See also Table S1.
The hypothalamic-pituitary-adrenal (HPA) axis secretes GCs in response to light-dark signals from the master clock in the brain, as well as in response to stress (Spiga et al., 2014). Brief spikes of GCs are beneficial for increasing overall catabolic activity to ready the body for a fight-or-flight response (Sapolsky et al., 1986a). However, if stress becomes chronic, then the continual secretion of GCs flattens the normal circadian oscillatory pattern, meaning that peak levels are decreased and trough levels are elevated, without the daily mean values significantly changing (Dallman et al., 2000) (Figure 1A, orange pattern). Such flattening of GC oscillations can occur from depression, jet lag, aging, and irregular eating and sleeping schedules (Adam et al., 2017; Leproult et al., 1997), as well as in disease conditions such as Cushing’s (Rossi et al., 2000), and has been shown to cause obesity and metabolic disease (Broussard and Van Cauter, 2016; Dallman et al., 2000; Joseph and Golden, 2017; Mazziotti et al., 2011).
When GC circadian oscillations are flattened, fat mass expands both from adipocyte hyperplasia, or the generation of new adipocytes from precursor cells, and adipocyte hypertrophy, or the enlargement of existing adipocytes (Bahrami-Nejad et al., 2018; Campbell et al., 2011). In previous work, we identified the mechanisms responsible for the adipocyte hyperplasia (Bahrami-Nejad et al., 2018). We found that preadipocytes have a transcriptional network with slow and fast positive feedback that triggers adipocyte hyperplasia only in response to flattened, but not oscillating circadian, GC signals. However, the molecular mechanisms underlying the significant adipocyte hypertrophy when GCs are flattened were not understood. Given increasing evidence that adipocyte hypertrophy is a primary driver of the harmful metabolic effects of obesity, it is of broad interest to understand how flattening of GC oscillations cause adipocyte hypertrophy and what the associated effects are on metabolic health.
How flattening of GC oscillations can cause adipocyte hypertrophy and increased fat mass (Bahrami-Nejad et al., 2018; Dallman et al., 2000; Karatsoreos et al., 2011; Rebuffé-Scrive et al., 1992; Shen et al., 2017) is puzzling since GCs are known to be catabolic and drive breakdown of tissue mass in animals and humans (Lee et al., 2018; Peckett et al., 2011). We hypothesized that carrying out time course measurements would allow us to resolve these controversial results. We also noted that the latter studies typically used high pharmacological doses of synthetically-stabilized GCs or administered GCs together with a high fat diet which introduces confounding factors (Dunford and Riddell, 2016). Thus, we used pellets containing corticosterone, the main physiological GC in mice (Sapolsky et al., 1986a), rather than synthetic GCs. We also avoided high pharmacological doses and instead used experimental conditions that maintained physiological mean circulating levels of GCs.
We designed time course experiments to flatten GC oscillations for a limited period that mimics chronic stress. Strikingly, we found that within 3 days of GC flattening, mice develop hyperinsulinemia and strongly upregulate de novo fatty acid synthesis genes in white (WAT) and brown adipose tissue (BAT), which is surprising since normally, adipocytes accumulate lipid primarily by uptaking fatty acids from the circulation and not by de novo synthesis (Santoro et al., 2021; Strawford et al., 2004). Surprisingly, even though BAT and WAT increased more than 2-fold within 21 days of GC flattening, no significant accumulation of lipid in the liver or increase in circulating fatty acids was observed. Surprisingly, the hyperinsulinemia and much of the adipocyte hypertrophy were reversed within 3 weeks after pellets stopped releasing GCs. By keeping fatty acids and glucose low in the circulation, the hyperinsulinemia and adipocyte hypertrophy resulting from GC flattening likely play a protective role in counteracting the potentially harmful side effects caused by increased GC-driven catabolic breakdown of tissues under periods of temporary, multi-week long stress.
RESULTS
Flattening of circadian GC oscillations causes rapid adipocyte hypertrophy in WAT and BAT
To mimic the chronic stress condition, we flattened daily GC oscillations in C57BL/6J male mice by implanting pellets that release corticosterone, the main GC in mice, at a constant rate over 21 days. The constant release of exogenous GCs into the mice alters the pattern of endogenous GCs, causing the troughs to become elevated and the peaks to be blunted since circulating GCs are a negative feedback signal and suppress secretion of endogenous GCs by the HPA axis (Figure 1A, orange) (Gjerstad et al., 2018). We specifically chose the dose of GC that was released each day such that the mean GC level was kept approximately at normal physiological levels (Bahrami-Nejad et al., 2018; Hodes et al., 2012). As shown in Figure 1B, blood serum ELISA measurements confirmed that mice implanted with corticosterone pellets (hereafter referred to as “cort-pellet mice”) had decreased peak levels and increased trough levels of GCs compared to placebo pellet-implanted mice (hereafter referred to as “placebo-pellet mice”). Even though they had similar circulating GC levels throughout the 21-day experiment, the cort-pellet mice ended up weighing 9.1% more than the placebo-pellet mice (Figure 1C).
As had been found in previous studies that administered continuous GCs (Bahrami-Nejad et al., 2018; Bowles et al., 2015; Luijten et al., 2019), the increased weight gain in the cort-pellet mice cannot be explained by higher food intake (Figure 1D). We thus conducted indirect calorimetry using metabolic cages. Similar to another study that used a mouse model of continuous corticosterone treatment (Luijten et al., 2019), we did not observe a significant increase in oxygen consumption rate, respiration rate, or total daily energy expenditure (Figures S1A–S1N). However, we did observe a significant decrease in lean mass and increase in fat mass (Figure S1O), suggesting an increase in nutrient absorption or different metabolic profile in the cort-pellet mice.
To determine whether the weight gain in the cort-pellet mice was due mainly to expansion of fat depots, we measured subcutaneous WAT (sWAT), visceral adipose tissue (vWAT), and BAT by dissecting and weighing the inguinal sWAT depot located in the flank region under the skin, the epididymal vWAT depot that surrounds the gonads, and the interscapular BAT depot (Cannon and Nedergaard, 2004; Chusyd et al., 2016). Strikingly, sWAT and vWAT weight increased more than 2-fold and BAT weight increased 3-fold within 21 days of GC oscillation flattening (Figures 1E–1G, left two panels). Significant expansions of sWAT and vWAT were apparent after 7 days of GC flattening, and an even earlier expansion was observed in BAT after only 3 days (Figures 1E–1G, left two panels). Hematoxylin and eosin (H&E) staining confirmed significantly increased adipocyte cell volumes in BAT and WAT starting at days 3 and 7, respectively (Figures 1E–1G, right two panels).
As a control to test whether the lipid accumulation in WAT and BAT shown in Figure 1 is indeed caused by flattening of GC oscillations and not simply due to an increased amount of corticosterone released into the mice by the pellets, we injected C57BL/6J male mice at 5 p.m. each day for 21 days with the same total amount of corticosterone that is released by the pellet each day (Figure 2A). Despite a more than 10-fold increase in the daily peak amplitude of corticosterone (Figure 2B), there was no significant increase in body weight (Figure 2C), food intake (Figure 2D), or sWAT, vWAT, or BAT fat pad size and morphology between cort- and placebo-injected mice (Figures 2E–2G and S2).
Figure 2. Increasing the peak amplitude of corticosterone in the evening phase does not affect body weight gain or lipid accumulation in WAT and BAT.
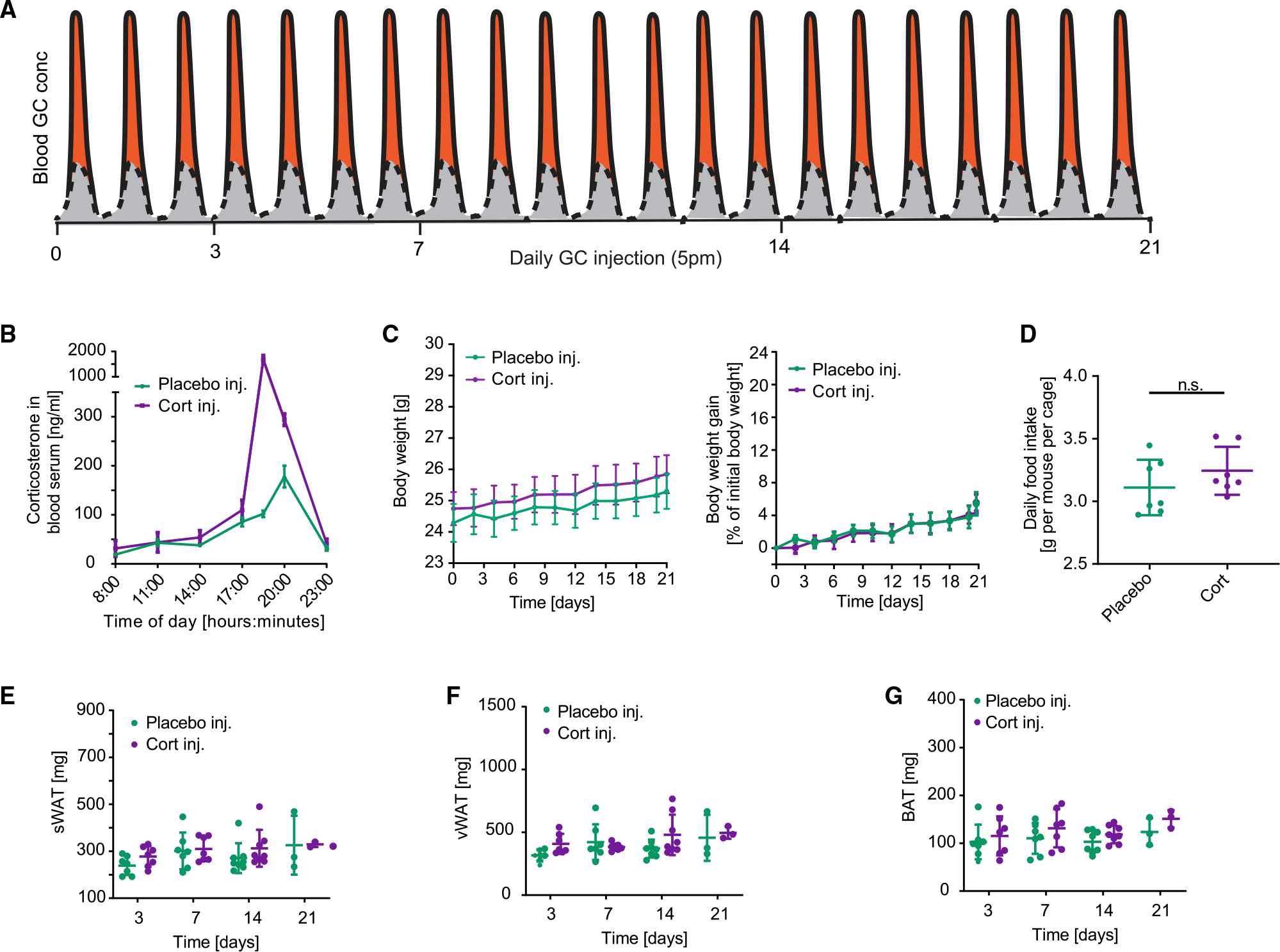
(A) Schematic of approach used to determine how flattening of GC oscillations affects adipocyte hypertrophy over time. The perturbation from the normal GC oscillation pattern is shown in orange.
(B) Time course of average corticosterone levels in mice injected with corticosterone compared to mice injected with solvent control (phosphate buffer) 7 days after start of injections. n = 3–4, means ± SEMs.
(C) Time course showing actual body weight and body weight gain of mice injected with corticosterone compared to mice injected with solvent control (phosphate buffer). n = 9, means ± SEMs.
(D) Average daily food intake of each mouse per cage. Up to 5 mice were housed in each cage. Food intake was monitored for 21 days, n = 7, means ± SDs. Unpaired t test; ns, not significant.
(E–G) sWAT, vWAT, and BAT fat pad weight measured at 3, 7, 14, and 21 days after start of daily injections. n = 3–8, means ± SDs.
Thus, as was previously shown (Bahrami-Nejad et al., 2018), short spurts of GCs do not promote weight gain as long as the increase in GCs occurs only when GCs are normally high and there are sufficiently long off-periods in which low GC levels are maintained. Furthermore, despite the same total amount of GC being introduced into both cort-pellet and cort-injected mice, significant adipocyte hypertrophy and fat mass increases were only observed in the cort-pellet mice, arguing that it is the GC flattening and not the total amount of added GCs that is responsible.
Flattening of daily GC oscillations does not result in fat deposition in the liver or increased fatty acids or triglycerides in the circulation
Chronic or dysregulated GC secretion has been shown to increase fat mass in rodents and humans, as well as to cause fat deposition in the liver and an increase in triglycerides and fatty acids in blood plasma (Bowles et al., 2015; Campbell et al., 2011; Chimin et al., 2014; Harris et al., 2013; Macfarlane et al., 2008). However, most in vivo studies administrated high pharmacological doses of GCs or administered GCs for several months, which does not provide insight into how variations in normal physiological levels of limited duration, such as are seen in chronic stress, jet lag, and nightshift work, affect body metabolism. Since our experimental conditions use physiological levels of GCs and a limited 3-week period of GC flattening, we tested to see whether there is still fat accumulation in the liver. Markedly, analysis of the livers from cort-pellet mice showed no increase in liver weight, upregulation of lipid metabolism genes (Figures S3A–S3J), or lipid accumulation as assessed by histology and triglyceride analysis (Figures 3A, S3K, and S3L).
Figure 3. Flattening of glucocorticoid oscillations does not result in fat deposition in the liver, elevated circulating triglycerides in blood serum, or defects in thermogenesis.
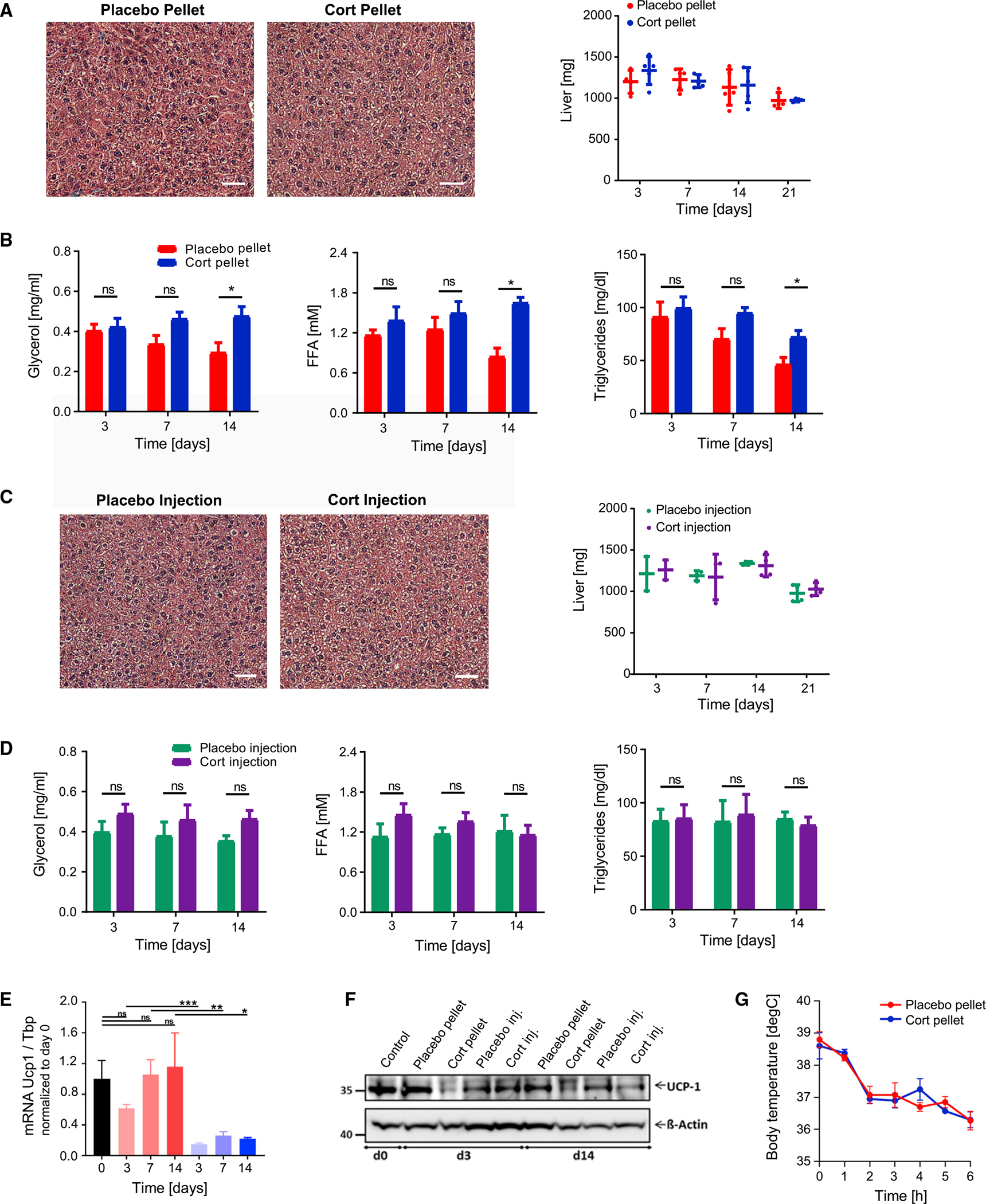
(A) Representative image of H&E staining of livers 21 days after placebo or cort-pellet implantation (n = 4). Weight of livers at different time points after pellet implantation. n = 4–5, means ± SDs. See also Table S1 and Figure S3.
(B) Time courses of changes in FFAs, glycerol, and triglycerides measured in blood serum after 14 days in fasted mice. n = 4–5, means ± SEMs, unpaired t test; *p < 0.05; ns, not significant.
(C and D) Same as in (A) and (B), but instead for daily placebo or cort-injected mice, n = 3–4.
(E) Flattening of daily GC oscillations results in downregulation of genes involved in thermogenesis. mRNA expression for UCP-1 measured by qPCR in BAT, normalized to the expression of Tbp, and presented as means ± SEMs (n = 3–4). Unpaired t test; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. Control group are mice at start of experiment (day 0).
(F) Western blot analysis of UCP-1 in BAT lysates (see Figure S3M for quantification).
(G) Acute cold exposure experiment of placebo and cort-pellet-implanted mice 21 days after pellet implantation. Means ± SEMs; n = 8.
Given that triglycerides in adipose tissue are thought to be generated primarily from fatty acids taken up from the circulation rather than from de novo synthesized fatty acids (Santoro et al., 2021; Strawford et al., 2004), we expected to find that the adipocyte hypertrophy in WAT and BAT was caused by higher circulating fatty acid levels. Surprisingly, we did not detect significant differences in circulating glycerol, free fatty acids (FFAs), or triglycerides between cort-pellet and placebo-pellet mice during the first 2 weeks after GC flattening (Figure 3B). Furthermore, what appeared as a small increase in these factors at day 14 was primarily due to reduced levels in controls. Taken together, these results support that even after 3 weeks of GC flattening and significant expansion of fat mass, metabolic health was not yet strongly impacted. In control experiments, we confirmed that no significant lipid accumulation in the liver (Figure 3C) or changes in triglyceride, glycerol, and FFA levels (Figure 3D) were detected when the same amount of corticosterone that was released each day by the pellet was injected during the GC peak period.
The expansion of WAT and BAT is not due to defective thermogenesis
Because we saw no lipid accumulation in the liver or significant lipid changes in the circulating blood plasma, we concluded that the adipocyte hypertrophy must originate from an adipose tissue-intrinsic mechanism. Thus, we next tested whether reduced thermogenesis due to defects in brown fat function could be contributing indirectly to white fat mass enlargement, as has been proposed (Strack et al., 1995; van den Beukel et al., 2014; Viengchareun et al., 2001). RT-PCR and western blot analysis showed that mRNA and protein expression of UCP1, a key protein regulator of adaptive thermogenesis, was reduced in the BAT of cort-pellet mice (Figures 3E and 3F). However, when the mice were placed at 4°C in an acute cold exposure experiment to compare their thermogenic capacities, there was no significant difference in how core body temperatures dropped (Figure 3G), supporting the idea that reduced thermogenesis is not responsible for the increased fat mass (Luijten et al., 2019) and indicating that UCP1-independent thermogenic pathways are likely compensating for the reduced levels of UCP1 (Brandão et al., 2021; Ikeda and Yamada, 2020).
Taken together, our results so far suggest that adipocyte hypertrophy in response to GC flattening is not caused by changes in the liver, plasma lipid levels, or thermogenesis, suggesting that other metabolic changes in WAT and BAT are responsible. Since the primary role of GCs is to regulate gene transcription, we next performed a transcriptome time course analysis of WAT and BAT tissues to understand whether transcription changes may cause the development of acute adipocyte hypertrophy in response to GC flattening.
Flattening of GC oscillations results in strong and rapid upregulation of de novo fatty acid synthesis genes in WAT and BAT
We focused on vWAT since its expansion has been recognized to be detrimental to metabolic health, while expansion of sWAT may be protective (Ghaben and Scherer, 2019; Shao et al., 2018). We also analyzed BAT because of increasing evidence that BAT activity can significantly affect whole-body energy expenditure and fat tissue mass (Cannon and Nedergaard, 2004; Saito, 2013). We harvested vWAT and BAT between 1 and 5 p.m. at four time points: 0, 3, 7, and 14 days after pellet implantation and carried out RNA sequencing (RNA-seq) analysis.
We determined significant differential gene expression by a false discovery rate (FDR) corrected p value (qval) less than 0.05 and found that several thousand transcripts were significantly changed in both vWAT and BAT (Figures 4, S4, S5 and Tables S4, S5, S6 and S7). RT-PCR was carried out to validate key genes identified in the RNA-seq analysis (Figure S4D). While principal-component analysis (PCA) showed that the cort-pellet and placebo-pellet samples were distinguishable from each other (Figures 4A and S4A), the samples obtained at different time points (3, 7, or 14 days) in the two treatment groups were largely indistinguishable from each other, supporting the notion that flattening GC oscillations reprograms the vWAT and BAT transcriptomes within 3 days and that the changes are sustained for at least 14 days thereafter. Significant changes in several thousand genes of cort-pellet mice were observed in vWAT already at day 3, whereas the expansion of vWAT was only observed at day 7 (Figure 1F), suggesting that the GC-mediated changes in the vWAT transcriptome help to drive the adipocyte hypertrophy in the vWAT, and not the other way around. Because the gene expression changes at 3, 7, or 14 days were largely indistinguishable from each other (Figure 4A), we combined the vWAT data from the 3 time points to increase statistical significance in the analysis of gene expression changes. Similarly, we found for the BAT that gene expression changes at 3, 7, or 14 days in GC flattened mice were largely indistinguishable from each other (Figure S5A), and thus, we also combined the data from the three time points in our analysis of BAT gene expression (Figure S5).
Figure 4. Flattening of glucocorticoid oscillations results in gene expression alterations in vWAT of genes involved in lipogenesis.
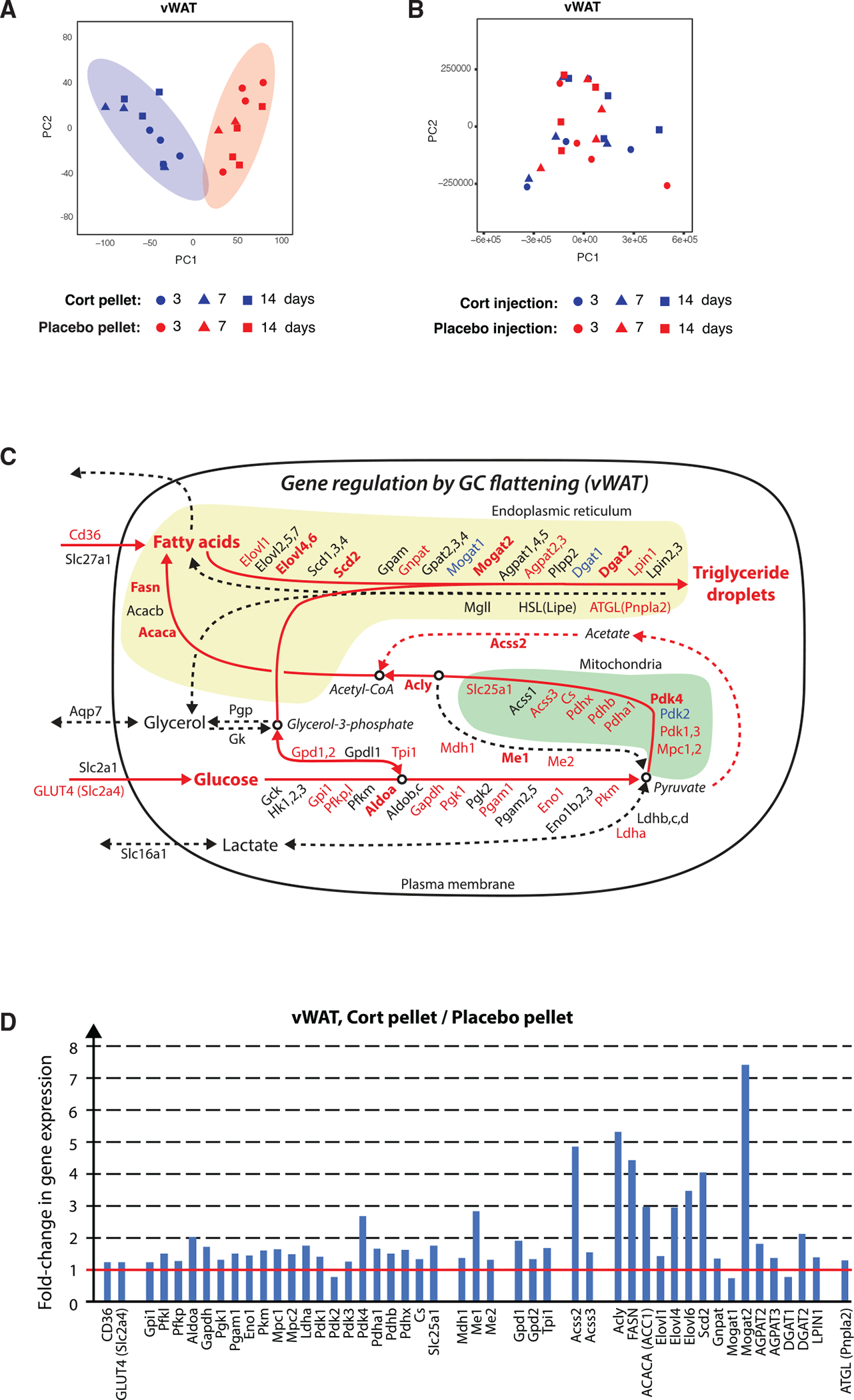
(A) Principal-component analysis of vWAT from cort- and placebo-pellet-implanted mice obtained at 3, 7, and 14 days after pellet implantation.
(B) Principal-component analysis of vWAT from cort- and placebo-injected mice obtained at 3, 7, and 14 days after the start of injections.
(C) Schematic of gene expression changes in triglyceride synthesis pathways in vWAT from cortversus placebo-pellet-implanted mice. Upregulated, unchanged, and downregulated genes are marked in red, black, and blue, respectively. Genes with a more than 2-fold change in gene expression are marked in bold.
(D) Bar plots showing fold change of significantly changed vWAT genes in cort- versus placebo-pellet-implanted mice.
See also Figures S4 and S5 and Tables S2, S3, S4, S5, S6, and S7.
In control experiments, we carried out RNA-seq analysis of cort-injected and placebo-injected mice. We sacrificed groups of mice between 1 and 5 p.m. at 4 time points: 0, 3, 7, or 14 days after the start of daily injections and collected and analyzed vWAT and BAT by RNA-seq. Remarkably, in comparing cort-injected versus placebo-injected mice, PCA analysis showed no significant differences between the two conditions (Figures 4B and S5B). Furthermore, no transcripts were changed in vWAT and only nine transcripts were changed in BAT between cort-injected and placebo-injected mice compared to the thousands of significant transcript changes observed in the vWAT and BAT between cort-pellet versus placebo-pellet mice (Figure S4A). Thus, even a large increase in the peak amplitude of corticosterone does not manifest as a significant change in gene expression in vWAT or BAT, consistent with our results in Figure 2 that showed no increase in fat mass in the cort-injected or placebo-injected mice. GCs have been shown to regulate hundreds of genes in adipose tissue (Lee et al., 2018). Interestingly, since the time between injection of corticosterone at 5 p.m. the day before until the mice were sacrificed between 1 and 5 p.m. was between 20 and 24 h, the fact that there are no significant changes in gene expression in the vWAT and BAT of cort-injected mice supports the idea that the effect of GCs on gene expression is short-lived—less than 24 h—in adipose tissue.
As an additional control, to validate that daily injections and pellet implantation do not on their own cause stress that triggers gene expression changes, we compared placebo-injected and placebo-pellet mice. No differently expressed genes were observed when comparing vWAT or BAT of these mice (Figures S4A and S4B), demonstrating that the procedure of pellet implantation and injections themselves do not alter gene expression and supporting that the injection and pellet-mediated delivery of GC results can be directly compared.
Gene Ontology (GO) analysis of the significantly regulated genes in vWAT of GC flattened mice identified “fatty acid biosynthetic process,” “acetyl-CoA metabolic process,” “pyruvate metabolic process,” and “glucose 6-phosphate metabolic process” as GO terms with high Z scores in vWAT and BAT (Tables S2 and S3). In addition, GO terms were enriched that were related to lipid uptake and utilization such as “lipid localization,” “fatty acid transport,” and “fatty acid metabolic process.” Similar metabolic GO terms were also identified in BAT. This analysis suggested that changes in adipocyte glucose and fatty acid metabolism may both contribute to the strong expansion of the BAT and WAT mass.
To better understand how flattening of GC oscillations could be changing glucose and fatty acid metabolism in vWAT and BAT, we mapped our RNA-seq data onto literature-curated maps that position metabolic genes into the main pathways that contribute to the synthesis and breakdown of triglycerides (Figures 4C, 4D, S5C, and S5D). We colored each gene name by whether the transcript is significantly increased (red), unchanged (black), or decreased (blue). A gene was considered unchanged if the fold change was between 0.8 and 1.2 (less than 20% change). We used bold type for the gene name if the increase or reduction was greater than 2-fold.
Due to the known catabolic role of GCs, we had initially expected that flattening oscillations would cause upregulation of the core lipolytic genes Pnpla2 (Atgl), Lipe (Hsl), and Mgll. However, these genes were either weakly or not upregulated in BAT or vWAT. In contrast, we observed a striking upregulation of rate-limiting enzymes in the de novo fatty acid synthesis pathway such as fatty acid synthase (Fasn), acetyl-coenzyme A (CoA) carboxylase (Acaca and Acacb), and ATP citrate lyase (Acly). We were surprised by this for two reasons. First, de novo fatty acid synthesis from glucose is not generally believed to be a main contributor to triglyceride synthesis in vWAT. Rather, the main source of fatty acids to make triglycerides in adipocytes is thought to be fatty acids taken up from the circulation (Santoro et al., 2021; Strawford et al., 2004). Second, expression of lipogenesis genes such as Fasn and Acaca are known to be regulated by insulin signaling (Krycer et al., 2020; Song et al., 2018). This raised the question of whether the transcriptome changes and adipocyte hypertrophy in mice with flattened GC oscillations may at least in part be due to an increase in the level of insulin in the blood plasma. Previous studies, for example, showed a link from insulin signaling to glucose anabolism in adipocytes (Krycer et al., 2020).
Notably, there was a smaller upregulation of de novo fatty acid synthesis genes in BAT (Figures S5C and S5D). Instead, BAT has a stronger upregulation of Cd36 expression compared to vWAT and an over 14-fold upregulation of Gck, which is thought to be important in capturing glucose in cells (Matschinsky and Wilson, 2019), suggesting that BAT may use a different strategy to increase fat mass than vWAT when GCs are flattened and offering a possible explanation for the more rapidly observed adipocyte hypertrophy in BAT versus WAT (Figures 1E–1G).
GC flattening results in rapidly induced hyperinsulinemia
At the outset of our study, we did not consider that insulin could be elevated in the cort-pellet since insulin is short-lived in the circulation and high or dysregulated GCs levels are commonly thought to inhibit insulin release from pancreatic beta cells (Delaunay et al., 1997). However, we realized that hyperinsulinemia is common in human patients with flattened circulating GC levels such as in subcritical hypercortisolism (Elhassan et al., 2019; Lopez et al., 2016; Rossi et al., 2000). Furthermore, other studies have shown that hyperinsulinemia, even when glucose was low, could originate from defects in insulin clearance (Bojsen-Møller et al., 2018; Protzek et al., 2016) and also that GC-stimulated pancreatic beta cells can in some cases still secrete insulin (Fine et al., 2018). Taken together, it was plausible that high insulin could be responsible for the gene expression profiles we had measured in mice with flattened GC oscillations.
We monitored insulin levels in blood plasma over 21 days, and strikingly, we found that in cort-pellet mice, insulin levels increased within 3 days to over 7 ng/mL compared to less than 1 ng/mL in placebo-pellet mice (Figure 5A). This unexpectedly initial high insulin level then slowly decreased during the 21 days of GC flattening to approximately 3 ng/mL, which was still several-fold higher than the insulin levels in placebo-pellet mice. High insulin levels in mice and humans are typically correlated with high glucose levels, such as occur after a meal, that stimulate pancreatic beta cells to secrete more insulin. We were thus surprised to find that glucose levels are low in GC-flattened mice, even slightly lower than in control mice (Figure 5B). Since glucose levels are not high, this suggests that it is not an increase in glucose levels that is signaling to pancreatic beta cells to secrete more insulin, as commonly occurs (Campbell and Newgard, 2021).
Figure 5. Flattening of glucocorticoid oscillations causes hyperinsulinemia within 3 days.
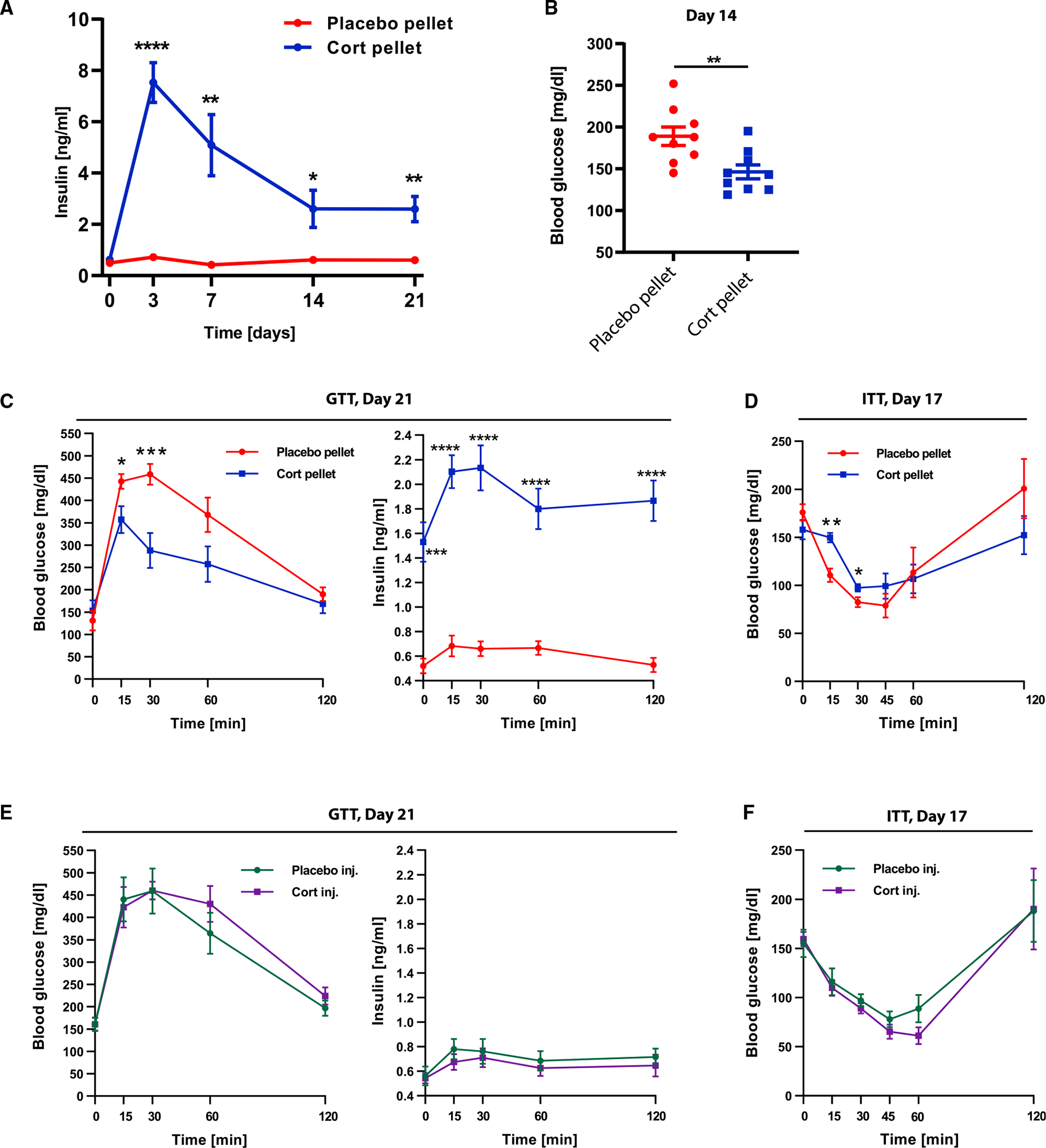
(A) Blood plasma insulin levels of 6 placebo and 6 cort-pellet-implanted mice (not fasted).
(B) Blood glucose levels of 9 placebo and 9 cort-pellet-implanted mice (not fasted) 14 days after pellet implantation.
(C) Glucose tolerance test (GTT) after 21 days of treatment of 5 placebo and 5 cort-pellet-implanted mice.
(D) Insulin tolerance test (ITT) after 17 days of treatment of 5 placebo and 6 cort-pellet-implanted mice. (A–D) Means ± SEMs, unpaired t test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(E) GTT of 6 placebo and 6 cort-injected mice after 21 days of treatment; means ± SEMs.
(F) ITT of 5 placebo and 6 cort-injected mice after 17 days of treatment; means ± SEMs.
Since previous studies have provided evidence that hyperinsulinemia in GC-treated rodents originates from increased β-cell proliferation (Courty et al., 2019; Rafacho et al., 2009), we assessed β-cell proliferation in GC flattened mice and found a significant increase in cort-pellet compared to placebo-pellet mice (Figure S6A). It is likely that other mechanisms also contribute to the hyperinsulinemia in GC-flattened mice, which should be tested in future work.
Given the persistent high levels of insulin, we also considered that high insulin for continuous periods of days and weeks may result in a loss of insulin sensitivity or ability to properly clear glucose from the bloodstream. Thus, we carried out insulin and glucose tolerance tests (ITT and GTT) at time points when insulin had been high for 17–21 days. Interestingly, as shown in the GTT test results (Figure 5C), an intraperitoneally injected bolus of glucose was taken up even more rapidly in GC pellet-implanted mice compared to placebo-pellet control mice. The faster drop in glucose was surprising but likely resulted from the higher level of insulin in the GC pellet-implanted mice and, furthermore, suggested that the GC flattened mice are still insulin sensitive. To test for this, we carried out ITTs (Figure 5D) and found that glucose levels dropped similarly as in control mice, albeit slightly slower, supporting the idea that cort-pellet mice are still insulin sensitive but may be developing insulin resistance (Janssen, 2021). The hyperinsulinemia together with insulin sensitivity can explain the observed relatively reduced glucose levels in GC-flattened mice since insulin promotes glucose uptake into fat and muscle (Leto and Saltiel, 2012).
In control experiments, mice injected with the same dose of GC as released by the pellet per day did not reveal any differences in glucose tolerance compared to placebo-injected mice and did not display higher insulin levels in the blood plasma (Figures 5E and 5F). Together, these results show that flattening GCs causes hyperinsulinemia rapidly within 3 days, before adipocyte hypertrophy is observed in WAT.
Metabolic changes caused by 3 weeks of flattening GCs are largely reversible
One of the goals of this study was to mimic a temporary 3-week-long stress condition. Accordingly, we chose a pellet that releases corticosterone over 3 weeks. During the 3 weeks of GC flattening, we observed significant weight gain, adipocyte hypertrophy, and hyperinsulinemia, which are typically considered to be harmful for metabolic health. It was therefore surprising that mice were able to maintain insulin sensitivity and normal fatty acid and triglyceride levels. Furthermore, there was no lipid accumulation in the liver or hyperglycemia (Figure 3), further suggesting that mice in which GCs are flattened even for up to 3 weeks are still metabolically healthy.
Strikingly, the hyperinsulinemia in GC-flattened mice was fully reversible. Insulin levels returned to the levels of the placebo-pellet mice at 42 days, which is 3 weeks after the time when the implanted pellets stop releasing corticosterone (Figures 6A, S6B, and S6C). These results support a direct role for flattening GC oscillations in driving the hyperinsulinemia. Furthermore, approximately half of the weight gain in WAT and all of the weight gain in BAT reverted back to control levels by 42 days (Figure 6B). Together, these results suggest that the greater than 2-fold increase in adipocyte hypertrophy is mostly reversible in WAT and fully reversible in BAT over a similar time period as it took to increase the fat mass.
Figure 6. The hyperinsulinemia and much of the adipocyte hypertrophy are reversible when the glucocorticoid flattening stimulus is removed.
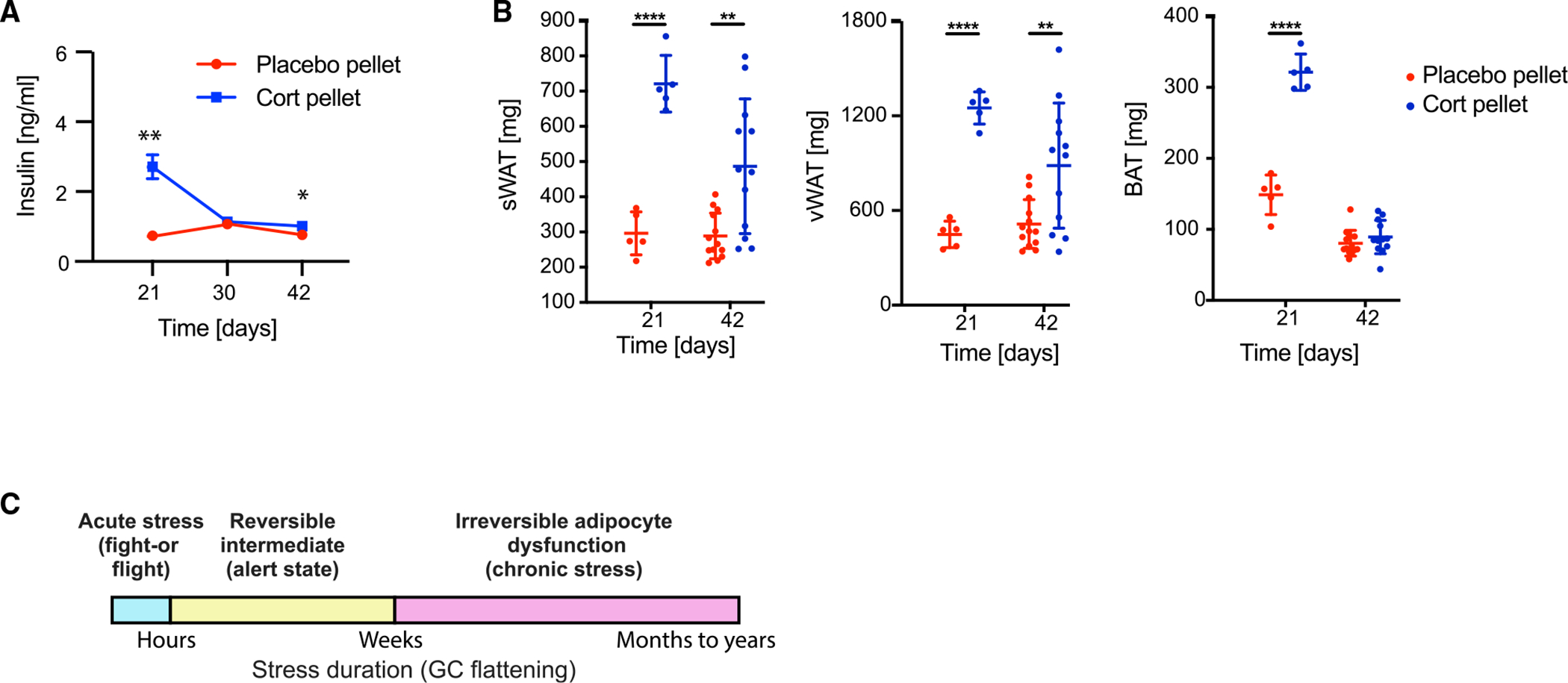
(A) Time course of insulin levels in blood plasma in 6 placebo and 6 cort-pellet-implanted mice (not fasted); means ± SEMs; unpaired t test; *p < 0.05, **p < 0.01.
(B) Fat pad weight measured 21 and 42 days after pellet implantation, n = 5–9, means ± SDs. Unpaired t test; **p < 0.01, ****p < 0.0001. Data from 21 days is the same as in Figures 1E and 1F, second panel.
(C) Schematic representation of the commonly discussed acute and chronic stress states, as well as a reversible intermediate state arising from 3 weeks of glucocorticoid flattening. Mice can mostly recover from this intermediate alert state without long-term adipocyte dysfunction. See also Figure S6.
These results support that there is a healthy “alert” state that mice can assume for weeks in response to temporary stress and continually activated GC signaling. This alert state is characterized by high circulating insulin and high glucose uptake rates and has a protective role by keeping the circulating levels of fatty acids and glucose in check despite possible catabolic effects on tissues due to dysregulated GC oscillation timing. The existence of this alert state is intriguing since most previous work has described stress as being either acute, “fight-or-flight response” stress, or chronic long-term stress that would cause irreversible adipocyte dysfunction and associated metabolic diseases (McEwen, 2000; Sapolsky, 2004; Sapolsky et al., 1986a). However, our data show that mice can recover from hyperinsulinemia and a more-than-doubling of fat mass, as long as the stress and GC flattening does not exceed a few weeks. Taken together, these results suggest that there should be a modification to the commonly discussed distinction between an acute, fight-or-flight-type GC response on the order of hours, and a chronic permanent GC dysregulation (McEwen, 2000; Sapolsky, 2004; Sapolsky et al., 1986b). Our study suggests that one should add a reversible alert state of intermediate duration of approximately 3 weeks. Based on previous studies that used longer GC flattening, this intermediate state would then transition to a chronic irreversible state if the stress lasts much longer or GC signals are elevated (Figure 6C, yellow box).
CD36ko mice are partially protected against weight gain induced by flattening of GC oscillations
Since insulin is known to mediate many of the transcriptome changes observed after 3 days, a likely order of events in developing GC-mediated obesity is that mice with dysregulated GCs rapidly develop hyperinsulinemia, which then causes insulin-driven upregulation of fatty acid synthesis genes such as Fasn, Acly, and Acaca in fat cells. Upregulation of these genes would increase the conversion of glucose into fatty acids and increase triglyceride synthesis in adipocytes. We nevertheless considered that the adipocyte hypertrophy could also be partially due to increased fatty acid uptake, especially since our RNA-seq analysis had shown that the upregulation of Cd36 is a common feature between WAT and BAT when GC oscillations are flattened (Figures 4C, 4D, S5C, and S5D). Cd36 mediates long-chain fatty acid uptake and has a role in lipid signaling, playing an important role in lipid metabolism (Pepino et al., 2014).
qRT-PCR confirmed 3-fold higher mRNA levels in vWAT and 1.5-fold higher mRNA levels in BAT of Cd36 upon flattening of corticosterone oscillations (Figure 7A). To investigate the role of Cd36 in in vivo, we implanted placebo and corticosterone pellets in Cd36 knockout (Cd36ko) mice. After 21 days, cort-pellet Cd36ko mice weighed 6.9% more than placebo-pellet Cd36ko mice, which is 2.2% less than observed in wild-type mice (Figure 7B). In wild-type mice, cort-pellet mice weighed 9.1% more than placebo-pellet mice (Figures 1B and 7B). In conclusion, Cd36ko mice are partially protected against body weight gain induced by flattening of corticosterone oscillations. Of note, during the experiment, Cd36ko mice gained less weight compared to wild-type mice (3.2% less comparing placebo-pellet mice and 5.4% less compared to cort-pellet mice). Furthermore, cort-pellet Cd36ko mice started to gain weight 2 days later than cort-pellet wild-type mice.
Figure 7. Cd36ko partially protects against adipocyte hypertrophy resulting from flattening of glucocorticoid oscillations.
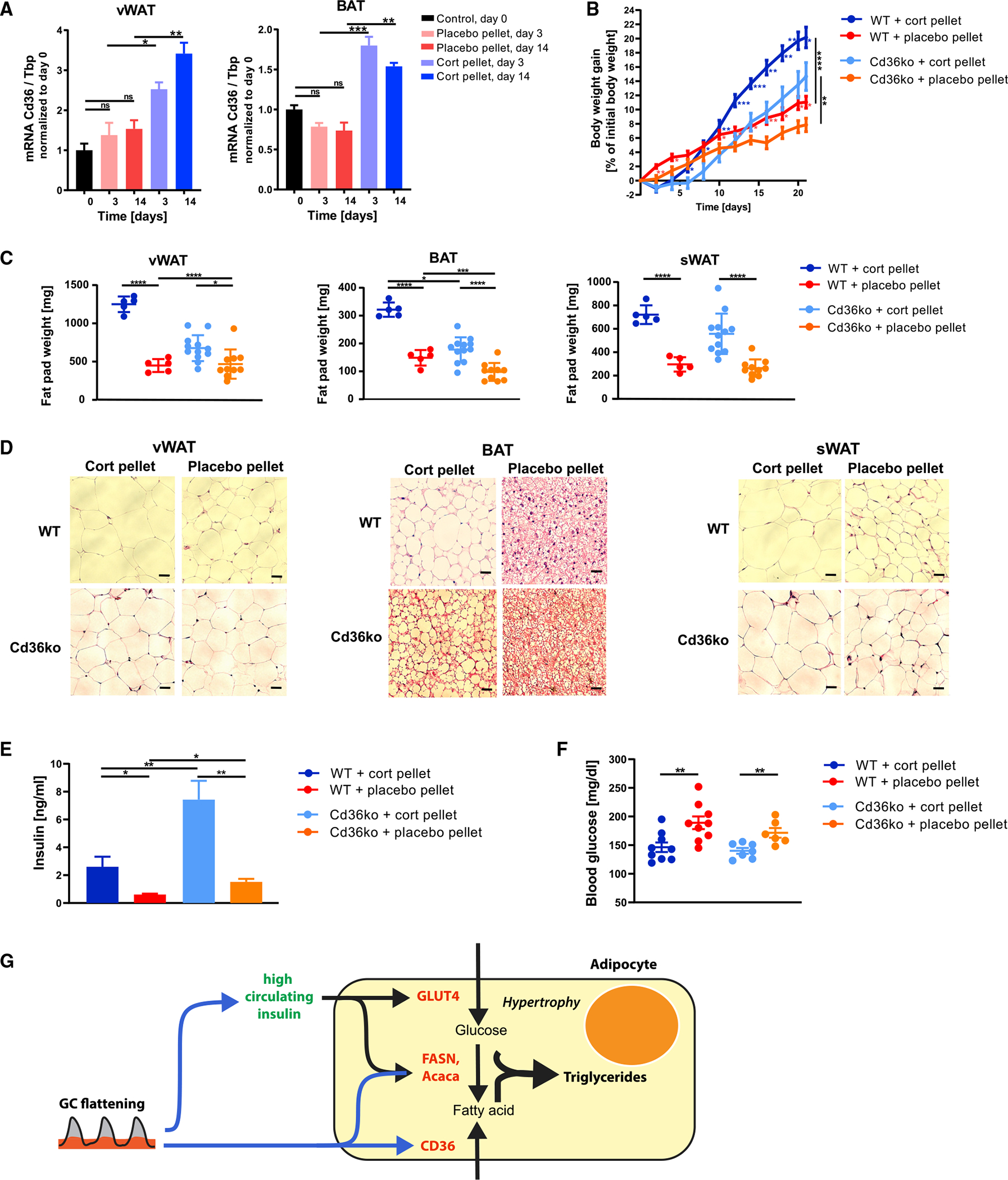
(A) qPCR of vWAT and BAT 3 and 14 days after cort- or placebo-pellet implantation. Data are normalized to the expression of Tbp and are presented as means ± SEMs (n = 3–4). Unpaired t test; *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. Control group are mice at start of experiment (day 0).
(B and C) Body weight gain (B) and (C) weight of vWAT, BAT, and sWAT fat depots 21 days after pellet implantation, n = 5–12, means ± SDs. Unpaired t test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Table S1.
(D) H&E staining of vWAT, BAT, and sWAT 21 days after pellet implantation. One representative staining is shown (n = 3–5). Scale bars, 50 μm. Quantification of lipid droplet size is shown in Figure S7.
(E) Insulin levels in blood plasma in cort- and placebo-pellet-implanted Cd36ko and wild-type mice (not fasted) 14 days after pellet implantation; means ± SEMs; n = 5–7; unpaired t test; *p < 0.05; **p < 0.01.
(F) Blood glucose levels in cort- and placebo-pellet-implanted Cd36ko and wild-type mice (not fasted) 14 days after pellet implantation; means ± SEMs; n = 6–9; unpaired t test; **p < 0.01.
(G) Scheme of how an initially protective adipocyte hypertrophy mechanism can be activated in response to flattening of circadian GC oscillations. GC flattening results in a distinct metabolic signature with high insulin, slightly lower-than-normal glucose, and normal FFA levels in the circulation.
To determine whether the lower body weight gain of corticosterone-implanted Cd36ko mice is manifested in lower organ weights of the fat depots, sWAT, vWAT, and BAT were dissected and weighed. As had been observed previously in Figures 1E–1G, flattening GC oscillations in wild-type mice increased vWAT, sWAT, and BAT weight more than 2-fold over control mice (dark blue compared to red; Figure 7C). Remarkably, knocking out Cd36 resulted in a much smaller increase in weight for all three fat tissue types (light blue compared to orange; Figure 7C). Histological analysis of the three fat tissue types showed that adipocytes of cort-pellet Cd36ko mice displayed lower increases in cell volume than cort-pellet wild-type mice (Figure 7D), further supporting the idea that at least part of the adipose hypertrophy in GC-flattened mice is caused by Cd36-mediated fatty acid uptake.
Loss of Cd36 did not protect against hyperinsulinemia when GCs were flattened, and in fact, insulin levels even increased in cort-pellet Cd36ko mice (Figure 7E), with a corresponding lowering of blood glucose levels (Figure 7F). Interestingly, higher insulin levels were also observed in placebo-pellet Cd36ko mice compared to placebo-pellet wild-type mice, suggesting that Cd36ko mice show in general higher insulin levels (Figure 7E). In conclusion, Cd36 loss partially protects vWAT and BAT from lipid accumulation induced by flattening GC oscillations. This protective effect is not observed for sWAT, pointing to depot-specific differences between the visceral and subcutaneous fat depot. That the rescue of adipocyte hypertrophy in Cd36ko mice is only partial is consistent with the interpretation that the remaining hypertrophy is the result of hyperinsulinemia. Together, these results suggest that adipocyte hypertrophy in response to GC flattening is caused by a combination of Cd36-mediated uptake of fatty acids and hyperinsulinemia-mediated increases in glucose uptake and de novo fatty acid synthesis from glucose.
DISCUSSION
Since circulating GCs are a negative feedback signal to the HPA axis, the exogenous GCs released by the corticosterone pellets suppress HPA axis activation and flatten the normal circadian GC levels (Bellavance and Rivest, 2014; Gjerstad et al., 2018). By implanting corticosterone pellets, we are mimicking flattened GCs that may occur with chronic stress or Cushing’s disease and that have been shown to cause obesity in rodents and humans (Bahrami-Nejad et al., 2018; Campbell et al., 2011; Chimin et al., 2014; Dallman et al., 2000; Joseph and Golden, 2017; Karatsoreos et al., 2011; Rossi et al., 2000), Here, we sought to understand the mechanisms underlying the obesity. Strikingly, within 3 days of GC flattening, circulating insulin levels became persistently high and thousands of transcripts in WAT and BAT changed. These effects preceded the adipocyte hypertrophy and expansion of WAT observed at 7 days.
Most lipid stored in adipocytes is thought to be synthesized from fatty acids taken up from the circulation, rather than being synthesized from glucose (Di Girolamo et al., 1971; Santoro et al., 2021). However, our analysis of Cd36ko mice showed that increased fatty acid uptake were only partially responsible for the adipocyte hypertrophy.
Since adipocyte hypertrophy is typically associated with increased fatty acids and triglycerides in the circulation and increased fat deposition in the liver (Campbell et al., 2011; Harris et al., 2013), we were surprised to find no increase in circulating fatty acid levels and no measurable fat deposition in the liver. Furthermore, because hyperinsulinemia is typically closely associated with high glucose levels, we were also surprised to find that circulating glucose levels were even slightly lower in mice with flattened GCs and hyperinsulinemia, compared to control animals.
Our GTT and ITT results showed that the cort-pellet mice remained insulin sensitive and able to uptake glucose from the circulation. The significant upregulation of de novo fatty acid synthesis genes observed in vWAT and BAT in our RNA-seq analysis support the idea that the percentage of glucose converted to triglycerides in these tissues is high. Furthermore, the lack of increase in lipolytic genes in vWAT and BAT and low circulating FFAs support that the newly synthesized triglycerides are retained in the adipocytes. Taken together, our results argue that increased insulin-mediated glucose uptake, de novo synthesis of fatty acids from glucose, and suppression of lipolysis in adipocytes drive the adipocyte hypertrophy in the GC-flattened mice.
The hyperinsulinemia we observed when GCs were flattened is consistent with observations in human patient studies of subcritical hypercortisolism (Papanastasiou et al., 2017; Rossi et al., 2000). We showed that pancreatic beta cell proliferation is increased when GCs are flattened (Figure S6A), but there could be other contributing factors such as increased beta cell neogenesis or transdifferentiation, a change in signaling in beta cells, or reduced insulin clearance in the liver (Bojsen-Møller et al., 2018; Protzek et al., 2016). Further studies are needed to determine all of the causes of hyperinsulinemia in the absence of increased glucose when GCs are flattened.
The implanted pellets were designed to release corticosterone at a constant rate for 21 days. Interestingly, analysis of mice at 42 days, 3 weeks after the GC-flattening stimulus ceased, showed that the hyperinsulinemia was completely reversed, GC levels during the trough period had returned to basal levels (Figures S6B and S6C), and approximately half of the gain in WAT mass and all of the gain in BAT mass were reversed as well (Figure 6B). We further found normal levels of circulating FFAs and no lipid accumulation in the liver after 2–3 weeks of GC flattening, supporting the idea that there is no significant loss of metabolic health during this period, despite a more than doubling of fat mass and adipocyte hypertrophy in WAT and BAT. Nevertheless, there were signs for longer term effects on the overall metabolism in mice after 3 weeks of GC flattening. Even after the pellet had ceased to release corticosterone for 3 weeks, both vWAT and sWAT mass remained 50% higher and the normally high peak levels of corticosterone during waking were not yet fully restored.
In summary, our study shows the cellular and physiological consequences of temporary flattening of GCs that is frequently observed during chronic stress. Loss of the normal circadian GC oscillation pattern results in acute hyperinsulinemia and a more-than-doubling of WAT and BAT within 3 weeks due to a combination of increased insulin-mediated glucose uptake and de novo fatty acid synthesis and increased Cd36-mediated fatty acid uptake (Figure 7G). Remarkably, for up to at least 3 weeks, fatty acids and glucose levels in the circulation remain near normal levels, and most of the WAT increase and all of the BAT increase can be reversed when GC flattening stops. Continued fatty acid and glucose uptake into fat tissue serves to minimize the damage that could result from high circulating lipid levels due to GC-mediated catabolism of tissues. Intriguingly, GC flattening induces an “alert” state marked by hyperinsulinemia and adipocyte hypertrophy, which is unexpectedly protective for metabolic health for a period of a few weeks.
Limitations of the study
One limitation of our study is that we used a whole-body knockout of Cd36, and knockout of Cd36 in other cell types and tissues such as skeletal muscle could have effects on the fat mass increases in our study. Furthermore, recent studies have shown that the metabolic actions of Cd36 cannot always be reduced to its role as a fatty acid transporter. For example, Cd36 deficiency has been shown to affect insulin signaling (Samovski et al., 2018) and to generate intracellular PPARG ligands, which activate PPARG signaling (Nagy et al., 1998; Tontonoz et al., 1998). Carrying out studies in an adipocyte-specific Cd36ko mouse model would help to better clarify the role of Cd36. Our results show that mice remain, for the most part, metabolically healthy up to a 3-week period of GC flattening, but future studies are needed to more precisely determine whether and when there is a transition from reversible to long-term loss of metabolic health. Furthermore, results from us and others suggest that the weight gain in mice with dysregulated GCs cannot be explained by reduced thermogenesis, increases in food intake, or reductions in energy expenditure (Luijten et al., 2019). Future studies are needed to elucidate the mechanisms that may involve increased nutrient absorption or a currently unexplained pathway that results in increased metabolic efficiency.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. Mary N. Teruel (mnt4002@med.cornell.edu).
Materials availability
This study did not generate any new reagents.
Data and code availability
RNAseq datasets have been deposited at GEO and are publically available as of the date of publication. Accession numbers are listed in the key resources table. Original western blots have been deposited at Weill Cornell Medicine eCommons and are publically available as of the data of publication. The web address is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited on github and is publically available as of the data of publication. The web address is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-Thermogenin (UCP1) | α-Diagnostics | Cat#UCP11-A; RRID:AB_1876090 |
| Mouse monoclonal β-Actin (HRP conjugated) | Santa Cruz | Cat#sc-47778 HRP; RRID:AB_2714189 |
| Goat anti-rabbit IgG | Cell Signaling | Cat#7074; RRID:AB_2099233 |
| Guinea pig anti-insulin IgG | Dako | Cat#IR002; RRID:AB_2800361 |
| Rabbit anti-Ki-67 IgG | Invitrogen | Cat#MA5-14520; RRID:AB_10979488 |
| Goat anti-guinea pig IgG | Jackson ImmunoResearch | Cat#106-545-003; RRID:AB_2337438 |
| Goat anti-rabbit IgG | Invitrogen | Cat#A11036; RRID:AB_10563566 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Corticosterone pellets (5mg, 21-day release) | Innovative Research of America | Cat#G-111 |
| Placebo pellets (5mg, 21-day release) | Innovative Research of America | Cat#C-111 |
| Corticosterone complexed with 2-hydroxypropyl-β-cyclodextrin | Sigma-Aldrich | Cat#C174 |
| Glucose | Sigma-Aldrich | Cat#G8270 |
| Insulin | Sigma-Aldrich | Cat#I9278 |
| Normal Goat Serum | Jackson ImmnuoResearch | Cat#005-000-121 |
| SlowFade™ Gold Antifade Mountant with DAPI | Thermo Scientific | Cat#S36939 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ELISA corticosterone | Arbor Assays | Cat#K014-H1 |
| ELISA insulin | Crystal Chem | Cat#90080 |
| Free Fatty Acid Quantitation Kit | Sigma | Cat#MAK044 |
| Triglyceride Determination Kit | Sigma | Cat#TR0100 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| iScript cDNA Synthesis | Bio-Rad | Cat#1708890 |
| PowerUp SYBR Green Master Mix | ThermoFisher | Cat#4368577 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| Qscript™ cDNA Synthesis Kit | VWR | Cat#101414-098 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | Cat#1725121 |
| L-Type Triglyceride M Enzyme Color A | Fujifilm Healthcare | Cat#994-02891 |
| L-Type Triglyceride M Enzyme Color B | Fujifilm Healthcare | Cat#990-02991 |
| Multi-Calibrator Lipid | Fujifilm Healthcare | Cat#464-01601 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq | This paper | NCBI Geo GSE202800 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mice: C57BL/6J | Jackson Labs | Cat#000664 |
| Mice: C57BL/6J; CD36ko | Jackson Labs | Cat#019006 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S8 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Fiji software plug-in Adiposoft | Galarraga et al. (2012) | https://imagej.net/plugins/adiposoft |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Custom RNAseq analysis codes | This paper | https://doi.org/10.5281/zenodo.6588146 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Seven-week-old C57BL/6J male mice were purchased from Jackson Laboratory (cat. 000664) and housed in the animal facility for 7 days prior to the start of experiments. Cd36ko mice were used as published before (Febbraio et al., 1999). Cd36ko breeding pairs were purchased from Jackson Laboratory (cat. 019006), and 8-week old male Cd36ko mice were used for experiments.
Mice were housed in small groups of five or fewer on a 12h light/dark cycle (lights on at 7:00 h) with food and water access ad libitum in the animal facility at Stanford University or at Weill Cornell Medicine. All mice were maintained on a normal chow diet and only male mice were used for experiments. All animal care and experimentation were conducted in accordance with current NIH and Institutional Animal Care and Use Committee guidelines. All mice were dissected between 13:00 and 18:00 alternating corticosterone-treated and control mice. A summary of all parameters measured in mice upon different treatments can be found in Table S1.
During the acute cold exposure experiments, mice were placed in individual cages with minimal bedding in order to prevent nesting or huddling behavior which would interfere with adaptive thermogenesis. The cages were placed in a 4°C deli fridge for the 6-h time course experiment. Core body temperatures of the mice were assessed with a rectal probe thermometer (Type J/K/T Thermocouple meter, Digisense, Kentscientific, USA; with rectal probe for mice RET-3, Physitemp Instruments, USA) before and at one-hour intervals during the 6-h cold exposure. Time point 0 was taken 1 min before the mice went into the cold.
METHOD DETAILS
Corticosterone administration in mice
To flatten circadian GC oscillations, mice were implanted subcutaneously with corticosterone releasing pellets (5mg, 21-day release; Innovative Research of America, Sarasota, FL, USA, Catalog number G-111). Placebo pellets (Catalog number C-111) were implanted as control. For pellet implantation mice were anesthetized via inhalation of isoflurane, when anesthetized an incision equal in diameter to that of the pellet was made on the lateral side of the neck and the pellet inserted using a trochar. Mice weighed an average of 24.1 ± 1.2 g, which results in a daily dose of 9 mg/kg/day.
For injections, corticosterone complexed with 2-hydroxypropyl-β-cyclodextrin (C174, Sigma) was dissolved in phosphate buffered saline (PBS) and injected subcutaneously once daily at 17:00 for up to 21 days with the same corticosterone dose (9 mg/kg/day) as released by the corticosterone pellets per day. PBS was injected as control (vehicle).
Measurement of corticosterone in blood serum
Blood was taken at multiple time points over a 15 h time period. At the first time point, blood was taken by nicking the tail vain. Blood samples collected on following time points were taken by removal of the crust formed after first blood withdrawal. The blood was allowed to clot by leaving it undisturbed at room temperature for 45 min. The clot was removed by centrifuging at 2000 × g for 15 min. The corticosterone concentration in blood serum was determined using the Enzyme Immunoassay kit (K014-H1, Arbor Assays, Michigan, USA) following the manufacturer’s instructions.
Glucose tolerance test
Before the procedure mice were fasted for 6 h in clean cages with no food or feces in the bedding. At 9 am blood glucose levels were measured with a glucometer (Diathrive, South Salt Lake, Utah, USA) by obtaining a drop of blood from the tail. Subsequently, mice received 2 g of glucose (Sigma G8270)/kg of body weight by intraperitoneal injection. Blood glucose levels were measured again after 15, 30, 60, and 120 min after injection.
To measure insulin levels, 20 μL of blood was collected from the tail before glucose injection and on every following time point and mixed with heparin. The blood plasma was prepared by centrifugation at 2000 × g for 15 min. The insulin concentration in blood plasma was determined using the Ultra-Sensitive Mouse Insulin ELISA Kit (Cat. 90080, Crystal Chem, Illinois, USA) following the manufacturer’s instructions.
Insulin tolerance test
Before the procedure mice were fasted for 6 h in clean cages with no food or feces in the bedding starting at 9am. Blood glucose levels were measured with a glucometer (Diathrive, South Salt Lake, Utah, USA) by obtaining a drop of blood from the tail. Subsequently, mice were injected with 0.75 units/kg body weight of human insulin (Sigma I9278) by intraperitoneal injection. Blood glucose levels were measured again after 15, 30, 45, 60, and 120 min after injection.
To measure insulin levels, 20 mL of blood was collected from the tail before glucose injection and on every following time point and mixed with heparin. The blood plasma was prepared by centrifugation at 2000 × g for 15 min. The insulin concentration in blood plasma was determined using the Ultra-Sensitive Mouse Insulin ELISA Kit (Cat. 90080, Crystal Chem, Illinois, USA) following the manufacturer’s instructions.
Measuring concentrations of FFA, glycerol, and triglycerides
FFA contents in blood serum were determined using the Free Fatty Acid Quantitation Kit (Sigma, MAK044) following the manufacturers’ protocols. Glycerol and triglyceride concentrations in blood serum were measured using the Triglyceride Determination Kit (Sigma, TR0100) following the manufacturer’s instructions.
Measurement of liver triglycerides
Livers were excised, snap frozen in liquid nitrogen, and stored at −80°C until analysis. Frozen livers (250 mg) were homogenized in a Potter-Elvehjem tube containing 1 mL methanol. Lipids were extracted into chloroform:methanol (2:1) and phases were separated using 0.05% H2SO4. The mixture was incubated overnight at room temperature and centrifuged at 1,000 × g for 20 min to ensure proper phase separation. 1 mL of the lower organic layer was collected and mixed with 0.5 mL 3% Triton X-100 in chloroform. This mixture was dried under an N2 stream then resuspended in 0.5 mL water. Concentration of triglycerides was measured using the Wako L-Type TG M test kit (Fujifilm Healthcare Aericas, Lexington, MA).
Histology
For histological examination interscapular brown adipose tissue (BAT), inguinal white adipose tissue (sWAT), and epididymal white adipose tissue (vWAT) were fixed in 4% formalin for 24 h directly after dissection. Tissues were rinsed and stored in 95% ethanol until embedding in paraffin. Paraffin blocks were sectioned (10μm) and sections mounted on glass slides and stained with hematoxylin and eosin (H & E). Images were taken at 20x magnification and the adipocyte area was calculated using the Fiji software plug-in Adiposoft (Galarraga et al., 2012).
Reverse transcription quantitative polymerase chain reaction
Total RNA was extracted from fat tissues using the RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany) and from liver using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Equal amounts of RNA were reverse transcribed to cDNA using the iScript cDNA Synthesis system (Bio-Rad, Hercules, CA, USA). Relative transcript amounts were measured by qPCR using PowerUp SYBR Green Master Mix (Appliedbiosystems, Vilnius, Lithuania). All expression data were normalized to TATA binding protein (Tbp) expression.
The PCR cycle at which PCR amplification begins its exponential phase was considered as the cycle threshold (Ct). The ΔCt value was obtained as a difference between the Ct of the target genes and Ct of reference gene (Tbp). The y axis of “Relative mRNA fold-change Gene-of-interest/Tbp” represents the difference between the ΔCt values (ΔCt Cort - ΔCt Placebo).
RNAseq
RNA was extracted from BAT or vWAT using the RNeasy Lipid Tissue Mini Kit (Qiagen, Hilden, Germany). For each condition (placebo pellet, corticosterone pellet, placebo injection, corticosterone injection) RNA was extracted from 4 mice per time point (3, 7, 14 days). RNA was also isolated from 4 mice before start of treatment (day 0). RNA quality of all samples was evaluated by Bioanalyzer 2100 High Sensitivity RNA Analysis chips (Agilent, Cat. 5067-1513) which displayed intact RNA integrity. mRNA samples were concentrated to ≤ 5 μL by MinElute column (QIAGEN, Cat. 74204). For generation of RNA-seq libraries, polyadenylated mRNA was isolated from 300 ng of total RNA by incubation with oligo-DT attached magnetic beads and followed by strand-specific library preparation using the TruSeq Stranded mRNA Library Preparation kit (Ilumina, Cat. 20020595). Briefly, isolated polyadenylated mRNA was fragmented using divalent cations under elevated temperature and first and second strands DNA were synthesized using SuperScript II Reverse Transcriptase (provided with Ilumina kit). A-tailing and adapter ligation was performed according to the manufacturer’s protocol; the resulting dsDNA was enriched in a PCR reaction based on predetermined CT values and cleaned using AMPure XP beads (provided with Ilumina kit). Concentrations of enriched dsDNA fragments with specific adapters were determined and base pair average size and library integrity were analyzed using the Bioanalyzer DNA High Sensitivity chips (Agilent, Cat. 5067-4626). Samples were pooled and sequenced on the Illumina NextSeq 500/550 High Output platform (Illumina, FC-404-2002) up to 18 samples per lane with 1% PhiX spike as a control.
To assess the read quality of the raw FASTQ files we used FastQC v0.11.7 (Babraham Bioinformatics (Hayashi et al., 2004)). Pseudo-alignment of the reads to the mouse reference transcriptome (Mus_musculus.GRCm38.cdna) was carried out using Kallisto v0.44.0 (Bray et al., 2016) with the quantification algorithm enabled, the number of bootstraps set to 100, and ran in paired-end mode. The Kallisto output files imported to R with Sleuth, and the transcripts per million (TPM) were used for downstream differential expression analysis (Pimentel et al., 2017).
We used the Sleuth package to identify differentially expressed genes with the full model consisting of treatment, lane, replicate, and day and the reduced model consisting of lane and replicate. This allowed us to identify genes that were differentially expressed based on treatment (i.e. corticosterone pellet vs placebo pellet) and time. All downstream analysis was done on transcripts that were considered significant from the DESeq2 analysis (likelihood ratio test with an FDR less than 0.05). Outlier samples were identified by both clustering and PCA analysis and removed. The number of differentially expressed genes after removing outliers increased by 1,668, with all but 31 genes common between the two models.
For more information, please view the analysis code repository on the Zenodo website at https://doi.org/10.5281/zenodo.6588146.
Gene ontology (GO) analysis
GO term analysis was done using the clusterProfiler package (Yu et al., 2012). GO terms of interest were identified by calculating the adjusted p value between two sample groups (i.e. corticosterone pellet vs. placebo pellet) as well as a Z score for general trends of increased or decreased differential expression of the genes within the GO terms. The Z score is defined as the number of genes within the GO term with an absolute value log2 fold change greater than or equal to 1.5 (fold change >1.5 or <0.67) divided by the square root of the total number of genes assigned to that GO term (Walter et al., 2015).
Western blot analysis
For Western blot analysis 10mg of fat tissue were lysed in 50 mM Tris-HCl, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate (SDS) and 0.15 M NaCl, pH 8, supplemented with protease inhibitors (cOmplete, Mini, EDTA-free, Sigma Aldrich). For preparation of tissue lysates, a homogenizer (Fisher Scientific) was used and cell debris was removed by centrifugation at max speed for 20 min at 4°C. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, USA). 50 μg of proteins of tissue lysates were boiled for 5 min at 95°C in NuPAGE LDS sample buffer containing 1x reducing agent (Invitrogen). Lysates were separated by SDS–PAGE using the XCell SureLock Electrophoresis Cell on NuPAGE Novex 4%–12% Bis-Tris Protein Gels (Invitrogen). Proteins were transferred onto polyvinylidene fluoride membranes (Thermo Scientific) using the XCell II Blot Module (Invitrogen). Membranes were blocked by incubating with TBS with 0.1% Tween 20 containing 3% non-fat milk for 2h at room temperature or overnight at 4°C. After blocking, membranes were exposed to the primary antibody (UCP-1; used 1:800) overnight at 4°C. After washing (TBS with 0.1% Tween 20), the membranes were incubated for 2 h with the secondary antibody. The membranes were washed (TBS with 0.1% Tween 20) and developed with the Supersignal West Femto chemiluminescence substrate (Pierce, Rockford, USA). Peroxidase activity was detected with a Lumi Imager device (ImageQuant LAS 4000mini, GE Healthcare). The primary antibody UCP-1 was purchased from α-Diagnostics (Catalog No. UCP11-A). The secondary antibody was purchased from Cell Signaling (a-rabbit: Catalog No. #7074).
Immunohistochemistry
After 21-days of GC flattening, mice were sacrificed, and the pancreas was harvested. To study β-cell proliferation [Insulin/Ki67], immunofluorescence staining was carried out as previously described (Patel et al., 2022). Briefly, the pancreas was fixed in 4% paraformaldehyde for histological processing and paraffin embedding. 5 μm sections were cut from the paraffin-embedded blocks. The sections were deparaffinized in xylene and rehydrated in a series of graded ethanol. Antigen retrieval was performed using 1 N HCL for 45 min at 37°C. Sections were blocked in 5% normal goat serum in PBST (PBS + 0.1% Tween 20) for 1-h at room temperature and incubated with primary antibodies prepared in blocking buffer at 37°C in a humidified chamber for 2-h. Sections were washed with PBS and were incubated with secondary antibodies prepared in blocking buffer at room temperature for 45 min in the dark. Sections were washed with PBS and distilled water and were mounted with Slowfade® Gold Antifade mountant with DAPI, and the coverslip was sealed with nail varnish. Stained sections were observed under a fluorescence microscope (Nikon Ti2E, Japan). Image analysis was carried out in ImageJ software. Observations were made from three pancreatic sections per group from five different areas.
Metabolic cages
Metabolic monitoring was conducted using a Promethion Metabolic Screening System (Promethion High-Definition Multiplexed Respirometry System for Mice; Sable Systems International, Las Vegas, NV, USA) as previously described (Krisko et al., 2020). Mice were placed in individual metabolic cages with a consistent volume of pine chip bedding, without nesting, for two days acclimation prior to the day of data recording. Rates of oxygen consumption (VO2) and carbon dioxide production (VCO2) were acquired by indirect calorimetry with a sampling frequency of 1 s. Values of respiratory exchange ratio (RER) were calculated as ratios of VCO2 to VO2. Food intake and body mass were recorded continuously by gravimetric measurements within the cages. Physical activity was determined according to beam breaks within a grid of infrared sensors built into each cage.
Energy expenditure, calculated using the Weir equation (Energy expenditure = 3.941 kcal/L × VO2 + 1.106 kcal/L × VCO2), is displayed as the total kcal per specified periods of time, with values adjusted by ANCOVA (Speakman, 2013) for body mass using VassarStats. Metabolic cages were contained within temperature-controlled environmental enclosures (DB034 Laboratory Incubator, Darwin Chambers, St. Louis, MO, USA).
Body compositions
Body composition (lean and fat) was measured by NMR spectroscopy using an EchoMRI 3-in-1 Body Composition Analyzer (Houston, TX, USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as mean ± SD or mean ± SEM and analyzed by unpaired t test using GraphPad Prism software. n indicates the number of animals per group or number of independent experiments. Results were considered significant if p < 0.05.
Supplementary Material
Highlights.
Flattening of circadian glucocorticoid oscillations causes rapid hyperinsulinemia
Hyperinsulinemia drives adipocyte hypertrophy and a lean-to-fat mass shift
Hyperinsulinemia occurs without hyperglycemia
The adipocyte hypertrophy and hyperinsulinemia are reversible even after 3 weeks
ACKNOWLEDGMENTS
This work was supported by NIH RO1-DK101743, RO1-DK106241, P50-GM107615, and R56-DK131432 and Stanford BioX Seed Grant funding (to M.N.T.), as well as the U.S. Department of Veterans Affairs I01BX000398 (to F.B.K.). We also acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) TH 2156/1-1 (to S.T.), the Stanford Bio-X Program and Novo Nordisk Foundation NNF16OC0018642 (to A.R.), and from NIDDK R01 DK103046, MPI–Cohen (to H.T.N.). We thank Tim McGraw, Fred Maxfield, Laura Alonso, Tobias Meyer (Weill Cornell Medicine), and the members of the Teruel lab for discussions and critical reading of the manuscript. We also thank the Stanford Animal Histology Services for paraffin embedding of fat tissue and performing H&E staining and the Weill Cornell Medicine Metabolic Phenotyping Center for performing the mouse phenotyping studies.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111018.
REFERENCES
- Adam EK, Quinn ME, Tavernier R, McQuillan MT, Dahlke KA, and Gilbert KE (2017). Diurnal cortisol slopes and mental and physical health outcomes: a systematic review and meta-analysis. Psychoneuroendocrinology 83, 25–41. 10.1016/j.psyneuen.2017.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrami-Nejad Z, Zhao ML, Tholen S, Hunerdosse D, Tkach KE, van Schie S, Chung M, and Teruel MN (2018). A transcriptional circuit filters oscillating circadian hormonal inputs to regulate fat cell differentiation. Cell Metab. 27, 854–868.e8. 10.1016/j.cmet.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellavance M-A, and Rivest S (2014). The HPA – immune axis and the immunomodulatory actions of glucocorticoids in the brain. Front. Immunol. 5, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojsen-Møller KN, Lundsgaard A-M, Madsbad S, Kiens B, and Holst JJ (2018). Hepatic insulin clearance in regulation of systemic insulin concentrations—role of carbohydrate and energy availability. Diabetes 67, 2129–2136. 10.2337/db18-0539. [DOI] [PubMed] [Google Scholar]
- Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood J-A, Li Z, Tamashiro KLK, Schwartz GJ, Makriyannis AM, Kunos G, et al. (2015). A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc. Natl. Acad. Sci. U S A 112, 285–290. 10.1073/pnas.1421420112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandão BB, Poojari A, and Rabiee A (2021). Thermogenic fat: development, physiological function, and therapeutic potential. Int. J. Mol. Sci. 22, 5906. 10.3390/ijms22115906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Broussard JL, and Van Cauter E (2016). Disturbances of sleep and circadian rhythms: novel risk factors for obesity. Curr. Opin. Endocrinol. Diabetes Obes. 23, 353–359. 10.1097/MED.0000000000000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, and Newgard CB (2021). Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat. Rev. Mol. Cell Biol. 22, 142–158. 10.1038/s41580-020-00317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JE, Peckett AJ, D’souza AM, Hawke TJ, and Riddell MC (2011). Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol. 300, C198–C209. 10.1152/ajpcell.00045.2010. [DOI] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359. 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Chimin P, Farias T. da S.M., Torres-Leal FL, Bolsoni-Lopes A, Campaña AB, Andreotti S, and Lima FB (2014). Chronic glucocorticoid treatment enhances lipogenic activity in visceral adipocytes of male Wistar rats. Acta Physiol. 211, 409–420. 10.1111/apha.12226. [DOI] [PubMed] [Google Scholar]
- Chusyd DE, Wang D, Huffman DM, and Nagy TR (2016). Relationships between rodent white adipose fat pads and human white adipose fat depots. Front. Nutr 3. 10.3389/fnut.2016.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courty E, Besseiche A, Do TTH, Liboz A, Aguid FM, Quilichini E, Buscato M, Gourdy P, Gautier J-F, Riveline J-P, et al. (2019). Adaptive β-cell neogenesis in the adult mouse in response to glucocorticoid-induced insulin resistance. Diabetes 68, 95–108. 10.2337/db17-1314. [DOI] [PubMed] [Google Scholar]
- Dallman M, Akana S, Bhatnagar S, Bell M, and Strack A (2000). Bottomed out: metabolic significance of the circadian trough in glucocorticoid concentrations. Int. J. Obes. Relat. Metab. Disord. 24, S40–S46. [DOI] [PubMed] [Google Scholar]
- Delaunay F, Khan A, Cintra A, Davani B, Ling ZC, Andersson A, Ostenson CG, Gustafsson J, Efendic S, and Okret S (1997). Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J. Clin. Invest. 100, 2094–2098. 10.1172/JCI119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Girolamo M, Mendlinger S, and Fertig J (1971). A simple method to determine fat cell size and number in four mammalian species. Am. J. Physiol. 221, 850–858. 10.1152/ajplegacy.1971.221.3.850. [DOI] [PubMed] [Google Scholar]
- Dunford EC, and Riddell MC (2016). The metabolic implications of glucocorticoids in a high-fat diet setting and the counter-effects of exercise. Metabolites 6, 44. 10.3390/metabo6040044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhassan YS, Alahdab F, Prete A, Delivanis DA, Khanna A, Prokop L, Murad MH, O’Reilly MW, Arlt W, and Bancos I (2019). Natural history of adrenal incidentalomas with and without mild autonomous cortisol excess: a systematic review and meta-analysis. Ann. Intern. Med. 171, 107. 10.7326/M18-3630. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SFA, and Silverstein RL (1999). A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J. Biol. Chem. 274, 19055–19062. 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Fine NHF, Doig CL, Elhassan YS, Vierra NC, Marchetti P, Bugliani M, Nano R, Piemonti L, Rutter GA, Jacobson DA, et al. (2018). Glucocorticoids reprogram β-cell signaling to preserve insulin secretion. Diabetes 67, 278–290. 10.2337/db16-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarraga M, Campión J, Muñoz-Barrutia A, Boqué N, Moreno H, Martínez JA, Milagro F, and Ortiz-de-Solórzano C (2012). Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res. 53, 2791–2796. 10.1194/jlr.D023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaben AL, and Scherer PE (2019). Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 20, 242–258. 10.1038/s41580-018-0093-z. [DOI] [PubMed] [Google Scholar]
- Gjerstad JK, Lightman SL, and Spiga F (2018). Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 21, 403–416. 10.1080/10253890.2018.1470238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris C, Roohk DJ, Fitch M, Boudignon BM, Halloran BP, and Hellerstein MK (2013). Large increases in adipose triacylglycerol flux in Cushingoid CRH-Tg mice are explained by futile cycling. Am. J. Physiol. Endocrinol. Metab. 304, E282–E293. 10.1152/ajpendo.00154.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi R, Wada H, Ito K, and Adcock IM (2004). Effects of glucocorticoids on gene transcription. Eur. J. Pharmacol. 500, 51–62. 10.1016/j.ejphar.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Hodes GE, Brookshire BR, Hill-Smith TE, Teegarden SL, Berton O, and Lucki I (2012). Strain differences in the effects of chronic corticosterone exposure in the hippocampus. Neuroscience 222, 269–280. 10.1016/j.neuroscience.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, and Yamada T (2020). UCP1 dependent and independent thermogenesis in Brown and beige adipocytes. Front. Endocrinol. 11, 498. 10.3389/fendo.2020.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen JAMJL (2021). Hyperinsulinemia and its pivotal role in aging, obesity, type 2 diabetes, cardiovascular disease and cancer. Int. J. Mol. Sci. 22, 7797. 10.3390/ijms22157797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JJ, and Golden SH (2017). Cortisol dysregulation: the bidirectional link between stress, depression, and type 2 diabetes mellitus: role of cortisol in stress, depression, and diabetes. Ann. N Y Acad. Sci. 1391, 20–34. 10.1111/nyas.13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatsoreos IN, Bhagat S, Bloss EB, Morrison JH, and McEwen BS (2011). Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc. Natl. Acad. Sci. U S A 108, 1657–1662. 10.1073/pnas.1018375108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisko TI, Nicholls HT, Bare CJ, Holman CD, Putzel GG, Jansen RS, Sun N, Rhee KY, Banks AS, and Cohen DE (2020). Dissociation of adaptive thermogenesis from glucose homeostasis in microbiome-deficient mice. Cell Metab. 31, 592–604.e9. 10.1016/j.cmet.2020.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krycer JR, Quek LE, Francis D, Zadoorian A, Weiss FC, Cooke KC, Nelson ME, Diaz-Vegas A, Humphrey SJ, Scalzo R, et al. (2020). Insulin signaling requires glucose to promote lipid anabolism in adipocytes. J. Biol. Chem. 295, 13250–13266. 10.1074/jbc.ra120.014907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RA, Harris CA, and Wang J-C (2018). Glucocorticoid receptor and adipocyte biology. Nucl. Receptor Res. 5, 101373. 10.32527/2018/101373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leproult R, Copinschi G, Buxton O, and van Cauter E (1997). Sleep loss results in an elevation of cortisol levels the next evening. Sleep 20, 865–870. 10.1093/sleep/20.10.865. [DOI] [PubMed] [Google Scholar]
- Leto D, and Saltiel AR (2012). Regulation of glucose transport by insulin: traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 13, 383–396. 10.1038/nrm3351. [DOI] [PubMed] [Google Scholar]
- Lopez D, Luque-Fernandez MA, Steele A, Adler GK, Turchin A, and Vaidya A (2016). “Nonfunctional” adrenal tumors and the risk for incident diabetes and cardiovascular outcomes: a cohort study. Ann. Intern. Med. 165, 533. 10.7326/M16-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijten IHN, Brooks K, Boulet N, Shabalina IG, Jaiprakash A, Carlsson B, Fischer AW, Cannon B, and Nedergaard J (2019). Glucocorticoid-induced obesity develops independently of UCP1. Cell Rep. 27, 1686–1698.e5. 10.1016/j.celrep.2019.04.041. [DOI] [PubMed] [Google Scholar]
- Macfarlane DP, Forbes S, and Walker BR (2008). Glucocorticoids and fatty acid metabolism in humans: fuelling fat redistribution in the metabolic syndrome. J. Endocrinol. 197, 189–204. 10.1677/JOE-08-0054. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM, and Wilson DF (2019). The central role of glucokinase in glucose homeostasis: a perspective 50 Years after demonstrating the presence of the enzyme in islets of langerhans. Front. Physiol. 10, 148. 10.3389/fphys.2019.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazziotti G, Gazzaruso C, and Giustina A (2011). Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol. Metab. 22, 499–506. [DOI] [PubMed] [Google Scholar]
- McEwen B (2000). Allostasis and allostatic load implications for neuropsychopharmacology. Neuropsychopharmacology 22, 108–124. 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- Nagy L, Tontonoz P, Alvarez JGA, Chen H, and Evans RM (1998). Oxidized LDL regulates macrophage gene expression through ligand activation of PPARg. Cell 93, 229–240. 10.1016/S0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- Oster H, Challet E, Ott V, Arvat E, de Kloet ER, Dijk D-J, Lightman S, Vgontzas A, and Van Cauter E (2017). The functional and clinical significance of the 24-hour rhythm of circulating glucocorticoids. Endocr. Rev. 38, 3–45. 10.1210/er.2015-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papanastasiou L, Alexandraki KI, Androulakis II, Fountoulakis S, Kounadi T, Markou A, Tsiavos V, Samara C, Papaioannou TG, Piaditis G, et al. (2017). Concomitant alterations of metabolic parameters, cardiovascular risk factors and altered cortisol secretion in patients with adrenal incidentalomas during prolonged follow-up. Clin. Endocrinol. 86, 488–498. 10.1111/cen.13294. [DOI] [PubMed] [Google Scholar]
- Patel R, Parmar N, Rathwa N, Palit SP, Li Y, Garcia-Ocaña A, and Begum R (2022). A novel therapeutic combination of sitagliptin and melatonin regenerates pancreatic b-cells in mouse and human islets. Biochim. Biophys. Acta Mol. Cell Res. 1869, 119263. [DOI] [PubMed] [Google Scholar]
- Peckett AJ, Wright DC, and Riddell MC (2011). The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism 60, 1500–1510. 10.1016/j.metabol.2011.06.012. [DOI] [PubMed] [Google Scholar]
- Pepino MY, Kuda O, Samovski D, and Abumrad NA (2014). Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 34, 281–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- Protzek AOP, Rezende LF, Costa-Júnior JM, Ferreira SM, Cappelli APG, Paula FMMD, Souza JCD, Kurauti MA, Carneiro EM, Rafacho A, et al. (2016). Hyperinsulinemia caused by dexamethasone treatment is associated with reduced insulin clearance and lower hepatic activity of insulin-degrading enzyme. J. Steroid Biochem. Mol. Biol. 155, 1–8. 10.1016/j.jsbmb.2015.09.020. [DOI] [PubMed] [Google Scholar]
- Rafacho A, Cestari TM, Taboga SR, Boschero AC, and Bosqueiro JR (2009). High doses of dexamethasone induce increased β-cell proliferation in pancreatic rat islets. Am. J. Physiol. Endocrinol. Metab. 296, E681–E689. 10.1152/ajpendo.90931.2008. [DOI] [PubMed] [Google Scholar]
- Rebuffé-Scrive M, Walsh UA, McEwen B, and Rodin J (1992). Effect of chronic stress and exogenous glucocorticoids on regional fat distribution and metabolism. Physiol. Behav. 52, 583–590. 10.1016/0031-9384(92)90351-2. [DOI] [PubMed] [Google Scholar]
- Rossi R, Tauchmanova L, Luciano A, Martino MD, Battista C, Viscovo LD, Nuzzo V, and Lombardi G (2000). Subclinical Cushing’s syndrome in patients with adrenal incidentaloma: clinical and biochemical features. J. Clin. Endocrinol. Metab. 85, 1440–1448. [DOI] [PubMed] [Google Scholar]
- Saito M (2013). Brown adipose tissue as a regulator of energy expenditure and body fat in humans. Diabetes Metab. J. 37, 22. 10.4093/dmj.2013.37.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samovski D, Dhule P, Pietka T, Jacome-Sosa M, Penrose E, Son NH, Flynn CR, Shoghi KI, Hyrc KL, Goldberg IJ, et al. (2018). Regulation of insulin receptor pathway and glucose metabolism by CD36 signaling. Diabetes 67, 1272–1284. 10.2337/db17-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A, McGraw TE, and Kahn BB (2021). Insulin action in adipocytes, adipose remodeling, and systemic effects. Cell Metab. 33, 748–757. 10.1016/j.cmet.2021.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapolsky RM (2004). Why Zebras Don’t Get Ulcers (Holt Paperbacks; ). [Google Scholar]
- Sapolsky RM, Krey LC, and McEwen BS (1986a). The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr. Rev. 7, 284–301. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, and McEwen BS (1986b). The adrenocortical axis in the aged rat: impaired sensitivity to both fast and delayed feedback inhibition. Neurobiol. Aging 7, 331–335. [DOI] [PubMed] [Google Scholar]
- Shao M, Vishvanath L, Busbuso NC, Hepler C, Shan B, Sharma AX, Chen S, Yu X, An YA, Zhu Y, et al. (2018). De novo adipocyte differentiation from Pdgfrβ+ preadipocytes protects against pathologic visceral adipose expansion in obesity. Nat. Commun. 9, 890. 10.1038/s41467-018-03196-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Roh HC, Kumari M, and Rosen ED (2017). Adipocyte glucocorticoid receptor is important in lipolysis and insulin resistance due to exogenous steroids, but not insulin resistance caused by high fat feeding. Mol. Metab. 6, 1150–1160. 10.1016/j.molmet.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Xiaoli AM, and Yang F (2018). Regulation and metabolic significance of De Novo lipogenesis in adipose tissues. Nutrients 10, 1–22. 10.3390/nu10101383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR (2013). Measuring energy metabolism in the mouse – theoretical, practical, and analytical considerations. Front. Physiol. 4. 10.3389/fphys.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiga F, Walker JJ, Terry JR, and Lightman SL (2014). HPA axisrhythms. In Comprehensive Physiology, Terjung R, ed. (Wiley; ), pp. 1273–1298. [DOI] [PubMed] [Google Scholar]
- Strack AM, Bradbury MJ, and Dallman MF (1995). Corticosterone decreases nonshivering thermogenesis and increases lipid storage in brown adipose tissue. Am. J. Physiol. 268, R183–R191. 10.1152/ajpregu.1995.268.1.R183. [DOI] [PubMed] [Google Scholar]
- Strawford A, Antelo F, Christiansen M, and Hellerstein MK (2004). Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. Am. J. Physiol. Endocrinol. Metab. 286, E577–E588. 10.1152/ajpendo.00093.2003. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JGA, Thomazy VA, and Evans RM (1998). PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93, 241–252. 10.1016/S0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- van den Beukel JC, Grefhorst A, Quarta C, Steenbergen J, Mastroberardino PG, Lombès M, Delhanty PJ, Mazza R, Pagotto U, Lely AJ, et al. (2014). Direct activating effects of adrenocorticotropic hormone (ACTH) on brown adipose tissue are attenuated by corticosterone. FASEB J. 28, 4857–4867. 10.1096/fj.14-254839. [DOI] [PubMed] [Google Scholar]
- Viengchareun S, Penfornis P, Zennaro M-C, and Lombès M (2001). Mineralocorticoid and glucocorticoid receptors inhibit UCP expression and function in brown adipocytes. Am. J. Physiol. Endocrinol. Metab. 280, E640–E649. 10.1152/ajpendo.2001.280.4.E640. [DOI] [PubMed] [Google Scholar]
- Walter W, Sánchez-Cabo F, and Ricote M (2015). GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. [DOI] [PubMed] [Google Scholar]
- Weitzman ED, Fukushima D, Nogeire C, Roffwarg H, Gallagher TF, and Hellman L (1971). Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J. Clin. Endocrinol. Metab. 33, 14–22. 10.1210/jcem-33-1-14. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 6, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq datasets have been deposited at GEO and are publically available as of the date of publication. Accession numbers are listed in the key resources table. Original western blots have been deposited at Weill Cornell Medicine eCommons and are publically available as of the data of publication. The web address is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited on github and is publically available as of the data of publication. The web address is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-Thermogenin (UCP1) | α-Diagnostics | Cat#UCP11-A; RRID:AB_1876090 |
| Mouse monoclonal β-Actin (HRP conjugated) | Santa Cruz | Cat#sc-47778 HRP; RRID:AB_2714189 |
| Goat anti-rabbit IgG | Cell Signaling | Cat#7074; RRID:AB_2099233 |
| Guinea pig anti-insulin IgG | Dako | Cat#IR002; RRID:AB_2800361 |
| Rabbit anti-Ki-67 IgG | Invitrogen | Cat#MA5-14520; RRID:AB_10979488 |
| Goat anti-guinea pig IgG | Jackson ImmunoResearch | Cat#106-545-003; RRID:AB_2337438 |
| Goat anti-rabbit IgG | Invitrogen | Cat#A11036; RRID:AB_10563566 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Corticosterone pellets (5mg, 21-day release) | Innovative Research of America | Cat#G-111 |
| Placebo pellets (5mg, 21-day release) | Innovative Research of America | Cat#C-111 |
| Corticosterone complexed with 2-hydroxypropyl-β-cyclodextrin | Sigma-Aldrich | Cat#C174 |
| Glucose | Sigma-Aldrich | Cat#G8270 |
| Insulin | Sigma-Aldrich | Cat#I9278 |
| Normal Goat Serum | Jackson ImmnuoResearch | Cat#005-000-121 |
| SlowFade™ Gold Antifade Mountant with DAPI | Thermo Scientific | Cat#S36939 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ELISA corticosterone | Arbor Assays | Cat#K014-H1 |
| ELISA insulin | Crystal Chem | Cat#90080 |
| Free Fatty Acid Quantitation Kit | Sigma | Cat#MAK044 |
| Triglyceride Determination Kit | Sigma | Cat#TR0100 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| iScript cDNA Synthesis | Bio-Rad | Cat#1708890 |
| PowerUp SYBR Green Master Mix | ThermoFisher | Cat#4368577 |
| RNeasy Lipid Tissue Mini Kit | Qiagen | Cat#74804 |
| Qscript™ cDNA Synthesis Kit | VWR | Cat#101414-098 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | Cat#1725121 |
| L-Type Triglyceride M Enzyme Color A | Fujifilm Healthcare | Cat#994-02891 |
| L-Type Triglyceride M Enzyme Color B | Fujifilm Healthcare | Cat#990-02991 |
| Multi-Calibrator Lipid | Fujifilm Healthcare | Cat#464-01601 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq | This paper | NCBI Geo GSE202800 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mice: C57BL/6J | Jackson Labs | Cat#000664 |
| Mice: C57BL/6J; CD36ko | Jackson Labs | Cat#019006 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S8 | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Fiji software plug-in Adiposoft | Galarraga et al. (2012) | https://imagej.net/plugins/adiposoft |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Custom RNAseq analysis codes | This paper | https://doi.org/10.5281/zenodo.6588146 |