

The NADPH oxidase DUOX1 contributes to epithelial production of alarmins, including interleukin (IL)-33, in response to injurious triggers such as airborne protease allergens, and mediates development of mucus metaplasia and airway remodeling in chronic allergic airways diseases. DUOX1 is also expressed in non-epithelial lung cell types, including macrophages that play an important role in airway remodeling during chronic lung disease. We therefore conditionally deleted DUOX1 in either lung epithelial or monocyte/macrophage lineages to address its cell-specific actions in innate airway responses to acute airway challenge with house dust mite (HDM) allergen, and in chronic HDM-driven allergic airway inflammation. As expected, acute responses to airway challenge with HDM, as well as type 2 inflammation and related features of airway remodeling during chronic HDM-induced allergic inflammation, were largely driven by DUOX1 with the respiratory epithelium. However, in the context of chronic HDM-driven inflammation, DUOX1 deletion in macrophages also significantly impaired type 2 cytokine production and indices of mucus metaplasia. Studies with isolated macrophages revealed a contribution of macrophage-intrinsic DUOX1 in macrophage recruitment upon chronic HDM challenge, as well as features of macrophage activation that impact on type 2 inflammation and remodeling.
The respiratory epithelium plays a key role in innate airway responses to diverse environmental hazards, pathogens and allergens. Indeed, airway responses to injurious airborne triggers or allergens largely involve activation of innate immune reactions initiated by early production of epithelial-derived cytokines, IL-33, IL-25 and TSLP, which orchestrate type 2 immune responses1, 2. The proximal mechanisms by which these early epithelial cytokines are generated are not fully elucidated, but recent studies from our laboratory implicated the important involvement of the NADPH oxidase Dual Oxidase 1 (DUOX1), an oxidant-generating enzyme that is prominently expressed within the airway epithelium and can mediate downstream epithelial signaling pathways by redox-dependent mechanisms3. Moreover, conditions of chronic allergic airways disease are characterized by increased epithelial expression and activation of DUOX1, and associated epithelial activation of redox-sensitive kinases such as Src/EGFR, leading to increased type 2 inflammation and airway remodeling evidenced by mucus metaplasia and subepithelial fibrosis4.
The most abundant leukocytes in the airspaces are resident macrophages, comprising both alveolar and interstitial lung macrophages, which are among the first cells that respond to environmental pathogens and/or tissue damage5. Macrophages also have diverse roles in the context of allergic airway inflammation, which include both pro-inflammatory functions of recruited monocyte-derived macrophages as well as protective and anti-inflammatory actions of resident alveolar macrophages6. While DUOX1 is most prominently expressed in epithelial cells at mucosal surfaces, emerging studies have highlighted apparent functional role(s) of DUOX1 in non-epithelial lineages, including macrophages4, 7, 8. Indeed, several studies have attempted to address the functions of macrophage DUOX1, by using macrophages from Duox1-deficient mice8, 9. However, a limitation of such studies is that observed differences may also be attributable to altered macrophage differentiation or polarization due to potential roles of DUOX1 in extrinsic macrophage regulation, either in the bone marrow or in the lung.
The present studies were conducted to dissect the cell-specific actions of DUOX1 in both innate airway responses to acute house dust mite (HDM) allergen challenge and in chronic HDM-induced allergic airway inflammation, by conditional deletion of DUOX1 in either airway/alveolar epithelial or monocyte/macrophage lineages. As expected3, 4, we observed a prominent role for epithelial-specific DUOX1 in these responses, but also uncovered a significant role of macrophage-intrinsic DUOX1 in type 2 inflammation and mucus metaplasia in the context of chronic HDM-induced allergic airway inflammation. Subsequent studies with isolated macrophages revealed intrinsic roles of DUOX1 in lung macrophage recruitment, as well as features of both classical (M1) and alternative (M2) activation and production of mediators that promote type 2 inflammation.
RESULTS
Epithelial DUOX1 mediates type 2 inflammation in response to acute or chronic airway HDM challenge.
We previously showed that DUOX1 contributes to airway secretion of the alarmin IL-33 and subsequent activation of type 2 cytokines in response to acute allergen challenge3. To address the specific involvement of epithelial DUOX1 in these responses, we used Duox1F/F mice and crossed them with Scgb1a1-CreERT mice to enable tamoxifen-inducible conditional deletion of DUOX1 within the airway epithelium (Duox1Δ/ΔScgb1a1-ERT) prior to allergen challenge (Supplementary Fig. S1A,B). This approach of inducible DUOX1 deletion was chosen to rule out a potential impact of DUOX1 deletion on epithelial differentiation or development. Consistent with earlier findings3, acute exposure to HDM in an unsensitized host provoked rapid release of the epithelial alarmin IL-33 and other innate cytokines (IL-1B, IL-17A) and growth factors (AREG) into the BAL, detectable 1 hr after exposure and in most cases declining thereafter (Fig. 1A). HDM also induced more delayed production of the type 2 cytokines IL-5 and IL-13 (Fig. 1A), presumably secondary to initial IL-33 activation. HDM challenge also induced rapid mRNA induction of Areg and Il1b, but not Il33 or Il17a, and more delayed induction of Il5 and Il13 mRNA (Fig. 1B and Supplementary Fig. S1A). Conditional deletion of DUOX1 within epithelial cells prior to HDM challenge dramatically attenuated most of these responses (Fig. 1A, B), although induction and production of IL-1B and IL-17A were not significantly affected. Thus, DUOX1 within the epithelium appears to be critical for early airway responses to HDM, by promoting secretion of IL-33 and AREG, and for more delayed induction and secretion of IL-5 and IL-15, most likely as a result of initially secreted IL-33.
Figure 1:
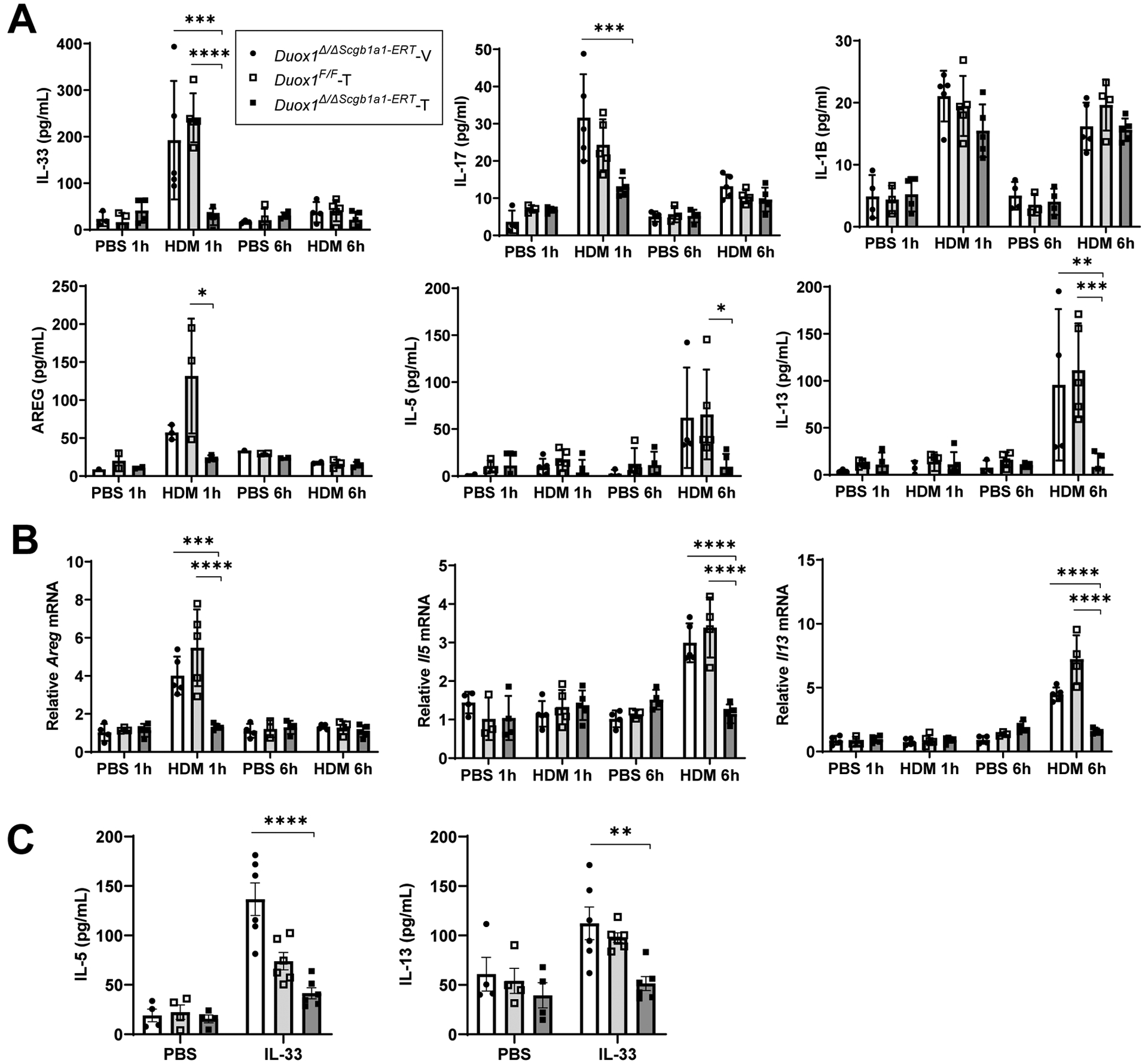
Epithelial DUOX1 deletion attenuates innate type 2 responses to acute HDM challenge. Duox1Δ/ΔScgb1a1-ERT mice, pretreated with either tamoxifen (T) or vehicle control (V), and control Duox1F/F mice were subjected to acute intranasal HDM or PBS, and BAL or tissues were harvested after 1 or 6 hrs. (A) BAL cytokine/growth factor analysis by ELISA. (B) Analysis of lung tissue cytokine/growth factor mRNA levels. (C) Analysis of BAL cytokine responses following 6-hr acute intranasal challenge with 1 μg of recombinant IL-33. Statistical significance was evaluated using 2-way ANOVA and Tukey’s post-hoc test. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001 (n=3–5 mice per group, from 2 independent experiments).
We recently reported that DUOX1 not only contributes to IL-33 secretion in response to HDM challenge, but also appears to participate in epithelial responses to IL-33 mediated by activation of IL33R/ST2 receptors on epithelial cells10. Therefore, we also exposed tamoxifen-treated Duox1Δ/ΔScgb1a1-ERT mice and corresponding controls to recombinant intranasal IL-33, which showed that IL-33-induced production of type 2 cytokines into the BAL are largely driven by epithelial DUOX1 (Fig. 1C). Collectively, these findings indicate that epithelial DUOX1 plays a key role in promoting innate airway responses to allergen (HDM), particularly with respect to IL-33 secretion and IL-33-mediated production of type 2 cytokines.
We previously showed that DUOX1 also mediates type 2 inflammation and airway remodeling during HDM-mediated allergic airway inflammation4. To assess the importance of epithelial DUOX1 for allergic inflammation and remodeling, we subjected Duox1Δ/ΔScgb1a1-ERT mice and appropriate controls to sensitization and repeated airway challenges with HDM. Tamoxifen-induced epithelial-specific deletion of DUOX1 in Duox1Δ/ΔScgb1a1-ERT mice was verified by immunohistochemical analysis (Supplementary Fig. S2A), and did not significantly alter total BAL cell counts after repeated HDM challenges, although macrophage, lymphocyte, eosinophil, and neutrophil counts were in some cases significantly lower compared to vehicle-treated Duox1Δ/ΔScgb1a1-ERT mice or tamoxifen-treated Duox1F/F mice (Fig. 2A and Supplementary Fig. S2B). Consistent with apparent reduction of HDM-induced macrophage recruitment in Duox1Δ/ΔScgb1a1-ERT mice, we also observed a trend toward lower BAL levels of the macrophage chemokine MCP-1 in these mice (Fig. 2B). Epithelial-specific deletion of DUOX1 significantly attenuated HDM-induced production of IL-5, IL-13 and AREG, and showed a trend toward attenuated IL-33 production (Fig. 2B and Supplementary Fig. 2C). Tamoxifen-treated Duox1Δ/ΔScgb1a1-ERT mice also had significantly reduced HDM-induced mucus metaplasia, as measured by Clca1, Muc5ac mRNA and by PAS staining (Fig. 2C and Supplementary Fig. S2D), and displayed significantly reduced expression of collagen genes associated with subepithelial fibrotic remodeling (Supplementary Fig. S2E). Subepithelial collagen production assessed by Masson’s Trichrome staining also tended to be reduced in Duox1Δ/ΔScgb1a1-ERT mice, but this was not statistically significant (Fig. 2D and Supplementary Fig. S2F). In aggregate, these findings are consistent with our previous findings using non-conditional Duox1−/− mice4 and indicate that HDM-induced features of type 2 inflammation, mucus metaplasia and airway remodeling, are largely driven by DUOX1 within the respiratory epithelium.
Figure 2:
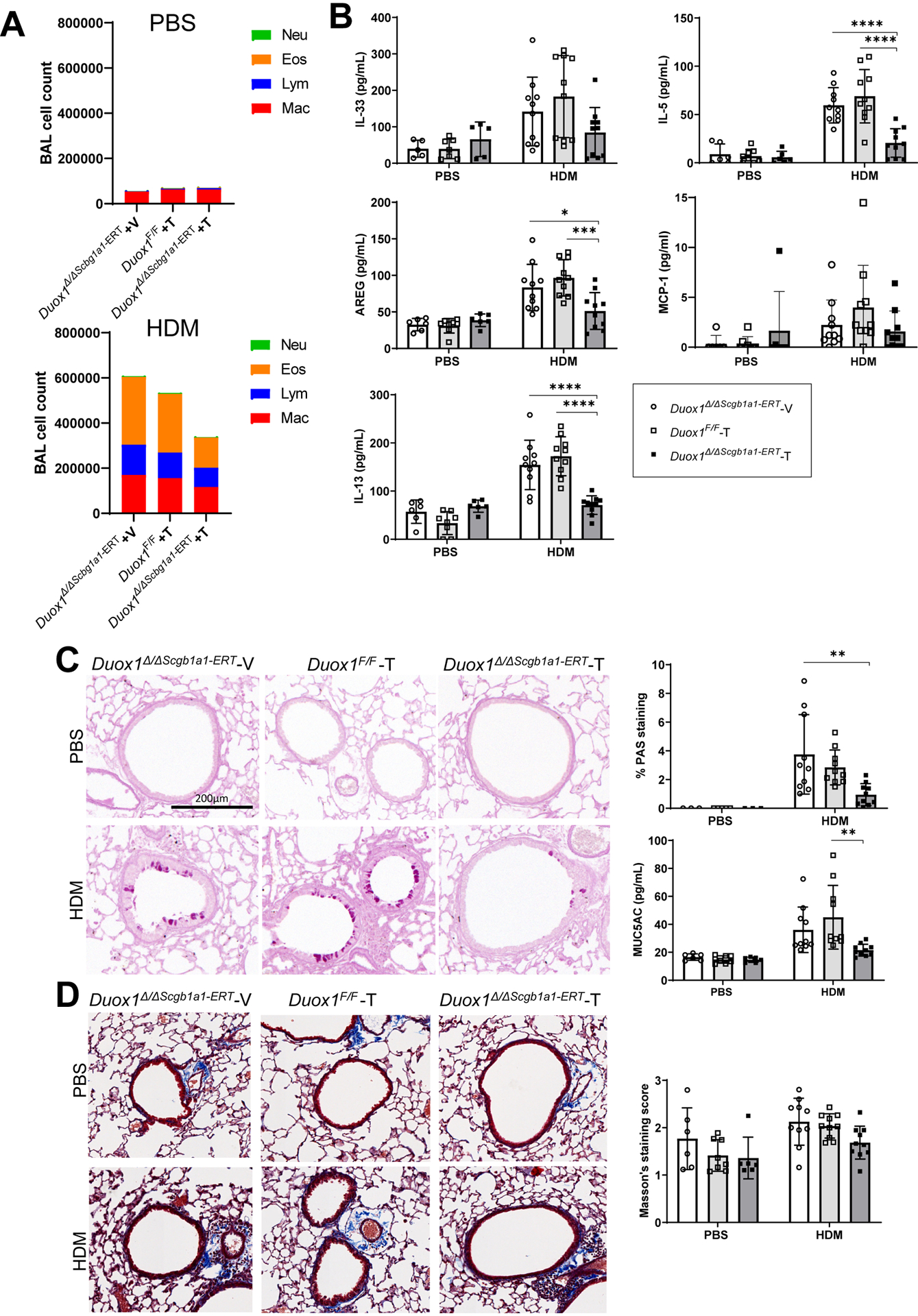
Epithelial DUOX1 deletion attenuates type 2 inflammation and airway remodeling during chronic HDM challenge. Duox1Δ/ΔScgb1a1-ERT mice, pretreated with tamoxifen (T) or vehicle control (V), and control Duox1F/F mice were subjected to chronic HDM to induce allergic airway disease. (A) Analysis of BAL cell differentials. (B) ELISA analysis of BAL cytokines/growth factors. (C) Analysis or mucus metaplasia by PAS staining and MUC5AC analysis in BAL. Representative PAS staining images (10x magnification) and quantified PAS staining are shown. (D) Analysis and quantification of collagen deposition by Masson’s trichrome staining (20x magnification). Statistical significance was evaluated using 2-way ANOVA and Tukey’s post-hoc test. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001 (n = 6–10 mice per group, from 2 independent experiments).
We also used Duox1F/F mice to confirm the therapeutic potential of DUOX1 inhibition in allergic airways disease, by first subjecting them to HDM sensitization and challenge, and subsequent administration of Adeno-Cre in mice immediately after the final HDM challenge to delete airway DUOX1 (Fig. S3A). As shown in Fig. S3B–D, administration of Adeno-Cre diminished HDM-induced increases in DUOX1 and attenuated type 2 inflammation as well as mucus metaplasia, but did not significantly reduce subepithelial fibrosis. These findings confirm that targeting of pulmonary DUOX1 in the context of ongoing allergic airways diseases can promote the resolution of allergic inflammation and remodeling, consistent with our earlier observations4.
Myeloid DUOX1 does not affect innate airway HDM responses but contributes to type 2 inflammation during chronic HDM challenge.
Intranasal administration of Adeno-Cre in the aforementioned studies is expected to target LoxP-flanked genes primary within the respiratory epithelium, but may also target other cell types in the lung lumen such as alveolar macrophages. Indeed, resident macrophages in the lung can also respond directly to inhaled allergens, or to early cytokines produced by other lung cells (e.g. IL-33), and may also express DUOX18, 9. Consistent with previous findings indicating DUOX1 protein expression in human monocyte-derived macrophages9, we also detected DUOX1 protein in BAL cells (representing >95% macrophages) from WT mice by western blot and immunohistochemistry (Fig. 3A,B), which was highly variable due to the heterogeneous nature of AMs11, 12, but undetectable in BAL cells from DUOX1-deficient mice. To examine a potential role for macrophage DUOX1 in acute and chronic airway HDM responses, we conditionally deleted DUOX1 in myeloid lineages (Duox1Δ/ΔLysM), primarily including monocytes and macrophages13 (Supplementary Figs. S1A and Fig. S4A). In contrast to findings of epithelial DUOX1 deletion, macrophage-specific deletion of DUOX1 did not affect acute HDM-induced secretion of IL-33, AREG, IL-1B or IL-17A, or more delayed production of type 2 cytokines (IL-5, IL-13) into the BAL fluid (Fig. 3C). Also, ablation of macrophage DUOX1 did not affect HDM-induced mRNA expression of these cytokines (results not shown). To assess whether macrophages could contribute to production of IL-33 or type 2 cytokines by HDM, we stimulated cultured BMDMs with HDM in vitro. Indeed, HDM exposure enhanced il33 and Areg mRNA in BMDMs, and enhanced AREG secretion, but this was not significantly attenuated in BMDMs from Duox1Δ/ΔLysM mice (Fig 3D). HDM exposure induced classical macrophage activation of BMDMs, indicated by increased Nos2 mRNA expression and accumulation of NO2− in the media, but these were also not significantly altered in BMDMs Duox1Δ/ΔLysM mice (Fig. 3E). In contrast, comparative studies revealed significantly impaired classical macrophage activation of BMDMs from non-conditional Duox1−/− mice (Fig 3F). This suggests that some functional alterations in macrophages obtained from non-conditional Duox1−/− mice (e.g.8, 9) are not necessary related to intrinsic DUOX1 function within macrophages, but may also reflect altered macrophage development or differentiation due to Duox1 deficiency in stromal cells in the bone marrow or in peripheral tissues.
Figure 3:
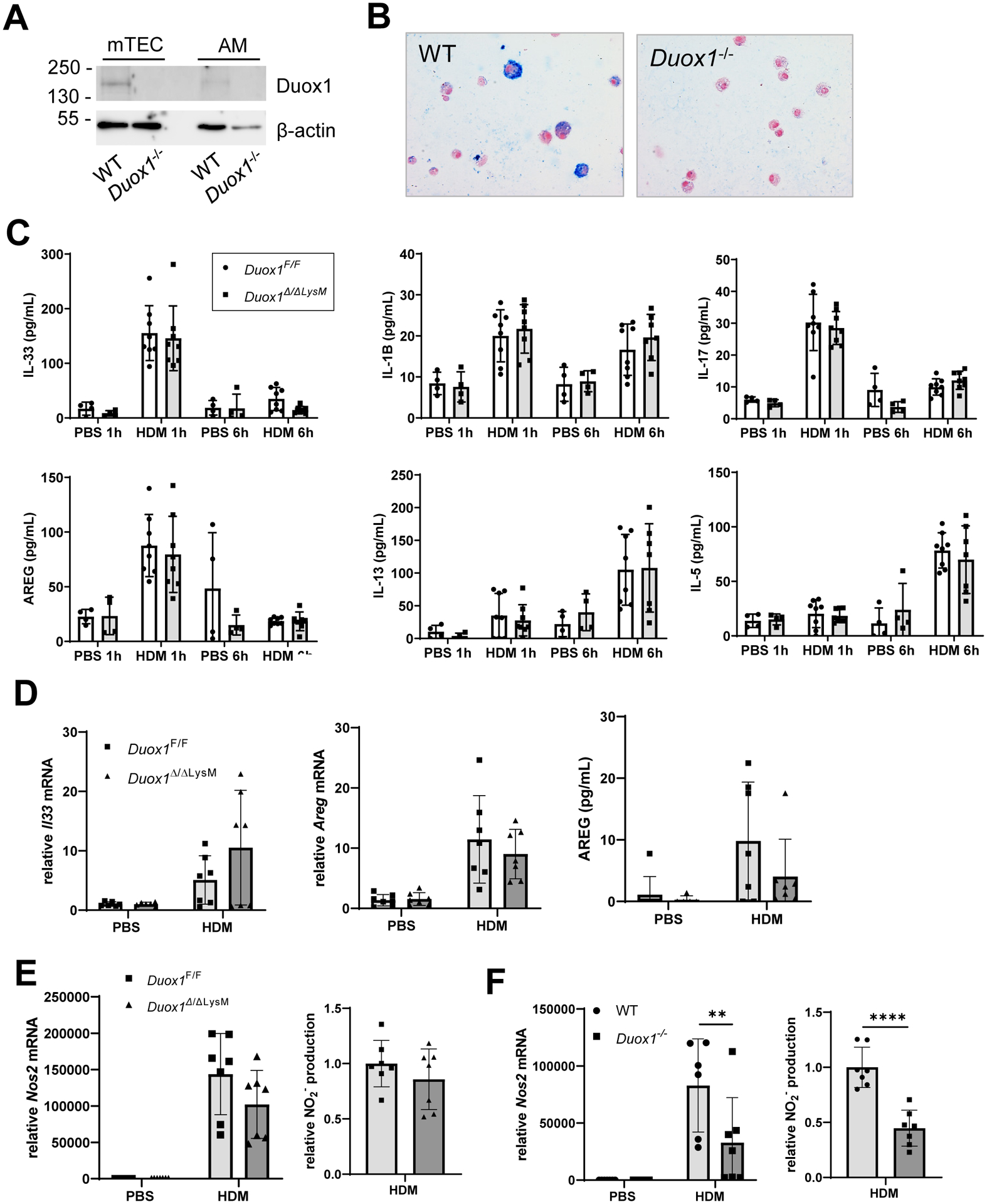
Macrophage DUOX1 does not affect innate type 2 responses to acute HDM challenge. Analysis of Duox1 protein in alveolar macrophages (AMs) from wild-type (WT) mice or Duox1−/− mice by immunohistochemistry (A) or by Western blot (B). Similar western blot analysis of mouse tracheal epithelial cell (mTEC) lysate is shown for comparison. (C) ELISA analysis of BAL cytokines/growth factors from Duox1Δ/ΔLysM or Duox1F/F mice after single intranasal HDM challenge (n = 4–8 mice per group, from 2 independent experiments). BMDMs from Duox1Δ/ΔLysM or Duox1F/F mice were stimulated for 24-hr with 50μg of HDM for RT-PCR analysis of Il33 or Areg (D) or for analysis of Nos2 mRNA or NO2− accumulation in the media (E). (F) Analysis of Nos2 mRNA or NO2− accumulation in the media from BMDMs from Duox1−/− or WT mice following 24-hr stimulation with HDM. NO2− production was normalized to levels in HDM-stimulated control cells (Duox1F/F or WT). Statistical significance was evaluated using 2-way ANOVA and Šidák’s post-hoc test. NO2− production was evaluated using unpaired t-test. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
Allergic airway inflammation is associated with increased lung macrophage recruitment, and macrophages are known to contribute to airway remodeling6. To examine the potential contribution of macrophage-associated DUOX1 to HDM-induced allergic inflammation, we subjected Duox1Δ/ΔLysM mice and corresponding controls to repeated HDM challenge. DUOX1 deletion in monocyte/macrophages tended to reduce HDM-induced recruitment of inflammatory cell types into the BAL, although this was not statistically significant (Fig. 4A and Fig. S5A). However, it markedly suppressed HDM-induced production of type 2 cytokines IL-5, IL-13, IL-33, and AREG (Fig. 4B), and significantly suppressed HDM-mediated induction of Duox1 and Il13 in the lung (Fig. S5B). Moreover, macrophage-specific deletion of DUOX1 also attenuated features of mucus metaplasia (Fig. 4C), but did not significantly affect subepithelial fibrosis (Fig. S5C). These collective findings indicate that macrophage-intrinsic DUOX1, either within resident or recruited macrophages, contributes to type 2 inflammation and mucus metaplasia in the context of allergic airway inflammation in sensitized mice induced by repeated HDM challenge.
Figure 4:
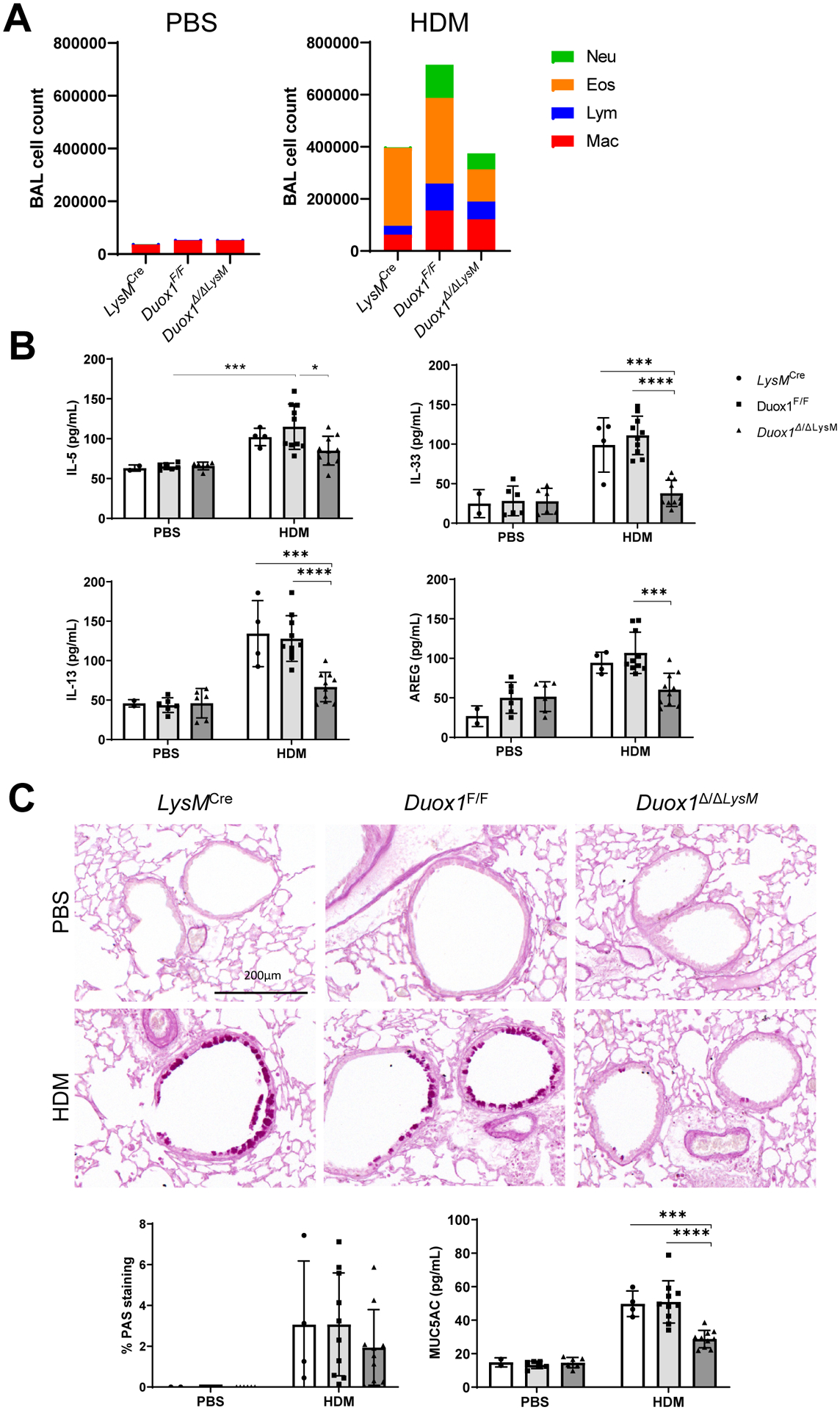
Macrophage DUOX1 contributes to type 2 inflammation and airway remodeling during chronic HDM challenge. Duox1Δ/ΔLysM mice and control Duox1F/F or LysMCre mice were subjected to chronic HDM to induce allergic airway disease. (A) Analysis of BAL cell differentials. (B) ELISA analysis of BAL cytokines/growth factors. (C) Analysis or mucus metaplasia by PAS staining (representative PAS staining images (10x magnification) and quantified PAS staining is shown) and by MUC5AC analysis in BAL. Statistical significance was evaluated using 2-way ANOVA and Tukey’s post-hoc test. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001 (n = 2–6 mice per PBS group and 4–10 mice per HDM group).
Intrinsic effects of DUOX1 on macrophage activation.
The role of macrophages in allergic airways disease is complex and not fully understood, and appears to differ between resident alveolar macrophages and recruited lung macrophages originating from circulating monocytes. Previous studies have indicated that recruited monocyte-derived macrophages contribute significantly to allergic inflammation and remodeling, largely due to their polarization to M2 pro-fibrotic phenotypes as a result of increased production of type 2 cytokines14. However, the M1/M2 paradigm does not fully clarify the overall involvement of macrophages in allergic airways disease, and allergen-induced injury can also induce innate macrophage M1 activation6. Moreover, classically activated macrophages can also be a significant source of AREG, as a mediator of airway remodeling and/or mucus metaplasia15, 16, 17. To address the intrinsic functional role(s) of DUOX1 in macrophage polarization to either classically activated (M1) or alternatively activated (M2) phenotypes, we first used the immortalized alveolar macrophage MH-S cell line in which DUOX1 was deleted by CRISPR gene editing (Fig. S6). First, we examined the potential impact of in vitro M1/M2 polarization on macrophage DUOX1 expression. Somewhat unexpectedly, and in apparent contrast to observations in epithelial cells (e.g.18), we did not observe induction of Duox1 in response to M2 polarization by IL-4/IL-13, but instead observed marked induction of Duox1 mRNA in response to M1 polarization of MH-S cells for 48 hrs (Fig. S7A,B). This was preceded by more rapid induction of Nos2, and pharmacological inhibition of NOS2 was found to prevent Duox1 induction under these polarizing conditions (Fig. S7B). Consistent with earlier reports15, M1 polarization also induced Areg mRNA (Fig. S7A). Similar induction of Nos2 or Areg were also observed in primary BMDMs or AMs upon M1 polarization, but in this case Duox1 expression was not affected, and Duox1 mRNA was also unaltered after M2 polarization of these primary macrophages (Fig. S7C,D). Although Duox1 editing in MH-S cells did not affect M1 polarization, based on induction of Nos2 mRNA and protein, it significantly attenuated induction of Areg (Fig. 5A). In addition, Duox1 editing also significantly attenuated expression of M2 markers Arg1 and Ym1 under M2-polarizing conditions (Fig. 5A). Qualitatively similar results were observed using primary BMDMs or AMs obtained from WT or Duox1Δ/ΔLysM mice, which showed no involvement of DUOX1 to induction of the M1 markers Nos2 or Il1b, but showed a marked effect of DUOX1 ablation on Areg induction in M1-stimulated BMDMs (Fig. 5B). Finally, DUOX1 also affected induction of Ym1 in BMDMs and Arg1 in AMs under M2 polarizing conditions (Fig. 5B,C). Overall, these findings indicate that DUOX1 contributes to features of both M1 and M2 activation, depending on the in vitro macrophage cell model used.
Figure 5:
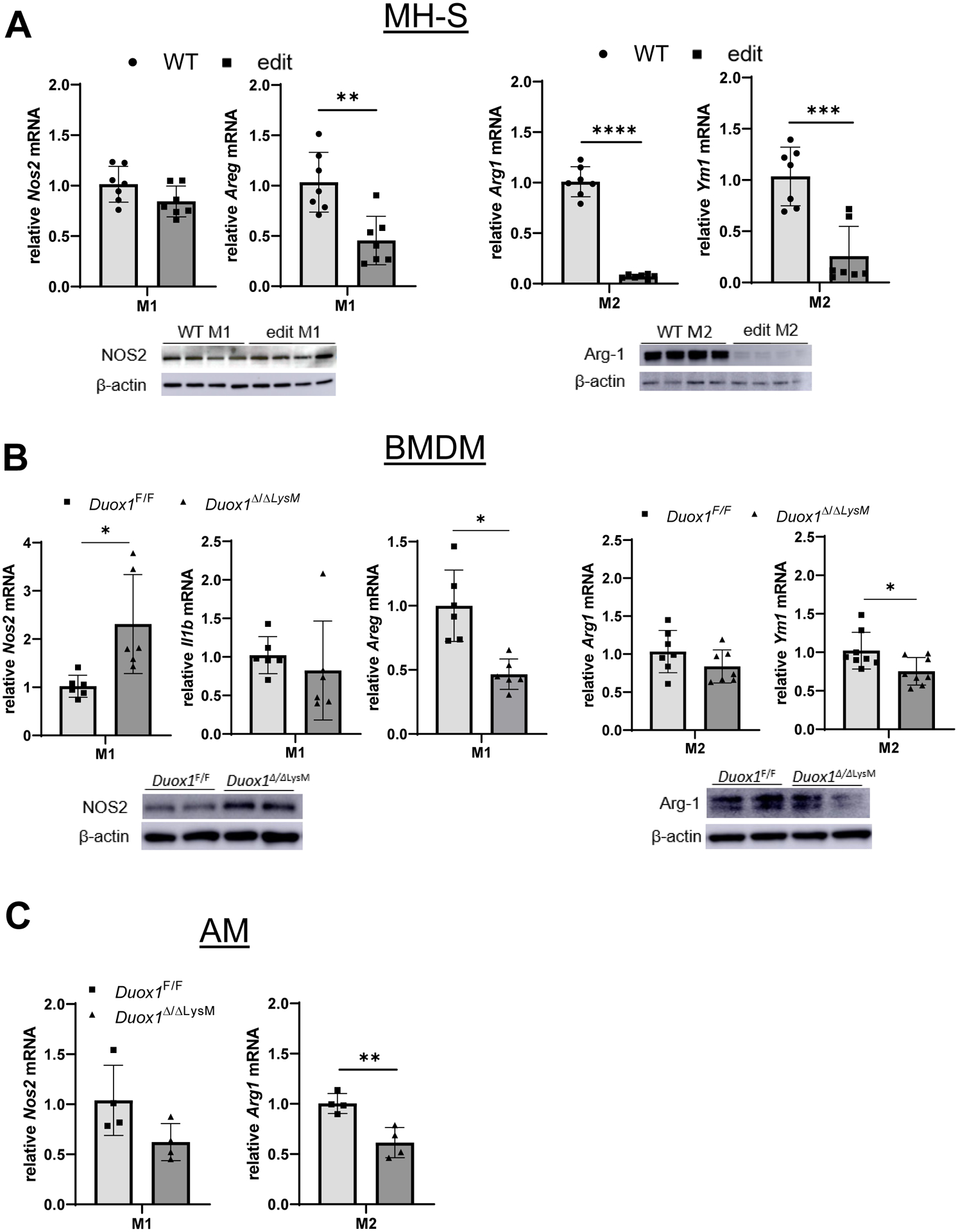
Macrophage-intrinsic DUOX1 contributes to M2 polarization. MH-S cells were CRISPR edited to delete DUOX1 and control and edited cells were subjected to M1 or M2 polarization for analysis of the M1 marker Nos2 and M2 markers Arg1 and Ym1 by RT-PCR or by Western blot (A). Analysis of M1 or M2 polarization by mRNA analysis of Nos2 or Arg1 of bone marrow-derived macrophages (BMDM’s; B) or alveolar macrophages (AM; C) obtained from Duox1Δ/ΔLysM or control Duox1F/F mice. Data represent 4–8 replicates from at least 2 independent experiments. Statistical significance was evaluated using t-test. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001.
To investigate a potential role for DUOX1 in lung macrophage polarization in vivo during HDM-induced inflammation, we generated lung single-cell suspensions after chronic HDM-induced inflammation for flow cytometry analysis of M1 and M2 macrophage populations19 (Fig. 6A). As expected14, HDM exposure resulted in an increase in the percentage of M2 polarized macrophages in lung tissues, whereas the proportion of M1-polarized macrophages was not affected. The relative proportion of M1/M2 polarized populations was not affected by epithelial-specific DUOX1 deletion, but the proportion of M2-polarized macrophages tended to be lower upon macrophage-specific DUOX1 deletion (Fig. 6B), consistent with results from in vitro macrophage polarization studies (Fig. 5).
Figure 6:
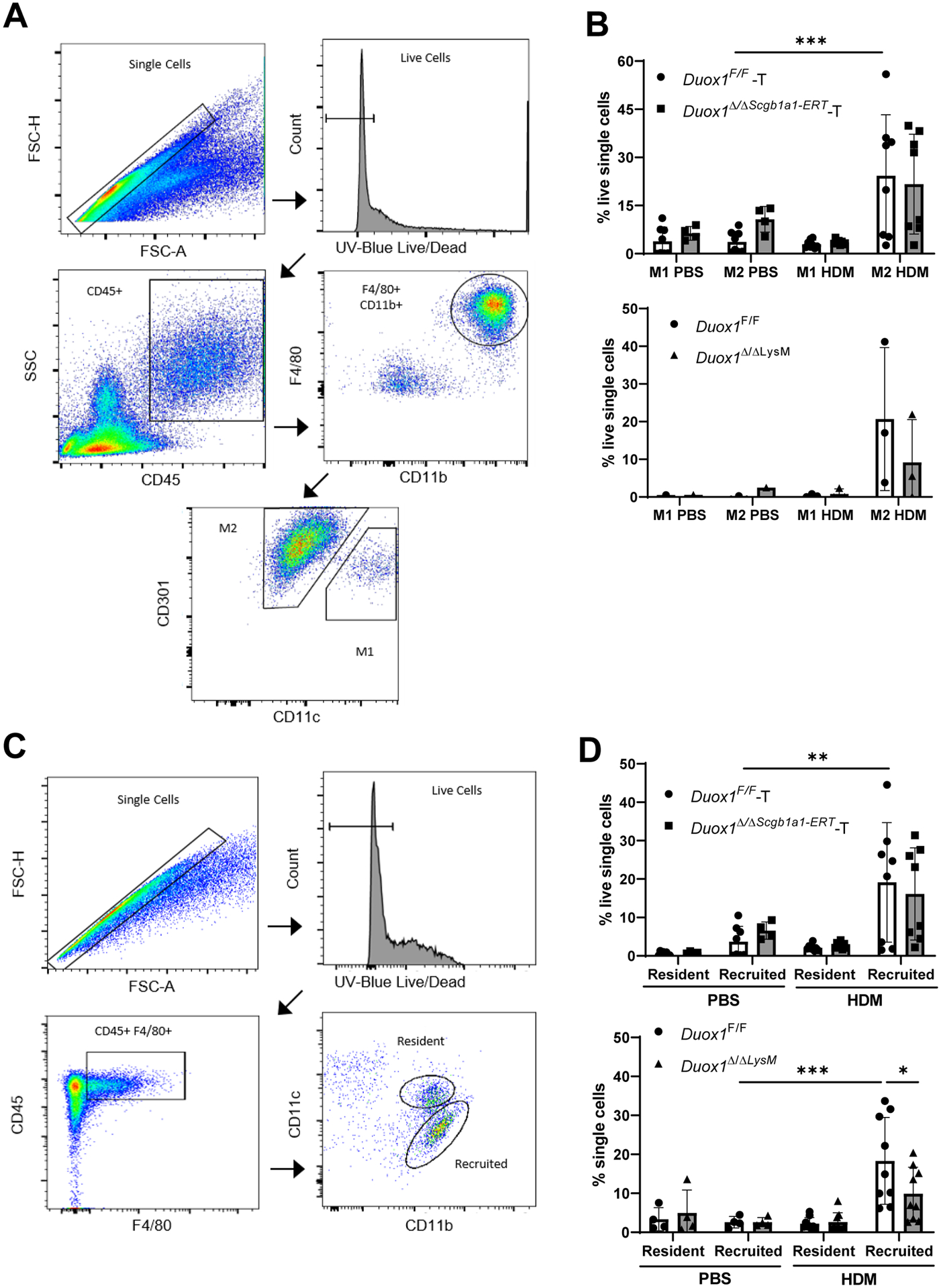
Macrophage-intrinsic DUOX1 contributes to macrophage recruitment during chronic HDM challenge. Mice were subjected to chronic HDM and lung single cell suspensions were evaluated for macrophage polarization. (A) Gating strategy to assess M1/M2 macrophage populations. (B) Quantification of M1 and M2 macrophage populations in lung tissues from tamoxifen-treated Duox1F/F and Duox1Δ/ΔScgb1a1-ERT mice (above) or Duox1F/F and Duox1Δ/ΔLysM mice (below) in response to chronic HDM challenge. Data are presented as a percentage of viable single cells. (C) Gating strategy for flow analysis of resident and recruited macrophage populations. (D) Quantification of resident and recruited macrophage populations in lung tissues from tamoxifen-pretreated Duox1F/F or Duox1Δ/ΔScgb1a1-ERT mice (above), or from Duox1F/F or Duox1Δ/ΔLysM mice (below), after chronic HDM challenge. Data are presented as a percentage of viable, single cells. Statistical significance was evaluated using 2-way ANOVA and Šidák’s post-hoc test. *: p<0.05**: p<0.01; ***: p<0.001 (n = 4–8 mice per group from 2 independent experiments, except bottom panel B which is one representative experiment in triplicate).
Since the contribution of macrophages to allergic asthma differs between resident macrophages or recruited (monocyte-derived) macrophages (e.g.6), we also wished to assess the potential impact of either epithelial or macrophage DUOX1 on macrophage recruitment during chronic HDM challenge. Using a flow cytometry gating strategy to distinguish resident and recruited macrophage populations20 (Fig. 6C), we confirmed that repeated HDM challenge results in increased numbers of recruited (monocyte-derived) macrophages in the lung (Fig. 6D). Intriguingly, while this was not affected by deleting epithelial DUOX1, macrophage recruitment was significantly reduced after myeloid-specific DUOX1 ablation, thus suggesting a role of macrophage-intrinsic DUOX1 in macrophage motility or responsiveness to chemotactic stimuli. Finally, we sorted resident and recruited macrophages from lung tissues of HDM-treated Duox1F/F mice and Duox1Δ/ΔLysM mice, to evaluate potential impact of macrophage-intrinsic DUOX1 on features of M1/M2 activation. As shown in Fig. 7, repeated HDM challenge dramatically induced expression of the M2 marker Arg1, primarily in resident macrophage populations and to a lesser extent in recruited macrophages, which tended to be attenuated in DUOX1-deficient macrophages, although this was not statistically significant. Moreover, consistent with in vitro studies, HDM induced relative increases in Areg mRNA, particularly in recruited macrophage populations, and this was significantly attenuated in DUOX1-deficient macrophages (Fig. 7B). We also observed greater relative Duox1 mRNA expression in recruited macrophages compared to resident subsets, although macrophage DUOX1 expression was not significantly altered in response to HDM (not shown). Collectively, our findings indicate a role for macrophage-intrinsic DUOX1 in recruitment of monocyte-derived macrophages during chronic allergic inflammation, as well as features of macrophage activation, particularly with respect to Areg production.
Figure 7:
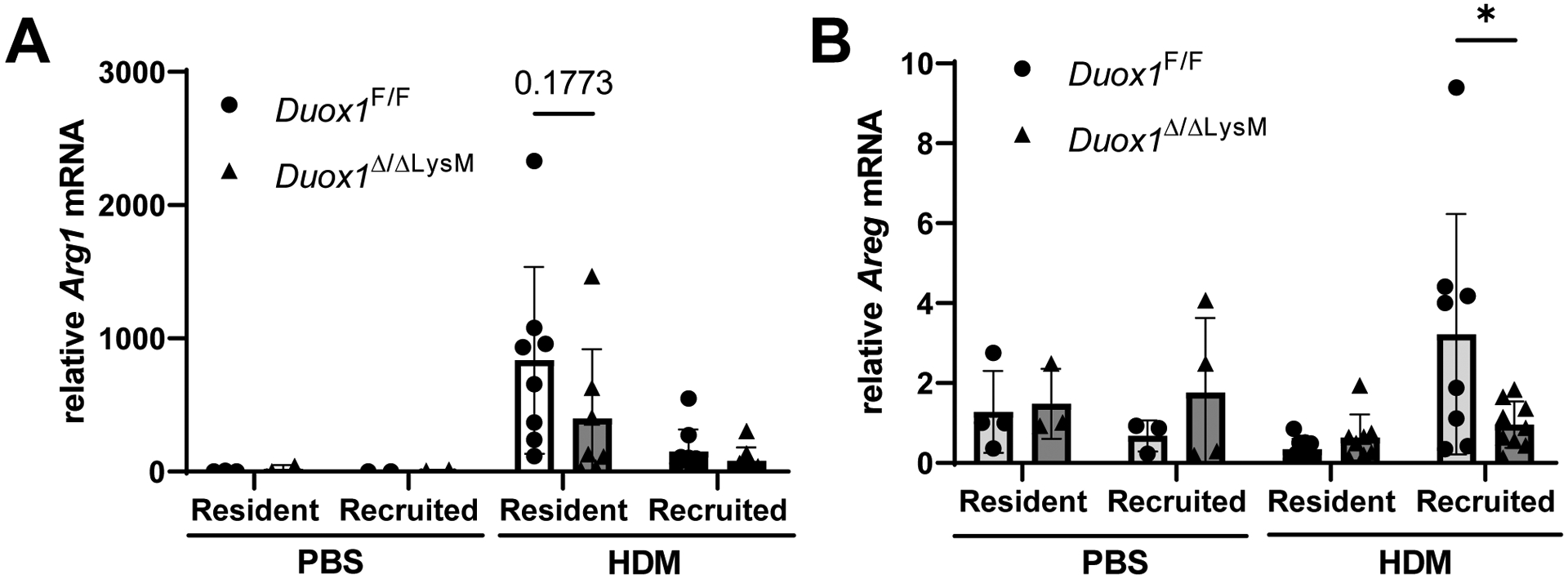
Macrophage-intrinsic DUOX1 affects to macrophage activation during chronic HDM challenge. RT-PCR analysis of the M2 marker Arg1 (A) and the putative M1 marker Areg (B) in sorted resident and recruited macrophage populations from lung tissues of HDM-stimulated Duox1F/F or Duox1Δ/ΔLysM mice. Statistical significance was evaluated using 2-way ANOVA and Šidák’s post-hoc test *: p<0.05 (n=2–4 for PBS groups and 8–9 for HDM groups, from 2 independent experiments).
DISCUSSION
Previous findings from our group identifying a role for the NOX homolog DUOX1 in airway responses to both acute and chronic challenge with HDM allergen3, 4. The present studies cell-specific contributions of DUOX1 in these outcomes, and identify a role for DUOX1 in epithelial cells, but also in monocyte-derived macrophages. Since DUOX1 is primarily expressed within epithelial cells in the lung21, and secretion of epithelial IL-33 is central early event in development of type 2 inflammation (e.g.10), it is not surprising that selective ablation of DUOX1 from the epithelium was found to attenuate these responses. Moreover, consistent with recent observations that DUOX1 may also participate in epithelial IL-33 signaling10, epithelial DUOX1 was found to contribute to induction of type 2 cytokines in response to direct instillation of IL-33. Epithelial deletion of DUOX1 also significantly suppressed mucus metaplasia, consistent with similar previous findings with non-conditional Duox1−/− mice4.
A more surprising outcome of our studies was that they also revealed a significant contribution of macrophage-intrinsic DUOX1 in the activation of type 2 inflammation and development of mucus metaplasia in the context of chronic HDM-induced airway inflammation. Previous studies have highlighted the presence of DUOX1 within macrophages and suggested its functional role in macrophage activation8, 9, but our studies are the first to firmly demonstrate a function of macrophage-intrinsic DUOX1 in the context of allergic airway inflammation. Macrophages have long been recognized to play various roles in asthma pathobiology, and while resident alveolar macrophages may help maintain lung homeostasis and suppress inflammation, recruitment of monocyte-derived macrophages and their polarization to an “alternatively activated” M2 phenotype during type 2 inflammation is thought to contribute to various aspects of allergic airway inflammation and remodeling6, 22. Our observations of macrophage-intrinsic DUOX1 contributing to production of type 2 cytokines such as IL-33 and IL-13 during chronic HDM-induced inflammation highlights a specific role of macrophage DUOX1 promoting type 2 inflammation in addition to the previously demonstrated involvement of epithelial DUOX14, 10, 23.
Previous studies have indicated that epithelial production of IL-33 can enhance type 2 inflammation and airway remodeling in part by enhancing alternative macrophage (M2) activation24, 25. Moreover, postnatal alveolar production of IL-33 can shape the local immune environment in the developing lung by sustaining local ILC2 homeostasis and alveolar macrophage development towards an M2 polarized phenotype26, 27. Based on this, we speculated that epithelial DUOX1, by mediating such IL-33 production3, 10, could contribute to promoting macrophage M2 polarization. However, we did not observe significant alterations in lung M1/M2 macrophage subpopulations in Duox1Δ/ΔScgb1a1-ERT mice, either at baseline or after chronic HDM challenge, but our findings instead suggested a contribution of macrophage-intrinsic DUOX1 to macrophage M2 polarization in response to chronic HDM challenge.
In our efforts to examine a role for DUOX1 in macrophage activation or polarization, we observed intriguing differences between macrophages obtained from non-conditional Duox1−/− mice and those from Duox1Δ/ΔLysM mice (in which case DUOX1 is still functional in stromal cells). Indeed, HDM-induced classical macrophage activation in vitro, as assessed by induction of Nos2, was markedly attenuated in BMDMs obtained from non-conditional Duox1−/− mice but was not significantly altered in BMDMs from Duox1Δ/ΔLysM mice. Similarly, although we observed impaired induction of M1 polarization markers (e.g. Nos2) by LPS/IFN-γ in AMs or BMDMs obtained from non-conditional Duox1−/− mice (results not shown), this was not observed in AMs or BMDMs from Duox1Δ/ΔLysM mice, nor in MH-S cells in which DUOX1 was deleted by CRISPR editing (Fig. 5). These various observations would suggest a significant impact of DUOX1 from stromal cells (e.g. epithelium) on macrophage development or differentiation, thereby affecting their ability to respond to proinflammatory triggers. These observations also justify our use of macrophages obtained from Duox1Δ/ΔLysM mice in attempts to address macrophage-intrinsic properties of DUOX1. Indeed, these studies did not indicate a significant contribution of DUOX1 to classical macrophage activation by LPS or HDM, but indicated a contribution of macrophage DUOX1 in M2 polarization in response to IL-13, illustrated by attenuated induction of Arg1 or Ym1. Also, deletion of macrophage DUOX1 appeared to attenuate macrophage M2 polarization in the context of chronic HDM-induced inflammation, although this could also be an indirect result of reduced production of IL-13 in this context (Fig. 4), thus leading to less M2 activation.
It is important to emphasize that macrophage M1 and M2 polarization states are defined primarily based on in vitro studies under strictly defined stimulation conditions, and that lung tissue macrophages in the context of allergic inflammation likely represent mixed phenotypes along a continuum between M1 or M2 states due to the presence of diverse mediators that variably impact on macrophage function6, 28. Indeed, the contribution of lung macrophages to allergic airway inflammation cannot be fully attributed to M2 activation and may also involve features of classical (M1) macrophage activation29. Also, inhibition of macrophage M2 polarization during allergic inflammation was found to attenuate eosinophilic inflammation, but actually worsened neutrophilic inflammation and airway hyperresponsiveness30. Our findings are consistent with this notion, particularly with respect to recruited (monocyte-derived) macrophage populations, which display features of both classical (M1) and alternative (M2) activation. Moreover, while macrophage DUOX1 may contribute to some aspects of macrophage M2 polarization, it also appears to participate in some features of classical M1 activation, illustrated by macrophage production of Areg15, 16, an important mediator of type 2 immune responses that promotes airway remodeling such as mucus metaplasia17. The fact that DUOX1 ablation impaired Areg induction under M1-polarizing conditions without affecting other classical M1 markers such as Nos2 suggest that DUOX1 involved in specific features of macrophage activation rather than in overall regulation of M1/M2 polarization.
Another important consideration with respect to the role(s) of macrophages in chronic lung disease is the diverse involvement of resident macrophage populations, which typically serve a homeostatic function and are largely sustained by local proliferation of resident macrophage populations, and recruited monocyte-derived macrophages that are phenotypically and functionally distinct31. Studies in rodents indicate that resident AMs persist over the entire lifespan via self-renewal with minimal contribution from circulating monocytes, although this may not be true in humans, in which case lung macrophages appear to be derived primarily from circulating monocytes32. Indeed, newly derived macrophages, originating from either proliferation or from recruited monocytes, are pathogenic in the context of allergic airway disease, whereas mature resident macrophages are not6, 14. Also, fate mapping and single-cell transcriptomics studies indicate that recruited monocyte-derived macrophage subsets display increased plasticity, which appears to be shut down during prolonged tissue residency33. Tissue recruitment of monocyte-derived macrophages during allergic inflammation may be driven by epithelial-derived chemokines such as MCP1/CCL214. Intriguingly, we observed that DUOX1 deletion from monocyte/macrophages, rather than epithelial cells, attenuated macrophage recruitment to the lung during chronic HDM-induced inflammation. This likely reflects a macrophage-intrinsic role for DUOX1 in macrophage chemotaxis or migratory properties, analogous to similarly observed functions in epithelial cells34, 35. In this regard, our findings expand on previous studies indicating the existence of mutual cross-talk between macrophages and T cells or type 2 innate lymphoid cells in driving disease pathology of allergic asthma36, 37, and suggest that macrophage DUOX1 may actively participate in such crosstalk.
An important limitation of our studies is the fact that they are based on deletion of loxP-flanked DUOX1 by targeted insertion of Cre cDNA into the endogenous lysozyme M (LysM) gene locus (LysM-Cre), which is expressed primarily in mature macrophages and granulocytes and not in lymphoid cells13, 38. However, some studies suggest that LysM-Cre expression is less prominent in immature macrophages derived from e.g. recruited monocyte precursors and thus would result in less effective deletion of loxP-flanked target genes in these cell populations (e.g.6). Nevertheless, our findings indicating effects of such LysM-Cre mediated DUOX1 deletion in recruited macrophages would argue against this. Also, since granulocytes (neutrophils) can also represent a source of type 2 cytokines (e.g.39), LysM-Cre mediated DUOX1 deletion could conceivably also affect neutrophils. However, we did not detect significant expression of DUOX1 in bone marrow derived neutrophils (not shown). Lastly, since LysM is also expressed in other cell lineages, including type 2 alveolar epithelial cells and also in neuronal cells in the brain40, a possible contribution of DUOX1 deletion in these cells cannot be completely ruled out. The potential role of DUOX1 in neuronal cells is intriguing, particularly if this also applies to peripheral sensory neurons which have been implicated in IL-33-mediated allergic responses in the lung41, and could contribute to type 2 inflammation and remodeling. Future studies using alternative Cre driver mice that target macrophages, e.g. CSF1R, CD11b, F4/80, or CXC3CR1, could more fully resolve this, although none of these are fully specific for macrophages13, 38.
In conclusion, the studies described herein provide additional insight into the cell-specific roles of DUOX1 in innate allergen responses and in chronic allergic airway inflammation, and reveal a previously unanticipated functional role for DUOX1 in macrophages, with respect to macrophage recruitment and production of mediators that contribute to type 2 inflammation and related aspects of airway remodeling. We did not explicitly address the downstream mechanism by which DUOX1 contributes to these various outcomes, but based on previous studies linking DUOX1-dependent cysteine oxidation of the non-receptor tyrosine kinase Src and activation of EGFR with epithelial cell migration or IL-33 secretion3, 10, we speculate that DUOX1 could similarly regulate Src activation in macrophages, and thereby affect its diverse regulatory roles in various aspects of macrophage biology, including cytokine production and migration42, 43. Finally, our findings further highlight the complex and intricate roles of diverse NOX enzymes in macrophage biology, adding to previously reported functions of alternative NOX enzymes (NOX2, NOX4) in macrophage differentiation and polarization44, 45. In light of recent advances with respect to our understanding of macrophage biology in the lung, it will be of interest to further define the specific functions of DUOX1 and/or other NOX homologs in these various aspects. Expanding on our previous suggestion of DUOX1 as an attractive therapeutic target in allergic diseases such as asthma23, our present studies would suggest that specific targeting of DUOX1 within monocyte-derived macrophages might be particularly useful, and would avoid compromising normal host defense functions of DUOX1 within the epithelium.
MATERIALS AND METHODS
Mice
Duox1−/− mice were originally generated by Lexicon Pharmaceuticals46, and were backcrossed to C57BL6/NJ mice (The Jackson Laboratory; stock #005304) for at least 5 generations. Duox1F/F mice (Duox1tm2a(EUCOMM)Hmgu) were generated on the C57BL6/NJ background at the University of Cologne using JM8A3.N1 ES cells with the targeting vector HTGR04001_Z_7_E03 from EUCOMM (http://www.mousephenotype.org/data/alleles/MGI:2139422/tm2a(EUCOMM)Hmgu). Correct targeting was confirmed by Southern blot. Duox1F/F mice were crossed with Scgb1a1-CreERT (B6N.129S6(Cg)-Scgb1a1tm1(cre/ERT)Blh/J; The Jackson Laboratory; stock #016225) or LysM-Cre (B6.129P2-Lyz2tm1(cre)Ifo/J; The Jackson Laboratory; stock #004781), to generate Duox1F/FScgb1a1-CreERT+/− mice (Duox1Δ/ΔScgb1a1-ERT) and Duox1F/FLysM-Cre+/− mice (Duox1Δ/ΔLysM) for selective ablation of DUOX1 in either airway epithelial cells or myeloid cells, respectively47, 48 (Supplementary Fig. S1A). Induction of epithelial DUOX1 deletion in Duox1Δ/ΔScgb1a1-ERT mice was achieved by intraperitoneal injection of 75 mg/kg tamoxifen (Sigma-Aldrich; #10540-29-1) for 5 consecutive days, at least one week prior to experimentation. Epithelial- or macrophage-selective DUOX1 deletion was verified by generating single-cell lung suspensions using enzymatic digestion (Miltenyi Biotec Lung dissociation Kit 130-095-927) and a GentleMACS™ Dissociator (Miltenyi Biotec Inc.), collection of hematopoietic cell types with CD45 Microbeads (Miltenyi Biotec, mouse CD45 Microbeads, #130-052-301), using LS Columns (Miltenyi Biotec, #130-042-401) and a QuadroMACS Separator (Miltenyi Biotec), and enrichment of CD326(EpCAM)-positive (epithelial) cells from CD45− fractions using Miltenyi Biotec CD326 EpCAM Microbeads (#130-105-958). Duox1 mRNA expression was assessed in CD45−/CD326+, CD45−/CD326− and CD45+ cell fractions by RNA extraction using QIAGEN RNeasy Micro kit and RT-PCR analysis. All animal studies were performed using mice at 9–12 weeks of age, including both male and female mice divided equally across treatment groups. All animal procedures were reviewed and approved by the University of Vermont Institutional Animal Care and Use Committee (protocol #: PROTO202000078).
Acute and chronic airway challenge to house dust mite (HDM) or IL-33
Mice were subjected to brief isofluorane anesthesia for intranasal administration of 50 μg of HDM extract (based on total weight) in 50 μl PBS (D. pteronyssinus; Greer Laboratories; lot # 269026 and 305470; 32–64 endotoxin units/mg; 6–9 μg DER P 1/mg), recombinant mouse IL-33 (BioLegend, 1.0 μg/mouse in 50 μl PBS), or 50 μl PBS vehicle control. In cases of acute challenge, BAL fluids and lung tissues were harvested 1 or 6 hours after a single HDM/IL-33 instillation.
In order to induce HDM-mediated allergic airway inflammation, mice were first sensitized by airway HDM instillation on days 1 and 8, followed by daily airway challenge with 50 μg HDM on days 15–19 to induce allergic airway inflammation. Bronchoalveolar lavage (BAL) fluids and lung tissues were collected three days after the final HDM challenge (day 22) for the various analyses described below. In some studies, Duox1F/F mice were subjected to the same HDM sensitization and challenge protocol, and were then administered an adenoviral vector containing Cre recombinase (Ad5CMVcre; Viral Vector Core of the University of Iowa; 108 pfu in 50 μl PBS) or control adenovirus (AdCMVempty) on day 20, to ablate Duox1 prior to analysis.
Analysis of lung inflammation
BAL fluids were harvested by 3 successive instillations of 500 μL of PBS into the lungs, and pooled BAL fluids were briefly centrifuged at 150 × g and supernatants was collected for analysis of cytokines and other secreted mediators. BAL levels of IL-33, IL-5, IL-13, IL-17A, IL-1β and amphiregulin (AREG) were assessed using DuoSet ELISA Kits (R&D Systems; DY3626, DY405, DY413, DY1399, DY401, DY989) according to manufacturer protocol and were read on a BioTek Synergy HT plate reader. MUC5AC in BAL was analyzed using a Mouse Mucin-5 Subtype AC ELISA kit (MyBioSource; MBS265057), according to the manufacturer’s instructions. Pelleted BAL cells were resuspended in PBS and counted using a hemocytometer prior to loading into an EZ Cytofunnel (Thermo Scientific) and centrifuged at 600 RPM for 10 minutes. Cytospin slides were fixed and stained using the Hema 3 kit (Thermo Scientific) and cell differentials were counted based on at least 200 cells per slide.
Lung tissue analyses
Tissue RNA was isolated from mouse inferior lobes using GeneJet RNA purification kits (Invitrogen; K0732) according to manufacturer protocol. cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad; 1708890) and/or Applied Biosystems High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; 4387406) according to manufacturer protocol. Quantitative RT-PCR was performed using iQ Sybr Green supermix (Bio-Rad; 1708882) 0.5 μL cDNA and 0.5 μM primer mix as previously described49. Primer sets used included: Il33 (F: GAT GGG AAG AAG GTG ATG GGT G, R: TTG TGA AGG ACG AAG AAG GC), Il13 (F: CCA CGG CCC CTT CTA ATG A, R: GCC TCT CCC CAG CAA AGT CT), Il5 (F: ATG GAG ATT CCC ATG AGC AC, R: CCC ACG GAC AGT TTG ATT CT), Areg (F: AAC GGT GTG GAG AAA AAT CC, R: TTG TCC TCA GCT AGG CAA TG), Duox1 (F: GAC CCC AGT ATC TCC CCA GA, R: ATG ACT GGG AAT CCC CTG GA), Muc5ac (F: AGT CTC TCT CCG CTC CTC TCA AT, R: CAG CCG AGA GGA GGG TTT GAT CT). Clca1 (F: CCC TCA TCC AAC TGA ACA AC, R: TCA GCG TTT TTG AAG GTT TC), Col1a1 (F: CAC CCT CAA GAG CCT GAG TC, R: AGA CGG CTG AGT AGG GAA CA), Col3a1 (F: CCG AAC TCA AGA GTG GAG AA, R: GGC CAT AGC TGA ACT GAA AA), and Gapdh (F: CTG GAG AAA CCT GCC AAG TA, R: TGT TGC TGT AGC CGT ATT CA).
Immunohistochemistry analyses were performed as previously described4. Briefly, left lung lobes were fixed in PFA and paraffin embedded, and 5-μm sections were stained with H&E, periodic acid-Schiff (PAS), or Masson’s trichrome using standardized protocols. PAS-positive staining areas were quantified using Meta-Morph imaging software (Molecular Devices) and Masson’s trichrome staining intensity around the small airways was scored by 2 blinded, independent investigators (0, no reactivity; 1, minimal staining; 2, moderate staining; 3, prominent staining).
Generation of lung single-cell suspensions for flow cytometry analysis and cell sorting
Mouse lungs were dissociated into single-cell suspensions using enzymatic digestion with the Lung Dissociation Kit (Miltenyi Biotec 130-095-927) and a GentleMACS Dissociator (Miltenyi Biotec) following the manufacturer’s protocol. One million cells per sample were incubated in Live/Dead Fix Blue (ThermoFisher L23105) for 20 minutes at 4°C and then washed and incubated with Fc Block (anti-mouse CD16/32) (BD Pharminogen 553142) for 10 minutes at room temperature and appropriate antibodies were added for 30 minutes at 4°C (Pacific Orange anti-CD45 (Invitrogen MCD4530), PE anti-F4/80 (BioLegend 123109), APC-eFluor780 anti-CD11b (Invitrogen 47-0112-82), PE-Cy7 anti CD11c (BioLegend 117317), and AlexaFluor647 anti-CD301 (BioLegend 145603)). Cells were then washed twice, fixed with 1% paraformaldehyde, and analyzed with the BD LSRII flow cytometer and FlowJo v10.7 software. For characterization of M1- and M2-polarized macrophage populations, CD45+F4/80highCD11bhigh populations were distinguished based on surface expression of CD11c (M1 marker) or CD301 (M2 marker)19. Resident and recruited macrophage populations were defined as F4/80+CD11b−CD11c+ and F4/80+CD11b+CD11c− populations, respectively, as described previously20.
Isolation and culture of alveolar and bone marrow-derived macrophages
Alveolar macrophages (AMs) were isolated by lavaging mouse lungs 5 times with 1 mL of PBS + 2 mM EDTA. Lavaged cells were pelleted, and red blood cells were lysed by briefly re-suspending cells in water. Alveolar macrophages were cultured in complete RPMI (RPMI 1640 containing 10% FBS, P/S, and plasmocin) plus Nystatin in 24-well culture plates for 2 hours to allow macrophages to adhere. Bone marrow was flushed from the femurs and tibias of mice and suspended in complete RPMI containing 5 ng/ml M-CSF overnight. The following day, non-adherent cells were collected, washed in PBS, and contaminating red blood cells were lysed by briefly re-suspending pelleted cells in water. Remaining cells were then differentiated into bone marrow-derived macrophages (BMDMs) by culturing in complete RPMI containing 30 ng/ml M-CSF for 7 days50, 51. For analysis of macrophage polarization to M1 or M2 phenotypes, AMs and BMDMs cultured for 48-hrs in the presence of either 100 ng/ml LPS + 5 ng/ml IFNγ (M1) or 20 ng/ml IL-4 + 10 ng/ml IL-13 (M2). Alternatively, MH-S (American Type Culture Collection (ATCC)) cells and BMDMs were also stimulated with 50 μg/ml HDM (Greer). Markers of macrophage activation/polarization were assessed by cytokine/mediator analysis in conditioned media, or by RT-PCR analysis of extracted RNA, using the following primer sets: Arg1 (F: CGA TTC ACC TGA GCT TTG AT, R: AAG CCA AGG TTA AAG CCA CT); Ym1 (F: CAT GAG CAA GAC TTG CGT GAC, R: GGT CCA AAC TTC CAT CCT CCA); Nos2 (F: GTT CTC AGC CCA ACA ATA CAA GA, R: GTGGAC GGG TCG ATGTCA C); Il1b (F: GCC CAT CCT CTG TGA CTC AT, R: AGG CCA CAG GTA TTT TGT CG); and Gnb2l1 (F: AAG TAC ACG GTC CAG GAT GA, R: AGG GAT CCA TCT GGA GAG AC) as housekeeping gene52.
Studies with MH-S macrophages
The murine alveolar macrophage cell line MH-S (ATCC; CRL-2019) was used in polarization studies as described above. Ribonuclear Proteins (RNPs) were made using the Alt-R CRIPSR-Cas9 system from Integrated DNA Technologies following the manufacturer’s instructions. sgRNAs were designed to target Duox1 exon 3 (5’-GtAGACACCAtCtGCAtAGC) using the design tool from synthego.com and Duox1 exon 4 (5’-TCAACATTTACATCCCGCAT) using the design tools at idtdna.com. Alt-R® CRISPR-Cas9 9 tracrRNA, ATTO™ 550 (IDT; 1075927) was combined with each gRNA to form a duplex. Duplexes were combined in a 1:1 molar ratio and Alt-R® S.p. Cas9 Nuclease (IDT; 1081058) was added to an RNP. The Neon transfection system (ThermoFisher) was used to electroporate RNPs into 50,000 MH-S cells with a voltage of 1720V, a pulse width of 10 ms, and 2 pulses. Electroporated cells were dispensed into 500 μl of RPMI media +10% FBS and expanded. Clones were generated and screened for a 1kb deletion between exons 3 and 4 using forward primer 5’- CCAGAGAAGCTCCTCAGAGTGT and reverse primer 5’- GGCTCATACACTCTCATTCACAG.
Nitrite measurements
Nitrite was measured by Griess reaction. Briefly, 50 μl of conditioned media from M1 polarized or HDM-treated cells were incubated with 50 μl of 2% sulfanilamide in 5% phosphoric acid in a 96-well plate for 5 min at room temperature followed by 50 μl of 0.2% naphtylethylenediamine in 5% phosphoric acid. After 5 minutes absorbance was read at 530nm using a BioTek Synergy HT plate reader.
Western blots
Cells were lysed in RIPA buffer (Cell Signaling Technology; 9806S) containing protease and phosphatase inhibitors (Thermo Scientific; 78442). Samples were separated by 10% SDS-PAGE gels and transferred to PVDF membranes. Blots were probed with antibodies against iNos (BD Biosciences; 610333), Arginase-1 (Santa Cruz; sc-18351), β-actin (Sigma; A5441) or Duox1 (kindly provided by F. Miot, Free University of Brussels, Belgium). Primary antibodies were probed with appropriate secondary antibodies conjugated with HRP (Cell Signaling; 7076S and 7074S) and detected by enhanced chemiluminescence (Pierce; 34578). Membranes were imaged with Amersham Imager 600 (GE Healthcare).
Statistical analysis
All quantitative data are presented as means and SEMs. Statistical significance was evaluated using 2-way ANOVA and Tukey’s or Šidák’s post-hoc test, unless otherwise noted, using GraphPad Prism Software (Version 7.0, GraphPad Software, La Jolla, CA), and differences were considered statistically significant if P<0.05.
Supplementary Material
Acknowledgements:
This work was supported by NIH grants R01 HL085646 and R01 HL138708 (to AvdV), F31 HL142221 (to CMD), R35 HL135828 (YMJ-H), R01 HL142081 and R01 HL133920 (to MEP). The authors also kindly thank Michael Schramm (University of Cologne) for his help with generating and characterizing Duox1F/F mice, and Roxana del Rio Guerra at the UVM Flow Cytometry and Cell Sorting facility for her technical assistance.
Footnotes
Disclosures: A.v.d.V. is inventor on U.S. Patent No. 10143718, “Covalent Inhibitors of Dual Oxidase 1 (DUOX1),” issued December 4, 2018.
References
- 1.Lambrecht BN & Hammad H Allergens and the airway epithelium response: gateway to allergic sensitization. J Allergy Clin Immunol 134, 499–507 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Holtzman MJ, Byers DE, Alexander-Brett J & Wang X The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol 14, 686–698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hristova M et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol 137, 1545–1556 e1511 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habibovic A et al. DUOX1 mediates persistent epithelial EGFR activation, mucous cell metaplasia, and airway remodeling during allergic asthma. JCI Insight 1, e88811 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrne AJ, Mathie SA, Gregory LG & Lloyd CM Pulmonary macrophages: key players in the innate defence of the airways. Thorax 70, 1189–1196 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Draijer C & Peters-Golden M Alveolar Macrophages in Allergic Asthma: the Forgotten Cell Awakes. Curr Allergy Asthma Rep 17, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon J et al. The nonphagocytic NADPH oxidase Duox1 mediates a positive feedback loop during T cell receptor signaling. Sci Signal 3, ra59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meziani L et al. Dual oxidase 1 limits the IFNgamma-associated antitumor effect of macrophages. J Immunother Cancer 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rada B, Park JJ, Sil P, Geiszt M & Leto TL NLRP3 inflammasome activation and interleukin-1beta release in macrophages require calcium but are independent of calcium-activated NADPH oxidases. Inflamm Res 63, 821–830 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustin CM et al. Oxidation-Dependent Activation of Src Kinase Mediates Epithelial IL-33 Production and Signaling during Acute Airway Allergen Challenge. J Immunol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu-Vanpala S et al. Functional heterogeneity of alveolar macrophage population based on expression of CXCL2. Sci Immunol 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mould KJ et al. Airspace Macrophages and Monocytes Exist in Transcriptionally Distinct Subsets in Healthy Adults. Am J Respir Crit Care Med 203, 946–956 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCubbrey AL, Allison KC, Lee-Sherick AB, Jakubzick CV & Janssen WJ Promoter Specificity and Efficacy in Conditional and Inducible Transgenic Targeting of Lung Macrophages. Front Immunol 8, 1618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YG et al. Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. Am J Respir Cell Mol Biol 52, 772–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng C et al. Amphiregulin may be a new biomarker of classically activated macrophages. Biochem Biophys Res Commun 466, 393–399 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Minutti CM et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manzo ND, Foster WM & Stripp BR Amphiregulin-dependent mucous cell metaplasia in a model of nonallergic lung injury. Am J Respir Cell Mol Biol 47, 349–357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harper RW et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett 579, 4911–4917 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Cho KW, Morris DL & Lumeng CN Flow cytometry analyses of adipose tissue macrophages. Methods Enzymol 537, 297–314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q et al. MCTR1 enhances the resolution of lipopolysaccharide-induced lung injury through STAT6-mediated resident M2 alveolar macrophage polarization in mice. J Cell Mol Med 24, 9646–9657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geiszt M, Witta J, Baffi J, Lekstrom K & Leto TL Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 17, 1502–1504 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Abdelaziz MH et al. Alternatively activated macrophages; a double-edged sword in allergic asthma. J Transl Med 18, 58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Vliet A, Danyal K & Heppner DE Dual oxidase: a novel therapeutic target in allergic disease. Br J Pharmacol 175, 1401–1418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurowska-Stolarska M et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 183, 6469–6477 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Li D et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol 134, 1422–1432 e1411 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Kleer IM et al. Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 45, 1285–1298 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Saluzzo S et al. First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep 18, 1893–1905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogger PP & Byrne AJ Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol 14, 282–295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Draijer C et al. Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J Allergy Clin Immunol 140, 280–283 e283 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Draijer C, Robbe P, Boorsma CE, Hylkema MN & Melgert BN Dual role of YM1+ M2 macrophages in allergic lung inflammation. Sci Rep 8, 5105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gundra UM et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 123, e110–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrne AJ et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med 217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guilliams M & Svedberg FR Does tissue imprinting restrict macrophage plasticity? Nat Immunol 22, 118–127 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Koff JL, Shao MX, Kim S, Ueki IF & Nadel JA Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J Immunol 177, 8693–8700 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Wesley UV, Bove PF, Hristova M, McCarthy S & van der Vliet A Airway epithelial cell migration and wound repair by ATP-mediated activation of dual oxidase 1. J Biol Chem 282, 3213–3220 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Mi LL, Zhu Y & Lu HY A crosstalk between type 2 innate lymphoid cells and alternative macrophages in lung development and lung diseases (Review). Mol Med Rep 23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu X et al. The Role of T Cells and Macrophages in Asthma Pathogenesis: A New Perspective on Mutual Crosstalk. Mediators Inflamm 2020, 7835284 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi J, Hua L, Harmer D, Li P & Ren G Cre Driver Mice Targeting Macrophages. Methods Mol Biol 1784, 263–275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun B et al. Characterization and allergic role of IL-33-induced neutrophil polarization. Cell Mol Immunol 15, 782–793 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orthgiess J et al. Neurons exhibit Lyz2 promoter activity in vivo: Implications for using LysM-Cre mice in myeloid cell research. Eur J Immunol 46, 1529–1532 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Liu B et al. IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc Natl Acad Sci U S A 113, E7572–E7579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abram CL & Lowell CA The diverse functions of Src family kinases in macrophages. Front Biosci 13, 4426–4450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byeon SE et al. The role of Src kinase in macrophage-mediated inflammatory responses. Mediators Inflamm 2012, 512926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helfinger V, Palfi K, Weigert A & Schroder K The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFkappaB-Dependent Manner. Oxid Med Cell Longev 2019, 3264858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu Q et al. NADPH Oxidases Are Essential for Macrophage Differentiation. J Biol Chem 291, 20030–20041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donko A et al. Urothelial cells produce hydrogen peroxide through the activation of Duox1. Free Radic Biol Med 49, 2040–2048 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Clausen BE, Burkhardt C, Reith W, Renkawitz R & Forster I Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8, 265–277 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Rawlins EL et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4, 525–534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiffers C et al. The Transient Receptor Potential Channel Vanilloid 1 Is Critical in Innate Airway Epithelial Responses to Protease Allergens. Am J Respir Cell Mol Biol 63, 198–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weischenfeldt J & Porse B Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008, pdb prot5080 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Ying W, Cheruku PS, Bazer FW, Safe SH & Zhou B Investigation of macrophage polarization using bone marrow derived macrophages. J Vis Exp (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ishii T et al. Stability of housekeeping genes in alveolar macrophages from COPD patients. Eur Respir J 27, 300–306 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.